Contents
What is chordoma
A chordoma is a rare primary bone and soft tissue tumor cancer (a type of sarcoma) that usually occurs along the spine from the base of skull to the tailbone or where the skull sits atop the spine (skull base). Chordoma is part of a group of malignant bone and soft tissue tumors called sarcomas. This type of bone tumor cancer most often occurs at the skull base, spine or bottom of the spine at the sacrum (sacral chordoma). Chordomas grow slowly, gradually extending into the bone and soft tissue around them. Chordomas often recur after treatment, and in about 40 percent of cases the cancer spreads (metastasizes) to other areas of the body, such as the lungs. Chordoma cancer forms from small remnants of a coil of cells in the embryo (cells of the notochord), a structure that is present in the developing embryo and is important for the development of the center of spinal disks of the spinal column. The notochord usually disappears before birth, though a few cells may remain embedded in the bones of the spine or at the base of the skull 1.
Chordomas account for about 3 percent of all bone tumors, about 20 percent of primary spinal tumors and fewer than 1 percent of tumors affecting the brain and spinal cord. They are the most common tumor of the sacrum and cervical spine. A chordoma tumor usually grows slowly, often without symptoms at first, and then might cause symptoms for years before doctors find it.
Approximately 50 percent of all chordomas occur at the base of the spine at the sacrum (sacral chordoma), about 33 percent occur in the base of the skull at the occiput – usually in a bone called the clivus (clival chordoma), and about 20 percent occur in the cervical (neck), thoracic (upper back), or lumbar (lower back) vertebrae of the spine. Very rarely, chordomas can start in more than one place along the spine. Extremely rare cases of chordoma occurring in bones away from the spine have been reported in the ribs, legs, and feet. Skull base chordomas occur more frequently in younger patients, while spinal chordomas are more common later in life. As the chordoma grows, it puts pressure on the adjacent areas of the brain or spinal cord, leading to the signs and symptoms of the disorder. A chordoma anywhere along the spine may cause pain, weakness, or numbness in the back, arms, or legs. A chordoma at the base of the skull (occipital chordoma) may lead to double vision (diplopia) and headaches. A chordoma that occurs in the tailbone (coccygeal chordoma) may result in a lump large enough to be felt through the skin and may cause problems with bladder and bowel function.
Chordomas are rare, occurring in approximately 1 per million individuals each year. That means that about 300 patients are diagnosed with chordoma each year in the United States, and about 700 in all of Europe. At any given time, fewer than one in 100,000 people are living with chordoma. Chordomas typically present in adults between the ages of 40 and 70, although can be seen at any age and can occur anywhere along the spine. About 5 percent of chordomas are diagnosed in children. For reasons that are unclear, males are affected about twice as often as females.
Figure 1. Notochord (embryo)
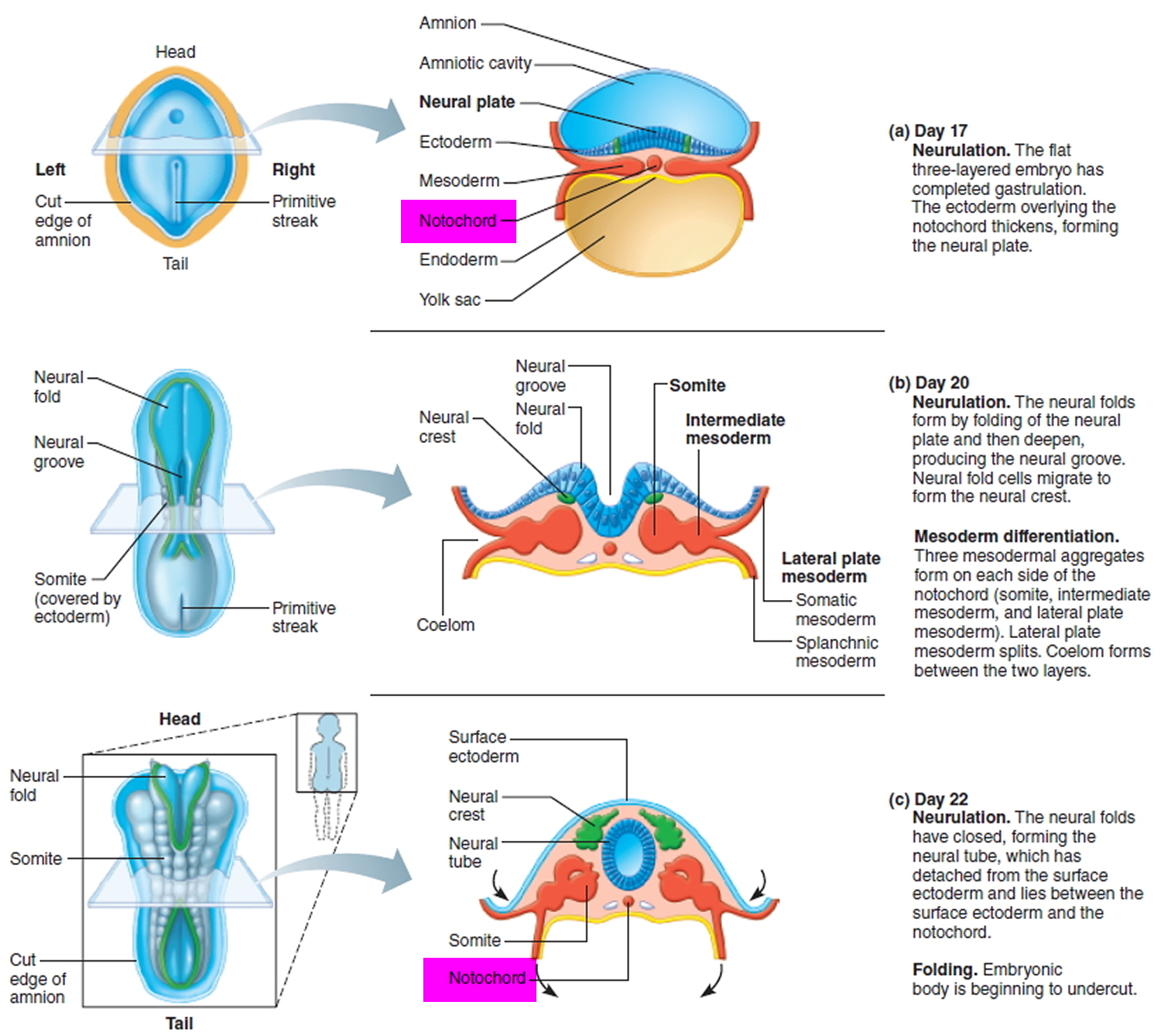
Figure 2. Vertebral column
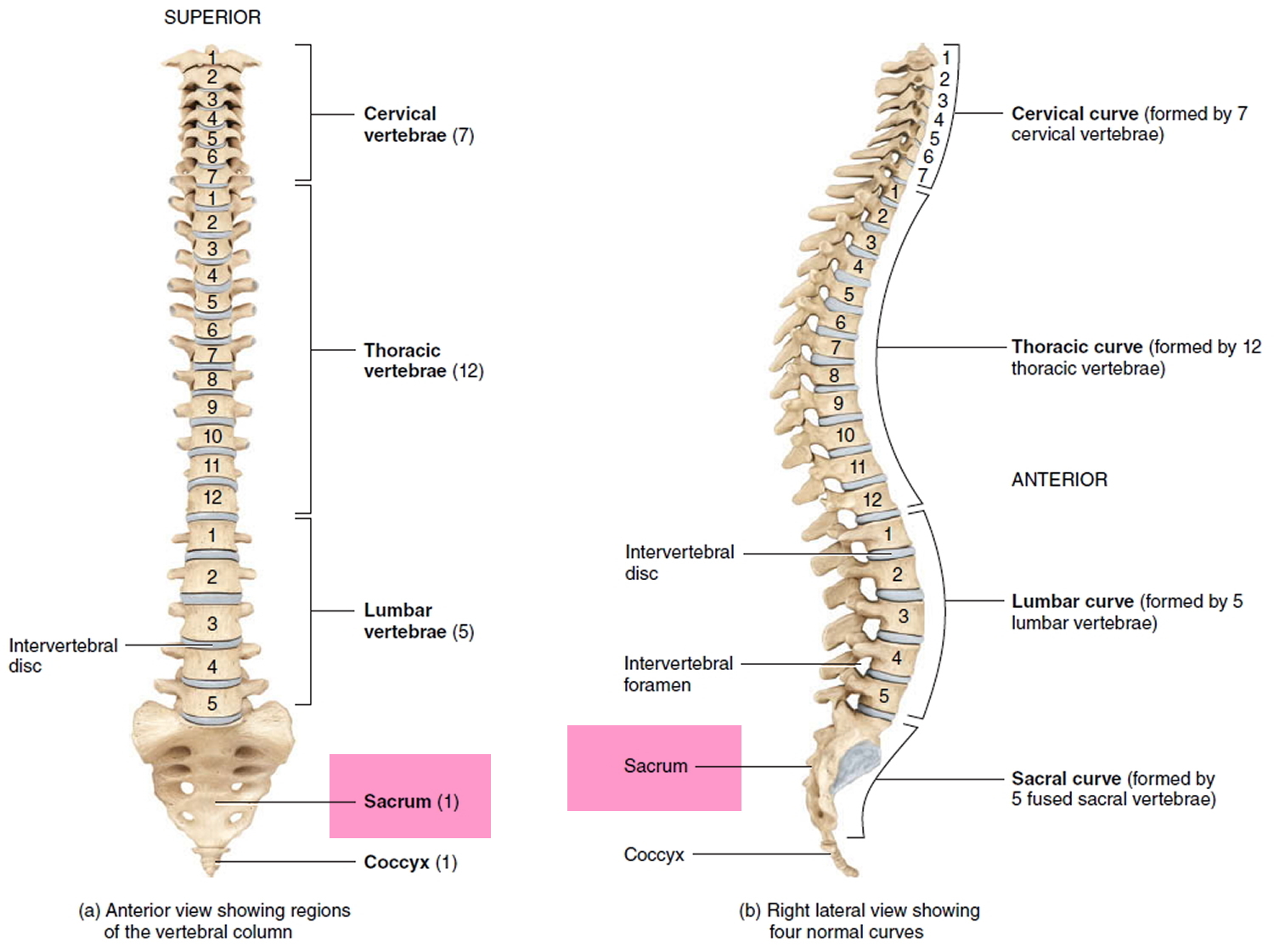
Figure 3. Base of skull (Clivus)
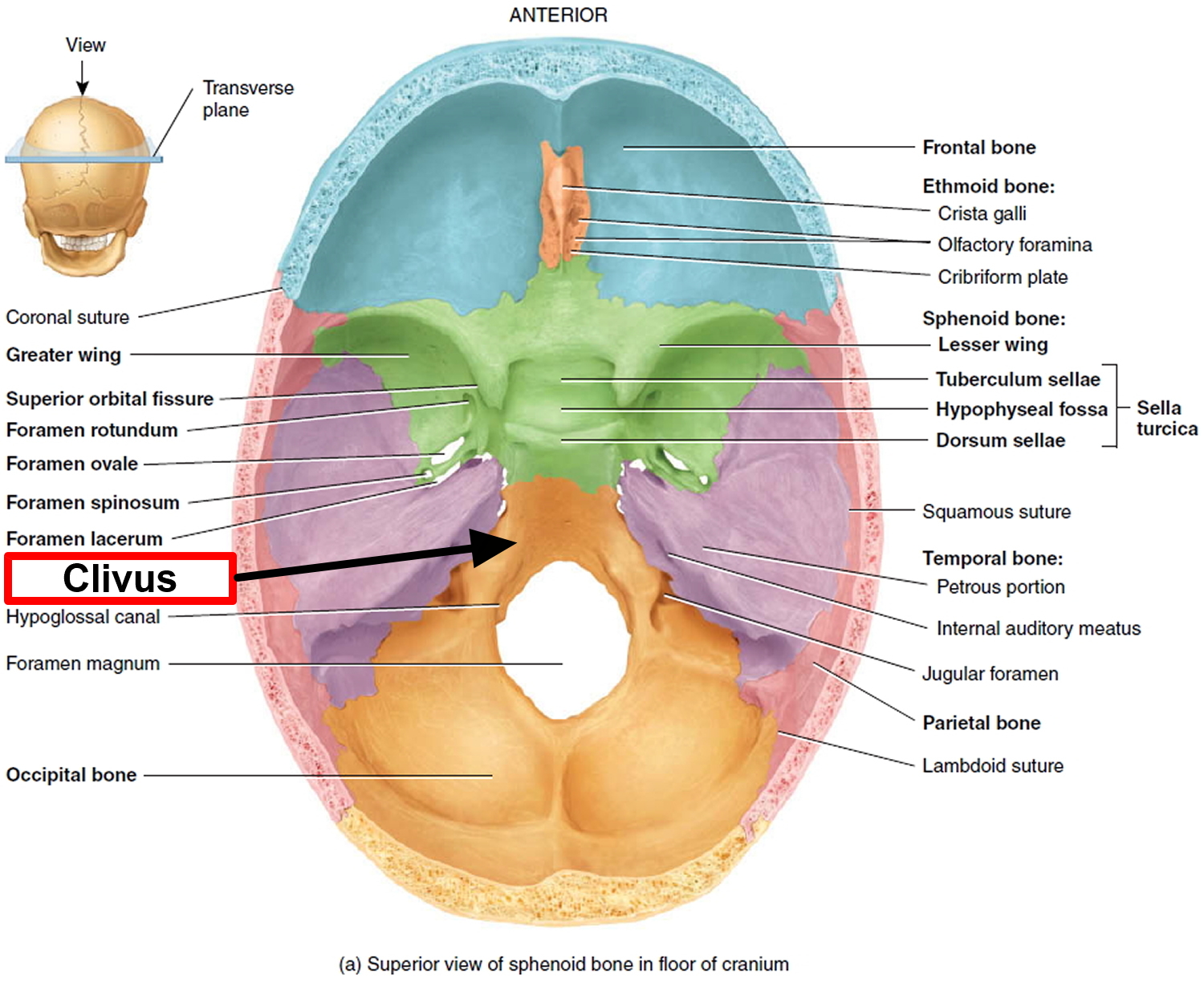
Figure 4. Clival chordoma (base of skull chordoma) – MRI scan
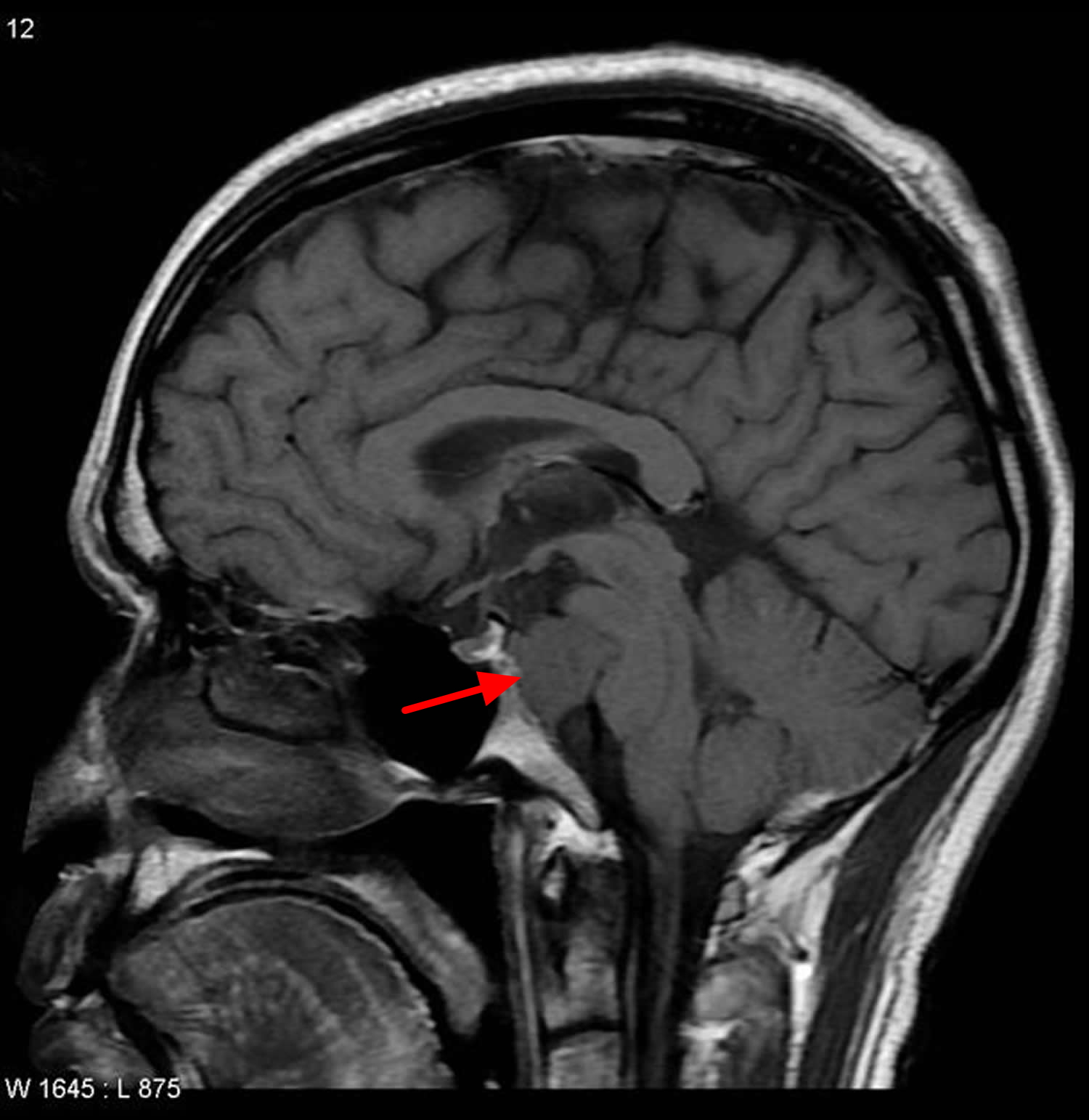
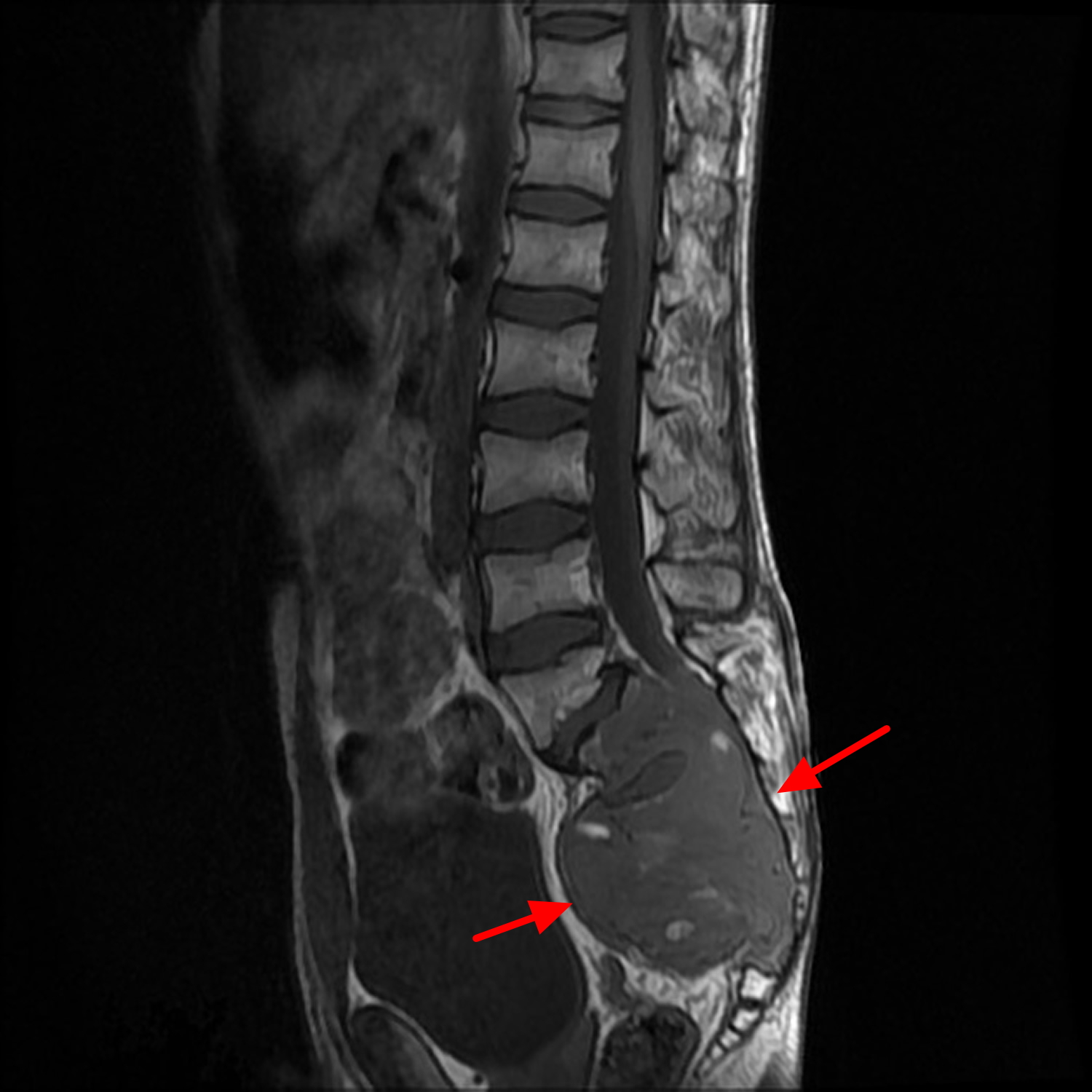
Figure 5. Sacral chordoma (MRI scan)
In many cases, the cause of the chordoma remains unknown. Recent studies have shown that changes in the TBXT gene have been associated with chordomas in a small set of families. In these families an inherited duplication of the TBXT gene is associated with an increased risk of developing a chordoma. Duplications of the TBXT gene have also been identified in people with chordoma who have no history of the tumor in their family, but in these cases the changes occur only in the tumor cells and are not inherited.
Though commonly slow growing, a chordoma is a difficult tumor to treat because it’s near the spinal cord or other critical structures, such as the carotid artery and brain tissue. The current treatment is often the surgical removal of the tumor, followed by radiotherapy 2. Chordomas can also come back, or recur, after treatment — usually in the same place as the first tumor. This is called a local recurrence. In about 30 to 40 percent of patients, the chordoma tumor eventually spreads, or metastasizes, to other parts of the body. If chordomas spread to other parts of the body (metastasize), the most common places they spread to are the lungs, liver, bones, or lymph nodes. Metastasis usually only occurs when the primary chordoma tumor has advanced and is rarely reported at the time of initial diagnosis.
Other names for chordoma
- CHDM
- chordocarcinoma
- chordoepithelioma
- notochordal sarcoma
- notochordoma
Chordoma subtypes
There are four subtypes of chordoma, which are classified based on how they look under a microscope:
- Conventional (or classic) chordoma is the most common form of chordoma. It is composed of a unique cell type that resembles notochordal cells and can have areas of chondroid appearance.
- Poorly differentiated chordoma is a recently identified subtype. It can be more aggressive and faster growing than conventional chordoma, and is characterized by a loss of a gene called INI-1. This type of chordoma is more common in pediatric and young adult patients, and in skull base tumors.
- Dedifferentiated chordoma is more aggressive and generally grows faster than the other types of chordoma, and is more likely to metastasize than conventional chordoma. It can also have a loss of the INI-1 gene. This type of chordoma is rare, occurring in only about 5 percent of patients, and is more common in pediatric patients.
- Chondroid chordoma is a term more commonly used in the past when it was difficult to distinguish conventional chordoma from chondrosarcoma. This is no longer a problem because brachyury is expressed in nearly all conventional chordomas, making them easier to distinguish from cartilaginous tumors like chondrosarcoma that do not express brachyury. There is no evidence that chordomas with a chondroid appearance behave differently than conventional types that do not have this appearance.
Chordoma cancer life expectancy
It is important to remember that the prognosis for each person is unique, and depends on many different factors. These include the patient’s age, type of chordoma, size and location of the tumor, method of treatment, extent of resection, and other factors. Only your doctors can advise about your individual prognosis and risks, and it’s very important that this advice come from doctors who have experience treating chordoma.
The prognosis of chordomas generally depends on the success of the surgery removing the tumor. Although chordomas are usually slow-growing tumors, they are locally aggressive and tend to infiltrate into adjacent tissues and organs and to have multiple local recurrences (return of the tumor). Local recurrence results in tissue destruction and generally is the cause of death. Spreading to distant places of the body (metastases) are recognized but are uncommon. In general, a more complete removal with wide removal delays the time between surgery and eventual recurrence.
One studied showed that about 67% of the patients with chordomas of the base of the skull were alive after 5 years and in 58% of the patients the tumor did not get worse (progression-free survival) compared with the time of the diagnosis; about 57% of the patients were alive after 10 years and, in about 44%, the tumor did not get worse. The prognosis was better if more of the tumor was removed, the patient had radiation therapy, and there was no invasion of the nose and pharynx. A large study confirmed that both 5-year progression-free survival and overall survival of skull chordomas are better when there is a complete resection of the tumor 3.
Knowledge of the completeness of the chordoma tumor resection helps predict patient outcome in terms of the length of the time that the patient will not have any tumor recurrence, and in determination of the need for radiotherapy.
In general, the chordomas may recur after 3.8 years for radically resected tumors, 2.1 years for subtotal resection followed by radiation therapy, and 8 months for subtotal excision without radiation therapy. Due to the high rate of recurrences, frequent follow-ups are required because when the recurrence is identified early it is easier to treat.
Survival among pediatric patients who go through surgery was significantly longer than for adults and overall survival was longer.
Chordoma causes
Chordoma tumors develop from cells of a tissue called the notochord, which is a structure in an embryo that helps in the development of the spine. The notochord disappears when the fetus is about 8 weeks old, but some notochord cells remain behind in the bones of the spine and skull base. Very rarely, these cells turn into cancer called chordoma. What causes notochord cells to become cancerous in some people is still not fully known, but researchers are working to learn the answer.
There are no known environmental, dietary, or lifestyle risk factors for chordoma. The vast majority of chordomas occur at random and not as a direct result of an inherited genetic trait; however, there are several genetic factors associated with chordoma. For example, more than 95 percent of individuals with chordoma have a single-letter variation, called a SNP (“snip”), in the DNA sequence of a gene called brachyury. This SNP causes an increase in the risk of developing chordoma, but does not by itself cause chordoma. In fact, a large fraction of the general population has this SNP, but individuals who have the SNP are still very unlikely to develop chordoma – the chances are less than two in a million.
Familial chordoma
There are a handful of known cases where multiple members of the same family are affected by chordoma. This indicates that in these very rare instances, a strong genetic predisposition for chordoma can be inherited. It is known that some of the families with familial chordoma have an extra copy of the TBXT gene (brachyury gene), but currently, there is no available test for the presence of extra copies of the TBXT gene. The National Cancer Institute is currently conducting a genetics study to identify additional hereditary causes of chordoma.
Changes in the TBXT gene have been associated with chordoma. An inherited duplication of the TBXT gene identified in a few families is associated with an increased risk of developing a chordoma. Duplications or increases in activity (expression) of the TBXT gene have also been identified in people with chordoma who have no history of the disorder in their family. In these individuals, the changes occur only in the tumor cells and are not inherited.
When development of a chordoma is associated with a duplication of the TBXT gene inherited from a parent, one copy of the altered gene in each cell is sufficient to increase the risk of the disorder, which is an inheritance pattern called autosomal dominant. People with this duplication inherit an increased risk of this condition, not the condition itself.
Other cases of chordoma are sporadic, which means they occur in people with no history of the condition in their family.
The TBXT gene provides instructions for making a protein called brachyury. Brachyury is a member of a protein family called T-box proteins, which play critical roles during embryonic development. T-box proteins regulate the activity of other genes by attaching (binding) to specific regions of DNA. On the basis of this action, T-box proteins are called transcription factors.
The brachyury protein is especially important for the early development of the spine. In human embryos, a structure called the notochord is the precursor of the spinal column. The notochord disappears before birth, but in a small percentage of individuals, some of its cells remain in the base of the skull or in the spine. In rare cases these cells begin to grow and divide uncontrollably, invading the nearby bone and soft tissue and resulting in the development of a chordoma.
Duplications and increases in expression of the TBXT gene both result in the production of excess brachyury protein. The specific mechanism by which excess brachyury protein contributes to the development of chordomas is unclear. Some people with chordoma do not have changes in the TBXT gene, and the cause of the disorder in these individuals is unknown.
Tuberous Sclerosis Complex
Chordomas have been reported at a higher incidence in children with the genetic disease Tuberous Sclerosis Complex (TSC). Changes in either of two genes involved in Tuberous Sclerosis Complex (TSC1 and TSC2) can cause a predisposition to developing chordoma.
Chordoma symptoms
The most common signs of chordoma are pain and neurological changes.
Skull base chordoma symptoms
Skull base chordomas most often cause headache, neck pain, or double vision. If large enough, they may affect facial sensation or movement, voice, speech, and swallowing function.
Sacral chordoma symptoms
Chordomas of the spine and sacrum can cause changes in bowel and/or bladder function, pain, aching, tingling, numbness, or weakness of the arms and legs. Often, sacral chordomas do not cause symptoms until the tumor is quite large, and sometimes a lump is the first sign of a sacral chordoma.
Chordoma diagnosis
Chordoma is not always easy to diagnose and can be confused with other diseases. It is important to go to a hospital, treatment center, or network of treatment centers where doctors have experience diagnosing and treating chordoma as soon as chordoma is suspected.
Getting the correct diagnosis can have a major impact on the treatment you have. For this reason, it is very important for your diagnosis to be made by doctors who have experience diagnosing and treating chordoma patients. Getting a second opinion to confirm the diagnosis can be helpful before making treatment decisions. It is always OK to ask for a second or even a third opinion. If you have not yet had treatment, ask whether any additional tests are needed to rule out other possible tumor types before going forward with treatment.
Tests and procedures used to diagnose a chordoma include:
- Removing a sample of cells for laboratory testing (biopsy). A biopsy is a procedure to remove a sample of suspicious cells for laboratory testing. In the lab, specially trained doctors called pathologists examine the cells under microscopes to determine whether cancer cells are present. Your pathologist may test your tumor tissue for the presence of a protein called brachyury. Nearly all chordomas have high levels of brachyury, which makes it helpful for diagnosis. It is important to seek the opinion of a doctor experienced with diagnosing and treating chordomas. The person performing your biopsy will ideally consult with experienced surgeons to plan the procedure so that it can be done in a way that won’t interfere with a later operation.
- Obtaining more detailed imaging. Your doctor may recommend imaging tests to help visualize your chordoma and determine whether it has spread beyond the spine or skull base. Tests may include an MRI or CT scan. An MRI is the best way to see a chordoma and how it is affecting the tissue around it, such as muscles, nerves, and blood vessels. No matter where the tumor is located, an MRI of the entire spine should be performed to see if the tumor may have spread to or developed in other areas of the spine. Chordoma is best seen on an MRI with a setting called T2 weighted imaging.Another imaging test called computed tomography, also called CT or CAT scan, is recommended in addition to MRI if it is not certain whether the tumor is chordoma. CT scans of the chest, abdomen, and pelvis are recommended to make sure there is no spread of tumor. Imaging tests should be interpreted by a radiologist who has experience diagnosing bone tumors.
After you receive a diagnosis of chordoma, your doctor will develop a treatment plan tailored to your needs in consultation with an expert in cancer and radiation therapy (radiation oncologist) and a surgical oncologist.
Are biopsies recommended for suspected chordomas?
For sacral and mobile spine tumors, a trocar CT-guided biopsy is recommended and should be done from the back. Trocar CT-guided biopsy uses a CT scan to precisely direct the biopsy needle to the correct location. The biopsy needle is enclosed in a tube to keep tumor cells from spreading along the path of the needle – this is often called seeding. Talk with your doctors to learn if they plan to use this method if a biopsy is recommended.
Skull base tumors can be difficult to reach safely for a biopsy, so your surgeon may opt for a biopsy during surgery. This means that a pathologist will be prepared to examine a sample of tumor tissue removed at the start of surgery and give an immediate diagnosis, and the surgical team will proceed with surgery based on that information.
If you have a biopsy before surgery, it is recommended that your surgeon take out the tissue around the area of the biopsy during surgery in order to remove any chordoma cells that might have spread when the biopsy disturbed the tumor.
Chordoma can be confused with other diseases, including:
- Benign notochordal cell tumors – These benign spine tumors can be seen on an MRI or CT scan and can sometimes look like chordoma. However, benign notochordal cell tumors stay confined within the bone and do not spread into other tissues like chordomas can. If you have a suspected benign notochordal cell tumor, you should have an MRI or CT scan from time to time to look for changes. Images should be reviewed by a radiologist with expertise in bone tumors.
- Chondrosarcoma – This type of bone cancer looks very similar to chordoma on CT and MRI. A specific type of MRI called diffusion MRI, or D-MRI, may help doctors tell the difference. Sometimes it is only possible to know a tumor is not chondrosarcoma after having a biopsy. Skull base chondrosarcomas usually respond better to radiation than skull base chordomas, and have a better prognosis.
- Giant cell tumor of the bone – These tumors look somewhat different on imaging tests than chordoma, and tend to be located in the upper part of the sacrum.
- Schwannoma – These tumors damage the bone differently than chordomas do, look different on imaging tests, and do not spread to nearby muscles or joints.
- Other tumors of the spine and skull base – These include other bone cancers such as Ewing sarcoma and osteosarcoma, as well as a type of nervous system tumor called a myxopapillary ependymoma. Lymphoma, a cancer of the body’s immune system, and multiple myeloma, a blood cancer, can also cause tumors in these areas.
- Metastasis (spread) of another cancer – Sometimes cancers in other places in the body can spread to the bones of the spine or skull base.
Chordoma treatment
Chordoma treatment depends on the size and its location as well as whether it has invaded nerves or other tissue. Options may include surgery, radiation therapy — including proton therapy — stereotactic radiosurgery, chemotherapy and targeted therapies. Tumors may recur after treatment. There are currently no drugs approved by the FDA to treat chordoma.
Chordomas are treated with surgery because these tumors continuously grow, although they grow slowly. If the chordoma is not removed, it may wear away the bone and adjacent soft tissue, causing destruction of surrounding tissues. The surgery aims to remove as much of the chordoma tumor as possible. The extent of the surgery, or the amount of chordoma tumor that may be removed, depends on the location of the tumor and how close it is to critical structures in the brain. In some cases, surgery is followed by radiation therapy to destroy any cancer cells that may remain after surgery, especially when the tumor cannot be removed completely 4. Several studies have shown that carbon ion therapy or proton beam radiation may control tumor growth and improve survival 5.
Radical resections of tumors (removal of all the tumor) with clean margins (with no remaining of the tumor) are associated with a longer period of being disease-free. If the tumor cannot be removed completely, because of the location and closeness to critical delicate structures, the addition of radiation therapy decreases the recurrence of the tumor. Frequent follow-up is needed because of the high rate of recurrence of these tumors. Tumor recurrence identified early is easier to treat. The time in between follow-up visits, including repeat MRI or CT scans, depends on the completeness of the resection. Because residual tumor shortens the recurrence time, patients with known or suspected residual tumor need to be evaluated more frequently 6.
If you decide to undergo chordoma surgery, ask about your doctor’s experience with complex cranial or spinal surgery. This type of surgery results in fewer complications when done by highly experienced, multidisciplinary surgical teams with expertise in chordomas.
Treatment for sacral chordoma
- Surgery. The goal of surgery for a sacral spine tumor is usually to remove the entire tumor in one piece, if possible. Surgery may be difficult to perform because the tumor is near critical structures in the spinal cord.
- Radiation therapy. Radiation therapy uses high-energy beams, such as X-rays or protons, to kill cancer cells. During radiation therapy, you lie on a table as a machine moves around you, directing the radiation beams to precise points on your body. Radiation therapy may be used before or after surgery or if surgery isn’t an option. Treatment with newer types of radiation treatment, such as proton therapy, allows doctors to use higher doses of radiation while protecting healthy tissue, which may be more effective in treating a chordoma.
- Radiosurgery. Stereotactic radiosurgery uses multiple beams of radiation to kill the cancer cells in a very small area. Each beam of radiation isn’t very powerful, but the point where all the beams meet — at the chordoma — receives a large dose of radiation to kill the cancer cells.
- Other treatments. Sometimes chemotherapy and targeted drug therapy are used to treat a chordoma.
Treatment for clival chordoma
Treatment for a skull base tumor usually involves surgery followed by radiation therapy. The goal of surgery is to remove as much of the tumor as possible without harming nearby healthy tissue or causing undue new problems. Complete resection may not be an option if it’s near critical structures, such as the carotid artery. Endoscopic surgery as well as traditional approaches may be needed or used together to remove as much of the tumor as possible at the lowest risk possible.
Surgery is usually followed by radiation therapy to kill any remaining cancer cells and help prevent recurrence.
A meta-analysis 7 of all published studies between 1999 and 2010 concluded that “complete resection is associated with improved progression free survival and overall survival in patients with skull base chordomas compared with subtotal resection.“
Specifically, patients who have a subtotal resection are 3.83 times more likely to experience a recurrence and 5.85 times more likely to die at 5 years versus patients with complete resection 7.
Due to the location of skull base tumors within the head, en bloc resection (removing the tumor in one piece) is rarely possible, however gross total resection can be achieved with an intralesional approach. When en-bloc resection or gross total resection is not possible, maximum safe resection is advocated.
There are a broad range of surgical approaches available to access skull base chordomas, and staged surgeries combining multiple approaches are often necessary. Selection of the surgical strategy for each patient is highly dependent on the location of the tumor within the skull base, and the surgeon’s preference. For each patient different approaches could have different benefits, and could carry different risks. For some approaches, a neurosurgeon may work in tandem with an otolaryngologist-head and neck surgeon (ear nose and throat [ENT] surgeon). Regardless of the approach, it is important that the surgeon(s) be experienced in skull base surgery, which is a distinct sub-specialty for neurosurgeons and ENT surgeons.
Sacral and Spinal Chordomas Surgery
For chordomas of the sacrum and mobile spine, complete en bloc resection (removing the tumor in one piece) with tumor-free margins is the goal of surgery. Intralesional resection (removing the tumor in pieces) is associated with a high rate of recurrence and diminished survival 8. In a 2009 review article 9 of the treatment of sacral and spinal chordomas in the Journal of the American Academy of Orthopaedic Surgeons, Scuibba and colleagues at Johns Hopkins University wrote, “The importance of obtaining wide tumor-free margins when possible cannot be underestimated. Numerous studies demonstrate a direct correlation between the extent of surgical resection and the length of recurrence-free survival.”
Surgery for sacral chordomas may involve removal a portion of the sacrum or the entire sacrum depending on the location and size of the tumor. Depending on the extent of the tumor, these procedures may require a combination of surgical approaches to access the tumor from different angles. In order to achieve wide margins, these procedures often involve the intentional sacrifice of sacral nerves, which may result in motor, sensory, sphincter, or sexual dysfunction. The extent of dysfunction depends on which sacral nerves must be sacrificed. In general, the higher the tumor extends in the sacrum, the more sacral nerves must be sacrificed, and the more dysfunction will be experienced.
Surgery for spinal chordomas involves removal of one or more vertebral bodies – a procedure called a “spondylectomy.” These procedures often require a combination of surgical approaches (from the front and back), and may need to be carried out in multiple stages.
For both spinal and sacral chordomas, mechanical and soft tissue reconstruction are usually required after removal of the tumor and involved vertebral bodies. Due to the complexity of surgery and reconstruction, a multidisciplinary surgical team may be needed, including specialists in surgical oncology, neurosurgery, orthopaedic surgery, vascular surgery, and plastic surgery. This operation should be led by a neurosurgeon or orthopaedic surgeon who specializes in complex spine surgery.
Radiation treatment
Once the tumor is removed, if complete resection is not achieved, radiation therapy is usually recommended to reduce the likelihood of re-growth of residual tumor 10. For patients who have a complete resection radiation is often advised, however the benefit of radiation for this group of patients is debated 11.
Chordomas can only be controlled with very high doses of radiation – doses that would permanently damage normal tissue. Because of their proximity to vital anatomy such as the brain and spinal cord, which cannot tolerate doses of radiation required to kill chordoma, radiotherapy for chordoma must be highly conformal, meaning focused on the tumor while avoiding surrounding tissue. Several different types of conformal radiotherapy are available.
A type of radiation called proton beam therapy is most often recommended for chordoma patients because it allows delivery of very high doses of radiation to the tumor while minimizing doses to tissues just millimeters away. Another type of particle beam radiation called carbon ion therapy has similar properties to proton beam therapy, but is only available at a small number of centers.
- Particle Therapy Centers list is available here: https://www.chordomafoundation.org/treatment/particle-therapy-centers/
Certain other types of conformal radiation, such as radiosurgery (including Gamma Knife and CyberKnife) and intensity modulated radiotherapy (IMRT) can also be effective, depending on the size and location of the tumor 11. No direct comparison trials have been performed to establish the optimal form of radiation. However, there is consensus that conventional photon radiation is not beneficial for chordoma patients 10.
While radiation is usually well tolerated, side effects are possible. For skull base tumors, pituitary dysfunction is common after radiation. Depending on the area radiated, there may also be a risk of damage to the optic nerve, cranial nerves and brain stem, which could cause blindness or paralysis. For tumors of the spine, radiation can also damage to the spinal cord, causing paralysis. Radiation for sacral tumors can cause radiation proctitis, which is damage or inflammation to the colon.
If a tumor returns after initial irradiation, it may or may not be possible to have radiation therapy again. This is because every tissue in the body has a certain life-time maximum tolerance for radiation, beyond which serious injury will occur. Often, delivering enough radiation to effectively treat chordoma exposes vital surrounding structures to a dose of radiation close to this lifetime maximum. Delivering more radiation to those structures after initial treatment could be very dangerous. The ability to have repeated radiation, therefore, depends on the location of the original tumor, the location of the recurrent tumor, the dose and distribution of radiation that was given initially, and other factors 12.
Systemic Therapy
If surgery and/or radiation are not possible, systemic therapy can be used to slow progression of the disease. Systemic therapy is the use of treatments that spread throughout the body to kill cancer cells wherever they are located. These treatments are prescribed by a medical oncologist, and sometimes by a neuro-oncologist. Systemic therapy includes different types of drugs and therapies, including cytotoxic chemotherapy drugs, targeted therapies, and immunotherapies.
Because chordomas tend to be slow growing, they are generally resistant to conventional cytotoxic chemotherapy drugs, which target rapidly dividing cells. For this reason, targeted therapies and immunotherapies are most often used for chordoma treatment. Learn more about these options for chordoma in this systemic therapy table here: https://www.chordomafoundation.org/systemic-therapy/
More information about the genes, proteins, and signaling pathways that are important in chordoma and may be promising targets for systemic therapies can be found here: https://www.chordomafoundation.org/targets/
Clinical Trials
Clinical trials are important treatment options for any cancer patient to consider. For chordoma patients, some clinical trials may provide an opportunity to receive new, experimental therapies that could be more effective than currently available options. Clinical trials may also provide an opportunity for patients to gain access to clinically available therapies at no cost. In addition, chordoma patients who participate in clinical trials contribute to knowledge that can guide therapy for future patients and potentially help identify new ways of treating this rare cancer. For information on clinical trials please go here: https://www.cancer.gov/about-cancer/treatment/clinical-trials
- National Cancer Institute. Chordoma research. https://dceg.cancer.gov/research/clinical-studies/chordoma[↩]
- http://emedicine.medscape.com/article/250902-overview[↩]
- Lanzino G, Dumont AS, Lopes MB & Laws ER Jr. Skull base chordomas: overview of disease, management options, and outcome. Neurosurgical Focus. 2001; 10:E12. http://www.ncbi.nlm.nih.gov/pubmed/16734404[↩]
- Lanzino G, Dumont AS, Lopes MB & Laws ER Jr. Skull base chordomas: overview of disease, management options, and outcome. Neurosurgical Focus. 2001; 10:E12. https://www.ncbi.nlm.nih.gov/pubmed/16734404[↩]
- Erdem E, Angtuaco EC, Van Hemert R, Park JS & Al-Mefty O. Comprehensive review of intracranial chordoma. Radiographics. 2003; 23:995-1009. https://www.ncbi.nlm.nih.gov/pubmed/12853676[↩]
- http://emedicine.medscape.com/article/250902-overview.[↩]
- S. Di Maio, N. Temkin, D. Ramanathan, and L.N. Sekhar, “Current comprehensive management of cranial base chordomas: 10-year meta-analysis of observational studies.”, Journal of neurosurgery, 2011. http://thejns.org/doi/full/10.3171/2011.7.JNS11355[↩][↩]
- S. Boriani, S. Bandiera, R. Biagini, P. Bacchini, L. Boriani, M. Cappuccio, F. Chevalley, A. Gasbarrini, P. Picci, and J.N. Weinstein, “Chordoma of the mobile spine: fifty years of experience.”, Spine, 2006. http://www.ncbi.nlm.nih.gov/pubmed/16481964[↩]
- D.M. Sciubba, J.J. Cheng, R.J. Petteys, K.L. Weber, D.A. Frassica, and Z.L. Gokaslan, “Chordoma of the sacrum and vertebral bodies.”, The Journal of the American Academy of Orthopaedic Surgeons, 2009. http://www.ncbi.nlm.nih.gov/pubmed/19880681[↩]
- M. Amichetti, M. Cianchetti, D. Amelio, R.M. Enrici, and G. Minniti, “Proton therapy in chordoma of the base of the skull: a systematic review.”, Neurosurgical review, 2009. https://link.springer.com/article/10.1007%2Fs10143-009-0194-4[↩][↩]
- S. Di Maio, N. Temkin, D. Ramanathan, and L.N. Sekhar, “Current comprehensive management of cranial base chordomas: 10-year meta-analysis of observational studies.”, Journal of neurosurgery, 2011. http://www.ncbi.nlm.nih.gov/pubmed/21819197[↩][↩]
- B.P. Walcott, B.V. Nahed, A. Mohyeldin, J. Coumans, K.T. Kahle, and M.J. Ferreira, “Chordoma: current concepts, management, and future directions.”, The Lancet. Oncology, 2012. http://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(11)70337-0/fulltext[↩]





{kind=link}