
Contents
What is Creutzfeldt Jakob disease
Creutzfeldt-Jakob disease (CJD) is a rare, fatal degenerative brain disorder that can be experimentally transmitted from one animal to another, as well as from human patients to other humans and animals 1. Creutzfeldt-Jakob disease (CJD) is classified as a transmissible spongiform encephalopathy (TSE) along with other prion diseases that occur in humans and animals. Creutzfeldt-Jakob disease (CJD) affects about one person in every one million people each year worldwide 2. The low random incidence of Creutzfeldt-Jakob disease (CJD) indicates that person-to-person transmission probably does not occur through normal contact. Spouses and other household members of people with sporadic Creutzfeldt-Jakob disease (CJD) appear to be at no higher risk of contracting the disorder than the general population. Creutzfeldt-Jakob disease (CJD) affects both men and women, and most often appears in people aged 50 to 75. In about 85% of patients occur on a sporadic basis via unknown mechanisms and no recognizable pattern of transmission. In a few rare families (7-10% of all cases), the disease is hereditary, because of inherited mutations of the prion protein gene. These inherited forms include Gerstmann-Straussler-Scheinker syndrome and fatal familial insomnia. In a smaller number of cases (<1% of all cases) it is transmitted iatrogenically (induced inadvertently by a physician or surgeon or by medical treatment or diagnostic procedures) through contaminated surgical instruments (e.g. implanted EEG depth electrodes) and human material (e.g. human growth hormone, dura mater or corneal transplants). Symptoms usually start around age 60. Memory problems, behavior changes, vision problems, and poor muscle coordination progress quickly to dementia, coma, and death. Most patients die within a year.
Classic Creutzfeldt-Jakob disease (CJD) has been recognized since the early 1920s. The most common form of classic Creutzfeldt-Jakob disease (CJD) is believed to occur sporadically, caused by the spontaneous transformation of normal prion proteins into abnormal prions. This sporadic disease occurs worldwide, including the United States, at a rate of roughly 1 to 1.5 cases per 1 million population per year, although rates of up to two cases per million are not unusual. The risk of Creutzfeldt-Jakob disease (CJD) increases with age, and in persons aged over 50 years of age, the annual rate is approximately 3.4 cases per million.
Physicians suspect a diagnosis of Creutzfeldt-Jakob disease on the basis of the typical signs and symptoms and progression of the disease. In most Creutzfeldt-Jakob disease patients, the presence of 14-3-3 protein in the cerebrospinal fluid and/or a typical electroencephalogram (EEG) pattern, both of which are believed to be diagnostic for Creutzfeldt-Jakob disease, have been reported 3. However, a confirmatory diagnosis of Creutzfeldt-Jakob disease requires neuropathologic and/or immunodiagnostic testing of brain tissue obtained either at biopsy or autopsy.
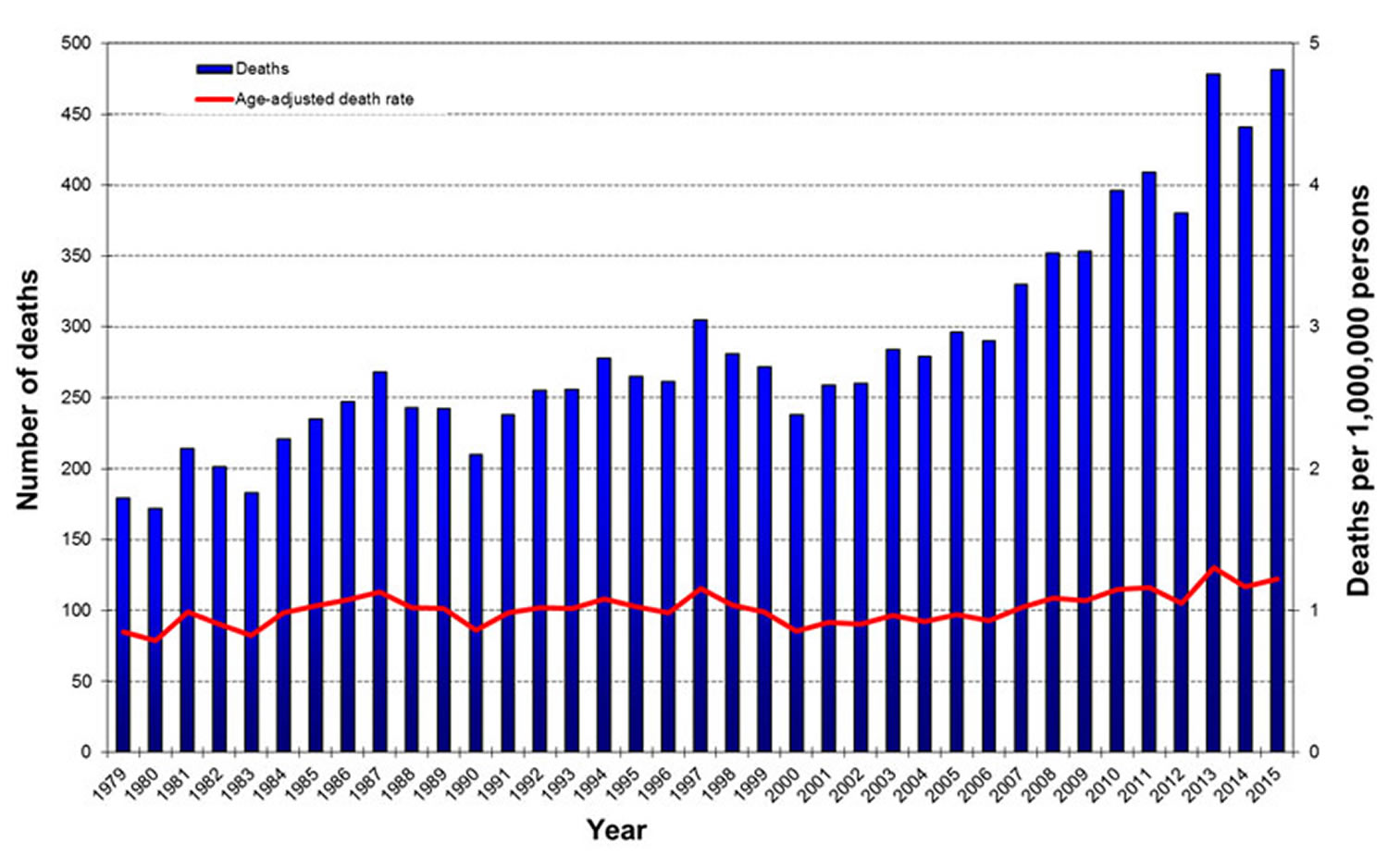
Figure 1. Creutzfeldt-Jakob Disease Deaths and Age-Adjusted Death Rate, United States, 1979-2015*
What is dementia ?
Dementia is a condition in which a person has significant difficulty with daily functioning because of problems with thinking and memory. Dementia is not a single disease. It’s an overall term —like heart disease—that covers a wide range of specific medical conditions, including Alzheimer’s disease. Disorders grouped under the general term “dementia” are caused by abnormal brain changes. These changes trigger a decline in thinking skills severe enough to impair daily life and independent function. They also affect behavior, feelings and relationships.
Brain changes that cause dementia may be temporary, but they are most often permanent and worsen over time, leading to increasing disability and a shortened life span. Survival can vary widely, depending on such factors as the cause of the dementia, age at diagnosis and coexisting health conditions.
- For more detail information on dementia please go here: Dementia
What are Prion diseases ?
Prion diseases or transmissible spongiform encephalopathies (TSEs) are a family of rare progressive neurodegenerative disorders that affect both humans and animals. They are distinguished by long incubation periods, characteristic spongiform changes associated with neuronal loss, and a failure to induce inflammatory response.
The term “prions” refers to abnormal, pathogenic agents that are transmissible and are able to induce abnormal folding of specific normal cellular proteins called prion proteins that are found most abundantly in the brain. The functions of these normal prion proteins are still not completely understood 5. The abnormal folding of the prion proteins leads to brain damage and the characteristic signs and symptoms of the disease. Prion diseases are usually rapidly progressive and always fatal.
Listed below are the prion diseases identified to date:
Human Prion Diseases
- Creutzfeldt-Jakob Disease (CJD)
- Variant Creutzfeldt-Jakob Disease (vCJD)
- Gerstmann-Straussler-Scheinker Syndrome
- Fatal Familial Insomnia
- Kuru
Animal Prion Diseases
- Bovine Spongiform Encephalopathy (BSE)
- Chronic Wasting Disease (CWD)
- Scrapie
- Transmissible mink encephalopathy
- Feline spongiform encephalopathy
- Ungulate spongiform encephalopathy
What causes Creutzfeldt Jakob disease
Creutzfeldt-Jakob disease occurs when prion protein, which is found throughout the body but whose normal function isn’t yet known, begins folding into an abnormal three-dimensional shape. This shape change gradually triggers prion protein in the brain to fold into the same abnormal shape.
Through a process scientists don’t yet understand, misfolded prion protein destroys brain cells 6. Resulting damage leads to rapid decline in thinking and reasoning as well as involuntary muscle movements, confusion, difficulty walking and mood changes.
Sporadic Creutzfeldt-Jakob disease has no known cause. Most scientists believe the disease begins when prion protein somewhere in the brain spontaneously misfolds, triggering a “domino effect” that misfolds prion protein throughout the brain. Genetic variation in the prion protein gene may affect risk of this spontaneous misfolding.
Mutations in the prion protein gene also may play a yet-to-be-determined role in making people susceptible to acquired Creutzfeldt-Jakob disease from external sources. Scientists don’t yet know the why acquired Creutzfeldt-Jakob disease seems to be transmitted through such a limited number of external sources. Researchers have found no evidence that the abnormal protein is commonly transmitted through sexual activity or blood transfusions.
Familial Creutzfeldt-Jakob disease is caused by variations in the prion protein gene that increases the risk of an individual developing Creutzfeldt-Jakob disease. Researchers have identified more than 50 prion protein mutations in those with inherited Creutzfeldt-Jakob disease. Genetic testing can determine whether family members at risk have inherited a Creutzfeldt-Jakob disease-causing mutation. Experts strongly recommend professional genetic counseling both before and after genetic testing for hereditary Creutzfeldt-Jakob disease.
Variant Creutzfeldt-Jakob disease
Experts generally recognize the following main types of Creutzfeldt-Jakob disease:
Sporadic Creutzfeldt-Jakob disease develops spontaneously for no known reason. It accounts for 85 percent of cases. On average, sporadic Creutzfeldt-Jakob disease first appears between ages 60 and 65.
Familial Creutzfeldt-Jakob disease is caused by certain changes in the chromosome 20 gene coding the biological blueprint for prion protein. People who develop familial Creutzfeldt-Jakob disease do so because they inherited the genetic changes from a parent. Familial Creutzfeldt-Jakob disease accounts for about 10 to 15 percent of cases. It develops, on average, at a younger age than sporadic Creutzfeldt-Jakob disease, with some genetic types appearing as early as ages 20 to 40.
Acquired Creutzfeldt-Jakob disease results from exposure to an external source of abnormal prion protein. These sources estimated to account for about 1 percent of Creutzfeldt-Jakob disease cases. The two most common outside sources are:
- Medical procedures involving instruments used in neurosurgery, growth hormone from human sources or certain transplanted human tissues. The risk of Creutzfeldt-Jakob disease from medical procedures has been greatly reduced by improved sterilization techniques, new single-use instruments and synthetic sources of growth hormone.
- Meat or other products from cattle infected with bovine spongiform encephalopathy (“mad cow disease”), recognized in the mid-1990s as the cause of variant-Creutzfeldt-Jakob disease (vCJD). Scientists traced this new type of Creutzfeldt-Jakob disease to consumption of beef from cattle whose feed included processed brain tissue from other animals.
Classic CJD versus Variant CJD
The variant form of Creutzfeldt-Jakob disease should not be confused with the classic form of Creutzfeldt-Jakob disease that is endemic throughout the world, including the United States. There are several important differences between these two forms of the disease. The median age at death of patients with classic Creutzfeldt-Jakob disease in the United States, for example, is 68 years, and very few cases occur in persons under 30 years of age. In contrast, the median age at death of patients with variant Creutzfeldt-Jakob disease in the United Kingdom is 28 years.
Variant Creutzfeldt-Jakob (vCJD) disease can be confirmed only through examination of brain tissue obtained by biopsy or at autopsy, but a “probable case” of variant Creutzfeldt-Jakob disease (vCJD) can be diagnosed on the basis of clinical criteria developed in the United Kingdom. The incubation period for variant Creutzfeldt-Jakob disease (vCJD) is unknown because it is a new disease. However, it is likely that ultimately this incubation period will be measured in terms of many years or decades. In other words, whenever a person develops variant Creutzfeldt-Jakob disease (vCJD) from consuming a Bovine Spongiform Encephalopathy (BSE)-contaminated product, he or she likely would have consumed that product many years or a decade or more earlier.
In contrast to classic Creutzfeldt-Jakob disease, vCJD in the United Kingdom predominantly affects younger people, has atypical clinical features, with prominent psychiatric or sensory symptoms at the time of clinical presentation and delayed onset of neurologic abnormalities, including ataxia within weeks or months, dementia and myoclonus late in the illness, a duration of illness of at least 6 months, and a diffusely abnormal non-diagnostic electroencephalogram.
The characteristic neuropathologic profile of variant Creutzfeldt-Jakob disease (vCJD) includes, in both the cerebellum and cerebrum, numerous kuru-type amyloid plaques surrounded by vacuoles and prion protein (PrP) accumulation at high concentration indicated by immunohistochemical analysis.
Recently published data indicate that the epidemic of variant CJD in the United Kingdom may have already reached a peak. A listing of monthly updated numbers of CJD and variant CJD cases in the United Kingdom 7.
Table 1. Clinical and Pathologic Characteristics Distinguishing Classic CJD from Variant CJD
| Characteristic | Classic CJD | Variant CJD |
|---|---|---|
| Median age at death | 68 years | 28 years |
| Median duration of illness | 4-5 months | 13-14 months |
| Clinical signs and symptoms | Dementia; early neurologic signs | Prominent psychiatric/behavioral symptoms; painful dyesthesiasis; delayed neurologic signs |
| Periodic sharp waves on electroencephalogram | Often present | Often absent |
| “Pulvinar sign” on MRI* | Not reported | Present in >75% of cases |
| Presence of “florid plaques” on neuropathology | Rare or absent | Present in large numbers |
| Immunohitochemical analysis of brain tissue | Variable accumulation | Marked accumulation of protease-resistance prion protein |
| Presence of agent in lymphoid tissue | Not readily detected | Readily detected |
| Increased glycoform ratio on immunoblot analysis of protease-resistance prion protein | Not reported | Marked accumulation of protease-resistance prion protein |
*An abnormal signal in the posterior thalami on T2- and diffusion-weighted images and fluid-attenuated inversion recovery sequences on brain magnetic resonance imaging (MRI); in the appropriate clinical context, this signal is highly specific for vCJD.
[Source 8]Creutzfeldt Jakob disease symptoms
Specific Creutzfeldt-Jakob disease symptoms experienced by an individual and the order in which they appear can differ significantly. Some common symptoms include:
- Depression
- Agitation, apathy and mood swings
- Rapidly worsening confusion, disorientation, and problems with memory, thinking, planning and judgment
- Difficulty walking
- Muscle stiffness, twitches and involuntary jerky movements
Creutzfeldt-Jakob disease Diagnosis
Rapid symptom progression is one of the most important clues that a person may have Creutzfeldt-Jakob disease.
There is no single test — or any combination of tests — that can conclusively diagnose sporadic Creutzfeldt-Jakob disease in a living person, but the following tests may help determine whether an individual has Creutzfeldt-Jakob disease:
- Electroencephalogram (EEG) measures the brain’s patterns of electrical activity similar to the way an electrocardiogram (ECG) measures the heart’s electrical activity.
- Brain magnetic resonance imaging (MRI) can detect certain brain changes consistent with Creutzfeldt-Jakob disease.
- Lumbar puncture (spinal tap) tests spinal fluid for the presence of certain proteins.
Centers for Disease Control and Prevention’s (CDC’s) Diagnostic Criteria for Creutzfeldt-Jakob Disease (CJD), 2010
1. Sporadic CJD 9
Definite:
- Diagnosed by standard neuropathological techniques; and/or immunocytochemically; and/or Western blot confirmed protease-resistant PrP; and /or presence of scrapie-associated fibrils.
Probable:
Rapidly progressive dementia; and at least two out of the following four clinical features:
- Myoclonus
- Visual or cerebellar signs
- Pyramidal/extrapyramidal signs
- Akinetic mutism
AND a positive result on at least one of the following laboratory tests:
- a typical EEG (periodic sharp wave complexes) during an illness of any duration; and/or
- a positive 14-3-3 cerebrospinal fluid (CSF) assay in patients with a disease duration of less than 2 years
- Magnetic resonance imaging (MRI) high signal abnormalities in caudate nucleus and/or putamen on diffusion-weighted imaging (DWI) or fluid attenuated inversion recovery (FLAIR)
AND without routine investigations indicating an alternative diagnosis.
Possible:
Progressive dementia; and at least two out of the following four clinical features:
- Myoclonus
- Visual or cerebellar signs
- Pyramidal/extrapyramidal signs
- Akinetic mutism
AND the absence of a positive result for any of the three laboratory tests that would classify a case as “probable” (see tests a-c above)
AND duration of illness less than two years
AND without routine investigations indicating an alternative diagnosis.
2. Iatrogenic CJD 9
Progressive cerebellar syndrome in a recipient of human cadaveric-derived pituitary hormone; or sporadic CJD with a recognized exposure risk, e.g., antecedent neurosurgery with dura mater implantation.
3. Familial CJD 9
Definite or probable CJD plus definite or probable CJD in a first degree relative; and/or Neuropsychiatric disorder plus disease-specific PrP gene mutation.
Treatment and outcomes of Creutzfeldt-Jakob disease
There is no treatment that can slow or stop the underlying brain cell destruction caused by Creutzfeldt-Jakob disease and other prion diseases. Various drugs have been tested but have not shown any benefit. Clinical studies of potential Creutzfeldt-Jakob disease treatments are complicated by the rarity of the disease and its rapid progression.
Current therapies focus on treating symptoms and on supporting individuals and families coping with Creutzfeldt-Jakob disease. Doctors may prescribe painkillers such as opiates to treat pain if it occurs. Muscle stiffness and twitching may be treated with muscle-relaxing medications or antiseizure drugs. In the later stages of the disease, individuals with Creutzfeldt-Jakob disease become completely dependent on others for their daily needs and comfort.
Creutzfeldt-Jakob disease progresses rapidly. Those affected lose their ability to move or speak and require full-time care to meet their daily needs. An estimated 90 percent of those diagnosed with sporadic Creutzfeldt-Jakob disease die within one year. Those affected by familial Creutzfeldt-Jakob disease tend to develop the disorder at an earlier age and survive somewhat longer than those with the sporadic form, as do those diagnosed with variant-Creutzfeldt-Jakob disease. Scientists have not yet learned the reason for these differences in survival.
Symptomatic treatment for Creutzfeldt-Jakob disease
Although there is no cure for CJD, treatments are available for a variety of symptoms that occur in patients with CJD. Often a treatment for a specific symptom is initially effective only to lose its effect over time. This particularly applies to behavioural disorders, rigidity and hallucinations. It is unclear why this occurs but a plausible explanation is that there is progressive loss of receptors for the different agents as the disease advances. Whether this is the explanation or not it is often necessary to change therapy in an attempt to control the breakthrough symptoms and use combinations of different treatments even though some combinations appear to be duplication of therapy. Sometimes it is better to use less specific drugs on the assumption that at least they can bind to multiple receptors some of which may be relatively spared.
Myoclonus
Myoclonus is common in many forms of Creutzfeldt-Jakob disease and is a characteristic feature of sporadic Creutzfeldt-Jakob disease. Positive myoclonus comprises irregular short duration jerk like episodes involving the somatic musculature. Conversely, negative myoclonus is seen in some neurological diseases, particularly hepatic failure, and is characterised by short lived inhibition of tone in a group of muscles but it is unusual in Creutzfeldt-Jakob disease. Myoclonus can be generated at several levels in the central nervous system. In the case of Creutzfeldt-Jakob disease it is either cortical or subcortical in origin. Cortical myoclonus is generated from the cerebral cortex grey matter and is thought to be due to loss of inhibitory gabanergic neurones. Typically cortical myoclonus is multifocal and involves those muscles with the greatest cortical representation i.e. the limbs, especially the digits and face, but no area is immune. It is stimulus sensitive especially to touch and on eliciting tendon reflexes. Action triggers the myoclonus. Proof of cortical origin can be obtained by back averaging the EEG when a discharge consistently precedes the movement by a short interval (10-40ms) this depending on the distance from the cortex to the muscles concerned. In contrast to propriospinal myoclonus there is no sequential progression of the muscles involved. Somatosensory evoked potentials are enhanced during the discharge.
Cortical myoclonus is often reduced by gabanergic drugs of which sodium valproate, an agent that increases cortical gamma amino butyric acid (gaba), is the most frequently prescribed. Most patients respond to 1-2.5G daily. Levetiracetam in a similar dose is useful but it’s mode of action is unclear. Benzodiazepines, which act on the gaba receptor complex, are also effective in many cases. Clonazepam can be dramatically effective in a small dose but many patients only respond to large doses (up to 15mg) which can result in excessive drowsiness. Combinations of therapy may have to be given and other anticonvulsants such as phenobarbitone are sometimes useful. Phenytoin, carbamazepine and lamotrigine have also been suggested although exacerbation, rather than improvement, of the myoclonus usually occurs with these drugs.
Subcortical myoclonus also occurs in Creutzfeldt-Jakob disease. If this arises from the thalamus the drugs used in cortical myoclonus are still effective but if the movement disorder arises from brain stem structures such as the giant reticular cell region of the brainstem as a result of loss of inhibitory input from the cerebellum treatment is more problematic. Clonazepam is the drug of choice and ethyl alcohol is sometimes beneficial. In general valproate and levetiracetam are not effective. Having said that, pathology in Creutzfeldt-Jakob disease involves multiple levels of the central nervous system raising the possibility that myoclonus may arise from more than one region so it is always worth trying different agents in combination.
Psychiatric Symptoms
Visual hallucinations occur in various forms of Creutzfeldt-Jakob disease although the true frequency is not known. They can be relatively benign e.g. seeing furry animals or terrifying ‘Dahliesque’ types. The pathophysiology of these is obscure although they occur in a wide range of other degenerative conditions and are particularly common in cortical Lewy body disease. A variety of antipsychotic drugs are used to control them but there are no firm data on efficacy. Quetiapine, a drug that has marked 5HT1A and 2 blocking activity, together with some activity against D1 and 2 and less against alpha adrenergic and H1 receptors, is most frequently prescribed. The advantage of this agent over conventional antipsychotic drugs, such as butyrophenones and phenothiazines, is that parkinsonian side effects are much less marked. This is potentially important in Creutzfeldt-Jakob disease patients who often have signs of basal ganglia dysfunction as part of the disease process. As with any degenerative disease it is wise to escalate the dose slowly beginning with 25mg daily. Adverse events are not usually a problem but concern has been raised over the long term use of this agent in elderly demented patients with Alzheimer’s disease whose cognitive impairment can worsen.
Because of the beneficial effect of centrally acting anticholinergic drugs upon visual hallucinations in cortical Lewy body disease it is reasonable to prescribe donepezil or rivastigmine to Creutzfeldt-Jakob disease patients with disturbing hallucinations and the former does seem to be effective in some patients with intractable visual hallucinations. If the patient has parkinsonian symptoms these do not become more severe.
Anxiety, Agitation, Insomnia and Depression
Agitation and anxiety are frequent symptoms in patients with Creutzfeldt-Jakob disease. Often the patient can’t settle and paces around. They can also become abusive and occasionally physically violent. Benzodiazepines such as diazepam, which act on benzodiazepine receptors associated with gaba receptors, are effective anxiolytic drugs and can promote sleep (see below). Dependence can be troublesome if used for long periods. They can have a paradoxical effect of increasing aggression but dosage adjustment usually ameliorates this. Olanzapine, which has its maximum receptor blocking activity against 5HT2 receptors, can be helpful although care is needed in the elderly because of an increased risk of stroke and diabetes. Weight gain is common although not usually a problem with Creutzfeldt-Jakob disease patients. Mirtazapine is an alternative anxiolytic. This drug is a potent inhibitor of 5HT2a and H1 receptors. Risperidone, primarily a D2 antagonist, is also used although there is a risk of exacerbating pre-existing parkinsonism which is also a major problem with the older anti-psychotic drugs such as phenothiazines and haloperidol. Akathisia and tardive dyskinesias are more common with this latter group of drugs. Physical symptoms of anxiety such as tremor can respond to beta-blockers which penetrate the blood brain barrier e.g. propranolol (10-120 mg). Anxiety induced sweating is helped by anticholinergic drugs such as oxybutynin (2.5-15 mg).
Sleep disturbance is characteristic of the fatal insomnias and some of the thalamic variants of Creutzfeldt-Jakob disease; it is also found in other forms of Creutzfeldt-Jakob disease and is often associated with anxiety. Unfortunately hypnotic agents are usually ineffective. Indeed these patients may seem to be excessively sleepy during the daytime which any long acting hypnotic could theoretically worsen. It is worth trying one of the benzodiazepines starting with agents that have a short duration of action such as temazepam (10-40 mg) before using longer duration drugs such as nitrazepam (5-10 mg). Non benzodiazepine hypnotics such as zopiclone (3.75-7.5 mg), which act on the diazepine receptor, are also worth trying as is mirtazapine (15-40 mg).
Depression occurs in some patients with Creutzfeldt-Jakob disease, particularly those with a family history, early in the evolution of the disorder. Cognitive behavioural therapy can be useful in these patients and SSRI’s are of value. If depression is accompanied by anxiety and agitation one of the sedating antidepressants are useful such as mirtazapine.
Pain
Pain is not usually a problem in patients with most types of Creutzfeldt-Jakob disease. However variant Creutzfeldt-Jakob disease is an exception. These patients characteristically have pain, probably of thalamic origin, in the limbs and hyperaesthesia. Gabapentin and amitriptyline are often effective and can be given in combination. Most analgesic drugs do not have a beneficial effect. The pain sometimes resolves as the disease progresses.
Patients with any type of Creutzfeldt-Jakob disease, but particularly variant Creutzfeldt-Jakob disease and kuru, appear to have headache. This sometimes responds to conventional analgesics.
Seizures
Epileptic fits are not that frequent in patients with Creutzfeldt-Jakob disease with the exception of patients with E220K mutation. Occasional epilepsia partialis continua (EPC) can occur. Levetiracetam (500-3000 mg) and sodium valproate (500-3000 mg) are useful for generalised tonic clonic seizures and is effective for seizures with myoclonus. Complex partial seizures are best treated with carbamazepine (400-2000 mg) or lamotrigine (25mg-300 mg) but these drugs can exacerbate myoclonus. Older anticonvulsants such as phenobarbitone (30-120 mg) are of value for generalised attacks and the side effects of drowsiness can be useful for the agitated patient and insomnia. Similarly, clobazam (10-40 mg) is effective for many seizure types and can help anxiety or insomnia. epilepsia partialis continua (EPC) is resistant to therapy.
Increased Muscle Tone
Increased tone is generally extrapyramidal. The rigidity can be severe making nursing difficult particularly late in the course of the disease. There is no experience of the best management of this rigidity. Theoretically dopamine replacement with agents such as sinemet might help but as behavioural disturbance is often a problem this might be contraindicated. Dantrolene (75-225 mg) acts directly upon the muscle membrane and can relieve rigidity even though the tone increase is centrally generated. At higher dose liver function may become deranged. Spasticity develops in many patients but is not usually a prominent feature. If troublesome baclofen (10-90 mg), tizanidine (8-24 mg) or diazepam (5-15 mg) can help as can dantrolene. Intramuscular botulinum toxin is effective in localised rigidity or spasticity although these symptoms are often too generalised to make this a feasible option. Further, botulinum toxin can exacerbate swallowing difficulties, frequent in the later stages of Creutzfeldt-Jakob disease, if given in large dosage.
Bladder Dysfunction
Incontinence occurs in most patients with Creutzfeldt-Jakob disease late in the course of the disease probably as a result of frontal disturbance. Retention rarely occurs. In general use of pads or catheterisation is required. Anticholinergic drugs such as tolterodine XL (4 mg) are helpful for frequency in some patients or if the bladder is irritable causing catheter extrusion. Bladder neck obstruction is not usually a problem but if it occurs can be treated with alpha adrenergic receptor blocking drugs (internal sphincter) such as prazosin (max 4 mg) and baclofen 10-60 mg(external sphincter).
Bowel Dysfunction
Terminally incontinence of stool occur requiring routine hygiene. Spurious diarrhoea with liquid motions is best treated with enemas. Constipating agents are not indicated in general.
Salivation Control
Occasionally patients who have severe swallowing problems require anticholinergic drugs such as atropine (subcutaneous injection 0.6-1.2 mg, or orally), hyoscine patches or oxybutynin tablets (5-15 mg) to reduce salivary flow. Blurring of vision and increase in mucus viscosity can be a problem.
Glycopyrronium bromide is effective in excessive salivation and can be administered orally or subcutaneously. It has a relativevly long duration of action and is said not to cross the blood brain barrier. Subcutaneous dosage ranges from 0.6-1.2 mg per day and oral adminstration from 1-3 mg per day in divided dosage.
Dysphagia
Difficulty in swallowing is a major problem with the severely disabled patient and a decision has to be made when nutritional intake is critical. Initially pureed food may help but ultimately this fails to maintain an adequate intake. If the patient is expected to survive for only a few days hydration can be maintained by nasal tube or subcutaneous administration of fluid. If the patient is likely to survive for a significant time or cannot tolerate a nasal tube, a gastrostomy will be necessary either by insertion of a tube parenterally (PEG) or radiologically (RIG). There are no trials to determine criteria for which technique is best suited to a particular patient. Invasive treatments should always be a mutual decision between the clinician and the patient or their relatives
Terminal Care
Patients should be on appropriate sedatives such as opiates. Nursing input and involvement of palliative care specialists is vital at this stage.
- Creutzfeldt-Jakob Disease. https://medlineplus.gov/creutzfeldtjakobdisease.html[↩]
- Creutzfeldt-Jakob Disease Fact Sheet for Healthcare Workers. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Creutzfeldt-Jakob-Disease-Fact-Sheet-Healthcare[↩]
- Creutzfeldt-Jakob Disease, Classic (CJD). https://www.cdc.gov/prions/cjd/about.html[↩]
- Creutzfeldt-Jakob Disease, Classic (CJD) Occurrence and Transmission. https://www.cdc.gov/prions/cjd/occurrence-transmission.html[↩]
- Prion Diseases. https://www.cdc.gov/prions/index.html[↩]
- Creutzfeldt-Jakob Disease. https://www.alz.org/dementia/creutzfeldt-jakob-disease-cjd-symptoms.asp[↩]
- https://www.gov.uk/government/collections/creutzfeldt-jakob-disease-cjd-guidance-data-and-analysis[↩]
- Belay E., Schonberger L. Variant Creutzfeldt-Jakob Disease and Bovine Spongiform Encephalopathy. Clin Lab Med 2002;22:849-862.[↩]
- Creutzfeldt-Jakob Disease, Classic (CJD) Diagnostic Criteria. https://www.cdc.gov/prions/cjd/diagnostic-criteria.html[↩][↩][↩]


{kind=link}