Contents
What is Li-Fraumeni syndrome
Li-Fraumeni syndrome is a rare inherited disorder that greatly increases the risk of developing several types of cancer, particularly in children and young adults. People with Li-Fraumeni syndrome often develop cancer at an earlier age than expected and may be diagnosed with more than one cancer during their lifetime. They also seem to have a higher risk of getting cancer from radiation therapy, so doctors treating these patients might try to avoid giving them radiation when possible.
The cancers most often associated with Li-Fraumeni syndrome include breast cancer, a form of bone cancer called osteosarcoma, and cancers of soft tissues (such as muscle) called soft tissue sarcomas. Other cancers commonly seen in Li-Fraumeni syndrome include brain tumors, cancers of blood-forming tissues (leukemias), and a cancer called adrenocortical carcinoma that affects the outer layer of the adrenal glands (small hormone-producing glands on top of each kidney). Several other types of cancer also occur more frequently in people with Li-Fraumeni syndrome 1.
A very similar condition called Li-Fraumeni-Like syndrome shares many of the features of classic Li-Fraumeni syndrome. Both conditions significantly increase the chances of developing multiple cancers beginning in childhood; however, the pattern of specific cancers seen in affected family members is different.
Li-Fraumeni syndrome is caused by changes (mutations) in the TP53 gene, which is a tumor suppressor gene and is inherited in an autosomal dominant manner 2. A normal TP53 gene makes a protein that helps stop abnormal cells from growing. Li-Fraumeni syndrome can also be caused by mutations in a tumor suppressor gene called CHEK2, which also normally helps stop cells with DNA damage from growing.
The exact prevalence of Li-Fraumeni is unknown. One U.S. registry of Li-Fraumeni syndrome patients suggests that about 400 people from 64 families have this disorder.
If someone has Li-Fraumeni syndrome, their close relatives (especially children) have an increased chance of having a mutation, too. They may wish to be tested, or even without testing they may wish to start screening for certain cancers early or take other precautions to help lower their risk of cancer.
There is no evidence of ethnic or geographic disparity in the occurrence of Li-Fraumeni syndrome, but a uniquely high prevalence of Li-Fraumeni syndrome has been reported in southern and southeastern Brazil. The population with Li-Fraumeni syndrome in this area has been associated with a highly specific mutation of the TP53 referred to as R337H. Having this particular alteration in the region led researchers to suspect one point of origin, and family lineages were traced to a common ancestor who migrated long ago from Portugal. Interestingly, though, as opposed to the 90% lifetime risk of developing cancer in most people with Li-Fraumeni syndrome, the population in Brazil with this “founder mutation” has roughly a 60% lifetime risk of cancers, which have relatively favorable survival rates.
Management may include high-risk cancer screening and/or prophylactic surgeries 3.
People with a strong family history of cancer may want to learn their genetic makeup. This may help the person or other family members plan their health care for the future. Since inherited mutations affect all cells of a person’s body, they can often be found by genetic testing done on blood or saliva (spit) samples. Still, genetic testing is not helpful for everyone, so it’s important to speak with a genetic counselor first to find out if testing might be right for you.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Li-Fraumeni syndrome cancers
Li-Fraumeni syndrome is associated with high risks of a diverse spectrum of childhood- and adult-onset malignancies 4.
The most common cancers observed in families with Li-Fraumeni syndrome per age group are:
- 0-10 yrs. Soft tissue sarcomas, brain tumors, and adrenocortical carcinoma
- 11-20 yrs. Bone sarcomas
- >20 yrs. Breast cancer and brain tumors
At-risk individuals who remain cancer free into their 50s and 60s are at lower risk of having the familial TP53 pathogenic variant. However, males who did not develop childhood cancers may present in their 50s with multiple primary tumors 5.
The tumors most closely associated with Li-Fraumeni syndrome are: soft tissue sarcoma, osteosarcoma, pre-menopausal breast cancer, brain tumors, and adrenocortical carcinoma. These core cancers, which are described below, account for about 70% of all Li-Fraumeni syndrome-related tumors 6:
Sarcomas
Individuals with Li-Fraumeni syndrome are at increased risk of developing soft tissue and bone sarcomas. The International Agency for Research on Cancer (IARC) TP53 database found that sarcomas represented 25% of the cancers reported in people with Li-Fraumeni syndrome. The most commonly occurring sarcomas in the IARC TP53 database were rhabdomyosarcomas before age five years and osteosarcomas at any age. Other forms of sarcoma included leiomyosarcomas, liposarcomas, and histiosarcomas; 16 other histology types were also noted 7. Li-Fraumeni syndrome-related sarcomas can occur in childhood or adulthood, with most Li-Fraumeni syndrome-associated sarcomas occurring before age 50 years. Sarcomas that were not reported in the IARC TP53 database, and are less likely to be features of Li-Fraumeni syndrome, include gastrointestinal stromal tumors, desmoid tumors/fibromatosis, Ewing sarcomas, and angiosarcomas 7.
Breast cancer
Women with Li-Fraumeni syndrome are at greatly increased risk of developing pre-menopausal breast cancer. The median age of breast cancer diagnosis in women with Li-Fraumeni syndrome is about 33 years 8. In one series of women with Li-Fraumeni syndrome-related breast cancers, 32% of the cancers occurred before age 30 years and none of the breast cancers occurred after age 50 years 9. Recent data suggest that Li-Fraumeni syndrome-related breast cancers are predominantly positive by immunohistochemistry and FISH (for HER2/neu) for hormone receptors and/or Her2/neu 10. In one series, 84% of the Li-Fraumeni syndrome-related breast tumors were positive for estrogen and/or progesterone hormone markers, and 63% of the invasive breast cancers and 73% of in situ breast cancers were Her2/neu positive 10. In another study, 67% of the Li-Fraumeni syndrome-related breast cancers were Her2/neu positive compared to 25% of the controls 11. Malignant phyllodes tumors of the breast may also be associated with Li-Fraumeni syndrome 12. To date, male breast cancer has rarely been reported in families with Li-Fraumeni syndrome.
Brain tumors
Individuals with Li-Fraumeni syndrome are at increased risk of developing many types of brain tumors (e.g., astrocytomas, glioblastomas, medulloblastomas, choroid plexus carcinomas). Li-Fraumeni syndrome-related brain tumors can occur in childhood or adulthood; the median age of onset is 16 years 8. The likelihood of germline TP53 pathogenic variants in children with choroid plexus carcinoma is high, even in the absence of a family history suggestive of Li-Fraumeni syndrome 13. A rare peripheral nerve sheath tumor termed malignant triton tumor has also been reported in a child with a germline TP53 pathogenic variant 14. Malignant triton tumors contain schwannoma cells and rhabdomyoblasts.
Adrenocortical carcinomas
Individuals with Li-Fraumeni syndrome are at increased risk of developing adrenocortical carcinoma. Children with adrenocortical carcinoma have a 50%-80% chance of having a germline TP53 pathogenic variant, even in the absence of additional family history 15. Individuals with adult-onset adrenocortical carcinoma may also be at increased risk for a germline TP53 pathogenic variant, especially if diagnosed before age 50 years 15. In one series, 5.8% of individuals diagnosed with adrenocortical carcinoma after age 18 years tested positive for a germline TP53 pathogenic variant 16.
Excess of early-onset cancers
Individuals with Li-Fraumeni syndrome are at increased risk of developing cancer at younger than typical ages. It is estimated that 50% of Li-Fraumeni syndrome-associated malignancies occur by age 30 years 17. In one series of individuals who have a germline TP53 pathogenic variant, the median age at diagnosis was 25 years 15.
When assessing the likelihood that a family could have Li-Fraumeni syndrome, the age at diagnosis is important 18. For example, one series found that in six individuals with germline TP53 pathogenic variants who had developed colorectal cancer, four occurred before age 21 years 19. Therefore, in assessing families with possible Li-Fraumeni syndrome, an unusually young age at cancer diagnosis may be as important as the specific type of malignancy observed.
Excess of multiple primary cancers
Individuals with Li-Fraumeni syndrome are also at increased risk of developing multiple primary tumors 15. A retrospective study on 200 affected members of families with Li-Fraumeni syndrome found that 15% had developed a second cancer, 4% a third cancer, and 2% a total of four cancers. In this cohort, survivors of childhood cancers were found to have the highest risks for developing additional malignancies 20. The risk to individuals with Li-Fraumeni syndrome of developing a second cancer has been estimated at 57%, and the risk of a third malignancy 38%. The subsequent malignancies are not all clearly related to the treatment of the previous neoplasms.
Additional cancers
Although consensus holds that sarcomas, breast cancer, brain tumors, and adrenocortical carcinomas constitute the core cancers of Li-Fraumeni syndrome, there is much less agreement about the non-core cancers which account for about 30% of malignancies in Li-Fraumeni syndrome.
The following malignancies have been found to occur excessively in at least some families who have met criteria for Li-Fraumeni syndrome and/or have tested positive for germline TP53 pathogenic variants 6:
- Gastrointestinal cancers. Colorectal, esophageal, pancreatic, and stomach cancers have all been reported in families with Li-Fraumeni syndrome. One series reported a 2.8% incidence of colorectal cancer in people with TP53 pathogenic variants with a mean age of diagnosis of 33 years 19. Another series reported that 22.6% of families with Li-Fraumeni syndrome had at least one relative with gastric cancer with a mean age of diagnosis of 43 years 21.
- Genitourinary cancers. Renal cell carcinomas have been reported in families with Li-Fraumeni syndrome. Endometrial, ovarian, prostate and gonadal germ cell tumors have all been reported in families with Li-Fraumeni syndrome.
- Leukemias and lymphomas. Leukemias, especially the acute form, were initially considered a cardinal feature of Li-Fraumeni syndrome; however, more recent studies have determined that leukemias are not a major feature of Li-Fraumeni syndrome. Hodgkin and non-Hodgkin lymphomas have also been reported in families with Li-Fraumeni syndrome.
- Lung cancers – Increased risks for lung cancers have been reported in individuals with Li-Fraumeni syndrome, especially in those who use tobacco products 22. A rare form of lung cancer, termed bronchoalveolar cancer, is associated with Li-Fraumeni syndrome 23.
- Neuroblastomas and other childhood cancers – Children with germlineTP53 pathogenic variants may be at increased risk of developing neuroblastoma as well as other cancers of early childhood.
- Skin cancers. Increased rates of melanoma and non-melanoma skin cancers have been reported in families with Li-Fraumeni syndrome.
- Thyroid cancers. Non-medullary thyroid cancers have been reported in families with Li-Fraumeni syndrome.
Cancer risk
Li-Fraumeni syndrome is associated with high lifetime risks of cancer. The risk of cancer is estimated at 50% by age 30 years and 90% by age 60 years 17.
Age-specific cancer rates have also been assessed. One study, based on five families with Li-Fraumeni syndrome, estimated age-specific cancer risks as 42% at ages 0-16 years, 38% at ages 17-45 years, and 63% after age 45 years; overall lifetime cancer risk was calculated at 85%. In another series, 56% of cancers in families with Li-Fraumeni syndrome occurred prior to age 30 years and 100% were diagnosed by age 50 years 24.
The cancer risks in Li-Fraumeni syndrome demonstrate significant gender differences. For women with Li-Fraumeni syndrome, the lifetime risk of cancer is nearly 100% and for men with Li-Fraumeni syndrome, the lifetime risk of cancer is about 73% 25. This gender difference in cancer risk is primarily the result of the high incidence of breast cancer among women with Li-Fraumeni syndrome 15. However, in one series, the excessive cancer risk in females with Li-Fraumeni syndrome was observed at all stages of life, including childhood 5.
Li-Fraumeni syndrome causes
The CHEK2 and TP53 genes are associated with Li-Fraumeni syndrome.
More than half of all families with Li-Fraumeni syndrome have inherited mutations in the TP53 gene. TP53 is a tumor suppressor gene, which means that it normally helps control the growth and division of cells. Mutations in this gene can allow cells to divide in an uncontrolled way and form tumors. Other genetic and environmental factors are also likely to affect the risk of cancer in people with TP53 mutations.
A few families with cancers characteristic of Li-Fraumeni syndrome and Li-Fraumeni-like syndrome do not have TP53 mutations, but have mutations in the CHEK2 gene. Like the TP53 gene, CHEK2 is a tumor suppressor gene. Researchers are uncertain whether CHEK2 mutations actually cause these conditions or are merely associated with an increased risk of certain cancers (including breast cancer).
Li-Fraumeni syndrome inheritance pattern
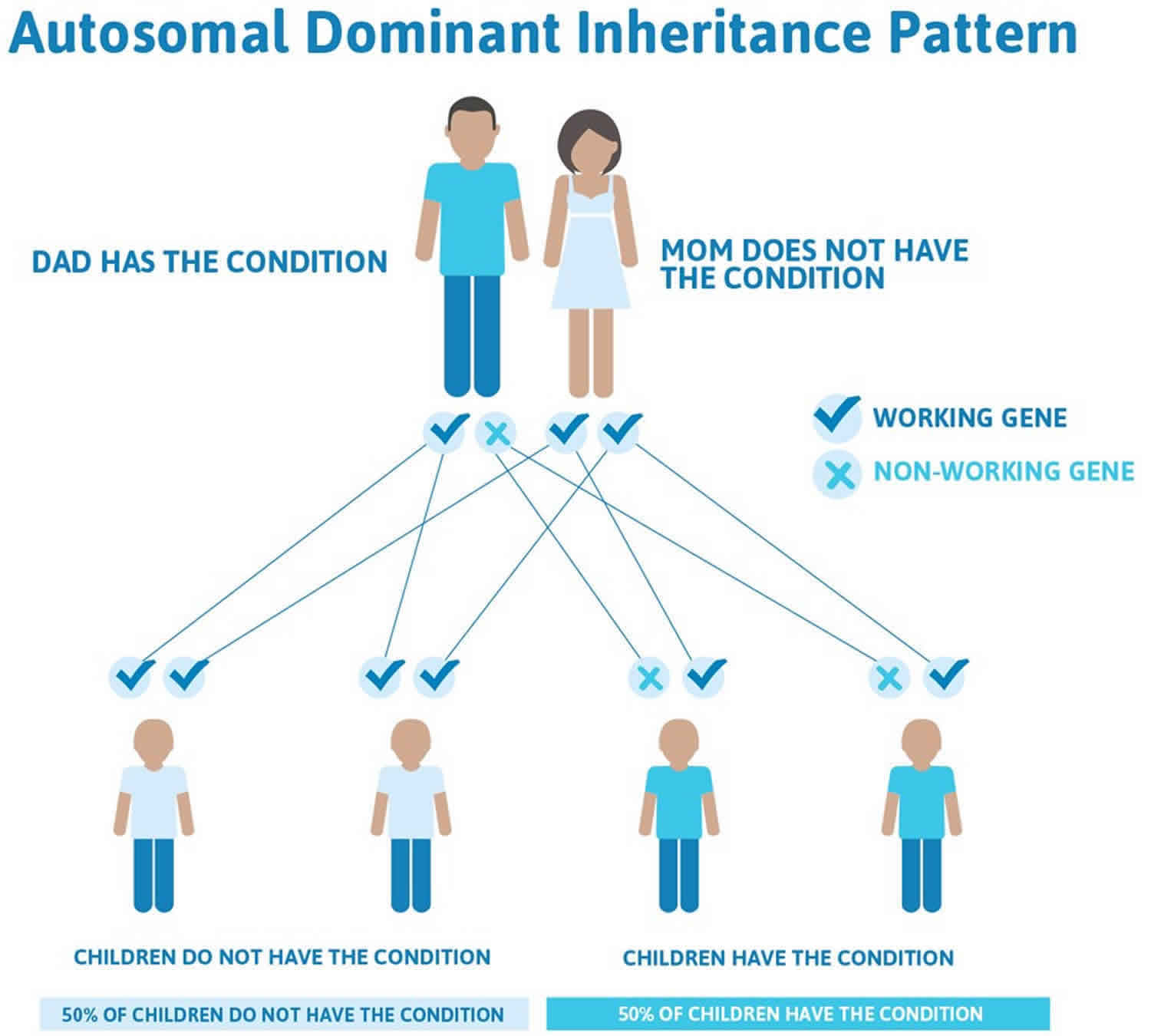
Li-Fraumeni syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to increase the risk of developing cancer. In most cases, an affected person has a parent and other family members with cancers characteristic of the condition.
In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 1 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 1. Li-Fraumeni syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Li-Fraumeni syndrome symptoms
Li-Fraumeni syndrome may be suspected if someone has a personal or family history of cancers featured in Li-Fraumeni syndrome. In addition, there are certain rare cancers that are characteristic of the syndrome that should alert clinicians to the potential of a diagnosis of Li-Fraumeni syndrome. Patients and families with multiple childhood cancers, or specific rare cancers such as adrenocortical or choroid plexus carcinoma, should alert practitioners to the potential of a hereditary cancer syndrome such as Li-Fraumeni syndrome. Although increasingly identified as a hereditary cancer syndrome, not all physicians are aware of the diagnosis of Li-Fraumeni syndrome.
Cancers most closely associated (core cancers) with Li-Fraumeni syndrome include:
- Soft tissue sarcoma
- Osteosarcoma
- Breast cancer
- Brain and CNS tumors (glioma, choroid plexus carcinoma, SHH subtype medulloblastoma, neuroblastoma)
- Adrenocortical carcinoma
- Acute leukemia
Other cancers may also appear, but risks are lower than for the core cancers:
- Lung adenocarcinoma
- Melanoma
- Gastrointestinal tumors (such as colon, pancreas)
- Kidney
- Thyroid
- Gonadal germ cells (such as ovarian, testicular, and prostate)
Individuals with Li-Fraumeni syndrome have an approximately 50% of developing cancer by age 40, and up to a 90% percent chance by age 60, while females have nearly a 100% risk of developing cancer in their lifetime due to their markedly increased risk of breast cancer. Many individuals with Li-Fraumeni syndrome develop two or more primary cancers over their lifetimes.
Li-Fraumeni syndrome diagnosis
Li-Fraumeni syndrome is diagnosed based on clinical criteria and/or genetic testing for the mutation in the TP53 gene. Genetic testing is typically considered with the below delineated criteria.
Clinical Testing (Clinical Screening & Genetic Testing)
The potential of genetic testing (and the implications of the results) should always involve discussions with a genetic counselor, medical providers, and family.
As delineated by the American Society of Clinical Oncology, the below criteria can be used in determining if genetic testing should be considered:
Classic Li-Fraumeni syndrome is diagnosed when a person has all of the following criteria:
- A sarcoma diagnosed before age 45
- A first-degree relative, meaning a parent, sibling or child, with any cancer before age 45
- A first-degree relative or second-degree relative, meaning a grandparent, aunt/uncle, niece/nephew, or grandchild, with any cancer before age 45 or a sarcoma at any age
Chompret Criteria for Clinical Diagnosis of Li-Fraumeni syndrome is a recent set of criteria that has been proposed to identify affected families beyond the Classic criteria listed above. A diagnosis of Li-Fraumeni syndrome and performing TP53 gene mutation testing is considered for anyone with a personal and family history that meets 1 of the following 3 criteria:
Criterion 1
- A tumor belonging to the Li-Fraumeni syndrome tumor spectrum, before the age of 46. This means any of the following diseases: soft-tissue sarcoma, osteosarcoma, pre-menopausal breast cancer, brain tumor, adrenal cortical carcinoma, leukemia, or lung cancer, and
- At least 1 first-degree or second-degree family member with an Li-Fraumeni syndrome-related tumor, except breast cancer if the individual has breast cancer before the age of 56 or with multiple tumors
Criterion 2
- A person with multiple tumors, except multiple breast tumors, 2 of which belonging to the Li-Fraumeni syndrome tumor spectrum and the first of which occurred before age 46
Criterion 3
- A person who is diagnosed with adrenocortical carcinoma or a tumor in the choroid plexus, meaning a membrane around the brain, regardless of family history.
Li-Fraumeni-Like Syndrome is another, similar set of criteria for affected families who do not meet Classic criteria (see above). There are 2 suggested definitions for Li-Fraumeni-Like Syndrome:
LFL Definition 1, called the Birch definition:
- A person diagnosed with any childhood cancer, sarcoma, brain tumor, or adrenal cortical tumor before age 45 and
- A first-degree or second-degree relative diagnosed with a typical Li-Fraumeni syndrome cancer, such as sarcoma, breast cancer, brain cancer, adrenal cortical tumor, or leukemia, at any age and
- A first-degree or second-degree relative diagnosed with any cancer before age 60
Li-Fraumeni-Like Syndrome Definition 2, called the Eeles definition:
- 2 first-degree or second-degree relatives diagnosed with a typical Li-Fraumeni syndrome cancer, such as sarcoma, breast cancer, brain cancer, adrenal cortical tumor, or leukemia, at any age
Other risk factors to consider, specific to breast cancer:
A woman who has a personal history of breast cancer at a younger age and does not have an identifiable mutation in breast cancer genes 1 or 2, called BRCA1 or BRCA2, may have a TP53 mutation.
A woman who is diagnosed with breast cancer before age 30 and is not found to have a BRCA mutation has an estimated 4% to 8% likelihood of having a TP53 mutation.
Women with breast cancer diagnosed between ages 30 and 39 may also have a small increased risk of having a TP53 mutation.
In younger woman with breast cancer, a TP53 mutation may also occur with any of the following features: a family history of cancer, especially Li-Fraumeni syndrome-related cancers, a personal history of a breast tumor that is positive for estrogen (ER), progesterone (PR), and HER2/neu markers, also known as “triple-positive” breast cancer, and a personal history of an additional Li-Fraumeni syndrome-related cancer.
Li-Fraumeni syndrome treatment
At this time, there is no standard treatment or cure for Li-Fraumeni syndrome or a germline TP53 gene mutation. With some exceptions, cancers in people with Li-Fraumeni syndrome are treated the same as for cancers in other patients, but research continues on how to best manage those cancers involved in Li-Fraumeni syndrome.
Research has indicated that those individuals with Li-Fraumeni syndrome appear to be an elevated risk for radiation-induced cancers, so the use of radiotherapy should be approached with caution. For this reason, computed tomography (CT) scans and other diagnostic techniques involving ionizing radiation should be limited. However, radiation therapy should not be avoided if the benefits outweigh the risks.
Since those living with Li-Fraumeni syndrome are susceptible to the development of a number of different cancers, individuals should ensure that they incorporate simple measures into a healthy lifestyle, such as sun protection and the avoidance of tobacco products.
It has been widely accepted that early cancer detection can greatly increase overall survival, and those diagnosed with Li-Fraumeni syndrome should seek to adhere to preventive screening. An expert panel of Li-Fraumeni syndrome researchers, oncologists, and genetic counselors has published surveillance recommendations that utilize whole body MRI screening for patients that fit the definition of Li-Fraumeni syndrome. This should be offered as soon as the diagnosis of Li-Fraumeni syndrome is established. In brief, the screening recommendations involve 26:
Children (birth to age 18 years)
General assessment
- Complete physical exam every 3-4 months
- Prompt assessment with primary care physician for any medical concerns
Adrenocortical carcinoma
- Ultrasound of abdomen and pelvis every 3-4 months
- In case of unsatisfactory ultrasound, blood tests every 3-4 months
Brain tumor
- Annual brain MRI (first MRI with contrast – thereafter without contrast if previous MRI normal with and no new abnormality
Soft tissue and bone sarcoma
- Annual whole body MRI
Adults
General assessment
- Complete physical exam every 6 months
- Prompt assessment with primary care physician for any medical concerns
Breast cancer
- Breast awareness (age 18 years and forward)
- Clinical breast exam twice a year (age 20 years and forward)
- Annual breast MRI screening (ages 20-75) – ideally, alternating with annual whole body MRI (one scan every 6 months)
- Consider risk-reducing bilateral mastectomy (Note that the use of ultrasound and mammography has been omitted)
Brain tumor (age 18 years and forward)
- Annual brain MRI (first MRI with contrast – thereafter without contrast if previous MRI normal)
Soft tissue and bone sarcoma (age 18 years and forward)
- Annual whole body MRI
- Ultrasound of abdomen and pelvis every 12 months
Gastrointestinal cancer (age 25 years and forward)
- Upper endoscopy and colonoscopy every 2-5 years)
Melanoma (age 18 years and forward)
- Annual dermatologic examination
Also noted, for families in which breast cancer has already made an appearance at or around age 20 – awareness and screening can be considered 5 to 10 years before the earliest age of onset known. The same is recommended for gastrointestinal cancers – consider screening 5 years before the earliest known onset of a gastrointestinal cancer in the family.
Li-Fraumeni syndrome life expectancy
Li-Fraumeni syndrome is associated with a high lifelong cancer risk. Individuals with Li-Fraumeni syndrome have an approximately 50% of developing cancer by age 40, and up to a 90% percent chance by age 60 17, while females have nearly a 100% risk of developing cancer in their lifetime due to their markedly increased risk of breast cancer. Many individuals with Li-Fraumeni syndrome develop two or more primary cancers over their lifetimes. It has been shown that TP53 mutation carriers enrolled in a surveillance program have an improved survival.
Children in families with Li-Fraumeni syndrome who survive an initial cancer have a relative risk of developing a second cancer that is 83 times greater than that of the general population. The risk for a second cancer increases with younger age at diagnosis of the first cancer.
A second cancer generally occurs 6-12 years after the first cancer. The cumulative probability of a person affected by Li-Fraumeni syndrome developing a second cancer is 57% at 30 years after developing the first cancer 27.
The risk for a second cancer increases with radiation exposure. Patients with Li-Fraumeni syndrome have a predilection for developing subsequent primary tumors (especially sarcomas) in prior radiation fields.
A study by Teepen et al evaluated individual chemotherapeutic agents and solid cancer risk in childhood cancer survivors and reported that compared to other cancers, the doxorubicin-breast cancer dose response was stronger in survivors of Li-Fraumeni syndrome-associated childhood cancers 28.
Individuals with Li-Fraumeni syndrome may also be prone to the carcinogenic risks associated with certain lifestyle or environmental exposures, such as tobacco smoking or radiation exposure. Li-Fraumeni syndrome patients should take preventive measures to reduce their exposures to behavioral risk factors and carcinogens.
- Li-Fraumeni Syndrome. https://emedicine.medscape.com/article/987356-overview[↩]
- Schneider K, Zelley K, Nichols KE, et al. Li-Fraumeni Syndrome. 1999 Jan 19 [Updated 2013 Apr 11]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1311[↩]
- Genetic Familial High-Risk Assessment: Colorectal Panel Members. Genetic Familial High-Risk Assessment: Colorectal. NCCN Clinical Practice Guidelines in Oncology. https://www.nccn.org/about/news/ebulletin/ebulletindetail.aspx?ebulletinid=294[↩]
- Lindor NM, McMaster ML, Lindor CJ, Greene MH, National Cancer Institute, Division of Cancer Prevention, Community Oncology and Prevention Trials Research Group (2008): Li-Fraumeni syndrome. In Concise Handbook of Familial Cancer Susceptibility Syndromes – Second Edition. J Natl Cancer Inst Monogr 38:80-5[↩]
- Wu CC, Shete S, Amos CI, Strong LC. Joint effects of germ-line p53 mutation and sex on cancer risk in Li-Fraumeni syndrome. Cancer Res. 2006;66:8287–92[↩][↩]
- Ruijs MW, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hagervorst EB, Kluijt I, Sijmons RH, Aalfs CM, Wagner A, Ausems MG, Hoogerbrugge N, van Asperen CJ, Gomez Garcia EB, Meijers-Heijboer H, Ten Kate LP, Menko FH, van’t Veer LJ. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: Mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47:421–8[↩][↩]
- Ognjanovic S, Olivier M, Bergemann TL, Hainaut P. Sarcomas in TP53 germline mutation carriers: A review of the IARC TP53 database. Cancer. 2012;118:1387–96[↩][↩]
- Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–50[↩][↩]
- Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM, Harries M, Jones PH, Binchy A, Crowther D, Craft A, Eden O, Evans D, Thompson E, Mann J, Martin J, Mitchell E, Santibanez-Koref M. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–304[↩]
- Masciari S, Dillon DA, Rath M, Robson M, Weitzel JN, Balmana J, Gruber SB, Ford JM, Euhus D, Lebensohn A, Telli M, Pochebit SM, Lypas G, Garber JE. Breast cancer phenotype in women with TP53 germline mutations: A Li-Fraumeni syndrome consortium effort. Breast Cancer Res Treat. 2012;133:1125–30[↩][↩]
- Melhem-Bertrandt A, Bojadzieva J, Ready KJ, Obeid E, Liu DD, Gutierrez-Barrera AM, Litton JK, Olopade OI, Hortobagyi GN, Strong LC, Arun BK. Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer. 2012;118:908–13[↩]
- Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, Eden OB, Varley JM. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001;20:4621–8[↩]
- Tabori U, Shlien A, Baskin B, Levitt S, Ray P, Alon N, Hawkins C, Bouffet E, Pienkowska M, Lafay-Cousin L, Gozali A, Zhukova N, Shane L, Gonzalez I, Finlay J, Malkin D. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol. 2010;28:1995–2001[↩]
- Chao MM, Levine JE, Ruiz RE, Kohlmann WK, Bower MA, Petty EM, Mody RJ. Malignant triton tumor in a patient with Li-Fraumeni syndrome and a novel TP53 mutation. Pediatr Blood Cancer. 2007;49:1000–4[↩]
- Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, Han JH, Lowstuter K, Longmate J, Sommer SS, Weitzel JN. Beyond Li-Fraumeni syndrome: Clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009b;27:1250–6[↩][↩][↩][↩][↩]
- Raymond VM, Else T, Everett JN, Long JM, Gruber SB, Hammer GD. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:E119–25[↩]
- Lustbader ED, Williams WR, Bondy ML, Strom S, Strong LC. Segregation analysis of cancer in families of childhood soft tissue sarcoma patients. Am J Hum Genet. 1992;51:344–56[↩][↩][↩]
- Nichols KE, Malkin D, Garber JE, Fraumeni JF Jr, Li FP. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev. 2001;10:83–7[↩]
- Wong P, Verselis SJ, Garber JE, Schneider K, DiGianni L, Stockwell DH, Li FP, Syngal S. Prevalence of early onset colorectal cancer in 397 patients with classic Li-Fraumeni syndrome. Gastroenterology. 2006;130:73–9[↩][↩]
- Hisada M, Garber JE, Fung CY, Fraumeni JF Jr, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90:606–11[↩]
- Masciari S, Dewanwala A, Stoffel EM, Lauwers GY, Zheng H, Achatz MI, Reigert-Johnson D, Foretova L, Silva EM, Digianni L, Verselis SJ, Schneider K, Li FP, Fraumeni J, Garber JE, Syngal S. Gastric cancer in individuals with Li-Fraumeni syndrome. Genet Med. 2011;13:651–7[↩]
- Hwang SJ, Cheng LS, Lozano G, Amos CI, Gu X, Strong LC. Lung cancer risk in germline p53 mutation carriers: association between an inherited cancer predisposition, cigarette smoking, and cancer risk. Hum Genet. 2003;113:238–43[↩]
- Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, Caron O, Bressac-de Paillerets B, Berthet P, Dugast C, Bonaiti-Pellie C, Stoppa-Lyonnet D, Frebourg T. 2009 Version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27:e108–9[↩]
- Varley JM, Evans DG, Birch JM. Li-Fraumeni syndrome: a molecular and cinical review. BR J Cancer. 1997;76:1–14[↩]
- Chompret A, Brugieres L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, Hua D, Ligot L, Dondon MG, Bressac-de Paillerets B, Frebourg T, Lemerle J, Bonaiti-Pellie C, Feunteun J. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000;82:1932–7[↩]
- Cancer Screening Recommendations forIndividuals with Li-Fraumeni Syndrome. Clin Cancer Res; 23(11) June 1, 2017, e38–e45. https://www.lfsassociation.org/wp-content/uploads/2017/06/e38.full_.pdf[↩]
- Hwang SJ, Lozano G, Amos CI, Strong LC. Germline p53 mutations in a cohort with childhood sarcoma: sex differences in cancer risk. Am J Hum Genet. 2003 Apr. 72(4):975-83.[↩]
- Teepen JC, van Leeuwen FE, Tissing WJ, et al. Long-Term Risk of Subsequent Malignant Neoplasms After Treatment of Childhood Cancer in the DCOG LATER Study Cohort: Role of Chemotherapy. J Clin Oncol. 2017 Jul 10. 35 (20):2288-2298[↩]


{kind=link}