Contents
What is progeria
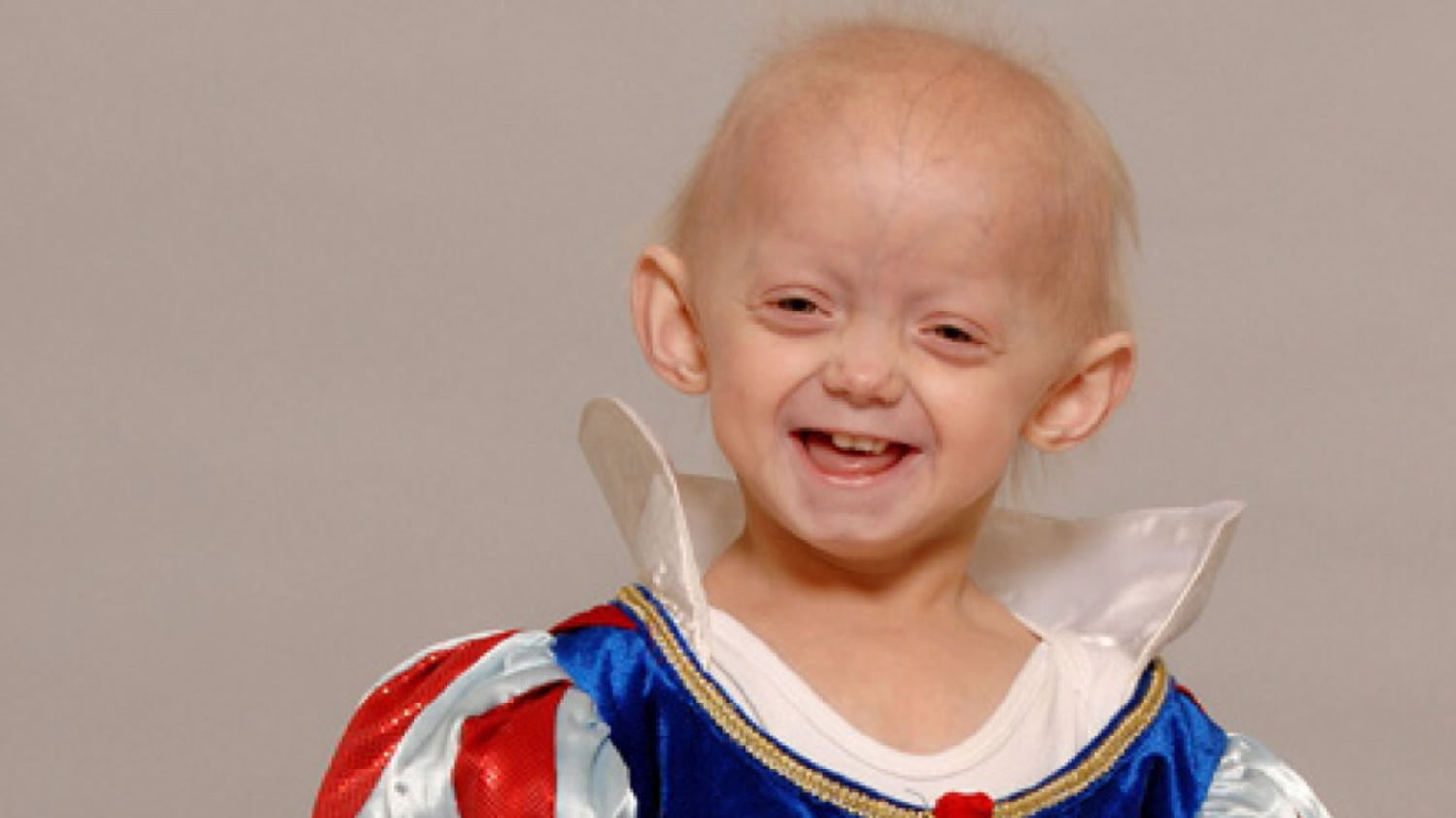
Hutchinson-Gilford progeria syndrome is an extremely rare genetic condition characterized by the dramatic, rapid appearance of aging beginning in childhood, starting in their first two years of life. Affected children typically look normal at birth and in early infancy, but then grow more slowly than other children and do not gain weight at the expected rate (failure to thrive). They develop a characteristic facial appearance including prominent eyes, a thin nose with a beaked tip, thin lips, a small chin, and protruding ears. Hutchinson-Gilford progeria syndrome also causes hair loss (alopecia), aged-looking skin, joint abnormalities, and a loss of fat under the skin (subcutaneous fat). Progeria signs include growth failure, loss of body fat and hair, aged-looking skin, stiffness of joints, hip dislocation, generalized atherosclerosis, cardiovascular (heart) disease and stroke. The children have a remarkably similar appearance, despite differing ethnic backgrounds. This condition does not affect intellectual development or the development of motor skills such as sitting, standing, and walking.
The term progeria is derived from the Greek word geras, meaning “prematurely old”. Significant morbidity and mortality result from accelerated atherosclerosis of the carotid and coronary arteries, leading to premature death during the first or second decade of life. Hutchinson Gilford progeria syndrome is considered a segmental aging syndrome, as affected patients do not manifest all of the typical features of aging, such as increased incidence of cancer and neurocognitive decline.
Hutchinson Gilford progeria syndrome is very rare; it is reported to occur in 1 in 20 million newborns worldwide. More than 130 cases have been reported in the scientific literature since the condition was first described in 1886 by Dr. Jonathan Hutchinson, and in 1897 by Dr. Hastings Gilford. There are an estimated 350-400 children living with Progeria worldwide at any one time. It affects both sexes equally and all races.
People with Hutchinson-Gilford progeria syndrome experience severe hardening of the arteries (atherosclerosis) beginning in childhood. This condition greatly increases the chances of having a heart attack or stroke at a young age. These serious complications can worsen over time and are life-threatening for affected individuals.
Heart problems or strokes are the eventual cause of death in most children with progeria. The average life expectancy for a child with progeria is about 13 years (with a range of about 8-21 years). Some with progeria may die younger and others may live longer, even up to 20 years.
There’s no cure for progeria, but ongoing research shows some promise for treatment.
Figure 1. Hutchinson-Gilford progeria syndrome
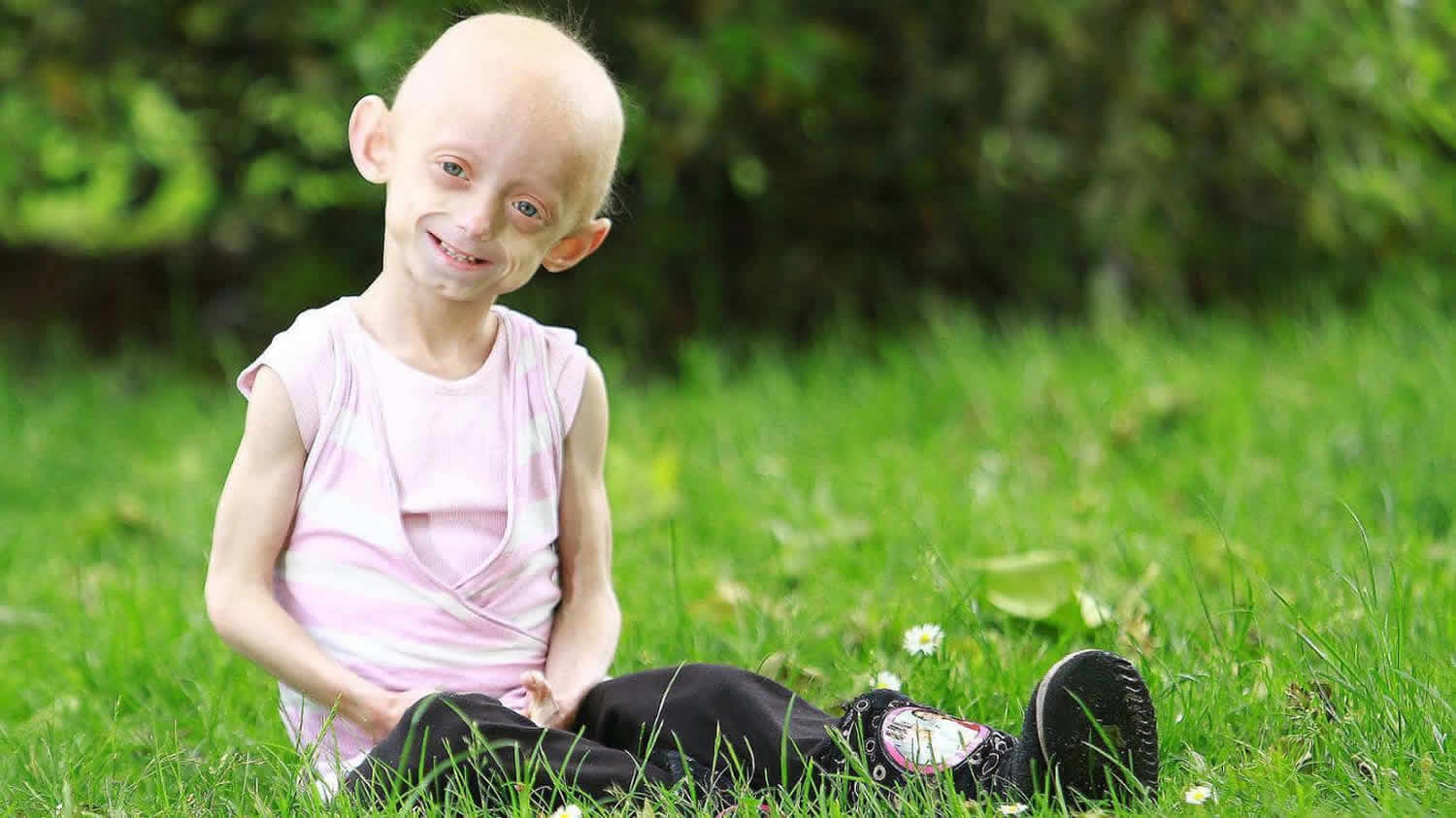
Hutchinson Gilford progeria syndrome is characterized by signs of premature aging most notable in the skin, cardiovascular system, and musculoskeletal systems. Hutchinson Gilford progeria syndrome is caused by mutations in LMNA gene that result in the production of an abnormal form of lamin A termed progerin.
Hutchinson Gilford progeria syndrome is not usually passed down in families. The gene change is almost always a chance occurrence that is extremely rare. Hutchinson Gilford progeria syndrome is a “sporadic autosomal dominant” mutation – sporadic because it is a new change in that family, and dominant because only one copy of the gene needs to be changed in order to have the syndrome. For parents who have never had a child with Progeria, the chances of having a child with Progeria are 1 in 20 million. But for parents who have already had a child with Progeria, the chances of it happening again to those parents is much higher – about 2-3%. Why the increase? This is due to a condition called “mosaicism”, where a parent has the genetic mutation for Progeria in a small proportion of their cells, but does not have Progeria. Prenatal testing is available to look for the LMNA genetic change that causes Hutchinson Gilford progeria syndrome.
Progeria life expectancy
The average life expectancy for a child with progeria is about 13 years. Some with progeria may die younger and others may live longer, even up to 20 years.
Progeria complications
Children with progeria usually develop severe hardening of the arteries (atherosclerosis). This is a condition in which the walls of the arteries — blood vessels that carry nutrients and oxygen from the heart to the rest of the body — stiffen and thicken, often restricting blood flow.
Most children with progeria die of complications related to atherosclerosis, including:
- Problems with blood vessels that supply the heart (cardiovascular problems), resulting in heart attack and congestive heart failure
- Problems with blood vessels that supply the brain (cerebrovascular problems), resulting in stroke
Other health problems frequently associated with aging — such as arthritis, cataracts and increased cancer risk — typically do not develop as part of the course of progeria.
Progeria causes
What causes progeria
Mutations in the LMNA gene cause Hutchinson-Gilford progeria syndrome. The LMNA gene provides instructions for making a protein called lamin A. This protein plays an important role in determining the shape of the nucleus within cells. It is an essential scaffolding (supporting) component of the nuclear envelope, which is the membrane that surrounds the nucleus. Mutations that cause Hutchinson-Gilford progeria syndrome result in the production of an abnormal version of the lamin A protein called progerin is produced and makes cells unstable. The altered protein (progerin) makes the nuclear envelope unstable and progressively damages the nucleus, making cells more likely to die prematurely. Researchers are working to determine how these changes lead to the characteristic features of Hutchinson-Gilford progeria syndrome.
Hutchinson-Gilford progeria syndrome is considered a “sporadic autosomal dominant” mutation – sporadic because it is a new change in that family, and dominant because only one copy of the gene needs to be changed in order to have the syndrome, which means one copy of the altered gene in each cell is sufficient to cause the disorder. The condition results from new mutations in the LMNA gene, and almost always occurs in people with no history of the disorder in their family.
For parents who have never had a child with Progeria, the chances of having a child with Progeria are 1 in 20 million. But for parents who have already had a child with Progeria, the chances of it happening again to those parents is much higher – about 2-3%. Why the increase? This is due to a condition called “mosaicism”, where a parent has the genetic mutation for Progeria in a small proportion of their cells, but does not have Progeria. Prenatal testing is available to look for the LMNA genetic change that causes Hutchinson Gilford progeria syndrome.
Other similar syndromes
There are other progeroid syndromes that do run in families. These inherited syndromes cause rapid aging and a shortened life span:
- Wiedemann-Rautenstrauch syndrome, also known as neonatal progeroid syndrome, starts in the womb, with signs and symptoms of aging apparent at birth.
- Werner syndrome, also known as adult progeria, begins in the teen years or early adulthood, causing premature aging and conditions typical of old age, such as cataracts and diabetes.
Risk factors for progeria
There are no known factors, such as lifestyle or environmental issues, which increase the risk of having progeria or of giving birth to a child with progeria. Progeria is extremely rare. For parents who have had one child with progeria, the chances of having a second child with progeria are about 2 to 3 percent.
Progeria symptoms
Usually within the first year of life, growth of a child with progeria slows markedly, but motor development and intelligence remain normal.
Signs and symptoms of this progressive disorder include a distinctive appearance:
- Slowed growth, with below-average height and weight
- Narrowed face, small lower jaw, thin lips and beaked nose
- Head disproportionately large for the face
- Prominent eyes and incomplete closure of the eyelids
- Hair loss, including eyelashes and eyebrows
- Thinning, spotty, wrinkled skin
- Visible veins
- High-pitched voice
Signs and symptoms also include health issues:
- Severe progressive heart and blood vessel (cardiovascular) disease
- Hardening and tightening of skin on the trunk and extremities (similar to scleroderma)
- Delayed and abnormal tooth formation
- Some hearing loss
- Loss of fat under the skin and loss of muscle mass
- Skeletal abnormalities and fragile bones
- Stiff joints
- Hip dislocation
- Insulin resistance
Although they are usually born looking healthy, most children with Progeria begin to display many characteristics of Progeria within the first 2 years of life. Progeria signs include growth failure, loss of body fat and hair, aged-looking skin and stiffness of joints. As children get older, they suffer from osteoporosis, generalized atherosclerosis, cardiovascular (heart) disease and stroke. The children have a remarkably similar appearance despite differing ethnic backgrounds. Children with Progeria die of atherosclerosis (heart attacks or strokes) at an average age of fourteen years.
Progeria diagnosis
Doctors may suspect progeria based on signs and symptoms characteristic of the syndrome. A genetic test for LMNA mutations can confirm the diagnosis of progeria.
A thorough physical exam of your child includes:
- Measuring height and weight
- Plotting measurements on a normal growth curve chart
- Testing hearing and vision
- Measuring vital signs, including blood pressure
- Looking for visible signs and symptoms that are typical of progeria
Don’t hesitate to ask questions. Progeria is a very rare disease, and it’s likely that your doctor will need to gather more information before determining next steps in caring for your child. Your questions and concerns can help your doctor develop a list of topics to investigate.
Progeria treatment
There’s no cure for progeria, but regular monitoring for heart and blood vessel (cardiovascular) disease may help with managing your child’s condition.
During medical visits, your child’s weight and height is measured and plotted on a chart of normal growth values. Additional regular evaluations, including electrocardiograms and dental, vision and hearing exams, may be recommended by your doctor to check for changes.
Certain therapies may ease or delay some of the signs and symptoms.
Treatments depend on your child’s condition and symptoms. These may include:
- Low-dose aspirin. A daily dose may help prevent heart attacks and stroke.
- Other medications. Depending on your child’s condition, the doctor may prescribe other medications, such as statins to lower cholesterol, drugs to lower blood pressure, anticoagulants to help prevent blood clots, and medications to treat headaches and seizures.
- Physical and occupational therapy. These therapies may help with joint stiffness and hip problems to help your child remain active.
- Nutrition. Nutritious, high-calorie foods and supplements can help maintain adequate nutrition.
- Dental care. Dental problems are common in progeria. Consultation with a pediatric dentist experienced with progeria is recommended.
Potential future treatment
Current research seeks to understand progeria and identify new treatment options. Some areas of research include:
- Studying genes and the course of the disease to understand how it progresses. This may help identify new treatments.
- Studying ways to prevent heart and blood vessel disease.
- Performing human clinical trials using drugs known as farnesyltransferase inhibitors (FTIs), such as lonafarnib, which were developed for treating cancer, but may be effective for treatment of progeria by helping with weight gain and increased flexibility of blood vessels.
- Testing other drugs for treatment of progeria.
Home remedies
Here are some steps you can take at home to help your child:
- Make sure your child stays well-hydrated. Dehydration can be more serious in children with progeria. Be sure your child drinks plenty of water, especially during an illness, with activity or in hot weather.
- Provide frequent, small meals. Because nutrition and growth can be an issue for children with progeria, giving your child smaller meals more often may help increase calorie intake. Add healthy, high-calorie foods and snacks or supplements as needed.
- Provide opportunities for regular physical activity. Check with your child’s doctor to learn which activities are appropriate for your child.
- Get cushioned shoes or shoe inserts for your child. The loss of body fat in the feet can cause discomfort.
- Use sunscreen. Use a broad-spectrum sunscreen with an SPF of at least 30+. Apply sunscreen generously, and reapply every two hours — or more often if your child is swimming or perspiring.
- Make sure your child is up to date on childhood immunizations. A child with progeria isn’t at increased risk of infection, but like all children, is at risk if exposed to infectious diseases.
- Provide learning and social opportunities. Progeria won’t affect your child’s intellect, so he or she can attend school at an age-appropriate level. Some adaptations for size and ability may be needed.
- Make adaptations. You may need to make some changes at home that enable your child to have some independence and to be comfortable. These can include household changes so that your child can reach items such as faucets or light switches, clothes with special closures or in special sizes, and extra padding for chairs and beds.
Coping and support
Learning that your child has progeria can be emotionally devastating. Suddenly you know that your child is facing many difficult challenges and a shortened life span. For you and your family, coping with the disorder involves a major commitment of physical, emotional and financial resources.
Some helpful resources include:
- Support network. Your health care team, family and friends can all be a valuable part of your support network. Also, ask your doctor about self-help groups or therapists in your community. Your local health department, public library and trustworthy sources on the internet may be helpful in finding resources.
- Support groups. In a support group, you’ll be with people who are facing challenges similar to yours. If you can’t find a progeria support group, you may be able to find a group for parents of children with chronic illness.
- Other families dealing with progeria. The Progeria Research Foundation 1 may be able to help you connect with other families coping with progeria.
- Therapists. If a group isn’t for you, talking to a therapist or clergy member may be beneficial.
Helping your child cope
If your child has progeria, he or she is also likely to increasingly feel different from others as the condition progresses. Over time, fear and grief will likely increase as awareness grows that progeria shortens life span. Your child will need your help coping with physical changes, special accommodations, other people’s reactions and eventually the concept of death.
Your child may have difficult but important questions about his or her condition, spirituality and religion. Your child may also ask questions about what will happen in your family after he or she dies. Siblings may have these same questions.
For such conversations with your child:
- Ask your doctor, therapist or clergy member to help you prepare.
- Consider input or guidance from friends you meet through support groups who’ve shared this experience.
- Talk openly and honestly with your child and his or her siblings, and offer reassurance that’s compatible with your belief system and appropriate to the child’s age.
- Recognize when your child or his or her siblings might benefit from talking to a therapist or clergy member.
- The Progeria Research Foundation. https://www.progeriaresearch.org/[↩]





{kind=link}