
Contents
Hemophilia A
Hemophilia A also called Haemophilia A or Classic Hemophilia is an inherited bleeding disorder that is inherited in X-linked recessive pattern caused by a deficiency in blood clotting factor VIII (factor 8) that results in the inability of the blood to clot properly with prolonged bleeding from injuries, tooth extractions, or surgery, and internal bleeding into joints and muscles, which can cause pain and swelling and delayed or recurrent bleeding prior to complete wound healing 1, 2, 3. Hemophilia A is caused by a defect in the F8 gene, which is located on the X chromosome. Because males only have one X-chromosome (males have XY sex chromosome), hemophilia A most commonly affects males, who inherit the X chromosome from their mother. The F8 gene provides instructions for making a protein called coagulation factor VIII (factor 8) 4. Coagulation factors are a group of related proteins that are essential for the formation of blood clots (see Figure 2). After an injury, blood clots protect your body by sealing off damaged blood vessels and preventing further blood loss. Coagulation factor VIII (factor 8) is made mainly by cells in your liver. Coagulation factor VIII (factor 8) protein circulates in your bloodstream in an inactive form, bound to another molecule called von Willebrand factor (vWF), until an injury that damages blood vessels occurs. Von Willebrand factor (vWF) is made within endothelial cells, which line the inside surface of blood vessels, and bone marrow cells 5. Von Willebrand factor (vWF) is made of several identical subunits. To facilitate binding to various cells and proteins, these subunits are cut into smaller pieces by an enzyme called ADAMTS13. Von Willebrand factor (vWF) helps platelets stick together and adhere to the walls of blood vessels at the site of a wound. These groups of platelets form temporary clots, plugging holes in blood vessel walls to help stop bleeding. Von Willebrand factor (vWF) also carries coagulation factor VIII (factor 8) to the area of clot formation. In response to an injury, coagulation factor VIII (factor 8) is activated and separates from von Willebrand factor. The active protein sometimes written as coagulation factor VIIIa (factor 8a) interacts with another coagulation factor called factor IX (factor 9) also known as Christmas factor. This interaction sets off a chain of additional chemical reactions that form a blood clot.
Table 1. Coagulation factors
| Coagulation factor | Other common name |
|---|---|
| I | Fibrinogen |
| II | Prothrombin |
| V | Proaccelerin or labile factor |
| VII | Proconvertin |
| VIII | Antihemophilic factor A |
| IX | Antihemophilic factor B or Christmas factor |
| X | Stuart-Prower factor |
| XI | Plasma thromboplastin antecendent |
| XIII | Fibrin stabilizing factor |
If you have hemophilia A, your blood doesn’t clot properly. This can lead to bleeding that is difficult to control.
The main signs and symptoms of hemophilia A are 6, 7, 8, 2, 3:
- Easy or excessive bruising from an early age. Bruises may form easily from minor bumps.
- Internal bleeding for no obvious reason, especially in your joints and muscles. That can result in chronic pain, swelling, joint damage, disability and joint deformity at an early age 9, 10, 11
- Greater than normal bleeding after injuries or surgery. Bleeding from cuts, surgery, or dental procedures may take a long time to stop.
- Unexplained nosebleeds.
- Bleeding into the urine or stool can also occur. Gastrointestinal tract and urinary tract bleeding.
- In more severe cases, bleeding can occur without a known cause.
The severity of hemophilia A symptoms vary between individuals. Prolonged bleeding is the main symptom. It is often first seen when an infant is circumcised. Other bleeding problems usually show up when the infant starts crawling and walking. Mild cases may go unnoticed until later in life. Hemophilia A symptoms may first occur after surgery or injury. Internal bleeding may occur anywhere.
Hemophilia A is caused by a defect in the F8 gene, which is located on the X chromosome and it’s passed down from parent in an X-linked recessive pattern. There are no genes for clotting factors on the Y chromosome. Males only have 1 X chromosome (XY sex chromosome). Males inherit an X chromosome from the mother and a Y chromosome from the father. Because males have only one X chromosome (males have XY chromosome). A single recessive F8 gene on that X chromosome will cause hemophilia A. This means that hemophilia A almost always occurs in males and is passed from mother to son through one of the mother’s genes. For this reason, most people with hemophilia A are males. Male hemophilia A patients do not transmit hemophilia A to their sons, but their female daughters will carry the hemophilia F8 gene 12, 13, 14, 15. All female children of men with of hemophilia A carry the mutated F8 gene whereas male children do not.
Females have 2 X chromosomes (XX chromosome), an X chromosome from the mother and an X chromosome from the father. Since a female has two X chromosomes (XX sex chromosome), in order to have hemophilia A, a female would have to inherit the F8 gene on the X chromosome from each parent. For this reason, it’s very rare for females to get hemophilia A. Usually, a female has only one mutated copy of the F8 gene in her X chromosome (one F8 gene with a mutation and one normal F8 gene), making her a carrier of hemophilia A. A female with one mutated F8 gene and one normal F8 gene is called heterozygous, or a carrier. This means the mutated F8 gene in her X chromosome can be passed down to her children. Boys born to a woman who is a carrier of hemophilia A (one F8 gene with a mutation and one normal F8 gene) have a 50% chance of having hemophilia A. Girls born to a woman who is a carrier of hemophilia A have a 50% chance of being a carrier of hemophilia A. Female hemophilia A F8 gene carriers (one F8 gene with a mutation and one normal F8 gene) do not have symptoms of hemophilia A but may have lower than usual quantities of Factor VIII (factor 8) in her blood. However, some females with hemophilia A can have heavy or long periods, which can cause iron deficiency or anemia and heavy bleeding after giving birth. While bleeding symptoms in females are usually milder than those in males with hemophilia A, in rare cases, a female with one hemophilia gene can have bleeding symptoms that are as serious as those of a male with hemophilia.
Newly recommended terminology for female carriers (one F8 gene with a mutation and one normal F8 gene) designates 5 clinical- and laboratory-based categories 16. For females with decreased (≤40%) factor VIII clotting activity, the terminology is the same as that used for males with hemophilia A:
- Mild hemophilia A (>5% to <40% factor VIII clotting activity)
- Moderate hemophilia A (1%-5% factor VIII clotting activity)
- Severe hemophilia A (<1% factor VIII clotting activity).
For female carriers (one F8 gene with a mutation and one normal F8 gene) with normal factor VIII clotting activity:
- Individuals with a bleeding phenotype: “symptomatic hemophilia carriers”
- Individuals who do not have a bleeding phenotype: “asymptomatic hemophilia carriers”
Note: 25% of females with normal factor VIII clotting activity have a bleeding phenotype.
Additionally, in rare cases females can inherit two abnormal F8 genes, one from each parent, or inherit one abnormal F8 gene but their other X chromosome is missing or does not work properly. Females with these inheritance patterns are as likely as males with hemophilia A to have severe bleeding symptoms.
Although most cases of hemophilia A are inherited (passed down) from a parent to a child, about 1/3 of cases have no previous family history.
Hemophilia A accounts for 80% of hemophilia cases. Hemophilia A is about three to four times as common as hemophilia B, and about half of those affected have the severe form 17. The birth prevalence of hemophilia A in the United States is approximately 1 per 6,500 live male births. Worldwide the birth prevalence for hemophilia A and B has been estimated at 1 in 10,000, although reports vary widely between countries 18. The birth prevalence of hemophilia A in males in Australia, Canada, France, Italy, New Zealand, and the United Kingdom is approximately 24.6 per 100,000 live male births, and 9.5 per 100,000 for severe hemophilia A 19. Hemophilia A occurs in more than 400,000 males worldwide, many of whom remain undiagnosed in the developing world 2, 19. According to the Annual Global Survey produced by the World Federation of Hemophilia for 2025, 459,606 people with hemophilia (244,353 with hemophilia A) were reported from 120 countries, including 10,945 females 20.
The birth prevalence is thought to be approximately the same in all countries and all races, presumably because of the high spontaneous mutation rate of clotting factor VIII gene (F8) and its presence on the X chromosome.
The age of diagnosis and frequency of bleeding episodes are related to the level of factor VIII clotting activity.
- Individuals with severe hemophilia A are usually diagnosed during the first two years of life following bleeding from minor mouth injuries and large “goose eggs” from minor head bumps. Without prophylactic treatment, they may average up to two to five spontaneous bleeding episodes each month including spontaneous joint bleeds or deep-muscle hematomas, and prolonged bleeding or excessive pain and swelling from minor injuries, surgery, and tooth extractions or renewed bleeding after initial bleeding has stopped 21.
- Individuals with moderate hemophilia A seldom have spontaneous bleeding; however, they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years; the frequency of bleeding episodes varies, usually from once a month to once a year.
- Individuals with mild hemophilia A do not have spontaneous bleeding episodes; however, without pre- and postoperative treatment, abnormal bleeding occurs with surgery or tooth extractions; the frequency of bleeding episodes varies widely, typically from once a year to once every ten years. Individuals with mild hemophilia A are often not diagnosed until later in life. Approximately 30% of heterozygous females have clotting activity below 40% and are at risk for bleeding (even if the affected family member is mildly affected). After major trauma or invasive procedures, prolonged or excessive bleeding usually occurs, regardless of severity.
If you are the first person in your family to have a suspected bleeding disorder, your doctor will ask about your bleeding episodes to determine if you have a lifelong, inherited bleeding disorder or an acquired (often transient) bleeding disorder. Increased bleeding with trauma, tonsillectomy, or for a few hours following tooth extraction may be seen in individuals without a bleeding disorder. In contrast, prolonged or intermittent bleeding that lasts several days following tooth extraction or mouth injury, renewed bleeding or increased pain and swelling several days after an injury, or development of a wound hematoma several days after surgery almost always indicates a coagulation problem. An older individual with severe or moderate hemophilia A may have joint deformities and muscle contractures. Large bruises and subcutaneous hematomas for which no trauma can be identified may be present. Individuals with a mild bleeding disorder have no outward signs except during an acute bleeding episode.
Some people develop hemophilia with no family history of the disorder. This is called acquired hemophilia. Acquired hemophilia is a variety of the condition that occurs when a person’s immune system attacks clotting factor 8 or 9 in the blood. It can be associated with:
- Pregnancy
- Autoimmune conditions
- Cancer
- Multiple sclerosis
- Drug reactions
Severe cases of hemophilia A usually are diagnosed within the first year of life. Mild forms might not be apparent until adulthood. Some people learn they have hemophilia A after they bleed excessively during a surgical procedure.
If you are the first person in your family to have a suspected bleeding disorder, your doctor will order a series of tests called a coagulation study.
Tests to diagnose hemophilia A include:
- Prothrombin time (PT). A prothrombin time (PT) test evaluates the coagulation factors VII, X, V, II, and I (fibrinogen).
- Partial thromboplastin time (PTT) also known as activated partial thromboplastin time (aPTT). The partial thromboplastin time (PTT) is used to evaluate the coagulation factors XII, XI, IX, VIII, X, V, II (prothrombin), and I (fibrinogen) as well as prekallikrein (PK) and high molecular weight kininogen (HK).
- Serum factor VIII activity
Clotting-factor tests can reveal a clotting-factor deficiency and determine how severe your hemophilia is. Most hemophilia A patients have a prolonged activated partial thromboplastin time (aPTT); however, a normal result does not rule out mild hemophilia. The activated partial thromboplastin time (APTT or PTT) is a functional measure of the intrinsic and common pathways of the coagulation cascade. The partial thromboplastin time (PTT) is used to evaluate the coagulation factors XII, XI, IX, VIII, X, V, II (prothrombin), and I (fibrinogen) as well as prekallikrein (PK) and high molecular weight kininogen (HK) 22. When blood vessel walls or body tissues are injured, bleeding occurs and a process known as hemostasis initiates. Platelets (small cell fragments) stick and clump at the site of the injury. When this happens, the coagulation cascade begins and coagulation factors are activated in a step-by-step process. The body uses the coagulation cascade to produce blood clots to seal off injuries to blood vessels and tissues, to prevent further blood loss, and to give the damaged areas time to heal. The cascade consists of a group of coagulation factors. These proteins are activated sequentially along either the extrinsic (tissue related) or intrinsic (blood vessel related) pathways. The branches of the pathway then come together into the common pathway, and complete their task with the formation of a stable blood clot. When a person starts bleeding, these three pathways have to work together.
Hemorrhage severity in hemophilia A correlates with scarcity of factor VIII. Factor VIII concentration is expressed in international units (IU); 1 IU is the concentration of factor VIII in 1 mL of pooled plasma or percentages of normal pooled plasma with normal levels ranging between 50% to 150% 2. Severe hemophilia A will have no measurable factor VIII, less than 0.01 IU/mL or less than 1%, and will bleed spontaneously 2. Moderate or mild hemophilia, 0.02 to 0.05 IU/mL (2% to 5%) or 0.06 IU/mL to 0.40 IU/mL (6% to 40%), respectively, will bleed excessively after relatively insignificant trauma 2.
For people with a family history of hemophilia, genetic testing might be used to identify carriers to make informed decisions about becoming pregnant.
It’s also possible to determine during pregnancy if your unborn child is affected by hemophilia. However, the testing poses some risks to your unborn child. Discuss the benefits and risks of testing with your doctor.
The main treatment for severe hemophilia A involves replacing the missing clotting factor VIII (factor 8) you need through a tube in a vein. You will receive factor VIII (factor 8) concentrates. How much you get depends on:
- Severity of bleeding
- Site of bleeding
- Your weight and height
Calculation of factor VIII (factor 8) replacement for bleeding in severe hemophilia A 23, 24, 25:
- Dose of factor VIII = percentage desired of factor x bodyweight (kg) x 0.5
- For severe, life-threatening hemorrhage, administer factor VIII to achieve a 100% desired factor VIII level.
- For mild to moderate hemorrhage, administer factor VIII to achieve a 30% to 50% desired factor VIII level.
- Accounting for the hemophilia A patient’s native factor VIII levels should be factored into factor VIII repletion if known.
This replacement therapy can be given to treat a bleeding episode in progress. It can also be given on a regular schedule at home to help prevent bleeding episodes. Some people receive continuous replacement therapy. Replacement clotting factor can be made from donated blood. Similar products, called recombinant clotting factors, are made in a laboratory, not from human blood.
Mild hemophilia A may be treated with a hormone called desmopressin (DDAVP or desamino-8-arginine vasopressin). This hormone helps the body to release more factor VIII (factor 8) and von Willebrand’s factor (vWF) that is stored within the lining of blood vessels. Desmopressin (DDAVP) can be injected slowly into a vein, subcutaneously or used as a nasal spray.
DDAVP (desmopressin) or factor VIII concentrate may also be needed before having dental extractions or surgery. Fibrin sealants can be applied directly to wound sites to promote clotting and healing. Fibrin sealants are especially useful for dental work.
To prevent a bleeding crisis, people with hemophilia A and their families can be taught to give factor VIII concentrates at home at the first signs of bleeding. People with severe hemophilia A may need regular preventive treatment. Periodic, prophylactic Factor VIII concentrates infusions for patients with severe Hemophilia A have benefits in preventing spontaneous bleeding. The intent of factor VIII prophylaxis aims to modify severe hemophilia A to a milder form by keeping the nadir level of factor VIII more than 1% of normal. The World Federation of Hemophilia recommends factor VIII prophylaxis initiation in hemophiliac children after their first or second episode of hemarthrosis to prevent joint destruction and preserve musculoskeletal function. Mild and moderate hemophilia A patients receive factor VIII concentrates or desmopressin (DDAVP) to prevent hemorrhage in anticipation of trauma or surgery 26, 27.
Some people with hemophilia A develop antibodies to factor VIII. These antibodies are called inhibitors. The inhibitors attack factor VIII so that it no longer works. Inhibitors are measured by the Bethesda assay or the Nijmegen-modified Bethesda assay 28, 29. The presence of a new inhibitor should be suspected in any patient with hemophilia who fails to respond clinically to clotting factor concentrate replacement therapy, particularly in previously responsive patients. Patients who develop inhibitors should have access to immune tolerance induction (ITI) therapy and to suitable hemostatic agents for control of bleeding as well as surgical interventions, if needed, at specialized centers with relevant experience 30, 31. In such cases, a man-made clotting factor called recombinant activated factor VIIa (rFVIIa) can be given. For patients with hemophilia A and persistent inhibitors who fail immune tolerance induction or never underwent immune tolerance induction, the World Federation of Hemophilia recommends emicizumab prophylaxis over bypass agent prophylaxis (recombinant activated factor VIIa [rFVIIa] or activated prothrombin complex concentrate [aPCC]), as emicizumab is more effective in bleed prevention and simpler to administer, as it is given weekly and subcutaneously 32, 33. Emicizumab (Hemlibra) is a newer drug that doesn’t include clotting factors 27, 34. Emicizumab (Hemlibra) can help prevent bleeding episodes in people with hemophilia A 27, 34. Emicizumab prophylaxis has been associated with a significantly greater reduction in bleeding rates than bypass agent prophylaxis 34. Emicizumab is not intended to treat acute bleeding episodes. Breakthrough bleeding is treated with doses of clotting factor concentrates (CFCs) or bypassing agents in the case of patients with inhibitors that are sufficient to achieve hemostasis. Caution is required when treating breakthrough bleeding episodes while on emicizumab as several patients have developed either venous thromboembolism or thrombotic microangiopathy with concomitant administration of activated prothrombin complex concentrate (aPCC) 32.
For first aid for minor cuts, using pressure and a bandage will generally take care of the bleeding. For small areas of bleeding beneath the skin, use an ice pack. Ice pops can be used to slow down minor bleeding in the mouth.
The combination of effective blood product screening with viral inactivation protocols and recombinant production of Factor VIII has enhanced factor VIII replacement products’ safety from viral transmission, such as HIV and hepatitis C. However, you should get the hepatitis B vaccine. People with hemophilia A are more likely to get hepatitis B because they may receive blood products.
Figure 1. Overview of blood coagulation
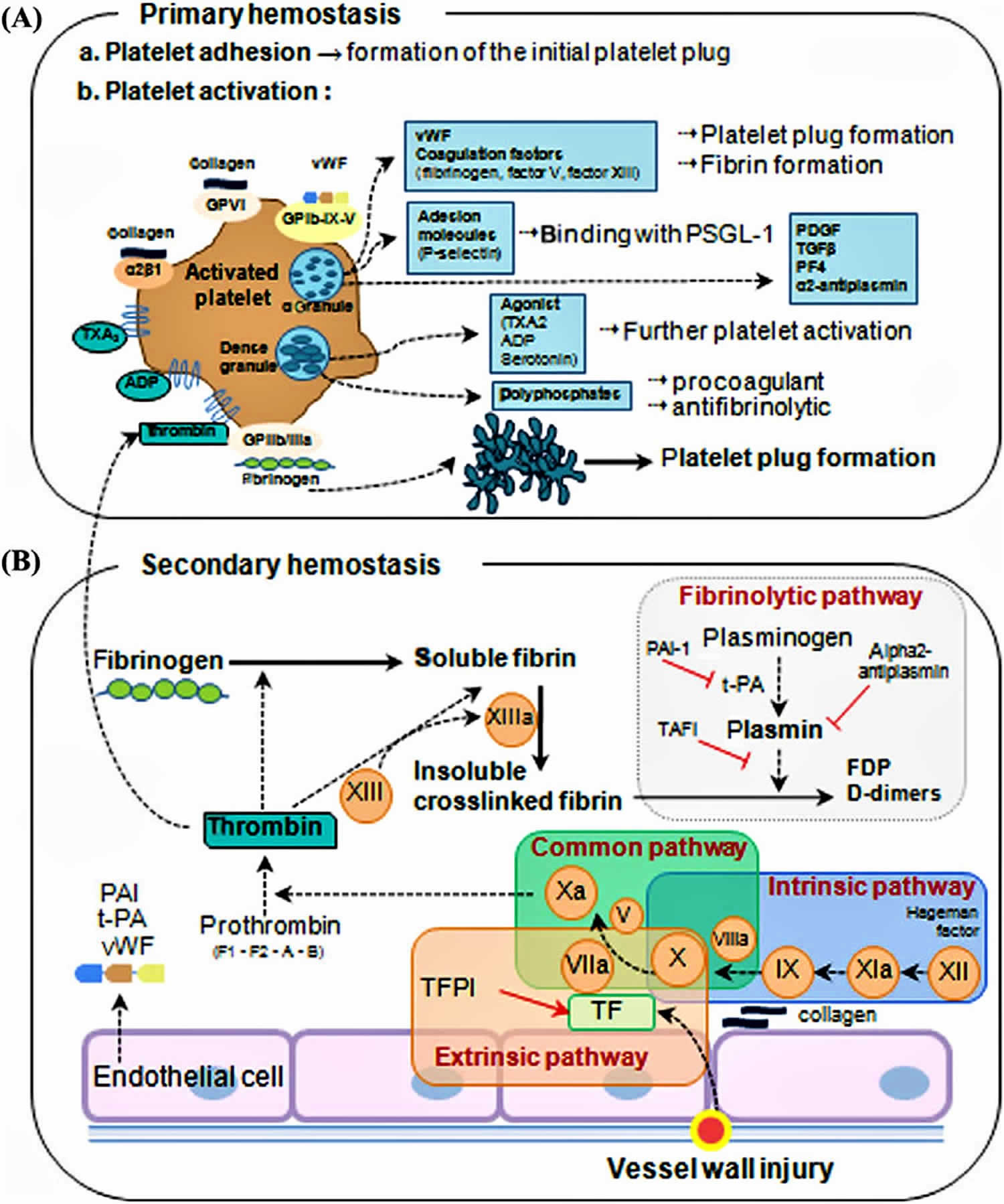
Figure 2. Coagulation cascade
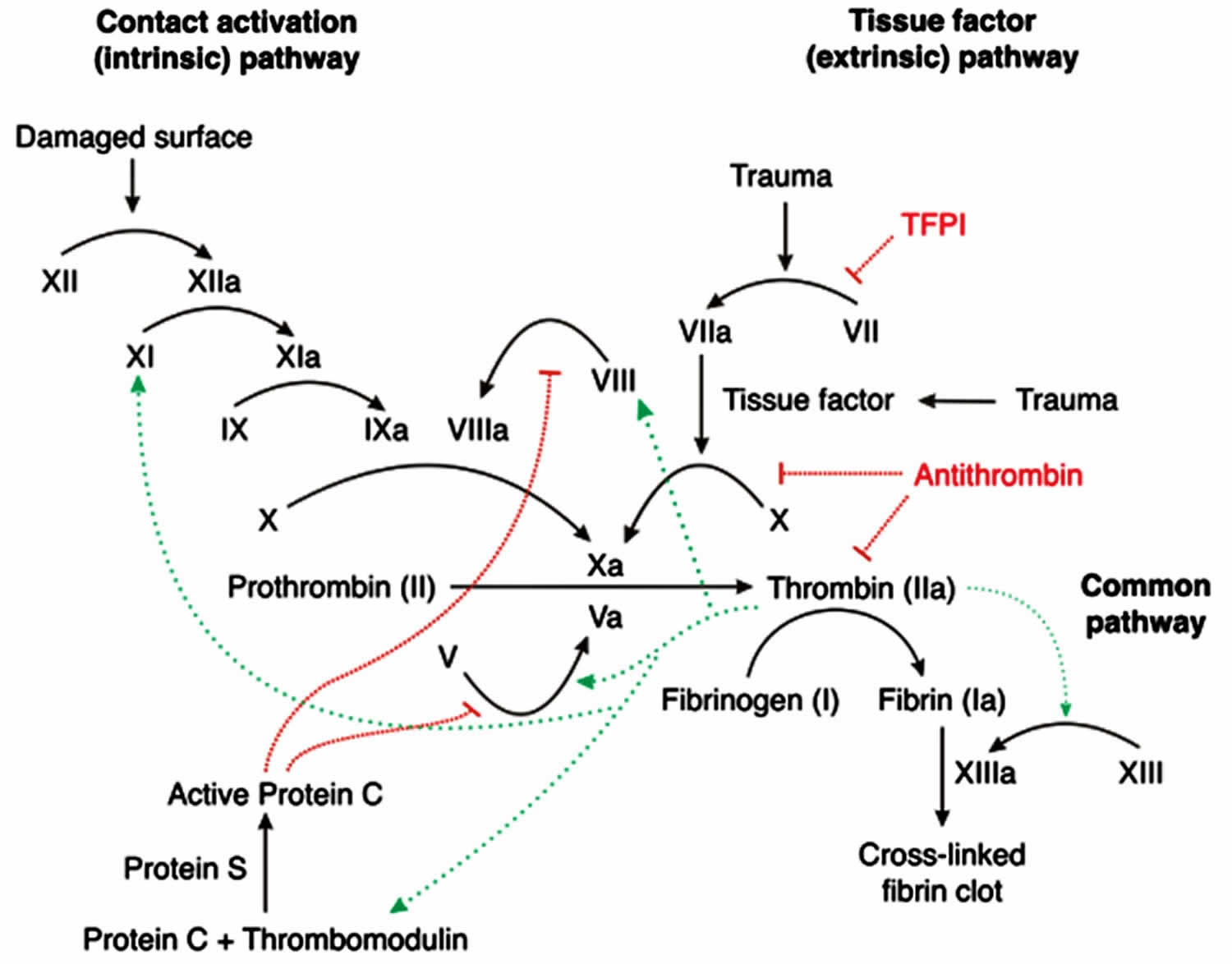
Figure 3. Hemophilia A – X-linked inheritance pattern
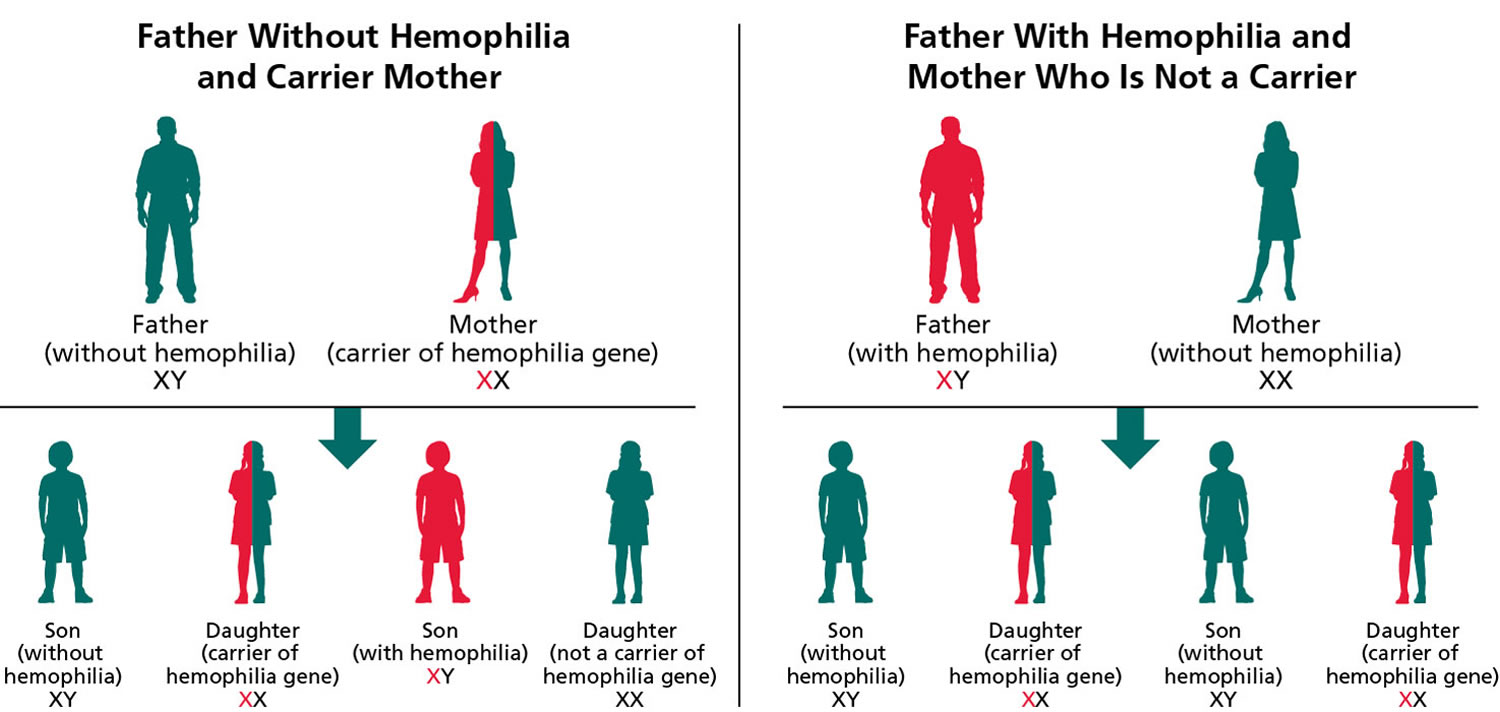
Note: Hemophilia A is inherited in an X-linked manner. Referring to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation.
Can people with hemophilia play sport?
Yes. People with hemophilia can safely participate in a wide range of sports 32, 33. Regular exercise and physical activity can strengthen muscles and protect joints, preventing injuries and bleeding episodes 35. As for everyone, physical activity can help people with hemophilia feel better generally, be with friends and have fun. Likewise, it is important that everyone uses the protective equipment that is appropriate to the sport. Generally, it is recommended that people with hemophilia do not do high contact sports like boxing. They can discuss their sporting goals with their Hemophilia Treatment Center and work together on their chosen sport. Ideally, individuals with hemophilia or their family caregivers should consult a physical therapist before engaging in new sports and physical activities to discuss their appropriateness, required protective gear, preventive measures, and required physical skills prior to beginning the activity. This is particularly important if the individual has any joint with recurrent bleeding (i.e., target joint) 36. Target joints can be protected with braces or splints during physical activity, especially in the absence of clotting factor coverage 37, 38.
Non-contact sports such as swimming, walking, jogging, golf, badminton, archery, cycling, rowing, sailing, and table tennis should be encouraged 32, 33. High-contact and collision sports such as soccer, hockey, rugby, boxing, and wrestling, and high-velocity activities such as motocross racing and skiing are not advised due to the potential for life-threatening injuries, unless the individual is on adequate prophylaxis to cover such activities and is well educated on the potential risks 32, 33. Custom-made dental mouthguards should be used by individuals with hemophilia for all contact sports to prevent trauma and injury to teeth and oral soft tissues 39.
For those with significant musculoskeletal dysfunction, weight-bearing activities that promote development and maintenance of good bone density should be encouraged to the extent their joint health permits 40. The choice of activities should reflect the individual’s preferences/interests, physical condition and ability, local contexts, and available resources. Organized sports programs should be encouraged over unstructured sports activities where protective equipment and supervision may be lacking.
Should a child with hemophilia wear protective gear?
Current treatments mean that protective gear for everyday living is not necessary. Wearing appropriate protective gear with some activities is recommended for everyone whether they have hemophilia or not. Examples of standard protective gear are helmets for cycling and motor bike riding, shin pads for soccer, helmets and pads for cricket and mouthgards for water polo and basketball.
Can people with hemophilia travel?
Yes. They just have a little more organizing and packing to do. They need to contact their Hemophilia Treatment Center to organize enough treatment product and equipment for the time they are away. They also need to find out where the nearest Hemophilia Treatment Centers are to where they will be staying. They may also need documentation to carry medication and treatment equipment through security and customs – it is important to talk to their HHemophilia Treatment Center about this well in advance and allow plenty of time to prepare the documentation.
Hemophilia A cause
Hemophilia A is caused by a defect in the F8 gene, which is located on the X chromosome and it’s passed down from parent in an X-linked recessive pattern. The F8 gene provides instructions for making a protein called coagulation factor VIII (factor 8) 4. Coagulation factors are a group of related proteins that are essential for the formation of blood clots (see Figure 2). After an injury, blood clots protect your body by sealing off damaged blood vessels and preventing further blood loss. Coagulation factor VIII (factor 8) is made mainly by cells in your liver. Coagulation factor VIII (factor 8) protein circulates in your bloodstream in an inactive form, bound to another molecule called von Willebrand factor (vWF), until an injury that damages blood vessels occurs. Von Willebrand factor (vWF) is made within endothelial cells, which line the inside surface of blood vessels, and bone marrow cells 5. Von Willebrand factor (vWF) is made of several identical subunits. To facilitate binding to various cells and proteins, these subunits are cut into smaller pieces by an enzyme called ADAMTS13. Von Willebrand factor (vWF) helps platelets stick together and adhere to the walls of blood vessels at the site of a wound. These groups of platelets form temporary clots, plugging holes in blood vessel walls to help stop bleeding. Von Willebrand factor (vWF) also carries coagulation factor VIII (factor 8) to the area of clot formation. In response to an injury, coagulation factor VIII (factor 8) is activated and separates from von Willebrand factor. The active protein sometimes written as coagulation factor VIIIa (factor 8a) interacts with another coagulation factor called factor IX (factor 9) also known as Christmas factor. This interaction sets off a chain of additional chemical reactions that form a blood clot.
Hemophilia A is inherited in an X-linked recessive manner. The F8 genes for factor VIII (factor 8) are located on the X chromosome. The X chromosome is one of two sex chromosomes, and it contains hundreds of genes that are essential for both male and female development. Females typically have two X chromosomes (XX sex chromosome), while males have one X and one Y chromosome (XY sex chromosome). There are no genes for clotting factors on the Y chromosome.
Males only have 1 X chromosome (XY sex chromosome). Males inherit an X chromosome from the mother and a Y chromosome from the father. Because males have only one X chromosome (males have XY chromosome). A single recessive F8 gene on that X chromosome will cause hemophilia A. This means that hemophilia A almost always occurs in males and is passed from mother to son through one of the mother’s genes. For this reason, most people with hemophilia A are males. Male hemophilia A patients do not transmit hemophilia A to their sons, but their female daughters will carry the hemophilia F8 gene 12, 13, 14, 15. All female children of men with of hemophilia A carry the mutated F8 gene whereas male children do not.
Females have 2 X chromosomes (XX chromosome), an X chromosome from the mother and an X chromosome from the father. Since a female has two X chromosomes (XX sex chromosome), in order to have hemophilia A, a female would have to inherit the F8 gene on the X chromosome from each parent. For this reason, it’s very rare for females to get hemophilia A. Usually, a female has only one mutated copy of the F8 gene in her X chromosome (one F8 gene with a mutation and one normal F8 gene), making her a carrier of hemophilia A. A female with one mutated F8 gene and one normal F8 gene is called heterozygous, or a carrier. This means the mutated F8 gene in her X chromosome can be passed down to her children. Boys born to a woman who is a carrier of hemophilia A (one F8 gene with a mutation and one normal F8 gene) have a 50% chance of having hemophilia A. Girls born to a woman who is a carrier of hemophilia A have a 50% chance of being a carrier of hemophilia A. Female hemophilia A F8 gene carriers (one F8 gene with a mutation and one normal F8 gene) do not have symptoms of hemophilia A but may have lower than usual quantities of Factor VIII (factor 8) in her blood. However, some females with hemophilia A can have heavy or long periods, which can cause iron deficiency or anemia and heavy bleeding after giving birth. While bleeding symptoms in females are usually milder than those in males with hemophilia A, in rare cases, a female with one hemophilia gene can have bleeding symptoms that are as serious as those of a male with hemophilia.
Additionally, in rare cases females can inherit two abnormal F8 genes, one from each parent, or inherit one abnormal F8 gene but their other X chromosome is missing or does not work properly. Females with these inheritance patterns are as likely as males with hemophilia A to have severe bleeding symptoms.
Although most cases of hemophilia A are inherited (passed down) from a parent to a child, about 1/3 of cases have no previous family history.
Risk factors for hemophilia A include:
- Family history of bleeding
- Being male.
Hemophilia A genetics
Hemophilia A is inherited in an X-linked recessive manner. The F8 genes for factor VIII (factor 8) are located on the X chromosome. The X chromosome is one of two sex chromosomes, and it contains hundreds of genes that are essential for both male and female development. Females typically have two X chromosomes (XX sex chromosome), while males have one X and one Y chromosome (XY sex chromosome). There are no genes for clotting factors on the Y chromosome.
Males only have 1 X chromosome (XY sex chromosome). Males inherit an X chromosome from the mother and a Y chromosome from the father. Because males have only one X chromosome (males have XY chromosome). A single recessive F8 gene on that X chromosome will cause hemophilia A. This means that hemophilia A almost always occurs in males and is passed from mother to son through one of the mother’s genes. For this reason, most people with hemophilia A are males. Male hemophilia A patients do not transmit hemophilia A to their sons, but their female daughters will carry the hemophilia F8 gene 12, 13, 14, 15. All female children of men with of hemophilia A carry the mutated F8 gene whereas male children do not.
Females have 2 X chromosomes (XX chromosome), an X chromosome from the mother and an X chromosome from the father. Since a female has two X chromosomes (XX sex chromosome), in order to have hemophilia A, a female would have to inherit the F8 gene on the X chromosome from each parent. For this reason, it’s very rare for females to get hemophilia A. Usually, a female has only one mutated copy of the F8 gene in her X chromosome (one F8 gene with a mutation and one normal F8 gene), making her a carrier of hemophilia A. A female with one mutated F8 gene and one normal F8 gene is called heterozygous, or a carrier. This means the mutated F8 gene in her X chromosome can be passed down to her children. Boys born to a woman who is a carrier of hemophilia A (one F8 gene with a mutation and one normal F8 gene) have a 50% chance of having hemophilia A. Girls born to a woman who is a carrier of hemophilia A have a 50% chance of being a carrier of hemophilia A. Female hemophilia A F8 gene carriers (one F8 gene with a mutation and one normal F8 gene) do not have symptoms of hemophilia A but may have lower than usual quantities of Factor VIII (factor 8) in her blood. However, some females with hemophilia A can have heavy or long periods, which can cause iron deficiency or anemia and heavy bleeding after giving birth. While bleeding symptoms in females are usually milder than those in males with hemophilia A, in rare cases, a female with one hemophilia gene can have bleeding symptoms that are as serious as those of a male with hemophilia.
Additionally, in rare cases females can inherit two abnormal F8 genes, one from each parent, or inherit one abnormal F8 gene but their other X chromosome is missing or does not work properly. Females with these inheritance patterns are as likely as males with hemophilia A to have severe bleeding symptoms.
Although most cases of hemophilia A are inherited (passed down) from a parent to a child, about 1/3 of cases have no previous family history.
Because most females who inherit hemophilia are heterozygous (one F8 gene with a mutation and one normal F8 gene) and have no bleeding symptoms or mild bleeding symptoms, hemophilia may be hidden in a family for many generations if it passes only through females.
If a mother is heterozygous (one F8 gene with a mutation and one normal F8 gene) for hemophilia and the father does not have hemophilia, each son has a 1 in 2 (50%) chance of getting his mother’s hemophilia allele and having hemophilia. Each daughter has a 1 in 2 (50%) chance of getting her mother’s hemophilia mutated F8 gene and being heterozygous (one F8 gene with a mutation and one normal F8 gene). Overall, there is a 1 in 4 (25%) chance for each pregnancy that the baby will be a son with hemophilia and a 1 in 4 (25%) chance that the baby will be hemophilia A carrier daughter. There is a 1 in 2 (50%) chance that the baby (either a son or a daughter) will not get the hemophilia A at all and, therefore, can’t pass it down to his or her children.
Note:
- Hemizygous refers to a gene normally present in only a single copy; usually an X-linked gene in a male.
- Heterozygous refers to a variant (distinct from the reference sequence) that comprises one of two alleles of a given gene. An individual with two different alleles at a particular locus (one on each chromosome of a pair), one of which is usually pathogenic.
- X-linked inheritance refers to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Penetrance
All males with a clotting factor VIII (F8) mutated gene will be affected and will have approximately the same severity of disease as other affected males in the family. However, other genetic and environmental effects may modify the clinical severity to some extent.
Approximately 30% of females with one clotting factor VIII gene (F8 gene) mutated gene and one normal F8 gene have a factor VIII clotting activity lower than 40% and a bleeding disorder; mild bleeding can occur in heterozygous (one F8 gene with a mutation and one normal F8 gene) females with low-normal factor VIII activity 41. Overall, female carriers have more bleeding than unaffected females 42.
Genotype-Phenotype Correlations
Evidence for an association between variant type and disease severity:
- F8 intron (noncoding sequence of DNA removed from mature messenger RNA prior to translation) 22 inversions are associated with severe hemophilia A and account for 45% of individuals with severe hemophilia A 43. Of these, 20% to 30% develop alloimmune inhibitors. Occasionally, individuals considered to have moderate hemophilia A have been found to have F8 inversions. Often their assays have contained either some residual factor VIII clotting activity from a prior transfusion or the assay methods used were inaccurate at low levels.
- An inversion between a 1-kb sequence in intron 1 and an inverted repeat 5′ to F8 44 is also associated with a severe phenotype, and some individuals have developed inhibitors.
- Single-nucleotide variants leading to new stop codons are essentially all associated with a severe phenotype, as are most frameshift variants. (An exception is the insertion or deletion of adenosine bases resulting in a sequence of eight to ten adenosines, which may result in moderate hemophilia A 45.
- Splice site variants often result in severe disease, but can result in mild or moderate disease, depending on the specific change and location.
- Missense variants occur in fewer than 20% of individuals with severe hemophilia A but are found in nearly all of those with a diagnosis of mild or moderate disease.
- A single-base change in the 5’ promoter region of F8 has been associated with mild hemophilia A 46.
Hemophilia A signs and symptoms
Hemophilia A should be suspected in an individual with any of the following clinical and/or laboratory features.
Hemophilia A clinical features:
- Bleeding into a joint (hemarthrosis), causing pain, swelling, and a decreased range of motion, especially with mild or no antecedent trauma
- Deep-muscle hematomas
- Intracranial bleeding in the absence of major trauma
- Neonatal cephalohematoma or intracranial bleeding
- Prolonged bleeding or renewed bleeding after initial bleeding stops following tooth extractions, mouth injury, or circumcision *
- Prolonged or delayed bleeding or poor wound healing following surgery or trauma *
- Unexplained gastrointestinal bleeding or blood in urine (hematuria) *
- Unusually heavy period (menorrhagia), especially with onset at menarche (a girl’s first menstrual period)
- Prolonged nosebleeds, especially recurrent and bilateral *
- Excessive bruising, especially with firm, subcutaneous hematomas
* Of any severity, or especially in more severely affected persons.
Sites of bleeding in hemophilia A 47:
- Serious
- Joints (hemarthrosis) ~ 70% to 80% approximate frequency of bleeding
- Muscles, especially deep compartments (iliopsoas, calf, forearm) ~ 10% to 20% approximate frequency of bleeding
- Mucous membranes of the mouth, nose, and genitourinary tract
- Life-threatening
- Intracranial < 5% approximate frequency of bleeding
- Neck/throat
- Gastrointestinal
Muscle hematomas or intracranial bleeding can occur four or five days after the original injury. Intermittent oozing may last for days or weeks after tooth extraction. Prolonged or delayed bleeding or wound hematoma formation after surgery is common. After circumcision, males with hemophilia A of any severity may have prolonged oozing, or they may heal normally without treatment. In severe hemophilia A, spontaneous joint bleeding is the most frequent symptom.
The age of diagnosis and frequency of bleeding episodes in the untreated individual are related to the factor VIII clotting activity (see Table 2). In any affected individual, bleeding episodes may be more frequent in childhood and adolescence than in adulthood. To some extent, this greater frequency is a function of both physical activity levels and vulnerability during more rapid growth. In two-thirds of cases, confirmation of the hemophilia A diagnosis occurs shortly after the delivery of an affected son to a mother who carries the susceptible gene.
Individuals with severe hemophilia A are usually diagnosed in the neonatal period due to birth- or neonatal-related procedures or during the first year of life 48. In untreated toddlers, bleeding from minor mouth injuries and large “goose eggs” from minor head bumps are common and are the most frequent presenting symptoms of severe hemophilia A. Intracranial bleeding may also result from head injuries. The untreated child almost always has subcutaneous hematomas; some have been referred for evaluation of possible non-accidental trauma.
As the child grows and becomes more active, spontaneous joint bleeds occur with increasing frequency unless the child is on a prophylactic treatment program. Spontaneous joint bleeds or deep-muscle hematomas initially cause pain or limping before swelling appears. Children and adults with severe hemophilia A who are not treated prophylactically have an average of two to five spontaneous bleeding episodes each month. While joints are the most common sites of spontaneous bleeding, other sites include the kidneys, gastrointestinal tract, and brain. Without prophylactic treatment, individuals with severe hemophilia A have prolonged bleeding or excessive pain and swelling from minor injuries, surgery, and tooth extractions.
Individuals with moderate hemophilia A seldom have spontaneous bleeding but bleeding episodes may be precipitated by relatively minor trauma. Without pretreatment (as for elective invasive procedures) they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years. The frequency of bleeding episodes requiring treatment with factor VIII concentrates varies from once a month to once a year. Signs and symptoms of bleeding are otherwise similar to those found in severe hemophilia A.
Individuals with mild hemophilia A do not have spontaneous bleeding. However, without treatment abnormal bleeding occurs with surgery, tooth extractions, and major injuries. The frequency of bleeding may vary from once a year to once every ten years. Individuals with mild hemophilia A are often not diagnosed until later in life when they undergo surgery or tooth extraction or experience major trauma.
Heterozygous females with a factor VIII clotting activity level lower than 40% are at risk for bleeding that is usually comparable to that seen in males with mild hemophilia. However, subtle abnormal bleeding may occur with a baseline factor VIII clotting activity between 35% and 60% or higher 42.
Table 2. Symptoms Related to Severity of Untreated Hemophilia A
| Severity | Factor VIII Clotting Activity 1 | Symptoms | Usual Age at Diagnosis |
|---|---|---|---|
| Severe | <1 IU/dL (<0.01 IU/mL) or <1% of normal | Frequent spontaneous bleeding. Spontaneous bleeding into joints or muscles, predominantly in the absence of identifiable hemostatic challenge. Abnormal bleeding after minor injuries, surgery, or tooth extractions. | Age ≤2 years |
| Moderate | 1-5 IU/dL (0.01-0.05 IU/mL) or 1% to 5% of normal | Occasional spontaneous bleeding. Prolonged bleeding after minor injuries, surgery, or tooth extractions. | Age <5-6 years |
| Mild | 5-40 IU/dL (0.05-0.40 IU/mL) or 5% to 40% of normal | Rare spontaneous bleeding. Severe bleeding after major injuries, surgery, or tooth extractions | Often later in life, depending on hemostatic challenges |
Footnote: 1 Clinical severity does not always correlate with the in vitro assay result.
[Source 49, 32, 33 ]Hemophilia A complications
Complications of untreated bleeding. The leading cause of death related to bleeding is intracranial hemorrhage. The major cause of disability from bleeding is chronic joint disease 50.
- Joint damage can result in chronic pain, swelling, stiffness, and reduced range of motion and disability and joint deformity at an early age. Individuals with Hemophilia A are more likely to suffer from arthritis and more likely to require knee/hip replacement compared with the general population 9, 51.
- Poor mobility, self‐care issues, and inability to perform usual daily activities 52, 53.
- Inability to participate in social or sporting activities 54.
- Higher pain levels and functional impairment associated with anxiety, depression and unemployment 55, 56. Pain/discomfort is an area where most individuals report experiencing ‘extreme’ issues 10. Individuals may experience anger and frustration due to the pain, inconvenience and erratic nature of bleeds 57.
- Anxiety/depression are the areas where most individuals report experiencing ‘extreme’ issues 10.
- Adverse impact on educational achievement and work productivity due to absence and difficulties due to functional impairments and pain 52, 58, 59.
Currently available treatment with clotting factor concentrates is normalizing life expectancy and reducing chronic joint disease for children and adults with hemophilia A. Prior to the availability of such treatment, the median life expectancy for individuals with severe hemophilia A was 11 years (the current life expectancy for affected individuals in several developing countries). Excluding death from HIV, life expectancy for severely affected individuals in the UK receiving adequate treatment was reported in 2007 as 63 years 60.
Since the mid-1960s, the mainstay of treatment of bleeding episodes has been factor VIII concentrates that initially were derived solely from donor plasma. Viral inactivation methods and donor screening of plasmas were introduced by the mid-1980s and recombinant factor VIII concentrates were introduced in the early 1990s, ending the risk of HIV transmission. Many individuals who received plasma-derived factor VIII concentrates from 1979 to 1985 contracted HIV. Approximately half of these individuals died of AIDS prior to the advent of effective HIV therapy.
Hepatitis B transmission from earlier plasma-derived concentrates was eliminated with donor screening and then vaccination in the 1970s. Most individuals exposed to plasma-derived concentrates prior to the late 1980s became chronic carriers of the hepatitis C virus. Viral inactivation methods implemented in concentrate preparation and donor screening assays developed by 1990 have eliminated this complication.
Approximately 30% of individuals with severe hemophilia A develop alloimmune inhibitors to factor VIII, usually within the first 20 exposures to infused factor VIII 61 and, less frequently, in those who have received more than 50 exposures 62. Among individuals with hemophilia A, inhibitors are more prevalent in blacks and Hispanics than whites. Reasons for these disparities are being actively investigated 63.
Hemophilia A diagnosis
The diagnosis of hemophilia A is established in an individual with low factor VIII (factor 8) clotting activity in the presence of a normal, functional von Willebrand factor (vWF) level. Identification of one mutated factor VIII (F8) gene on molecular genetic testing in a male confirms the diagnosis. Identification of one F8 gene with a mutation and one normal F8 gene on molecular genetic testing in a symptomatic female confirms the diagnosis.
Laboratory features
- Normal platelet count
- Prolonged activated partial thromboplastin time (aPTT)
- Normal prothrombin time (PT)
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hemophilia A, the following evaluations are recommended if they have not already been completed 3:
- A personal and family history of bleeding to help predict disease severity
- A joint and muscle evaluation, particularly if the individual describes a history of hemarthrosis or deep-muscle hematomas
- Screening for hepatitis A, B, and C as well as HIV if blood products or plasma-derived clotting factor concentrates were administered prior to 1990
- Baseline complete blood count (CBC) with a platelet count, especially if there is a history of nose bleeds, GI bleeding, mouth bleeding, or (in females) menorrhagia or postpartum hemorrhage
- Referral to a hemophilia treatment center. For locations:
- Worldwide, see World Federation of Haemophilia 64 at https://wfh.org/find-local-support/#HTCs
- US only, see National Hemophilia Foundation 65 at https://www.bleeding.org/
- Identification of the specific clotting factor VIII gene (F8) pathogenic variant in an individual to aid in determining disease severity, the likelihood of inhibitor development, and the chance that immune tolerance will be successful if an inhibitor does develop
- Consultation with a clinical geneticist and/or genetic counselor, particularly for a new diagnosis in the family and for females of childbearing years.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Hemophilia A Molecular Genetic Testing
Molecular genetic testing approaches can include single-gene testing, use of a multi-gene panel, and more comprehensive genomic testing:
- Single-gene testing. Targeted analysis for the intron 22 or intron 1 inversion is frequently performed first in (a) individuals with severe hemophilia A, (b) females with a family history of severe hemophilia A, or (c) females with a family history of hemophilia A of unknown severity in whom the family-specific pathogenic variant is not known. Sequence analysis of clotting factor VIII gene (F8) followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found is performed if the common intron 22 or intron 1 inversion is not detected.
- A multi-gene panel that includes clotting factor VIII gene (F8) and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and over time. (2) Some multi-gene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multi-gene panel provides the best opportunity to identify the genetic cause of the condition at the most reasonable cost while limiting secondary findings. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing based tests. (4) The ability of panels to detect structural variants in clotting factor VIII gene (F8), a common cause of hemophilia A, should be confirmed.
- More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered if serial single-gene testing [and/or use of a multi-gene panel that includes clotting factor VIII gene (F8)] fails to confirm a diagnosis in an individual with features of hemophilia A. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
Male proband (index case). The diagnosis of hemophilia A is established in a male proband by identification of decreased factor VIII clotting activity and a normal, functional von Willebrand factor level.
- Severe hemophilia A. <1% factor VIII
- Moderate hemophilia A. 1%-5% factor VIII
- Mild hemophilia A. 6%-40% factor VIII
Note: Rarely, in individuals with mild hemophilia A, a standard “one-stage” factor VIII clotting activity assay shows near-normal or low-normal factor VIII clotting activity (40%-80%), whereas in a “two-stage” or chromogenic assay, factor VIII activity is low. Thus, low-normal in vitro clotting activity does not always exclude the presence of mild hemophilia A.
Identification of a hemizygous pathogenic variant in clotting factor VIII gene (F8) by molecular genetic testing can help predict the clinical phenotype, assess the risk of developing a factor VIII inhibitor, and allow family studies.
Female proband (index case). The diagnosis of hemophilia A is established by determination of low factor VIII clotting activity. Carrier status is determined by identification of a heterozygous pathogenic variant in clotting factor VIII gene (F8) by molecular genetic testing (see Table 2). Factor VIII clotting activity is unreliable in the detection of heterozygous females; only approximately 30% of hemophilia A heterozygous females have factor VIII clotting activity lower than 40% 41.
Table 3. Molecular Genetic Testing Used in Hemophilia A
| Gene | Test Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by This Method | |
|---|---|---|---|
| Severe Hemophilia A | Moderate or Mild Hemophilia A | ||
| F8 | Targeted mutation analysis | ~48% | 0% |
| Sequence analysis | ~43%-51% | 76%-99% | |
| Gene-targeted deletion/duplication analysis | 1.5% | 0.2% | |
Hemophilia A differential diagnosis
Hemophilia A differential diagnosis include 2, 3:
- Acquired hemophilia
- Ehlers-Danlos syndrome
- Factor XI deficiency
- Glanzmann thrombasthenia
- Hemophilia B
- Hemophilia C
- Physical child abuse
- Platelet disorders
- Von Willebrand disease
Table 4. Inherited bleeding disorders with low factor VIII clotting activity
| Gene(s) | Disorder | Mode of inheritance | Clinical Features | Laboratory Findings / Comment |
|---|---|---|---|---|
| von Willebrand factor (vWF) | Type 1 von Willebrand disease | Autosomal dominant | Mucous membrane bleeding incl epistaxis, bleeding with dental extractions, heavy menstrual & postpartum bleeding, & spontaneous bruises. Also may have trauma- & procedure-related bleeding. | Partial quantitative deficiency of von Willebrand factor (vWF) (low von Willebrand factor (vWF) antigen, low factor VIII clotting activity, & low von Willebrand factor (vWF) activity). von Willebrand factor (vWF) levels can differentiate mild hemophilia A from von Willebrand disease (persons with hemophilia A have normal von Willebrand factor (vWF) antigen level). |
| Type 2A & 2B von Willebrand disease | Autosomal dominant | In type 2A, bleeding as in type 1 von Willebrand disease or may be more severe. In type 2B, bleeding as in type 1 von Willebrand disease or may be more severe. Also may have thrombocytopenia. | Qualitative deficiency of von Willebrand factor (vWF) w/↓ of high-molecular-weight multimers (more loss in type 2A). Measures of von Willebrand factor (vWF) platelet or collagen binding activity are ↓, while von Willebrand factor (vWF) antigen & factor VIII clotting activity may be low-normal to mildly ↓. | |
| Type 2M von Willebrand disease | Autosomal dominant | Bleeding as in type 2A von Willebrand disease | Qualitative deficiency of von Willebrand factor (vWF) w/similar ↓ in function as seen in type 2A, but assoc w/normal multimer pattern | |
| Type 2N von Willebrand disease | Autosomal recessive | Clinically indistinguishable from mild/moderate hemophilia A | von Willebrand factor (vWF) platelet binding is completely normal. Type 2N von Willebrand disease is biochemically indistinguishable from mild hemophilia A. Mild hemophilia A can be distinguished from type 2N von Willebrand disease by molecular genetic testing or measuring binding of factor VIII to von Willebrand factor (vWF) using enzyme-linked immunosorbent assay (ELISA) or column chromatography. | |
| Type 3 von Willebrand disease | Autosomal recessive | Frequent episodes of mucous membrane bleeding; joint & muscle bleeding similar to that seen in hemophilia A but usually with more mucosal bleeding symptoms | Complete or near-complete quantitative deficiency of von Willebrand factor (vWF). von Willebrand factor (vWF) level is often <1% & factor VIII clotting activity is most commonly 2%-8%. | |
| LMAN1 MCFD2 | Combined factor V & factor VIII deficiency (OMIM 613625 & 227300) | Autosomal recessive | Very rare disorder with mucocutaneous & bleeding with trauma & surgery | Low factor VIII and factor V clotting activity levels (usually 10%-20%); prolonged prothrombin time (PT) & activated partial thromboplastin time (aPTT). Mild hemophilia A does not result in prolonged prothrombin time (PT). |
Table 5. Inherited bleeding disorders with Normal factor VIII clotting activity
| Gene(s) | Disorder | Mode of inheritance | Clinical Features | Laboratory Findings / Comment |
|---|---|---|---|---|
| F9 | Hemophilia B | X-linked | Clinically indistinguishable from hemophilia A | Diagnosis is based on factor IX clotting activity <40%. |
| F11 | Factor XI deficiency (OMIM 612416) | Autosomal recessive & Autosomal dominant | Compound heterozygous & homozygous persons may exhibit bleeding similar to mild/moderate hemophilia A. Some heterozygotes have mucocutaneous bleeding symptoms. | Heterozygotes have factor XI coagulant activity 25%-75% of normal; homozygotes have activity <1%-15%. 1 A specific factor XI clotting assay establishes diagnosis. |
| F12 KLKB1 KNG1 | Factor XII (OMIM 234000), prekallikrein (OMIM 612423), & high-molecular-weight kininogen (OMIM 228960) deficiencies | Autosomal recessive | Not associated with clinical bleeding | Can cause long activated partial thromboplastin time (aPTT) |
| F2 F5 F7 F10 | Prothrombin (factor II) (OMIM 613679), factor V (OMIM 227400), factor X (OMIM 227600), & factor VII (OMIM 227500) deficiencies | Autosomal recessive | Rare bleeding disorders. Persons may have easy bruising & hematoma formation, epistaxis, heavy menstrual bleeding, & bleeding after trauma & surgery. Hemarthroses are less common. Spontaneous intracranial bleeding can occur. | Factor VII deficiency should be suspected if prothrombin time (PT) is prolonged & activated partial thromboplastin time (aPTT) normal. Persons w/deficiency of factors II, V, or X usually have prolonged prothrombin time (PT) & activated partial thromboplastin time (aPTT). Specific coagulation factor assays establish diagnosis.2 |
| FGA FGB FGG | Afibrinogenemia (OMIM 202400), hypofibrinogenemia (OMIM 202400), dysfibrinogenemia (OMIM 616004) | Autosomal recessive & Autosomal dominant 3 | Afibrinogenemia is assoc w/manifestations similar to hemophilia A except that bleeding from minor cuts is prolonged due to lack of fibrinogen to support platelet aggregation. Hypofibrinogenemia & dysfibrinogenemia can be assoc w/mild-to-moderate bleeding symptoms. Rarely, persons w/dysfibrinogenemia are at risk for thrombosis. | In dysfibrinogenemia there is discordance between functional & antigenic level, w/latter usually in normal range. For all fibrinogen disorders, thrombin & reptilase times are almost always prolonged & functional measurements of fibrinogen are ↓. |
| F13A1 F13B | Factor XIII deficiency (OMIM 613225, 613235) | Autosomal recessive | Umbilical stump bleeding in >80% of persons. Intracranial bleeding that occurs spontaneously or following minor trauma in 30% of persons. Subcutaneous hematomas, muscle hematomas, defective wound healing, & recurrent spontaneous abortion are also seen. Joint bleeding is rare. | All coagulation screening tests are normal; screening test for clot solubility or specific assay for factor XIII activity can confirm diagnosis. Bleeding symptoms are reported in persons w/levels <13% by quantitative assay.4 |
| GP1BA GP1BB GP9 ITGA2B | Platelet function disorders: Bernard-Soulier syndrome (OMIM 231200) & Glanzmann thrombasthenia (OMIM 273800) | Autosomal recessive | In Bernard-Soulier syndrome, Glanzmann thrombasthenia, & storage pool & nonspecific secretory defects: skin & mucous membrane bleeding, recurring epistaxis, gastrointestinal bleeding, heavy menstrual bleeding, & excessive bleeding during or immediately after trauma & surgery. Joint, muscle, & intracranial bleeding is rare. | Diagnosis is established using platelet aggregation assays, flow cytometry, & platelet electron microscopy. |
Footnotes:
1 Duga S, et al. 66
2 Combined (multiple) deficiencies are usually acquired disorders, although a few families have hereditary deficits of the vitamin K-dependent factors, often resulting from deficiency of gamma-carboxylase..
3 Afibrinogenemia is inherited in an autosomal recessive manner. Hypofibrinogenemia can be inherited in either an autosomal dominant or an autosomal recessive manner. Dysfibrinogenemia is inherited in an autosomal dominant manner.
4 Menegatti M et al. 67
[Source 3 ]Inhibitor screening
Approximately 1 in 5 people with hemophilia A will develop an antibody called an inhibitor to the treatment product (medicine) used to treat or prevent their bleeding episodes 68. Developing an inhibitor is one of the most serious and costly medical complications of a bleeding disorder, because having an inhibitor makes it more difficult to treat bleeds. Inhibitors make it more difficult to stop a bleeding episode because they prevent the treatment from working.
Inhibitors are measured by the Bethesda assay or the Nijmegen-modified Bethesda assay 28, 29.
- The definition of a positive inhibitor is a Bethesda titer of >0.6 Bethesda units (BU) for factor VIII 69
Inhibitor measurement may be performed during replacement therapy by assays utilizing heat treatment techniques 70. - A low-responding inhibitor is an inhibitor <5.0 BU, whereas a high-responding inhibitor is an inhibitor ≥5.0 BU.
- Low-responding inhibitors tend to be transient; a transient inhibitor is defined as a positive inhibitor that drops below the definition threshold within 6 months of initial documentation without any change in treatment regimen and despite antigenic challenge with clotting factor concentrates (CFCs) 71. A suspected inhibitor should be confirmed by repeat laboratory testing, documenting poor factor recovery and/or shortened half-life (t½) of less than 6 hours in hemophilia A (in the case of standard half-life factor VIII clotting factor concentrates (CFCs) 72.
- High-responding inhibitors tend to be persistent and may fall or become undetectable after a long period without clotting factor concentrate exposure; however, they increase 3-5 days after re-
challenge with clotting factor concentrates (CFCs) (anamnestic response) 73. - It is critical to detect inhibitors early to ensure appropriate treatment. At least half of inhibitor cases are detected by routine inhibitor screening after initial exposures to clotting factor concentrates (CFCs), while the rest are detected after there is poor clinical response to clotting factor concentrate replacement therapy (i.e., when factor recovery and/or half-life are not as expected) when treating or preventing a bleed 30.
- Inhibitor testing should be performed before major surgery and if there is suboptimal response to clotting factor concentrate replacement therapy in the post-operative period and in any patient who fails to respond to adequate clotting factor concentrate replacement therapy after past responsiveness 74, 75, 76, 77, 78, 79.
- It is particularly important to perform routine inhibitor screening during the time of greatest risk for inhibitor development, at least every 6-12 months after clotting factor concentrate replacement therapy is initiated, and annually thereafter. While some advocate more frequent screening, this remains controversial with few data to support the benefit of this approach 73.
- Screening should be performed in any patient, regardless of age or disease severity, who is intensively treated (i.e., for more than 5 consecutive days) and within 4 weeks of the last infusion 74, 80.
Hemophilia A treatment
The World Federation of Hemophilia has published treatment guidelines for the management of individuals with hemophilia 32, 33. Treatment should be coordinated through a hemophilia treatment center. Individuals in the USA see National Bleeding Disorders Foundation 65; individuals worldwide see World Federation of Hemophilia for locations 81
- Treatment of Hemophilia A manifestations: Referral to a hemophilia treatment center to facilitate treatment; intravenous infusion of factor VIII concentrate is most effective when infused within one hour of the onset of bleeding; training to facilitate home infusions administered by parents; immune tolerance therapy. For those with mild disease, including most symptomatic females, immediate treatment of bleeding with intravenous or nasal desmopressin acetate or factor VIII concentrate.
- Intravenous infusion of plasma-derived or recombinant factor VIII for bleeding episodes within an hour of noticing symptoms:
- Dosing is weight based and target levels and duration of treatment vary by the severity of bleeding and/or the risk associated with the surgery or procedure.
- Staff members who are expert in performing venipunctures in infants and toddlers should be identified, as frequent venipunctures may be necessary.
- Parents of children age two to five years with severe hemophilia A should be trained to administer the infusions. Home treatment allows for prompt treatment and facilitates prophylactic therapy.
- Pediatric issues. Special considerations for care of infants and children with hemophilia A include the following 82:
- Infant males with a family history of hemophilia A should not be circumcised unless hemophilia A is either excluded or, if present, is treated with factor VIII concentrate directly before and after the procedure.
- Immunizations should be administered preferably subcutaneously; intramuscular injections should be avoided unless under factor coverage.
- Effective dosing of factor VIII requires an understanding of different pharmacokinetics in young children.
- DDAVP® (desmopressin acetate). For individuals with mild hemophilia A, including symptomatic females, immediate treatment of bleeding can be achieved with DDAVP®. A single intravenous dose often doubles or triples factor VIII clotting activity. Alternatively, a multi-use nasal formulation of DDAVP® Nasal may be more convenient.Note: Hemophilia genotype influences DDAVP® response 83.
- Immune tolerance therapy. Alloimmune inhibitors to factor VIII greatly compromise the ability to manage bleeding episodes 21. High titer inhibitors can often be eliminated by immune tolerance therapy 84. Individuals with large gene deletions are less likely to respond to immune tolerance than individuals with other types of variants 85.
- Prevention of primary manifestations: For those with severe disease, prophylactic infusions of factor VIII concentrate three times a week or every other day to maintain factor VIII clotting activity higher than 1% nearly eliminates spontaneous bleeding and prevents chronic joint disease. Newer modified recombinant products with longer half-lives allow less frequent infusions.
- Prophylactic treatment is recommended by the National Bleeding Disorders Foundation and the World Federation of Hemophilia for children with severe hemophilia and is usually administered as infusions of factor VIII concentrate three times a week or every other day to maintain factor VIII clotting activity above 1%, although a less intense regimen may provide protection for some affected boys 86. Newer modified recombinant products with longer half-lives allow less frequent infusions.
- Factor VIII concentrate infusions given prophylactically in young boys before or just after their first few joint bleeds can nearly eliminate spontaneous bleeding and prevent chronic joint disease 87.
- The greatest benefit is seen in affected individuals who start therapy before age 2.5 to three years. Routine prophylaxis begun later in childhood or in adults significantly decreases bleeding episodes 88.
- “Secondary” prophylaxis, started after some joint damage has occurred, can be given on a long-term basis or around periods of increased activity or surgical procedures. An increasing number of adults with hemophilia are on long-term prophylaxis and clinical benefit is being documented in short- and long-term studies.
- Prevention of secondary complications: Reduction of bleeding and chronic joint disease is achieved by prophylactic treatment and prompt effective treatment of bleeding, including by use of home therapy. Many recombinant products are now created without human- or animal-derived proteins in the process or final product. Virucidal treatment of plasma-derived concentrates has eliminated the risk of HIV transmission since 1985, and of hepatitis B and C viruses since 1990.
- Surveillance: Persons with hemophilia who are followed at hemophilia treatment centers have lower mortality than those who are not 89. Young children with severe or moderate hemophilia A should be evaluated at a hemophilia treatment center (accompanied by their parents) every six to 12 months to review their history of bleeding episodes and adjust treatment plans as needed. Early signs and symptoms of possible bleeding episodes are reviewed. The assessment should also include a joint and muscle evaluation, an inhibitor screen, and a discussion of any other problems related to the individual’s hemophilia and family and community support.
- For individuals with severe or moderate hemophilia A, assessments including inhibitor screen every six to 12 months at a hemophilia treatment center are recommended; for individuals with mild hemophilia A, assessment at a hemophilia treatment center every one to two years. Comorbidities may require more frequent visits.
- Screening for alloimmune inhibitors is indicated at least once during the first ten to 20 treatment days in children with severe hemophilia, and then every three to six months after treatment with factor VIII concentrates has been initiated either for bleeding or prophylaxis. After 50 to 100 exposure days, annual screening and screening prior to elective surgical procedures is sufficient. Testing for inhibitors should be performed in any individual with hemophilia whenever a suboptimal clinical response to treatment is suspected, regardless of disease severity.
- Older children and adults with severe or moderate hemophilia A benefit from at least yearly contact with a hemophilia treatment center and periodic assessments to review bleeding episodes and treatment plans, evaluate joints and muscles, screen for an inhibitor, perform viral testing if indicated, provide education, and discuss other issues relevant to the individual’s hemophilia.
- Individuals with mild hemophilia A can benefit from an assessment at a hemophilia treatment center every one to two years. Affected individuals with comorbidities and other complications or treatment challenges may require more frequent visits.
- Agents/circumstances to avoid: Circumcision of at-risk males until hemophilia A is either excluded or treated with factor VIII concentrate regardless of severity; intramuscular injections; activities with a high risk of trauma, particularly head injury; cautious, if any, use of medications and herbal remedies that affect platelet function, including aspirin. Medications and herbal remedies that affect platelet function, including aspirin, should be avoided unless there is strong medical indication, such as in individuals with atherosclerotic cardiovascular disease. Individuals with severe hemophilia usually require clotting factor prophylaxis to allow aspirin and other platelet inhibitory drugs to be used safely 90.
- Avoid the following:
- Intramuscular injections
- Activities that involve a high risk of trauma, particularly of head injury.
- Evaluation of relatives at risk: To clarify genetic status of females at risk before pregnancy or early in pregnancy and to facilitate management.
- Identification of at-risk relatives. A thorough family history may identify other relatives who are at risk but have not been tested (particularly in families with mild hemophilia A).
- Early determination of the genetic status of males at risk. Either assay of factor VIII clotting activity from a cord blood sample obtained by venipuncture of the umbilical vein (to avoid contamination by amniotic fluid or placenta tissue) or molecular genetic testing for the family-specific F8 pathogenic variant can establish or exclude the diagnosis of hemophilia A in newborn males at risk. Infants with a family history of hemophilia A should not be circumcised unless hemophilia A is either excluded or, if present, factor VIII concentrate is administered immediately before and after the procedure to prevent delayed oozing and poor wound healing.Note: Ideally, the cord blood for factor VIII clotting activity assay should be drawn into a syringe containing one-tenth volume of sodium citrate to avoid clotting and to provide an optimal mixing of the sample with the anticoagulant. If not available a standard blue top tube can be used.
- Determination of genetic status of females at risk. Approximately 30% of heterozygous females have factor VIII activity lower than 40% and may have abnormal bleeding. In a survey of Dutch heterozygous females, bleeding symptoms correlated with baseline factor clotting activity; there was suggestion of a very mild increase in bleeding even in those with 40% to 60% factor VIII activity [Plug et al 2006]. Therefore, all daughters and mothers of an affected male and other at-risk females should have a baseline factor VIII clotting activity assay to determine if they are at increased risk for bleeding (unless they are known to be non-carriers based on molecular genetic testing). Very occasionally, a female will have particularly low factor VIII clotting activity that may result from heterozygosity for an F8 pathogenic variant associated with skewed X-chromosome inactivation or, on rare occasion, compound heterozygosity for two F8 pathogenic variants 91.It is recommended that the carrier status of a female at risk be established prior to pregnancy or as early in a pregnancy as possible.
Hemophilia A pregnancy management
Hemophilia A pregnancy management requires a multidisciplinary team approach, including preconception counseling, careful planning for delivery at a specialized center, and postpartum care to address the risks of bleeding for both the mother and the baby 92, 93, 94, 95, 96, 97. A key component is the management of factor VIII levels, which can be inconsistent during pregnancy, necessitating regular monitoring and planned factor replacement therapy, especially around delivery. Genetic counseling is crucial to determine fetal status, especially for male fetuses, and invasive procedures must be carefully considered and managed by experts.
Preconception and prenatal planning
- Genetic counseling: Discuss family planning with a hemophilia specialist and a genetic counselor to understand potential risks for the fetus. It is recommended that the carrier status of a female at risk be established prior to pregnancy or as early in a pregnancy as possible. If the female is symptomatic (i.e., has baseline factor VIII clotting activity <40%), she will be somewhat protected by the natural rise of factor VIII clotting activity during pregnancy, which may even double by the end of the third trimester. The factor VIII level should be measured in the third trimester to confirm that the level is in the normal range, and if it is not, a plan for factor replacement therapy should be developed. Postpartum factor VIII clotting activity can return to baseline within 48 hours, and postpartum hemorrhage may ensue 98.
- Multidisciplinary team: Assemble a team including a hematologist, an obstetrician, a pediatrician, and an anesthesiologist early in the pregnancy.
- Fetal diagnosis: If the fetus is male, prenatal diagnostic tests like chorionic villus sampling or amniocentesis may be offered to confirm hemophilia, but only after specific mutations have been identified in the family.
- Factor monitoring: Check factor VIII levels early in pregnancy and again between 28 and 34 weeks to plan for delivery, as levels can rise but are often inconsistent.
- Delivery plan: Create a detailed, written plan for delivery that outlines obstetric care, the planned mode of delivery, and immediate newborn care. This plan should be shared with the entire care team.
During labor and delivery
- Delivery mode: A spontaneous vaginal delivery is usually preferred unless there are obstetric complications. Controversy remains as to indications for cesarean section versus vaginal delivery 99. In retrospective data analysis of 580 males age 0-2 years with hemophilia, 17 suffered intracranial hemorrhages with delivery, and all but one were delivered vaginally 48. This finding supports the recommendation of cesarean section for hemophilic infants, however, 12 of the 17 were born to women not known to be carriers, suggesting that a planned delivery may mitigate risks. In anticipation of delivery, the relative risks of cesarean section versus vaginal delivery should be considered and discussed with the family and obstetrician in anticipation of delivery so that a coordinated plan can be developed.
- Anesthesia: Epidural or spinal anesthesia should only be considered in close consultation with the hematology and anesthesiology teams.
- Factor replacement: The mother may need factor VIII infusion just before delivery to ensure adequate levels, depending on the plan established by the multidisciplinary team.
- Avoidance: Avoid invasive procedures like fetal scalp electrodes or operative vaginal delivery if the fetus is potentially severely affected.
Postpartum care
- Maternal care: Continue factor replacement therapy to maintain normal levels for several days after delivery (up to 7 days for a C-section).
- Monitoring: Closely monitor factor levels for up to 21 days postpartum, as they can drop significantly as they return to baseline.
- Postpartum hemorrhage: Active management of the third stage of labor is recommended to prevent postpartum hemorrhage.
- Fetal care: The newborn requires specific care based on fetal status, including a thorough evaluation by a pediatric hematologist.
Table 6. Products available to treat people with hemophilia A
| Product | Proper name | Manufacturer | Indication | Route of administration | Strengths (International Units (IU)) | Storage |
|---|---|---|---|---|---|---|
| Human plasma-derived concentrates that contain factor VIII and von Willebrand factor to treat hemophilia A | ||||||
| Alphanate | Antihemophilic factor/von Willebrand factor complex (human) | Grifols Biologicals Inc | Control and prevention of bleeding in patients with hemophilia A or acquired FVIII deficiency. Surgical and/or invasive procedures in adult and pediatric patients with von Willebrand disease in whom desmopressin (DDVAP) is either ineffective or contraindicated. It is not indicated for patients with severe von Willebrand disease (type 3) undergoing major surgery. | Intravenous, 5 or 10 mL of sterile water for injection | 250, 500, 1000, and 1500 International Units (IU) factor VIII in a single-use vial | 2 and 8 °C or may be stored at room temperature not to exceed 30 °C for up to 2 months. |
| Koate-DVI | Antihemophilic factor (human) | Kedrion Biopharma | Control and prevention of bleeding episodes or in order to perform emergency and elective surgery in patients with hemophilia A (hereditary factor VIII deficiency). | Intravenous, 5 or 10 mL of sterile water for injection | 250, 500, and 1000 International Units (IU) factor VIII in a single-use vial | 2 and 8 °C or may be stored at room temperature not to exceed 30 °C for up to 6 months. |
| Humate P | Antihemophilic factor/von Willebrand factor complex (human) | CSL Behring GmbH | Hemophilia A–treatment and prevention of bleeding in adults. von Willebrand disease–in adults and pediatric patients in the (1) treatment of spontaneous and trauma-induced bleeding episodes and (2) prevention of excessive bleeding during and after surgery. | Intravenous, 5, 10, or 15 mL of sterile water for injection | 600 International Units (IU) von Willebrand Factor Ristocetin Cofactor and 250 International Units (IU) factor VIII; 1200 International Units (IU) von Willebrand Factor Ristocetin Cofactor and 500 International Units (IU) factor VIII 2400 International Units (IU) von Willebrand Factor Ristocetin Cofactor and 1000 International Units (IU) factor VIII per vial | Stored at temperatures up to 25 °C (77 °F) for 36 months up to the expiry date. |
| Human plasma-derived immunoaffinity-purified factor VIII concentrates to treat hemophilia A | ||||||
| Hemofil M | Antihemophilic factor human, method M, monoclonal purified | Takeda Pharmaceuticals U.S.A., Inc | For the prevention and control of hemorrhagic episodes in hemophilia A. | Intravenous, 10 mL of sterile water for injection | 250, 500, 1000, and 1700 International Units (IU) vials | 2 to 8 °C (36-46 °F) or at room temperature, not to exceed 30 °C (86 °F) until the expiry date. |
| standard half-life recombinant products to treat hemophilia A | ||||||
| Advate | Antihemophilic factor (recombinant), plasma/albumin-free method | Takeda Pharmaceuticals U.S.A., Inc | Control and prevention of bleeding episodes in adults and children (0-16 y), perioperative management in adults and children (0-16 y), and routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children (0-16 y) | Intravenous, 2 or 5 mL of sterile water for injection | 250, 500, 1000, 1500, 2000, and 3000 International Units (IU) vials | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 6 mo |
| NovoEight | Antihemophilic factor (recombinant) | Novo Nordisk | For adults, adolescents, and children with hemophilia A for the control and prevention of bleeding episodes, perioperative management, and routine prophylaxis to prevent or reduce the frequency of bleeding episodes. | Intravenous, 4 mL prefilled sodium chloride diluent in a syringe | 250, 500, 1000, 1500, 2000, and 3000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 12 mo |
| NUWIQ | Antihemophilic factor (recombinant) | Octapharma USA, Inc | Indicated in adults and children with hemophilia A for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, 2.5 mL prefilled syringe with water for injection | 250, 500, 1000, and 2000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 24 mo |
| Recombinate | Antihemophilic factor (recombinant) | Baxter Healthcare Corporation | Indicated in adults and children with hemophilia A for the prevention and control of hemorrhagic and perioperative management of patients with hemophilia A. | Intravenous, 5 mL of sterile water for injection | 250, 500, 1000, 1500, and 2000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) |
| Xyntha | Antihemophilic factor (recombinant), plasma/albumin free | Wyeth Pharmaceuticals, LLC | Indicated in adults and children with hemophilia A for on-demand treatment and control of bleeding episodes, perioperative management, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, 4 mL prefilled syringe with 0.9% sodium chloride | 250, 500, 1000, or 2000 International Units (IU). | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 3 mo |
| Extended half-life recombinant products to treat hemophilia A | ||||||
| Adynovate | Antihemophilic factor, recombinant, PEGylated | Takeda Pharmaceuticals U.S.A., Inc | On-demand treatment and control of bleeding episodes, perioperative management, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, 2 or 5 mL of sterile water for injection in a BAXJECT III system | 250, 500, 750, 1000, 1500, 2000, or 3000 International Units (IU) vials | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 3 mo |
| Afstyla | Antihemophilic factor (recombinant), single chain | CSL Behring GmbH | On-demand treatment and control of bleeding episodes, routine prophylaxis to reduce the frequency of bleeding episodes, and perioperative management of bleeding. | Intravenous, 2.5 or 5 mL of sterile water for injection | 250, 500, 1000, 1500, 2000, 2500, or 3000 International Units (IU) | 2 to 8 °C and room temperature (up to 25 °C [77 °F]) for a period of up to 3 mo |
| Altuviiio | Antihemophilic factor (recombinant), Fc-VWF-XTEN fusion protein-ehtl | Bioverativ Therapeutics Inc | Routine prophylaxis to reduce the frequency of bleeding episodes, on-demand treatment and control of bleeding episodes, and perioperative management of bleeding. | Intravenous, prefilled syringe with 3 mL of sterile water for injection | 250, 500, 750, 1000, 2000, 3000, or 4000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 6 mo |
| Eloctate | Antihemophilic factor (recombinant), Fc fusion protein | Biogen Idec, Inc | For adults and children with hemophilia A for (1) on-demand treatment and control of bleeding episodes, perioperative management, and routine prophylaxis to prevent or reduce the frequency of bleeding episodes. | Intravenous, prefilled syringe with 3 mL of sterile water for injection | 250, 500, 750, 1000, 1500, 2000, or 3000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 12 mo |
| Esperoct | Antihemophilic factor (recombinant), GlycoPEGylated-exei | Novo Nordisk, Inc | Adults and children with hemophilia A for control and prevention of bleeding, perioperative management, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, MixPro (NovoNordisk) prefilled diluent syringe with 4 mL of 0.9% saline solution | 500, 1000, 1500, 2000, and 3000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 6 mo |
| Jivi | Antihemophilic factor (recombinant), PEGylated-aucl | Bayer Healthcare, Inc | Indicated for use in previously treated adults and adolescents (12 y of age and older) with hemophilia A (congenital FVIII deficiency) for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, prefilled diluent syringe with 2.5 mL of sterile water for injection | 500, 1000, 2000, or 3000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 6 mo |
| Kovaltry | Antihemophilic factor (recombinant), full length | Bayer HealthCare, LLC | For use in adults and children with hemophilia A for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, prefilled syringe with 2.5 or 5 mL of sterile water for injection | 250, 500, 1000, 2000, or 3000 International Units (IU) | 2 to 8 °C and room temperature (up to 30 °C [86 °F]) for a period of up to 12 mo |
| Recombinant humanized bispecific activated factor IX (FIXa) and factor X-directed monoclonal antibody to treat hemophilia A | ||||||
| Hemlibra | emicizumab-kxwh | Genentech, Inc | Routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients ages newborn and older with hemophilia A with or without factor VIII inhibitors. | Subcutaneous injection | 12 (0.4), 30 (1.0), 60 (0.4), 105 (0.7), 150 (1.0), and 300 (2.0 mL) mg per vial | 36 to 46 °F (2-8 °C) |
| Gene therapy products to treat hemophilia A | ||||||
| Roctavian | valoctocogene roxaparvovec-rvox | BioMarin Pharmaceutical Inc | Treatment of adults with severe hemophilia A (congenital factor VIII deficiency with factor VIII activity < 1 International Units (IU)/dL) without preexisting antibodies to adeno-associated virus serotype 5 detected by an US Food and Drug Administration (FDA)-approved test. | Intravenous injection of 6 × 10e13 vector genomes per kg of body weight | 2 × 10e13 ector genomes per mL, each vial contains an extractable volume of not less than 8 mL (16 × 10e13 vector genomes). | Frozen at ≤−60 °C (−76 °F) |
Hemophilia A prognosis
With treatment, most people with hemophilia A are able to lead a fairly normal life with a normal lifespan. Treatments currently available allow people with hemophilia born today to live a normal lifespan. If you have hemophilia A, you should have regular checkups with a hematologist. Key factors for a positive prognosis for people with hemophilia A include regular checkups, access to treatment centers, proper management, and a healthy lifestyle. Receiving regular, adequate treatment is essential for a good prognosis. Without it, life expectancy can be significantly reduced.
A positive hemophilia A prognosis also depends on a proactive approach to health, including:
- Regular checkups with a hematologist.
- Participating in activities and exercise that are safe for people with bleeding disorders.
- Managing pain and maintaining a healthy weight to reduce joint strain.
- Having vaccinations such as for hepatitis B, particularly if receiving donated clotting factors.
Potential complications of hemophilia A:
- Inhibitors: Some individuals may develop inhibitors, which are antibodies that make clotting factor treatments less effective. These can often be managed with specific treatments, but may lead to ongoing problems.
- Joint damage: Repeated bleeds can lead to joint damage over time.
- Serious bleeds: Despite treatment, serious bleeding into the brain can still occur in some severe cases, and it is important to be aware of the symptoms and seek immediate medical attention if they appear.
- Valentino LA, Santaella ME, Carlson SA, Recht M. Contemporary approaches to treat people with hemophilia: what’s new and what’s not? Res Pract Thromb Haemost. 2025 Jan 31;9(1):102696. doi: 10.1016/j.rpth.2025.102696[↩][↩]
- Salen P, Babiker HM. Hemophilia A. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470265[↩][↩][↩][↩][↩][↩][↩]
- Konkle BA, Nakaya Fletcher S. Hemophilia A. 2000 Sep 21 [Updated 2025 Aug 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1404[↩][↩][↩][↩][↩][↩][↩]
- F8 gene. https://medlineplus.gov/genetics/gene/f8[↩][↩]
- VWF gene. https://medlineplus.gov/genetics/gene/vwf[↩][↩]
- López Fernández MF. Limitations of prophylactic treatment in patients with hemophilia. Blood Coagul Fibrinolysis. 2019 Sep;30(1S Suppl 1):S22-S24. doi: 10.1097/MBC.0000000000000825[↩]
- Mahlangu J. Emicizumab for the prevention of bleeds in hemophilia A. Expert Opin Biol Ther. 2019 Aug;19(8):753-761. doi: 10.1080/14712598.2019.1626370[↩]
- Oldenburg J, Hay CRM, Jiménez-Yuste V, Peyvandi F, Schved JF, Szamosi J, Winding B, Lethagen S. Design of a prospective observational study on the effectiveness and real-world usage of recombinant factor VIII Fc (rFVIIIFc) compared with conventional products in haemophilia A: the A-SURE study. BMJ Open. 2019 May 30;9(5):e028012. doi: 10.1136/bmjopen-2018-028012[↩]
- O’Hara J, Hughes D, Camp C, Burke T, Carroll L, Diego DG. The cost of severe haemophilia in Europe: the CHESS study. Orphanet J Rare Dis. 2017 May 31;12(1):106. doi: 10.1186/s13023-017-0660-y[↩][↩]
- O’Hara J, Walsh S, Camp C, Mazza G, Carroll L, Hoxer C, Wilkinson L. The relationship between target joints and direct resource use in severe haemophilia. Health Econ Rev. 2018 Jan 16;8(1):1. doi: 10.1186/s13561-018-0185-7[↩][↩][↩]
- Manco-Johnson MJ, Abshire TC, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007 Aug 9;357(6):535-44. doi: 10.1056/NEJMoa067659[↩]
- Ulrich-Merzenich G, Hausen A, Zeitler H, Goldmann G, Oldenburg J, Pavlova A. The role of variant alleles of the mannose-binding lectin in the inhibitor development in severe hemophilia A. Thromb Res. 2019 Jul;179:140-146. doi: 10.1016/j.thromres.2019.05.005[↩][↩][↩]
- Tegenge MA, Mahmood I, Forshee R. Clinical Pharmacology Review of Plasma-derived and Recombinant Protein Products: CBER Experience and Perspectives on Model-Informed Drug Development. Haemophilia. 2019 Jul;25(4):e240-e246. doi: 10.1111/hae.13767[↩][↩][↩]
- Miesbach W, O’Mahony B, Key NS, Makris M. How to discuss gene therapy for haemophilia? A patient and physician perspective. Haemophilia. 2019 Jul;25(4):545-557. doi: 10.1111/hae.13769[↩][↩][↩]
- Schep SJ, Boes M, Schutgens REG, van Vulpen LFD. An update on the ‘danger theory’ in inhibitor development in hemophilia A. Expert Rev Hematol. 2019 May;12(5):335-344. doi: 10.1080/17474086.2019.1604213[↩][↩][↩]
- van Galen KPM, d’Oiron R, James P, Abdul-Kadir R, Kouides PA, Kulkarni R, Mahlangu JN, Othman M, Peyvandi F, Rotellini D, Winikoff R, Sidonio RF. A new hemophilia carrier nomenclature to define hemophilia in women and girls: Communication from the SSC of the ISTH. J Thromb Haemost. 2021 Aug;19(8):1883-1887. doi: 10.1111/jth.15397[↩]
- Soucie JM, Miller CH, Dupervil B, Le B, Buckner TW. Occurrence rates of haemophilia among males in the United States based on surveillance conducted in specialized haemophilia treatment centres. Haemophilia. 2020; 26: 487–493. https://doi.org/10.1111/hae.13998[↩]
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, Ludlam CA, Mahlangu JN, Mulder K, Poon MC, Street A. Guidelines for the management of hemophilia. World Federation of Hemophilia. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2516.2012.02909.x/pdf[↩]
- Iorio A, Stonebraker JS, Chambost H, Makris M, Coffin D, Herr C, Germini F; Data and Demographics Committee of the World Federation of Hemophilia. Establishing the Prevalence and Prevalence at Birth of Hemophilia in Males: A Meta-analytic Approach Using National Registries. Ann Intern Med. 2019 Oct 15;171(8):540-546. doi: 10.7326/M19-1208[↩][↩]
- World Federation of Hemophilia 2025. World Federation of Hemophilia Report on the annual global survey. https://www1.wfh.org/publications/files/pdf-2588.pdf[↩]
- Fogarty PF, Kessler CM. Hemophilia A and B. In: Kitchens CS, Kessler CM, Konkle BA, eds. Consultative Hemostasis and Thrombosis. 3 ed. Philadelphia, PA: Elsevier Saunders; 2013:45-59.[↩][↩]
- Activated partial thromboplastin time. https://pathologytestsexplained.org.au/ptests-pro.php?q=Activated+partial+thromboplastin+time[↩]
- Samuelson Bannow B, Recht M, Négrier C, Hermans C, Berntorp E, Eichler H, Mancuso ME, Klamroth R, O’Hara J, Santagostino E, Matsushita T, Kessler C. Factor VIII: Long-established role in haemophilia A and emerging evidence beyond haemostasis. Blood Rev. 2019 May;35:43-50. doi: 10.1016/j.blre.2019.03.002[↩]
- Top O, Geisen U, Decker EL, Reski R. Critical Evaluation of Strategies for the Production of Blood Coagulation Factors in Plant-Based Systems. Front Plant Sci. 2019 Mar 7;10:261. doi: 10.3389/fpls.2019.00261[↩]
- Preijers T, Schütte LM, Kruip MJHA, Cnossen MH, Leebeek FWG, van Hest RM, Mathôt RAA. Strategies for Individualized Dosing of Clotting Factor Concentrates and Desmopressin in Hemophilia A and B. Ther Drug Monit. 2019 Apr;41(2):192-212. doi: 10.1097/FTD.0000000000000625[↩]
- Lasne D, Pouplard C, Nougier C, et al. Groupe d’études de la biologie des maladies hémorragiques du Groupe français d’études de l’hémostase et la thrombose. Dosage de l’activité du facteur VIII chez les patients hémophiles A substitués [Factor VIII assays in treated hemophilia A patients]. Ann Biol Clin (Paris). 2019 Feb 1;77(1):53-65. French. doi: 10.1684/abc.2019.1413[↩]
- Emicizumab for haemophilia A. Aust Prescr. 2019 Feb;42(1):42. doi: 10.18773/austprescr.2019.010[↩][↩][↩]
- Meijer P, Verbruggen B. The between-laboratory variation of factor VIII inhibitor testing: the experience of the external quality assessment program of the ECAT foundation. Semin Thromb Hemost. 2009 Nov;35(8):786-93. doi: 10.1055/s-0029-1245111[↩][↩]
- Verbruggen B, van Heerde WL, Laros-van Gorkom BA. Improvements in factor VIII inhibitor detection: From Bethesda to Nijmegen. Semin Thromb Hemost. 2009 Nov;35(8):752-9. doi: 10.1055/s-0029-1245107[↩][↩]
- Ragni MV, Ojeifo O, Feng J, Yan J, Hill KA, Sommer SS, Trucco MN, Brambilla DJ; Hemophilia Inhibitor Study. Risk factors for inhibitor formation in haemophilia: a prevalent case-control study. Haemophilia. 2009 Sep;15(5):1074-82. doi: 10.1111/j.1365-2516.2009.02058.x[↩][↩]
- Eckhardt CL, Menke LA, van Ommen CH, van der Lee JH, Geskus RB, Kamphuisen PW, Peters M, Fijnvandraat K. Intensive peri-operative use of factor VIII and the Arg593–>Cys mutation are risk factors for inhibitor development in mild/moderate hemophilia A. J Thromb Haemost. 2009 Jun;7(6):930-7. doi: 10.1111/j.1538-7836.2009.03357.x[↩]
- Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020: 26(Suppl 6): 1-158. https://doi.org/10.1111/hae.14046[↩][↩][↩][↩][↩][↩][↩]
- World Federation of Hemophilia: GUIDELINES FOR THE MANAGEMENT OF HEMOPHILIA 3rd Edition. https://www1.wfh.org/publications/files/pdf-1863.pdf[↩][↩][↩][↩][↩][↩]
- Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N Engl J Med. 2017 Aug 31;377(9):809-818. doi: 10.1056/NEJMoa1703068[↩][↩][↩]
- Gomis M, Querol F, Gallach JE, González LM, Aznar JA. Exercise and sport in the treatment of haemophilic patients: a systematic review. Haemophilia. 2009 Jan;15(1):43-54. doi: 10.1111/j.1365-2516.2008.01867.x[↩]
- Seuser A, Boehm P, Kurme A, Schumpe G, Kurnik K. Orthopaedic issues in sports for persons with haemophilia. Haemophilia. 2007 Sep;13 Suppl 2:47-52. doi: 10.1111/j.1365-2516.2007.01507.x[↩]
- Philpott J, Houghton K, Luke A. Physical activity recommendations for children with specific chronic health conditions: Juvenile idiopathic arthritis, hemophilia, asthma and cystic fibrosis. Paediatr Child Health. 2010 Apr;15(4):213-25. doi: 10.1093/pch/15.4.213[↩]
- Querol F, Aznar JA, Haya S, Cid A. Orthoses in haemophilia. Haemophilia. 2002 May;8(3):407-12. doi: 10.1046/j.1365-2516.2002.00637.x[↩]
- ADA Council on Access, Prevention and Interprofessional Relations; ADA Council on Scientific Affairs. Using mouthguards to reduce the incidence and severity of sports-related oral injuries. J Am Dent Assoc. 2006 Dec;137(12):1712-20; quiz 1731. doi: 10.14219/jada.archive.2006.0118[↩]
- Iorio A, Fabbriciani G, Marcucci M, Brozzetti M, Filipponi P. Bone mineral density in haemophilia patients. A meta-analysis. Thromb Haemost. 2010 Mar;103(3):596-603. doi: 10.1160/TH09-09-0629[↩]
- Plug I, Mauser-Bunschoten EP, Brocker-Vriends AH, van Amstel HK, van der Bom JG, van Diemen-Homan JE, Willemse J, Rosendaal FR. Bleeding in carriers of hemophilia. Blood. 2006;108:52–6. http://www.bloodjournal.org/content/108/1/52.long[↩][↩]
- Paroskie A, Gailani D, DeBaun MR, Sidonio RF Jr. A cross-sectional study of bleeding phenotype in haemophilia A carriers. Br J Haematol. 2015;170:223–8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4490107/[↩][↩]
- Kaufman RJ, Fay PJ, Popolo L, Ortel TL. Factor V and factor VIII. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, eds. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 6 ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2013:179-96.[↩]
- Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99:168–74. http://www.bloodjournal.org/content/99/1/168.long[↩]
- Nakaya S, Liu ML, Thompson AR. Some factor VIII exon 14 frameshift mutations cause moderately severe haemophilia A. Br J Haematol. 2001;115:977–82. http://onlinelibrary.wiley.com/doi/10.1046/j.1365-2141.2001.03173.x/full[↩]
- Riccardi F, Rivolta GF, Franchini M, Pattacini C, Neri TM, Tagliaferri A. Characterization of a novel mutation in the F8 promoter region associated with mild hemophilia A and resistance to DDAVP therapy. J Thromb Haemost. 2009;7:1234–5. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2009.03468.x/full[↩]
- Alabek M, Mohan R, Raia M. Genetic Counselling for Hemophilia. Treatment of Hemophilia Monograph No. 25. Montreal, QC: World Federation of Hemophilia; 2015. https://www1.wfh.org/publications/files/pdf-1160.pdf[↩]
- Kulkarni R, Soucie JM, Lusher J, Presley R, Shapiro A, Gill J, Manco-Johnson M, Koerper M, Mathew P, Abshire T, Dimichele D, Hoots K, Janco R, Nugent D, Geraghty S, Evatt B., Haemophilia Treatment Center Network Investigators. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from The Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia. 2009;15:1281–90. https://www.ncbi.nlm.nih.gov/pubmed/19637999[↩][↩]
- Konkle BA, Huston H, Nakaya Fletcher S. Hemophilia A. 2000 Sep 21 [Updated 2017 Jun 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1404/[↩]
- Luck JV Jr, Silva M, Rodriguez-Merchan EC, Ghalambor N, Zahiri CA, Finn RS. Hemophilic arthropathy. J Am Acad Orthop Surg. 2004;12:234–45. https://www.ncbi.nlm.nih.gov/pubmed/15473675[↩]
- Chu WM, Ho HE, Wang JD, Chan WC, Liou YS, Ho WC, Hu SY, Tsan YT. Risk of major comorbidities among workers with hemophilia: A 14-year population-based study. Medicine (Baltimore). 2018 Feb;97(6):e9803. doi: 10.1097/MD.0000000000009803[↩]
- Kodra Y, Cavazza M, Schieppati A, De Santis M, Armeni P, Arcieri R, Calizzani G, Fattore G, Manzoli L, Mantovani L, Taruscio D. The social burden and quality of life of patients with haemophilia in Italy. Blood Transfus. 2014 Apr;12 Suppl 3(Suppl 3):s567-75. doi: 10.2450/2014.0042-14s[↩][↩]
- Skinner MW, Chai-Adisaksopha C, Curtis R, Frick N, Nichol M, Noone D, O’Mahony B, Page D, Stonebraker JS, Iorio A. The Patient Reported Outcomes, Burdens and Experiences (PROBE) Project: development and evaluation of a questionnaire assessing patient reported outcomes in people with haemophilia. Pilot Feasibility Stud. 2018 Feb 27;4:58. doi: 10.1186/s40814-018-0253-0[↩]
- Baumann K, Hernandez G, Witkop M, Peltier S, Dunn S, Cutter S, Frick N, Haugstad K, Guelcher C, Frey MJ, Rotellini D, Clark DB, Iyer NN, Cooper DL. Impact of mild to severe hemophilia on engagement in recreational activities by US men, women, and children with hemophilia B: The Bridging Hemophilia B Experiences, Results and Opportunities into Solutions (B-HERO-S) study. Eur J Haematol. 2017 Apr;98 Suppl 86:25-34. doi: 10.1111/ejh.12852[↩]
- Batt K, Boggio L, Neff A, Buckner TW, Wang M, Quon D, Witkop M, Recht M, Kessler C, Iyer NN, Cooper DL. Patient-reported outcomes and joint status across subgroups of US adults with hemophilia with varying characteristics: Results from the Pain, Functional Impairment, and Quality of Life (P-FiQ) study. Eur J Haematol. 2018 Apr;100 Suppl 1:14-24. doi: 10.1111/ejh.13028[↩]
- Kempton CL, Buckner TW, Fridman M, Iyer NN, Cooper DL. Factors associated with pain severity, pain interference, and perception of functional abilities independent of joint status in US adults with hemophilia: Multivariable analysis of the Pain, Functional Impairment, and Quality of Life (P-FiQ) study. Eur J Haematol. 2018 Apr;100 Suppl 1:25-33. doi: 10.1111/ejh.13025[↩]
- Flood E, Pocoski J, Michaels LA, McCoy A, Beusterien K, Sasanè R. Patient-reported experience of bleeding events in haemophilia. Eur J Haematol. 2014 Jun;93 Suppl 75:19-28. doi: 10.1111/ejh.12329[↩]
- Curtis R, Baker J, Riske B, Ullman M, Niu X, Norton K, Lou M, Nichol MB. Young adults with hemophilia in the U.S.: demographics, comorbidities, and health status. Am J Hematol. 2015 Dec;90 Suppl 2:S11-6. doi: 10.1002/ajh.24218[↩]
- Cutter S, Molter D, Dunn S, Hunter S, Peltier S, Haugstad K, Frick N, Holot N, Cooper DL. Impact of mild to severe hemophilia on education and work by US men, women, and caregivers of children with hemophilia B: The Bridging Hemophilia B Experiences, Results and Opportunities into Solutions (B-HERO-S) study. Eur J Haematol. 2017 Apr;98 Suppl 86:18-24. doi: 10.1111/ejh.12851[↩]
- Darby SC, Kan SW, Spooner RJ, Giangrande PL, Hill FG, Hay CR, Lee CA, Ludlam CA, Williams M. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110:815–25. http://www.bloodjournal.org/content/110/3/815.long[↩]
- Hay CR, Palmer B, Chalmers E, Liesner R, Maclean R, Rangarajan S, Williams M, Collins PW. Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood. 2011;117:6367–70. http://www.bloodjournal.org/content/117/23/6367.long[↩]
- Eckhardt CL, van Velzen AS, Peters M, Astermark J, Brons PP, Castaman G, Cnossen MH, Dors N, Escuriola-Ettingshausen C, Hamulyak K, Hart DP, Hay CR, Haya S, van Heerde WL, Hermans C, Holmström M, Jimenez-Yuste V, Keenan RD, Klamroth R, Laros-van Gorkom BA, Leebeek FW, Liesner R, Mäkipernaa A, Male C, Mauser-Bunschoten E, Mazzucconi MG, McRae S, Meijer K, Mitchell M, Morfini M, Nijziel M, Oldenburg J, Peerlinck K, Petrini P, Platokouki H, Reitter-Pfoertner SE, Santagostino E, Schinco P, Smiers FJ, Siegmund B, Tagliaferri A, Yee TT, Kamphuisen PW, van der Bom JG, Fijnvandraat K, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122:1954–62. http://www.bloodjournal.org/content/122/11/1954.long[↩]
- Schwarz J, Astermark J, Menius ED, Carrington M, Donfield SM, Gomperts ED, Nelson GW, Oldenburg J, Pavlova A, Shapiro AD, Winkler CA, Berntorp E, et al. F8 haplotype and inhibitor risk: results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Haemophilia. 2013;19:113–8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3521089/[↩]
- Search the Global Treatment Centre Directory. World Federation of Hemophilia https://wfh.org/find-local-support/#HTCs[↩]
- National Bleeding Disorders Foundation https://www.bleeding.org/[↩][↩]
- Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013 Sep;39(6):621-31. doi: 10.1055/s-0033-1353420[↩]
- Menegatti M, Palla R, Boscarino M, Bucciarelli P, Muszbek L, Katona E, Makris M, Peyvandi F; PRO-RBDD study group. Minimal factor XIII activity level to prevent major spontaneous bleeds. J Thromb Haemost. 2017 Sep;15(9):1728-1736. doi: 10.1111/jth.13772[↩]
- Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003 Jul;9(4):418-35. doi: 10.1046/j.1365-2516.2003.00780.x[↩]
- Miller CH. Laboratory testing for factor VIII and IX inhibitors in haemophilia: A review. Haemophilia. 2018 Mar;24(2):186-197. doi: 10.1111/hae.13424[↩]
- Miller CH, Platt SJ, Rice AS, Kelly F, Soucie JM; Hemophilia Inhibitor Research Study Investigators. Validation of Nijmegen-Bethesda assay modifications to allow inhibitor measurement during replacement therapy and facilitate inhibitor surveillance. J Thromb Haemost. 2012 Jun;10(6):1055-61. doi: 10.1111/j.1538-7836.2012.04705.x[↩]
- Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A; Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014 Nov;12(11):1935-9. doi: 10.1111/jth.12672[↩]
- Peyvandi F, Mannucci PM, Garagiola I, et al. A Randomized Trial of Factor VIII and Neutralizing Antibodies in Hemophilia A. N Engl J Med. 2016 May 26;374(21):2054-64. doi: 10.1056/NEJMoa1516437[↩]
- de Moerloose P, Fischer K, Lambert T, Windyga J, Batorova A, Lavigne-Lissalde G, Rocino A, Astermark J, Hermans C. Recommendations for assessment, monitoring and follow-up of patients with haemophilia. Haemophilia. 2012 May;18(3):319-25. doi: 10.1111/j.1365-2516.2011.02671.x[↩][↩]
- Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br J Haematol. 2006 Jun;133(6):591-605. doi: 10.1111/j.1365-2141.2006.06087.x[↩][↩]
- Astermark J, Altisent C, Batorova A, Diniz MJ, Gringeri A, Holme PA, Karafoulidou A, Lopez-Fernández MF, Reipert BM, Rocino A, Schiavoni M, von Depka M, Windyga J, Fijnvandraat K; European Haemophilia Therapy Standardisation Board. Non-genetic risk factors and the development of inhibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010 Sep 1;16(5):747-66. doi: 10.1111/j.1365-2516.2010.02231.x[↩]
- Teitel JM, Carcao M, Lillicrap D, Mulder K, Rivard GE, St-Louis J, Smith F, Walker I, Zourikian N. Orthopaedic surgery in haemophilia patients with inhibitors: a practical guide to haemostatic, surgical and rehabilitative care. Haemophilia. 2009 Jan;15(1):227-39. doi: 10.1111/j.1365-2516.2008.01840.x[↩]
- Kempton CL, Soucie JM, Miller CH, Hooper C, Escobar MA, Cohen AJ, Key NS, Thompson AR, Abshire TC. In non-severe hemophilia A the risk of inhibitor after intensive factor treatment is greater in older patients: a case-control study. J Thromb Haemost. 2010 Oct;8(10):2224-31. doi: 10.1111/j.1538-7836.2010.04013.x[↩]
- Berntorp E, Collins P, D’Oiron R, Ewing N, Gringeri A, Négrier C, Young G. Identifying non-responsive bleeding episodes in patients with haemophilia and inhibitors: a consensus definition. Haemophilia. 2011 Jan;17(1):e202-10. doi: 10.1111/j.1365-2516.2010.02377.x[↩]
- McMillan CW, Shapiro SS, Whitehurst D, Hoyer LW, Rao AV, Lazerson J. The natural history of factor VIII:C inhibitors in patients with hemophilia A: a national cooperative study. II. Observations on the initial development of factor VIII:C inhibitors. Blood. 1988 Feb;71(2):344-8. https://doi.org/10.1182/blood.V71.2.344.344[↩]
- Sharathkumar A, Lillicrap D, Blanchette VS, Kern M, Leggo J, Stain AM, Brooker L, Carcao MD. Intensive exposure to factor VIII is a risk factor for inhibitor development in mild hemophilia A. J Thromb Haemost. 2003 Jun;1(6):1228-36. doi: 10.1046/j.1538-7836.2003.00230.x[↩]
- Search the Global Treatment Centre Directory. World Federation of Haemophilia https://wfh.org/find-local-support/#HTCs[↩]
- Chalmers E, Williams M, Brennand J, Liesner R, Collins P, Richards M., Paediatric Working Party of the United Kingdom Haemophilia Doctors’ Organization. Guideline on the management of haemophilia in the fetus and neonate. Br J Haematol. 2011;154:208–15. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2010.08545.x/full[↩]
- Nance D, Fletcher SN, Bolgiano DC, Thompson AR, Josephson NC, Konkle BA. Factor VIII mutation and desmopressin-responsiveness in 62 patients with mild hemophilia A. Haemophilia. 2013;19:720–6. https://www.ncbi.nlm.nih.gov/pubmed/23711294[↩]
- Hay CR, DiMichele DM. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119:1335–44. http://www.bloodjournal.org/content/119/6/1335.long[↩]
- Coppola A, Margaglione M, Santagostino E, Rocino A, Grandone E, Mannucci PM, Di Minno G., AICE PROFIT Study Group. Factor VIII gene (F8) mutations as predictors of outcome in immune tolerance induction of hemophilia A patients with high-responding inhibitors. J Thromb Haemost. 2009;7:1809–15. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2009.03615.x/full[↩]
- Feldman BM, Pai M, Rivard GE, Israels S, Poon MC, Demers C, Robinson S, Luke KH, Wu JK, Gill K, Lillicrap D, Babyn P, McLimont M, Blanchette VS. Tailored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J Thromb Haemost. 2006;4:1228–36. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2006.01953.x/full[↩]
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. http://www.nejm.org/doi/full/10.1056/NEJMoa067659[↩]
- Manco-Johnson MJ, Kempton CL, Reding MT, Lissitchkov T, Goranov S, Gercheva L, Rusen L, Ghinea M, Uscatescu V, Rescia V, Hong W. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART). J Thromb Haemost. 2013;11:1119–27. http://onlinelibrary.wiley.com/doi/10.1111/jth.12202/full[↩]
- Pai M, Key NS, Skinner M, Curtis R, Feinstein M, Kessler C, Lane SJ, Makris M, Riker E, Santesso N, Soucie JM, Yeung CHT, Iorio A, Schünemann HJ. NHF-McMaster guideline on care models for haemophilia management.[↩]
- Angelini D, Konkle BA, Sood SL. Aging among persons with hemophilia: contemporary concerns. Semin Hematol. 2016;53:35–9. https://www.ncbi.nlm.nih.gov/pubmed/26805905[↩]
- Pavlova A, Brondke H, Müsebeck J, Pollmann H, Srivastava A, Oldenburg J. Molecular mechanisms underlying hemophilia A phenotype in seven females. J Thromb Haemost. 2009;7:976–82. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2009.03346.x/full[↩]
- Leebeek FWG, Duvekot J, Kruip MJHA. How I manage pregnancy in carriers of hemophilia and patients with von Willebrand disease. Blood. 2020 Nov 5;136(19):2143-2150. doi: 10.1182/blood.2019000964[↩]
- Dunkley SM, Russell SJ, Rowell JA, Barnes CD, Baker RI, Sarson MI, Street AM; Australian Haemophilia Centre Directors’ Organisation. A consensus statement on the management of pregnancy and delivery in women who are carriers of or have bleeding disorders. Med J Aust. 2009 Oct 19;191(8):460-3. doi: 10.5694/j.1326-5377.2009.tb02887.x[↩]
- Moorehead PC, Chan AKC, Lemyre B, Winikoff R, Scott H, Hawes SA, Shroff M, Thomas A, Price VE. A Practical Guide to the Management of the Fetus and Newborn With Hemophilia. Clin Appl Thromb Hemost. 2018 Dec;24(9_suppl):29S-41S. doi: 10.1177/1076029618807583[↩]
- How do I prepare for pregnancy? https://www.haemophilia.org.au/bleeding-disorders/women-with-bleeding-disorders/carrying-the-haemophilia-gene/pregnancy/[↩]
- James AH, Pacheco LD, Konkle BA. Management of pregnant women who have bleeding disorders. Hematology Am Soc Hematol Educ Program. 2023 Dec 8;2023(1):229-236. doi: 10.1182/hematology.2023000475[↩]
- Rodrigues-Martins D, Buchner G, Braga JS. Symptomatic haemophilia A: management during pregnancy and the postpartum period. BMJ Case Rep. 2022 Jan 7;15(1):e245474. doi: 10.1136/bcr-2021-245474[↩]
- Lee CA, Chi C, Pavord SR, Bolton-Maggs PH, Pollard D, Hinchcliffe-Wood A, Kadir RA. The obstetric and gynaecological management of women with inherited bleeding disorders–review with guidelines produced by a taskforce of UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2006;12:301–36. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2516.2006.01314.x/full[↩]
- James AH, Hoots WK. The optimal mode of delivery for the haemophilia carrier expecting an affected infant is caesarean delivery. Haemophilia. 2010;16:420–4. https://www.ncbi.nlm.nih.gov/pubmed/20028425[↩]
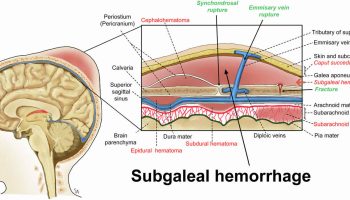
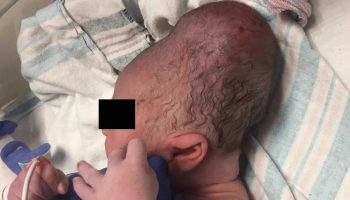

{kind=link}