Contents
- What is Fronto Temporal Lobar Degeneration/Dementia
- Clinical Features of Frontotemporal Dementia (Frontotemporal Degeneration Subtypes)
- Table 1. Types of Frontotemporal Dementia (FTD) or Frontotemporal Lobar Degeneration (FLTD)
- Changes in the Brain
- Causes of frontotemporal dementia
- Is Frontotemporal dementia hereditary ?
- Symptoms of frontotemporal dementia
- Tests for frontotemporal dementia
- Treatments for frontotemporal dementia
- Outlook for frontotemporal dementia
What is Fronto Temporal Lobar Degeneration/Dementia
Frontotemporal dementia, is an uncommon cause of dementia, is a group of disorders that occur when nerve cells in the frontal and temporal lobes of the brain are lost 1. This causes the frontal and temporal lobes to shrink — the parts of the brain that control “executive functions” such as decision-making, personality, social behavior and language. Frontotemporal dementia can affect behavior, personality, language, and movement.
Frontotemporal degeneration is also commonly referred to as frontotemporal dementia, fronto-temporal lobar degeneration (FTLD), or Picks disease. Originally known as Pick’s disease, the name and classification of frontotemporal dementia has been a topic of discussion for over a century.
For many years, scientists and physicians used the term frontotemporal dementia (FTD) to describe this group of illnesses 2. After further research, FTD is now understood to be just one of several possible variations and is more precisely called behavioral variant frontotemporal dementia, or bvFTD 2.
The current designation of the syndrome groups together Pick’s disease, primary progressive aphasia, and semantic dementia as Frontotemporal dementia 3. Some doctors propose adding corticobasal degeneration and progressive supranuclear palsy to Frontotemporal dementia and calling the group Pick Complex. These designations will continue to be debated.
Frontotemporal dementia is distinct from other forms of dementia in two important ways 4:
- The hallmark of Frontotemporal dementia is a gradual, progressive decline in behavior and/or language (with memory usually relatively preserved). As the disease progresses, it becomes increasingly difficult for people to plan or organize activities, behave appropriately in social or work settings, interact with others, and care for oneself, resulting in increasing dependency on caregivers.
- Onset of Frontotemporal dementia often occurs in a person’s 50s and 60s, but has been seen as early as 21 and as late as 80 years. Roughly 60% of cases occur in people 45-64 years old 5, thus Frontotemporal dementia can affect work and family in a way dementia in older patients does not.
These disorders are among the most common dementias that strike at younger ages. Scientists estimate that FrontoTemporal Lobar Degeneration may cause up to 10 percent of all cases of dementia and is the second most common cause of dementia, after Alzheimer’s disease, in people younger than age 65 6. Roughly 60 percent of people with FrontoTemporal Lobar Degeneration symptoms typically start between the ages of 40 and 65, but FrontoTemporal Lobar Degeneration can strike young adults and those who are older.
There is a strong genetic component to the disease; Frontotemporal dementia often runs in families, but sporadic cases suggest that environmental triggers also exist. Several genetic etiologies are known but environmental triggers remain largely undefined. Frontotemporal dementia affects men and women equally.
The underlying biology of the Frontotemporal dementia variants is diverse. There are several clinical variants that involve cognition, behavior/affect and language. One major subgroup involves affect and social comportment (behavioral variant or bvFrontotemporal degeneration, also commonly referred to as frontotemporal dementia), and another is the syndrome of primary progressive aphasia (PPA). A dysexecutive syndrome may also be seen. Many or most affected individuals have overlapping symptoms, particularly as the disease progresses. The following descriptions of Frontotemporal degeneration subtypes present the most classic features of these subtypes. However, it is important to remember that the phenotype is highly variable and that many patients present with only some of the features or even with a memory disorder.
Clinical Features of Frontotemporal Dementia (Frontotemporal Degeneration Subtypes)
The most common types of Frontotemporal dementia are:
- Behavior Variant Frontotemporal Degeneration. Progressive behavior/personality decline—characterized by changes in personality, behavior, emotions, and judgment (called
behavioral variant frontotemporal dementia). - Primary Progressive Aphasia. Progressive language decline — Aphasia means difficulty communicating. This form has two subtypes: Progressive nonfluent aphasia, which affects the ability to speak. Semantic dementia, which affects the ability to use and understand language.
- Progressive Motor Decline. A less common form of frontotemporal dementia affects movement—characterized by various difficulties with physical movement, including the use of one or more limbs, shaking, difficulty walking, frequent falls, and poor coordination (called corticobasal syndrome, supranuclear palsy, or amyotrophic lateral sclerosis), causing symptoms similar to Parkinson disease or amyotrophic lateral sclerosis (Lou Gehrig’s disease).
In the early stages it can be hard to know which of these disorders a person has because symptoms and the order in which they appear can vary widely from one person to the next. Also, the same symptoms can appear later in different disorders. For example, language problems are most typical of primary progressive aphasia but can also appear later in the course of behavioral variant frontotemporal dementia.
Changes in Behavior (Behavior Variant Frontotemporal Degeneration)
The first type features behavior that can be either impulsive (disinhibited) or bored and listless (apathetic) and includes inappropriate social behavior; lack of social tact; lack of empathy; distractability; loss of insight into the behaviors of oneself and others; an increased interest in sex; changes in food preferences; agitation or, conversely, blunted emotions; neglect of personal hygiene; repetitive or compulsive behavior, and decreased energy and motivation.
In 2011, an international consortium developed revised guidelines for the diagnosis of behavioral variant frontotemporal dementia based on recent literature and collective experience 7. The following criteria delineates the new criteria for bvFTD (Behavior Variant Frontotemporal Degeneration or Frontotemporal Dementia).
Note: *As a general guideline ‘early’ refers to symptom presentation within the first 3 years.
I. Neurodegenerative disease
The following symptom must be present to meet criteria for bvFTD
A. Shows progressive deterioration of behaviour and/or cognition by observation or history (as provided by a knowledgeable informant).
II. Possible bvFTD
Three of the following behavioural/cognitive symptoms (A–F) must be present to meet criteria. Ascertainment requires that symptoms be persistent or recurrent, rather than single or rare events.
A. Early* behavioural disinhibition [one of the following symptoms (A.1–A.3) must be present]:
A.1. Socially inappropriate behaviour
A.2. Loss of manners or decorum
A.3. Impulsive, rash or careless actions
B. Early apathy or inertia [one of the following symptoms (B.1–B.2) must be present]:
B.1. Apathy
B.2. Inertia
C. Early loss of sympathy or empathy [one of the following symptoms (C.1–C.2) must be present]:
C.1. Diminished response to other people’s needs and feelings
C.2. Diminished social interest, interrelatedness or personal warmth
D. Early perseverative, stereotyped or compulsive/ritualistic behaviour [one of the following symptoms (D.1–D.3) must be present]:
D.1. Simple repetitive movements
D.2. Complex, compulsive or ritualistic behaviours
D.3. Stereotypy of speech
E. Hyperorality and dietary changes [one of the following symptoms (E.1–E.3) must be present]:
E.1. Altered food preferences
E.2. Binge eating, increased consumption of alcohol or cigarettes
E.3. Oral exploration or consumption of inedible objects
F. Neuropsychological profile: executive/generation deficits with relative sparing of memory and visuospatial functions [all of the following symptoms (F.1–F.3) must be present]:
F.1. Deficits in executive tasks
F.2. Relative sparing of episodic memory
F.3. Relative sparing of visuospatial skills
III. Probable bvFTD
All of the following symptoms (A–C) must be present to meet criteria.
A. Meets criteria for possible bvFTD
B. Exhibits significant functional decline (by caregiver report or as evidenced by Clinical Dementia Rating Scale or Functional Activities Questionnaire scores)
C. Imaging results consistent with bvFTD [one of the following (C.1–C.2) must be present]:
C.1. Frontal and/or anterior temporal atrophy on MRI or CT
C.2. Frontal and/or anterior temporal hypoperfusion or hypometabolism on PET or SPECT
IV. Behavioural variant FTD with definite FTLD Pathology
Criterion A and either criterion B or C must be present to meet criteria.
A. Meets criteria for possible or probable bvFTD
B. Histopathological evidence of FTLD on biopsy or at post-mortem
C. Presence of a known pathogenic mutation
V. Exclusionary criteria for bvFTD
Criteria A and B must be answered negatively for any bvFTD diagnosis. Criterion C can be positive for possible bvFTD but must be negative for probable bvFTD.
A. Pattern of deficits is better accounted for by other non-degenerative nervous system or medical disorders
B. Behavioural disturbance is better accounted for by a psychiatric diagnosis
C. Biomarkers strongly indicative of Alzheimer’s disease or other neurodegenerative process.
Dysexecutive Syndrome
Another frontotemporal dementia subtype is the dysexecutive syndrome. Here, an affected individual suffers from problems with complex reasoning and problem solving. Planning, sequencing and goal- directed behavior are involved. Problems with controlling attention and disinhibition underlie the syndrome; individuals can become stimulus-bound. These individuals cannot multitask, and they need guidance to remain on simple tasks including basic activities of daily living. In contrast, memory testing remains close to normal and these individuals may do well on simple mental status tests.
Problems with Language (Primary Progressive Aphasia)
The second type primarily features symptoms of language disturbance characterized by progressive loss of oral and written language skills, including difficulty making or understanding speech including difficulty reading and writing, often in conjunction with the behavioral variant FTD symptoms. Problems with spatial skills, memory and judgment are not apparent at first but can develop over time.
There are three types of Primary Progressive Aphasia, categorized by the kind of language problems seen at first. Researchers do not fully understand the biological basis of the different types of Primary Progressive Aphasia. But they hope one day to link specific language problems with the abnormalities in the brain that cause them.
In semantic PPA, also called semantic dementia, a person slowly loses the ability to understand single words and sometimes to recognize the faces of familiar people and common objects. Here, the meaning of specific words is lost, and both comprehension of the word and the ability to retrieve the name of an object may be lost. Speech remains fluent and grammar is good, however, paraphasias (word substitution errors) are common. This subtype of PPA is usually associated with TDP-43 pathology.
In agrammatic PPA, also called progressive nonfluent aphasia, a person has more and more trouble producing speech. Eventually, the person may no longer be able to speak at all. He or she may eventually develop movement symptoms similar to those seen in corticobasal syndrome. In this case, individuals lose grammar (the small connecting words) but have preserved language comprehension for specific items/objects. This causes speech to become effortful, hesitant, and sentence length becomes progressively truncated. Writing and language comprehension may be affected in the same manner. This subtype of PPA is usually associated with tau pathology.
Logopenic PPA is the third major PPA variant, a person has trouble finding the right words during conversation but can understand words and sentences. The person does not have problems with grammar. In these individuals, speech is slow, but grammar and comprehension are less affected. Impairment in the repetition of multisyllabic words and particularly phrases is a key feature. Sound substitution (phonemic) paraphasias are also seen in this group, as in a false word that rhymes with the intended word. Logopenic aphasia is usually associated with an underlying Alzheimer’s pathology.
Movement Disorders
Two rare neurological disorders associated with frontotemporal lobar degeneration, corticobasal syndrome (CBS) and progressive supranuclear palsy (PSP), occur when the parts of the brain that control movement are affected 2. The disorders may affect thinking and language abilities, too.
Corticobasal syndrome (CBS) can be caused by corticobasal degeneration—gradual atrophy and loss of nerve cells in specific parts of the brain. This degeneration causes progressive loss of the ability to control movement, typically beginning around age 60. The most prominent symptom may be the inability to use the hands or arms to perform a movement despite normal strength (called apraxia). Symptoms may appear first on one side of the body, but eventually both sides are affected. Occasionally, a person with Corticobasal syndrome (CBS) first has language problems or trouble orienting objects in space and later develops movement symptoms.
Progressive supranuclear palsy (PSP) causes problems with balance and walking. People with the disorder typically move slowly, experience unexplained falls, lose facial expression, and have body stiffness, especially in the neck and upper body—symptoms similar to those of Parkinson’s disease. A hallmark sign of PSP is trouble with eye movements, particularly looking down. These symptoms may give the face a fixed stare. Behavior problems can also develop.
Other movement-related frontotemporal disorders include frontotemporal dementia with parkinsonism and frontotemporal dementia with amyotrophic lateral sclerosis (FTD-ALS).
Frontotemporal dementia with parkinsonism can be an inherited disease caused by a genetic tau mutation. Symptoms include movement problems similar to those of Parkinson’s disease, such as slowed movement, stiffness, and balance problems, and changes in behavior or language.
Frontotemporal dementia and Motor Neuron Disease
FTD-ALS is a combination of bvFTD and ALS, commonly called Lou Gehrig’s disease. Symptoms include the behavioral and/or language changes seen in bvFTD as well as the progressive muscle weakness seen in ALS. Symptoms of either disease may appear first, with other symptoms developing over time. Mutations in certain genes have been found in some patients with FTD-ALS.
As many as 20% of frontotemporal dementia patients develop signs of motor neuron disease (MND). Likewise approximately half of amyotrophic lateral sclerosis (ALS) patients develop some fronto-executive problems 8. A smaller number of these patients develop full-blown FTD-ALS. Many patients that have the combination syndrome carry an expansion in the C9orf72 gene, and thus a hereditary form of the disease (see Genetics section).
With or without the gene expansion, the addition of motor neuron disease to frontotemporal dementia is a compromising factor which greatly reduces median survival to less than 3 years 9. Behavioral symptoms complicate the management of dysphagia as well as respiratory dysfunction as respiration therapy and percutaneous endoscopic gastrostomy (PEG) feeding tubes are not well tolerated by the patient. PEG shows little survival benefit 10.
Although significant phenotypic heterogeneity exists among C9orf72 carriers, the majority present with either bvFTD or FTD-ALS. PPA variants including nonfluent/agrammatic and semantic are rare. Prominent psychosis with delusions and hallucinations are relatively common 11). Research into the clinical features associated with the C9orf72 mutation is active and on-going.
OTHER VARIANTS
FTD subtypes may also be associated with the Parkinson’s plus syndromes of progressive supranuclear palsy (PSP) or corticobasal syndrome (CBS). CBS-like symptoms of asymmetrical movement problems, abnormal muscle tone, complex tremors, myoclonus and limb apraxia form the core CBS pheonotype. A recently appreciated speech/language variant known as primary progressive apraxia of speech (PPAOS) tends to precede the evolution into the nonfluent/agrammatic subtype; these patients experience articulatory difficulties but are not truly aphasic.
Table 1. Types of Frontotemporal Dementia (FTD) or Frontotemporal Lobar Degeneration (FLTD)
Changes in the Brain
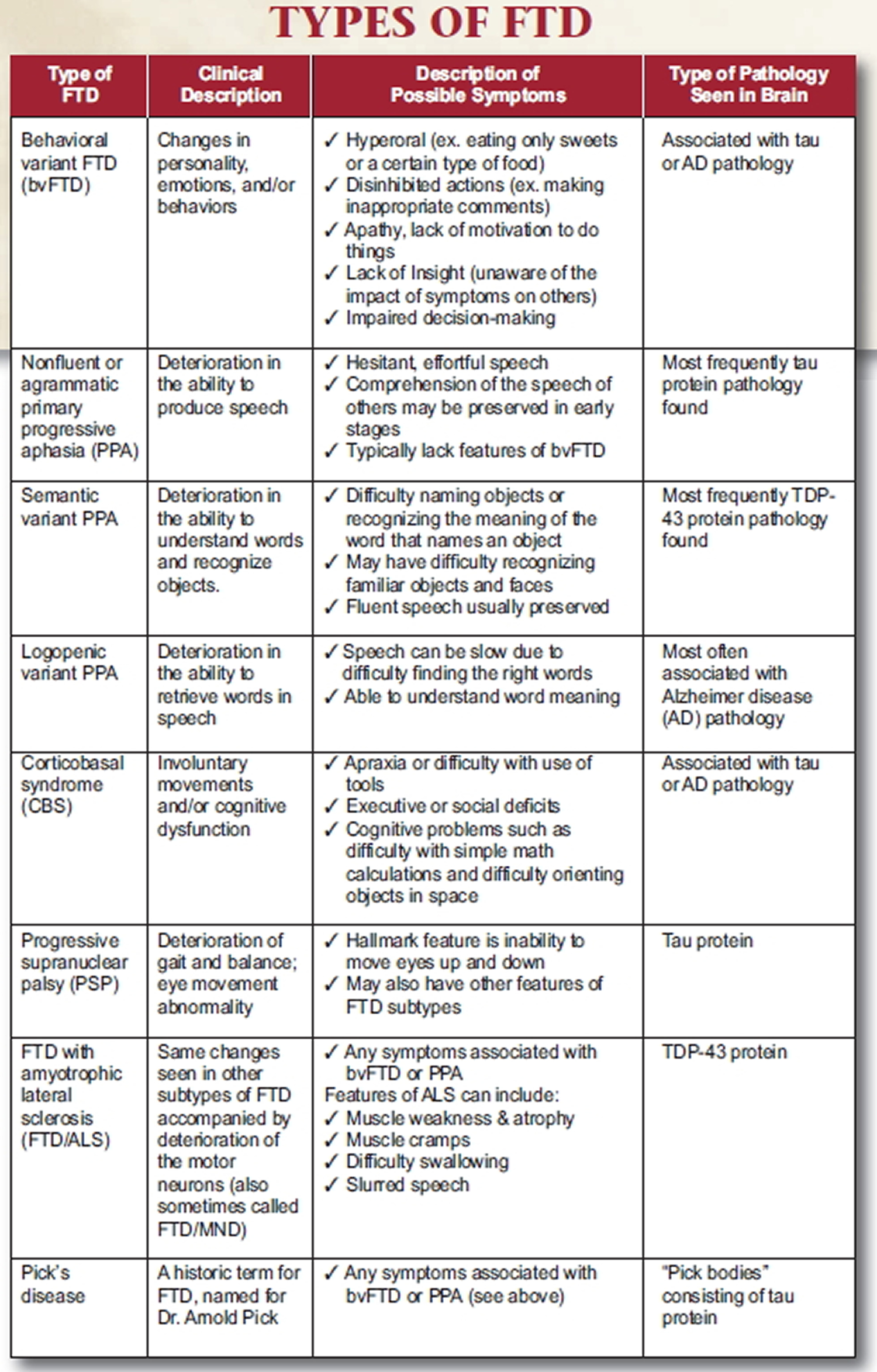
Frontotemporal disorders affect the frontal and temporal lobes of the brain (see Brain image below). They can begin in the frontal lobe, the temporal lobe, or both. Initially, frontotemporal disorders leave other brain regions untouched, including those that control short-term memory 2.
The frontal lobes, situated above the eyes and behind the forehead both on the right and left sides of the brain, direct executive functioning. This includes planning and sequencing (thinking through which steps come first, second, third, and so on), prioritizing (doing more important activities first and less important activities last), multitasking (shifting from one activity to another as needed), and monitoring and correcting errors.
When functioning well, the frontal lobes also help manage emotional responses. They enable people to avoid inappropriate social behaviors, such as shouting loudly in a library or at a funeral. They help people make decisions that make sense for a given situation. When the frontal lobes are damaged, people may focus on insignificant details and ignore important aspects of a situation or engage in purposeless activities. The frontal lobes are also involved in language, particularly linking words to form sentences, and in motor functions, such as moving the arms, legs, and mouth.
The temporal lobes, located below and to the side of each frontal lobe on the right and left sides of the brain, contain essential areas for memory but also play a major role in language and emotions. They help people understand words, speak, read, write, and connect words with their meanings. They allow people to recognize objects and to relate appropriate emotions to objects and events. When the temporal lobes are dysfunctional, people may have difficulty recognizing emotions and responding appropriately to them.
Which lobe—and part of the lobe—is affected first determines which symptoms appear first. For example, if the disease starts in the part of the frontal lobe responsible for decision-making, then the first symptom might be trouble managing finances. If it begins in the part of the temporal lobe that connects emotions to objects, then the first symptom might be an inability to recognize potentially dangerous objects—a person might reach for a snake or plunge a hand into boiling water, for example.
Causes of frontotemporal dementia
Frontotemporal lobar degeneration (frontotemporal dementia) is not a single brain disease but rather a family of brain diseases that share some common molecular features. Scientists are beginning to understand the biological and genetic basis for the changes observed in brain cells that lead to frontotemporal dementia.
Scientists describe frontotemporal dementia or frontotemporal lobar degeneration in terms of patterns of change in the brain seen in an autopsy after death. These changes include loss of neurons and abnormal amounts or forms of proteins called tau and TDP-43. These proteins occur naturally in the body and help cells function properly. When the proteins don’t work properly and accumulate in cells, for reasons not yet fully understood, neurons in specific brain regions are damaged.
In most cases, the cause of a frontotemporal disorder is unknown. In about 15 to 40 percent of people, a genetic (hereditary) cause can be identified. Individuals with a family history of frontotemporal disorders are more likely to have a genetic form of the disease than those without such a history.
Familial and inherited forms of frontotemporal disorders are often related to mutations (permanent changes) in certain genes. Genes are basic units of heredity that tell cells how to make the proteins the body needs to function. Even small changes in a gene may produce an abnormal protein, which can lead to changes in the brain and, eventually, disease.
Scientists have discovered several different genes that, when mutated, can lead to frontotemporal disorders:
- Tau gene (also called the MAPT gene)—A mutation in this gene causes abnormalities in a protein called tau, which forms tangles inside neurons and ultimately leads to the destruction of brain cells. Inheriting a mutation in this gene means a person will almost surely develop a frontotemporal disorder, usually the bvFTD form, but the exact age of onset and symptoms cannot be predicted.
- PGRN gene— A mutation in this gene can lead to lower production of the protein progranulin, which in turn causes TDP-43, a cellular protein, to go awry in brain cells. Many frontotemporal disorders can result, though bvFTD is the most common. The PRGRN gene can cause different symptoms in different family members and cause the disease to begin at different ages.
- VCP, CHMP2B, TARDBP, and FUS genes— Mutations in these genes lead to very rare familial types of frontotemporal disorders. TARDBP and FUS gene mutations are more often associated with hereditary ALS.
- C9ORF72 gene— An unusual mutation in this gene appears to be the most common genetic abnormality in familial frontotemporal disorders and familial ALS. It also occurs in some cases of sporadic ALS. This mutation can cause a frontotemporal disorder, ALS, or both conditions in a person.
Scientists are continuing to study these genes and to search for other genes and proteins, as well as nongenetic risk factors, that may play a role in frontotemporal disorders. They are trying to understand, for example, how mutations in a single gene lead to different frontotemporal disorders in members of the same family. Environmental factors that may influence risk for developing the disorders are also being examined.
If you have a family history of frontotemporal dementia, you may want to consider talking to your doctor about being referred to a geneticist and possibly having a genetic test to see if you’re at risk.
There’s a lot of research being done to try to improve understanding of the causes of frontotemporal dementia so treatments can be discovered.
Doctors sometimes use different names to describe frontotemporal dementia. These include:
- FTD
- Pick’s disease
- frontal dementia
- frontotemporal lobar degeneration
- behavioural variant frontotemporal dementia
- primary progressive aphasia
- semantic dementia
- progressive non-fluent aphasia.
Is Frontotemporal dementia hereditary ?
In the majority of cases, frontotemporal dementia is not being inherited within the family 13. However, people with both inherited and sporadic (not inherited) frontotemporal lobe dementia exhibit the same clinical symptoms, which makes evaluation of the family history the most sensitive tool for determining the likelihood of a genetic cause.
While scientists have found gene mutations that are linked to frontotemporal lobe dementia, most cases of frontotemporal lobe dementia are sporadic, meaning that there is no known family history of frontotemporal lobe dementia. Current genetic research shows that if a diagnosed individual has no family history of frontotemporal lobe dementia or related neurological conditions, there is less than a 10% chance that they carry a mutation in a currently known frontotemporal lobe dementia gene.
However, approximately 40% of individuals with frontotemporal lobe dementia do have a family history that includes at least one other relative who also has or had a neurodegenerative disease. In approximately 15-40% of all frontotemporal lobe dementia cases, a genetic cause (e.g., a gene mutation) can be identified as the likely cause of the disease and in most cases it is an inherited mutation.
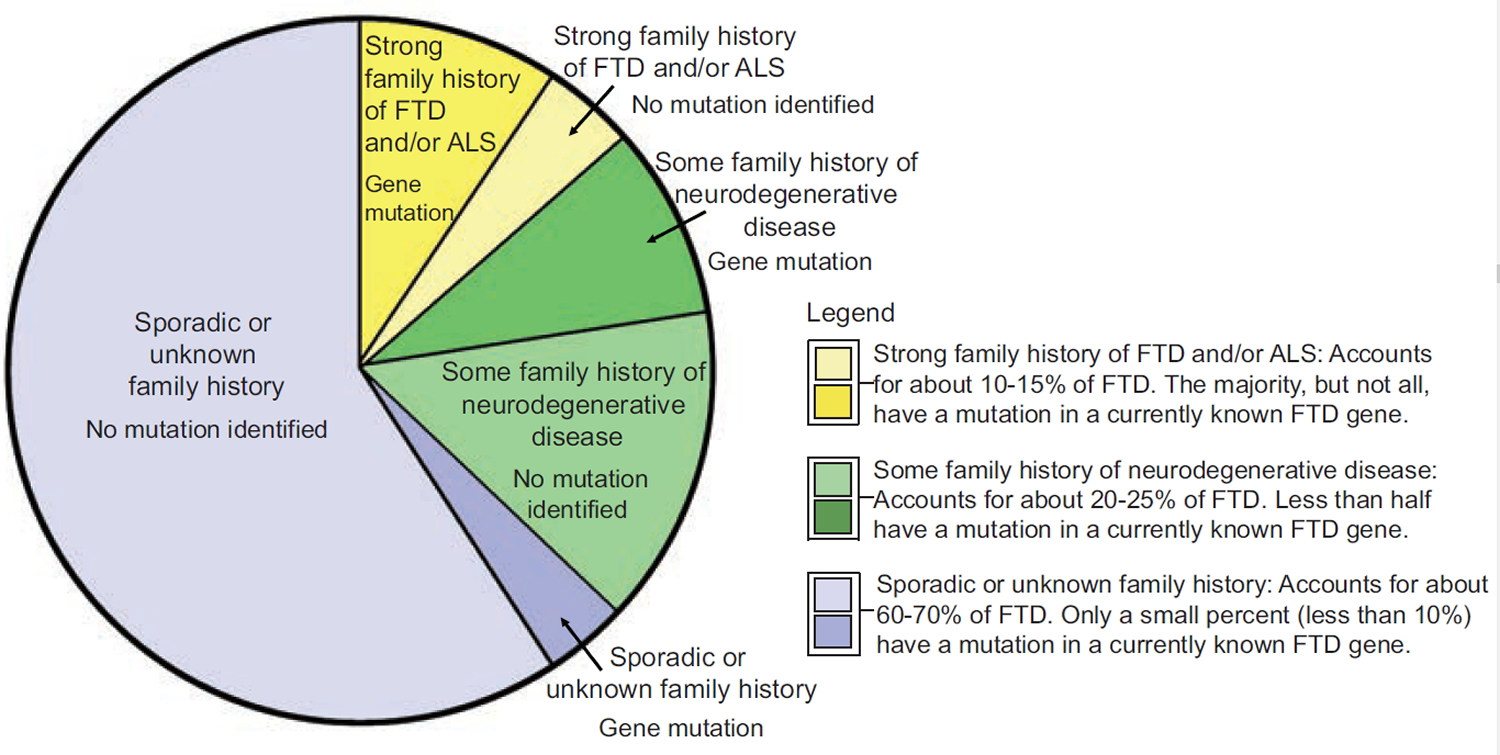
As seen in the pie chart image, families that include a history of multiple relatives with FTD (frontotemporal dementia) and/or ALS are the most likely to have an underlying genetic cause, while cases with sporadic or unknown family histories are the least likely. There are also families that fall in the middle, meaning that there is at least one relative who has or had a related neurodegenerative disease like Alzheimer’s disease or Parkinson disease, but not a clear-cut family history of FTD or ALS in close relatives. In short, the greater the family history of FTD or an associated disease like ALS, the more likely genetics plays a role. It is important to note that not all individuals with a family history of neurodegenerative disease will have an identifiable gene mutation. This could be because the responsible gene has not yet been identified, or because the disease is not actually due to a gene mutation.
There are many descriptive words used in the medical setting when discussing family history. These words are often used interchangeably, making it potentially confusing to understand each term’s meaning and significance. For example, the term “hereditary” indicates that a trait or disease can be directly transmitted between parent and offspring. “Familial” is a very broad term used to indicate that more than one person in a family has a trait or disease. “Familial” denotes that
there is a possibility of a genetic cause due to apparent clustering of affected individuals in a family. This may occur by chance or due to a genetic mutation (hereditary cause). When a genetic counselor looks at a person’s family history and draws a pedigree, they look for patterns of disease in the family and depending on the number and type of affected individuals, a “pattern of inheritance” is assigned.
A classic pattern of genetic inheritance is called autosomal dominant. “Autosomal” refers to the first 22 pairs of chromosomes, which are identical in both males
and females. Therefore, both genders have an equal chance of being affected if a mutation is present on a gene in an autosomal chromosome. “Dominant” inheritance means that only one copy of a gene has to have a mutation to cause disease. Typically, the copy of the gene with the mutation is inherited from a parent, and most individuals with an autosomal dominant condition will have a family history of the disease. A child of a person with a dominant mutation has a
50% chance of inheriting the copy of the gene with the mutation and a 50% chance of inheriting the copy of the gene without the mutation.
At this time, the only proven genetic causes of FTD fall under the pattern of autosomal dominant inheritance. It is believed that there are likely other factors, both genetic and environmental, that influence an individual’s risk of developing FTD. There may be genes or specific mutations that do not directly cause FTD, but rather increase susceptibility to disease. This type of genetic finding is referred to as a risk factor. It is also likely that a combination of both genetic and environmental factors influence the risk of disease; this is often referred to as multifactorial inheritance. Research is underway to determine other risk factors that play a role in the development of FTD.
Scientists have discovered several different genes that, when mutated, can lead to frontotemporal disorders:
- Tau gene (also called the MAPT gene)—A mutation in this gene causes abnormalities in a protein called tau, which forms tangles inside neurons and ultimately leads to the destruction of brain cells. Inheriting a mutation in this gene means a person will almost surely develop a frontotemporal disorder, usually the bvFTD form, but the exact age of onset and symptoms cannot be predicted.
- PGRN gene— A mutation in this gene can lead to lower production of the protein progranulin, which in turn causes TDP-43, a cellular protein, to go awry in brain cells. Many frontotemporal disorders can result, though bvFTD is the most common. The PRGRN gene can cause different symptoms in different family members and cause the disease to begin at different ages.
- VCP, CHMP2B, TARDBP, and FUS genes— Mutations in these genes lead to very rare familial types of frontotemporal disorders. TARDBP and FUS gene mutations are more often associated with hereditary ALS.
- C9ORF72 gene— An unusual mutation in this gene appears to be the most common genetic abnormality in familial frontotemporal disorders and familial ALS. It also occurs in some cases of sporadic ALS. This mutation can cause a frontotemporal disorder, ALS, or both conditions in a person. Individuals with the C9ORF72 mutation have abnormal accumulations of the TDP-43 protein in affected neurons.
- Not all genetic causes are known. Scientists are still working to find other genes that contribute to frontotemporal dementia. As with any human
disease, there may be environmental or genetic risk factors that could increase susceptibility to a disease. At this time, there are no known risk factors that could indicate a susceptibility to FTD, but ongoing research is working to better understand risk factors for FTD.
Clinical genetic testing is available to look for mutations in MAPT, GRN, VCP, TARDBP, C9orf72 and FUS. A doctor or genetic counselor would be able to
provide more details about the specific genetic testing options that are most appropriate based on an individual’s symptoms and family history.
If an individual has a mutation in one of the known FTD associated genes, there is a 50% chance of passing on the mutation and a 50% chance of NOT passing on the mutation to his/her child. This same 50/50 risk applies to every child of that individual. If the child does inherit the mutation, he or she will most likely develop symptoms of the disease at some point in adulthood, but the age of onset, severity, and type of symptoms cannot be predicted at this time.
Genetic testing can be done either with blood or saliva being collected and used to isolate a sample of DNA. This DNA sample can then be analyzed for mutations in the specific genes of interest. For individuals with an unknown genetic cause, a complete analysis for any known disease-causing genetic changes would need to be performed. However, for individuals that already know the specific gene mutation that is present in their family, the genetic test can be targeted to just check for that known family mutation.
Who can get genetic testing for frontotemporal dementia ?
Any individual with frontotemporal dementia who is interested in genetic testing may get tested, although a genetic cause is more likely in those individuals who also have a family history of frontotemporal dementia. Anyone considering genetic testing or who has a family history of frontotemporal dementia should seek genetic counseling prior to genetic testing. The test is most beneficial when a family member with frontotemporal dementia is the first person tested. Once a mutation has been identified, other members of the family may choose to be tested for the same mutation even if they don’t have symptoms of the disease (presymptomatic genetic testing).
Identifying a gene that causes FTD in an affected individual will not provide an answer for curing the disease, but may help prevent the patient from being treated for the wrong diagnosis. Additionally, if an unaffected person finds out that they inherited a FTD gene mutation, there is no treatment available at this time to prevent the onset of disease.
Testing a person who is currently not showing symptoms but who is at risk for inheriting a known family mutation is called presymptomatic (or predictive) testing. For pre-symptomatic testing, a mutation must have been first identified in the family so that the clinician knows the appropriate test to order and can properly interpret the results. Individuals in the same family may have differing opinions on whether or not to pursue pre symptomatic testing. The pursuit of pre-symptomatic testing is a very personal decision that may be confusing and emotionally difficult. Therefore, genetic counseling and psychological evaluation
are usually required to help the individual throughout the testing process.
The interpretation of genetic test results can be confusing. It is recommended that anyone who has genetic testing for frontotemporal dementia discuss the results with a genetic counselor or another medical genetics professional.
Symptoms of frontotemporal dementia
Symptoms of frontotemporal disorders vary from person to person and from one stage of the disease to the next as different parts of the frontal and temporal lobes are affected. In general, changes in the frontal lobe are associated with behavioral symptoms, while changes in the temporal lobe lead to language and emotional disorders. These come on gradually and get worse slowly over time. Eventually, most people will experience problems in both of these areas. Some people also develop physical problems and difficulties with their mental abilities.
Symptoms are often misunderstood. Family members and friends may think that a person is misbehaving, leading to anger and conflict. For example, a person with bvFTD may neglect personal hygiene or start shoplifting. It is important to understand that people with these disorders cannot control their behaviors and other symptoms. Moreover, they lack any awareness of their illness, making it difficult to get help.
Behavior and personality changes
Many people with frontotemporal dementia develop a number of unusual behaviors they’re not aware of.
These can include Behavioral Symptoms:
- being insensitive or rude
- acting impulsively or rashly – difficulty resisting impulses to use or touch objects that one can see and reach. For example, a person picks up the telephone receiver while walking past it when the phone is not ringing and the person does not intend to place a call.
- loss of inhibitions
- seeming subdued
- losing interest in people and things
- losing drive and motivation
- inability to empathise with others, seeming cold and selfish
- repetitive behaviors, such as humming, hand-rubbing and foot-tapping, or routines such as walking exactly the same route repetitively
- overeating, a change in food preferences, such as suddenly liking sweet foods, and poor table manners like taking food from other people’s plates.
- neglecting personal hygiene
As the condition progresses, people with frontotemporal dementia may become socially isolated and withdrawn.
Language problems
Some people experience problems with speech and language, including:
- using words incorrectly – for example, calling a sheep a dog
- loss of vocabulary
- repeating a limited number of phrases – a tendency to repeat the same activity or to say the same word over and over, even when it no longer makes sense.
- forgetting the meaning of common words
- slow, hesitant speech
- difficulty making the right sounds to say words
- getting words in the wrong order
- automatically repeating things other people have said
Some people gradually lose the ability to speak, and can eventually become completely mute.
Problems with mental abilities
Problems with thinking don’t tend to occur in the early stages of frontotemporal dementia, but these often develop as the condition progresses.
These can include:
- difficulty working things out and needing to be told what to do
- poor planning, judgement and organisation
- becoming easily distracted
- thinking in a rigid and inflexible way
- losing the ability to understand abstract ideas
- difficulty recognising familiar people or objects
- memory difficulties, although this isn’t common early on
Emotional Symptoms
- Apathy—A lack of interest, drive, or initiative. Apathy is often confused with depression, but people with apathy may not be sad. They often have trouble starting activities but can participate if others do the planning.
- Compulsive eating—Gorging on food, especially starchy foods like bread and cookies, or taking food from other people’s plates.
- Emotional changes—Emotions are flat, exaggerated, or improper. Emotions may seem completely disconnected from a situation or are expressed at the wrong times or in the wrong circumstances. For example, a person may laugh at sad news.
- Social-interpersonal changes—Difficulty “reading” social signals, such as facial expressions, and understanding personal relationships. People may lack empathy—the ability to understand how others are feeling—making them seem indifferent, uncaring, or selfish. For example, the person may show no emotional reaction to illnesses or accidents that occur to family members.
Physical and Movement problems
In the later stages, some people with frontotemporal dementia develop physical problems and difficulties with movement.
These can include:
- slow, stiff movements, similar to Parkinson’s disease
- difficulty swallowing
- loss of bladder control
- loss of bowel control
- Dystonia—Abnormal postures of body parts such as the hands or feet. A limb may be bent stiffly or not used when performing activities that are normally done with two hands.
- Gait disorder—Abnormalities in walking, such as walking with a shuffle, sometimes with frequent falls.
- Tremor—Shakiness, usually of the hands.
- Clumsiness—Dropping of small objects or difficulty manipulating small items like buttons or screws.
- Apraxia—Loss of ability to make common motions, such as combing one’s hair or using a knife and fork, despite normal strength.
- Neuromuscular weakness—Severe weakness, cramps, and rippling movements in the muscles.
Some people have frontotemporal dementia overlapping with other neurological (nerve and brain) problems, including:
- motor neurone disease – causes increasing weakness, usually with muscle wasting
- corticobasal degeneration – causes problems controlling limbs, loss of balance and co-ordination, slowness and reduced mobility
- progressive supranuclear palsy – causes problems with balance, movement, eye movements and swallowing
Damage to the frontal lobe of the brain may impact important functions. Common symptoms involve dramatic changes in behavior and personality. These may include:
- An increased tendency to make socially inappropriate comments or actions
- Decreased empathy, or new difficulties understanding how one’s actions may impact others
- Difficulties with logical judgments or understanding the relationship between cause and effect
- Changes in sexual behaviors
- Aggressive behaviors or actions
- Decline in personal hygiene, toileting habits, etc.
- Severe mental rigidity
- Language abnormalities, such as being unable to express language, find words or understand the meaning of words
- Inattention, increased distractibility or a tendency to jump from one topic to another
- Difficulty initiating or completing tasks
- Significant changes in eating patterns
In addition to cognitive impairments, neurologic symptoms may occur including:
- Parkinsonism: slowness of movement (bradykinesia), increased rigidity in the arms and/or legs, problems with walking (short stride length or a “shuffling” gait)
- Tremor
- Muscle spasms and/or rippling of the muscles underneath the skin
- Seizures
Tests for frontotemporal dementia
No single test, such as a blood test, can be used to diagnose a frontotemporal disorder. A definitive diagnosis can be confirmed only by a genetic test in familial cases or a brain autopsy after a person dies. To diagnose a probable frontotemporal disorder in a living person, a doctor— usually a neurologist, psychiatrist, or psychologist—will:
Require a thorough history, verified by a caregiver, and a neurological examination.
The following may be needed to make a diagnosis:
- an assessment of symptoms – it’s normally helpful to have somebody who knows the person well to give an account of their symptoms, especially as someone with frontotemporal dementia may not be aware of changes in their behaviour
- an assessment of mental abilities – this will usually involve a number of tasks and questions
- blood tests – to rule out conditions with similar symptoms
- brain scans – such as an MRI scan, a CT scan or a PET scan; these can detect signs of dementia and help identify which parts of the brain are most affected, or help rule out other problems with the brain
- lumbar puncture – to test the spinal fluid; this may be useful to rule out Alzheimer’s disease as the cause of symptoms
- All patients should be screened for obstructive sleep apnea (OSA), as executive dysfunction and behavior changes are common in OSA. If the classic features of OSA are present (e.g., loud disruptive snoring, snorts and apneic pauses while sleeping, crowded oropharynx, excessive daytime sleepiness, repetitive desaturations on overnight oximetry), then referral to a sleep medicine specialist and polysomnography is indicated. Vascular risk factors should be assessed. Infections (including HIV), immune based dementias and neoplastic/paraneoplastic etiologies are occasionally causative or significant contributors, and should be considered. When a family history is positive, genetic testing can be undertaken.
Frontotemporal disorders can be hard to diagnose because their symptoms—changes in personality and behavior and difficulties with speech and movement—are similar to those of other conditions. For example, bvFTD is sometimes misdiagnosed as a mood disorder, such as depression, or as a stroke, especially when there are speech or movement problems. To make matters more confusing, a person can have both a frontotemporal disorder and another type of dementia, such as Alzheimer’s disease. Also, since these disorders are rare, physicians may be unfamiliar with the relevant symptoms and signs.
Getting the wrong diagnosis can be frustrating. Without knowing their true condition, people with frontotemporal disorders may not get appropriate treatment to manage their symptoms. Families may not get the help they need. People lose valuable time needed to plan treatment and future care.
Researchers are studying ways to diagnose frontotemporal disorders earlier and more accurately. One area of research involves biomarkers, such as proteins or other substances in the blood or cerebrospinal fluid, which can be used to measure the progress of disease or the effects of treatment. Also being studied are ways to improve brain imaging, including seeing the tau protein, and neuropsychological testing, which assesses learning, language, problem solving, memory, and other thinking skills.
Treatments for frontotemporal dementia
No treatment has been shown to slow the progression of frontotemporal dementia. Behavior modification may help control unacceptable or dangerous behaviors. Aggressive, agitated, or dangerous behaviors could require medication. Anti-depressants have been shown to improve some symptoms.
Managing Behavior
The behaviors of a person with bvFTD can upset and frustrate family members and other caregivers. It is natural to grieve for the “lost person,” but it is also important to learn how to best live with the person he or she has become. Understanding changes in personality and behavior and knowing how to respond can reduce caregivers’ frustration and help them cope with the challenges of caring for a person with a frontotemporal disorder.
Managing behavioral symptoms can involve several approaches. To ensure the safety of a person and his or her family, caregivers may have to take on new responsibilities or arrange care that was not needed before. For example, they may have to drive the person to appointments and errands, care for young children, or arrange for help at home.
It is helpful, though often difficult, to accept rather than challenge people with behavioral symptoms. Arguing or reasoning with them will not help because they cannot control their behaviors or even see that they are unusual or upsetting to others. Instead, be as sensitive as possible and understand that it’s the illness “talking.”
Frustrated caregivers can take a “timeout”—take deep breaths, count to 10, or leave the room for a few minutes.
To deal with apathy, limit choices and offer specific choices. Open-ended questions (“What would you like to do today?”) are more difficult to answer than specific ones (“Do you want to go to the movies or the shopping center today?”).
Maintaining the person’s schedule and modifying the environment can also help. A regular schedule is less confusing and can help people sleep better. If compulsive eating is an issue, caregivers may have to supervise eating, limit food choices, lock food cabinets and the refrigerator, and distract the person with other activities. To deal with other compulsive behaviors, caregivers may have to change schedules or offer new activities.
Medications are available to treat certain behavioral symptoms. Antidepressants called selective serotonin reuptake inhibitors are commonly prescribed to treat social disinhibition and impulsive behavior. Patients with aggression or delusions sometimes take low doses of antipsychotic medications. The use of Alzheimer’s disease medications to improve behavioral and cognitive symptoms in people with bvFTD and related disorders is being studied, though results so far have been mixed, with some medications making symptoms worse. If a particular medication is not working, a doctor may try another. Always consult a doctor before changing, adding, or stopping a drug.
Treating Language Problems
Treatment of primary progressive aphasia (PPA) has two goals—maintaining language skills and using new tools and other ways to communicate. Treatment tailored to a person’s specific language problem and stage of PPA generally works best. Since language ability declines over time, different strategies may be needed as the illness progresses.
To communicate without talking, a person with PPA may use a communication notebook (an album of photos labeled with names of people and objects), gestures, and drawings. Some people find it helpful to use or point to lists of words or phrases stored in a computer or personal digital assistant.
Caregivers can also learn new ways of talking to someone with PPA. For example, they can speak slowly and clearly, use simple sentences, wait for responses, and ask for clarification if they don’t understand something.
A speech-language pathologist who knows about PPA can test a person’s language skills and determine the best tools and strategies to use. Note that many speech-language pathologists are trained to treat aphasia caused by stroke, which requires different strategies from those used with PPA. (See the Resources section starting on page 27 to find speech-language pathologists and other experts who know about frontotemporal disorders.)
Managing Movement Problems
No treatment can slow down or stop frontotemporal-related movement disorders, though medications and physical and occupational therapy may provide modest relief.
For people with corticobasal syndrome (CBS), movement difficulties are sometimes treated with medications for Parkinson’s disease. But these medicines offer only minimal or temporary improvement. Physical and occupational therapy may help people with CBS move more easily. Speech therapy may help them manage language symptoms.
For people with progressive supranuclear palsy (PSP), sometimes Parkinson’s disease drugs provide temporary relief for slowness, stiffness, and balance problems. Exercises can keep the joints limber, and weighted walking aids— such as a walker with sandbags over the lower front rung—can help maintain balance. Speech, vision, and swallowing difficulties usually do not respond to any drug treatment. Antidepressants have shown modest success. For people with abnormal eye movements.
People with FTD-ALS typically decline quickly over the course of 2 to 3 years. During this time, physical therapy can help treat muscle symptoms, and a walker or wheelchair may be useful. Speech therapy may help a person speak more clearly at first. Later on, other ways of communicating, such as a speech synthesizer, can be used. The ALS symptoms of the disorder ultimately make it impossible to stand, walk, eat, and breathe on one’s own.
For any movement disorder caused by FTLD, a team of experts can help patients and their families address difficult medical and caregiving issues. Physicians, nurses, social workers, and physical, occupational, and speech therapists who are familiar with frontotemporal disorders can ensure that people with movement disorders get appropriate medical treatment and that their caregivers can help them live as well as possible.
Care Plans
Before treatment starts, your current and future health and social care needs will be assessed, and a care plan drawn up.
This is a way of ensuring you receive the right treatment for your needs. It involves identifying areas where you may need some assistance.
These may be:
- what support you or your carer need for you to remain as independent as possible – including whether you might need care at home or in a nursing home
- whether there are any changes that need to be made to your home to make it easier to live in
- whether you need any financial assistance
Medication
Medication can’t stop frontotemporal dementia getting worse, but it can help reduce some of the symptoms for some people.
The following medicines may help:
- antidepressants – antidepressants called selective serotonin reuptake inhibitors (SSRIs) may help control the loss of inhibitions, overeating and compulsive behaviours seen in some people
- antipsychotics – these are rarely used, but if needed they can help control severely challenging behaviour that’s putting the person with dementia or others around them at risk of harm
The medications for Alzheimer’s disease aren’t effective for frontotemporal dementia.
Support and other therapies
In addition to medication, there are a number of therapies and practical measures that can help make everyday living easier for someone with dementia.
These include:
- occupational therapy – to identify problem areas in everyday life, such as getting dressed, and help work out practical solutions
- speech and language therapy – to help improve any communication or swallowing problems
- physiotherapy – to help with movement difficulties
- relaxation techniques – such as massage, and music or dance therapy
- social interaction, leisure activities and other dementia activities – such as memory cafés, which are drop-in sessions for people with memory problems and their carers to get support and advice
- strategies for challenging behaviour – such as distraction techniques, a structured daily routine, and activities like doing puzzles or listening to music
incontinence products if needed
It may also be helpful to get in touch with a support group, such as Rare Dementia Support, the Alzheimer’s Society or Dementia Organization.
End of life and legal issues
If you’ve been diagnosed with dementia, you might want to make arrangements for your care that take into account the decline in your mental abilities.
This may include ensuring your wishes are upheld if you’re not able to make decisions for yourself.
You may want to consider:
- drawing up an advance decision – this makes your treatment preferences known in case you’re unable to do this in the future
- having a plan for where you want to receive treatment as your condition becomes more advanced
- giving a relative lasting power of attorney – this is the power to make decisions about you if you’re unable to.
Outlook for frontotemporal dementia
How quickly frontotemporal dementia gets worse varies from person to person and is very difficult to predict.
People with the condition can become socially isolated as the illness progresses. They may not want to spend time in the company of others, or may behave in rude or insulting ways. The disease progresses steadily and often rapidly, ranging from less than 2 years in some individuals to more than 10 years in others.
Eventually some individuals with frontotemporal dementia will need 24-hour care and monitoring at home or in an institutionalized care setting. Home-based help will usually be needed at some stage, and some people will eventually need care in a nursing home.
The average survival time after symptoms start is around eight years. But this is highly variable and some people live much longer than this.
If you or a loved one has been diagnosed with dementia, remember you’re not alone. The National Institutes of Health, National Institute on Aging and social services, as well as voluntary organisations and specialist support groups, can provide advice and support for you and your family.
- UC Gardner Neuroscience Institute. Understanding Memory Disorders. http://uchealth.com/memory-disorders/conditions/#frontotemporal-dementia[↩]
- National Institute of Neurological Disorders and Stroke. Frontotemporal Disorders: Hope Through Research. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Hope-Through-Research/Frontotemporal-Disorders[↩][↩][↩][↩]
- The National Institute of Neurological Disorders and Stroke. Frontotemporal Dementia Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Frontotemporal-Dementia-Information-Page[↩]
- The Association for Frontotemporal Degeneration. Disease Overview. https://www.theaftd.org/understandingftd/ftd-overview[↩]
- Knopman DS. Alzheimer’s disease and other dementias. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier Saunders; 2011:chap 409[↩]
- National Institutes of Health. National Institute on Aging. Frontotemporal Disorders. https://order.nia.nih.gov/sites/default/files/2017-07/ADEAR_FTD_508.pdf[↩]
- Rascovsky K et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011 Sep, 134(pt9): 2456-77. Epub 2011 Aug 2. https://www.theaftd.org/wp-content/uploads/2013/01/bvFTD-criteria-article.pdf[↩]
- Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003: 60 (7), 1094–1097.[↩]
- Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, et al. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. 2005; 65:1774–7.[↩]
- Alagiakrishnan K, Bhanji RA, Kurian M, Evaluation and management of oropharyngeal dysphagia in different types of dementia: a systematic review. Arch Gerontol Geriatr. 2013 Jan-Feb; 56(1):1-9.[↩]
- Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR Jr, Parisi JE, Lucas JA, Petersen RC. Rademakers R. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012 Mar;135 (Pt 3):765-83.[↩]
- The Association for Frontotemporal Degeneration. Genetics of FTD. https://www.theaftd.org/wp-content/uploads/2009/03/Final-FTD-Genetics-Brochure-with-Cover-8.2.2012.pdf[↩][↩]
- The Association for Frontotemporal Degeneration. Genetics of FTD. https://www.theaftd.org/understandingftd/genetics[↩]





{kind=link}