
Contents
What is Sanfilippo syndrome
Sanfilippo syndrome also known as mucopolysaccharidosis type III (MPS 3), is a rare progressive genetic disorders caused by the deficiency of one of the enzymes needed to break down complex sugar molecules called mucopolysaccharides or called glycosaminoglycans (GAGs). Sanfilippo syndrome (mucopolysaccharidosis type III) is caused by a lack of enzymes to break down a particular mucopolysaccharide called heparan sulfate. Mucopolysaccharides get their name from their thick jelly-like consistency (‘muco’), ‘poly’ meaning many and ‘saccharide’ meaning sugar. Mucopolysaccharides are long chains of sugar molecules, which are used in building bones, cartilage, skin, and connective tissues. When the body is finished using these molecules, it breaks them down, with enzymes, and disposes of them. Children with Sanfilippo syndrome (mucopolysaccharidosis type III) are missing or are deficient in the enzyme to break down the molecules. Instead, the body stores these glycosaminoglycans (GAGs) in the cells. This storage causes progressive damage. High concentrations of mucopolysaccharides or glycosaminoglycans (GAGs) in the cells of the central nervous system, including the brain, cause the neurological and developmental problems that accompany these disorders.
Mucopolysaccharidoses are part of a larger group of disorders known as “lysosomal storage disorders” because the enzymes that break down mucopolysccharides are found within a compartment of the cell called the lysosome. The lysosome is often called the “waste disposal plant” of the cell. There are more than 50 lysosomal storage disorders.
Sanfilippo syndrome (mucopolysaccharidosis type III) is divided into four types IIIA, IIIB, IIIC, and IIID, which are distinguished by their genetic cause. Sanfilippo syndrome is the most common form of mucopolysaccharidosis; the estimated incidence of all four types combined is 1 in 70,000 newborns. Sanfilippo syndrome type A (MPS IIIA) and Sanfilippo syndrome type B (MPS IIIB) are much more common than Sanfilippo syndrome type C (MPS IIIC) and Sanfilippo syndrome type D (MPS IIID). The different types of Sanfilippo syndrome (mucopolysaccharidosis type III) have similar signs and symptoms, although the features of Sanfilippo syndrome type A (MPS IIIA) typically appear earlier in life and progress more rapidly. People with Sanfilippo syndrome (mucopolysaccharidosis type III) usually live into adolescence or early adulthood.
- Sanfilippo Type A is the most common. It is considered the most severe type with earlier death than the others. These children are deficient in the enzyme Heparan N-sulfatase.
- Sanfilippo Type B is the second most common. This is the result of a deficiency in N-acetyl-alpha-D-glucosaminidase.
- Sanfilippo Type C is caused by a deficiency in Acetyl-CoAlpha-glucosaminide acetyltransferase.
- Sanfilippo Type D is caused by a deficiency in N-acetylglucosamine 6-sulfatase.
Sanfilippo syndrome (mucopolysaccharidosis type III) is classified as a rare disease with incidence reported to be between 0.28 and 4.1 cases per 100,000 births 1. Sanfilippo syndrome type A (MPS IIIA) is the most common subtype affecting around 1 in 100 000 births, closely followed by type B at 1 in 200,000 1. In some countries in Southern Europe, Sanfilippo syndrome type B has been reported to be more common than A. Sanfilippo syndrome type C (MPS IIIC) and Sanfilippo syndrome type D (MPS IIID) are rarer with reported incidences of approximately 1 in 1.5 million and 1 in 1 million births respectively.
Sanfilippo syndrome (mucopolysaccharidosis type III) primarily affects the brain and spinal cord (central nervous system). Other body systems can also be involved. People with Sanfilippo syndrome (mucopolysaccharidosis type III) generally do not display any features of the condition at birth, but they begin to show signs and symptoms of the disorder during early childhood. Affected children often initially have delayed speech and behavior problems. They may become restless, destructive, anxious, or aggressive, and some display features of autism spectrum disorder, which is a condition characterized by difficulty with social interactions and communication. Sleep disturbances are also very common in children with Sanfilippo syndrome (mucopolysaccharidosis type III). This condition causes progressive intellectual disability and the loss of previously acquired skills (developmental regression). In later stages of the disorder, people with Sanfilippo syndrome (mucopolysaccharidosis type III) may develop seizures and movement disorders.
The physical features of Sanfilippo syndrome (mucopolysaccharidosis type III) are less pronounced than those of other types of mucopolysaccharidosis. Individuals with Sanfilippo syndrome (mucopolysaccharidosis type III) typically have mildly “coarse” facial features, a large head (macrocephaly), a slightly enlarged liver (mild hepatomegaly), and a soft out-pouching around the belly-button (umbilical hernia) or lower abdomen (inguinal hernia). Some people with Sanfilippo syndrome (mucopolysaccharidosis type III) have short stature, joint stiffness, or mild dysostosis multiplex, which refers to multiple skeletal abnormalities seen on x-ray. Affected individuals often experience chronic diarrhea and recurrent upper respiratory and ear infections. People with Sanfilippo syndrome (mucopolysaccharidosis type III) may also have hearing loss and vision problems.
Sanfilippo syndrome causes
Mutations in the GNS, HGSNAT, NAGLU, and SGSH genes cause Sanfilippo syndrome (mucopolysaccharidosis type III). These genes provide instructions for making enzymes involved in the breakdown of large sugar molecules called glycosaminoglycans (GAGs). Glycosaminoglycans (GAGs) were originally called mucopolysaccharides, which is where this condition gets its name. The GNS, HGSNAT, NAGLU, and SGSH enzymes are involved in the step-wise breakdown of a subset of glycosaminoglycans called heparan sulfate.
TYPE..……….GENE…………ENZYME
MPS IIIA……..SGSH…………heparan N-sulfatase
MPS IIIB……..NAGLU……….alpha-N-acetylglucosaminidase
MPS IIIC……..HGSNAT……..heparan-alpha-glucosaminide N-Acetyltransferase
MPS IIID……..GNS………….N-acetylglucosamine 6-sulfatase
Sanfilippo syndrome type A (MPS IIIA) is caused by mutations in the SGSH gene, and Sanfilippo syndrome type B (MPS IIIB) is caused by NAGLU gene mutations. Mutations in the HGSNAT gene result in Sanfilippo syndrome type C (MPS IIIC) and GNS gene mutations cause Sanfilippo syndrome type D (MPS IIID). Mutations in these genes reduce or eliminate enzyme function. A lack of any one of these enzymes disrupts the breakdown of heparan sulfate. As a result, partially broken down heparan sulfate accumulates within cells, specifically inside the lysosomes. Lysosomes are compartments in the cell that digest and recycle different types of molecules. Conditions such as MPS III that cause molecules to build up inside the lysosomes are called lysosomal storage disorders. Researchers believe that the accumulation of glycosaminoglycans (GAGs) interferes with the functions of other proteins inside the lysosomes and disrupts the normal functions of cells. It is unknown why the buildup of heparan sulfate mostly affects the central nervous system in Sanfilippo syndrome (mucopolysaccharidosis type III).
Sanfilippo syndrome inheritance pattern
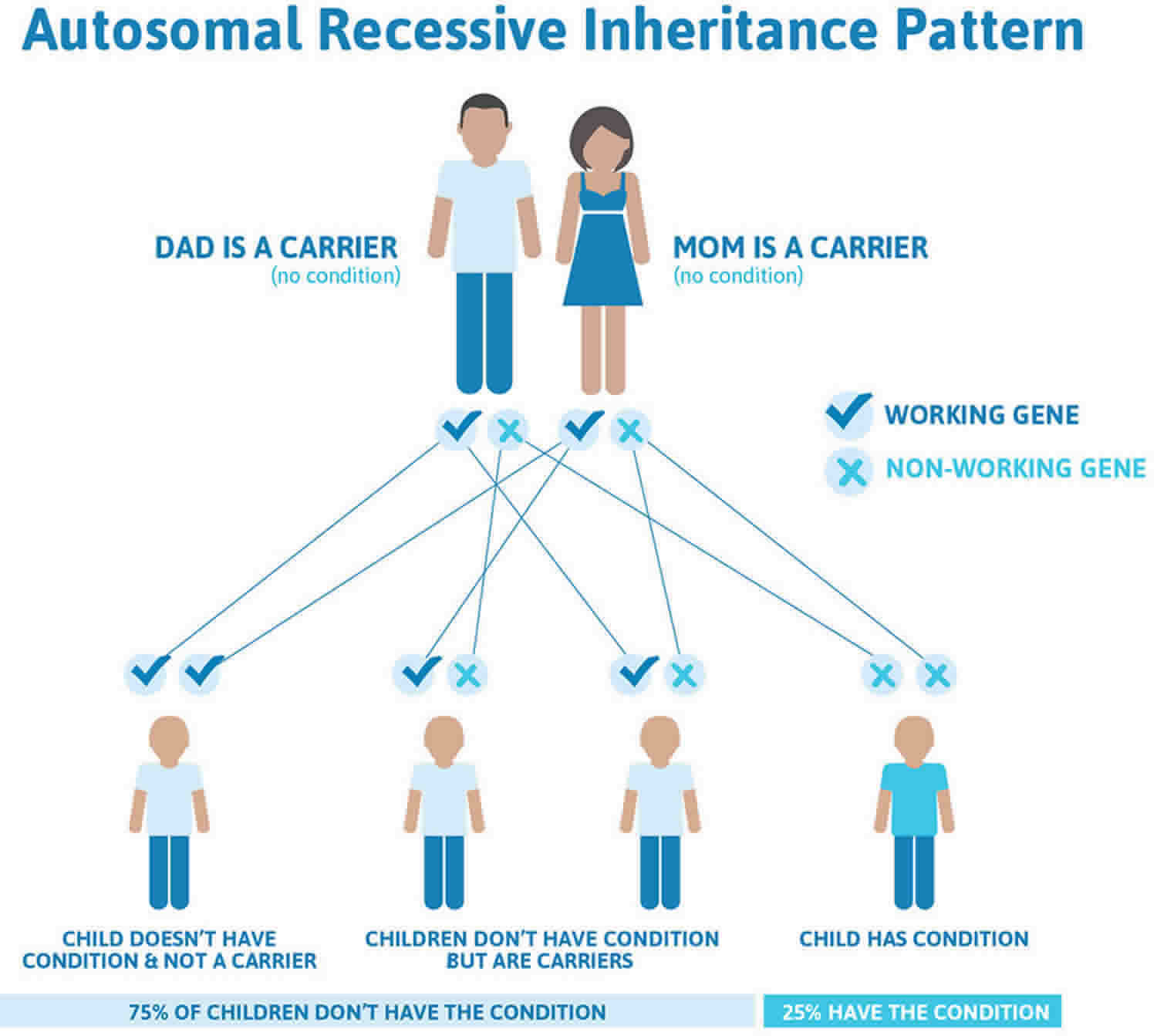
Sanfilippo syndrome (mucopolysaccharidosis type III) is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. This means that both parents have one copy of the altered gene and one normal copy – they are known as carriers and do not show signs of the condition. A child with Sanfilippo syndrome (mucopolysaccharidosis type III) inherits two copies of the altered gene, one from each parent.
In autosomal recessive inheritance, in each pregnancy of a couple who are both carriers, there is a:
- 25% (1 in 4) chance of having an affected child
- 50% (1 in 2) chance of a child receiving only one copy of the altered gene and therefore being a carrier
- 25% (1 in 4) chance that a child will be neither affected nor a carrier.
- The risk is the same for males and females.
Figure 1. Sanfilippo syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Sanfilippo syndrome symptoms
Children with Sanfilippo syndrome (mucopolysaccharidosis type III) usually appear normal at birth, but developmental delay is usually evident by age 2-5 years. Mental and motor development reach a peak by 3-6 years of age after which behavioral disturbances and intellectual decline usually occur. However, behavioral problems such as hyperactivity and irritability may become obvious earlier. Severe behavioral disturbance is a very common feature of Sanfilippo syndrome, and one of the more difficult aspects of the disorder to manage.
Sanfilippo syndrome early symptoms:
- Speech delay
- Recurrent ear/sinus infections
- Large head size
- Diarrhea/chronic loose stool
- Poor sleep
- Speech/developmental delays
- “Autistic” behaviors
- Significant hyperactivity
- Respiratory issues at birth
Other symptoms can be coarse hair, excess hair growth (hirsutism), slightly coarse facial features, sleeping problems, mildly enlarged liver and/or spleen, speech delay, respiratory and ear infections, diarrhea, hernias, seizures, and a wobbly and erratic walk. Hearing loss and vision impairment may develop in some children.
Children with Sanfilippo syndrome usually start to lose their intellectual functions, especially speech before their motor function declines. There is great variability in the rate of regression, even among siblings, with some showing rapid loss of function and others who exhibit a much slower progression of the disease. Death can occur from before the age of 10 until the third or fourth decades of life, with the average being around 15 to 20 years of age.
Sanfilippo syndrome is often diagnosed when parents find their child missing developmental milestones, between ages 2 to 6, and undertake formal assessment. Because of its rarity, the disease remains largely unfamiliar to most medical practitioners. It is not uncommon that parents search for years to find the cause of their child’s difficulties before a correct diagnosis is made. Sanfilippo syndrome can masquerade as ADHD (attention deficit hyperactivity disorder), autism spectrum disorder, and intellectual disability. Because of this delay in finding the underlying diagnosis, many families have had other children born in the meantime only to find out that their younger children are also affected with Sanfilippo disease.
As Sanfilippo syndrome progresses, children will typically develop extreme hyperactivity and behavioral problems. Sleep problems become so severe that they may not sleep for days or for only a few hours per night. They gradually lose all the abilities and skills they had acquired, such as speaking, walking, and the ability to eat by mouth.
Sanfilippo syndrome later features:
- Progressive intellectual disability
- Seizures
- Enlarged liver/spleen
- Prominent/thick eyebrows
- Hearing loss (may occur earlier)
- Loss of mobility – walking, running, sitting unsupported
- Loss of ability to eat by mouth
- Movement disorders, dystonia
Sanfilippo syndrome diagnosis
To diagnose Sanfilippo syndrome (mucopolysaccharidosis type III), glycosaminoglycans (GAGs) are usually first measured in urine, followed by measurement of enzyme activity in blood or a small skin sample. Increased heparan sulfate in urine, and a decrease in the activity of any one of the four enzymes (shown in the table above) is usually consistent with a diagnosis of Sanfilippo syndrome and will identify the Sanfilippo syndrome type A, B, C or D. It is important to know the Sanfilippo syndrome type as this will make genetic testing easier and importantly, many of the treatments being developed are only for specific types.
Genetic testing of a blood sample will allow the identification of the exact changes in the DNA. It is important to attend genetic counseling to learn the implications for other children in the family, future pregnancies and extended family members. The counselor will explain the inheritance pattern and help advise who should be tested.
If the genetic diagnosis is known, this information can be used to test other at risk members of the family. It can also be used for prenatal testing of future pregnancies (testing a fetus while still in the womb) and/or preimplantation diagnosis (testing of embryos created through IVF to select those that do not carry the relevant gene mutation).
Sanfilippo syndrome treatment
Treatment of Sanfilippo syndrome is symptomatic and supportive. It is important for children with Sanfilippo syndrome (mucopolysaccharidosis type III) to be managed by a multidisciplinary team of specialists to give these children the best quality of life. At different stages this could include a combination of the following: a neurologist, developmental pediatrician, metabolic/genetics specialist, orthopedics, gastroenterologist, ophthalmologist, cardiologist, endocrinologist, allied health (e.g. physiotherapy, OT, behavioral therapists) and an ENT (ear, nose and throat) specialist.
Sanfilippo syndrome cure
Sanfilippo syndrome does not yet have a cure yet. At this time, only supportive or “palliative” care is available. For children living with Sanfilippo today, participation in a clinical trial is their only opportunity to try a treatment. Unfortunately, these trials only accept an extremely small number of patients, and time is not something these children have on their side.
Clinical trials designed to gauge the safety and efficacy of several different approaches are under way. Therapeutic approaches include:
- Gene therapy which involves using a harmless virus to deliver a functional copy of the altered gene into the body
- Enzyme replacement therapy, where the missing enzyme is administered
- Substrate reduction therapy: researchers are searching for drugs which could reduce the amount of mucopolysaccharides produced by the body
- Stem cell therapy, which although promising is in the early stages of lab based research
Information on current clinical trials is posted on the Internet at https://clinicaltrials.gov . All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu
Sanfilippo syndrome prognosis
Sanfilippo syndrome has its most devastating effects on the brain. Children experience hyperactivity, sleeplessness, loss of speech, loss of toileting skills, intellectual disability, heart problems, vision loss, seizures, loss of mobility, dementia and finally death.
The disease progression can vary significantly from one child to the next, making it particularly difficult to predict. Symptoms cover a wide spectrum and some children may experience particular symptoms more than others.
Sanfilippo syndrome life expectancy
People with Sanfilippo syndrome (mucopolysaccharidosis type III) usually live into adolescence or early adulthood. Death can occur from before the age of 10 until the third or fourth decades of life, with the average being around 15 to 20 years of age. Life expectancy is typically around 15 years 2.


{kind=link}