Contents
- Glomerular disease
- Glomerular disease causes
- Glomerular disease prevention
- Glomerular disease signs and symptoms
- Glomerular disease complications
- Glomerular disease diagnosis
- Glomerular disease treatment
- Glomerular disease diet
- Focal Segmental Glomerulosclerosis (FSGS)
- Focal segmental glomerulosclerosis types
- Focal segmental glomerulosclerosis causes
- Focal segmental glomerulosclerosis pathophysiology
- Focal segmental glomerulosclerosis histopathology
- Focal segmental glomerulosclerosis signs and symptoms
- Focal segmental glomerulosclerosis complications
- Focal segmental glomerulosclerosis diagnosis
- Focal segmental glomerulosclerosis treatment
- Focal segmental glomerulosclerosis prognosis
- Focal segmental glomerulosclerosis life expectancy
- Membranous nephropathy
- Minimal change disease
- IgA Nephropathy
- Collapsing glomerulopathy
Glomerular disease
Glomerular diseases damage the tiny blood vessels called glomerulus (more than one glomerulus are called glomeruli) where your blood is cleaned within your kidneys, letting protein (mainly albumin) and sometimes red blood cells to leak into your urine. Glomerular disease can damage your kidneys and, in some cases, lead to kidney failure. Sometimes a glomerular disease also interferes with the clearance of waste products by the kidney, so they begin to build up in your blood. Furthermore, loss of blood proteins like albumin in the urine can result in a fall in their level in the bloodstream. In normal blood, albumin acts like a sponge, drawing extra fluid from the body into the bloodstream, where it remains until the kidneys remove it. But when albumin leaks into your urine, the blood loses its capacity to absorb extra fluid from your body. Fluid can accumulate outside your circulatory system, leading to swelling in your face, hands, feet, or ankles and cause swelling (edema). Glomerular disease affects men and women of all ages and all racial and ethnic groups. Having a family member who has glomerular disease increases your risk.
Many diseases and conditions can damage the glomeruli. Two broad terms used to describe many forms of damage to the glomeruli are:
- Glomerulonephritis = inflammation (swelling) of the glomeruli. The reason glomerulonephritis appears is often unknown. Glomerulonephritis causes may include:
- A complication of bacterial endocarditis, an infection in your heart valves.
- A complication of infections like strep throat, HIV or hepatitis C.
- Problems with your immune system attacking healthy parts of your body, such as with lupus.
- Anti-GBM disease (Goodpasture syndrome), a group of autoimmune diseases that affect your lungs and kidneys.
- IgA nephropathy, a kidney disease caused by a buildup of abnormal IgA antibody (immunoglobulin A).
- Rare diseases that inflame blood vessels like granulomatosis with polyangiitis (Wegener’s disease), microscopic polyangiitis, Henoch-Schönlein purpura, or eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome).
- Genetics, meaning it runs in your biological family (this is rare).
- Certain types of cancer (like multiple myeloma).
- Glomerulosclerosis = scarring or hardening of the glomeruli. This condition is a scarring (sclerosis) of the glomeruli. Lupus and diabetes are two examples of diseases that can cause glomerulosclerosis.
The structural and functional unit of your kidney, the ‘nephron,’ consists of a renal corpuscle (glomerulus surrounded by a Bowman capsule) and a renal tubule (Figure 1 and 2) 1. Each kidney in an adult human contains around 1 million nephrons 2. A fenestrated endothelium forms the inner glomerular layer, followed by a layer composed of various extracellular proteins forming a meshwork called the glomerular basement membrane (GBM). The outer layer has visceral epithelial cells, podocytes, and mesangial cells. The intricate arrangement provides the basis for continuous plasma volume filtration at the glomerular level.
The glomeruli filter your blood as it passes through your kidneys, separating things your body needs from those it doesn’t. Healthy glomeruli keep blood protein (mainly albumin) — which is needed to maintain the right amount of fluid in your body — from seeping into your urine. When damaged, glomeruli allow too much blood protein to leave your body, leading to nephrotic syndrome also called nephrosis. Some people with glomerular disease have nephrotic syndrome. Nephrotic syndrome is a group of symptoms that indicate your kidneys are not working properly. Nephrotic syndrome happens when your kidneys lose large amounts of protein in your urine and causes extra fluids and salt build up in your body. This causes you to have swelling (edema), high blood pressure, and higher levels of cholesterol. Nephrotic syndrome may come from kidney diseases or from other illnesses such as diabetes and lupus. Some medicines, IV drug abuse, and HIV (the AIDS virus) may also cause it. In some people, nephrotic syndrome goes away after treatment. But for others, this condition may last for many years and over time, lead to kidney failure.
Other people with glomerular disease may not have nephrotic syndrome, but still have some signs of it, such as protein in their urine, edema, and high blood pressure. They may also have other signs that are not part of nephrotic syndrome, such as blood in the urine, inflammation of glomeruli, and lower kidney function because of kidney damage. If you have all of these extra signs, then you may have nephritic syndrome, which is caused by inflammation in the filters of the kidney called glomerulonephritis.
Signs and symptoms of nephrotic syndrome include:
- Severe swelling (edema), particularly around your eyes and in your ankles and feet
- Foamy urine, a result of excess protein in your urine (proteinuria)
- Low levels of a protein called albumin in your blood (hypoalbuminemia)
- Weight gain due to fluid retention
- High levels of cholesterol and other lipids (fats) in your blood, called hyperlipidemia or hypercholesterolemia
- Fatigue
- Loss of appetite
Signs of nephritic syndrome can include:
- Blood in the urine (dark, rust-colored, or brown urine)
- Foamy urine (due to excess protein in the urine)
- Swelling (edema) of the face, eyes, ankles, feet, legs, or abdomen
- Anemia
- High blood pressure
- Signs of reduced kidney function
Possible complications of nephrotic syndrome include:
- Blood clots. The inability of the glomeruli to filter blood properly can lead to loss of blood proteins that help prevent clotting. This increases your risk of developing a blood clot in your veins.
- High blood cholesterol and elevated blood triglycerides. When the level of the protein albumin in your blood falls, your liver makes more albumin. At the same time, your liver releases more cholesterol and triglycerides.
- Poor nutrition. Loss of too much blood protein can result in malnutrition. This can lead to weight loss, which can be masked by edema. You may also have too few red blood cells (anemia), low blood protein levels and low levels of vitamin D.
- High blood pressure. Damage to your glomeruli and the resulting buildup of excess body fluid can raise your blood pressure.
- Acute kidney injury. If your kidneys lose their ability to filter blood due to damage to the glomeruli, waste products can build up quickly in your blood. If this happens, you might need emergency dialysis — an artificial means of removing extra fluids and waste from your blood — typically with an artificial kidney machine (dialyzer).
- Chronic kidney disease (CKD). Nephrotic syndrome can cause your kidneys to lose their function over time. If kidney function falls low enough, you might need dialysis or a kidney transplant.
- Infections. People with nephrotic syndrome have an increased risk of infections.
Many diseases and conditions can cause glomerular damage and lead to nephrotic syndrome, including:
- Diabetic kidney disease. Diabetic kidney disease affects more than 1 in 3 U.S. adults who have diabetes 3. Diabetic kidney disease is also the leading cause of end-stage kidney disease, which is kidney failure that is treated with dialysis or a kidney transplant. Diabetes can lead to kidney damage (diabetic nephropathy) that affects the glomeruli.
- Minimal change disease. This is the most common cause of nephrotic syndrome in children. Minimal change disease results in abnormal kidney function, but when the kidney tissue is examined under a microscope, it appears normal or nearly normal. The cause of the abnormal function typically can’t be determined.
- Focal segmental glomerulosclerosis (FSGS). Characterized by scarring of some of the glomeruli, this condition can result from another disease, a genetic defect or certain medications or occur for no known reason.
- Membranous nephropathy. This kidney disorder is the result of thickening membranes within the glomeruli. The thickening is due to deposits made by the immune system. It can be associated with other medical conditions, such as lupus, hepatitis B, malaria and cancer, or it can occur for no known reason.
- Systemic lupus erythematosus. This chronic inflammatory disease can lead to serious kidney damage called lupus nephritis.
- Amyloidosis. This disorder occurs when amyloid proteins accumulate in your organs. Amyloid buildup often damages the kidneys’ filtering system.
Factors that can increase your risk of nephrotic syndrome include:
- Medical conditions that can damage your kidneys. Certain diseases and conditions increase your risk of developing nephrotic syndrome, such as diabetes, lupus, amyloidosis, reflux nephropathy and other kidney diseases.
- Certain medications. Medications that might cause nephrotic syndrome include nonsteroidal anti-inflammatory drugs and drugs used to fight infections.
- Certain infections. Infections that increase the risk of nephrotic syndrome include HIV (human immunodeficiency virus), hepatitis B, hepatitis C and malaria.
Because glomerular disease symptoms may develop slowly, the disorder may be discovered when you have an abnormal urinalysis during a routine physical or examination for another condition.
Patients with glomerular disease have significant amounts of protein in the urine (proteinuria), which may be referred to as “nephrotic range” if levels are very high. Blood in the urine (hematuria) are a frequent finding as well, particularly in some forms of glomerular disease. Urinalysis provides information about kidney damage by indicating levels of protein and red blood cells in your urine. Blood tests measure the levels of waste products such as creatinine and urea nitrogen to determine whether the filtering capacity of the kidneys is impaired. If these lab tests indicate kidney damage, your doctor may recommend ultrasound or an X-ray to see whether the shape or size of your kidneys is abnormal. These tests are called renal imaging. But since glomerular disease causes problems at the cellular level, your doctor will probably also recommend a kidney biopsy—a procedure in which a needle is used to extract small pieces of tissue for examination with different types of microscopes, each of which shows a different aspect of the tissue. A kidney biopsy may be helpful in confirming glomerular disease and identifying the cause.
Health care professionals diagnose glomerular disease by ordering tests, such as:
Blood tests:
- Blood tests can measure the levels of products in your blood, such as creatinine, urea nitrogen, and a protein called cystatin C, to find out how well your kidneys are working. Blood tests can also check for low levels of a protein in your blood, called albumin, which can happen when too much of that protein passes from your blood into your urine.
- Other blood test include:
- Antiglomerular basement membrane antibody test
- Antineutrophil cytoplasmic antibodies (ANCAs)
- Antinuclear antibodies
- Complement levels
A simple test of your urine can confirm if there is blood or protein in your urine.
Urinalysis, which examines a sample of your urine to find out if levels of protein and red blood cells are too high:
- Creatinine clearance
- Examination of the urine under a microscope
- Urine total protein
- Uric acid in the urine
- Urine concentration test
- Urine creatinine
- Urine protein
- Urine red blood cell
- Urine specific gravity
- Urine osmolality
Imaging tests that may be done include:
- Abdominal CT scan
- Kidney ultrasound
- Chest x-ray
- Intravenous pyelogram (IVP)
In some cases, a test called a kidney biopsy may be needed. In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope. A kidney biopsy can confirm you have glomerular disease and help find the cause in order to help your doctor plan the best treatment for you.
Treatment for glomerular disease varies by symptoms, causes, and how badly your kidneys are damaged. In some cases, glomerular disease may go away once its cause has been treated. In other cases, the disease may go away but later return. Less often, glomerular disease may not respond to treatment and lead to kidney failure over time.
How your kidneys work
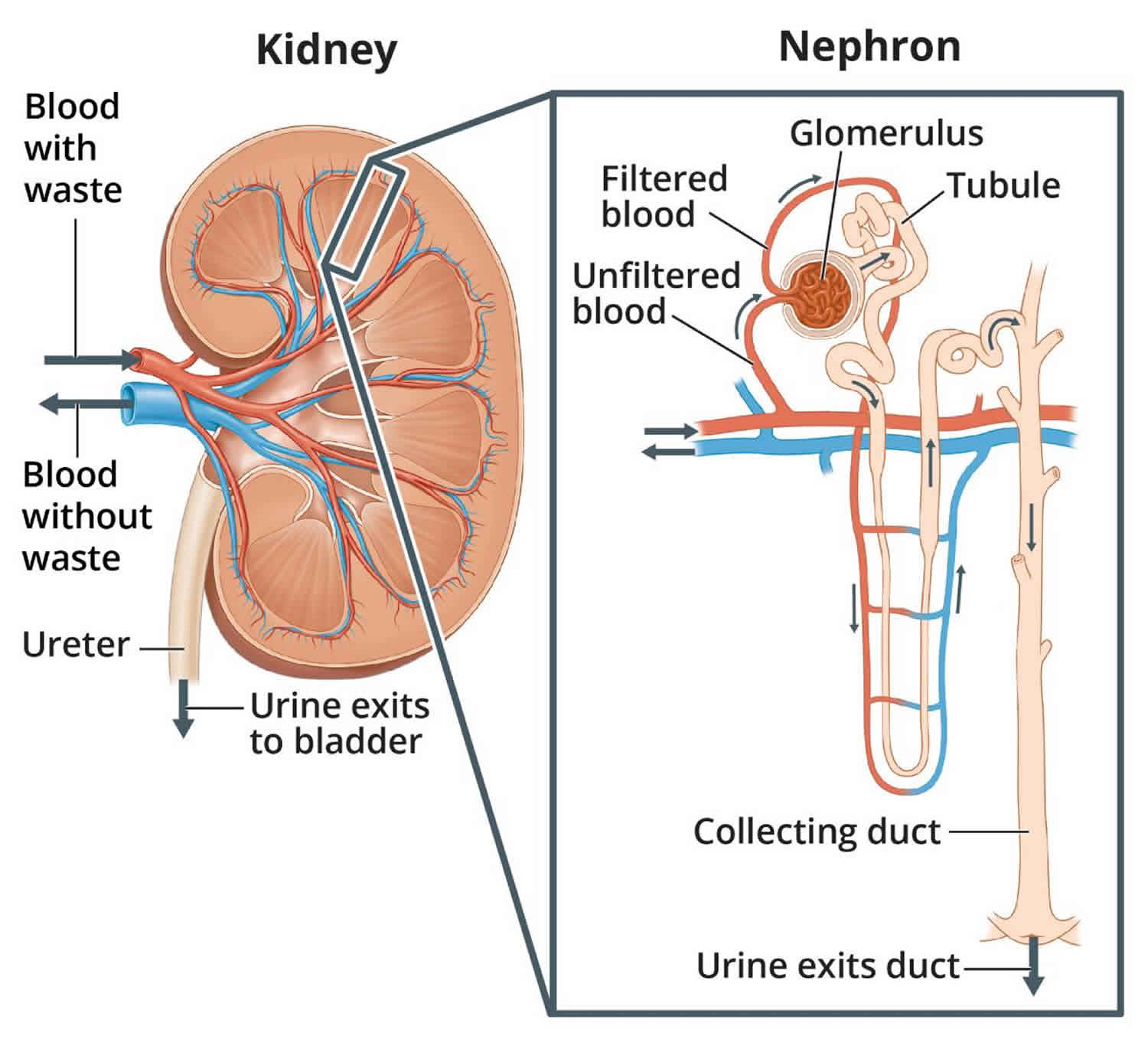
You have two kidneys, each about the size of an adult fist, located on either side of the spine just below the rib cage. Although they are small, your kidneys perform many complex and vital functions that keep the rest of the body in balance. Your kidneys remove waste and excess fluid from your blood through filtering units called nephrons. Each nephron contains a filter (glomerulus) that has a network of tiny blood vessels called capillaries. When blood flows into a glomerulus, tiny molecules — water, essential minerals and nutrients, and wastes — pass through the capillary walls. Large molecules, such as proteins and red blood cells, do not. The filtered solution then passes into another part of the nephron called the tubule. The water, nutrients and minerals your body needs are transferred back to the bloodstream. The excess water and waste become urine that flows to the bladder.
Kidney functions:
- Help remove waste and excess fluid
- Filter the blood, keeping some compounds while removing others
- Control the production of red blood cells
- Make vitamins that control growth
- Release hormones that help regulate blood pressure
- Help regulate blood pressure, red blood cells, and the amount of certain nutrients in the body, such as calcium and potassium.
Here’s how kidneys perform their important work:
- Blood enters the kidneys through an artery from the heart
- Blood is cleaned by passing through millions of tiny blood filters
- Waste material passes through the ureter and is stored in the bladder as urine
- Newly cleaned blood returns to the bloodstream by way of veins
- Bladder becomes full and urine passes out of the body through the urethra.
The kidneys perform their life-sustaining job of filtering and returning to the bloodstream about 200 quarts of fluid every 24 hours. Approximately two quarts are eliminated from the body in the form of urine, while the remainder, about 198 quarts, is retained in the body. The urine we excrete has been stored in the bladder for approximately one to eight hours.
Figure 1. How kidneys work
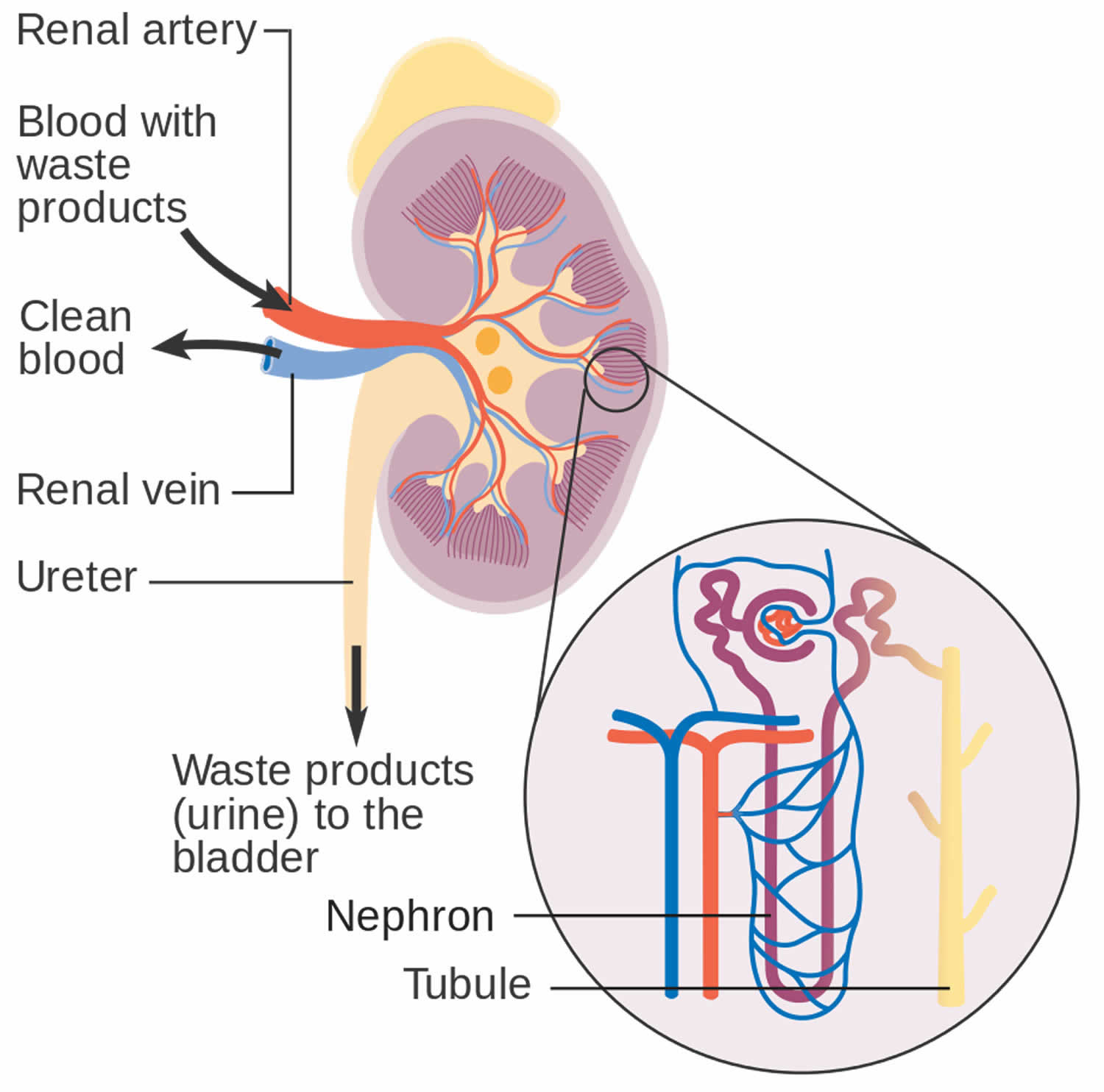
Figure 2. Glomerulus
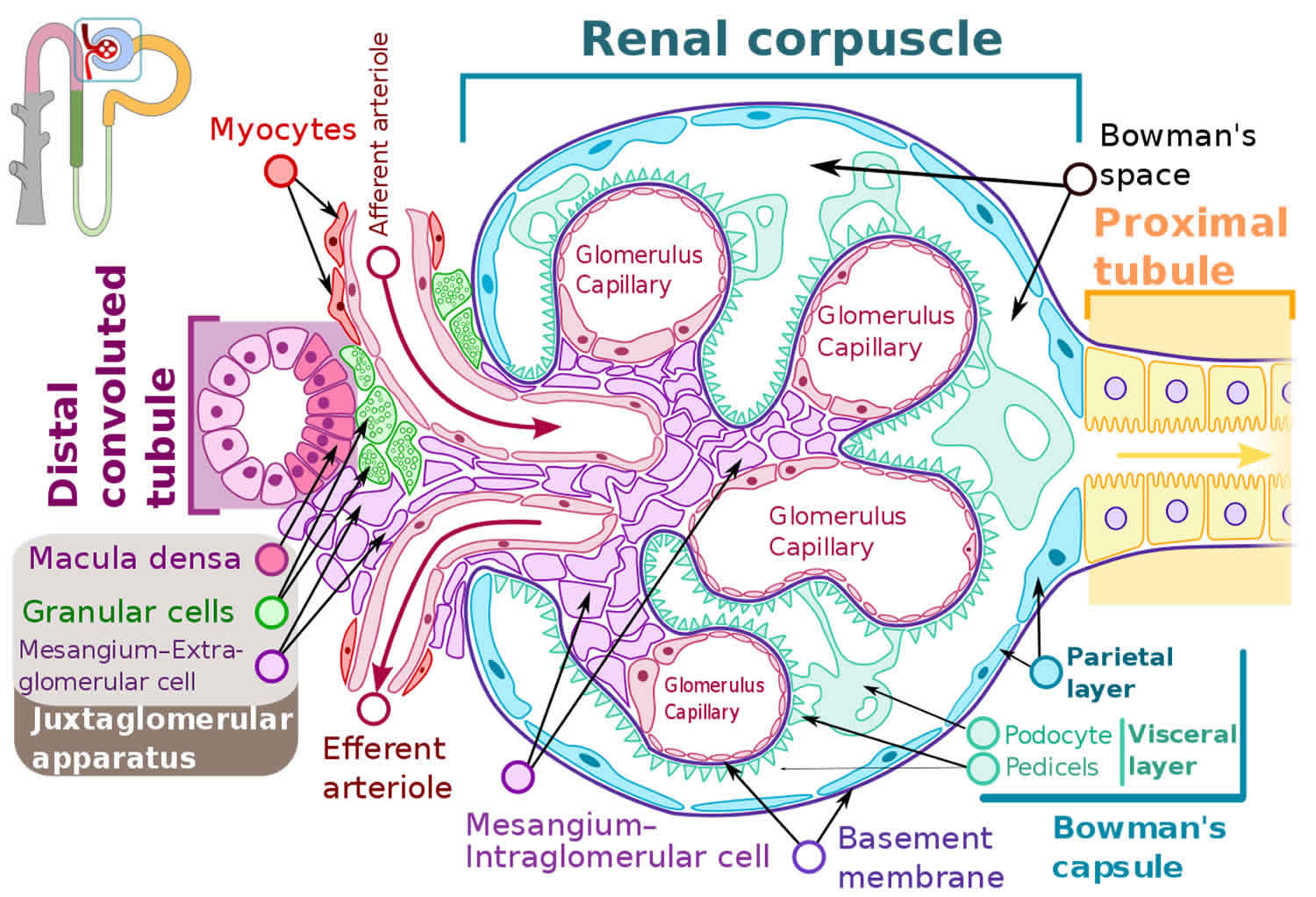
How do glomerular diseases interfere with kidney function?
Glomerular diseases damage the glomeruli, letting protein and sometimes red blood cells leak into the urine. Sometimes a glomerular disease also interferes with the clearance of waste products by the kidney, so they begin to build up in the blood. Furthermore, loss of blood proteins like albumin in the urine can result in a fall in their level in the bloodstream. In normal blood, albumin acts like a sponge, drawing extra fluid from the body into the bloodstream, where it remains until the kidneys remove it. But when albumin leaks into the urine, the blood loses its capacity to absorb extra fluid from the body. Fluid can accumulate outside the circulatory system in the face, hands, feet, or ankles and cause swelling.
What are renal failure and end-stage renal disease?
Renal failure is any acute or chronic loss of kidney function and is the term used when some kidney function remains. Total kidney failure, sometimes called end-stage renal disease (ESRD), indicates permanent loss of kidney function. Depending on the form of glomerular disease, kidney function may be lost in a matter of days or weeks or may deteriorate slowly and gradually over
the course of decades.
Acute renal failure (acute kidney failure)
A few forms of glomerular disease cause very rapid deterioration of kidney function. For example, post-streptococcal glomerulonephritis (PSGN) can cause severe symptoms (hematuria, proteinuria, edema) within 2 to 3 weeks after a sore throat or skin infection develops. The patient may temporarily require dialysis to replace kidney function. This rapid loss of kidney function is called acute renal failure (acute kidney failure). Although acute renal failure (acute kidney failure) can be life-threatening while it lasts, kidney function usually returns after the cause of the kidney failure has been treated. In many patients, acute kidney failure is not associated with any permanent damage. However, some patients may recover from acute renal failure and subsequently develop chronic kidney disease (CKD).
Chronic kidney disease (CKD)
Most forms of glomerular disease develop gradually, often causing no symptoms for many years. Chronic kidney disease (CKD) is the slow, gradual loss of kidney function. Some forms of chronic kidney disease (CKD) can be controlled or slowed down. For example, diabetic nephropathy can be delayed by tightly controlling blood glucose levels and using angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs) to reduce proteinuria and control blood pressure. But chronic kidney disease (CKD) cannot be cured. Partial loss of kidney function means that some portion of the patient’s nephrons have been scarred, and scarred nephrons cannot be repaired. In many cases, CKD leads to total kidney failure.
Total kidney failure
To stay alive, a patient with total kidney failure must go on dialysis, either hemodialysis or peritoneal dialysis or receive a new kidney through kidney transplantation. Patients with chronic kidney disease (CKD) who are approaching total kidney failure should learn as much about their treatment options as possible so they can make an informed decision when the time comes. With the help of dialysis or kidney transplantation, many people continue to lead full, productive lives after reaching total kidney failure.
Glomerular disease causes
Most of the diseases that cause glomerular disease are rare. Glomerular disease may be caused by an infection or a drug that is harmful to your kidneys. In other cases, it may be caused by a disease that affects the entire body, like diabetes or lupus. Many different diseases can cause swelling (inflammation) or scarring (sclerosis) of the glomerulus. Sometimes glomerular disease is idiopathic, meaning it happens without any cause that can be found.
The most common causes of glomerular disease include 4, 5, 6:
- Diabetic kidney disease, a type of kidney disease caused by diabetes. Over several years, high levels of blood glucose, also called blood sugar, can damage your glomeruli.
- Focal segmental glomerulosclerosis (FSGS), a disease that causes scar tissue to form in some of your kidneys’ glomeruli. FSGS has several causes, including genes, autoimmune diseases, and diseases that cause pressure to build in the glomeruli, such as obesity and sleep apnea.
- Lupus nephritis, a kidney disease caused by systemic lupus erythematosus (lupus). Lupus is an autoimmune disease that affects many parts of the body. The disease can cause antibodies to build up in your glomeruli, causing inflammation that can keep your kidneys from working properly and lead to scarring over time.
- Membranous nephropathy, a disease that causes antibodies to build up in a part of your kidney called the glomerular basement membrane. As a result, your glomeruli can become thick and inflamed. Causes can include infection, cancer, and autoimmune diseases.
- IgA nephropathy also called Berger’s disease, an autoimmune disease that causes an antibody called immunoglobulin A (IgA) to build up in your glomeruli, causing inflammation and damage.
- Minimal change disease also called nil disease. This kidney disease causes changes to your glomeruli that can only be seen under a very powerful microscope, called an electron microscope.
- Anti-glomerular basement membrane (anti-GBM) also known as Goodpasture’s disease, an autoimmune disease in which antibodies attack your glomeruli, causing damage and inflammation. The disease can also affect your lungs.
Other causes of glomerular disease include 7:
- Infections, including strep throat, bacterial endocarditis, HIV/AIDS, hepatitis B, and hepatitis C
- Some drugs and medicines that can harm the kidneys, such as nonsteroidal anti-inflammatory drugs (NSAIDs)
- Genetic disorders that affect the kidneys, such as Alport syndrome and Fabry disease
In some cases, the exact cause of a glomerular disease is unknown (idiopathic).
Nephrotic syndrome subtypes
Nephrotic syndrome is a kidney disorder that causes your body to pass too much protein in your urine (proteinuria). Nephrotic syndrome is usually caused by damage to the clusters of small blood vessels in your kidneys called glomeruli that filter waste and excess water from your blood. Nephrotic syndrome causes swelling, particularly in your feet and ankles, and increases the risk of other health problems.
Nephrotic syndrome is a set of symptoms that happen together and affect your kidneys. These include:
- Swelling in body parts like your legs, ankles, or around your eyes (edema)
- Large amounts of protein in your urine (proteinuria)
- Foamy urine, a result of excess protein in your urine
- Loss of protein in your blood
- High levels of fat lipids in your blood (high cholesterol)
- High blood pressure (in some cases)
- Severe swelling (edema), particularly around your eyes and in your ankles and feet
- Weight gain due to fluid retention
- Fatigue
- Loss of appetite
Glomerular disease with nephrotic syndrome:
- Focal segmental glomerulosclerosis (FSGS).
- Focal segmental glomerulosclerosis (FSGS) describes scarring in scattered regions of the kidney, typically limited to one part of the glomerulus and to a minority of glomeruli in the affected region. Focal segmental glomerulosclerosis (FSGS) may result from a systemic disorder or it may develop as an idiopathic kidney disease, without a known cause. Proteinuria is the most common symptom of FSGS, but, since proteinuria is associated with several other kidney conditions, the doctor cannot diagnose FSGS on the basis of proteinuria alone. Biopsy may confirm the presence of glomerular scarring if the tissue is taken from the affected section of the kidney. But finding the affected section is a matter of chance, especially early in the disease process, when lesions may be scattered. Confirming a diagnosis of focal segmental glomerulosclerosis (FSGS) may require repeat kidney biopsies. Arriving at a diagnosis of idiopathic FSGS requires the identification of focal scarring and the elimination of possible systemic causes such as diabetes or an immune response to infection. Since idiopathic FSGS is, by definition, of unknown cause, it is difficult to treat. No universal remedy has been found, and most patients with FSGS progress to total kidney failure over 5 to
20 years. Some patients with an aggressive form of FSGS reach total kidney failure in 2 to 3 years. Treatments involving steroids or other immunosuppressive drugs appear to help some patients by decreasing proteinuria and improving kidney function. But these treatments are beneficial to only a minority of those in whom they are tried, and some patients experience even poorer kidney function as a result. Angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs) may also be used in FSGS to
decrease proteinuria. Treatment should focus on controlling blood pressure and blood cholesterol levels, factors that may contribute to kidney scarring.
- Focal segmental glomerulosclerosis (FSGS) describes scarring in scattered regions of the kidney, typically limited to one part of the glomerulus and to a minority of glomeruli in the affected region. Focal segmental glomerulosclerosis (FSGS) may result from a systemic disorder or it may develop as an idiopathic kidney disease, without a known cause. Proteinuria is the most common symptom of FSGS, but, since proteinuria is associated with several other kidney conditions, the doctor cannot diagnose FSGS on the basis of proteinuria alone. Biopsy may confirm the presence of glomerular scarring if the tissue is taken from the affected section of the kidney. But finding the affected section is a matter of chance, especially early in the disease process, when lesions may be scattered. Confirming a diagnosis of focal segmental glomerulosclerosis (FSGS) may require repeat kidney biopsies. Arriving at a diagnosis of idiopathic FSGS requires the identification of focal scarring and the elimination of possible systemic causes such as diabetes or an immune response to infection. Since idiopathic FSGS is, by definition, of unknown cause, it is difficult to treat. No universal remedy has been found, and most patients with FSGS progress to total kidney failure over 5 to
- Diabetic glomerulosclerosis also called diabetic nephropathy.
- Diabetic nephropathy or diabetic glomerulosclerosis is the leading cause of glomerular disease and of total kidney failure in the United States. Kidney disease is one of several problems caused by elevated levels of blood glucose, the central feature of diabetes. In addition to scarring the kidney, elevated glucose levels appear to increase the speed of blood flow into the kidney, putting a strain on the filtering glomeruli and raising blood pressure. Diabetic nephropathy usually takes many years to develop. People with diabetes can slow down damage to their kidneys by controlling their blood glucose through healthy eating with moderate protein intake, physical activity, and medications. People with diabetes should also be careful to keep their blood pressure at a level below 140/90 mm Hg, if possible. Blood pressure medications called angiotensin-converting enzyme inhibitors (ACE inhibitors) or an angiotensin 2 receptor blocker (ARB) are particularly effective at minimizing kidney damage and are now frequently prescribed to control blood pressure in patients with diabetes and in patients with many forms of kidney disease.
- Membranous nephropathy
- Membranous nephropathy also called membranous glomerulopathy, is the second most common cause of the nephrotic syndrome (proteinuria, edema, high cholesterol) in U.S. adults after diabetic nephropathy. Diagnosis of membranous nephropathy requires a kidney biopsy, which reveals unusual deposits of immunoglobulin G and complement C3, substances created by the body’s immune system. 75 percent of membranous nephropathy cases are idiopathic, which means that the cause of the membranous nephropathy is unknown. The remaining 25 percent of cases are the result of other diseases like systemic lupus erythematosus (SLE), hepatitis B or C infection, or some forms of cancer. Drug therapies involving
penicillamine, gold, or captopril have also been associated with membranous nephropathy. About 20 to 40 percent of patients with membranous nephropathy progress, usually over decades, to total kidney failure, but most patients experience either complete remission or continued symptoms without progressive kidney failure. Doctors disagree about how aggressively to treat this condition, since about 20 percent of patients recover without treatment. Angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs) are generally used to reduce proteinuria. Additional medication to control high blood pressure and edema is frequently required. Some patients benefit from steroids, but this treatment does not work for everyone. Additional immunosuppressive medications are helpful for some patients with progressive disease.
- Membranous nephropathy also called membranous glomerulopathy, is the second most common cause of the nephrotic syndrome (proteinuria, edema, high cholesterol) in U.S. adults after diabetic nephropathy. Diagnosis of membranous nephropathy requires a kidney biopsy, which reveals unusual deposits of immunoglobulin G and complement C3, substances created by the body’s immune system. 75 percent of membranous nephropathy cases are idiopathic, which means that the cause of the membranous nephropathy is unknown. The remaining 25 percent of cases are the result of other diseases like systemic lupus erythematosus (SLE), hepatitis B or C infection, or some forms of cancer. Drug therapies involving
- Minimal change disease also called “nil disease“.
- Minimal change disease also called “nil disease” is the diagnosis given when a patient has the nephrotic syndrome and the kidney biopsy reveals little or no change to the structure
of glomeruli or surrounding tissues when examined by a light microscope. Tiny drops of a fatty substance called a lipid may be present, but no scarring has taken place within the kidney. Minimal change disease may occur at any age, but it is most common in childhood. A small percentage of patients with idiopathic nephrotic syndrome do not respond to
steroid therapy. For these patients, the doctor may recommend a low-sodium diet and prescribe a diuretic to control edema. Your doctor may recommend the use of nonsteroidal anti-inflammatory drugs (NSAIDs) to reduce proteinuria. ACE inhibitors and ARBs have also been used to reduce proteinuria in patients with steroid-resistant minimal change disease. These patients may respond to larger doses of steroids, more prolonged use of steroids, or steroids in combination with immunosuppressant drugs, such as chlorambucil, cyclophosphamide, or cyclosporine.
- Minimal change disease also called “nil disease” is the diagnosis given when a patient has the nephrotic syndrome and the kidney biopsy reveals little or no change to the structure
- Membranoproliferative glomerulopathy (MPGN) (form of glomerulonephritis due to abnormal buildup of antibodies in the kidneys)
- Amyloidosis
- Monoclonal immune deposition disease (MIDD)
- Dense deposit disease
- Fabry disease
- Collagenofibrotic glomerulopathy
Treatment for nephrotic syndrome includes treating the condition that’s causing it and taking medications. Nephrotic syndrome can increase your risk of infections and blood clots. Your doctor might recommend medications and dietary changes to prevent complications.
Nephritic subtypes
Common symptoms of glomerulonephritis are:
- Blood in the urine (dark, rust-colored, or brown urine)
- Foamy urine (due to excess protein in the urine)
- Swelling (edema) of the face, eyes, ankles, feet, legs, or abdomen
Symptoms may also include the following:
- Abdominal pain
- Blood in the vomit or stools
- Cough and shortness of breath
- Diarrhea
- Excessive urination
- Fever
- General ill feeling, fatigue, and loss of appetite
- Joint or muscle aches
- Nosebleed
The symptoms of chronic kidney disease may develop over time. Chronic kidney failure symptoms may gradually develop.
Glomerular disease with nephritic syndrome subtypes:
- Lupus nephritis (kidney complication of lupus)
- IgA nephropathy also called Berger’s disease (disorder in which antibodies called immunoglobulin A (IgA) build up in kidney tissue).
- IgA nephropathy is a form of glomerular disease that results when immunoglobulin A (IgA) forms deposits in the glomeruli, where it creates inflammation. IgA nephropathy was not recognized as a cause of glomerular disease until the late 1960s, when sophisticated biopsy techniques were developed that could identify IgA deposits in kidney tissue. The most common symptom of IgA nephropathy is blood in the urine (hematuria), but it is often a silent disease that may go undetected for many years. The silent nature of IgA nephropathy makes it difficult to determine how many people are in the early stages of IgA nephropathy, when specific medical tests are the only way to detect it. IgA nephropathy is estimated to
be the most common cause of primary glomerulonephritis—that is, glomerular disease not caused by a systemic disease like lupus or diabetes mellitus. IgA nephropathy appears to affect men more than women. Although IgA nephropathy is found in all age groups, young people rarely display signs of kidney failure because the disease usually takes several years to progress to the stage where it causes detectable complications. No treatment is recommended for early or mild cases of IgA nephropathy when the patient has normal blood pressure and less than 1 gram of protein in a 24-hour urine output. When proteinuria exceeds 1 gram/day, treatment is aimed at protecting kidney function by reducing proteinuria and controlling blood pressure. Blood pressure medicines such as angiotensin-converting enzyme inhibitors (ACE inhibitors) or angiotensin 2 receptor blockers (ARBs) that block a hormone called angiotensin are most effective at achieving those two goals simultaneously.
- IgA nephropathy is a form of glomerular disease that results when immunoglobulin A (IgA) forms deposits in the glomeruli, where it creates inflammation. IgA nephropathy was not recognized as a cause of glomerular disease until the late 1960s, when sophisticated biopsy techniques were developed that could identify IgA deposits in kidney tissue. The most common symptom of IgA nephropathy is blood in the urine (hematuria), but it is often a silent disease that may go undetected for many years. The silent nature of IgA nephropathy makes it difficult to determine how many people are in the early stages of IgA nephropathy, when specific medical tests are the only way to detect it. IgA nephropathy is estimated to
- Anti-neutrophil cytoplasmic antibody associated glomerulonephritis (ANCA-GN) or pauci-immune glomerulopathy
- Thin basement membrane lesion (disorder that affects the glomerular basement membrane, the part of the kidney that helps filter waste and extra fluid from the blood)
- Fibrillary glomerulopathy
- Anti-glomerular basement membrane nephritis (disorder in which the immune system attacks the glomeruli)
- Alport syndrome.
- The primary indicator of Alport syndrome is a family history of chronic glomerular disease, although it may also involve hearing or vision impairment. Alport syndrome affects both men and women, but men are more likely to experience chronic kidney disease and sensory loss. Men with Alport syndrome usually first show evidence of renal insufficiency while in their twenties and reach total kidney failure by age 40. Women rarely have significant renal impairment, and hearing loss may be so slight that it can be detected only through testing with special equipment. Usually men can pass the disease only to their daughters. Women can transmit the disease to either their sons or their daughters. Treatment focuses on controlling blood pressure to maintain kidney function.
- Immunotactoid glomerulopathy
- Infection-related glomerular disease. Glomerular disease sometimes develops rapidly after an infection in other parts of the body.
- Acute post-streptococcal glomerulonephritis (PSGN) can occur after an episode of strep throat or, in rare cases, impetigo (a skin infection). The Streptococcus bacteria do not attack the kidney directly, but an infection may stimulate the immune system to overproduce antibodies, which are circulated in the blood and finally deposited in the glomeruli, causing damage. Post-streptococcal glomerulonephritis (PSGN) can bring on sudden symptoms of swelling (edema), reduced urine output (oliguria), and blood in the urine (hematuria). Tests will show large amounts of protein in the urine and elevated levels of creatinine and urea nitrogen in the blood, thus indicating reduced kidney function. High blood pressure frequently accompanies reduced kidney function in post-streptococcal glomerulonephritis (PSGN). Acute post-streptococcal glomerulonephritis (PSGN) is most common in children between the ages of 3 and 7, although it can strike at any age, and it most often affects boys. It lasts only a brief time and usually allows the kidneys to recover. In a few cases, however, kidney damage may be permanent, requiring dialysis or transplantation to replace renal function.
- Bacterial endocarditis, infection of the tissues inside the heart, is also associated with subsequent glomerular disease. Researchers are not sure whether the kidney lesions that form after a heart infection are caused entirely by the immune response or whether some other disease mechanism contributes to kidney damage. Treating the heart infection is the most
effective way of minimizing kidney damage. Endocarditis sometimes produces chronic kidney disease (CKD). - HIV (human immunodeficiency virus), the virus that leads to AIDS, can also cause glomerular disease. Between 5 and 10 percent of people with HIV experience kidney failure, even before developing full-blown AIDS. HIV-associated nephropathy usually begins with heavy proteinuria and progresses rapidly (within a year of detection) to total kidney failure. Researchers are looking for therapies that can slow down or reverse this rapid deterioration of kidney function, but some possible solutions involving immunosuppression are risky because of the patients’ already compromised immune system.
- Analgesic nephropathy syndrome (kidney disease due to heavy use of pain relievers, especially NSAIDs)
- Blood vessel diseases, such as vasculitis or polyarteritis
- Henoch-Schönlein purpura (disease that involves purple spots on the skin, joint pain, gastrointestinal problems and glomerulonephritis)
Glomerular disease prevention
It is important to pay attention to signs and symptoms of kidney disease and let your healthcare provider know as early as possible when you notice them. Treating conditions that can cause glomerular disease or kidney disease may help prevent it from getting worse and slow down the damage to your kidneys.
But you may reduce your risk of glomerular disease by taking care of your kidneys:
- Pay attention to labels when taking over-the-counter (OTC) pain medications. Follow the instructions for OTC pain medications, such as aspirin, acetaminophen (Tylenol, others), ibuprofen (Advil, Motrin IB, others) and naproxen sodium (Aleve, others). Taking too much of these medications may increase your risk of kidney injury. This is especially true if you have pre-existing kidney disease, diabetes or high blood pressure.
- Work with your doctor to manage kidney and other chronic conditions. If you have kidney disease or another condition that increases your risk of acute kidney failure, such as diabetes or high blood pressure, stay on track with treatment goals and follow your doctor’s recommendations to manage your condition.
- Make a healthy lifestyle a priority. Be active; eat a sensible, balanced diet; and drink alcohol only in moderation — if at all.
Glomerular disease signs and symptoms
Symptoms of glomerular disease vary and are related to the type of damage to your glomeruli. Some people with less damage may have few or no symptoms, whereas people with more severe damage may have more noticeable symptoms.
Glomerular disease symptoms can include:
- High blood pressure (hypertension), which may develop or get worse.
- Swelling (edema). Glomerular disease can cause fluid to build up in your body. The extra fluid can cause swelling in body parts like your hands, ankles, legs, or around your eyes.
- Weight gain. As fluid builds up in your body, your weight can increase.
- Too much protein in your urine (proteinuria). Glomerular disease can cause your glomeruli to leak protein into your urine. Your urine may be foamy or bubbly because of the protein.
- Blood in your urine (hematuria). Glomerular disease can cause your glomeruli to leak blood into your urine. Blood in your urine can make your urine look pink or light brown. The blood can usually only be seen under a microscope.
One or more of these symptoms can be the first sign of kidney disease.
If you have one or all of these symptoms, be sure to see your doctor right away.
Glomerular disease complications
Glomerular disease often progresses slowly, causing no symptoms for many years. But, over time, it can cause serious health problems such as:
- High blood pressure (hypertension).
- Nephrotic syndrome, a group of symptoms that indicate your kidneys are not working properly. These symptoms include too much protein in your urine, low levels of albumin in your blood, swelling in parts of your body, and high levels of cholesterol in your blood.
- Chronic kidney disease (CKD), the gradual loss of kidney function, when your kidneys are no longer able to process and remove toxic waste products from your body.
- Kidney failure, the loss of about 85% or more of kidney function, which often leads to symptoms such as appetite loss, nausea, vomiting, and worsening swelling.
In some cases, glomerular disease can cause rapid kidney failure that may lead to confusion and death if not treated immediately.
Glomerular disease diagnosis
The first clues are the signs and symptoms. Health care professionals diagnose glomerular disease by ordering tests, such as:
Your doctor may order the following blood tests:
- Blood tests can measure the levels of products in your blood, such as creatinine, urea nitrogen, and a protein called cystatin C, to find out how well your kidneys are working. Blood tests can also check for low levels of a protein in your blood, called albumin, which can happen when too much of that protein passes from your blood into your urine.
- Other blood test include:
- Antiglomerular basement membrane antibody test
- Antineutrophil cytoplasmic antibodies (ANCAs)
- Antinuclear antibodies
- Complement levels
A simple test of your urine can confirm if there is blood or protein in your urine.
Urinalysis, which examines a sample of your urine to find out if levels of protein and red blood cells are too high:
- Creatinine clearance
- Examination of the urine under a microscope
- Urine total protein
- Uric acid in the urine
- Urine concentration test
- Urine creatinine
- Urine protein
- Urine red blood cell
- Urine specific gravity
- Urine osmolality
Imaging tests that may be done include:
- Abdominal CT scan
- Kidney ultrasound
- Chest x-ray
- Intravenous pyelogram (IVP)
In some cases, a test called a kidney biopsy may be needed. In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope. A kidney biopsy can confirm you have glomerular disease and help find the cause in order to help your doctor plan the best treatment for you.
Glomerular disease treatment
Your doctor will need to find what is causing your glomerular disease and if possible, treat the cause of the underlying disease. The goals of treatment are to help your symptoms, avoid complications, and slow down the damage to your kidneys. Your healthcare provider will work to understand the type and severity of your symptoms and help find the best treatment for you. Sometimes you may need medication or temporary treatment with an artificial kidney machine to remove extra fluid, control high blood pressure and treat kidney failure. Controlling high blood pressure is usually the most important part of treatment.
Overall, there is no one specific treatment that works for all glomerular diseases, but your doctor may tell you to:
- Control your blood pressure and stop protein loss in your urine with drugs called angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs)
- Take diuretics (water pills) to treat swelling in your ankles and feet
- Make certain changes in your diet such as eating less salt
- Take medications that make your immune system less active for example corticosteroids
If your glomerular disease is getting worse very quickly, you may need high doses of medicine that affect your immune system. Sometimes your doctor may order plasmapheresis, a special blood filtering process to remove harmful proteins from your blood caused by immune problems. The fluid part of the blood that contains antibodies is removed and replaced with intravenous fluids or donated plasma (that does not contain antibodies). Removing antibodies may reduce inflammation in your kidney tissues.
People with glomerular disease should be closely watched for signs of kidney failure. Dialysis or a kidney transplant may eventually be needed.
Medicines
Health care professionals often treat glomerular disease with medicines, such as:
- An angiotensin-converting enzyme (ACE) inhibitor or an angiotensin 2 receptor blocker (ARB). These medicines can reduce protein loss, lower blood pressure, and slow the progression of kidney disease.
- Diuretics (water pills), which reduce swelling by helping the kidneys remove sodium—or salt—and water from your blood.
- Statins, which lower blood cholesterol and can reduce the risk for certain types of heart disease that can develop among people with glomerular disease.
- Sodium-glucose transporter 2 (SGLT2) inhibitors, medicines originally used to treat diabetes, can also slow the progression of kidney disease.
- Medicines that make your immune system less active such as corticosteroids, antimetabolites, alkylating agents, or monoclonal antibodies, which can reduce inflammation.
Other treatments
If your kidney disease advances to kidney failure, your doctor may suggest treatment options such as:
- Hemodialysis, a procedure that filters your blood outside of your body, using an external filter called a dialyzer
- Peritoneal dialysis, a procedure that filters your blood inside your body, using the lining of your abdomen, or belly
- Kidney transplant, a surgery that places a healthy kidney from another person inside your body.
Glomerular disease diet
Eating, diet, and nutrition have not been shown to play a role in causing or preventing glomerular disease. But if you have glomerular disease, your health care professional may recommend you
- Limit salt (sodium) intake
- Reduce calories if your doctor advises you to lose excess weight
- Limit saturated fats if your cholesterol is high
- Make a healthy lifestyle a priority. Be active; eat a sensible, balanced diet; and drink alcohol only in moderation — if at all.
Focal Segmental Glomerulosclerosis (FSGS)
Focal segmental glomerulosclerosis also called FSGS is one of the most common causes of primary glomerular diseases in adults 8, 9. Focal segmental glomerulosclerosis (FSGS) is scar tissue in the filtering unit of the kidney called the glomerulus 10. The glomeruli serve as filters that help the body get rid of harmful substances. Each kidney has thousands of glomeruli. “Focal” means that some of the glomeruli become scarred. Others remain normal. “Segmental” means that only part of an individual glomerulus is damaged. Focal segmental glomerulosclerosis is a pathologic term, referring to glomerulosclerosis (glomerular scarring, representing an increase in collagens and other proteins) that is found in a focal distribution (initially some glomeruli are affected while most glomeruli are entirely normal) and a segmental distribution (early in disease, the affected glomerular show sclerosis in a segmental pattern while other portions of that glomerulus remains normal). As the disease progresses, this pattern may shift so that some glomeruli are globally (totally) sclerosed and that all glomeruli have at least some sclerosis. Focal segmental glomerulosclerosis causes asymptomatic proteinuria or nephrotic syndrome with or without renal insufficiency. Histologically, focal segmental glomerulosclerosis is characterized by segmental scarring, involving a part of the glomerulus, and affects some but not all glomeruli sampled. Recent research has shed light on the pathogenesis of focal segmental glomerulosclerosis which is podocyte injury and damage, leading to protein loss and subsequent development of focal sclerosing lesions 11. Generally, focal segmental glomerulosclerosis is a progressive form of kidney disease, accounting for 2.3% of end-stage renal disease (ESRD).
Patients with focal segmental glomerulosclerosis (FSGS) may present in different ways. First some present with swelling (edema), either a sudden onset similar to that seen in patients with minimal change disease or slower onset over weeks to months, in their face, hands, feet, legs or ankles. Second, some patients have no symptoms and are found to have no symptoms (asymptomatic) protein in the urine (proteinuria), found on routine urinalysis performed as part of a physical examination. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin.
Focal segmental glomerulosclerosis affects both children and adults. It occurs slightly more often in men and boys. Focal segmental glomerulosclerosis is also more common in African American men. Focal segmental glomerulosclerosis causes up to 40% of cases in adults and 20% in children 12. Focal segmental glomerulosclerosis is the most common cause of idiopathic (or primary) nephrotic syndrome among adults in the US. Though usually idiopathic (unknown cause), focal segmental glomerulosclerosis can occur in association with other factors (secondary focal segmental glomerulosclerosis), including drugs (e.g., heroin, lithium, interferon alfa, pamidronate, cyclosporine, or NSAIDs [causing analgesic nephropathy]), atheroembolic disease affecting the kidneys, obesity, HIV infection (HIV-associated nephropathy), and disorders causing nephron loss (eg, reflux nephropathy, subtotal nephrectomy, renal dysgenesis [eg oligomeganephronia: renal hypoplasia with a decreased number of nephrons]). Familial cases exist.
In focal segmental glomerulosclerosis, because charge as well as size ultrafiltration barriers are defective, protein in the urine (proteinuria) is typically nonselective, affecting high molecular-weight proteins (eg, immunoglobulins [IGs] or antibodies) as well as albumin. Kidneys tend to be small.
A blood test, urine test, and a kidney biopsy will determine if you have FSGS.
- Urine test: A urine test will help find protein and blood in your urine.
- Blood test: A blood test will help find levels of protein, cholesterol, and wastes in your blood.
- Glomerular filtration rate (GFR): A blood test will be done to know how well your kidneys are filtering the wastes from your body.
- Kidney ultrasound. Earlier in the course of illness, kidney ultrasound will reveal normal or enlarged kidneys with increased echogenicity, indicating diffuse intrinsic medical renal disease 13. In advanced kidney failure, kidneys are shrunken and small, suggesting severe interstitial fibrosis and glomerular scarring. In HIV-associated FSGS, ultrasound study generally reveals large echogenic kidneys.
- Kidney biopsy: In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope.
- Genetic testing: A genetic test may be done to see if you were born with genes that caused your kidney disease. This information may help your doctor decide what type of treatment is best for you.
FSGS is a serious condition that can lead to kidney failure, which can only be treated with dialysis or kidney transplant. Treatment options for focal segmental glomerulosclerosis (FSGS) depend on the type you have.
Types of focal segmental glomerulosclerosis (FSGS) include:
- Primary focal segmental glomerulosclerosis. Many people diagnosed with FSGS have no known cause for their condition. This is called primary (idiopathic) FSGS.
- Secondary focal segmental glomerulosclerosis. Several factors, such as infection, drug toxicity, diseases including diabetes or sickle cell disease, obesity, and even other kidney diseases can cause secondary FSGS. Controlling or treating the underlying cause often slows ongoing kidney damage and might lead to improved kidney function over time.
- Genetic focal segmental glomerulosclerosis. This is a rare form of FSGS caused by genetic changes. It is also called familial FSGS. It’s suspected when several members of a family show signs of FSGS. Familial FSGS can also occur when neither parent has the disease but each one carries a copy of an altered gene that can be passed on to the next generation.
- Unknown focal segmental glomerulosclerosis. In some cases, the underlying cause of FSGS cannot be determined despite the evaluation of clinical symptoms and extensive testing.
Usually, treatments for FSGS include:
- Corticosteroids (often called “steroids”)
- Immunosuppressive drugs
- Corticosteroids and immunosuppressive drugs: These medications are used to calm your immune system (your body’s defense system) and stop it from attacking your glomeruli.
- Plasmapheresis. Sometimes your doctor may order plasmapheresis, a special blood filtering process to remove harmful proteins from your blood caused by immune problems. The fluid part of the blood that contains antibodies is removed and replaced with intravenous fluids or donated plasma (that does not contain antibodies). Removing antibodies may reduce inflammation in your kidney tissues.
- Angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs). These are blood pressure medications used to reduce protein loss and control blood pressure.
- Diuretics (water pills). These medications help your body get rid of excess fluid and swelling. These can be used to lower your blood pressure too.
- Diet changes. Some diet changes may be needed, such as reducing salt (sodium) and protein in your food choices to lighten the load of wastes on the kidneys.
Figure 3. Focal segmental glomerulosclerosis
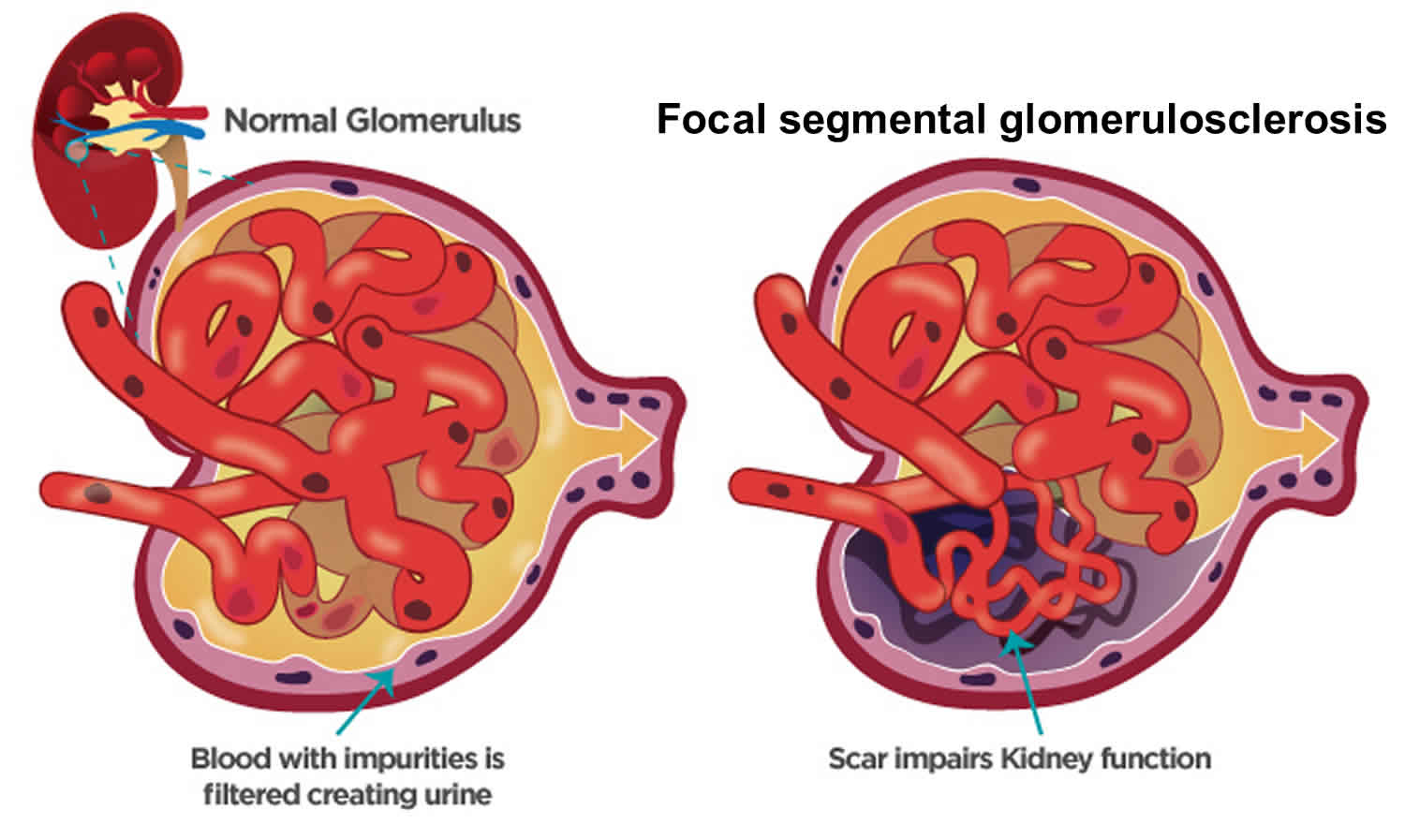
Will I have kidney failure because of focal segmental glomerulosclerosis (FSGS)?
You should talk with your doctor about your condition because the progression of the disease depends on many factors. FSGS is a chronic disease, because the scarred glomeruli cannot be repaired. Treatment can slow the process of kidney disease. Everyone is different in how they respond to treatment. Over time, some patients with FSGS gradually get worse until they reach kidney failure, If this occurs, they will need a kidney transplant or dialysis to stay alive. Some people respond well to treatment and may live with the disease for many years while being monitored for any signs of change.
Focal segmental glomerulosclerosis types
Focal segmental glomerulosclerosis has different types based on the cause. Focal segmental glomerulosclerosis is broadly categorized into primary (idiopathic) and secondary forms, and such distinction carries both prognostic and therapeutic implications 14. Idiopathic focal segmental glomerulosclerosis is of unknown cause and probably is the most common form. Primary focal segmental glomerulosclerosis has long been thought to be due to the presence of circulating permeability factors/cytokines which causes foot process effacement and proteinuria. These include cardiotrophin-like cytokine factor 1, apoA1b, anti-CD40 Ab and suPAR 15.
Below are the types of focal segmental glomerulosclerosis:
Primary focal segmental glomerulosclerosis
Many people diagnosed with focal segmental glomerulosclerosis have no known cause for their condition. This is called primary (idiopathic) focal segmental glomerulosclerosis.
Secondary focal segmental glomerulosclerosis
Several factors, such as infection, drug toxicity, diseases such as diabetes or sickle cell disease, obesity, and even other kidney diseases can cause secondary focal segmental glomerulosclerosis. Controlling or treating the underlying cause often halts ongoing kidney damage and might lead to improved kidney function over time.
Genetic or familial focal segmental glomerulosclerosis
This rare form of focal segmental glomerulosclerosis is caused by genetic mutations. It’s suspected when several members of a family show signs of focal segmental glomerulosclerosis. Familial focal segmental glomerulosclerosis can also occur when neither parent has the disease, but each carries one copy of an abnormal gene that can be passed on to the next generation.
Unknown focal segmental glomerulosclerosis
In some cases, the underlying cause of FSGS cannot be determined despite the evaluation of clinical symptoms and extensive testing.
Focal segmental glomerulosclerosis causes
Focal segmental glomerulosclerosis is not caused by a single disease. It can have many different causes. The scarring may happen because of an infection, or drug, or a disease that affects the entire body, like diabetes, HIV infection, sickle cell disease or lupus. focal segmental glomerulosclerosis can also be caused by another glomerular disease that you had before you got focal segmental glomerulosclerosis. Focal segmental glomerulosclerosis has different types based on the cause.
Below are the types of focal segmental glomerulosclerosis:
- Primary focal segmental glomerulosclerosis. Many people diagnosed with focal segmental glomerulosclerosis have no known cause for their condition. This is called primary (idiopathic) focal segmental glomerulosclerosis.
- Secondary focal segmental glomerulosclerosis. Several factors, such as infection, drug toxicity, diseases such as diabetes or sickle cell disease, obesity, and even other kidney diseases can cause secondary focal segmental glomerulosclerosis. Controlling or treating the underlying cause often halts ongoing kidney damage and might lead to improved kidney function over time.
- Genetic or familial focal segmental glomerulosclerosis. This rare form of focal segmental glomerulosclerosis is caused by genetic mutations. It’s suspected when several members of a family show signs of focal segmental glomerulosclerosis. Familial focal segmental glomerulosclerosis can also occur when neither parent has the disease, but each carries one copy of an abnormal gene that can be passed on to the next generation.
- Unknown focal segmental glomerulosclerosis. In some cases, the underlying cause of FSGS cannot be determined despite the evaluation of clinical symptoms and extensive testing.
Focal segmental glomerulosclerosis is broadly categorized into primary (idiopathic) and secondary forms, and such distinction carries both prognostic and therapeutic implications 14. Idiopathic focal segmental glomerulosclerosis is of unknown cause and probably is the most common form. Primary focal segmental glomerulosclerosis has long been thought to be due to the presence of circulating permeability factors/cytokines which causes foot process effacement and proteinuria. These include cardiotrophin-like cytokine factor 1, apoA1b, anti-CD40 Ab and suPAR 15.
Known causes also called acquired focal segmental glomerulosclerosis include:
- Drugs such as heroin, bisphosphonates, anabolic steroids, interferon, lithium, pamidronate and mTOR inhibitors 16
- Infection
- Viral causes include HIV, parvo B19, CMV, EBV, hepatitis C, and Simian virus 40 17.
- Inherited genetic problems
- Diabetes mellitus,
- Hypertension,
- Renal aplasia, hypoplasia or dysplasia,
- Renal artery stenosis,
- Cholesterol emboli 18
- Obesity
- Reflux nephropathy (a condition in which urine flows backward from the bladder to the kidney)
- Sickle cell disease
- Some medicines
- Vascular disease 18
Post-adaptive focal segmental glomerulosclerosis arises after a phase in which the glomeruli are exposed to higher than normal blood flow for a number of years. Increased glomerular blood flow is typical of two settings. First, it occurs when the number of glomeruli is reduced, as happens following bilateral kidney surgery or congenital kidney abnormalities or chronic scarring of the kidney tubules (for example, due to reflux of urine from the bladder back into the kidney). Second, it occurs with obesity, sickle cell anemia and a few other conditions; the reasons for the increase in glomerular blood flow in these settings are unknown.
Occasionally other glomerular diseases, such as IgA nephropathy or membranous nephropathy or lupus nephritis may show focal and segmental glomerular scarring which may be referred by some pathologists as focal segmental glomerulosclerosis. These patients should be treated according to their underlying glomerular disease and the term focal segmental glomerulosclerosis is best not applied in these situations.
Most focal segmental glomerulosclerosis is sporadic (no family history). Some focal segmental glomerulosclerosis is familial (there is more than one affected family member). Genetic mutations may be seen in sporadic focal segmental glomerulosclerosis and familial focal segmental glomerulosclerosis, but is more common in the latter. Several genes encoding slit diaphragm proteins, cell membrane proteins, cytoskeleton proteins, nuclear proteins, mitochondrial proteins, and lysosomal proteins have been identified to be abnormal/mutated leading to loss of integrity of glomerular filtration barrier resulting in focal segmental glomerulosclerosis 19. To date eight genes have been associated with focal segmental glomerulosclerosis. This is an area of intense research and more genes are likely to be identified soon. Several inheritance patterns have been identified and the five most commonly-mutated genes will be discussed below.
- Autosomal recessive inheritance. The inheritance pattern skips generations; parents each carry one copy of the mutant gene and are themselves normal. In children with focal segmental glomerulosclerosis patients who also have a family history of focal segmental glomerulosclerosis, perhaps 35% will have two mutations in podocin. When families are small, podocin mutations can also appear as sporadic focal segmental glomerulosclerosis (no family history); perhaps 20% of children with sporadic focal segmental glomerulosclerosis will have podocin mutations. Reasons for pursuing a diagnosis of podocin mutation include the following: 1) steroid-resistance is typical; 2) the risk of focal segmental glomerulosclerosis recurrence after kidney translantation is lower than with idiopathic focal segmental glomerulosclerosis; 3) family counseling. Podocin mutations almost never cause adult-onset focal segmental glomerulosclerosis
- Autosomal dominant inheritance. This inheritance pattern affects every generation, with one parent and approximately 50% of children affected. Two genes are have been reported (alpha actinin-4 and TRPC6); in both cases focal segmental glomerulosclerosis appears during adulthood.
- Other patterns. WT-1 mutations cause focal segmental glomerulosclerosis appearing in the teenage years. Due to the effects of this mutation on sex development, these individuals may appear to be female but may lack ovaries and a uterus and consequently do not begin menstruation. Mitochondrial DNA mutations may be associated with isolated focal segmental glomerulosclerosis or the focal segmental glomerulosclerosis may be accompanied by other symptoms, include acidosis, stroke-like episodes, muscle weakness, deafness and diabetes.
Individuals of African descent are at increased risk for focal segmental glomerulosclerosis, by a factor of approximately 4-fold compared to those of other races and ethnic groups. It is likely that this has a genetic component but the specific gene or genes have not been identified.
Finally certain medications have been associated with focal segmental glomerulosclerosis.
Focal segmental glomerulosclerosis pathophysiology
The pathogenesis of focal segmental glomerular sclerosis involves a complex interplay of several cell types including podocytes, endothelial cells, and the basement membrane. Podocytes are terminally differentiated cells that provide structural support to the glomerulus and are essential in maintaining an intact glomerular filtration barrier essential to prevent nephrotic range proteinuria. Injury and loss of podocytes result in podocyte hypertrophy of remaining podocytes to cover the glomerular capillary surface resulting in effacement and protein loss 20, 21. Foot process effacement and the proliferation of mesangial, endothelial, and epithelial cells earlier in the course of illness, followed by collapse/shrinkage of glomerular capillaries, all result in scarring (glomerulosclerosis) 22.
The proposed mechanism for podocyte injury includes viral- or toxin-mediated insult and intrarenal hemodynamic alterations, such as high intraglomerular capillary pressure and glomerular hyperperfusion. Many morphologic subsets, such as a collapsing variant (FSGS with mesangial hypercellularity), a cellular variant (endocapillary and extracapillary hypercellularity), and FSGS with tip lesions, are known 23.
Understanding the pathophysiology of FSGS has improved with the discovery that mutations in several proteins responsible for maintaining podocyte structure, function, or both not only result in FSGS but can predict disease characteristics, such as steroid responsiveness 24. For instance, FSGS with mutations in NPHS2 or TRPC6 is challenging to treat with immunosuppressive therapy; however, when such patients undergo kidney transplantation, the disease does not usually recur. APOL1 G1/G2 variants have been associated with a poor renal prognosis and steroid resistance in nephrotic syndrome or FSGS 25.
Proposed circulating factors linked to the development of FSGS include candidate molecules, such as hemopexin, cardiotrophin-like cytokine 1, and vascular endothelial growth factor. One molecule that has been extensively studied is a form of urokinase receptor (suPAR) 26.
Focal segmental glomerulosclerosis histopathology
Histologically, focal segmental glomerular sclerosis (FSGS) is characterized by sclerosis, hyalinosis, adhesions or synechiae formation, resulting in segmental obliteration of glomerular capillaries 10. On electron microscopy, foot process effacement is the predominant finding without significant basement membrane abnormalities. Immunofluorescence shows staining for IgM and C3 in sclerotic areas. Juxtamedullary nephrons are affected first and hence inadequate sampling may miss focal lesions.
Histologically, focal segmental glomerular sclerosis (FSGS) is classified into five variants: perihilar, tip, cellular, collapsing and not otherwise specified (NOS) 27, 28, 29.
Perihilar focal segmental glomerulosclerosis
The sclerosing lesion is located at the vascular pole of the glomerulus. Perihilar focal segmental glomerulosclerosis is commonly seen in adaptive FSGS due to increased pressure in the glomerulus which is in close proximity to the afferent arteriole. Foot process effacement is mild, resulting in subnephrotic proteinuria and relatively normal serum albumin levels 10.
Tip focal segmental glomerulosclerosis
The segmental lesion involves the tubular pole of the glomerulus 10. Tip focal segmental glomerulosclerosis is commonly seen in Caucasians, presenting with diffuse foot process effacement and abrupt onset of nephrotic syndrome. These patients have lower baseline creatinine, have an excellent response to treatment, and the lowest rate of progression 30.
Cellular focal segmental glomerulosclerosis
Cellular focal segmental glomerulosclerosisis the least common variant of focal segmental glomerulosclerosis, characterized by hypercellular glomerulus including endocapillary and glomerular epithelial cell hyperplasia. It presents with diffuse foot process effacement and full-blown nephrotic syndrome 31.
Collapsing focal segmental glomerulosclerosisis
Collapsing focal segmental glomerulosclerosisisis is characterized by hyperplasia and hypertrophy of visceral glomerular epithelial cells leading to the collapse of the glomerular tuft 10. This is commonly seen in viral (parvovirus B19, CMV, HIV) and drug-associated forms of focal segmental glomerulosclerosis (interferon-alpha, interferon-beta, interferon-gamma & pamidronate) and presents with diffuse effacement of foot processes, heavy proteinuria with the lowest rate of remission, and the worst prognosis 32, 33, 34.
Not otherwise specified (NOS) focal segmental glomerulosclerosisisis
NOS focal segmental glomerulosclerosisisis is the most common subtype of focal segmental glomerulosclerosis and does not fit into any other morphological forms of focal segmental glomerulosclerosis 10. NOS focal segmental glomerulosclerosisisis presents with a variable degree of effacement and proteinuria. Histopathology may sometimes resemble nodular sclerosis as in diabetes and other conditions 35.
Focal segmental glomerulosclerosis signs and symptoms
Children with focal segmental glomerular sclerosis (FSGS) typically present with the full-blown nephrotic syndrome (edema, massive proteinuria, hypoalbuminemia, hypercholesterolemia). Adults can have nephrotic or sub-nephrotic proteinuria, hypertension, microscopic hematuria, or present with renal insufficiency. Patients with primary focal segmental glomerulosclerosis often have profound hypoalbuminemia and edema, but these are rare in secondary forms.
Patients with focal segmental glomerulosclerosis may present in different ways. First some present with edema, either a sudden onset similar to that seen in patients with minimal change disease or slower onset over weeks to months. Second, some patients have no symptoms and are found to have asymptomatic (no symptoms) proteinuria, found on routine urinalysis performed as part of a physical examination. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin.
Generally, edema develops over a few weeks; however, the onset may be abrupt, with sudden weight gain of 15-20 lbs (6.8 to 9 kg) or more. Frequently, a recent upper respiratory tract infection precedes edema.
Pleural effusion and ascites could be present, although pericardial effusions are rare. Gross edema could predispose patients to infections and ulcerations in dependent areas, such as the lower extremities. Abdominal pain may be a sign of peritonitis, a common finding in children. Rarely, xanthomas may be seen in cases of severe hyperlipidemia. In many patients, physical examination is normal except for edema. Severe hypertension is not uncommon, particularly in Black patients with renal impairment 36. Rarely do patients experience severe kidney failure with features of advanced uremia, such as nausea, vomiting, seizures, bleeding, or altered mental status. Patients with FSGS secondary to conditions such as reflux nephropathy, massive obesity, and renal dysplasia/agenesis usually present with non-nephritic proteinuria. These patients may often experience worsening renal function over the course of months to years.
Focal segmental glomerulosclerosis signs and symptoms include:
- Foamy urine caused by high protein levels in your urine (called proteinuria)
- Poor appetite
- Swelling in body parts like your legs, ankles and around your eyes (called edema)
- Weight gain due to extra fluid building in your body
- High fat levels in the blood (high cholesterol)
- Low levels of protein in your blood
Focal segmental glomerulosclerosis can cause nephrotic syndrome.
- Nephrotic syndrome: A set of symptoms that happen together and affect your kidneys. These include:
- Swelling in body parts like your legs, ankles, or around your eyes (edema)
- Large amounts of protein in your urine (proteinuria)
- Loss of protein in your blood
- High levels of fat lipids in your blood (high cholesterol)
- High blood pressure (in some cases)
If the condition is advanced, the symptoms may be like those of kidney failure. People may report fatigue, a poor appetite, headache, itchy skin, shortness of breath and/or nausea.
Focal segmental glomerulosclerosis complications
Focal segmental glomerulosclerosis cmplications may include:
- Chronic kidney failure
- End-stage kidney disease
- Infection
- Malnutrition
- Nephrotic syndrome
Focal segmental glomerulosclerosis diagnosis
Your doctor will perform a physical exam. This exam may show tissue swelling (edema) and high blood pressure. Signs of kidney (renal) failure and excess fluid may develop as the condition gets worse.
Tests may include:
- Kidney biopsy. In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope.
- Kidney function tests (blood and urine)
- Glomerular filtration rate (GFR): A blood test will be done to know how well your kidneys are filtering the wastes from your body.
- 24-hour urine collection for protein quantification
- Urinalysis
- Urine microscopy
- Urine protein
- Hepatitis and HIV serology
- Complement levels
- Serum and urine protein electrophoresis in elderly to rule out paraproteinemias
- Genetic testing: A genetic test may be done to see if you were born with genes that caused your kidney disease. This information may help your doctor decide what type of treatment is best for you.
Ultimately, a kidney biopsy is required to confirm the diagnosis of focal segmental glomerulosclerosis. The characteristic finding in FSGS is segmental solidification of the glomeruli, typically in the perihilar region and occasionally in the peripheral areas, such as the tubular pole 37. In the diseased glomeruli, the accumulation of acellular matrix and hyaline deposits obliterates capillaries in a segmental fashion. Coarsely granular deposits of C3 and IgM are often seen in these areas. Diffuse foot process fusion is seen predominantly in the sclerotic segments, whereas partial effacement is observed overlying normal-appearing lobules. In HIV-associated FSGS, electron microscopy of the kidney shows tubuloreticular inclusions in mesangial and endothelial cells, an indirect indication of viral disease 38.
In patients with focal segmental glomerulosclerosis, urinalysis shows large amounts of protein and casts (hyaline and broad waxy), although red blood cell casts are usually absent. In advanced cases, broad casts may be evident. Serum creatinine (SCr) and creatinine clearance (CrCl) are usually within the reference range in the early stages. Features of nephrotic syndrome (proteinuria >3.5 g/day, serum albumin <30 g/L, with or without edema) may or may not be present.
In idiopathic FSGS, investigations for an underlying cause are usually negative. Such conditions include the following:
- Systemic lupus erythematosus (antinuclear antibody/anti-DNA titers, serum complement C4/C3 levels)
- Hepatitis B or C infection
- Vasculitis (serum protein electrophoresis, antineutrophil cytoplasmic antibody titers)
In patients suspected to have secondary FSGS, the following should be obtained:
- HIV antibody, CD4, and viral load
- Serology for hepatitis B and C
- Parvovirus testing
FSGS in morbidly obese patients is diagnosed by excluding other causes. The common features in obesity-related FSGS include glomerular hyperfiltration and activation of the renin-angiotensin-aldosterone system 39. FSGS may be considered in patients with proteinuria; however, in younger patients with no red blood cell casts and negative serologic studies, the definitive diagnosis is made on a kidney biopsy.
Ultrasonography
Earlier in the course of illness, ultrasonography will reveal normal or enlarged kidneys with increased echogenicity, indicating diffuse intrinsic medical renal disease 13. In advanced kidney failure, kidneys are shrunken and small, suggesting severe interstitial fibrosis and glomerular scarring. In HIV-associated FSGS, ultrasound study generally reveals large echogenic kidneys.
Focal segmental glomerulosclerosis treatment
The type of treatment you get depends on the cause. Everyone is different and your doctor will make a treatment plan that is right for your type of focal segmental glomerulosclerosis. Corticosteroids (daily or every other day) are first line of treatment in children and adults with focal segmental glomerular sclerosis (FSGS) with the best chance of inducing a sustained complete remission; they may also induce a partial remission. While some patients with a complete remission experience relapse, they often undergo a second remission with a second course of therapy. Steroids may also induced a sustained partial remission. Standard courses of steroid therapy include daily or alternate day steroids for period of 2 to 4 months, possibly extended longer with a taper off if the response has been good.
In patients with subnephrotic proteinuria (adaptive focal segmental glomerulosclerosis), a trial of renin-angiotensin system (RAS) inhibition (e.g., angiotensin-converting enzyme inhibitors [ACE inhibitors] and angiotensin 2 receptor blockers [ARBs]), and sodium restriction can be tried. ACE inhibitors and angiotensin 2 receptor blockers are blood pressure medications used to reduce protein loss and control blood pressure. In other secondary forms of focal segmental glomerulosclerosis, removing the offending agent or treating the underlying disorder is recommended. In addition, optimization of blood pressure, treatment of edema with diuretics, statin therapy for hypercholesterolemia and anticoagulation in select patients at risk for thrombosis/embolization are indicated.
Children respond within a few weeks, but adults may take months to respond. Glucocorticoids are associated with a remission rate of approximately 30% compared to about 50% in patients treated with calcineurin inhibitor. Rituximab, mTOR inhibitors, and plasmapheresis have been tried in select patients with varied results.
Corticosteroids
- Oral prednisone: 2 mg/kg/day for 6 weeks followed by 1 mg/kg/day on alternate days for 6 weeks in children and 1mg/kg/day for 3 to 6 months in adults
- High-dose corticosteroid therapy with prednisolone is started at an initial dose of 1 mg/kg/day daily single dose (maximum 80 mg) or alternate day dose of 2 mg/kg/day (maximum 120 mg) for at least four weeks and until complete remission is achieved or a maximum of 16 weeks treatment, whichever is earlier 40, 41. Patients with the potential to remit would likely show some decline in proteinuria before 16 weeks of high-dose steroid therapy. Hence, it is unnecessary to persist with high-dose steroids treatment until 16 weeks if proteinuria persists or worsens. This becomes more important in patients who are experiencing side effects of steroids 42.
- Tapering off steroids should start after at least four weeks of achieving remission with high-dose therapy or after two weeks of the disappearance of proteins, whichever is longer. Prednisolone is reduced by 5 mg every one to two weeks to complete a total duration of six months. If partial remission is achieved within eight to twelve weeks of high-dose steroid therapy, continue until 16 weeks to ensure complete remission. After that, the prednisolone dose is reduced by 5 mg every one to two weeks to complete a total six-month duration of 6 months.
Immunosuppressive drugs
Patients who are resistant or intolerant to steroids are treated with immunosuppressive drugs with calcineurin inhibitors, mycophenolate mofetil, or rituximab 43, 44, 45.
- Calcineurin inhibitors:
- Cyclosporine 3 to 5 mg/kg/day (target trough levels 100 to 175 ng/ml)
- Tacrolimus 0.05 to 0.1 mg/kg/day (target trough levels 5 to 10 ng/ml) 46
- Trough levels should be monitored to prevent drug toxicity. The duration of determining the efficacy of cyclosporin or tacrolimus is at least six months, after which the patient can be labeled calcineurin inhibitor-resistant. Calcineurin inhibitorss should be continued for at least 12 months in patients with partial or complete remission to prevent relapses. The dose of calcineurin inhibitors is to be slowly tapered over 6-12 months as tolerated.
- In patients resistant or intolerant to calcineurin inhibitors, there is a lack of evidence regarding any particular agent. Mycophenolate mofetil, high-dose dexamethasone, rituximab, and adrenocorticotropic hormone (ACTH) have been studied 47. In addition, rituximab, mTOR inhibitors, and plasmapheresis have been tried in select patients with varied results.
- For those who are steroid-responsive but experience one or more relapses when steroids are stopped, other therapies include cyclophosphamide or cyclosporine.
- For those who are steroid-resistant, other therapies include cyclosporine or (less well studied) mycophenolate mofetil.
- Mycophenolate mofetil: 25 to 35 mg/kg/day +/- dexamethasone
- Edema is managed by dietary salt restriction (low sodium diet) and diuretics, either administered orally or intravenously. Diuretics are medications help your body get rid of excess fluid and swelling. These can be used to lower your blood pressure too.
Focal segmental glomerulosclerosis prognosis
Several features predict outcome in focal segmental glomerulosclerosis including, race (Blacks have worse outcomes), degree of proteinuria, presence of renal insufficiency, histological variant (tip variant had the best outcome and collapsing variant had the worst outcome), degree of interstitial fibrosis or tubular atrophy and response to treatment with patients attaining partial or complete remission having better prognosis 10. Also, patients with primary focal segmental glomerulosclerosis did worse when compared to those with adaptive or secondary causes of focal segmental glomerulosclerosis 48, 41.
Focal segmental glomerulosclerosis life expectancy
At ten years after diagnosis, approximately 50% of individuals with focal segmental glomerulosclerosis will developed end-stage kidney disease (ESRD), requiring dialysis or kidney transplant. This statistic includes both individuals who received treatment and did not respond favorably and individuals who came to medical attention too late for effective therapy to be attempted. This prognosis makes focal segmental glomerulosclerosis the most serious primary glomerular disease. The prognosis in those who enter a complete remission is excellent, even when they experience a relapse. The long-term prognosis in those who enter a partial remission is less good, but is substantially better than those who did not respond to therapy or did not take therapy.
Recurrence after kidney transplant
Approximately 25% of patients who have focal segmental glomerulosclerosis in their own kidneys will experience recurrent focal segmental glomerulosclerosis after kidney transplant. When focal segmental glomerulosclerosis recurs, it typically does so within days to weeks of transplant, and essentially always within one year of transplant. The cause of recurrent focal segmental glomerulosclerosis is unknown but is believed to be a circulating protein. Patients at increased risk for recurrent focal segmental glomerulosclerosis include the following: rapid progression (from diagnosis to end-stage kidney disease in less than 3 years, recurrent focal segmental glomerulosclerosis in a prior transplant (risk of recurrent focal segmental glomerulosclerosis is approximately 70%), and white race (weak risk factor). In patients with focal segmental glomerulosclerosis, it is prudent to check the urine weekly by dipstick for protein or every 2 to 4 weeks by measuring a random urine protein/creatinine ratio. The appearance of proteinuria should prompt a transplant kidney biopsy, which would initially show just podocyte foot process effacement and after weeks to months of proteinuria will show focal segmental glomerulosclerosis. If the diagnosis can be made within 2 to 4 weeks of proteinuria onset, therapy may include plasma exchange and possibly cyclophosphamide.
Membranous nephropathy
Membranous nephropathy also known as membranous glomerulopathy, is one of the many glomerular diseases causing nephrotic syndrome 49. Nephrotic syndrome includes significant amounts of protein in the urine (at least 3.5 grams per day), low blood protein (albumin) levels, and swelling (edema). Membranous nephropathy (membranous glomerulopathy) is the second most common glomerular disease in adults after focal segmental glomerulosclerosis (FSGS) 50. Membranous nephropathy (membranous glomerulopathy) is the most common cause of primary nephrotic syndrome in White adults 51. Membranous nephropathy most commonly occurs above 40 years of age, with the peak incidence between 50 to 60 years in the USA 52. It is more common in males than females and tends to show a better outcome in females. The incidence is 8 to 10 cases per 1 million population worldwide, and 12 per 1 million population per year in the USA. Membranous nephropathy is less common in children and mostly due to secondary causes 53. Membranous nephropathy can occur by itself (primary) or due to another disease or underlying cause (secondary). Membranous nephropathy can also present in conjunction with other types of glomerulonephritis like IgA nephropathy, FSGS, and lupus nephritis 49.
Membranous nephropathy is considered an autoimmune disease, which means that it caused by your body’s immune system turning against you and harming your body when it should be protecting you. Membranous nephropathy is caused by the build-up of immune complexes within the filters (glomeruli) of the kidney itself. Figure 4 below is a diagram of how the immune complexes deposit in the kidney. The immune system normally creates antibodies to recognize and attach to something called an antigen. When an antibody attaches to an antigen, this is called an immune complex. Antigens are normally foreign to the body, like a virus or bacteria. However, sometimes, the body can make antibodies that recognize and attach to something in the body itself (not foreign) – these types of antibodies are called autoantibodies. Immune complexes are normally cleared from the blood before causing any problems, but under certain conditions they can accumulate in different parts of the body. In membranous nephropathy these immune complexes (antibodies made by the immune system attached to antigens) get caught in the kidney filters (glomeruli). In most cases of membranous nephropathy, antibodies are made to an antigen that is part of the kidney filter (glomerulus) itself. When your immune system attacks the glomeruli in membranous nephropathy, it causes changes to the filters that lead you to lose large amount of protein into the urine. Together these antibodies and antigens create immune complexes that get stuck in the kidney filter (glomerulus) and it can eventually lead to kidney failure.
Recently the antibody that causes most cases of membranous nephropathy was discovered and identified 54. In about 70-80% of patients with primary membranous nephropathy (meaning their membranous nephropathy is not associated with or due to other diseases or causes), an antibody called anti‐phospholipase‐A2‐receptor‐antibodies (anti‐PLA2R) is found in the kidney and/or bloodstream 55, 56. The anti-PLA2R antibody (short for anti-phospholipase A2 receptor antibody) attaches to the phospholipase A2 receptor (the antigen). The phospholipase A2 receptor is a protein found in the kidney filter (glomerulus), specifically within a cell called the podocyte which makes up part of this filter (see below). Another antibody called anti‐thrombospondin type‐1 domain‐containing protein 7A‐antibodies (anti‐THSD7A) was also discovered but is found in a much smaller number of patients with primary membranous nephropathy, only about 2-3% 57. This is an antibody to a different antigen, THSD7A, that is also found in the kidney filter (another protein in the podocyte) 56. Identifying presence or absence of PLA2R antibody and the subclass of IgG deposits may help to differentiate idiopathic from secondary membranous nephropathy. For example, the deposits in idiopathic membranous nephropathy are PLA2R antibody positive and predominantly IgG 4, whereas PLA2R antibody is typically negative and IgG 1 and 2 predominate in cancer-associated membranous nephropathy 55.
Patients with membranous nephropathy typically present with edema and nephrotic-range proteinuria (greater than 3.5 g/day) and occasionally with microscopic hematuria and hypertension. Symptoms and signs of a disorder causing membranous nephropathy (eg, a cancer) may be present initially.
Membranous nephropathy is characterized by proteinuria (greater than 3.5 g/day) found on routine urinalysis performed as part of a physical examination, or may present with peripheral edema and frothy urine 49. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol), low serum albumin (hypoalbuminemia) and acute kidney injury with elevated creatinine. The lack of active sediments with the absence of blood in urine (hematuria) and red cell casts in urine microscopy differentiates it from nephritic syndromes. The nephrotic range proteinuria is attributed to podocyte injury and loss of membrane anionic charge barrier, causing albuminuria. This is in contrast to a nephritic syndrome, which involves an inflammatory process in the glomerular basement membrane 58.
Early in the course of disease, light microscopy may be nearly normal or may show thickening of the glomerular capillary loops. Immunofluorescence microscopy shows deposition of IgG (antibody) along capillary loops. Electron microscopy shows immune deposits (antibody) along the outside of the glomerular basement membrane, facing the urinary space and located underneath the podocyte).
The cause of membranous nephropathy can be primary or secondary. A kidney biopsy is used to confirm the diagnosis of membranous nephropathy. Immunosuppressive therapy plays a major role in the treatment of this disease.
Figure 4. Membranous nephropathy
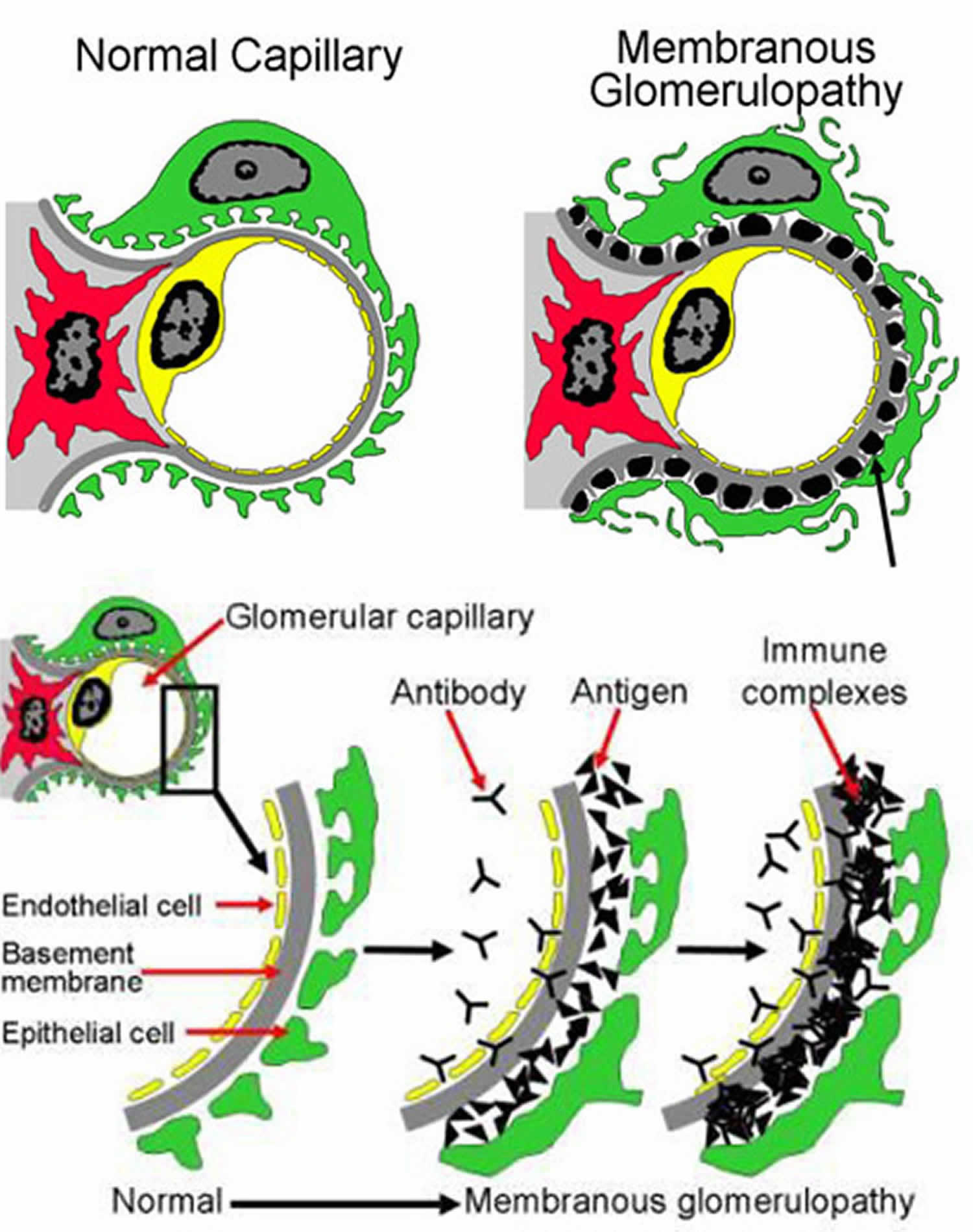
Footnotes: Top image shows part of a glomerulus, comparing a normal to one affected by membranous nephropathy. On the right, the black spots or lumps (there is an arrow pointing to one) are collections of immune complexes (antigen-antibody complexes). As more of these immune complexes build up between the layers of the filters, it becomes thickened. The kidney cells (green in this picture, called podocytes) that make up part of the filter become damaged from the immune complexes and the inflammation caused by the immune system, and stop working properly. You can see in the picture on the right that the grey layer (basement membrane) has become thicker and has started filling in the spaces between the black spots/lumps. You can also see that the green cell does not look the same as in the normal healthy filtering loop (capillary loop) on the left. Under a microscope, the filters (glomeruli) in the kidney become thickened, which is where the name membranous nephropathy comes from. The bottom image shows a cross section of part of the kidney filter (glomerulus). This includes different layers, including the cells making up the capillary blood vessel (endothelial cell, in yellow), the basement membrane (gray), and layer of kidney cells (podocyte, in green). Blood inside the capillary blood vessel is filtered across these layers and becomes urine. Antibodies (Y-shaped, black in the picture) in the blood stream attach to antigens (triangles, black in the picture) and form immune complexes that get stuck and build up between the layers of the filter (glomerulus). These immune complexes also activate the immune system causing inflammation. Buildup of these immune complexes as well as the inflammation cause the filter to stop working properly and can lead to kidney damage. Normally the filter (glomerulus) allows water, electrolytes and some waste materials through to become urine, and larger things like blood cells and proteins are too big to pass through the filter – so they stay in the blood. However, in this disease, protein and blood cells can leak into the urine because the filter is not working properly.
[Source 56 ]Figure 5. Membranous nephropathy pathology
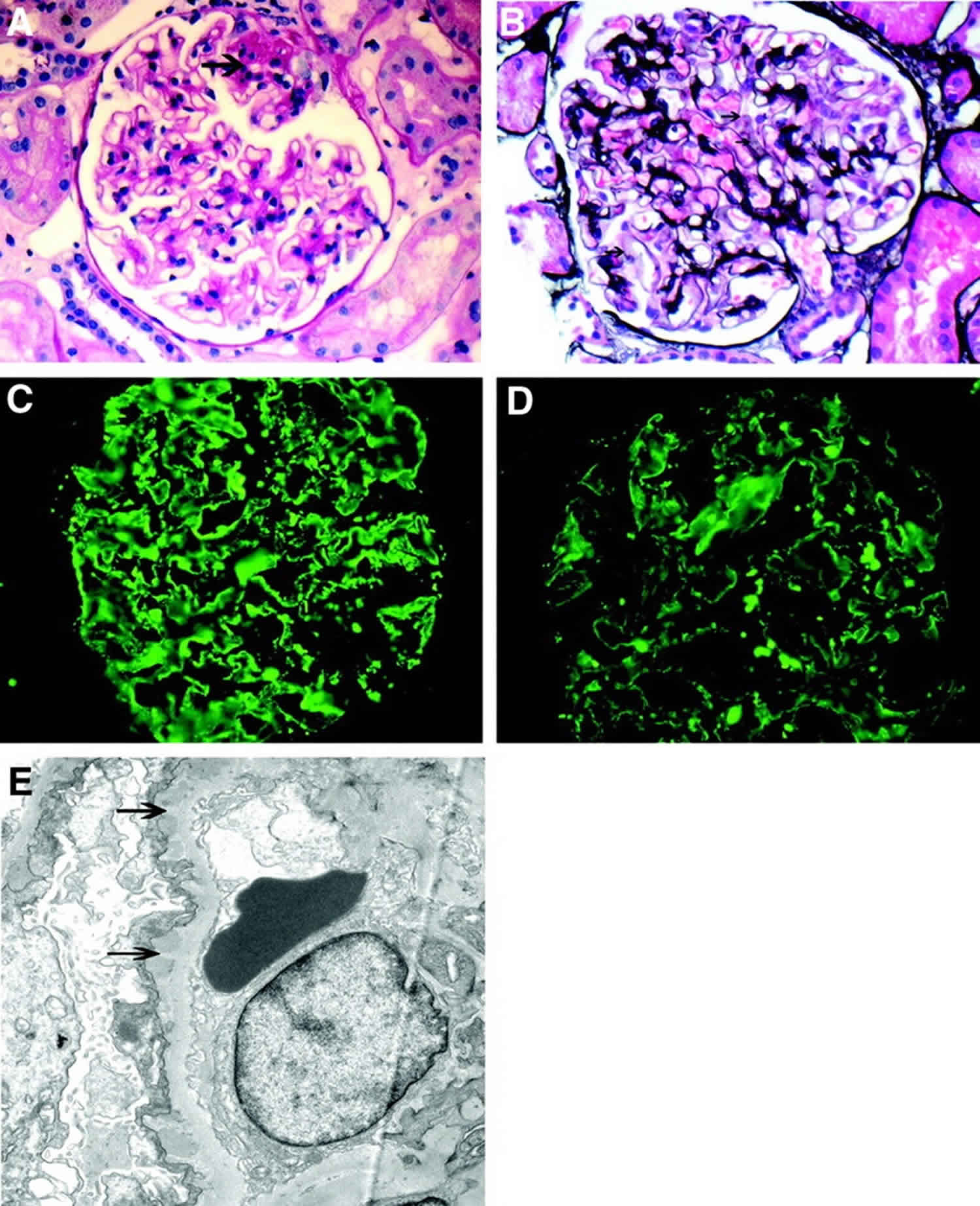
Footnotes: (A and B) Light microscopy showing segmental sclerosis (arrow; A, periodic acid Schiff stain) and pinholes (arrow) along the glomerular basement membranes (B, silver stain). (C and D) Immunofluorescence microscopy showing granular IgG (C) and C3 (D) along the capillary walls. (E) Electron microscopy showing subepithelial electron-dense deposits and basement membrane spikes (arrows) separating the deposits. Magnification, ×40 in A, B, C, and D; ×7400 in E.
[Source 50 ]Will I have kidney failure because of membranous nephropathy?
You should talk with your doctor about your condition because the progression of the disease depends on many factors. Treatment can slow the process of kidney disease. Everyone is different in how they respond to treatment. Over time, some patients with membranous nephropathy gradually get worse until they reach kidney failure, If this occurs, they will need a kidney transplant or dialysis to stay alive. Some people respond well to treatment and may live with the disease for many years while being monitored for any signs of change.
Membranous nephropathy causes
Membranous nephropathy may be idiopathic (of unknown cause) also known as Primary membranous nephropathy (75% to 80% of cases) or associated with infections (hepatitis B) or stem cell transplant also known as Secondary membranous nephropathy (20% to 25% of cases) 49, 59.
Primary membranous nephropathy
Idiopathic (unknown cause) membranous nephropathy can attributed to the presence of one of the following antibodies and the absence of a secondary cause 60, 61:
- Antibodies against phospholipase A2 receptor (PLA2R) antigen, associated with HLA DQA1 (70% to 80% of primary cases)
- Antibodies against neural epidermal growth factor-like 1 (NELL-1) (15% to 20% of primary cases) 62
- Antibodies against thrombospondin (THSD7A) (1% to 5% of primary cases)
- Others: Antibodies against neutral endopeptidase (NEP) and exostosin (EXT1/EXT2)
Membranous nephropathy is considered an autoimmune disease, which means that it caused by the body’s own immune system. membranous nephropathy is caused by the build-up of immune complexes within the filters (glomeruli) of the kidney itself.The immune system normally creates antibodies to recognize and attach to something (called an antigen). When an antibody attaches to an antigen, this is called an immune complex. Antigens are normally foreign to the body, like a virus or bacteria. However, sometimes, the body can make antibodies that recognize and attach to something in the body itself (not foreign) – these types of antibodies are called autoantibodies. Immune complexes are normally cleared from the blood before causing any problems, but under certain conditions they can accumulate in different parts of the body. In membranous nephropathy these immune complexes (antibodies made by the immune system attached to antigens) get caught in the kidney filters (glomeruli). In most cases of membranous nephropathy, antibodies are made to an antigen that is part of the kidney filter (glomerulus) itself. Together these antibodies and antigens create immune complexes that get stuck in the kidney filter (glomerulus) and cause disease.
As with other kinds of autoimmune diseases (such as lupus, rheumatoid arthritis, or Crohn’s disease), scientists think that there are probably multiple things that contribute to membranous nephropathy developing – meaning multiple things that must occur for the immune system to target and attack/damage the body (rather than just targeting foreign things like infections). Some people may have a gene or genes that make them more likely to have an autoimmune disease (more susceptible to developing them). Though some people may be more likely to get autoimmune diseases if they have family members with autoimmune diseases, membranous nephropathy is not a genetic disease and it is not passed down from a parent to their child. In people who may be at higher risk of an autoimmune disease, certain events or triggers may cause the disease to ultimately develop – such as an infection or other inflammation in the body that might activate the immune system. However, these are hypotheses, and we do not understand at this point why one person gets membranous nephropathy when others do not.
Recently the antibody that causes most cases of membranous nephropathy was discovered and identified 54. In about 70-80% of patients with primary membranous nephropathy (meaning their membranous nephropathy is not associated with or due to other diseases or causes), an antibody called anti-PLA2R is found in the kidney and/or bloodstream 55. The anti-PLA2R antibody (short for anti-phospholipase A2 receptor antibody) attaches to the phospholipase A2 receptor (the antigen). The phospholipase A2 receptor is a protein found in the kidney filter (glomerulus), specifically within a cell called the podocyte which makes up part of this filter. Another antibody called anti‐thrombospondin type‐1 domain‐containing protein 7A‐antibodies (anti‐THSD7A) was also discovered but is found in a much smaller number of patients with primary membranous nephropathy, only about 2-3% 57. This is an antibody to a different antigen, THSD7A, that is also found in the kidney filter (another protein in the podocyte).
Secondary membranous nephropathy
- Infections (e.g., hepatitis B, hepatitis C, syphilis, malaria, human immunodeficiency virus [HIV], schistosomiasis, leishmaniasis)
- Neoplasms (e.g., adenocarcinoma and squamous cell carcinoma of lungs and gastrointestinal tract, hematological malignancies)
- Medications (e.g., non-steroidal anti-inflammatory drugs [NSAIDs], anti-tumor necrosis factor-alpha inhibitors, gold therapy, penicillamine, probenecid)
- Heavy metal poisoning (gold, mercury)
- Autoimmune diseases (e.g., systemic lupus erythematosus [SLE], Sjogren syndrome, rheumatoid arthritis, IgG4 related nephropathy)
- Thyroiditis
- Miscellaneous (hematopoietic stem cell transplant, graft versus host disease, diabetes mellitus)
Depending on the patient’s age, 4 to 20% have an underlying cancer, including solid cancers of the lung, colon, stomach, breast, or kidney; Hodgkin or non-Hodgkin lymphoma; chronic lymphocytic leukemia; and melanoma.
Because hepatitis and cancer can be associated with membranous nephropathy, anyone who is found to have membranous nephropathy should be tested for hepatitis and make sure they are up to date on age-appropriate cancer screening. A blood test can be done to check for hepatitis. Age appropriate cancer screening can include tests such as a pap smear, mammogram, colonoscopy, or CT scan of the lungs (in people who smoke or have smoked). Your doctor can clarify which of these tests are appropriate or necessary for you.
Membranous nephropathy is rare in children and, when it occurs, is usually due to hepatitis B virus infection, SLE, or autoimmune thyroid disease.
Membranous nephropathy pathology
Recent findings of anti‐phospholipase‐A2‐receptor‐antibodies (anti‐PLA2R) 55 and anti‐thrombospondin type‐1 domain‐containing protein 7A‐antibodies (anti‐THSD7A) 57 have improved understanding of the autoimmune pathophysiology of primary membranous nephropathy. Primary membranous nephropathy is caused by the subepithelial formation of immune complex deposits in the kidney’s glomerular basement membrane (GBM) 63. The exact mechanisms behind this remain unclear, however, there are a number of presumptive hypotheses. Firstly, systemically pre‐formed immune‐complexes may deposit in the glomerular basement membrane, suggesting a similar pathophysiological mechanism as in lupus‐associated nephritis 63. Secondly, circulating antigens (such as during infection) might be targeted by antibodies, thus forming immune complexes that deposit in this site. this has especially been observed in infection‐related (i.e. secondary) forms of membranous nephropathy, such as during infection with hepatitis B virus 63, 64, 65. Thirdly, a rat model of Heymann nephritis 66, podocyte‐antigens (such as megalin) may lead to binding of autoantibodies to the glomerular basement membrane’s podocytes which cause the subepithelial deposits that are present in primary membranous nephropathy 67. However, thus far, this connection has not been clearly established through the extraction of anti‐megalin‐antibodies in primary membranous nephropathy. Finally, the complement system and genetic factors might contribute to the autoimmune etiology of primary membranous nephropathy. So far, two associated genomic loci have been identified: chromosome 2q24 encodes for the anti‐PLA2R‐receptor auto‐antibody and chromosome 6p21 encodes for HLADQA1, which might play pivotal roles in the pathogenesis of primary membranous nephropathy 68, 69.
Membranous nephropathy is caused by the deposition of antigen-antibody complexes between the glomerular basement membrane (GBM) and podocytes. These complexes mainly consist of immunoglobulin IgG4, complement C3, and C5b-C9 membrane attack complexes (MAC). In the case of secondary membranous nephropathy (like systemic lupus erythematosus) complexes may also include IgG1/IgG3, IgA, IgM, or C1q, and rarely involve the mesangium (“full house pattern”) 53. The immune complexes activate the complement system and generate membrane attack complex (MAC), which releases proteases, cytokines, and oxidants, causing cellular and tissue damage. This leads to disruption of podocyte structure, hampering of slit diaphragm integrity, and loss of membrane anionic charge barrier, resulting in proteinuria. This nephrotic range proteinuria leads to hyperlipidemia, prothrombotic state, vitamin D deficiency, and hypertension. Studies have also suggested that an unknown cytokine in membranous nephropathy leads to decreased nephrin synthesis, a protein responsible for glomerular filtration barrier integrity 70.
Membranous nephropathy signs and symptoms
Membranous nephropathy happens slowly over time, so you may not notice anything is wrong. You may only see some signs on your own, while others may be found by your healthcare provider.
Signs and symptoms of membranous nephropathy include:
- Swelling in body parts like your legs, ankles and around your eyes (called edema)
- Weight gain
- Fatigue
- Foaming of the urine caused by high protein levels in the urine (called proteinuria)
- High fat levels in the blood (high cholesterol)
- Low levels of protein in the blood
The most common symptom of membranous nephropathy is swelling (edema). This can range from mild to severe. Most people with membranous nephropathy have some swelling and it is often the first symptom people notice. In membranous nephropathy (as opposed to some other diseases that cause protein in the urine and nephrotic syndrome), swelling usually comes on slowly (over weeks to months) but it can sometimes come on more quickly. It typically starts in the feet, ankles, or legs, but can occur anywhere in the body, including the abdomen, hands or arms, and face.
The swelling in membranous nephropathy occurs because of fluid building up in the body, and specifically in different tissues. When fluid builds up, it can sometimes go into the lungs (pulmonary edema) and cause difficulty breathing or shortness of breath. Pulmonary edema (excess fluid in the lungs) is less common than swelling (edema) but in people who have this it may be most noticeable when walking or exerting yourself or when lying down flat.
Some people with membranous nephropathy- in particular, people who have nephrotic syndrome (with large amounts of protein in the urine and low blood protein levels)- feel very tired or run-down. Doctors do not know exactly how or why this happens but some people with membranous nephropathy (and other diseases that cause nephrotic syndrome) notice this.
When protein gets through the filter in your kidney and into your urine, the urine can become foamy or bubbly. Some people may notice this change in their urine before they have other symptoms.
Renal vein thrombosis is more frequent in membranous nephropathy and is usually asymptomatic but may manifest with flank pain, hematuria, and hypertension. It may progress to pulmonary embolism.
There are other symptoms that can occur with membranous nephropathy, but the ones above are some of the most common and the ones that people with membranous nephropathy often notice first.
Nephrotic syndrome
Membranous nephropathy often causes nephrotic syndrome. Nephrotic syndrome is a group of symptoms or changes that often occur together in someone that is losing a lot of protein into the urine. Nephrotic syndrome can also happen in other diseases that cause a lot of protein to be lost into the urine. Although a lot of people with membranous nephropathy have nephrotic syndrome, though not everybody does. Nephrotic syndrome includes these findings:
- At least 3.5 grams of protein in the urine per day (proteinuria). This can be measured on a 24 hour urine collection but can also be estimated on a single urine sample. To estimate the amount of proteinuria from a single urine sample, the urine protein to creatinine ratiois used – this gives an estimate of how many grams of protein would be in a 24 hour urine sample.
- Low blood protein (albumin) levels
- Swelling (sometimes called edema)
- High cholesterol
- Increased risk for blood clots
Membranous nephropathy complications
- Complications from a pulmonary embolism, deep vein thrombosis, renal vein thrombosis, and other systemic thromboembolic phenomena as well as increased risk of bleeding from systemic anticoagulation
- Membranous nephropathy leading to hyperlipidemia, hypertension, and chronic kidney disease, compromising cardiovascular health
- Progression of membranous nephropathy to chronic kidney disease (CKD) with reduced eGFR. CKD-related complications like anemia, bone-mineral disorders, and vitamin D deficiency
- Complications and side effects of immunotherapy:
- Increased risk of infections (fungal, viral, and bacterial)
- Increased risk of malignancies over time, like bladder cancer, specifically with cyclophosphamide
- Renal tubular acidosis, stones from chemotherapy (proximal renal tubular acidosis)
- Cytopenias due to bone marrow suppression
- Infertility risk with cyclophosphamide
- Mycophenolate mofetil: gastrointestinal side effects, cytopenias, teratogenicity, and hence the need for dual contraception during therapy
- Reactivation of infections like tuberculosis and hepatitis B with rituximab
- Infusion hypersensitivity reactions
- Calcineurin inhibitors associated nephrotoxicity and neurotoxicity, renal parenchymal fibrosis, hair loss, and pancreatic toxicity
- Steroids increase the risk of infections, bone diseases like avascular necrosis of large joints, metabolic syndrome, hypertension from salt and fluid retention, psychosis, and gastrointestinal irritation, to name a few
- End-stage kidney disease (ESKD) requiring renal replacement therapies and concomitant complications inherent to the procedure
- Risk of catheter-associated bacteremia, hypotension, neurological side effects, and accelerated cardiovascular morbidity and mortality
- Post-transplant recurrent membranous nephropathy 71:
- Prevalent in 30% to 50% of patients with positive anti-PLA2R antibodies on initial presentation
- Pathophysiology of recurrent (de novo) membranous nephropathy is different from primary membranous nephropathy
Membranous nephropathy diagnosis
Membranous nephropathy is an uncommon disease and diagnosis can sometime be delayed. Since swelling, the most common symptom, can be caused by a lot of different diseases or problems (including kidney, heart, or liver problems), the kidneys may not be identified right away as the cause. Most often it is diagnosed when evaluating someone for protein in the urine (normally there should not be protein in the urine). Some people go to their doctor because of symptoms (such as swelling) and urine tests reveal protein in the urine. Other times, a urine test may be done for another reason (on a routine physical for example) and protein in the urine is discovered. Protein levels can be measured (or quantified) on a 24 hour urine collection or estimated from a single urine sample. Other evaluation usually includes measuring kidney function (from a blood test called creatinine) and doing other bloodwork.
These tests are done to find out if you have membranous nephropathy:
- Urine test: A urine analysis will help find protein (proteinuria) and blood in your urine (hematuria). Urine microscopy for cellular casts, fatty casts, and oval fat bodies.
- Spot urine protein or urine albumin to creatinine ratio (UPCR). A 24-hour urine collection is rarely indicated and/or is based on clinician preference. It is done serially and aids in monitoring the clinical response.
- Blood test: A blood test will help find levels of protein, cholesterol, and wastes in your blood.
- Serum metabolic panel to assess blood urea nitrogen, creatinine, uric acid, electrolytes, vitamin D levels, and estimated glomerular filtration rate (eGFR) 72.
- Serum albumin and total protein to evaluate hypoalbuminemia and hypoproteinemia.
- Lipid profile to assess for dyslipidemia.
- Serum IgG with subtypes, for decreased serum levels 73.
- For primary membranous nephropathy, serum anti-PLA2R antibody and anti-thrombospondin by Western blot and indirect immunofluorescence.
- Detailed studies for respective secondary causes based on history and presentation (like heavy metal levels, infectious etiology like HIV, parasitic infections, autoimmune panel).
- Glomerular filtration rate (GFR): A blood test will be done to know how well your kidneys are filtering the wastes from your body.
- Kidney biopsy: In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope. The kidney biopsy may show if you have a certain type of a protein that helps your body fight infection, called an antibody. Your body usually makes this antibody when you have membranous nephropathy. The pathological specimen should be sent for light microscopy, silver stain, immunofluorescence, electron microscopy, and immunohistochemistry for anti- PLA2R antibody.
Imaging tests that may be done include:
- Abdominal CT scan
- Kidney ultrasound. Ultrasonography study of the kidneys to assess for radiological kidney disease, evidence of obstruction, and renal vein thromboembolism.
- Renal vein doppler and computed tomography (CT)/magnetic resonance angiography (MRA) to rule out renal vein thrombosis.
- CT angiogram of the chest to rule out pulmonary embolism.
- Chest x-ray
- Intravenous pyelogram (IVP)
- Lower extremity doppler to assess for deep vein thrombosis.
- The benefit from a contrast study outweighs the risk of contrast-induced nephropathy and should be based on the acuity of presentation.
Different kidney diseases – not just membranous nephropathy – can cause protein in the urine, and a kidney biopsy is ultimately needed to diagnose the specific disease causing the protein in the urine. A kidney biopsy is a procedure that involves using a needle to get a sample of kidney tissue to look at under the microscope. This allows the individual glomeruli (kidney filters) to be seen under high magnification. Additional tests can be done on the kidney tissue from the biopsy to help make a diagnosis. Blood tests and measuring protein in the urine can be helpful to understand the severity of the disease and to rule out or look for certain causes, so these tests are often done as part of the workup, but a biopsy is needed to diagnose membranous nephropathy.
Since most people with membranous nephropathy have anti-PLA2R antibodies, bloodwork can be done to check for this antibody. If it is positive, it is very likely that someone has membranous nephropathy. However, a negative test does not mean that someone does not have membranous nephropathy, and a kidney biopsy is important to confirm the diagnosis as well as to provide more information to guide management.
Membranous nephropathy histopathology
A kidney biopsy is the gold standard in confirming the diagnosis of membranous nephropathy. It is used either exclusively or in combination with antibody assay, depending on the ease of availability. Antibody assay is replacing biopsy mostly due to the temporal relationship between proteinuria and circulating anti-PLA2R antibodies. It is a non-invasive method to monitor disease progression and outcomes. Since clinical disease lags behind immunological outcomes, antibody assay is an excellent biomarker for clinical management. Nevertheless, the following kidney biopsy findings specific for membranous nephropathy should be noted 53:
- Light microscopy: Diffuse capillary and glomerular basement thickening. These might be absent in earlier stages of membranous nephropathy.
- Immunofluorescence: Granular pseudo-linear IgG4 deposits are present all along the glomerular capillary wall.
- Silver stain: Spikes (represent staining of basement membrane between the deposits)
- Electron microscopy: Effaced podocytes. Ehrenreich and Churg classification based on the appearance of electron-dense deposits 50:
- Stage 1: Small, sparse, electron-dense deposits on the epithelial side of glomerular basement membrane (GBM)
- Stage 2: Larger deposits causing glomerular basement membrane (GBM) thickening, along with foot process effacement giving the characteristic “spike and dome” appearance
- Stage 3: Stage 2 plus intramembranous coarse granular deposits with “neomembrane” formation
- Stage 4: Irregular thickening, dissolution of deposits (holes), and sclerosis of glomerular basement membrane (GBM)
- Variants of membranous nephropathy with crescents have also been found 74.
- Chronic membranous nephropathy may show tubular atrophy, interstitial fibrosis, and nephrosclerosis on biopsy 75.
- Excessive protein reabsorption can lead to the deposition of hyaline droplets in the proximal convoluted tubules as seen in light microscopy 76.
The diagnosis of membranous nephropathy is based on the following findings 50:
- Thickened glomerular basement membrane (GBM), often showing pinholes and spikes on silver and periodic acid-Schiff stains, and occasionally subepithelial fuchsinophilic deposits on trichome stains;
- Immunofluorescence microscopy showing granular Ig, usually IgG and C3, along the capillary walls; and
- Subepithelial deposits on electron microscopy.
Membranous nephropathy treatment
Treatment of membranous nephropathy is usually managed by a nephrologist (kidney specialist). After your nephrologist (kidney specialist) finds that you have membranous nephropathy, your doctor will follow up with you very closely without treatment. This waiting period allows time to see if you go into remission (the disease stops being active and causes no symptoms) without having to use strong drugs. During this waiting time you will be given supportive care (treatments that have little or no risk).
Supportive care includes:
- Angiotensin-converting enzyme inhibitors (ACE inhibitors) or angiotensin 2 receptor blockers (ARBs): These drugs help reduce high blood pressure and the amount of protein in the urine (proteinuria). These medications are generally the first step in treating membranous nephropathy. Though they were designed as blood pressure medicines, in membranous nephropathy they are used to lower the amount of protein leaking into the urine, and may have the added benefit of helping to control blood pressure if it is elevated. Sometimes these medications cannot be used if someone’s blood pressure is too low (since they can lower blood pressure) or if someone has high potassium levels in the blood (since they can raise potassium levels). However in most people, if there is no contraindication, one of these medications should be used.
- ACE-inhibitors (angiotensin converting enzyme inhibitors) include lisinopril (Zestril, Prinivil), enalapril (Vasotec), ramipril (Altace), benazepril (Lotensin), and quinapril (Accupril).
- Angiotensin 2 receptor blockers (ARBs) include losartan (Cozaar), valsartan (Diovan), irbesartan (Avapro), telmisartan (Micardis), olmesartan (Benicar), and candesartan (Atacand).
- Diuretics or water pills that cause you to urinate more. Diuretics can be used to treat swelling if it occurs. Swelling can be a very bothersome or problematic symptom of membranous nephropathy and so these medications can be an important part of managing the disease. However, they do not treat the membranous nephropathy itself. Diuretics include furosemide (Lasix), torsemide, bumetanide (Bumex), and sometimes others.
- Low-salt diet: Lowering salt may help to reduce edema (swelling in your body parts like your legs, and around your eyes)
If your symptoms do not go away after supportive care, you may be given treatments that affect your immune system (immunosuppressive medication). The goal of your treatment is to manage your symptoms. If you have nephrotic syndrome, your healthcare provider may give you pills to reduce the water in your body (diuretics).
Immunosuppressive medication– not all people with membranous nephropathy will need medication to suppress their immune system, but it is an important part of the treatment for many people who have membranous nephropathy. Reasons that someone might need immunosuppresive medication include: worsening kidney function, high levels of protein in the urine (especially if they do not get better after a period of observation), or complications from the nephrotic syndrome (such as a blood clot). Because membranous nephropathy is an autoimmune disease, in which the body’s immune system targets and injury the body’s own tissue (in this case, part of the glomerulus), medications to suppress or decrease the immune system are needed to treat the disease in many people. There are different immune suppressing medications that can be used to treat membranous nephropathy. Since these medications all suppress the immune system, all of them increase the risk for infection. However, they have different individual side effects besides this, and the different possible side effects can be a factor in deciding which one to use. It is very important to discuss the side effects of your treatments with your healthcare provider. If you are a woman, and are considering having children, be sure to speak with your doctor about how your treatments may affect this process.
Various immunosuppressive medications have been shown to induce remission in patients with membranous nephropathy, including steroids, cyclophosphamide, and cyclosporine. Edema is managed by dietary salt restriction and diuretics, either administered orally or intravenously.
- Cyclophosphamide (Cytoxan)– this is a medication that can be given as a monthly infusion or as a daily pill (by mouth). One of the most common regimens for treating membranous nephropathy includes alternating months of cyclophosphamide and corticosteroids for a total of 6 months (Ponticelli protocol).
- Rituximab (Rituxan)– this is a medication that is given as an infusion, either 4 weekly doses or 2 doses spaced 2 weeks apart. Its effects last about 6 months in the body, and sometimes a maintenance (repeat) dose can be given after 6 months.
- Calcineurin inhibitors (cyclosporine or tacrolimus)– these medications are taken as a pill by mouth, usually twice a day.
- Corticosteroids (prednisone)– this is a medication that is often used with one of other immunosuppressive medications above (most commonly with cyclophosphamide or with one of the calcineurin inhibitors). Corticosteroids are not effective by themselves for treating membranous nephropathy but can be used as part of other regimens.
Because cholesterol levels can be elevated in people with nephrotic syndrome, treatment may include a medication for this. The most common type of cholesterol medication is called a “statin” and these include atorvastatin (Lipitor), lovastatin, pravastatin (Pravachol), and rosuvastatin (Crestor).
Blood pressure management – blood pressure is often elevated in people with kidney disease, including membranous nephropathy. It is important to keep blood pressure well-controlled to prevent damage to the kidneys. In addition to an ACE-inhibitor (angiotensin converting enzyme inhibitor) or angiotensin 2 receptor blocker (ARB) that can help with protein in the urine, there are many other medications that can be used to treat high blood pressure. Limiting salt intake can help with blood pressure control as well as with decreasing swelling.
Blood thinners – because people with nephrotic syndrome (protein in the urine, low blood protein levels, and swelling) are at higher risk for blood clots, treatment may also include a blood thinner to prevent blood clots. The decision of whether to start a blood thinner is based on balancing the risk of having a clot with the risk of having bleeding from being on a blood thinner. There is a website that you can use with your doctor to help decide if a blood thinner would be beneficial for you. In most cases, the blood thinner should be stopped when the nephrotic syndrome gets better and blood protein levels go up.
Therapies of unproven long-term value include IV immuneglobulin and nonsteroidal anti-inflammatory drugs (NSAIDs).
Kidney transplantation is an option for patients with end-stage kidney disease. Membranous nephropathy recurs in about 10% of patients, with loss of graft in up to 50%.
Finally, if the membranous nephropathy is considered secondary to something else, then it is most important to treat the underlying disease (infection, cancer, etc.) or to stop the causative drug. This is often enough and treatment with immune suppressing medications can be avoided.
General management
Symptomatic management of membranous nephropathy is done with diuretics, statins, angiotensin-converting enzyme inhibitors (ACE inhibitors) OR angiotensin receptor blockers (ARBs), systemic anticoagulant therapy (newer direct oral anticoagulant agents or vitamin K antagonist therapy), antihypertensives, and dietary salt restriction. 1/3rd of the patients respond to these conservative measures and another 1/3rd will need one of the following immunosuppressive therapies 77, 78, 79.
Immunosuppressive therapy
Immunosuppressive therapy should be considered only for patients with symptomatic idiopathic membranous nephropathy and for those most at risk of progressive disease. While there is no strong evidence that immunosuppressive therapy has a long-term benefit for patient or kidney survival, it appears to improve rates of remission and possibly progression to end-stage kidney disease (ESKD) 54, 80, 81. Older and chronically ill patients are at greater risk of infectious complications due to immunosuppressants.
No consensus protocol exists, but historically a common regimen included corticosteroids, followed by chlorambucil. However, chlorambucil is no longer preferred because of other therapeutic options with better safety profiles. Most experts favor use of a combination of rituximab and corticosteroids 80. Alternatives to rituximab include a calcineurin inhibitor and cyclophosphamide. The choice of agent is guided by disease severity and anti-PLA2R antibody levels.
Ponticelli regimen (6 months):
- Months 1, 3, and 5: Methylprednisolone (1 g) daily for 3 days followed by prednisolone (0.4 mg/kg/day) or prednisone (0.5mg/kg/day) for 27 days.
- Months 2, 4, and 6: Cyclophosphamide orally 2 mg/kg/day for 30 days.
Modified Ponticelli regimen (6 months):
- Months 1, 3, and 5: Methylprednisolone (1 g) daily for 3 days followed by oral prednisone (0.5mg/kg/day) for 27 days.
- Months 2, 4, and 6: Cyclophosphamide orally 2 to 2.5 mg/kg/day for 30 days.
To be given with appropriate pneumocystis pneumonia (PCP) and antiviral prophylaxis (trimethoprim-sulfamethoxazole and valganciclovir).
Rituximab a monoclonal antibody to CD20 antigen of B cell 82:
- Usually a second-line therapy after failure of steroids.
- As first-line therapy or in refractory cases, or after 6 months of conservative management.
- Dose: 375 mg/m² intravenously once weekly × 4. The maintenance dose is similar but is given every six months.
Alternate therapy with calcineurin inhibitors are less popular due to the high relapse rate 83. Either of the following can be used:
- Tacrolimus ( 0.025 to 0.040 mg/kg divided into twice a day ) for six months.
- Cyclosporine (1.5 to 2.5 mg/kg divided into twice a day) +/- 0.15 mg/kg prednisone for six months.
Other drug options are:
- Chlorambucil
- Mycophenolate mofetil
- Adrenocorticotropic hormone (ACTH) analogs
The various immunosuppressive treatments come with their inherent side effects like increased risk of infections, cancers, reduction in the number of red blood cells, white blood cells and platelets (cytopenias), side effects of long-term steroids like cataract, metabolic syndrome, avascular necrosis of joints, etc.
Management of progressive disease
In advanced oliguric (urinary output less than 400 ml per day or less than 20 ml per hour) or anuric (no urine or your kidneys aren’t producing urine) kidney injury, renal replacement therapy may be required.
Patients may need vitamin D and calcium supplementation, especially if treated with steroids.
Kidney transplant for patients with advanced chronic kidney disease (CKD Stage 5) or end-stage kidney disease (ESKD).
Membranous nephropathy treatment in children
It is very rare for a child to have membranous nephropathy, so it is important to check for anything that might be causing the disease, especially lupus. If your child needs treatment for membranous nephropathy, the treatment is usually the same as it is for adults. It is important to know that some people with membranous nephropathy go into spontaneous remission, which means it suddenly goes away without any treatments.
Will the membranous nephropathy come back in my kidney transplant?
There is a 40% chance that membranous nephropathy will return in a transplanted kidney with loss of graft in up to 50% of cases. Unfortunately, there are no factors that have been identified to give doctors an idea of patients at risk for this problem. Generally, recurrence of the disease will occur in the first 2 years after the kidney transplant.
Is there any treatment for membranous nephropathy that comes back in a kidney transplant?
There have not been any trials to evaluate different therapies for membranous nephropathy that comes back in a transplanted kidney. However, Rituximab is one option that has been used successfully for treatment.
Membranous nephropathy prognosis
Definition of membranous nephropathy prognosis or outcomes 84:
- Complete remission: Proteinuria <0.3 g/d or 300 mg/g on spot urine albumin to creatinine ratio (UPCR).
- Partial remission: Greater than 50% reduction of proteinuria from baseline or between 0.3 and 3.5 g/day, with relatively stable estimated glomerular filtration rate (eGFR).
- Relapse: Recurrence of greater than 3.5 g/day of proteinuria after remission.
- End-stage kidney disease (ESKD): GFR less than 15 ml/min or requirement for kidney dialysis or kidney transplant.
Rule of One-thirds 50
- Spontaneous remission in one-third of the patients with conservative management only.
- One-third have symptomatic proteinuria without progressing into renal failure and might benefit from immunosuppressive therapies.
- The remaining one-third are refractory to treatment and require dialysis due to end-stage kidney disease (ESKD). These should be evaluated for renal transplantation.
Risk Factors for Poor Prognosis 75:
- Male gender, White race
- Old age
- Hypertension on presentation 85
- Massive proteinuria (greater than 8 g/day) for 6 months
- Elevated creatinine or acute kidney injury at the time of presentation
- Extensive tubulointerstitial fibrosis on biopsy
Glomerular filtration rate is typically normal at the outset of membranous nephropathy, but in some patients glomerular filtration rate will decline after months or years. Risk factors for progressive loss of kidney function include heavy proteinuria and male sex.
Up to a third of patients diagnosed with primary membranous nephropathy will go into remission spontaneously within 5 years, even without immunosuppressive therapy 86. With treatment, most patients’ disease will go into remission. Complete remission means stable (or improved) kidney function and protein levels in the urine decreased to normal. Partial remission means stable or improved kidney function plus protein levels in the urine decreased to less than half of original levels and not in the nephrotic syndrome range (less than 3.5 grams/day). Despite this, 15% to 50% of patients who do not receive immunosuppressive treatment progress to end‐stage kidney disease (ESKD) within 10 years 87, 88, 89.
However, relapse is common in membranous nephropathy, and patients that go into remission either spontaneously or in response to immunosuppressive can have the disease return later. Long term (over decades) about 1/3 of patients will progress to end-stage kidney disease (kidney failure needing dialysis), about 1/3 will have continued protein in the urine without kidney failure (persistent nephrotic syndrome), and about 1/3 will be in remission.
Women, children, and young adults with non–nephrotic-range proteinuria and patients with persistently normal renal function 3 years after diagnosis tend to have little disease progression. More than 50% of patients with nephrotic-range proteinuria who are asymptomatic or who have edema that can be controlled with diuretics will have a partial or complete remission within 3 to 4 years.
Risk of progression to renal failure is highest among patients with 90:
- Persistent proteinuria ≥ 8 g/day, particularly men age > 50 years
- An elevated serum creatinine level at presentation or diagnosis
- Biopsy evidence of substantial interstitial inflammation
Things that predict a better long-term outcome in membranous nephropathy include lower levels of protein in the urine, being female, being younger (less than 60), and achieving remission.
Minimal change disease
Minimal change disease also called “nil disease“, minimal change lesion or lipoid nephrosis (a description of lipid droplets in urine seen on light microscopy) is a kidney disorder that involves damage to the filtering units of the kidney (glomeruli) that can be seen only with an electron microscope and minimal change disease is the most common cause of idiopathic childhood nephrotic syndrome 91, 92, 93, 94. Minimal change disease accounts for 70% to 90% of children that present with nephrotic syndrome who are older than one year old as opposed to 10-15% of adults who present with nephrotic syndrome 95. Minimal change disease is the third most common cause of idiopathic nephrotic syndrome in adults after focal segmental glomerular sclerosis (FSGS) and membranous nephropathy (membranous glomerulopathy) 95. The reported incidence of minimal change disease in children varies between 2 to 7 new cases per 100,000 children 96, 97. The exact prevalence is unknown; however, it is estimated to be about 10 to 50 cases per 100,000 children 92. Minimal change disease is slightly more common in Asia and has a male predominance (approximately 2:1) in young children that disappears in adolescents and adults 96, 98. Minimal change disease is much less frequent in adults, but the exact incidence in this population is less well documented 93.
Nephrotic syndrome is a group of symptoms that include protein in the urine (proteinuria >40 mg/m2 per hour in children or urine protein-to-creatinine ratio >200 mg/mmol in children, >3.5 g/day in adults), low protein levels in the blood (hypoalbuminemia (<2.5 g/dl), high cholesterol levels, high triglyceride levels, an increased risk for blood clots, and swelling 99, 95. Other features of this disease include weight gain and a foamy appearance of the urine. Those with minimal change disease experience the signs and symptoms of nephrotic syndrome much quicker than they would with other glomerular diseases.
Minimal change disease gets its name because this damage is not visible under a regular microscope. It can only be seen under a very powerful microscope called an electron microscope.
On light microscopy of renal biopsy specimens from patients with minimal change disease, the kidney appears normal or nearly normal. Older pathologic terms for minimal change disease include nil disease (i.e, no abnormality) and lipoid nephrosis. Immunofluorescence microscopy may be normal or may show deposition of immunoglobulin M (IgM) antibody within the glomerulus. Electron microscopy shows podocyte foot process effacement, which is common to nearly all proteinuric diseases. Minimal change disease typically shows, prior to therapy, very extensive foot process effacement, often covering >90% of the glomerular capillary surface.
Each kidney is made of more than a million units called nephrons, which filter blood and produce urine.
In minimal change disease, there is damage to the glomeruli. These are the tiny blood vessels inside the nephron where blood is filtered to make urine and waste is removed.
Patients with minimal change typically present with the sudden onset of edema (swelling), either affecting the legs or, particularly in children, affecting the whole body, including facial edema (which may be the first manifestation). Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin. In children, the peak age of onset is age 2, with the disease being uncommon before age 1 and less common in during the teenage years. Minimal change disease also occurs in adulthood, even in the elderly, but is less common with advancing age.
The cause or causes of minimal change disease are largely unknown, but it may occur following an allergic reaction or infection 93, 92, 91, 100, 101, 102. Treatment may involve the use of steroids. The fluid retention and high blood pressure that often accompanies minimal change disease may be treated with the use of water pills (diuretics) in combination with a low sodium diet and blood pressure medications such as angiotensin-converting enzyme inhibitors (ACE inhibitors), angiotensin 2 receptor blocker (ARB) andcalcium channel antagonists.
There is an increased risk for the formation of blood clots (thromboembolic events) and infection in individuals with minimal change disease. It is recommended that individuals with minimal change disease stay active and should a blood clot occur, they may be treated with blood thinners. Infections, such as cellulitis, peritonitis, and pneumonia are common in individuals with minimal change disease and should be treated quickly 103.
Will minimal change disease cause kidney failure?
Kidney failure is rare if you have minimal change disease. Almost all children and adults recover from minimal change disease and avoid relapses over the long term. However, some may experience relapses of the protein in the urine, which can often be treated in the same way as the first episode. Progression to renal failure occurs in < 5 % of patients and is more common among those who do not initially respond to corticosteroids 104.
Minimal change disease causes
Scientists do not know the exact cause of minimal change disease 93, 92, 91, 100, 101, 102. In adults, minimal change disease is usually secondary (it is caused by another disease) to medication use (nonsteroidal anti-inflammatory drugs [NSAIDS], gold injection, interferon-alpha, lithium, pamidronate) or may follow insect stings. In children, minimal change disease is usually primary or idiopathic, which means the exact cause is not known. Minimal change disease is the most common cause of nephrotic syndrome in children. It is also seen in adults with nephrotic syndrome, but is less common.
The cause of minimal change disease is unknown, but if you have secondary causes for minimal change disease, the disease may occur or be related to 105, 106, 92:
- Allergic reactions (e.g., bee and medusa stings, cat fur, fungi, poison ivy, ragweed pollen, house dust, food allergens such as cow’s milk & egg)
- Drugs: nonsteroidal anti-inflammatory drugs (NSAIDs), lithium, antibiotics (ampicillin, cephalosporins), salazopyrin, D-penicillamine, mercury, gold, tiopronin, tyrosine-kinase inhibitors, immunizations, and gamma interferon
- Neoplasms: blood cancers, including leukemia, Hodgkin and non-Hodgkin lymphoma, multiple myeloma, thymoma, bronchogenic cancer, colon cancer, eosinophilic lymphoid granuloma (Kimura disease)
- Vaccinations (flu and pneumococcal, though rare)
- Infections: tuberculosis, syphilis, mycoplasma, ehrlichiosis, hepatitis C virus, HIV
- Autoimmune disorders: systemic lupus erythematosus (SLE), type 1 diabetes mellitus, myasthenia gravis, autoimmune pancreatitis, celiac disease, allogeneic stem cell transplantation
- Other glomerular diseases associated with IgA nephropathy
Minimal change disease pathophysiology
Minimal change disease presents with a nephrotic syndrome characterized by an increased renal membrane permeability and loss of protein (primarily albumin) due to damage to the glomerular filtration barrier 92. The glomerular filtration barrier is composed of fenestrated endothelium (inner layer), the glomerular basement membrane (middle layer), and an outer epithelial layer composed of podocytes 92. Podocytes are epithelial cells with large cell bodies and long foot processes that run parallel along the outside of glomerular capillaries. The space between foot processes is interspersed by cell-to-cell junctions called slit diaphragms 107.
Glomerular filtration is both size-specific and charge-specific. The actin cytoskeleton of podocytes provides support to the glomerular basement membrane (GBM) and regulates flow across the basement membrane depending on hydrostatic pressures, molecular size, and molecular charge 107. The apical and luminal membrane of the slit diaphragms and podocytes are coated with a sialoglycoprotein (podocalyxin) which contributes to repels negatively charged molecules such as albumin. The two outer layers of the glomerular basement membrane are composed of heparin sulfate proteoglycans that are also negatively charged and contribute to the charge selectivity of the barrier 108. Disruption of this barrier leads to the proteinuria seen in nephrotic syndrome.
The pathogenesis of minimal change disease is not exactly known, but it is thought to be multifactorial. Several studies have focused on the integrity and biology of podocytes. Because the actin cytoskeleton of podocytes maintains the integrity of the podocytes by supporting the cell body and foot processes, regulation of flow across the basement membrane is controlled by a series of interactions. As a result, multiple theories have been proposed to explain the cause of proteinuria in minimal change disease. Some of the proposed theories published include T cell dysfunction/dysregulation that leads to cytokine release and upregulation of proteins, such as CD80 and C-mip that affects the integrity of podocytes, systemic circulating factors that disrupt podocyte function, and B- cell activation (suspected due to the efficacy of anti- CD-20 monoclonal antibodies, such as rituximab) 109, 110, 111.
Minimal change disease symptoms
Minimal change disease causes nephrotic syndrome, usually without hypertension or azotemia (a condition in which you have too much nitrogen, creatinine and other waste products in your blood) 112. Microscopic blood in urine (hematuria) occurs in about 10% to 30% of patients, mainly adults 113, 112. Albumin is lost in the urine of patients with minimal change disease more so than larger serum proteins, probably because the disease causes changes in the charge barrier that affect albumin selectively.
There may be symptoms of nephrotic syndrome, including:
- Foamy appearance of the urine due to large amounts of protein leaking into your urine, called proteinuria
- Poor appetite
- Swelling called edema (especially around the eyes, feet, legs and ankles, and in the abdomen), due to fluid building up in your body
- Weight gain (from fluid retention), due to the fluid your body is not able to get rid of
Always speak with your doctor if you experience any of these signs and symptoms.
Minimal change disease does not reduce the amount of urine produced. It rarely progresses to kidney failure. Progression to renal failure occurs in < 5 % of patients and is more common among those who do not initially respond to corticosteroids 104.
Minimal change disease diagnosis
Your health care provider may not be able to see any signs of minimal change disease, other than swelling. Blood and urine tests reveal signs of nephrotic syndrome, including:
- High cholesterol
- High levels of protein in the urine (proteinuria)
- Low levels of albumin in the blood (hypoalbuminemia)
Tests and procedures used to diagnose nephrotic syndrome include:
- Urine tests. A urinalysis can reveal abnormalities in your urine, such as large amounts of protein, if you have nephrotic syndrome. You may be asked to collect urine samples over 24 hours for an accurate measure of the protein in your urine.
- Blood tests. If you have nephrotic syndrome, a blood test may show low levels of the protein albumin (hypoalbuminemia) specifically and often decreased levels of blood protein overall. Albumin is lost in the urine of patients with minimal change disease more so than larger serum proteins. Loss of albumin is often associated with an increase in blood cholesterol and blood triglycerides. Serum creatinine, blood urea and glomerular filtration rate (GFR) also may be measured to assess your overall kidney function and to know how well your kidneys are filtering the wastes from your body.
- Complete metabolic panel (CMP): Complete metabolic panel (CMP) demonstrates a low total protein, low albumin (frequently <2.5 g/dl), and low total calcium (ionized calcium binds to albumin, and albumin is low).
- Complete blood count (CBC): Complete blood count (CBC) will show hemoconcentration and thrombocytosis. This is seen due to the intravascular volume contraction from fluid sequestration into the interstitial space.
- Total cholesterol and triglyceride levels: Total cholesterol and triglyceride are increased due to an increase in hepatic lipoprotein synthesis as a result of low oncotic pressures 107.
- A kidney biopsy and examination of the tissue with an electron microscope can show signs of minimal change disease. During a kidney biopsy, a special needle is inserted through your skin and into your kidney.
If a kidney biopsy shows little or no damage under a regular microscope, then a diagnosis of minimal change disease may be made if other symptoms, such as protein in the urine and swelling, are noticed. Because minimal change disease is the most common cause of nephrotic syndrome in children, they first get treated for minimal change disease before getting a kidney biopsy. Most people will have a response in fewer than 8 weeks. If the protein in the urine disappears, the doctors may call the disease steroid-sensitive nephrotic syndrome instead of minimal change disease. If treatment does not improve their symptoms over the course of several months a kidney biopsy is done to see if there is another cause for their symptoms.
In children, minimal change disease is primarily a clinical diagnosis, and biopsy is only required in the presence of atypical clinical features 92, 93:
- Age of onset before 1 or after 12 years old
- Gross hematuria
- Low serum C3
- Marked Hypertension
- Elevated creatinine
- Renal failure without hypovolemia
- Positive history or serology for secondary causes
- Steroid resistance
Minimal change disease treatment
Medicines called corticosteroids can cure minimal change disease in most children. Some children may need to stay on steroids to keep the disease from returning. It is very important to not stop treatment suddenly. By sticking to the full treatment plan, your child will be less likely to relapse (experience the signs and symptoms again).
- Children. Initial prednisone therapy is 60 mg/m²(or 2 mg/kg) administered daily for 4-6 weeks (maximum dose, 60 mg/day), or 40 mg/m²/(or 1.5 mg/kg) on alternate days for 2-5 months taper. Reduce the dose by 5 mg/m² to 10 mg/m² each week for another four weeks, then stop, with a minimum duration of 12 weeks 114.
- Adults. Initial prednisone treatment is 1 mg/kg per day or 2 mg/kg/kg every other day (max 80mg/day or 120 mg every other day) for 4-16 weeks. Taper slowly over a course of 6 months after remission 113.
Most children (>90%) and adults (>70%) enter a complete remission with a course of steroids. Adults may have more frequent relapses and become dependent on steroids.
Patients who relapse demonstrate the following 92:
- Steroid resistance is noted as the persistence of proteinuria in children after 4 weeks of prednisone and after 16 weeks for adults.
- Frequent relapses occur, which are defined as two or more relapses in the first six months of presentation or four or more relapses within any 12 months
- Steroid dependency is defined as relapses that occur during the tapering phase of steroid therapy or less than two weeks after discontinuing steroids
- Relapse nephrosis – > 2+ proteinuria on 3 consecutive days
- Prednisolone should be restarted if there is a relapse: 2 mg/kg daily (maximum 60 mg) until in remission for 3 days, then 1.5 mg/kg alternate days for 4 weeks, then stop or taper the dose over 4-8 weeks.
Mortality at present is very low. Many steroid regimens have been used. The most common regimens involve the use prednisone daily or every other day. Unfortunately, relapses are common. When patients respond to steroids but experience frequent relapses, therapeutic options include cyclophosphamide, chlorambucil, and cyclosporine.
If steroids are not effective (you do not enter complete remission with steroid therapy), your doctor will likely suggest other medicines that include cyclosporine. For children who do not respond to traditional treatment they have what is called steroid-resistant nephrotic syndrome or SRNS. Treatment for steroid-resistant nephrotic syndrome includes other combinations of drugs. It is recommended that children with steroid-resistant nephrotic syndrome take a blood pressure medication (ACE inhibitor or angiotensin 2 receptor blocker [ARB]). These two drugs control high blood pressure and reduce the amount of protein in the urine.
For frequent relapses/ steroid-dependent (steroid-sparing agents) 92:
- Cyclophosphamide: the dose of 2 mg/kg/ day for 8 to 12 weeks (should be started after reaching remission with the steroid)- potential gonadal toxicity, alopecia, bone marrow suppression.
- Cyclosporine: At a dose of 4 to 5 mg/kg/day, usually for 1-2 years. Levels should be monitored after 1 to 2 weeks. Aim for a trough of 70 to 150. Can cause nephrotoxicity, hirsutism, hypertension, and gingival hyperplasia.
If intolerant to the above-mentioned drugs, can give 92:
- Mycophenolate mofetil (MMF): Doses of 500 to 1000 mg two times a day for 1 to 2 years. Should be monitored for leukopenia 114.
- Rituximab (chimeric monoclonal antibody): 375 mg/weekly for 1 to 4 doses 115. Side effects such as fulminant myocarditis, pulmonary fibrosis, fatal Pneumocystis jirovecii infections, ulcerative colitis, and allergic reactions 93.
Edema is managed by dietary salt restriction and diuretics, either administered orally or intravenously.
- Cholesterol-reducing medications. Medications called statins can help lower cholesterol levels. However, it’s currently unclear whether or not cholesterol-lowering medications can specifically improve the outcomes of people with nephrotic syndrome, such as avoiding heart attacks or decreasing the risk of early death. Statins include atorvastatin (Lipitor), fluvastatin (Lescol), lovastatin (Altoprev), pravastatin (Pravachol), rosuvastatin (Crestor) and simvastatin (Zocor).
- Blood thinners. Medications called anticoagulants help decrease your blood’s ability to clot and may be prescribed if you’ve had a blood clot to reduce your risk of future blood clots. Anticoagulants include heparin, warfarin (Coumadin, Jantoven), dabigatran (Pradaxa), apixaban (Eliquis) and rivaroxaban (Xarelto).
Swelling may be treated with:
- Angiotensin-converting-enzyme inhibitor (ACE inhibitor) medicines. Medications in this category include benazepril (Lotensin), captopril and enalapril (Vasotec). Another group of drugs that works in a similar way is called angiotensin 2 receptor blockers (ARBs) and includes losartan (Cozaar) and valsartan (Diovan). Other medications, such as renin inhibitors, also may be used, though ACE inhibitors and angiotensin 2 receptor blockers are generally used first.
- Dietary salt restriction. Limit sodium (salt) in your diet
- Blood pressure control
- Diuretics (water pills). Water pills (diuretics) help control swelling by increasing your kidneys’ fluid output. Diuretic medications typically include furosemide (Lasix). Others may include spironolactone (Aldactone) and thiazides, such as hydrochlorothiazide.
How is minimal change disease treated in adults?
The treatment for nephrotic syndrome in adults with minimal change disease is usually with a type of drug called a corticosteroid, often called steroids. You may notice that you start getting better within weeks, or less, although it may take an adult longer to respond than a child. It is important to stick with your treatment plan until all medications are finished; even if your symptoms go away sooner
If you are a woman and want to have children, you should speak with your healthcare provider to see how the medicines you are given affect this process.
For symptoms of swelling (edema), your healthcare provider may give you:
- ACE inhibitor or angiotensin 2 receptor blocker (ARB) medicines
- Diuretics (water pills)
- Limit sodium (salt) in your diet
Minimal change disease diet
Changes to your diet may help you cope with nephrotic syndrome. Your doctor may refer you to a dietitian to discuss how what you eat can help you cope with the complications of nephrotic syndrome. A dietitian may recommend that you:
- Choose lean sources of protein
- Reduce the amount of fat and cholesterol in your diet to help control your blood cholesterol levels
- Eat a low-salt diet to help control the swelling (edema) you experience
- Limit foods that increase blood sugar levels when taking medications that can lead to weight gain, such as steroids
Some people with nephrotic syndrome may also be deficient in the mineral zinc. A recent study showed treatment with zinc supplements in children under 18 improved nephrotic syndrome. But always check with your doctor before giving your child a supplement or taking one yourself to avoid any potential adverse interactions.
Minimal change disease relapse
Other medications that may be used in instances of minimal change disease recurrence include those that are used to treat certain types of cancer (cyclophosphamide, chlorambucil, rituximab) and those that suppress the immune system (cyclosporine, tacrolimus, azathioprine, mycophenolate mofetil) 103.
Minimal change disease prognosis
Before the use of steroids became standard in the 1960’s, there was a high mortality in children with minimal change disease due to infection, as antibodies are lost in urine and this produces immunodeficiency. As discussed below, most patients respond to steroids and have an excellent long-term prognosis. In those who are steroid-resistant, a renal biopsy may subsequently show focal segmental glomerulosclerosis, which has a worse prognosis (more likely to progress to end-stage kidney disease). Spontaneous remissions (occurring without specific therapy) also occur.
Children usually respond better to corticosteroids than adults. Children often respond within the first month 93.
A relapse can occur. The condition may improve after long-term treatment with corticosteroids and medicines that suppress the immune system (immunosuppressive medicines).
In individuals who are not treated, there is an increased risk for infection and blood clotting events. About 5-10% of untreated adults may have spontaneous remission (resolution) of disease within a few months.
One major indication of the long-term outcome of minimal change disease is the initial response to corticosteroid treatment. About 80-95% of adults with minimal change disease who receive treatment via corticosteroids experience complete remission of symptoms. About half of all adults treated for minimal change disease have remission within four weeks, while 10-25% require longer treatment. minimal change disease may recur or relapse in about half of all adults. This usually occurs within one year of treatment 116.
Despite the potential for minimal change disease to recur, the occurrence of kidney failure and end stage renal disease is rare 103. Progression to renal failure occurs in < 5 % of patients and is more common among those who do not initially respond to corticosteroids 104.
IgA Nephropathy
IgA nephropathy also known as Berger’s disease, is a common kidney disease that occurs when an antibody called immunoglobulin A (IgA) builds up in your kidneys, causing local inflammation that damages kidney tissues, over time, can hamper your kidneys’ ability to filter waste from your blood 117, 118, 119. IgA nephropathy is one of the most common kidney diseases, other than those caused by diabetes or high blood pressure 120. About 1 in 10 kidney biopsies in the United States show IgA nephropathy 117. IgA nephropathy can occur at any age, although the first evidence of kidney disease most frequently appears when people are in their teens to late 30s 121. IgA nephropathy is rare in children younger than five years of age 122. The incidence of IgA nephropathy peaks in the second and third decades of life 123, 124. The incidence of IgA nephropathy varies substantially between ethnic or racial groups, being highest in East Asians and Caucasians where IgA nephropathy accounts for about 40% of all native-kidney biopsies in Japan, 25% in Europe, 12% in the United States, and less than 5% in central Africa 125, 126. IgA nephropathy in the United States and Europe is twice as likely to appear in men than in women but about 1:1 in East Asia 127, 128, 129, 130, 131, 132. A systematic study of biopsy-based literature from multiple countries reveals an overall IgA nephropathy incidence of over 2.5 per 100,000 133. In another study, IgA nephropathy is more common in Asian individuals (45 cases per million population/year in Japan) than in Whites (31 cases per million population/year in France) 134. Compelling data suggests a higher burden of IgA nephropathy in East and Pacific Asian countries 135.
IgA nephropathy usually progresses slowly over years, but the course of the disease varies from person to person. Some people leak blood in their urine (hematuria) without developing problems, some eventually achieve complete remission and others develop chronic kidney disease (CKD) and end-stage kidney failure 136. IgA nephropathy is a common cause of chronic kidney disease (CKD), particularly for patients with proteinuria persistently more than 1 g/day 137, 138, 139. In a study of 669 patients, the multivariable analysis revealed that people of Pacific Asian origin have a higher risk of end-stage renal disease (ESRD) 140.
In children and adolescents, painless visible blood in urine (hematuria), often at the same time with an infection of the upper respiratory or gastrointestinal tract, frequently heralds the onset of clinical IgA nephropathy disease 122. This sign and symptom of blood in urine (hematuria) may also accompany intense physical activity. Most patients with macroscopic (visible) hematuria have additional episodes over several years 141. Visible hematuria due to IgA nephropathy rarely begins after age 40 years 122. For patients in their 30s and 40s, microscopic hematuria, with or without, proteinuria may be discovered at the time of routine health screenings 122. The magnitude of proteinuria varies widely between patients, although proteinuria without microscopic hematuria is uncommon.
IgA (immunoglobulin A) is an antibody, a protein made by your immune system to protect your body from foreign substances such as bacteria or viruses. IgA nephropathy affects the kidneys by attacking the glomerulus (more than one glomerulus are called glomeruli) 117, 118. The glomeruli are sets of looping blood vessels in nephrons, the tiny working units of your kidneys that filter wastes and remove extra fluid from the blood. The buildup of IgA deposits inflames and damages the glomeruli, causing the kidneys to leak blood and protein into the urine. The damage may lead to scarring of the nephrons that progresses slowly over many years. Eventually, IgA nephropathy can lead to end-stage kidney disease, sometimes called ESRD, which means the kidneys no longer work well enough to keep a person healthy. When a person’s kidneys fail, he or she needs a kidney transplant or blood-filtering treatments called dialysis.
A person may be more likely to develop IgA nephropathy if 136, 117, 118:
- he or she has a family history of IgA nephropathy, IgA vasculitis or Henoch-Schönlein purpura—a disease that causes small blood vessels in the body to become inflamed and leak
- he is a male in his teens to late 30s
- he or she is Asian or white European ancestry
- he or she has certain health conditions, such as celiac disease, hepatitis, cirrhosis, and HIV infection
IgA nephropathy often becomes worse slowly over years. But the course of the disease varies from person to person. Some people leak blood into their urine (hematuria) without having other problems. Others might have complications such as losing kidney function and spilling protein into the urine. Still others develop kidney failure, which means the kidneys stop working well enough to filter the body’s waste on their own.
Sometimes, routine medical tests find signs of IgA nephropathy, such as protein and red blood cells in the urine that are seen under a microscope.
Most people with IgA nephropathy receive care from a nephrologist, a doctor who specializes in treating people with kidney disease.
No cure exists for IgA nephropathy and no sure way of knowing what course your disease will take, but certain medications can slow its course. Some people need treatment to lower inflammation, reduce the spilling of protein into the urine and prevent the kidneys from failing. Such treatments may help the disease become not active, a state called remission. Keeping your blood pressure under control and reducing your cholesterol levels also slow the disease.
Some people need only monitoring to determine whether the disease is getting worse. For others, a number of medications can slow disease progress and help manage symptoms.
Medications to treat IgA nephropathy include:
- High blood pressure medications. Taking angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) can lower your blood pressure and reduce protein loss.
- Omega-3 fatty acids. These fats, available in dietary fish oil supplements, might reduce inflammation in the glomeruli without harmful side effects. Talk to your doctor before you start supplements.
- Immunosuppressants. In some cases, corticosteroid medications, such as prednisone, and other potent drugs that suppress the immune response (immunosuppressants) might keep your immune system from attacking your glomeruli. These drugs can cause serious side effects, such as high blood pressure, high blood sugar and increased risk of infection.
- Statin therapy. If you have high cholesterol, cholesterol-lowering medications can help control it and slow the progression of kidney damage.
- Diuretics. These help remove extra fluid from your blood. Removing extra fluid can help improve blood pressure control.
The ultimate goal is to avoid the need for kidney dialysis or kidney transplantation. But in some cases, dialysis or transplantation is necessary.
IgA nephropathy key points
- Immunoglobulin A (IgA) nephropathy, also known as Berger’s disease, is a kidney disease that occurs when IgA deposits build up in the kidneys, causing inflammation that damages kidney tissues.
- Scientists think that IgA nephropathy is an autoimmune kidney disease, meaning that the disease is due to the body’s immune system attacking tissues in the kidney.
- IgA nephropathy is one of the most common kidney diseases, other than those caused by diabetes or high blood pressure.
- In its early stages, IgA nephropathy may have no symptoms; it can be silent for years or even decades.
- Once symptoms appear, the most common one is hematuria, or blood in the urine.
- Another symptom of IgA nephropathy is albuminuria—when a person’s urine contains an increased amount of albumin, a protein typically found in the blood, or large amounts of protein in the urine.
- Currently, health care providers do not use blood or urine tests as reliable ways to diagnose IgA nephropathy; therefore, the diagnosis of IgA nephropathy requires a kidney biopsy.
- Researchers have not yet found a specific cure for IgA nephropathy.
Who is more likely to have IgA nephropathy?
IgA nephropathy is more common in people who 136, 117, 118:
- have a family history of IgA nephropathy or of IgA vasculitis (Henoch-Schönlein purpura)
- have certain health conditions, such as celiac disease, hepatitis, cirrhosis, and HIV infection
- are ages 10 to 40
- are of East Asian or white European ancestry
- are male
Is IgA nephropathy genetic?
For some people, this condition runs in families. Researchers have discovered some genetic markers, meaning that a genetic mutation (change) may cause IgA nephropathy.
What’s the difference between IgA nephropathy and selective IgA deficiency?
Both conditions have to do with the protein IgA. People with selective IgA deficiency either don’t have enough IgA or have low levels of it.
What is the connection between IgA nephropathy and end-stage renal disease (ESRD)?
IgA nephropathy attacks the glomeruli. This type of glomerular disease occurs when IgA deposits build up and damage the glomeruli. The damage causes your kidneys to leak blood (hematuria) and protein (proteinuria) into your urine.
Eventually, the nephrons may scar, causing kidney disease. As the scarring progresses, you may develop end-stage kidney (renal) disease (ESRD) or total kidney failure. This process can happen quickly over the course of months or can take as long as 20 years after your initial diagnosis.
If you develop end-stage renal disease (ESRD), your kidneys can’t work well enough to keep you healthy. You may need:
- Dialysis, a machine that helps filter your blood.
- Kidney transplant, surgery to remove your damaged kidney and replace it with a healthy donor kidney.
IgA nephropathy causes
The exact cause of IgA nephropathy is unknown. Scientists think that IgA nephropathy is an autoimmune kidney disease causing antibody-mediated destruction of the glomerular basement membrane, meaning that the disease is due to the body’s immune system harming the kidneys 142. Usually, there is an infectious disease preceding IgA nephropathy, which leads to the dysregulated immune response, but IgA nephropathy per se is not of an infectious origin. There has been no evidence to suggest that IgA nephropathy is secondary to any specific infectious agents despite the association between macroscopic hematuria and mucosal inflammation 117.
More than 90% of IgA nephropathy cases are sporadic, and the underlying cause is unknown 143.
People with IgA nephropathy have an increased blood level of IgA that contains less of a special sugar, galactose, than normal circulating IgA1 in healthy persons 143, 144, 145, 146. This galactose-deficient IgA (Gd-IgA1) is considered “foreign” by other antibodies circulating in the blood. As a result, these other antibodies attach to the galactose-deficient IgA and form a clump 143, 146. This clump is also called an immune complex. Some of the clumps become stuck in the glomerulus of the nephron and cause inflammation and damage 143. The immune proteins in the glomeruli of patients with IgA nephropathy generally include complement C3; IgG, IgM, or both, are often present 147, 123. Light microscopy typically shows glomerular injury as mesangial hypercellularity and increased mesangial matrix 148.
Less than 10% of IgA nephropathy cases are due to familial IgA nephropathy where IgA nephropathy runs in families. Scientists have recently found several genetic markers that may play a role in the development of the disease 149. IgA nephropathy may also be related to respiratory or intestinal infections and the immune system’s response to these infections. In some people, the first signs or symptoms of IgA nephropathy may become noticeable after a cold, sore throat, or other respiratory infection.
The following things might be linked with IgA nephropathy 150:
- Genes. IgA nephropathy is more common in some families and in certain ethnic groups, such as people of Asian and European descent.
- Genome-wide linkage analysis revealed an association of IgA nephropathy to 6q22-23 and gene locus IGAN1 149
- There are no obvious genes within the linked interval
- It is unclear that genetic findings in these families will have a direct bearing on typical sporadic cases of IgA nephropathy
- Liver diseases. These include scarring of the liver called cirrhosis and chronic hepatitis B and C infections.
- Celiac disease. Eating gluten, a protein found in most grains, triggers this digestive condition.
- Infections. These include hepatitis, cirrhosis, human immunodeficiency virus (HIV) infection and some bacterial infections 142, 151
Risk factors for IgA nephropathy
Although the exact cause of IgA nephropathy is unknown, these factors might increase your risk of developing IgA nephropathy 150:
- Sex. In North America and Western Europe, IgA nephropathy affects at least twice as many men as it does women.
- Ethnicity. IgA nephropathy is more common in whites and Asians than it is in blacks.
- Age. IgA nephropathy most often develops between the late teens and late 30s.
- Family history. In some cases, IgA nephropathy appears to run in families, indicating that genetic factors contribute to the disease.
IgA nephropathy pathophysiology
The current understanding is that IgA nephropathy occurs due to a multi-hit mechanism (Figure 5) 152, 122, 153. The first ‘hit’ is a genetically susceptible host predisposed to developing a dysregulated immune response. The next ‘hit’ is a precipitating factor producing the immunological attack. Infections are potential precipitants of IgA nephropathy 142. Trivial mucosal infections, chronic exposure to pathogens, and abnormal handling of commensals in the gut have all been hypothesized to trigger the abnormal immune response in IgA nephropathy 142. The damage to the basement membrane results in the ultrafiltration of larger molecules and produces hematuria. The pathophysiology of how some develop asymptomatic hematuria while some develop rapidly progressive glomerulonephritis, culminating in kidney failure, is poorly understood 154.
Susceptibility to IgA nephropathy is dependent on many genetic and environmental factors. The pathogenesis of this disease is a multi-“hit” process 152, 122, 153. These “hits” are understood from the IgA moieties found in biopsies and the circulation of patients with IgA nephropathy. A central finding in patients with IgA nephropathy is the presence of immune complexes in circulation and glomeruli comprised of galactose-deficient IgA1, which is an IgG autoantibody against C3 and the hinge region O-glycans. The presence of abnormally glycosylated IgA1 is a heritable trait 155. In a quarter of blood relatives of IgA nephropathy patients, galactose-deficient IgA1 levels are elevated, and segregation analysis reveals a major dominant gene with a polygenic background 156. A recent study outlines the cellular mechanisms causing IgA glycosylation 157.
The levels of galactose-deficient IgA1 could also be influenced by environmental factors. For instance, these antibodies are prone to bacteria-derived proteases 158. Recent studies suggest that anti-glycan autoantibodies could target the IgA VH gene segment due to somatic hypermutation and not sequences found in the host germline 159. Glomerular inflammation and mesangial proliferation are thought to occur because these immune complexes are nephritogenic. Activation of the renin-angiotensin and complement systems also leads to glomerulosclerosis and tubulointerstitial fibrosis, causing deranged renal function. Other risk factors, such as smoking and hypertension, contribute to disease progression through microvascular injury 160. Glmoerulomegaly and maladaptive hyperfiltration injury attributed to obesity may also be implicated in the nonimmunologic progression of the disease 161.
Experimental studies in mice suggest that exposure to bacteria is needed for excess IgA production, which is enabled by the mediators of B cell differentiation and proliferation. Although the application of this theory to IgA nephropathy in humans must be made cautiously, this idea is further aided by genome-wide association studies and studies of disease progression 162.
It is hypothesized that cytokine APRIL (a proliferation-inducing ligand) contributes to IgA nephropathy by propagating B cell class switch to IgA-producing plasma cells through actions on the TACI receptor. APRIL gene polymorphism confers IgA nephropathy susceptibility, and various risk alleles linked to IgA nephropathy are also associated with several diseases of mucosal immunity 163.
Activation of complement is described as a significant pathogenic contributor to IgA nephropathy, particularly the lectin pathway. Polymeric IgA1 can activate this pathway, and the mannose-binding lectin pathway is detected in glomerular deposits 164, 165. Immune complexes contain C3, as seen in the immunofluorescence study of kidney biopsies 166. Complement factor H (CFH) and properdin are also observed in immune deposits 167. Genome-wide association studies report an allele localized to the CFH gene conferring protection against the development of IgA nephropathy. Further analysis indicates that the deletion of complement factor H–related (CFHR) genes is in linkage disequilibrium with the observed risk allele. The CFHR1 and CFHR3 titrate CFH activity, and their absence leads to altered CFH levels and increased CFH activity 168.
Figure 5. IgA nephropathy pathogenesis
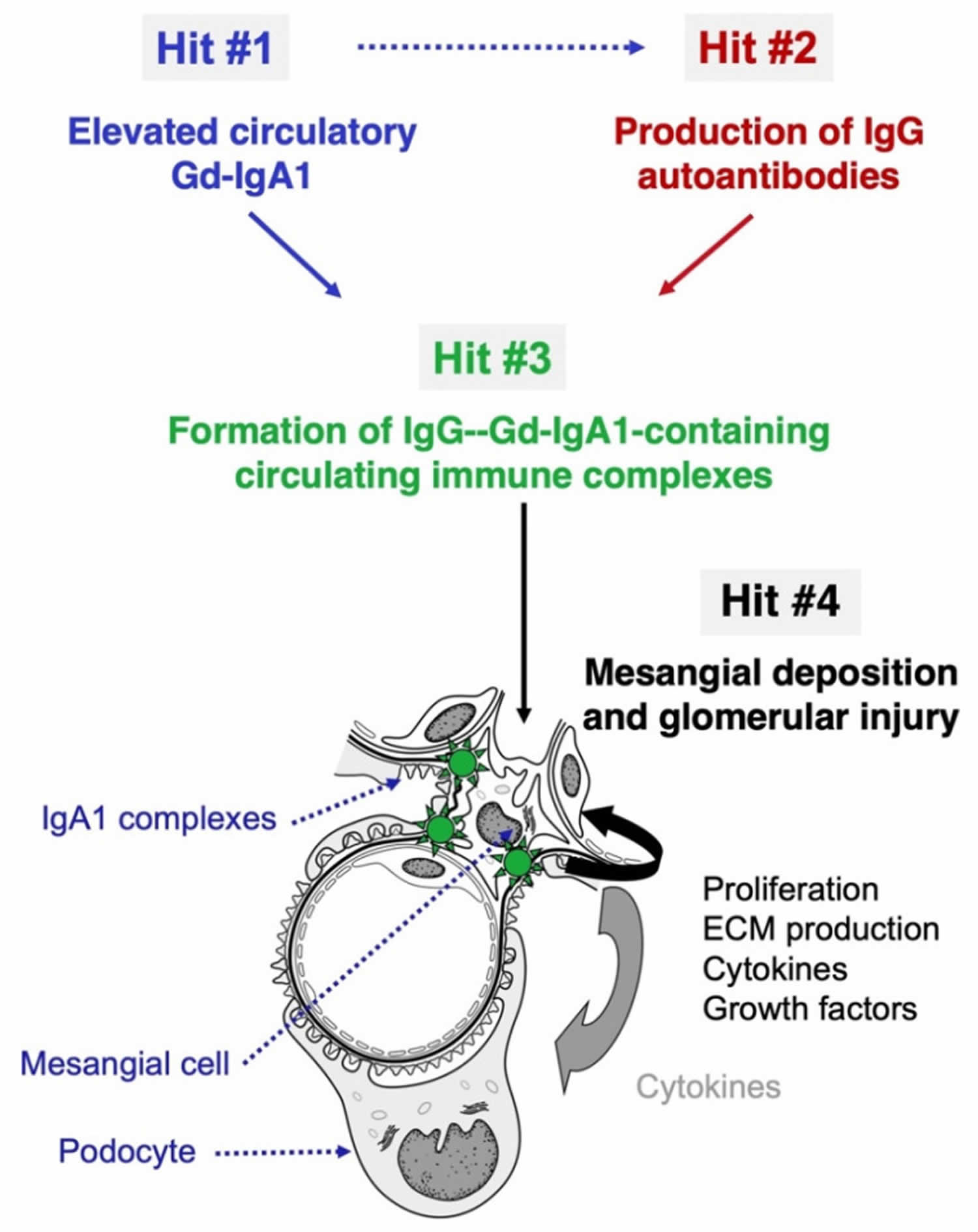
Footnotes: The “four hit” hypothesis of IgA nephropathy. IgA nephropathy (IgAN) is an autoimmune disease with a genetically and environmentally co-determined multi-hit process 152. (Hit #1) Appearance in the circulation of increased levels poorly O-galactosylated IgA1 (galactose-deficient IgA1; Gd-IgA1). (Hit #2) Generation of IgG and IgA autoantibodies directed against Gd-IgA1. (Hit #3) Formation of anti-Gd-IgA1-Gd-IgA1 immune complexes with other serum proteins being added (e.g., complement). Blood levels of the autoantigen (Gd-IgA1) and the corresponding IgG autoantibodies correlate in IgA nephropathy patients, suggesting that elevated circulating levels of Gd-IgA1 are associated with the production of IgG autoantibodies specific for Gd-IgA1 (dashed arrow). Some of the immune complexes formed in the circulation deposit in the kidneys, activate mesangial cells, and induce glomerular injury (Hit #4).
[Source 169 ]IgA nephropathy prevention
Researchers have not found a way to prevent IgA nephropathy. People with a family history of IgA nephropathy should talk with their health care provider to find out what steps they can take to keep their kidneys healthy, such as controlling their blood pressure and keeping their blood cholesterol at healthy levels.
You may reduce your risk of kidney disease by taking care of your kidneys:
- Pay attention to labels when taking over-the-counter (OTC) pain medications. Follow the instructions for OTC pain medications, such as aspirin, acetaminophen (Tylenol, others), ibuprofen (Advil, Motrin IB, others) and naproxen sodium (Aleve, others). Taking too much of these medications may increase your risk of kidney injury. This is especially true if you have pre-existing kidney disease, diabetes or high blood pressure.
- Work with your doctor to manage kidney and other chronic conditions. If you have kidney disease or another condition that increases your risk of acute kidney failure, such as diabetes or high blood pressure, stay on track with treatment goals and follow your doctor’s recommendations to manage your condition.
- Make a healthy lifestyle a priority. Be active; eat a sensible, balanced diet; and drink alcohol only in moderation — if at all.
Eating, diet, and nutrition have not been shown to play a role in causing or preventing glomerular disease. But if you have glomerular disease, your health care professional may recommend you
- Limit salt (sodium) intake
- Reduce calories if your doctor advises you to lose excess weight
- Limit saturated fats if your cholesterol is high
- Make a healthy lifestyle a priority. Be active; eat a sensible, balanced diet; and drink alcohol only in moderation — if at all.
IgA nephropathy symptoms
In its early stages, IgA nephropathy may have no symptoms; it can be silent for years or even decades. Once symptoms appear, the most common one is hematuria, or blood in the urine. Hematuria can be a sign of damaged glomeruli. Blood in the urine may appear during or soon after a cold, sore throat, or other respiratory infection. The amount of blood may be:
- visible with the naked eye. The urine may turn pink or the color of tea or cola. Sometimes a person may have dark or bloody urine.
- so small that it can only be detected using special medical tests.
Another symptom of IgA nephropathy is albuminuria (proteinuria), where a person’s urine contains an increased amount of albumin, a protein typically found in the blood, or large amounts of protein in the urine. Albumin is the main protein in the blood. Healthy kidneys keep most proteins in the blood from leaking into the urine. However, when the glomeruli are damaged, large amounts of protein leak out of the blood into the urine.
When albumin leaks into the urine, the blood loses its capacity to absorb extra fluid from the body. Too much fluid in the body may cause edema, or swelling, usually in the legs, feet, or ankles and less often in the hands or face. Foamy urine is another sign of albuminuria. Some people with IgA nephropathy have both hematuria and albuminuria (proteinuria).
When IgA nephropathy causes symptoms, they might include:
- Cola- or tea-colored urine caused by blood. You might notice these color changes after a cold, sore throat or respiratory infection.
- Blood that can be seen in the urine.
- Foamy urine from protein leaking into the urine. This is called proteinuria.
- Pain on one or both sides of the back below the ribs.
- Swelling in the hands and feet called edema.
- High blood pressure.
- Weakness and tiredness.
After 10 to 20 years with IgA nephropathy, about 20 to 40 percent of adults develop end-stage kidney disease 170. Signs and symptoms of end-stage kidney disease may include:
- high blood pressure
- little or no urination
- edema
- feeling tired
- drowsiness
- confusion
- generalized itching or numbness
- dry skin
- headaches
- weight loss
- appetite loss
- metallic taste in the mouth
- nausea
- upset stomach and vomiting
- sleep problems
- trouble concentrating
- darkened skin
- rashes and itchy skin
- muscle cramps.
Kidney failure is life-threatening without treatment. But dialysis or a kidney transplant can help people live for many more years.
IgA nephropathy complications
The course of IgA nephropathy varies from person to person. Some people have IgA nephropathy for years with few or no problems. Many don’t get diagnosed. Complications of IgA nephropathy include 117, 118:
- High blood pressure (hypertension). Damage to your kidneys from IgA deposits can raise your blood pressure, and high blood pressure can cause further damage to your kidneys.
- Acute kidney failure—sudden and temporary loss of kidney function. If your kidneys lose their filtering ability due to IgA deposits, waste products build up quickly in your blood. And if kidney function gets worse very quickly, health care professionals may use the term rapidly progressive glomerulonephritis.
- Chronic kidney failure (CKD)—reduced kidney function over a period of time. IgA nephropathy can cause your kidneys to gradually stop functioning. Then permanent dialysis or a kidney transplant is needed to live.
- Nephrotic syndrome—a collection of symptoms that indicate kidney damage; symptoms include albuminuria (high urine protein levels or proteinuria), low blood protein levels, and high blood cholesterol or lipids levels (hypercholesterolemia or hyperlipidemia) and fluid can accumulate outside your circulatory system, leading to swelling in your face, hands, feet, or ankles and cause swelling also called edema. About 1 in 5 people with IgA nephropathy develop kidney failure within 10 years of diagnosis 117.
- Heart or cardiovascular problems —the slow loss of kidney function over many years—which can lead to heart disease or stroke.
- Henoch-Schönlein purpura or IgA vasculitis
- High cholesterol. High levels of cholesterol can increase your risk of a heart attack.
Side effects and complications of steroid and steroid-sparing therapy are common 117. Increased risk of infections, hypertension, fluid retention, weight gain, diabetes mellitus, osteoporosis, and iatrogenic Cushing syndrome is the most frequent side effects of steroid therapy 171. Immunosuppression, anaphylaxis, renal, and hepatotoxicity are complications of steroid-sparing agents.
IgA nephropathy diagnosis
IgA nephropathy is often detected after you notice blood in your urine or when a routine test shows that you have protein or blood in your urine. Currently, doctors do not use blood or urine tests as reliable ways to diagnose IgA nephropathy; therefore, the diagnosis of IgA nephropathy requires a kidney biopsy. A kidney biopsy is a procedure that involves taking a small piece of kidney tissue for examination with a microscope. A doctor performs a kidney biopsy in a hospital or an outpatient center with light sedation and a local anesthetic. The health care provider uses imaging techniques such as ultrasound or a computerized tomography scan to guide the biopsy needle into the kidney. A pathologist—a doctor who specializes in examining tissues to diagnose diseases—examines the kidney tissue with a microscope. Only a biopsy can show the IgA deposits in the glomeruli. The biopsy can also show how much kidney damage has already occurred. The biopsy results can help the health care provider determine the best course of treatment.
These tests can help identify which kidney disease you have:
- Urine tests. Blood or protein in the urine, a possible first sign of IgA nephropathy, might be discovered during a routine checkup. If your doctor suspects that you have problems with your kidneys, you might be asked to collect your urine for 24 hours for additional kidney function tests.
- Dipstick test for albumin and blood. A dipstick test performed on a urine sample can detect the presence of albumin and blood. The patient provides a urine sample in a special container in a health care provider’s office or a commercial facility. A nurse or technician can test the sample in the same location, or he or she can send it to a lab for analysis. The test involves placing a strip of chemically treated paper, called a dipstick, into the patient’s urine sample. Patches on the dipstick change color when albumin or blood is present in urine.
- Urine albumin-to-creatinine ratio (UACR). A health care provider uses this measurement, which compares the amount of albumin with the amount of creatinine in a urine sample, to estimate 24-hour albumin excretion. A patient may have chronic kidney disease if the urine albumin-to-creatinine ratio is greater than 30 milligrams (mg) of albumin for each gram (g) of creatinine (30 mg/g).
- Blood tests. If you have kidney disease, a blood test might show increased blood levels of the waste product creatinine or the protein cystatin C. A blood test involves having blood drawn at a health care provider’s office or a commercial facility and sending the sample to a lab for analysis. A health care provider may order a blood test to estimate how much blood a patient’s kidneys filter each minute—a measurement called the estimated glomerular filtration rate (eGFR). Depending on the results, the test can indicate the following:
- eGFR of 60 or above is in the normal range
- eGFR below 60 may indicate kidney disease
- eGFR of 15 or below may indicate kidney failure
- Iothalamate clearance test. Your doctor may also recommend this test, which uses a special contrast agent to track how well your kidneys are filtering wastes.
Histologically, IgA nephropathy is characterized by the following 117:
- The diffuse proliferation of mesangial cells and matrix
- Hypercellular or normal glomeruli with diffuse necrotizing crescentic glomerulonephritis
- Mesangial involvement resembling focal and segmental glomerulosclerosis
- Immunofluorescence will reveal a diffuse granular pattern of IgA deposits in the mesangium
Light Microscopy
The most commonly seen light microscopy findings are focal or diffuse mesangial proliferation and expansion of the extracellular matrix 172. Morphology shows intracapillary and extracapillary proliferative lesions. Occasionally, focal glomerular sclerosis is seen as indistinguishable from focal segmental glomerulosclerosis. In advanced diseases, interstitial fibrosis, vascular sclerosis, and tubular atrophy can be seen. A few patients show segmental necrotizing areas with crescent formation because of extensive disruption of the capillaries.
Electron Microscopy
Electron microscopy reveals mesangial hypercellularity and excess mesangial matrix. An important finding is the presence of mesangial electron-dense deposits of IgA; however, subepithelial and subendothelial deposits of the glomerular capillary wall are seen in a minority of patients, particularly those with the more severe form of the disease 173.
Immunofluorescence
Immunofluorescence reveals mesangial IgA deposits in a diffuse granular pattern. These deposits are majorly polymeric IgA of the IgA1 subclass. Additionally, IgG is found in 43% of patients and IgM in 54% 153. C3 is also often present. The presence of C4d imparts a worse prognosis.
IgA nephropathy treatment
Researchers have not yet found a specific cure for IgA nephropathy. The amount of proteinuria, eGFR, blood pressure, and histological appearance is important in formulating the management plan. Once the kidneys are scarred, they cannot be repaired. Therefore, the ultimate goal of IgA nephropathy treatment is to prevent or delay end-stage kidney disease 174. Your kidney specialist may prescribe medications to:
- Control your blood pressure and slow the progression of kidney disease. Angiotensin-converting enzyme inhibitors (ACE inhibitors) or angiotensin 2 receptor blockers (ARBs) are used to manage proteinuria and lower blood pressure. Salt intake is restricted to control blood pressure. A sodium-glucose cotransporter 2 (SGLT2) inhibitor may be added for persistent proteinuria despite ACE inhibitors or ARBs 175. In a pre-specified subgroup analysis of patients with IgA nephropathy, sodium-glucose cotransporter 2 (SGLT2) inhibitors (which improve outcomes in patients with proteinuria due to diabetic kidney disease) reduced the risk of chronic kidney disease progression 176.
- Remove extra fluid from your blood
- Control your immune system. Immunosuppression with corticosteroids or steroid-sparing agents is used to reduce the rate of progression, including increasing proteinuria, especially into the nephrotic range, and increasing serum creatinine level 177. Steroids have the most benefit if there is heavy proteinuria. Various regimens of oral prednisolone and methylprednisolone are available 171. If there are contraindications for steroids or if the risks of therapy outweigh the anticipated benefits of steroid therapy, steroid-sparing agents may be an option. Cyclophosphamide, azathioprine, and cyclosporine are potential steroid-sparing agents 178, 179. Corticosteroids and cyclophosphamide for proliferative injury or rapidly progressive glomerulonephritis 175
- Lower your blood cholesterol levels
- Weight reduction may reduce proteinuria in IgA nephropathy 180.
- Given the association between smoking and IgA nephropathy progression, smoking cessation should also be advised 160.
Normotensive patients with intact renal function (serum creatinine < 1.2 mg/dL [106.08 micromol/L]) and only mild proteinuria (< 0.5 g/day) usually are not treated beyond angiotensin inhibition (with an ACE inhibitor or ARB) and an SGLT2 inhibitor 175. Patients with renal insufficiency or more severe proteinuria and hematuria are usually offered corticosteroids, which ideally should be started before significant renal insufficiency develops 175.
For the few who progress to develop end-stage renal disease (ESRD), renal transplantation is an option. There is still the risk of IgA nephropathy in the transplanted kidney. Treatment with an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker may delay the progression of recurrent disease in allografts 181.
High blood pressure drugs
People with IgA nephropathy that has high blood pressure may need to take high blood pressure medications to lower the blood pressure which can also significantly slow the progression of kidney disease 182. Two types of blood pressure-lowering medications, angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs)—have proven effective in slowing the progression of kidney disease 182. Many people require two or more medications to control their blood pressure. A person may also need beta-blockers, calcium channel blockers, sodium-glucose cotransporter 2 inhibitors and other blood pressure medications. The blood pressure target is 130/80 mmHg 183.
Diuretics (water pills)
Your doctor may prescribe a diuretic or water pill, a medication that helps the kidneys remove extra fluid from your blood. Removing the extra fluid can control swelling in your hands and feet called edema and also improve the control of your blood pressure. Taking a diuretic along with an ACE inhibitor or an angiotensin receptor blocker often increases the effectiveness of these medications.
Corticosteroids and immunosuppressants
Doctors sometimes use medications called immunosuppressants to control a person’s immune system. Since inflammation is the immune system’s normal response, controlling the immune system can decrease inflammation. Health care providers may prescribe the following medications:
- corticosteroids, such as prednisone
- cyclophosphamide
There are many studies supporting that corticosteroids reduce proteinuria in IgA nephropathy. In an old study, patients received corticosteroid therapy or supportive therapy alone. The use of renin-angiotensin system (RAS) blockade was random, and the run-in period of optimizing conservative treatment was not carried out. In this trial, 5-year renal survival was greater in the corticosteroid group 184. This finding suggested that corticosteroids for a period of 6 months have a “legacy effect” with a sustained risk reduction in progressive renal dysfunction. In another similar study of patients with biopsy-proven IgA nephropathy, corticosteroids were reported to have reduced proteinuria 185.
Corticosteroids, in combination with an additional agent, are generally reserved for progressive disease. A single-center, randomized, prospective trial with “high-risk” IgA nephropathy demonstrated better renal survival in patients receiving prednisone in combination with cyclophosphamide/azathioprine as opposed to no immunotherapy 186. In contrast, combination therapy does not always provide an advantage. A multicenter trial of 207 patients with IgA nephropathy demonstrated no difference between patients receiving corticosteroids alone and those taking combination therapy 187.
There is no consensus among experts regarding the optimal corticosteroid regimen. One protocol uses methylprednisolone 1 g IV once a day for 3 days at the beginning of months 1, 3, and 5 plus prednisone 0.5 mg/kg orally every other day for 6 months. Another regimen uses prednisone beginning 1 mg/kg orally once a day with dose gradually tapered over 6 months.
Because of the risk of adverse effects, corticosteroids should probably be reserved for patients with any of the following 175:
- Worsening or persistent proteinuria (> 1 g/day), especially if in the nephrotic range despite maximal ACE inhibitor or ARB therapy
- Increasing serum creatinine level
Combinations of IV corticosteroids and cyclophosphamide plus oral prednisone are used for severe disease, such as proliferative or crescentic (rapidly progressive) nephropathy. Evidence for mycophenolate mofetil is conflicting; it should not be used as first-line treatment. None of these medications, however, prevents recurrence in transplant patients. Immunosuppressive therapy should also be avoided in patients with advanced fibrotic kidney disease, which is not reversible.
A novel potential therapy for IgA nephropathy is a formulation of oral budesonide. This agent can potentially act locally at the lymphoid tissue of the mucosa in the distal ileum and proximal large intestine to modulate IgA production. Higher first-pass metabolism theoretically minimizes systemic effects.
Although evidence for rituximab efficacy in other glomerular diseases is significant, early results in IgA nephropathy are not encouraging. A pilot study assessed the efficacy of rituximab versus conservative management in patients with proteinuria. No favorable effects on proteinuria or renal function were observed 188.
Data around the efficacy of mycophenolate are indefinite, such that current guidelines recommend against using mycophenolate in IgA nephropathy 189. The majority of studies are limited by small sample size. There is also possible race-specific variation in response to mycophenolate, as in lupus nephritis 190. However, systematic reviews and meta-analyses of the randomized trials of mycophenolate and systematic reviews suggest mixed results 191, 192.
Lower blood cholesterol levels
People with IgA nephropathy may develop high blood cholesterol levels. Cholesterol is a type of fat found in the body’s cells, in blood, and in many foods. People who take medications for high blood cholesterol levels can lower their blood cholesterol levels. A health care provider may prescribe one of several cholesterol-lowering medications called statins.
IgA nephropathy diet
Researchers have not found that eating, diet, and nutrition play a role in causing or preventing IgA nephropathy. Health care providers may recommend that people with kidney disease, such as IgA nephropathy, make dietary changes such as:
- limiting dietary sodium, often from salt, to help reduce edema and lower blood pressure
- decreasing liquid intake to help reduce edema and lower blood pressure
- eating a diet low in saturated fat and cholesterol to help control high levels of lipids, or fats, in the blood
Health care providers may also recommend that people with kidney disease eat moderate or reduced amounts of protein, although the benefit of reducing protein in a person’s diet is still being researched. Proteins break down into waste products the kidneys must filter from the blood. Eating more protein than the body needs may burden the kidneys and cause kidney function to decline faster. However, protein intake that is too low may lead to malnutrition, a condition that occurs when the body does not get enough nutrients. People with kidney disease on a restricted protein diet should receive blood tests that can show nutrient levels.
Some researchers have shown that fish oil supplements containing omega-3 fatty acids may slow kidney damage in some people with kidney disease by lowering blood pressure. Omega-3 fatty acids may help reduce inflammation and slow kidney damage due to IgA nephropathy. To help ensure coordinated and safe care, people should discuss their use of complementary and alternative medical practices, including their use of dietary supplements and probiotics, with their health care provider. Ask your doctor if prescription fish oil supplements might help you.
People with IgA nephropathy should talk with a health care provider about dietary changes to best manage their individual needs.
Other treatments
Although other interventions have been tried to lower IgA overproduction and to inhibit mesangial proliferation, data supporting any of these are limited or absent, and none can be recommended for routine treatment. These interventions include elimination of gluten, dairy products, eggs, and meat from the diet; tonsillectomy; and immune globulin 1 g/kg IV 2 days a month for 3 months followed by immune globulin 0.35 mL/kg of 16.5% solution IM every 2 weeks for 6 months all theoretically reduce IgA production. Heparin, dipyridamole, and statins are just a few examples of in vitro mesangial cell inhibitors.
For patients who progress to end-stage renal disease (ESRD), kidney transplantation is preferred over dialysis because of improved long-term disease-free survival. The condition recurs in approximately 30% of graft recipients 193.
IgA nephropathy prognosis
Frequently IgA nephropathy patients have a benign course 194. Infrequently, IgA nephropathy patients may gradually progress to end-stage renal disease (ESRD), with the frequency of ESRD increasing with age 195, 196. In general, about 1 in 4 adults with IgA nephropathy eventually get end-stage renal disease (ESRD). About 1 in every 10 to 20 children with IgA nephropathy develop end-stage renal disease (ESRD). About 20% of patients will progress to end-stage renal disease (ESRD) within ten years.
Efforts have been made to determine clinical and histological features associated with progression to end-stage renal disease 197. IgA nephropathy prognosis is predictable to some extent, based on the Oxford classification called the MEST-C score 198. Additionally, nephrotic range proteinuria, hypertension, high serum creatinine level, and widespread intestinal fibrosis of the kidneys on presentation indicate a poor prognosis 199.
Currently, the MEST-C score published in 2009, comprises four histological features is often used to predict IgA nephropathy prognosis 198. The IgA Nephropathy Classification Working Group added crescents to the Oxford classification, to form the MEST-C score 200.
The Oxford classification of IgA nephropathy or MEST score includes the following features 198, 200, 117:
- M = Mesangial cellularity, defined as greater than 4 mesangial cells in any mesangial segment of the glomerulus. M0 is mesangial cellularity in <50% of glomeruli; M1 ≥50%
- E = Endocapillary proliferation is the degree of hypercellularity due to an increased number of cells within glomerular capillary lumina: E0 is absence of hypercellularity; E1 is hypercellularity in any glomeruli
- S = Segmental glomerulosclerosis is defined as sclerosis or adhesions in the glomerular (obliteration of capillary lumina by matrix) in part of but not the whole glomerular tuft: S0 is absence of segmental glomerulosclerosis, S1 is presence of segmental glomerulosclerosis in any glomerulus
- T = Tubular atrophy or interstitial fibrosis, defined as the estimated percentage of cortical area showing tubular atrophy or interstitial fibrosis, whichever is greater: T0 is 0-25%; T1 is 25-50%; T2 is >50%
- C = Presence or absence of crescents. C0 (no crescents), C1 (crescents in less than one-fourth of glomeruli), and C2 (crescents in over one-fourth of glomeruli).
The clinical significance of the individual MEST-C features is as follows:
- M1 – Worse outcomes than M0
- E1 – Worse renal survival in patients not on immunosuppression and improved renal survival with immunosuppression
- S1 – Predictive of worse outcomes
- T – Strongest predictor of worse outcomes
- C1 – Predictive of worse outcomes if no immunosuppression is given, but not if immunosuppression is used; C2 is predictive of worse outcomes regardless of Immunosuppression
Other predictors of poor renal outcomes include the following:
- High serum creatinine level (>120 mmol/L) at presentation
- Hypertension (diastolic >95 mm hg or need for antihypertensive treatment)
- Proteinuria: Urinary protein excretion 3.5 g/24 hr with 7% renal survival 201
- Extensive interstitial fibrosis and tubular atrophy on renal biopsy
- C4d staining on biopsy
A calculator for estimating the risk of progression to end-stage renal disease (ESRD) in patients with IgA nephropathy has been developed by Xie et al 202, based on a cohort of 619 Chinese patients. It has yet to be validated in other ethnic groups. The calculator uses four variables: glomerular filtration rate, hemoglobin level, serum albumin level, and systolic blood pressure.
In general, if any of the above features are seen (i.e., GFR of less than 60 mL/minute, proteinuria greater than 0.5 g/day, hypertension greater than 140/90 mm Hg, more than 50% glomeruli affected by mesangial hypercellularity), then the prognosis is poor. Other factors that determine outcomes include elevated creatinine, hypertension, need for antihypertensive treatment, proteinuria, decreased eGFR at diagnosis, significant interstitial fibrosis, and CD4 staining 203, 204. Isolated microscopic hematuria with mild proteinuria is considered favorable, particularly in Whites. Race may also be a significant determinant of outcome 140.
Will I need a kidney transplant?
Everyone’s IgA nephropathy disease progression is different. Some people respond well to treatment and can live with IgA nephropathy for a long time.
If IgA nephropathy progresses to kidney failure, you may need to consider a kidney transplant. Your care team will discuss dialysis and kidney transplant with you.
In patients with kidney failure due to IgA nephropathy, IgA deposits can recur in a subsequent kidney transplant. If only patients who had undergone a post-transplant kidney biopsy are analyzed, the recurrence rate was 42% after 10 years 205. Cumulative risk of IgA nephropathy recurrence increased after transplant and was associated with a 3.7-fold greater risk of graft loss 205.
If you have kidney disease such as IgA nephropathy, it’s important to:
- Limit sodium (salt) in your foods to help lower blood pressure and reduce swelling.
- Decrease how much liquid you drink, another way to lower blood pressure and reduce swelling.
- Eat foods low in saturated fat and cholesterol to reduce levels of fat in your blood.
- Lifestyle modification such as quitting smoking, diet and exercise, if needed, can also be beneficial.
- Some healthcare providers recommend fish oil supplements that contain omega-3 fatty acids. Research suggests this approach may lower blood pressure and slow the progress of the disease. Speak to your healthcare provider before you start taking any supplements.
IgA nephropathy life expectancy
Although IgA nephropathy usually follows a benign course, end-stage renal disease (ESRD) develops in 15-20% of patients within 10 years of onset and in about 25-30% of patients by 20 years 132. 30–40% of IgA nephropathy patients progress to kidney failure that reduces life expectancy by about 10 years 206. Cumulative incidence of recurrent IgA nephropathy was 19% at 10 years and 23% at 15 years after kidney transplantation 205.
Collapsing glomerulopathy
Collapsing glomerulopathy also known as collapsing variant of focal segmental glomerulosclerosis is a pattern of kidney injury from multiple causes that include HIV infection (HIV-associated nephropathy) and APOL1 gene mutations in African American patients 207, 208, 209, 210, 211, 212, 213, 214, 215, 216. Collapsing glomerulopathy is characterized by segmental or global collapse of the glomerular capillaries and hypertrophy and hyperplasia of podocytes migrating to the tuft to give the appearance of ‘pseudocrescents’ 217, 218, 211. The Columbia classification of 2004 classified collapsing glomerulopathy as a histological subtype of focal segmental glomerulosclerosis (FSGS) 219, 220.
The most common signs and symptoms of collapsing glomerulopathy is massive, severe proteinuria associated with pure nephrotic syndrome. Patients may present with high blood pressure, lipiduria, and hematuria on urinalysis 211. A significantly high number of patients present with renal failure on admission and rapidly progress to end-stage renal disease (ESRD) 221, 222, 223, 224, 225.
The normal kidney dimensions are usually preserved in collapsing glomerulopathy and presents as in other conditions, such as diabetic nephropathy, polycystic kidney disease, and amyloidosis. Ultrasonography may demonstrate normal or increased renal size and hyperechogenicity, which may be due to edema, fibrosis, and tubulointerstitial infiltrates resulting from the rapid progression to end-stage renal disease (ESRD) and absence of renal parenchymal contraction 222.
Collapsing glomerulopathy is the most common morphological pattern in HIV nephropathy 226. The increased incidence of HIV infection has been identified as a cause for the proportionate increase in the cases of collapsing glomerulopathy 222. Disease activity characterized by a high viral load and low CD4 lymphocyte count results in greater kidney damage 216; however, the virus has also been detected in the kidney tissue of patients with undetectable viral loads 227. Non-structural HIV proteins, such as viral protein R and negative factor, promote cell cycle dysregulation, which stimulates podocyte proliferation. In some studies, the prevalence of HIV infection in patients with collapsing glomerulopathy ranged from 30 to 55% 221, 228.
Other factors such as chronic inflammation are associated with the genetic predisposition for APOL1 and MHY9 mutations, which result in collapsing glomerulopathy 227.
Cytomegalovirus infection is associated with immunosuppression, but it can also occur in immunocompetent patients and progress to collapsing glomerulopathy, even in the acute phase of the disease. Specific treatment is associated with several benefits for prognosis in this population 229, 230, 231, 232.
Arboviruses were recently identified as an important causative factor for collapsing glomerulopathy, particularly in Brazil 233, 234. Eight of 13 collapsing glomerulopathy biopsy samples obtained from the first half of 2016 from a large Brazilian kidney biopsy center were positive for arbovirus, wherein six samples were positive for dengue, one for Zika, and one for a concomitant infection; only one case had APOL1 mutations. These findings suggested that direct viral action in tissues may be associated with other risk factors, such as G1 and G2 mutations 233, 234.
The association between SARS-CoV-2 (COVID-19) infection and collapsing glomerulopathy was initially demonstrated in a series of autopsies 235. A recent systematic review of 59 studies reporting COVID-19 related histopathological diagnoses from kidney biopsy identified collapsing glomerulopathy as the most common finding, followed by acute tubular injury and trombotic microangiopathy 236. Various mechanisms of acute kidney injury secondary to COVID-19 have been proposed—from direct intrarenal infection to dysregulation of the renin-angiotensin-aldosterone system, to altered hemodynamic control, coagulation and cytokine homeostasis 236. Although the association is multifactorial, it has been emphasized the influence of the hyperactive inflammatory process and participation of circulating interferons 236 and direct infection appears highly unlikely to play a significant pathogenic role.
APOL1 mutations have been detected in many patients with COVID-19, which suggested that the SARS-CoV-2 virus is a potential secondary trigger for glomerular damage 237. Studies have confirmed the strong association of collapsing glomerulopathy and non-collapsing podocytopathies with concurrent or recent COVID-19 in patients with APOL1 high-risk alleles 236, 237, 238.
Systemic lupus erythematosus (SLE) can trigger collapsing glomerulopathy as an extreme form of lupus podocytopathy in the absence of other lupus nephritis patterns 239. Collapsing glomerulopathy may also result from the association of SLE with other risk factors, such as black ethnicity and APOL1 mutations. collapsing glomerulopathy is occasionally present during the diagnosis of SLE, with low levels of therapeutic response 239.
Collapsing glomerulopathy can be drug-induced; bisphosphonates, especially pamidronate and zoledronic acid, inhibit the mevalonate synthesis pathways, which are essential for cell differentiation. This triggers podocyte proliferation and progression to collapsing glomerulopathy 240. Synthetic interferons (which are used to treat some infectious, autoimmune, and neoplastic diseases) can result in APOL1 overexpression in the glomerular epithelium, which triggers podocyte damage. This effect is evidenced by the presence of tubuloreticular inclusions on electron microscopy (48). Illicit drugs, such as cocaine and heroin, are also associated with collapsing glomerulopathy. The most probable mechanism involves ischemic glomerular damage from oxidative endothelial damage, accelerated atheromatosis, and direct vasoconstriction 241.
Acquired Hemophagocytic Syndrome results from a hyperactive immune system that develops secondary to infectious diseases or lymphatic hematological neoplasms. Excessive T lymphocyte and circulating cytokine activation can promote podocyte proliferation and progression to collapsing glomerulopathy 242. Monoclonal gammopathies can also encourage progression toward collapsing glomerulopathy. Histological analysis demonstrates diffuse Ig chain deposits, with glomerular collapse resulting from the deposition of extracellular elements instead of from podocyte proliferation 243. Once triggered, collapsing glomerulopathy progresses independently of the underlying disease. Further, while the underlying gammopathy may go into remission, collapsing glomerulopathy can continue to worsen until kidney dialysis becomes necessary 244.
Collapsing glomerulopathy is also associated with diseases characterized by microvascular damage, such as sickle cell anemia, intravascular hemolysis syndromes, drug reactions (calcineurin inhibitors), malignant arterial hypertension, and antiphospholipid syndrome. A total of 53 histological samples that demonstrated thrombotic microangiopathy were evaluated. These samples were acquired from 33 patients with focal segmental glomerulosclerosis, 19 of whom had collapsing glomerulopathy 245. Glomerular ischemia resulted in the loss of podocyte differentiation and reduced cell proliferation. The population in this study was predominantly of white ethnicity and was characterized by a lower incidence of nephrotic proteinuria.
Other conditions, such as hypertensive disorders of pregnancy, have also been implicated as a trigger for collapsing glomerulopathy. Hypertensive disorders of pregnancy are characterized by diffuse endothelial damage and possible glomerular ischemia. Kidney biopsy often shows association between collapsing glomerulopathy and thrombotic microangiopathy 238.
Collapsing glomerulopathy demonstrates no sex differences, but preferentially affects patients of African descent. Collapsing glomerulopathy also often affects young adult patients and has a varying predominance in the pediatric age group 246, 221, 222, 247, 223, 248.
Population-based studies in the United States, India, Pakistan, Macedonia, and Portugal proposed prevalence rates of collapsing glomerulopathy to 1.7, 0.75, 0.38, 1.7, and 0.29%, respectively 249, 246, 250, 251, 221. A study from Brazil, the São Paulo Registry of Glomerulopathies, indicates that 36% of focal segmental glomerulosclerosis cases comprise patients with collapsing glomerulopathy 252.
Figure 6. Collapsing glomerulopathy
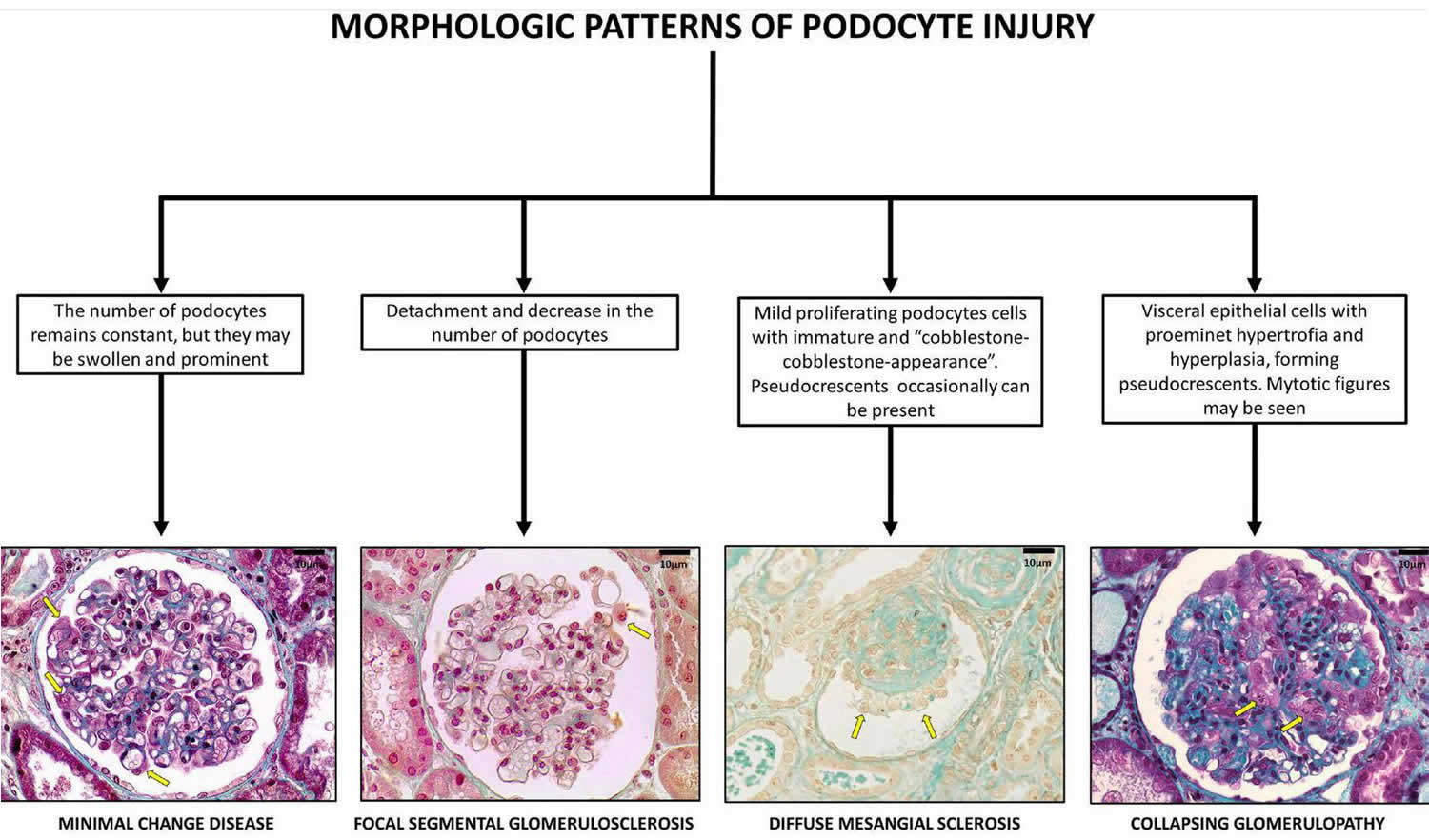
Footnote: Glomerular pattern and podocytes morphological changes in human podocytopathies (bar = 10 μm). Podocytopathies are kidney diseases in which direct or indirect podocyte injury drives proteinuria or nephrotic syndrome 253.
[Source 211 ]Figure 7. Collapsing glomerulopathy causes
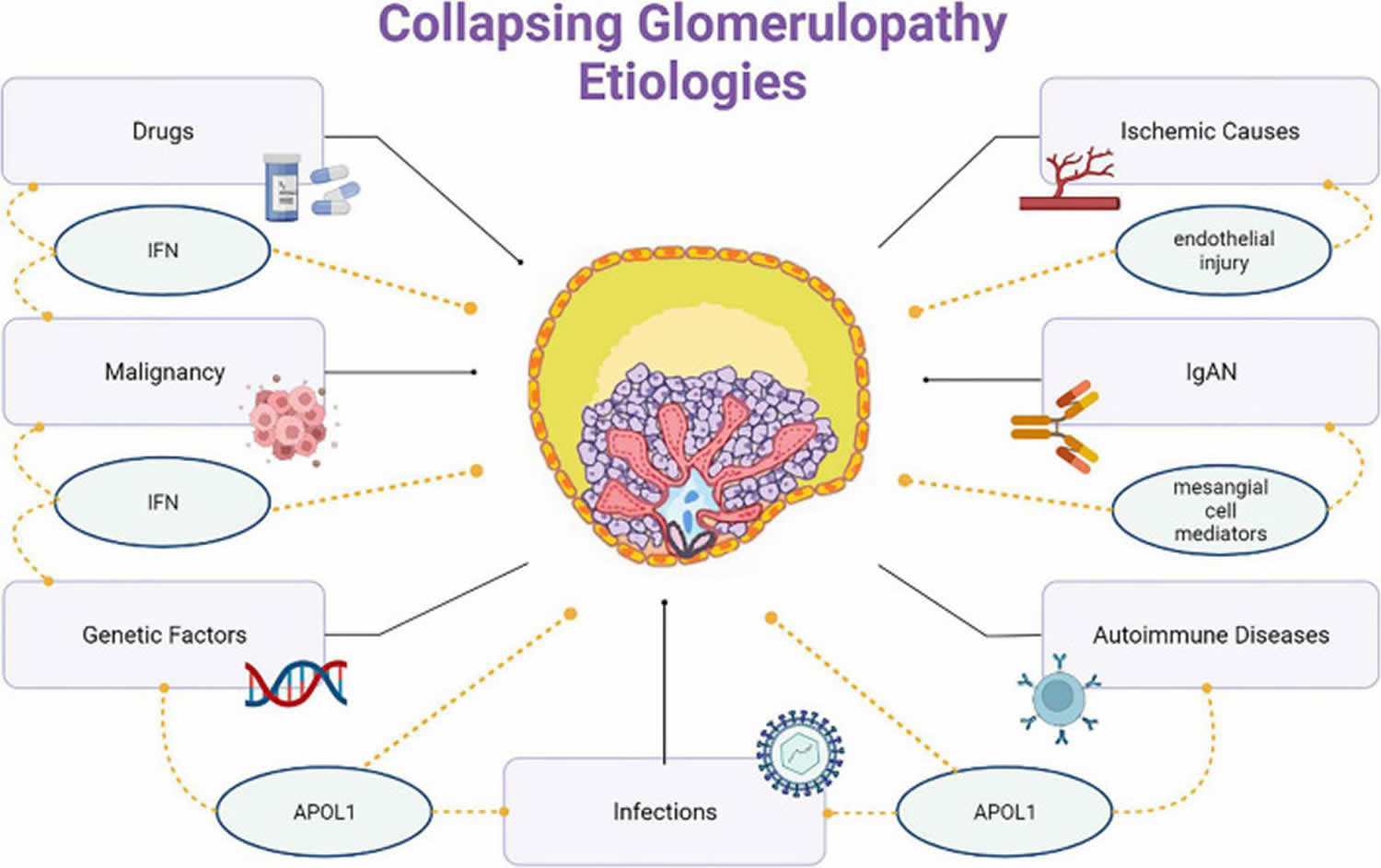
Figure 8. Collapsing glomerulopathy diagnosis
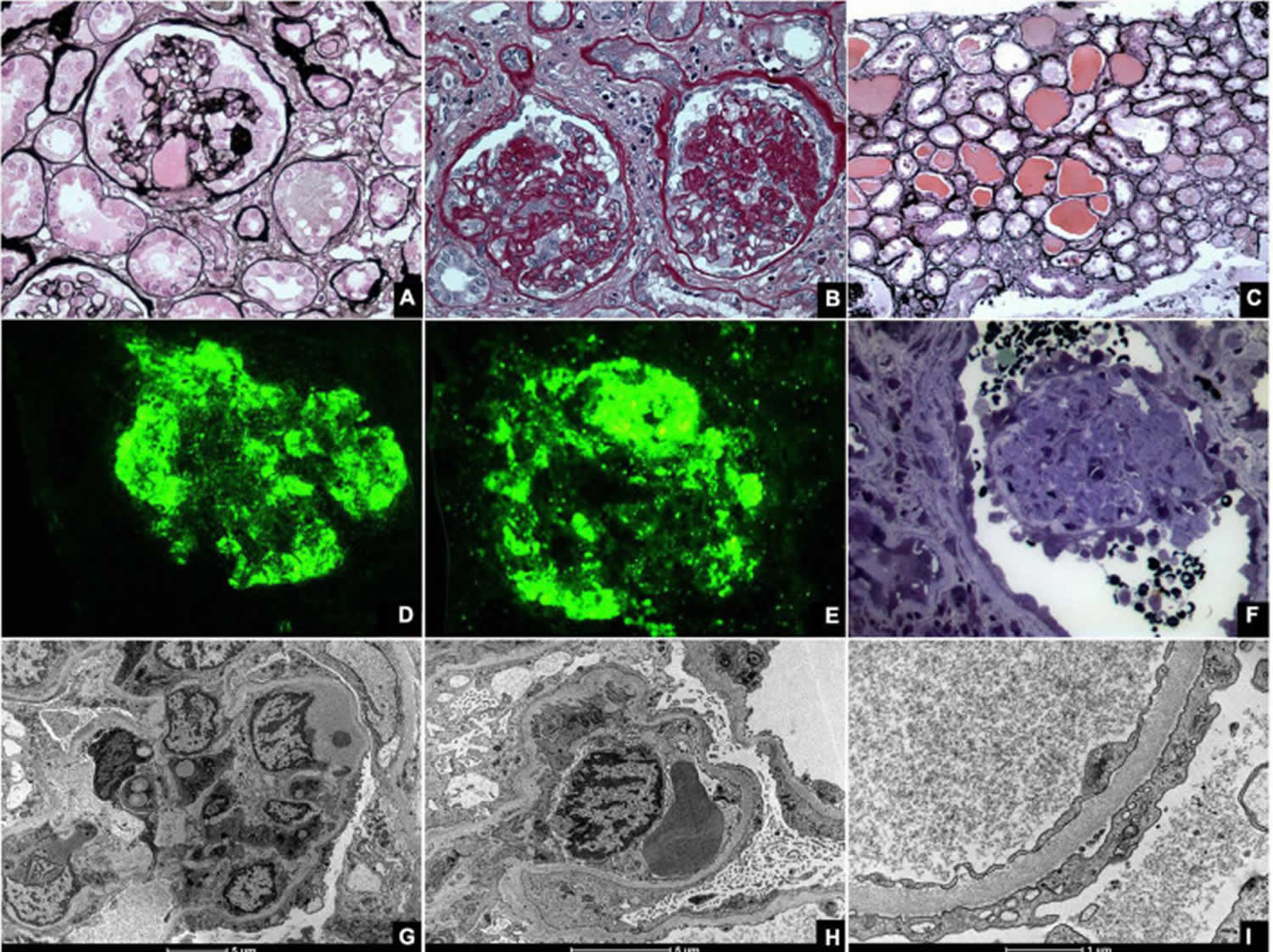
Footnotes: Kidney biopsies of collapsing glomerulopathy. (A,B) Periodic Acid Schiff (PAS) and Jones Methenamine Silver (JMS) (40×), respectively show intense podocyte hyperplasia and glomerular tuft collapse. (C) Jones Methenamine Silver (JMS) (20×) exhibits microcytic transformation of distal convoluted tubules with accumulations of hyaline material inside of those. (D,E) Fluorescence microscopy (40×) shows, respectively, IgM and C3 trapping in areas of collapse/sclerosis. (F) Semi-fine stained in Toluidine Blue (63×) with collapse of the entire glomerular tuft and hyperplasia of podocytes and dilated Bowman’s space. (G,H) Transmission electron microscopy contrasted with Osmium Tetroxide, Lead Citrate and Uranyl in block shows capillary loop collapse with hyalinosis in addition to diffuse fusion and flattening of the pedicels associated with microvillous transformation. (I) Electron microscopy tubes contrasted with osmium tetroxide, lead citrate, and uranyl in block with detail of disorganization of the cytoskeleton in the podocyte cytoplasm, with extensive effacement of the pedicels.
[Source 211 ]Collapsing glomerulopathy causes
Barisoni proposed a classification scheme for podocytopathies and characterized collapsing glomerulopathy as idiopathic/primary, genetic, and reactive/secondary 218. Podocytopathies are kidney diseases in which direct or indirect podocyte injury drives proteinuria or nephrotic syndrome 253. The primary conditions that cause collapsing glomerulopathy are highlighted here and classified according to the nature of injury:
- Infectious disease like HIV, T-cell lymphotropic virus, hepatitis B virus (HBV), hepatitis C virus (HCV), Epstein-Barr virus, cytomegalovirus (CMV), parvovirus B19, dengue, Zika virus, Chikungunya, tuberculosis, visceral leishmaniasis, filariasis, and SARS-COV-2 (COVID-19).
- Autoimmune diseases including systemic lupus erythematosus (SLE), adult Still’s disease, temporal arteritis, mixed connective tissue disease, and Behçet’s disease.
- Neoplastic causes, such as multiple myeloma, acute monoblastic leukemia, natural killer cell leukemia, and acquired hemophagocytic syndrome.
- Drug-related causes like bisphosphonates, interferons, anabolic steroids, heroin, valproic acid, and anthracyclines.
- Ischemic causes, that include thrombotic microangiopathy, atheroembolic disease, sickle cell anemia, and thromboembolism by hydrophilic polymers.
Collapsing glomerulopathy can also coexist with other nephropathies, such as immunoglobulin A (IgA) nephropathy, membranous glomerulopathy, and other histological types of focal segmental glomerulosclerosis and diffuse mesangial sclerosis.
Several case reports have linked collapsing glomerulopathy to infectious diseases. Chandra proposed the following criteria to assess the causal link between infection and collapsing glomerulopathy 229:
- Collapsing glomerulopathy demonstration in multiple cases of viral disease;
- Clear demonstration of collapsing glomerulopathy lesions, including glomerular tuft collapse and changed podocyte phenotype;
- Demonstration of viral proteins or nucleic acids in glomerular cells, especially in podocytes; and
- Experimental demonstration (in animal models) of some (or all) collapsing glomerulopathy findings.
Collapsing glomerulopathy pathogenesis
Regarding the origin of the hyperplastic podocytes in the collapsing glomerulopathy, one key study using immunofluorescent techniques indicates that cellular crescents in crescentic glomerulonephritis and hyperplastic podocytes in collapsing glomerulopathy all come from the same parietal epithelium of the glomeruli 254. The study also finds that there are 3 populations of hyperplastic podocytes in the collapsing glomerulopathy. Using confocal microscopy to evaluate several merged marker expressions, the study determined that there are approximately 60% immature podocytes (CD133+, CD24+, podocalyxin− [PDX, a podocyte marker] nestin− [another podocyte marker]), 30% mature podocytes (CD133−, CD24−, PDX+, nestin+), and 10% transitional podocytes (CD133+, CD24+, PDX+, nestin+) 254. The finding indicates that glomerular hyperplastic lesions are derived from the proliferation of renal progenitors at different stages of their differentiation toward mature podocytes 254.
Cell Behavior
From a histopathologic perspective, collapsing glomerulopathy falls within the spectrum of podocytopathies (see Figure 6). Podocytes are terminal cells that constitute an essential part of the glomerular structure. They consist of a cell body, primary, secondary, and tertiary podocyte processes, and interdigitations between these processes 255. The spaces between pedicels are filled with slit diaphragm proteins, which function as mechanical and electrical barriers for a plasma ultrafiltrate 256, 257, 258. The proteins that constitute the diaphragm are strongly anchored to the cytoskeleton of the podocytes 256 and comprise a strong functional part of the glomerular filtration barrier.
Barisoni 218 classified podocytopathies based on their morphological characteristics and pathogenesis. Minimal change disease presents with slit diaphragm involvement instead of podocyte damage, whereas FSGS presents with marked podocytopenia 224. In contrast, diffuse mesangial sclerosis and collapsing glomerulopathy are characterized by high proliferation rates 249. The proliferation rate is higher in collapsing glomerulopathy than in diffuse mesangial sclerosis 218.
Although podocytes are usually considered as terminal differentiation cells, they have a marked loss of differentiation and high mitotic rates in collapsing glomerulopathy. Shkeri et al 259 utilized the western blot technique to analyze the gene expressions of several glomerulopathies and demonstrated that collapsing glomerulopathy was associated with increased cell proliferation markers, such as T-telomerase, Ki-67 protein, and beta-cadherin 1; however, these findings were not observed in the other types of FSGS.
The human Wilms tumor 1 (WT1) gene is a tumor suppressor gene that spans ∼50 kb and consists of 10 exons. It encodes a protein that shares a high degree of structural homology with the early growth response family of transcription factors. Several lines of evidence suggest that Wilms tumor 1 (WT1) protein is important for normal podocyte function 260. Overexpression of the Wilms tumor 1 (WT1) protein results in an increased telomerase function, which promotes podocyte cell differentiation and renal histological organization. Inhibition of the WT1 protein is associated with terminal phenotype loss and podocyte hyperplasia 259. In collapsing glomerulopathy, WT1 inhibition results in podocytes that have similar gene expressions patterns with those of the glomerular parietal epithelium, which may represent a phenotypic return to the original cells that yielded the Bowman’s capsule epithelium 259. Wilms tumor 1 (WT1) protein has been demonstrated to activate transcription of the podocalyxin gene. The integral membrane protein podocalyxin connects to the cytoskeleton of the podocytes and is implicated in maintaining the complex three-dimensional shape of the cells 260. Gene inhibition of podocalyxin and other proteins that contribute to the structure of podocytes and slit diaphragms by determining protein binding and local electrostatic forces as synaptopodin, glomerular epithelial protein 1, common acute lymphoblastic leukemia antigen, and the C3b receptor has also been documented in collapsing glomerulopathy 257.
Other cells may also be associated with the pathophysiology of collapsing glomerulopathy. Under hypoxic conditions, glomerular endothelial cells have been reported to secrete paracrine factors that modify podocyte structure 245. HIV-affected T lymphocytes are also involved in cell proliferation 227.
Genetics
APOL1 gene, which is located on the long arm of chromosome 22 (22q12 region), contributes to the pathogenesis of collapsing glomerulopathy. APOL1 gene is responsible for forming high-density lipoproteins in different cell membranes and is associated with innate immunity. APOL1 gene confers resistance to Trypanosoma brucei rhodesiense, the etiologic agent for African sleeping sickness. APOL1 improves gene function and fights against infections through G1 (missense) and G2 (deletion of two amino acids) mutations. APOL1 is also associated with several cell damage mechanisms, such as mitochondrial damage, lysosomal degranulation, and cell pore formation 208. Lysosomal degranulation plays a particular role in destroying the cell membranes of foreign pathogens 261.
APOL1 gene mutations are correlated with the distribution of sleeping sickness. As such, these mutations are quite prevalent in sub-Saharan Africa, reaching over 40% in countries such as Ghana and Nigeria 208. In several countries including Brazil, APOL1 mutations are considered as risk factors and determinants of chronic kidney disease (CKD). A national case-control study demonstrated that these mutations are associated with an odds ratio of 10.95 for progression to chronic kidney disease compared to controls. APOL1 mutations have also been associated with an increased indication for hemodialysis within a mean period of 12 years or earlier 262.
Other genes have been implicated in the pathogenesis of collapsing glomerulopathy. The COQ2 gene (4q21.23) encodes the mitochondrial protein CoQ1, which plays a role in some neurologic, muscle, and renal syndromes. Mitochondrial gene mutations contribute to the proliferation of a poorly differentiated podocyte profile, which results in collapsing glomerulopathy 263, 264. In contrast, the MHY9 gene is responsible for synthesizing myosin microfilaments that maintain the podocyte structure and filtration barrier. Among patients with HIV, MHY9 gene mutations increase the risk of progression to collapsing glomerulopathy by 4–8-fold 265.
Collapsing glomerulopathy signs and symptoms
The most common signs and symptoms of collapsing glomerulopathy is massive, severe proteinuria associated with pure nephrotic syndrome. Nephrotic syndrome is a kidney disorder that causes your body to pass too much protein in your urine (proteinuria), low levels of a protein called albumin in your blood (hypoalbuminemia), swelling in parts of your body (edema) and high levels of cholesterol and other lipids (fats) in your blood (hyperlipidemia) 266. Patients may present with high blood pressure (hypertension), presence of lipids in the urine (lipiduria) and blood in urine (hematuria) on urinalysis 211. A significantly high number of patients present with kidney failure on admission and rapidly progresses to end-stage renal disease (ESRD) 221, 222, 223, 224, 225.
Signs and symptoms of nephrotic syndrome include:
- Severe swelling (edema), particularly around your eyes and in your ankles and feet
- Foamy urine, a result of excess protein in your urine
- Weight gain due to fluid retention
- Fatigue
- Loss of appetite
Collapsing glomerulopathy complications
Possible complications of nephrotic syndrome include:
- Blood clots. The inability of the glomeruli to filter blood properly can lead to loss of blood proteins that help prevent clotting. This increases your risk of developing a blood clot in your veins.
- High blood cholesterol and elevated blood triglycerides. When the level of the protein albumin in your blood falls, your liver makes more albumin. At the same time, your liver releases more cholesterol and triglycerides.
- Poor nutrition. Loss of too much blood protein can result in malnutrition. This can lead to weight loss, which can be masked by edema. You may also have too few red blood cells (anemia), low blood protein levels and low levels of vitamin D.
- High blood pressure. Damage to your glomeruli and the resulting buildup of excess body fluid can raise your blood pressure.
- Acute kidney injury. If your kidneys lose their ability to filter blood due to damage to the glomeruli, waste products can build up quickly in your blood. If this happens, you might need emergency dialysis — an artificial means of removing extra fluids and waste from your blood — typically with an artificial kidney machine (dialyzer).
- Chronic kidney disease. Nephrotic syndrome can cause your kidneys to lose their function over time. If kidney function falls low enough, you might need dialysis or a kidney transplant.
- Infections. People with nephrotic syndrome have an increased risk of infections.
Collapsing glomerulopathy diagnosis
Health care professionals diagnose glomerular disease by ordering tests, such as:
Blood tests:
- Blood tests can measure the levels of products in your blood, such as creatinine, urea nitrogen, and a protein called cystatin C, to find out how well your kidneys are working. Blood tests can also check for low levels of a protein in your blood, called albumin, which can happen when too much of that protein passes from your blood into your urine.
- Other blood test include:
- Antiglomerular basement membrane antibody test
- Antineutrophil cytoplasmic antibodies (ANCAs)
- Antinuclear antibodies
- Complement levels
A simple test of your urine can confirm if there is blood or protein in your urine.
Urinalysis, which examines a sample of your urine to find out if levels of protein and red blood cells are too high:
- Creatinine clearance
- Examination of the urine under a microscope
- Urine total protein
- Uric acid in the urine
- Urine concentration test
- Urine creatinine
- Urine protein
- Urine red blood cell
- Urine specific gravity
- Urine osmolality
Imaging tests that may be done include:
- Abdominal CT scan
- Kidney ultrasound
- Chest x-ray
- Intravenous pyelogram (IVP)
In some cases, a test called a kidney biopsy may be needed. In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope. A kidney biopsy can confirm you have glomerular disease and help find the cause in order to help your doctor plan the best treatment for you.
Treatment for glomerular disease varies by symptoms, causes, and how badly your kidneys are damaged. In some cases, glomerular disease may go away once its cause has been treated. In other cases, the disease may go away but later return. Less often, glomerular disease may not respond to treatment and lead to kidney failure over time.
Collapsing glomerulopathy histology
Although initially treated as a single entity, focal segmental glomerulosclerosis (FSGS) encompasses several distinct histological patterns. FSGS is heterogenous in terms of etiology, histological characteristics, clinical presentation, treatment response, and prognosis. For this reason, a classification of FSGS histological subtypes was proposed in 2004 27. The diagnosis of collapsing glomerulopathy requires the presence of at least one glomerulus with segmental or global collapse and podocyte hypertrophy and hyperplasia (Figure 8). Collapsing glomerulopathy can occur with other morphological subtypes, and a single characteristic lesion is adequate for a diagnosis of collapsing glomerulopathy.
Light microscope shoes partial or total obliteration of the lumen of the glomerular capillaries secondary to podocyte hypertrophy and hyperplasia 267, 268. Podocyte crowns may contain intracytoplasmic deposits, which denote protein reabsorption. Pseudocrescents, which resemble glomerular crescents, may temporally precede glomerular sclerosis 224. Other findings, such as glomerulomegaly, hyalinosis, hypercellularity, and adhesions, are unusual but more common in the final stages of the disease 269.
As a rule, tubulointerstitial damage in collapsing glomerulopathy is more intense than in other FSGS subtypes 222. Important specific findings include tubular dilatation with molding and the formation of tubular microcysts 222, 246. Tissue and lymphatic macrophages, specifically CD4 and CD8, can also be found in interstitial infiltrates 246.
Immunofluorescence (IF) findings are non-specific and, in many cases, negative. If present, they usually comprise granular or mesangial IgM and C3 deposits. IF may suggest associated conditions such as Berger’s disease, which is characterized by mesangial IgA deposits 268, or SLE, which is associated with several antibody deposits.
Electron microscopy (EM) is mostly unnecessary; however, it can demonstrate collapsed and ruptured capillary membranes and swollen, hypertrophic, and/or hyperplastic podocytes 268. Electron microscopy may also identify intracellular inclusions that are often associated with HIV or SLE. In these conditions, the podocyte processes are greatly compromised with loss of glomerular filtration barrier integrity.
Collapsing glomerulopathy treatment
The treatment of collapsing glomerulopathy consists of the following:
- Targeted therapy for disorders resulting from nephrotic syndrome, such as dyslipidemia, hypertension, and edema;
- Treatment of the underlying disease when collapsing glomerulopathy is associated with other conditions; and
- Immunosuppressive therapy.
Symptoms of nephrotic syndrome are most often treated with these medicines 270, 271:
- Blood pressure medications. Drugs called angiotensin-converting enzyme (ACE) inhibitors reduce blood pressure and the amount of protein released in urine. Medications in this category include lisinopril (Prinivil, Qbrelis, Zestril), benazepril (Lotensin), captopril and enalapril (Vasotec). Another group of drugs that works similarly is called angiotensin 2 receptor blockers (ARBs) and includes losartan (Cozaar) and valsartan (Diovan). Other medications, such as renin inhibitors, also might be used, though angiotensin-converting enzyme (ACE) inhibitors and angiotensin 2 receptor blockers (ARBs) are generally used first.
- Water pills (diuretics). These help control swelling by increasing your kidneys’ fluid output. Diuretic medications typically include furosemide (Lasix). Others include spironolactone (Aldactone, Carospir) and thiazides, such as hydrochlorothiazide or metolazone (Zaroxolyn).
- Cholesterol-reducing medications. Statins can help lower cholesterol levels. However, it’s not clear whether cholesterol-lowering medications can improve the outcomes for people with nephrotic syndrome, such as avoiding heart attacks or decreasing the risk of early death. Statins include atorvastatin (Lipitor), fluvastatin (Lescol XL), lovastatin (Altoprev), pravastatin (Pravachol), rosuvastatin (Crestor, Ezallor) and simvastatin (Zocor).
- Blood thinners (anticoagulants). These might be prescribed to decrease your blood’s ability to clot, especially if you’ve had a blood clot. Anticoagulants include heparin, warfarin (Coumadin, Jantoven), dabigatran (Pradaxa), apixaban (Eliquis) and rivaroxaban (Xarelto).
- Immune system-suppressing medications. Medications to control the immune system, such as corticosteroids, can decrease the inflammation that accompanies some of the conditions that can cause nephrotic syndrome. Medications include rituximab (Rituxan), cyclosporine and cyclophosphamide.
According to the Kidney Disease Improving Global Outcomes (KDIGO) guidelines on the management of glomerulopathies 272, 273, the measures to control complications associated with nephrotic syndrome consist of edema control through a low-sodium diet (<2 g/day), fluid restriction, diuretic therapy (water pill), and, if necessary, hemodialysis or ultrafiltration. Systolic blood pressure should be maintained at <120 mmHg with angiotensin-converting enzyme inhibitors or angiotensin 2 receptor blockers, unless renal function worsens on these medications 270.
The severity of dyslipidemia in patients with nephrotic syndrome is proportional to the degree of proteinuria. Once the proteinuria in patients with collapsing glomerulopathy is very high, the serum levels of cholesterol and its fractions as well as lipoproteins can reach high elevated values. The potential contributors to dyslipidemia in patients with nephrotic syndrome are the patient’s diet, use of drugs (such as corticosteroids, calcineurin inhibitors, and mTOR inhibitors), in addition to genetic predisposition 274, 275. According to the new KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases 270 in patients with nephrotic syndrome the dyslipidemia should be controlled, especially in those ones with other cardiovascular risk factors (e.g., hypertension and diabetes) and/or diseases that respond poorly to stabilized therapy.
As non-pharmacological measures, lifestyle changes (diet, smoking cessation, weight loss, and physical activity) must be encouraged in all patients. The tratment with statins as a first-line treatment should be indicated in patients at increased risk of atherosclerotic cardiovascular disease, such as those with GFR <60 ml/min/1.73m² and/or albuminuria >30 mg/g, in addition to treating infectious/inflammatory diseases that contribute to increased cardiovascular risk. For patients who do not tolerate the use of statins or cannot achieve the recommended lipid levels targets, other drugs as bile acid chelators, fibrates, nicotinic acid, ezetimibe, PCSK9 inhibitors and lipid apheresis can be used.
Special attention should also be given to the risk of thrombotic events; prophylactic anticoagulation should be considered for susceptible populations. Immunocompromised patients should be given full immunizations; tested for HIV, hepatisis B (HBV), hepatisis C (HCV), tuberculosis (TB), and syphilis; and initiated on strongyloidiasis and pneumocystis prophylaxis.
When an underlying cause is identified, such as drug-related, infectious, genetic, autoimmune, and other causes, the patient should undergo specific therapy for the underlying cause, when available. In drug-induced collapsing glomerulopathy, the offending drug must be discontinued whenever possible 276. In cases associated with infections, specific antimicrobial treatments can reduce collapsing glomerulopathy progression or even promote remission. Some HIV-associated cases demonstrate 38% delay in the progression to ESRD following the initiation of antiretroviral therapy (ART) 277; however, other studies proposed that up to 50% of patients progress toward the initiation of kidney dialysis despite adequate treatment 278. Nevertheless, HIV-associated nephropathy should always be treated with ART because it is very effective at controlling the disease 279.
Intravenous immunoglobulin (IVIg) and cidofovir can be used to treat parvovirus B19 infection, which is particularly important from the perspective of renal transplantation 280. CMV infections should be treated with ganciclovir, which also promotes collapsing glomerulopathy remission and renal function improvement 230.
Immunosuppressant therapy was administered to most patients. While there are no specific recommendations for collapsing glomerulopathy, the immunotherapy guidelines for collapsing glomerulopathy are extrapolated from the FSGS protocol 223. Several types of immunosuppressants have been proposed 281, 222, 228, 223, but the initial therapeutic regimen consists of high-dose of oral corticosteroids for 4–16 weeks or until remission is achieved. Calcineurin inhibitors or cyclophosphamide are considered second-line medications for patients that exhibit corticosteroid resistance, corticosteroid dependence, or frequent relapses; they should be given for at least 12 weeks. Other immunosuppressants (such as rituximab and mycophenolate mofetil) are indicated for patients who do not respond to other immunosuppressive regimens. Patients with extensive kidney damage may choose to forego treatment for collapsing glomerulopathy because of the lack of benefits and risks associated with immunosuppression therapy.
Collapsing glomerulopathy self care
If you have developed nephrotic syndrome, your health care professional may recommend that you change your diet. Your doctor might refer you to a dietitian, who might recommend that you do the following:
- Choose lean sources of protein. Plant-based protein is helpful in kidney disease.
- Reduce the amount of fat and cholesterol in your diet to help control your blood cholesterol levels.
- Eat a low-salt diet to help control swelling.
- Reduce the amount of liquid in your diet.
Collapsing glomerulopathy prognosis
Collapsing glomerulopathy has a poor prognosis 211. Most collapsing glomerulopathy cases present with refractory proteinuria, severe loss of renal function, and progression to permanent kidney dialysis 282, 267, 268. While other FSGS subtypes have a kidney survival time of 62.5 months, collapsing glomerulopathy has a kidney survival time of 13 months 222.
- Kazi AM, Hashmi MF. Glomerulonephritis. [Updated 2023 Jun 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560644[↩]
- Stahl RA, Hoxha E. Glomerulonephritis [Glomerulonephritis]. Dtsch Med Wochenschr. 2016 Jul;141(13):960-8. German. doi: 10.1055/s-0042-107410[↩]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2020. US Department of Health and Human Services; 2020. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf[↩]
- O’Shaughnessy MM, Hogan SL, Poulton CJ, et al. Temporal and demographic trends in glomerular disease epidemiology in the southeastern United States, 1986–2015. Clinical Journal of the American Society of Nephrology. 2017;12(4):614–623. doi: 10.2215/CJN.10871016[↩]
- O’Shaughnessy MM, Hogan SL, Thompson BD, Coppo R, Fogo AB, Jennette JC. Glomerular disease frequencies by race, sex, and region: results from the International Kidney Biopsy Survey. Nephrology, Dialysis, Transplantation. 2018;33(4):661–669. doi: 10.1093/ndt/gfx189[↩]
- Mariani LH, Bomback AS, Canetta PA, et al. CureGN Study rationale, design, and methods: establishing a large prospective observational study of glomerular disease. American Journal of Kidney Diseases. 2019;73(2):218–229. doi: 10.1053/j.ajkd.2018.07.020[↩]
- Glomerular Disease. https://www.niddk.nih.gov/health-information/kidney-disease/glomerular-disease[↩]
- Rosenberg AZ, Kopp JB. Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2017 Mar 7. 12 (3):502-517.[↩]
- Focal Segmental Glomerulosclerosis (FSGS). https://www.kidney.org/atoz/content/focal[↩]
- Guruswamy Sangameswaran KD, Hashmi MF, Baradhi KM. Focal Segmental Glomerulosclerosis. [Updated 2023 Feb 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532272[↩][↩][↩][↩][↩][↩][↩]
- Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007 Jun;71(12):1205-14.[↩]
- McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol. Dial. Transplant. 2011 Feb;26(2):414-30.[↩]
- Hogan JJ. A Case of Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2021 Aug;16(8):1272-1274. doi: 10.2215/CJN.19591220[↩][↩]
- D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am. J. Kidney Dis. 2004 Feb;43(2):368-82[↩][↩]
- Königshausen E, Sellin L. Circulating Permeability Factors in Primary Focal Segmental Glomerulosclerosis: A Review of Proposed Candidates. Biomed Res Int. 2016;2016:3765608.[↩][↩]
- Petersen CE, Amaral S, Frosch E. Lithium-induced nephrotic syndrome in a prepubertal boy. J Child Adolesc Psychopharmacol. 2008 Apr;18(2):210-3[↩]
- Chandra P, Kopp JB. Viruses and collapsing glomerulopathy: a brief critical review. Clin Kidney J. 2013 Feb;6(1):1-5.[↩]
- Kriz W, Lemley KV. Mechanical challenges to the glomerular filtration barrier: adaptations and pathway to sclerosis. Pediatr. Nephrol. 2017 Mar;32(3):405-417[↩][↩]
- Yu H, Artomov M, Brähler S, Stander MC, Shamsan G, Sampson MG, White JM, Kretzler M, Miner JH, Jain S, Winkler CA, Mitra RD, Kopp JB, Daly MJ, Shaw AS. A role for genetic susceptibility in sporadic focal segmental glomerulosclerosis. J. Clin. Invest. 2016 Apr 01;126(4):1603[↩]
- Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol. 2015 Feb;26(2):258-69. doi: 10.1681/ASN.2014030278[↩]
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998 Sep;54(3):687-97. doi: 10.1046/j.1523-1755.1998.00044.x[↩]
- Barisoni L, Schnaper HW, Kopp JB. Advances in the biology and genetics of the podocytopathies: implications for diagnosis and therapy. Arch Pathol Lab Med. 2009 Feb;133(2):201-16. doi: 10.5858/133.2.201[↩]
- Shabaka A, Tato Ribera A, Fernández-Juárez G. Focal Segmental Glomerulosclerosis: State-of-the-Art and Clinical Perspective. Nephron. 2020;144(9):413-427. doi: 10.1159/000508099[↩]
- Shankland SJ, Pollak MR. A suPAR circulating factor causes kidney disease. Nat Med. 2011 Aug 4;17(8):926-7. doi: 10.1038/nm.2443[↩]
- Gribouval O, Boyer O, Knebelmann B, Karras A, Dantal J, Fourrage C, Alibeu O, Hogan J, Dossier C, Tête MJ, Antignac C, Servais A. APOL1 risk genotype in European steroid-resistant nephrotic syndrome and/or focal segmental glomerulosclerosis patients of different African ancestries. Nephrol Dial Transplant. 2019 Nov 1;34(11):1885-1893. doi: 10.1093/ndt/gfy176[↩]
- McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010 Nov;5(11):2115-21. doi: 10.2215/CJN.03800609[↩]
- D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004 Feb;43(2):368-82. doi: 10.1053/j.ajkd.2003.10.024[↩][↩]
- Stokes MB, D’Agati VD. Morphologic variants of focal segmental glomerulosclerosis and their significance. Adv Chronic Kidney Dis. 2014 Sep;21(5):400-7. doi: 10.1053/j.ackd.2014.02.010[↩]
- Thomas DB, Franceschini N, Hogan SL, Ten Holder S, Jennette CE, Falk RJ, Jennette JC. Clinical and pathologic characteristics of focal segmental glomerulosclerosis pathologic variants. Kidney Int. 2006 Mar;69(5):920-6. doi: 10.1038/sj.ki.5000160[↩]
- Stokes MB, Markowitz GS, Lin J, Valeri AM, D’Agati VD. Glomerular tip lesion: a distinct entity within the minimal change disease/focal segmental glomerulosclerosis spectrum. Kidney Int. 2004 May;65(5):1690-702. doi: 10.1111/j.1523-1755.2004.00563.x[↩]
- Stokes MB, Markowitz GS, Lin J, Valeri AM, D’Agati VD. Glomerular tip lesion: a distinct entity within the minimal change disease/focal segmental glomerulosclerosis spectrum. Kidney Int. 2004 May;65(5):1690-702[↩]
- Moudgil A, Nast CC, Bagga A, Wei L, Nurmamet A, Cohen AH, Jordan SC, Toyoda M. Association of parvovirus B19 infection with idiopathic collapsing glomerulopathy. Kidney Int. 2001 Jun;59(6):2126-33. doi: 10.1046/j.1523-1755.2001.00727.x[↩]
- Tomlinson L, Boriskin Y, McPhee I, Holwill S, Rice P. Acute cytomegalovirus infection complicated by collapsing glomerulopathy. Nephrol Dial Transplant. 2003 Jan;18(1):187-9. doi: 10.1093/ndt/18.1.187[↩]
- Markowitz GS, Nasr SH, Stokes MB, D’Agati VD. Treatment with IFN-{alpha}, -{beta}, or -{gamma} is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010 Apr;5(4):607-15. doi: 10.2215/CJN.07311009. Epub 2010 Mar 4. Erratum in: Clin J Am Soc Nephrol. 2010 Jul;5(7):1353.[↩]
- Baradhi KM, Gary Abuelo J, Stillman IE. The Case: diabetic nephropathy in a nondiabetic smoker? Kidney Int. 2012 Nov;82(10):1141-2. doi: 10.1038/ki.2012.333[↩]
- Freedman BI, Hicks PJ, Bostrom MA, Cunningham ME, Liu Y, Divers J, Kopp JB, Winkler CA, Nelson GW, Langefeld CD, Bowden DW. Polymorphisms in the non-muscle myosin heavy chain 9 gene (MYH9) are strongly associated with end-stage renal disease historically attributed to hypertension in African Americans. Kidney Int. 2009 Apr;75(7):736-45. doi: 10.1038/ki.2008.701[↩]
- Chen YM, Liapis H. Focal segmental glomerulosclerosis: molecular genetics and targeted therapies. BMC Nephrol. 2015 Jul 9;16:101. doi: 10.1186/s12882-015-0090-9[↩]
- Winston JA, Burns GC, Klotman PE. The human immunodeficiency virus (HIV) epidemic and HIV-associated nephropathy. Semin Nephrol. 1998 Jul;18(4):373-7.[↩]
- Rüster C, Wolf G. The role of the renin-angiotensin-aldosterone system in obesity-related renal diseases. Semin Nephrol. 2013 Jan;33(1):44-53. doi: 10.1016/j.semnephrol.2012.12.002[↩]
- Nagai R, Cattran DC, Pei Y. Steroid therapy and prognosis of focal segmental glomerulosclerosis in the elderly. Clin Nephrol. 1994 Jul;42(1):18-21.[↩]
- Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC; Toronto Glomerulonephritis Registry Group. Focal and segmental glomerulosclerosis: definition and relevance of a partial remission. J Am Soc Nephrol. 2005 Apr;16(4):1061-8. doi: 10.1681/ASN.2004070593[↩][↩]
- Costello R, Patel R, Humphreys J, McBeth J, Dixon WG. Patient perceptions of glucocorticoid side effects: a cross-sectional survey of users in an online health community. BMJ Open. 2017 Apr 3;7(4):e014603. doi: 10.1136/bmjopen-2016-014603[↩]
- Banfi G, Moriggi M, Sabadini E, Fellin G, D’Amico G, Ponticelli C. The impact of prolonged immunosuppression on the outcome of idiopathic focal-segmental glomerulosclerosis with nephrotic syndrome in adults. A collaborative retrospective study. Clin Nephrol. 1991 Aug;36(2):53-9.[↩]
- Cattran DC, Appel GB, Hebert LA, Hunsicker LG, Pohl MA, Hoy WE, Maxwell DR, Kunis CL. A randomized trial of cyclosporine in patients with steroid-resistant focal segmental glomerulosclerosis. North America Nephrotic Syndrome Study Group. Kidney Int. 1999 Dec;56(6):2220-6. doi: 10.1046/j.1523-1755.1999.00778.x[↩]
- Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, Burke GW 3rd. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. 2011 Jun 1;3(85):85ra46. doi: 10.1126/scitranslmed.3002231[↩]
- Ramachandran R, Kumar V, Rathi M, Nada R, Jha V, Gupta KL, Sakhuja V, Kohli HS. Tacrolimus therapy in adult-onset steroid-resistant nephrotic syndrome due to a focal segmental glomerulosclerosis single-center experience. Nephrol Dial Transplant. 2014 Oct;29(10):1918-24. doi: 10.1093/ndt/gfu097[↩]
- Canetta PA, Radhakrishnan J. Impact of the National Institutes of Health Focal Segmental Glomerulosclerosis (NIH FSGS) clinical trial on the treatment of steroid-resistant FSGS. Nephrol Dial Transplant. 2013 Mar;28(3):527-34. doi: 10.1093/ndt/gfs563[↩]
- Chun MJ, Korbet SM, Schwartz MM, Lewis EJ. Focal segmental glomerulosclerosis in nephrotic adults: presentation, prognosis, and response to therapy of the histologic variants. J Am Soc Nephrol. 2004 Aug;15(8):2169-77. doi: 10.1097/01.ASN.0000135051.62500.97[↩]
- Alok A, Yadav A. Membranous Nephropathy. [Updated 2023 Jun 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559169[↩][↩][↩][↩]
- Fervenza FC, Sethi S, Specks U. Idiopathic membranous nephropathy: diagnosis and treatment. Clin J Am Soc Nephrol. 2008 May;3(3):905-19. doi: 10.2215/CJN.04321007[↩][↩][↩][↩][↩]
- Keri KC, Blumenthal S, Kulkarni V, Beck L, Chongkrairatanakul T. Primary membranous nephropathy: comprehensive review and historical perspective. Postgrad Med J. 2019 Jan;95(1119):23-31. doi: 10.1136/postgradmedj-2018-135729[↩]
- Cattran DC. Idiopathic membranous glomerulonephritis. Kidney Int. 2001 May;59(5):1983-94. doi: 10.1046/j.1523-1755.2001.0590051983.x[↩]
- Menon S, Valentini RP. Membranous nephropathy in children: clinical presentation and therapeutic approach. Pediatr Nephrol. 2010 Aug;25(8):1419-28. doi: 10.1007/s00467-009-1324-5[↩][↩][↩]
- von Groote TC, Williams G, Au EH, Chen Y, Mathew AT, Hodson EM, Tunnicliffe DJ. Immunosuppressive treatment for primary membranous nephropathy in adults with nephrotic syndrome. Cochrane Database Syst Rev. 2021 Nov 15;11(11):CD004293. doi: 10.1002/14651858.CD004293.pub4[↩][↩][↩]
- Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009 Jul 2;361(1):11-21. doi: 10.1056/NEJMoa0810457[↩][↩][↩][↩]
- Membranous Nephropathy. https://unckidneycenter.org/kidneyhealthlibrary/glomerular-disease/membranous-nephropathy[↩][↩][↩]
- Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, Dolla G, Hoxha E, Helmchen U, Dabert-Gay AS, Debayle D, Merchant M, Klein J, Salant DJ, Stahl RAK, Lambeau G. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med. 2014 Dec 11;371(24):2277-2287. doi: 10.1056/NEJMoa1409354[↩][↩][↩]
- Ramachandran R, Kaundal U, Girimaji N, Rakha A, Rathi M, Gupta KL, Kohli HS, Jha V. Regulatory B Cells Are Reduced and Correlate With Disease Activity in Primary Membranous Nephropathy. Kidney Int Rep. 2020 Mar 30;5(6):872-878. doi: 10.1016/j.ekir.2020.03.023[↩]
- Abe S, Amagasaki Y, Konishi K, Kato E, Iyori S, Sakaguchi H. Idiopathic membranous glomerulonephritis: aspects of geographical differences. J Clin Pathol. 1986 Nov;39(11):1193-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1140761/pdf/jclinpath00319-0029.pdf[↩]
- Ronco P, Debiec H. Molecular Pathogenesis of Membranous Nephropathy. Annu Rev Pathol. 2020 Jan 24;15:287-313. doi: 10.1146/annurev-pathol-020117-043811[↩]
- Couser WG. Primary Membranous Nephropathy. Clin J Am Soc Nephrol. 2017 Jun 7;12(6):983-997. doi: 10.2215/CJN.11761116. Epub 2017 May 26. Erratum in: Clin J Am Soc Nephrol. 2017 Sep 7;12 (9):1528.[↩]
- Sethi S, Debiec H, Madden B, Charlesworth MC, Morelle J, Gross L, Ravindran A, Buob D, Jadoul M, Fervenza FC, Ronco P. Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int. 2020 Jan;97(1):163-174. doi: 10.1016/j.kint.2019.09.014[↩]
- Lai WL, Yeh TH, Chen PM, Chan CK, Chiang WC, Chen YM, Wu KD, Tsai TJ. Membranous nephropathy: a review on the pathogenesis, diagnosis, and treatment. J Formos Med Assoc. 2015 Feb;114(2):102-11. doi: 10.1016/j.jfma.2014.11.002[↩][↩][↩]
- Bhimma R, Coovadia HM. Hepatitis B virus-associated nephropathy. Am J Nephrol. 2004 Mar-Apr;24(2):198-211. doi: 10.1159/000077065[↩]
- Lai FM, To KF, Wang AY, Choi PC, Szeto CC, Li PK, Leung CB, Lai KN. Hepatitis B virus-related nephropathy and lupus nephritis: morphologic similarities of two clinical entities. Mod Pathol. 2000 Feb;13(2):166-72. doi: 10.1038/modpathol.3880031[↩]
- HEYMANN W, HACKEL DB, HARWOOD S, WILSON SG, HUNTER JL. Production of nephrotic syndrome in rats by Freund’s adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med. 1959 Apr;100(4):660-4. doi: 10.3181/00379727-100-24736[↩]
- Tramontano A, Knight T, Vizzuso D, Makker SP. Nested N-terminal megalin fragments induce high-titer autoantibody and attenuated Heymann nephritis. J Am Soc Nephrol. 2006 Jul;17(7):1979-85. doi: 10.1681/ASN.2005101144[↩]
- Bullich G, Ballarín J, Oliver A, Ayasreh N, Silva I, Santín S, Díaz-Encarnación MM, Torra R, Ars E. HLA-DQA1 and PLA2R1 polymorphisms and risk of idiopathic membranous nephropathy. Clin J Am Soc Nephrol. 2014 Feb;9(2):335-43. doi: 10.2215/CJN.05310513[↩]
- Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, Voinescu C, Patel N, Pearce K, Hubank M, Stephens HA, Laundy V, Padmanabhan S, Zawadzka A, Hofstra JM, Coenen MJ, den Heijer M, Kiemeney LA, Bacq-Daian D, Stengel B, Powis SH, Brenchley P, Feehally J, Rees AJ, Debiec H, Wetzels JF, Ronco P, Mathieson PW, Kleta R. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med. 2011 Feb 17;364(7):616-26. doi: 10.1056/NEJMoa1009742[↩]
- Akankwasa G, Jianhua L, Guixue C, Changjuan A, Xiaosong Q. Urine markers of podocyte dysfunction: a review of podocalyxin and nephrin in selected glomerular diseases. Biomark Med. 2018 Aug;12(8):927-935. doi: 10.2217/bmm-2018-0152[↩]
- Lim WH, Shingde M, Wong G. Recurrent and de novo Glomerulonephritis After Kidney Transplantation. Front Immunol. 2019 Aug 14;10:1944. doi: 10.3389/fimmu.2019.01944[↩]
- Zhang J, Pan M, Zhang J, You X, Li D, Lin F, Lu G. Serum uric acid is an independent predictor of renal outcomes in patients with idiopathic membranous nephropathy. Int Urol Nephrol. 2019 Oct;51(10):1797-1804. doi: 10.1007/s11255-019-02254-7[↩]
- Hou J, Cheng Y, Hou Y, Wu H. Lower Serum and Higher Urine Immunoglobulin G Are Associated with an Increased Severity of Idiopathic Membranous Nephropathy. Ann Clin Lab Sci. 2019 Nov;49(6):777-784[↩]
- Alawieh R, Brodsky SV, Satoskar AA, Nadasdy T, Parikh SV, Rovin B, Cassol CA. Membranous Nephropathy With Crescents. Kidney Int Rep. 2020 Jan 30;5(4):537-541. doi: 10.1016/j.ekir.2020.01.010[↩]
- Zhang XD, Cui Z, Zhang MF, Wang J, Zhang YM, Qu Z, Wang X, Huang J, Wang F, Meng LQ, Cheng XY, Wang SX, Liu G, Zhao MH. Clinical implications of pathological features of primary membranous nephropathy. BMC Nephrol. 2018 Aug 28;19(1):215. doi: 10.1186/s12882-018-1011-5[↩][↩]
- Sato S, Kitamura H, Ghazizadeh M, Adachi A, Sasaki Y, Ishizaki M, Inoue K, Wakamatsu K, Sugisaki Y. Occurrence of hyaline droplets in renal biopsy specimens: an ultrastructural study. Med Mol Morphol. 2005 Mar;38(1):63-71. doi: 10.1007/s00795-004-0272-1[↩]
- Mirkovic K, Frenay AS, van den Born J, van Goor H, Navis G, de Borst MH; NIGRAM consortium. Sodium restriction potentiates the renoprotective effects of combined vitamin D receptor activation and angiotensin-converting enzyme inhibition in established proteinuric nephropathy. Nephrol Dial Transplant. 2017 Aug 1;32(8):1293-1301. doi: 10.1093/ndt/gfv304[↩]
- Bomback AS, Fervenza FC. Membranous Nephropathy: Approaches to Treatment. Am J Nephrol. 2018;47 Suppl 1:30-42. doi: 10.1159/000481635[↩]
- Cattran D, Brenchley P. Membranous nephropathy: thinking through the therapeutic options. Nephrol Dial Transplant. 2017 Jan 1;32(suppl_1):i22-i29. doi: 10.1093/ndt/gfw404[↩]
- Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, Jefferson JA, Gipson PE, Rizk DV, Sedor JR, Simon JF, McCarthy ET, Brenchley P, Sethi S, Avila-Casado C, Beanlands H, Lieske JC, Philibert D, Li T, Thomas LF, Green DF, Juncos LA, Beara-Lasic L, Blumenthal SS, Sussman AN, Erickson SB, Hladunewich M, Canetta PA, Hebert LA, Leung N, Radhakrishnan J, Reich HN, Parikh SV, Gipson DS, Lee DK, da Costa BR, Jüni P, Cattran DC; MENTOR Investigators. Rituximab or Cyclosporine in the Treatment of Membranous Nephropathy. N Engl J Med. 2019 Jul 4;381(1):36-46. doi: 10.1056/NEJMoa1814427[↩][↩]
- Ponticelli C, Altieri P, Scolari F, Passerini P, Roccatello D, Cesana B, Melis P, Valzorio B, Sasdelli M, Pasquali S, Pozzi C, Piccoli G, Lupo A, Segagni S, Antonucci F, Dugo M, Minari M, Scalia A, Pedrini L, Pisano G, Grassi C, Farina M, Bellazzi R. A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol. 1998 Mar;9(3):444-50. doi: 10.1681/ASN.V93444[↩]
- Bomback AS, Derebail VK, McGregor JG, Kshirsagar AV, Falk RJ, Nachman PH. Rituximab therapy for membranous nephropathy: a systematic review. Clin J Am Soc Nephrol. 2009 Apr;4(4):734-44. doi: 10.2215/CJN.05231008[↩]
- Goździk M, Płuciennik A, Zawiasa-Bryszewska A, Nowicka M, Nowicka Z, Wągrowska-Danilewicz M, Kurnatowska I. Acute Kidney Injury Following Exposure to Calcineurin Inhibitors in a Patient with Idiopathic Membranous Nephropathy. Drug Saf Case Rep. 2019 Oct 5;6(1):9. doi: 10.1007/s40800-019-0103-x[↩]
- Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC; Toronto Glomerulonephritis Registry Group. Idiopathic membranous nephropathy: definition and relevance of a partial remission. Kidney Int. 2004 Sep;66(3):1199-205. doi: 10.1111/j.1523-1755.2004.00873.x[↩]
- Lu W, Gong S, Li J, Wang Y. Clinicopathological features and prognosis in patients with idiopathic membranous nephropathy with hypertension. Exp Ther Med. 2020 Apr;19(4):2615-2621. doi: 10.3892/etm.2020.8506[↩]
- Polanco N, Gutiérrez E, Covarsí A, Ariza F, Carreño A, Vigil A, Baltar J, Fernández-Fresnedo G, Martín C, Pons S, Lorenzo D, Bernis C, Arrizabalaga P, Fernández-Juárez G, Barrio V, Sierra M, Castellanos I, Espinosa M, Rivera F, Oliet A, Fernández-Vega F, Praga M; Grupo de Estudio de las Enfermedades Glomerulares de la Sociedad Española de Nefrología. Spontaneous remission of nephrotic syndrome in idiopathic membranous nephropathy. J Am Soc Nephrol. 2010 Apr;21(4):697-704. doi: 10.1681/ASN.2009080861[↩]
- Deegens JK, Wetzels JF. Diagnosis and treatment of primary glomerular diseases. Membranous nephropathy, focal segmental glomerulosclerosis and IgA nephropathy. Minerva Urol Nefrol. 2005 Sep;57(3):211-36.[↩]
- Ponticelli C, Passerini P. Management of idiopathic membranous nephropathy. Expert Opin Pharmacother. 2010 Sep;11(13):2163-75. doi: 10.1517/14656566.2010.494599[↩]
- Waldman M, Austin HA 3rd. Controversies in the treatment of idiopathic membranous nephropathy. Nat Rev Nephrol. 2009 Aug;5(8):469-79. doi: 10.1038/nrneph.2009.101[↩]
- Membranous Nephropathy. https://www.msdmanuals.com/professional/genitourinary-disorders/glomerular-disorders/membranous-nephropathy[↩]
- Teh YM, Lim SK, Jusoh N, Osman K, Mualif SA. CD80 Insights as Therapeutic Target in the Current and Future Treatment Options of Frequent-Relapse Minimal Change Disease. Biomed Res Int. 2021 Jan 6;2021:6671552. doi: 10.1155/2021/6671552[↩][↩][↩]
- Zamora G, Pearson-Shaver AL. Minimal Change Disease. [Updated 2023 Jul 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560639[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal Change Disease. Clin J Am Soc Nephrol. 2017 Feb 7;12(2):332-345. doi: 10.2215/CJN.05000516[↩][↩][↩][↩][↩][↩][↩]
- Nephrotic Syndrome. https://www.merckmanuals.com/home/kidney-and-urinary-tract-disorders/kidney-filtering-disorders/nephrotic-syndrome[↩]
- Waldman M, Crew RJ, Valeri A, Busch J, Stokes B, Markowitz G, D’Agati V, Appel G. Adult minimal-change disease: clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol. 2007 May;2(3):445-53. doi: 10.2215/CJN.03531006[↩][↩][↩]
- Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003 Aug 23;362(9384):629-39. doi: 10.1016/S0140-6736(03)14184-0[↩][↩]
- Hogg RJ, Portman RJ, Milliner D, Lemley KV, Eddy A, Ingelfinger J. Evaluation and management of proteinuria and nephrotic syndrome in children: recommendations from a pediatric nephrology panel established at the National Kidney Foundation conference on proteinuria, albuminuria, risk, assessment, detection, and elimination (PARADE). Pediatrics. 2000 Jun;105(6):1242-9. doi: 10.1542/peds.105.6.1242[↩]
- Gdadegesin R, Smoyer WE: Nephrotic syndrome. In: Comprehensive Pediatric Nephrology, edited by Geary DF, Scaefer F, Philadelphia, Mosby Elsevier, 2008, pp 205–218.[↩]
- Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein SL: Idiopathic Nephrotic Syndrome in Children: Clinical Aspects. Pediatric Nephrology, 7th Ed., Berlin, Springer, 2016, pp 2730[↩]
- Mathieson PW. Immune dysregulation in minimal change nephropathy. Nephrol Dial Transplant. 2003 Aug;18 Suppl 6:vi26-9. doi: 10.1093/ndt/gfg1066[↩][↩]
- Elie V, Fakhoury M, Deschênes G, Jacqz-Aigrain E. Physiopathology of idiopathic nephrotic syndrome: lessons from glucocorticoids and epigenetic perspectives. Pediatr Nephrol. 2012 Aug;27(8):1249-56. doi: 10.1007/s00467-011-1947-1[↩][↩]
- Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell function. Lancet. 1974 Sep 7;2(7880):556-60. doi: 10.1016/s0140-6736(74)91880-7[↩][↩]
- Minimal-Change Disease. https://emedicine.medscape.com/article/243348-overview[↩][↩][↩]
- Tarshish P, Tobin JN, Bernstein J, Edelmann CM Jr. Prognostic significance of the early course of minimal change nephrotic syndrome: report of the International Study of Kidney Disease in Children. J Am Soc Nephrol. 1997 May;8(5):769-76. doi: 10.1681/ASN.V85769[↩][↩][↩]
- Nachman PH, Jennette JC, Falk RJ: Primary glomerular disease. In: The Kidney, 8th Ed., edited by Brenner BM, Philadelphia, Saunders Elsevier, 2008, pp 987–1066.[↩]
- Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein SL: Idiopathic Nephrotic Syndrome in Children: Clinical Aspects. Pediatric Nephrology, 7th Ed., Berlin, Springer, 2016, pp 2730.[↩]
- Downie ML, Gallibois C, Parekh RS, Noone DG. Nephrotic syndrome in infants and children: pathophysiology and management. Paediatr Int Child Health. 2017 Nov;37(4):248-258. doi: 10.1080/20469047.2017.1374003[↩][↩][↩]
- Bertelli R, Bonanni A, Caridi G, Canepa A, Ghiggeri GM. Molecular and Cellular Mechanisms for Proteinuria in Minimal Change Disease. Front Med (Lausanne). 2018 Jun 11;5:170. doi: 10.3389/fmed.2018.00170[↩]
- Frank C, Herrmann M, Fernandez S, Dirnecker D, Böswald M, Kolowos W, Ruder H, Haas JP. Dominant T cells in idiopathic nephrotic syndrome of childhood. Kidney Int. 2000 Feb;57(2):510-7. doi: 10.1046/j.1523-1755.2000.00870.x[↩]
- Fiser RT, Arnold WC, Charlton RK, Steele RW, Childress SH, Shirkey B. T-lymphocyte subsets in nephrotic syndrome. Kidney Int. 1991 Nov;40(5):913-6. doi: 10.1038/ki.1991.293[↩]
- Cara-Fuentes G, Clapp WL, Johnson RJ, Garin EH. Pathogenesis of proteinuria in idiopathic minimal change disease: molecular mechanisms. Pediatr Nephrol. 2016 Dec;31(12):2179-2189. doi: 10.1007/s00467-016-3379-4[↩]
- Minimal Change Disease. https://www.msdmanuals.com/professional/genitourinary-disorders/glomerular-disorders/minimal-change-disease[↩][↩]
- Hogan J, Radhakrishnan J. The treatment of minimal change disease in adults. J Am Soc Nephrol. 2013 Apr;24(5):702-11. doi: 10.1681/ASN.2012070734[↩][↩]
- Lombel RM, Gipson DS, Hodson EM; Kidney Disease: Improving Global Outcomes. Treatment of steroid-sensitive nephrotic syndrome: new guidelines from KDIGO. Pediatr Nephrol. 2013 Mar;28(3):415-26. doi: 10.1007/s00467-012-2310-x[↩][↩]
- Munyentwali H, Bouachi K, Audard V, Remy P, Lang P, Mojaat R, Deschênes G, Ronco PM, Plaisier EM, Dahan KY. Rituximab is an efficient and safe treatment in adults with steroid-dependent minimal change disease. Kidney Int. 2013 Mar;83(3):511-6. doi: 10.1038/ki.2012.444[↩]
- Treatment of minimal change disease in adults. https://www.uptodate.com/contents/treatment-of-minimal-change-disease-in-adults[↩]
- Rawla P, Limaiem F, Hashmi MF. IgA Nephropathy. [Updated 2023 Jul 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538214[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Tashakkorinia N, Muco E, Tudor ME. Berger Disease. [Updated 2023 Feb 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK499998[↩][↩][↩][↩][↩]
- IgA Nephropathy. https://www.niddk.nih.gov/health-information/kidney-disease/iga-nephropathy[↩]
- Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013 Jun 20;368(25):2402-14. doi: 10.1056/NEJMra1206793[↩]
- IgA nephropathy. https://medlineplus.gov/ency/article/000466.htm[↩]
- Knoppova B, Reily C, King RG, Julian BA, Novak J, Green TJ. Pathogenesis of IgA Nephropathy: Current Understanding and Implications for Development of Disease-Specific Treatment. J Clin Med. 2021 Sep 29;10(19):4501. doi: 10.3390/jcm10194501[↩][↩][↩][↩][↩][↩]
- Wyatt R.J., Julian B.A. IgA nephropathy. N. Engl. J. Med. 2013;368:2402–2414. doi: 10.1056/NEJMra1206793[↩][↩]
- D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Q J Med. 1987 Sep;64(245):709-27.[↩]
- Woo KT, Lau YK, Chan CM, Wong KS. Angiotensin-converting enzyme inhibitor versus angiotensin 2 receptor antagonist therapy and the influence of angiotensin-converting enzyme gene polymorphism in IgA nephritis. Ann Acad Med Singap. 2008 May;37(5):372-6. https://annals.edu.sg/pdf/37VolNo5May2008/V37N5p372.pdf[↩]
- Satpathy HK. IgA nephropathy. In: Ferri FF, ed. Ferri’s Clinical Advisor 2013. 1st ed. St. Louis: Mosby; 2012: 570–571.[↩]
- Geddes C.C., Rauta V., Gronhagen-Riska C., Bartosik L.P., Jardine A.G., Ibels L.S., Pei Y., Cattran D.C. A tricontinental view of IgA nephropathy. Nephrol. Dial. Transplant. 2003;18:1541–1548. doi: 10.1093/ndt/gfg207[↩]
- Shen A.Y., Brar S.S., Khan S.S., Kujubu D.A. Association of race, heart failure and chronic kidney disease. Future Cardiol. 2006;2:441–454. doi: 10.2217/14796678.2.4.441[↩]
- Wyatt R.J., Julian B.A., Baehler R.W., Stafford C.C., McMorrow R.G., Ferguson T., Jackson E., Woodford S.Y., Miller P.M., Kritchevsky S. Epidemiology of IgA nephropathy in central and eastern Kentucky for the period 1975 through 1994. Central Kentucky Region of the Southeastern United States IgA Nephropathy DATABANK Project. J. Am. Soc. Nephrol. 1998;9:853–858. doi: 10.1681/ASN.V95853[↩]
- Schena F.P. A retrospective analysis of the natural history of primary IgA nephropathy worldwide. Am. J. Med. 1990;89:209–215. doi: 10.1016/0002-9343(90)90300-3[↩]
- Wyatt R.J., Kritchevsky S.B., Woodford S.Y., Miller P.M., Roy S., Holland N.H., Jackson E., Bishof N.A. IgA nephropathy: Long-term prognosis for pediatric patients. J. Pediatr. 1995;127:913–919. doi: 10.1016/S0022-3476(95)70027-7[↩]
- IgA Nephropathy. https://emedicine.medscape.com/article/239927-overview[↩][↩]
- McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol Dial Transplant. 2011 Feb;26(2):414-30. doi: 10.1093/ndt/gfq665[↩]
- Schena FP, Nistor I. Epidemiology of IgA Nephropathy: A Global Perspective. Semin Nephrol. 2018 Sep;38(5):435-442. doi: 10.1016/j.semnephrol.2018.05.013[↩]
- Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6):e1002765. doi: 10.1371/journal.pgen.1002765[↩]
- Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 clinical practice guideline for the management of glomerular diseases. Kidney International. 2021;100(4S):S1–S276. doi: 10.1016/j.kint.2021.05.021[↩][↩][↩]
- D’Amico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin. Nephrol. 2004;24:179–196. doi: 10.1016/j.semnephrol.2004.01.001[↩]
- Barratt J., Feehally J. IgA nephropathy. J. Am. Soc. Nephrol. 2005;16:2088–2097. doi: 10.1681/ASN.2005020134[↩]
- Reich H.N., Troyanov S., Scholey J.W., Cattran D.C., Registry T.G. Remission of proteinuria improves prognosis in IgA nephropathy. J. Am. Soc. Nephrol. 2007;18:3177–3183. doi: 10.1681/ASN.2007050526[↩]
- Barbour SJ, Cattran DC, Kim SJ, Levin A, Wald R, Hladunewich MA, Reich HN. Individuals of Pacific Asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney Int. 2013 Nov;84(5):1017-24. doi: 10.1038/ki.2013.210. Epub 2013 Jun 5. Erratum in: Kidney Int. 2015 Jan;87(1):242.[↩][↩]
- D’Amico G, Colasanti G, Barbiano di Belgioioso G, Fellin G, Ragni A, Egidi F, Radaelli L, Fogazzi G, Ponticelli C, Minetti L. Long-term follow-up of IgA mesangial nephropathy: clinico-histological study in 374 patients. Semin Nephrol. 1987 Dec;7(4):355-8.[↩]
- Rollino C, Vischini G, Coppo R. IgA nephropathy and infections. J Nephrol. 2016 Aug;29(4):463-8. doi: 10.1007/s40620-016-0265-x[↩][↩][↩][↩]
- Lai KN, Tang SC, Schena FP, Novak J, Tomino Y, Fogo AB, Glassock RJ. IgA nephropathy. Nat Rev Dis Primers. 2016 Feb 11;2:16001. doi: 10.1038/nrdp.2016.1[↩][↩][↩][↩]
- Allen A.C., Bailey E.M., Brenchley P.E., Buck K.S., Barratt J., Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001;60:969–973. doi: 10.1046/j.1523-1755.2001.060003969.x[↩]
- Hiki Y., Odani H., Takahashi M., Yasuda Y., Nishimoto A., Iwase H., Shinzato T., Kobayashi Y., Maeda K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59:1077–1085. doi: 10.1046/j.1523-1755.2001.0590031077.x[↩]
- Conley M.E., Cooper M.D., Michael A.F. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J. Clin. Investig. 1980;66:1432–1436. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC371631/pdf/jcinvest00696-0242.pdf[↩][↩]
- Jennette J.C. The immunohistology of IgA nephropathy. Am. J. Kidney Dis. 1988;12:348–352. doi: 10.1016/S0272-6386(88)80022-2[↩]
- Roberts I.S. Pathology of IgA nephropathy. Nat. Rev. Nephrol. 2014;10:445–454. doi: 10.1038/nrneph.2014.92[↩]
- Woo KT, Lau YK, Choong HL, Tan HK, Foo MW, Lee EJ, Anantharaman V, Lee GS, Yap HK, Yi Z, Fook-Chong S, Wong KS, Chan CM. Genomics and disease progression in IgA nephritis. Ann Acad Med Singap. 2013 Dec;42(12):674-80. https://annals.edu.sg/pdf/42VolNo12Dec2013/V42N12p674.pdf[↩][↩]
- IgA nephropathy (Berger disease). https://www.mayoclinic.org/diseases-conditions/iga-nephropathy/symptoms-causes/syc-20352268[↩][↩]
- Han SH, Kang EW, Kie JH, Yoo TH, Choi KH, Han DS, Kang SW. Spontaneous remission of IgA nephropathy associated with resolution of hepatitis A. Am J Kidney Dis. 2010 Dec;56(6):1163-7. doi: 10.1053/j.ajkd.2010.08.018[↩]
- Suzuki H., Kiryluk K., Novak J., Moldoveanu Z., Herr A.B., Renfrow M.B., Wyatt R.J., Scolari F., Mestecky J., Gharavi A.G., et al. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011;22:1795–1803. doi: 10.1681/ASN.2011050464[↩][↩][↩]
- Magistroni R, D’Agati VD, Appel GB, Kiryluk K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015 Nov;88(5):974-89. doi: 10.1038/ki.2015.252[↩][↩][↩]
- Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, Julian BA. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011 Oct;22(10):1795-803. doi: 10.1681/ASN.2011050464[↩]
- Kiryluk K, Moldoveanu Z, Sanders JT, Eison TM, Suzuki H, Julian BA, Novak J, Gharavi AG, Wyatt RJ. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney Int. 2011 Jul;80(1):79-87. doi: 10.1038/ki.2011.16[↩]
- Gharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, Mestecky J, Novak J, Julian BA. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008 May;19(5):1008-14. doi: 10.1681/ASN.2007091052[↩]
- Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, Raska M, Renfrow MB, Julian BA, Novak J. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front Immunol. 2016 Apr 12;7:117. doi: 10.3389/fimmu.2016.00117[↩]
- Lamm ME, Emancipator SN, Robinson JK, Yamashita M, Fujioka H, Qiu J, Plaut AG. Microbial IgA protease removes IgA immune complexes from mouse glomeruli in vivo: potential therapy for IgA nephropathy. Am J Pathol. 2008 Jan;172(1):31-6. doi: 10.2353/ajpath.2008.070131[↩]
- Huang ZQ, Raska M, Stewart TJ, Reily C, King RG, Crossman DK, Crowley MR, Hargett A, Zhang Z, Suzuki H, Hall S, Wyatt RJ, Julian BA, Renfrow MB, Gharavi AG, Novak J. Somatic Mutations Modulate Autoantibodies against Galactose-Deficient IgA1 in IgA Nephropathy. J Am Soc Nephrol. 2016 Nov;27(11):3278-3284. doi: 10.1681/ASN.2014101044[↩]
- Yamamoto R, Nagasawa Y, Shoji T, Iwatani H, Hamano T, Kawada N, Inoue K, Uehata T, Kaneko T, Okada N, Moriyama T, Horio M, Yamauchi A, Tsubakihara Y, Imai E, Rakugi H, Isaka Y. Cigarette smoking and progression of IgA nephropathy. Am J Kidney Dis. 2010 Aug;56(2):313-24. doi: 10.1053/j.ajkd.2010.02.351[↩][↩]
- Kataoka H, Ohara M, Honda K, Mochizuki T, Nitta K. Maximal glomerular diameter as a 10-year prognostic indicator for IgA nephropathy. Nephrol Dial Transplant. 2011 Dec;26(12):3937-43. doi: 10.1093/ndt/gfr139[↩]
- Zhai YL, Zhu L, Shi SF, Liu LJ, Lv JC, Zhang H. Increased APRIL Expression Induces IgA1 Aberrant Glycosylation in IgA Nephropathy. Medicine (Baltimore). 2016 Mar;95(11):e3099. doi: 10.1097/MD.0000000000003099[↩]
- Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014 Nov;46(11):1187-96. doi: 10.1038/ng.3118[↩]
- Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, Stahl GL, Matsushita M, Fujita T, van Kooten C, Daha MR. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. 2006 Jun;17(6):1724-34. doi: 10.1681/ASN.2005090923[↩]
- Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. 2001 Sep 1;167(5):2861-8. doi: 10.4049/jimmunol.167.5.2861[↩]
- Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, Novak J. Current Understanding of the Role of Complement in IgA Nephropathy. J Am Soc Nephrol. 2015 Jul;26(7):1503-12. doi: 10.1681/ASN.2014101000[↩]
- Gharavi AG, Kiryluk K, Choi M, Li Y, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011 Mar 13;43(4):321-7. doi: 10.1038/ng.787[↩]
- Zhu L, Zhai YL, Wang FM, Hou P, Lv JC, Xu DM, Shi SF, Liu LJ, Yu F, Zhao MH, Novak J, Gharavi AG, Zhang H. Variants in Complement Factor H and Complement Factor H-Related Protein Genes, CFHR3 and CFHR1, Affect Complement Activation in IgA Nephropathy. J Am Soc Nephrol. 2015 May;26(5):1195-204. doi: 10.1681/ASN.2014010096[↩]
- Novak J., Julian B.A., Mestecky J., Renfrow M.B. Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin. Immunopathol. 2012;34:365–382. doi: 10.1007/s00281-012-0306-z[↩]
- IgA Nephropathy. https://www.kidney.org/atoz/content/iganeph[↩]
- Lv J, Xu D, Perkovic V, Ma X, Johnson DW, Woodward M, Levin A, Zhang H, Wang H; TESTING Study Group. Corticosteroid therapy in IgA nephropathy. J Am Soc Nephrol. 2012 Jun;23(6):1108-16. doi: 10.1681/ASN.2011111112[↩][↩]
- Hassler JR. IgA nephropathy: A brief review. Semin Diagn Pathol. 2020 May;37(3):143-147. doi: 10.1053/j.semdp.2020.03.001[↩]
- Kusaba G, Ohsawa I, Ishii M, Inoshita H, Takagi M, Tanifuji C, Takahashi K, Nakamoto J, Yoshida M, Ohi H, Horikoshi S, Kurihara H, Tomino Y. Significance of broad distribution of electron-dense deposits in patients with IgA nephropathy. Med Mol Morphol. 2012 Dec;45(1):29-34. doi: 10.1007/s00795-011-0538-3[↩]
- Pozzi C. Treatment of IgA nephropathy. J Nephrol. 2016 Feb;29(1):21-5. doi: 10.1007/s40620-015-0248-3[↩]
- Immunoglobulin A Nephropathy. https://www.msdmanuals.com/professional/genitourinary-disorders/glomerular-disorders/immunoglobulin-a-nephropathy[↩][↩][↩][↩][↩]
- Wheeler DC, Toto RD, Stefánsson BV, Jongs N, Chertow GM, Greene T, Hou FF, McMurray JJV, Pecoits-Filho R, Correa-Rotter R, Rossing P, Sjöström CD, Umanath K, Langkilde AM, Heerspink HJL; DAPA-CKD Trial Committees and Investigators. A pre-specified analysis of the DAPA-CKD trial demonstrates the effects of dapagliflozin on major adverse kidney events in patients with IgA nephropathy. Kidney Int. 2021 Jul;100(1):215-224. doi: 10.1016/j.kint.2021.03.033[↩]
- Gharavi AG, Yan Y, Scolari F, Schena FP, Frasca GM, Ghiggeri GM, Cooper K, Amoroso A, Viola BF, Battini G, Caridi G, Canova C, Farhi A, Subramanian V, Nelson-Williams C, Woodford S, Julian BA, Wyatt RJ, Lifton RP. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nat Genet. 2000 Nov;26(3):354-7. doi: 10.1038/81677[↩]
- Coppo R. Treatment of IgA nephropathy: Recent advances and prospects. Nephrol Ther. 2018 Apr;14 Suppl 1:S13-S21. doi: 10.1016/j.nephro.2018.02.010[↩]
- Lai KN, Leung JC, Tang SC. The Treatment of IgA Nephropathy. Kidney Dis (Basel). 2015 May;1(1):19-26. doi: 10.1159/000381508[↩]
- Kittiskulnam P, Kanjanabuch T, Tangmanjitjaroen K, Chancharoenthana W, Praditpornsilpa K, Eiam-Ong S. The beneficial effects of weight reduction in overweight patients with chronic proteinuric immunoglobulin a nephropathy: a randomized controlled trial. J Ren Nutr. 2014 May;24(3):200-7. doi: 10.1053/j.jrn.2014.01.016[↩]
- Courtney AE, McNamee PT, Nelson WE, Maxwell AP. Does angiotensin blockade influence graft outcome in renal transplant recipients with IgA nephropathy? Nephrol Dial Transplant. 2006 Dec;21(12):3550-4. doi: 10.1093/ndt/gfl506[↩]
- Rauen T, Eitner F, Fitzner C, Sommerer C, Zeier M, Otte B, Panzer U, Peters H, Benck U, Mertens PR, Kuhlmann U, Witzke O, Gross O, Vielhauer V, Mann JF, Hilgers RD, Floege J; STOP-IgAN Investigators. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy. N Engl J Med. 2015 Dec 3;373(23):2225-36. doi: 10.1056/NEJMoa1415463[↩][↩]
- Appel GB, Waldman M. The IgA nephropathy treatment dilemma. Kidney Int. 2006 Jun;69(11):1939-44. doi: 10.1038/sj.ki.5000434[↩]
- Pozzi C, Bolasco PG, Fogazzi GB, Andrulli S, Altieri P, Ponticelli C, Locatelli F. Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet. 1999 Mar 13;353(9156):883-7. doi: 10.1016/s0140-6736(98)03563-6[↩]
- Manno C, Torres DD, Rossini M, Pesce F, Schena FP. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrol Dial Transplant. 2009 Dec;24(12):3694-701. doi: 10.1093/ndt/gfp356. Erratum in: Nephrol Dial Transplant. 2010 Apr;25(4):1363-4.[↩]
- Ballardie FW, Roberts ISD. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol. 2002 Jan;13(1):142-148. doi: 10.1681/ASN.V131142[↩]
- Pozzi C, Andrulli S, Pani A, Scaini P, Roccatello D, Fogazzi G, Pecchini P, Rustichelli R, Finocchiaro P, Del Vecchio L, Locatelli F. IgA nephropathy with severe chronic renal failure: a randomized controlled trial of corticosteroids and azathioprine. J Nephrol. 2013 Jan-Feb;26(1):86-93. doi: 10.5301/jn.5000110[↩]
- Lafayette RA, Canetta PA, Rovin BH, Appel GB, Novak J, Nath KA, Sethi S, Tumlin JA, Mehta K, Hogan M, Erickson S, Julian BA, Leung N, Enders FT, Brown R, Knoppova B, Hall S, Fervenza FC. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. J Am Soc Nephrol. 2017 Apr;28(4):1306-1313. doi: 10.1681/ASN.2016060640[↩]
- Beck L, Bomback AS, Choi MJ, Holzman LB, Langford C, Mariani LH, Somers MJ, Trachtman H, Waldman M. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis. Am J Kidney Dis. 2013 Sep;62(3):403-41. doi: 10.1053/j.ajkd.2013.06.002. Erratum in: Am J Kidney Dis. 2017 Mar;69(3):485.[↩]
- Appel GB, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, Li LS, Mysler E, Sánchez-Guerrero J, Solomons N, Wofsy D; Aspreva Lupus Management Study Group. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. 2009 May;20(5):1103-12. doi: 10.1681/ASN.2008101028[↩]
- Chen Y, Li Y, Yang S, Li Y, Liang M. Efficacy and safety of mycophenolate mofetil treatment in IgA nephropathy: a systematic review. BMC Nephrol. 2014 Dec 5;15:193. doi: 10.1186/1471-2369-15-193[↩]
- Tian L, Shao X, Xie Y, Wang L, Wang Q, Che X, Ni Z, Mou S. The long-term efficacy and safety of immunosuppressive therapy on the progression of IgA nephropathy: a meta-analysis of controlled clinical trials with more than 5-year follow-up. Expert Opin Pharmacother. 2015 Jun;16(8):1137-47. doi: 10.1517/14656566.2015.1038238[↩]
- Jäger C, Stampf S, Molyneux K, Barratt J, Golshayan D, Hadaya K, Huynh-Do U, Binet FI, Mueller TF, Koller M, Kim MJ. Recurrence of IgA nephropathy after kidney transplantation: experience from the Swiss transplant cohort study. BMC Nephrol. 2022 May 10;23(1):178. doi: 10.1186/s12882-022-02802-x[↩]
- Walsh M, Sar A, Lee D, Yilmaz S, Benediktsson H, Manns B, Hemmelgarn B. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clin J Am Soc Nephrol. 2010 Mar;5(3):425-30. doi: 10.2215/CJN.06530909[↩]
- Shen PC, He LQ, Tang Y, Wang Q, Wang W, Li J. Clinicopathological characteristics and prognostic factors of asymptomatic IgA nephropathy. J Investig Med. 2010 Mar;58(3):560-5. doi: 10.231/JIM.0b013e3181d20aa1[↩]
- Herlitz LC, Bomback AS, Stokes MB, Radhakrishnan J, D’Agati VD, Markowitz GS. IgA nephropathy with minimal change disease. Clin J Am Soc Nephrol. 2014 Jun 6;9(6):1033-9. doi: 10.2215/CJN.11951113[↩]
- Walsh M, Sar A, Lee D, et al. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clin J Am Soc Nephrol. 2010 Mar. 5(3):425-30.[↩]
- Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Cattran DC, Coppo R, Cook HT, Feehally J, Roberts IS, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009 Sep;76(5):534-45. doi: 10.1038/ki.2009.243[↩][↩][↩]
- Soares MF, Roberts IS. IgA nephropathy: an update. Curr Opin Nephrol Hypertens. 2017 May;26(3):165-171. doi: 10.1097/MNH.0000000000000312[↩]
- Haas M, Verhave JC, Liu ZH, Alpers CE, Barratt J, Becker JU, Cattran D, Cook HT, Coppo R, Feehally J, Pani A, Perkowska-Ptasinska A, Roberts IS, Soares MF, Trimarchi H, Wang S, Yuzawa Y, Zhang H, Troyanov S, Katafuchi R. A Multicenter Study of the Predictive Value of Crescents in IgA Nephropathy. J Am Soc Nephrol. 2017 Feb;28(2):691-701. doi: 10.1681/ASN.2016040433. Epub 2016 Sep 9. Erratum in: J Am Soc Nephrol. 2017 May;28(5):1665.[↩][↩]
- Le W, Liang S, Hu Y, Deng K, Bao H, Zeng C, et al. Long-term renal survival and related risk factors in patients with IgA nephropathy: results from a cohort of 1155 cases in a Chinese adult population. Nephrol Dial Transplant. 2012 Apr. 27 (4):1479-85.[↩]
- Xie J, Kiryluk K, Wang W, Wang Z, Guo S, Shen P, Ren H, Pan X, Chen X, Zhang W, Li X, Shi H, Li Y, Gharavi AG, Chen N. Predicting progression of IgA nephropathy: new clinical progression risk score. PLoS One. 2012;7(6):e38904. doi: 10.1371/journal.pone.0038904[↩]
- Barbour SJ, Reich HN. Risk stratification of patients with IgA nephropathy. Am J Kidney Dis. 2012 Jun;59(6):865-73. doi: 10.1053/j.ajkd.2012.02.326[↩]
- D’Amico G. Natural history of idiopathic IgA nephropathy: role of clinical and histological prognostic factors. Am J Kidney Dis. 2000 Aug;36(2):227-37. doi: 10.1053/ajkd.2000.8966[↩]
- Uffing A, Pérez-Saéz MJ, Jouve T, Bugnazet M, et al. Recurrence of IgA Nephropathy after Kidney Transplantation in Adults. Clin J Am Soc Nephrol. 2021 Aug;16(8):1247-1255. doi: 10.2215/CJN.00910121[↩][↩][↩]
- Hastings M.C., Bursac Z., Julian B.A., Villa Baca E., Featherston J., Woodford S.Y., Bailey L., Wyatt R.J. Life expectancy for patients from the southeastern United States with IgA nephropathy. Kidney Int. Rep. 2018;3:99–104. doi: 10.1016/j.ekir.2017.08.008[↩]
- Kaufman L, Yang G, Hayashi K, Ashby JR, Huang L, Ross MJ, et al.. The homophilic adhesion molecule sidekick-1 contributes to augmented podocyte aggregation in HIV-associated nephropathy. FASEB J. (2007) 21:1367–75. 10.1096/fj.06-7191com[↩]
- Freedman BI, Limou S, Ma L, Kopp JB. APOL1-associated nephropathy: a key contributor to racial disparities in CKD. Am J Kidney Dis. (2018) 72(5 Suppl. 1):S8–16. 10.1053/j.ajkd.2018.06.020[↩][↩][↩]
- Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int. 1999 Dec;56(6):2203-13. https://doi.org/10.1046/j.1523-1755.1999.00769.x[↩]
- Smith KD, Prince DK, Henriksen KJ, Nicosia RF, Alpers CE, Akilesh S. Digital spatial profiling of collapsing glomerulopathy. Kidney Int. 2022 May;101(5):1017-1026. doi: 10.1016/j.kint.2022.01.033[↩]
- Cutrim ÉMM, Neves PDMM, Campos MAG, Wanderley DC, Teixeira-Júnior AAL, Muniz MPR, Ladchumananandasivam FR, Gomes OV, Vasco RFV, Brito DJA, Lages JS, Salgado-Filho N, Guedes FL, de Almeida JB, Magalhães M, Araújo SA, Silva GEB. Collapsing Glomerulopathy: A Review by the Collapsing Brazilian Consortium. Front Med (Lausanne). 2022 Mar 3;9:846173. doi: 10.3389/fmed.2022.846173[↩][↩][↩][↩][↩][↩][↩][↩]
- Amoura A, Moktefi A, Halfon M, Karras A, Rafat C, Gibier JB, Gleeson PJ, Servais A, Argy N, Maillé P, Belenfant X, Gueutin V, Delpierre A, Tricot L, El Karoui K, Jourde-Chiche N, Houze S, Sahali D, Audard V. Malaria, Collapsing Glomerulopathy, and Focal and Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2020 Jul 1;15(7):964-972. doi: 10.2215/CJN.00590120[↩]
- Wu H, Larsen CP, Hernandez-Arroyo CF, Mohamed MMB, Caza T, Sharshir M, Chughtai A, Xie L, Gimenez JM, Sandow TA, Lusco MA, Yang H, Acheampong E, Rosales IA, Colvin RB, Fogo AB, Velez JCQ. AKI and Collapsing Glomerulopathy Associated with COVID-19 and APOL1 High-Risk Genotype. J Am Soc Nephrol. 2020 Aug;31(8):1688-1695. doi: 10.1681/ASN.2020050558[↩]
- Weiss MA, Daquioag E, Margolin EG, Pollak VE. Nephrotic syndrome, progressive irreversible renal failure, and glomerular “collapse”: a new clinicopathologic entity? Am J Kidney Dis. (1986) 7:20–8. 10.1016/s0272-6386(86)80052-x[↩]
- Cohen AH, Nast CC. HIV-associated nephropathy. A unique combined glomerular, tubular, and interstitial lesion. Mod Pathol. 1988 Mar;1(2):87-97.[↩]
- Wyatt CM, Klotman PE, D’Agati VD. HIV-associated nephropathy: clinical presentation, pathology, and epidemiology in the era of antiretroviral therapy. Semin Nephrol. (2008) 28:513–22. 10.1016/j.semnephrol.2008.08.005[↩][↩]
- Rosenberg AZ, Kopp JB. Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2017 Mar 7;12(3):502-517. doi: 10.2215/CJN.05960616. Epub 2017 Feb 27. Erratum in: Clin J Am Soc Nephrol. 2018 Dec 7;13(12):1889.[↩]
- Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol. 2007 May;2(3):529-42. doi: 10.2215/CJN.04121206[↩][↩][↩][↩]
- D’Agati V. D., Fogo A. B., Bruijn J. A., Jennette J. C. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. American Journal of Kidney Diseases. 2004;43(2):368–382. doi: 10.1053/j.ajkd.2003.10.024[↩]
- Stokes M. B., D’Agati V. D. Morphologic variants of focal segmental glomerulosclerosis and their significance. Advances in Chronic Kidney Disease. 2014;21(5):400–407. doi: 10.1053/j.ackd.2014.02.010[↩]
- Ferreira AC, Carvalho D, Carvalho F, Galvo MJ, Nolasco F. Collapsing glomerulopathy in Portugal: a review of the histological and clinical findings in HIV and non-HIV patients. Nephrol Dial Transplant. (2011) 26:2209–15. 10.1093/ndt/gfq686[↩][↩][↩][↩][↩]
- Valeri A, Barisoni L, Appel GB, Seigle R, D’Agati V. Idiopathic collapsing focal segmental glomeruloscierosis: a clinicopathologic study. Kidney Int. (1996) 50:1734–46. 10.1038/ki.1996.493[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Laurin LP, Gasim AM, Derebail VK, McGregor JG, Kidd JM, Hogan SL, et al. Renal survival in patients with collapsing compared with not otherwise specified FSGS. Clin J Am Soc Nephrol. (2016) 11:1752–9. 10.2215/CJN.13091215[↩][↩][↩][↩][↩]
- Albaqumi M, Soos TJ, Barisoni L, Nelson PJ. Collapsing glomerulopathy. J Am Soc Nephrol. (2006) 17:2854–63. 10.1681/ASN.2006030225[↩][↩][↩][↩]
- Albaqumi M, Barisoni L. Current views on collapsing glomerulopathy. J Am Soc Nephrol. (2008) 19:1276–81. 10.1681/ASN.2007080926[↩][↩]
- Husain NE, Ahmed MH, Almobarak AO, Noor SK, Elmadhoun WM, Awadalla H, et al. HIV-associated nephropathy in Africa: pathology, clinical presentation and strategy for prevention. J Clin Med Res. (2018) 10:1–8. 10.14740/jocmr3235w[↩]
- Ross MJ. Advances in the pathogenesis of HIV-associated kidney diseases. Kidney Int. (2014) 86:266–74. 10.1038/ki.2014.167[↩][↩][↩]
- Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int. (1999) 56:2203–13. 10.1046/j.1523-1755.1999.00769[↩][↩]
- Chandra P, Kopp JB. Viruses and collapsing glomerulopathy: a brief critical review. Clin Kidney J. (2013) 6:1–5. 10.1093/ckj/sft002[↩][↩]
- Grover V, Gaiki MR, DeVita MV, Schwimmer JA. Cytomegalovirus-induced collapsing focal segmental glomerulosclerosis. Clin Kidney J. (2013) 6:71–3. 10.1093/ckj/sfs097[↩][↩]
- Freitas GRR, Praxedes MRG, Malheiros D, Testagrossa L, Dias CB, Woronik V. Collapsing variant of focal segmental glomerulosclerosis by parvovirus B19: case report. J Bras Nefrol. (2015) 37:121–6. 10.5935/0101-2800.20150017[↩]
- Besse W, Mansour S, Jatwani K, Nast CC, Brewster UC. Collapsing glomerulopathy in a young woman with APOL1 risk alleles following acute parvovirus B19 infection: a case report investigation. BMC Nephrol. (2016) 17:125. 10.1186/s12882-016-0330-7[↩]
- Araújo SDA, Cordeiro TM, Belisário AR, Araújo RFA, Marinho PES, Kroon EG, et al. First report of collapsing variant of focal segmental glomerulosclerosis triggered by arbovirus: dengue and zika virus infection. Clin Kidney J. (2019) 12:355–61. 10.1093/ckj/sfy104[↩][↩]
- Queiroz PC, Jorge AES, Mourão PHV, Penido MGMG. Collapsing focal segmental glomerulosclerosis probably triggered by dengue virus infection – two case reports. J Bras Nefrol. (2020) 42:489–93. 10.1590/2175-8239-JBN-2019-0237[↩][↩]
- Nasr SH, Kopp JB. COVID-19–associated collapsing glomerulopathy: an emerging entity. Kidney Int Rep. (2020) 5:759–61. 10.1016/j.ekir.2020.04.030[↩]
- Oliveira P, Cunha K, Neves P, Muniz M, Gatto G, Salgado-Filho N, et al. Renal morphology in coronavirus disease: a literature review. Medicina (Kaunas). (2021) 57:258. 10.3390/medicina57030258[↩][↩][↩][↩]
- Teixeira Júnior AAL, Neves PDMM, Lages JS, Cunha KA, Muniz MRP, Brito DJA, et al. Brazilian consortium for the study on renal diseases associated with COVID-19: a multicentric effort to understand SARS-CoV-2-related nephropathy. Front Med. (2020) 7:584235. 10.3389/fmed.2020.584235[↩][↩]
- Gopalakrishnan N, Dhanapriya J, Padmakumar C, Dineshkumar T, Kurien AA, Sakthirajan R, et al. Collapsing glomerulopathy and thrombotic microangiopathy in postpartum period: two case reports. Indian J Nephrol. (2018) 28:157–9. 10.4103/ijn.IJN_242_16[↩][↩]
- Salvatore SP, Barisoni LMC, Herzenberg AM, Chander PN, Nickeleit V, Seshan SV. Collapsing glomerulopathy in 19 patients with systemic lupus erythematosus or lupus-like disease. Clin J Am Soc Nephrol. (2012) 7:914–25. 10.2215/CJN.11751111[↩][↩]
- Markowitz GS, Bomback AS, Perazella MA. Drug-induced glomerular disease: direct cellular injury. Clin J Am Soc Nephrol. (2015) 10:1291–9. 10.2215/CJN.00860115[↩]
- Jaffe JA, Kimmel PL. Chronic nephropathies of cocaine and heroin abuse: a critical review. Clin J Am Soc Nephrol. (2006) 1:655–67. 10.2215/CJN.00300106[↩]
- Gebregeorgis W, Patel I, Thakur M, Bhutani D, Woldie I. Collapsing glomerulopathy associated with hemophagocytic syndrome in a patient with NK/T cell lymphoma. Clin Nephrol Case Stud. (2016) 4:11. 10.5414/CNCS108586[↩]
- Korbet SM, Schwartz MM. Multiple myeloma. J Am Soc Nephrol. (2006) 17:2533–45. 10.1681/ASN.2006020139[↩]
- Bhowmik D, Dinda AK, Gupta S, Agarwal SK, Tiwari SC, Dash SC. Multiple myeloma presenting as collapsing glomerulopathy. Indian J Pathol Microbiol. 2003 Apr;46(2):233-4.[↩]
- Buob D, Decambron M, Gnemmi V, Frimat M, Hoffmann M, Azar R, et al. Collapsing glomerulopathy is common in the setting of thrombotic microangiopathy of the native kidney. Kidney Int. (2016) 90:1321–31. 10.1016/j.kint.2016.07.021[↩][↩]
- Kanodia KV, Vanikar AV, Patel RD, Shutar KS, Nigan LK, Patel HV, et al. Collapsing glomerulopathy: a single centre clinicopathologic study of seven years. J Clin Diagn Res. (2016) 10:EC15–7. 10.7860/JCDR/2016/17297.7646[↩][↩][↩][↩]
- Ahuja A, Gupta R, Sharma A, Bagga A, Bohwmik DN, Agarwal SK, et al. Idiopathic collapsing glomerulopathy: a clinicopathologic analysis of 30 cases. Indian J Nephrol. (2014) 24:239–42. 10.4103/0971-4065.133009[↩]
- Watanabe A, Guaragna MS, Belangero VMS, Casimiro FMS, Pesquero JB, Feltran LS, et al. APOL1 in an ethnically diverse pediatric population with nephrotic syndrome: implications in focal segmental glomerulosclerosis and other diagnoses. Pediatr Nephrol. (2021) 36:1–10. 10.1007/s00467-021-04960-w[↩]
- Cossey NL, Larsen CP, Liapis H. Collapsing glomerulopathy: a 30-year perspective and single, large center experience. Clin Kidney J. (2017) 10:443–9. 10.1093/ckj/sfx029[↩][↩]
- Mubarak M, Kazi JI. Collapsing FSGS: a clinicopathologic study of 10 cases from Pakistan. Clin Exp Nephrol. (2010) 14:222–7. 10.1007/s10157-010-0275-2[↩]
- Grcevska L, Polenakovik M. Collapsing glomerulopathy: clinical characteristics and follow-up. Am J Kidney Dis. (1999) 33:652–7. 10.1016/s0272-6386(99)70215-5[↩]
- De Abreu Testagrossa L, Malheiros DMAC. Estudo brasileiro das variantes morfológicas da glomerulosclerose segmentar e focal. J Bras Patol Med Lab. (2012) 48:211–5. 10.1590/S1676-24442012000300009[↩]
- Kopp JB, Anders HJ, Susztak K, Podestà MA, Remuzzi G, Hildebrandt F, Romagnani P. Podocytopathies. Nat Rev Dis Primers. 2020 Aug 13;6(1):68. doi: 10.1038/s41572-020-0196-7[↩][↩]
- Smeets B, Angelotti ML, Rizzo P, et al. Renal progenitor cells contribute to hyperplastic lesions of podocytopathies and crescentic glomerulonephritis. J Am Soc Nephrol. 2009;20(12):2593–2603. doi: 10.1681/ASN.2009020132[↩][↩][↩]
- Kopp JB, Anders HJ, Susztak K, Posteda MA, Remuzzi G, Friedhelm H, et al. Podocytopathies. Nat Rev Dis Primers. (2020) 6:1–24. 10.1038/s41572-020-0196-7[↩]
- Cheng H, Harris RC. The glomerulus – a view from the outside – the podocyte. Int J Biochem Cell Biol. (2010) 42:1380–7. 10.1016/j.biocel.2010.05.014[↩][↩]
- Barisoni L, Kriz W, Mundel P, D’agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (1999) 10:51–61. 10.1681/ASN.V10151[↩][↩]
- Chugh SS, Clement LC. Telomerase at the center of collapsing glomerulopathy. Nat Med. (2012) 18:26–7. 10.1038/nm.2602[↩]
- Shkreli M, Sarin KY, Pech MF, Papeta N, Chang W, Brockmann SA, et al. Reversible cell-cycle entry in adult kidney podocytes through regulated control of telomerase and Wnt signaling. Nat Med. (2012) 18:111–9. 10.1038/nm.2550[↩][↩][↩]
- Scholz H, Kirschner KMA. Role for the Wilms’ tumor protein WT1 in organ development. Physiology. (2005) 20:54–9. 10.1152/physiol.00048.2004[↩][↩]
- Siemens TA, Riella MC, Moraes TP, Riella CV. APOL1 risk variants and kidney disease: what we know so far. J Braz Nephrol. (2018) 40:388–402. 10.1590/2175-8239-JBN-2017-0033[↩]
- Riella C, Siemens TA, Wang M, Campos RP, Moraes TP, Riella LV, et al. APOL1-associated kidney disease in Brazil. Kidney Int Rep. (2019) 4:923–9. 10.1016/j.ekir.2019.03.006[↩]
- Barisoni L, Diomedi-camassei F, Santorelli FM, Caridi G, Thomas DB, Emma F, et al. Collapsing glomerulopathy associated with inherited mitochondrial injury. Kidney Int. (2008) 74:237–43. 10.1038/sj.ki.5002767[↩]
- Starr MC, Chang IJ, Finn LS, Sun A, Larson AA, Goebel J, et al. COQ2 nephropathy: a treatable cause of nephrotic syndrome in children. Pediatr Nephrol. (2018) 33:1257–61. 10.1007/s00467-018-3937-z[↩]
- Winkler CA, Nelson G, Oleksyk TK, Nava MB, Kopp JB. Genetics of focal segmental glomerulosclerosis and human immunodeficiency virus-associated collapsing glomerulopathy: the role of MYH9 genetic variation. Semin Nephrol. (2010) 30:111–25. 10.1016/j.semnephrol.2010.01.003[↩]
- Nephrotic Syndrome in Adults. https://www.niddk.nih.gov/health-information/kidney-disease/nephrotic-syndrome-adults[↩]
- D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. (2004) 43:368–82. 10.1053/j.ajkd.2003.10.024[↩][↩]
- Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD Atlas of renal pathology: collapsing glomerulopathy. Am J Kidney Dis. (2015) 66:e3–4. 10.1053/j.ajkd.2015.04.009[↩][↩][↩][↩]
- Nieto Ríos JF, Milena Brand S, Serna Higuita LM, Fernando Arias L. The collapsing variant of focal segmental glomerulosclerosis in children. Iatreia. (2013) 26:481–6.[↩]
- Cheung AK, Chang TI, Cushman WC, Furth SL, Hou FF, Ix JH, et al. Executive summary of the KDIGO 2021 clinical practice guideline for the management of blood pressure in chronic kidney disease. Kidney Int. (2021) 99:559–69. 10.1016/j.kint.2020.10.026[↩][↩][↩]
- Kodner C. Diagnosis and Management of Nephrotic Syndrome in Adults. Am Fam Physician. 2016 Mar 15;93(6):479-85. https://www.aafp.org/pubs/afp/issues/2016/0315/p479.html[↩]
- Floege J, Barbour SJ, Cattran DC, Hogan JJ, Nachman PH, Tang SCW, et al. Management and treatment of glomerular diseases (part 1): conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2019) 95:268–80. 10.1016/j.kint.2018.10.018[↩]
- Rovin BH, Caster DJ, Cattran DC, Gibson KL, Hogan JJ, Moeller MJ, et al. Management and treatment of glomerular diseases (part 2): conclusions from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. (2019) 95:268–80. 10.1016/j.kint.2018.11.008[↩]
- Vaziri ND. Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int. (2016) 90:41–52. 10.1016/j.kint.2016.02.026[↩]
- Agrawal S, Zaritsky JJ, Fornoni A, Smoyer WE. Dyslipidaemia in nephrotic syndrome: mechanisms and treatment. Nat Rev Nephrol. (2018) 14:57–70. 10.1038/nrneph.2017.155[↩]
- Morales E, Alonso M, Gutiérrez E. Collapsing glomerulopathy: update. Med Clin (Barc). (2019) 152:361–7. 10.1016/j.medcli.2018.10.021[↩]
- Schwartz EJ, Szczech LA, Ross MJ, Klotman ME, Winston JA, Klotman PE. Highly active antiretroviral therapy and the epidemic of HIV end-stage renal disease. J Am Soc Nephrol. (2005) 16:2412–20. 10.1681/ASN.2005040340[↩]
- Szczech LA, Gupta SK, Habash R, Guasch A, Kalayjian R, Appel A, et al. The clinical epidemiology and course of the spectrum of renal diseases associated with HIV infection. Kidney Int. (2004) 66:1145–52. 10.1111/j.1523-1755.2004.00865.x[↩]
- Atta MG, Gallant JE, Rahman MH, Nagajothi N, Racusen LC, Scheel PL, et al. Antiretroviral therapy in the treatment of HIV-associated nephropathy. Nephrol Dial Transplant. (2006) 21:2809–13. 10.1093/ndt/gfl337[↩]
- Nair V, Jandovitz N, Jhaveri KD, Hirschwerk D, Grodstein L, Bijol V, et al. Treatment of parvovirus B19 viremia to facilitate kidney transplantation in a patient with collapsing glomerulopathy. Clin Nephrol Case Stud. (2020) 8:41–5. 10.5414/CNCS110113[↩]
- Detwiler RK, Falk RJ, Hogan SL, Jennette JC. Collapsing glomerulopathy: a clinically and pathologically distinct variant of focal segmental glomeruloscierosis. Kidney Int. (1994) 45:1416–24. 10.1038/ki.1994.185[↩]
- Haas M, Spargo BH, Coventry S. Increasing incidence of focal-segmental glomerulosclerosis among adult nephropathies: a 20-year renal biopsy study. Am J Kidney Dis. (1995) 26:740–50. 10.1016/0272-6386(95)90437-9[↩]



{kind=link}