Contents
5 alpha reductase deficiency
5-alpha reductase deficiency also called 5 alpha-reductase type 2 deficiency, is a rare inherited condition that primarily affects male sexual development before birth and during puberty. 5 alpha reductase deficiency (5 alpha-reductase type 2 deficiency) is one of the important causes of ambiguous genitalia in children. The phenotype of children with 5 alpha reductase deficiency can vary from under-developed male genitalia to a complete female phenotype 1. People with 5-alpha reductase deficiency are genetically male, with one X and one Y chromosome in each cell, and they have male gonads (testes). Their bodies, however, do not produce enough of a hormone called dihydrotestosterone (DHT). DHT (dihydrotestosterone) has a critical role in male sexual development, and a shortage of this hormone disrupts the formation of the external sex organs before birth.
Many people with 5-alpha reductase deficiency are born with external genitalia that appear female. In other cases, the external genitalia do not look clearly male or clearly female sometimes called ambiguous genitalia. Still other affected infants have genitalia that appear predominantly male, often with an unusually small penis (micropenis) and the urethra opening on the underside of the penis (hypospadias).
During puberty, an increase in the levels of male sex hormones leads to the development of some secondary sex characteristics, such as increased muscle mass, deepening of the voice, development of pubic hair, and a growth spurt. The penis and scrotum (the sac of skin that holds the testes) grow larger. Unlike many men, people with 5-alpha reductase deficiency do not develop much facial or body hair. Most affected individuals are unable to have biological children without assisted reproduction.
Children with 5-alpha reductase deficiency are often raised as girls. Some of these individuals adopt a male gender role in adolescence or early adulthood, while others adopt a female gender role.
5-alpha reductase deficiency is a rare condition with the prevalence of 1 in 4500 live births 2. Even with the recent advances in the technology like genetic workup, assay of hormones, karyotyping, only 20 to 40% of the time can the diagnosis be made in these children 3. 5 alpha reductase deficiency is very rare among Caucasians. Due to a large number of consanguineous marriages, there is a high prevalence of 5-alpha reductase deficiency in the population of the Dominican Republic; the first reported case of 5-alpha reductase deficiency was also from the same island. Apart from the Caribbean, 5-alpha reductase deficiency is also present in southern Lebanon, Turkey, Egypt and Papua New Guinea 4.
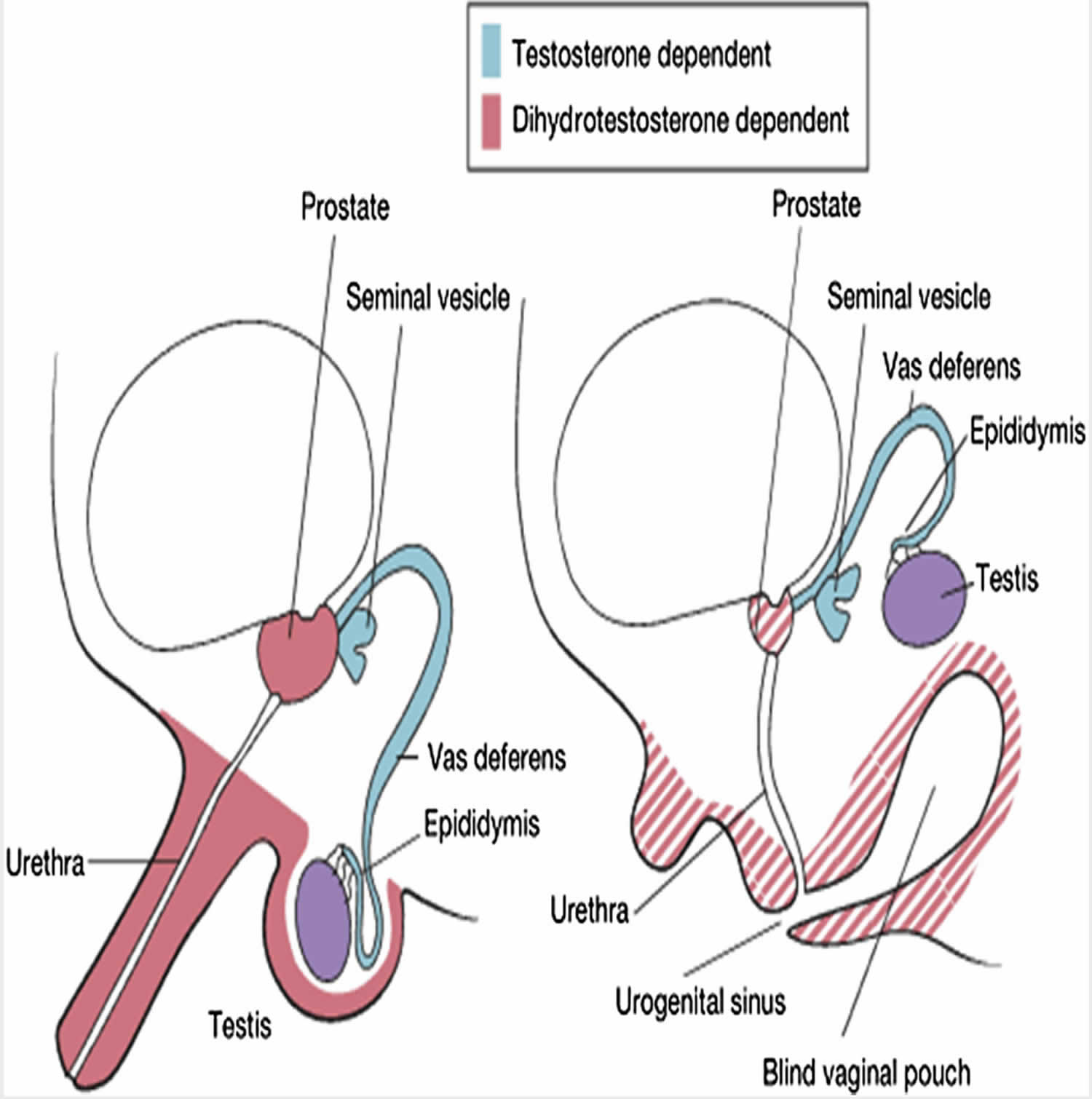
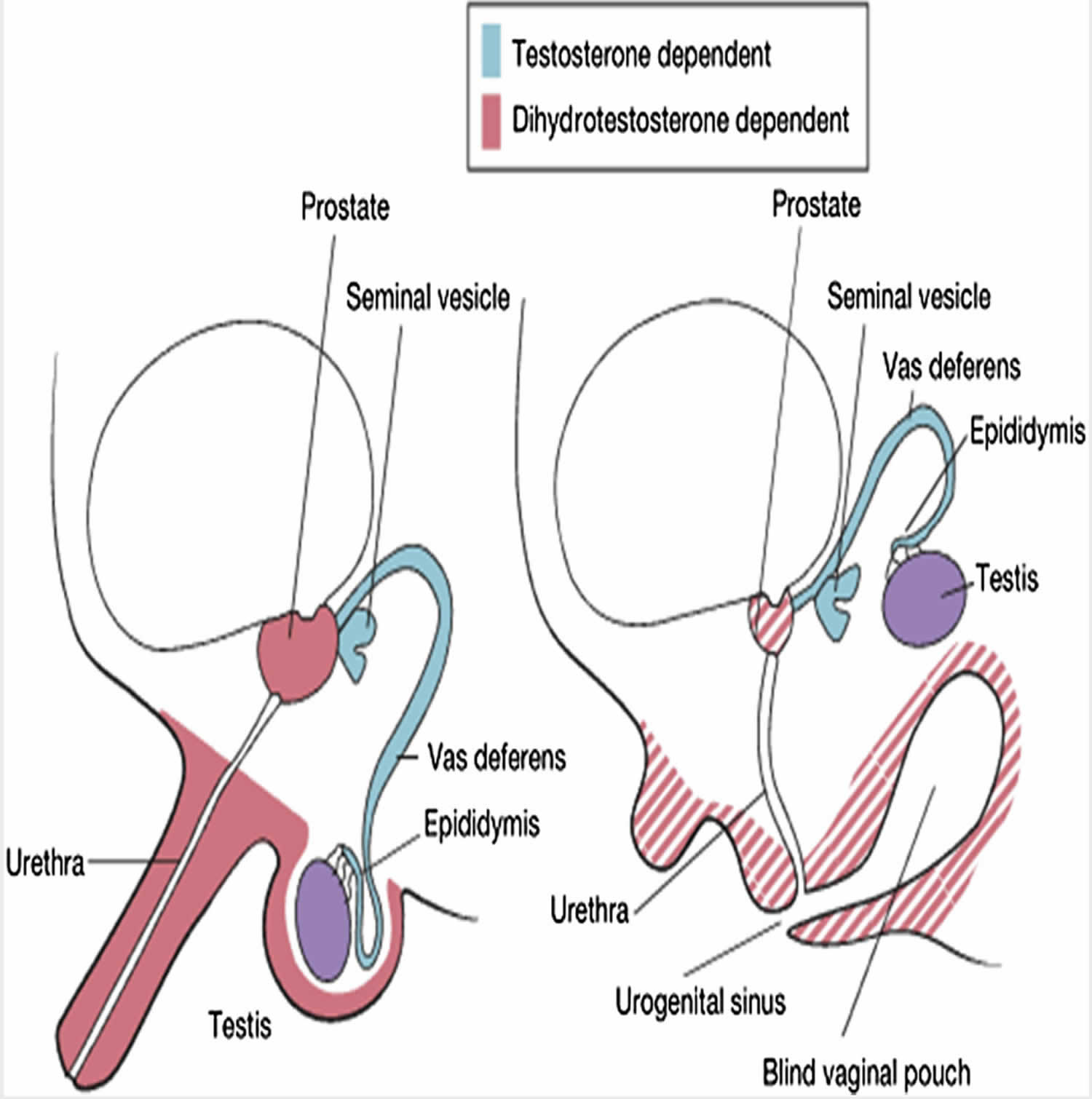
Figure 1. 5 alpha reductase deficiency
5 alpha reductase deficiency causes
Mutations in the SRD5A2 gene cause 5-alpha reductase deficiency. The SRD5A2 gene provides instructions for making an enzyme called steroid 5-alpha reductase 2. This enzyme is involved in processing androgens, which are hormones that direct male sexual development. Specifically, the enzyme is responsible for a chemical reaction that converts the hormone testosterone to dihydrotestosterone. dihydrotestosterone is essential for the normal development of male sex characteristics before birth, particularly the formation of the external genitalia.
Mutations in the SRD5A2 gene prevent steroid 5-alpha reductase 2 from effectively converting testosterone to dihydrotestosterone in the developing reproductive tissues. These hormonal factors underlie the changes in sexual development seen in infants with 5-alpha reductase deficiency.
During puberty, the testes produce more testosterone. Researchers believe that people with 5-alpha reductase deficiency develop secondary male sex characteristics in response to higher levels of this hormone. Some affected people also retain a small amount of 5-alpha reductase 2 activity, which may produce dihydrotestosterone and contribute to the development of secondary sex characteristics during puberty.
5 alpha reductase deficiency inheritance pattern
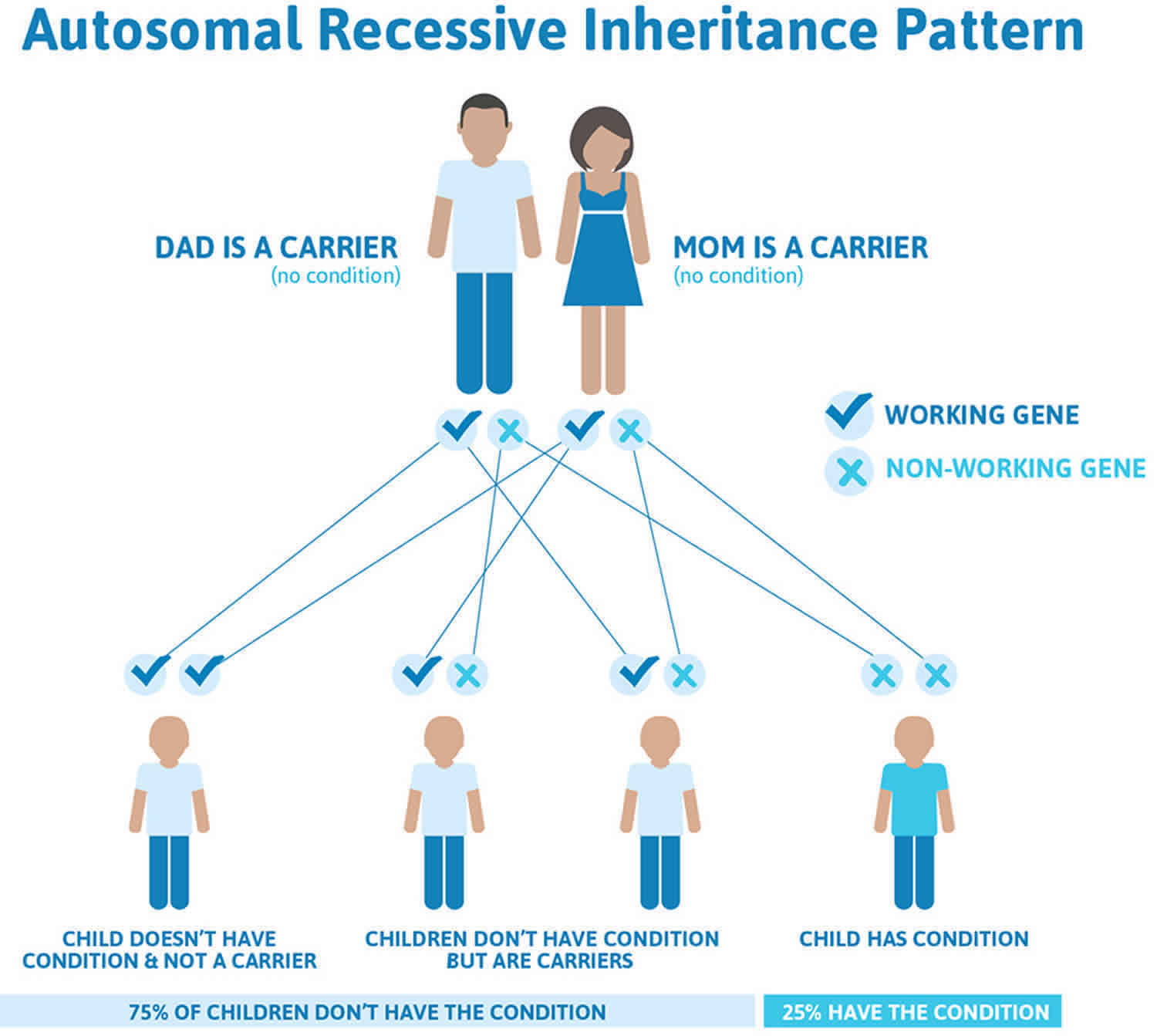
5 alpha reductase deficiency is inherited in an autosomal recessive pattern, which means both copies of the SRD5A2 gene in each cell have a mutation. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they do not show signs and symptoms of the condition.
Although people who are genetically female (with two X chromosomes in each cell) may inherit mutations in both copies of the SRD5A2 gene, their sexual development is not affected. The development of female sex characteristics does not require dihydrotestosterone (DHT), so a lack of steroid 5-alpha reductase 2 activity does not cause physical changes in these individuals. Only people who have mutations in both copies of the SRD5A2 gene and are genetically male (with one X and one Y chromosome in each cell) have the characteristic signs of 5-alpha reductase deficiency.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. 5 alpha reductase deficiency autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
5 alpha reductase deficiency pathophysiology
The human embryo possesses the capability to develop and differentiate into both male and female reproductive systems. By the timeline of the 6th week of gestation, the fetus would have developed the primordial structures that could either form a male or female reproductive system. In the absence of testosterone, the fetus would develop the female reproductive system. Differentiation of the primordial undifferentiated gonadal ridge into the male reproductive system initiates during the embryonic life by SRY gene located on the short arm of the Y chromosome. Testosterone produced by the testes leads to differentiation of Wolffian duct in males to form the male internal genitalia including seminal vesicles, epididymis and vas deferens. The overlying skin on the external genitalia has the activity of 5alpha-reductase deficiency activity which helps in conversion of testosterone to di-hydro testosterone. Dihydrotestosterone will then start forming the external genitalia. By 12 weeks of gestation, the male external genitalia differentiates. After 12 weeks the penis starts to grow in size and testes begins its descent into the scrotum 5.
5alpha-reductase deficiency deficiency is an inherited autosomal recessive disease. The deficiency is due to mutations in the gene which encodes for the enzyme 5α-reductase deficiency on the short arm of chromosome 2. Up to 40 different types of mutations have been reported leading to this disease. The levels of dihydrotestosterone will be low in these children, which will affect the sexual differentiation of male external genitalia. Although the genotype of these children is 46 XY, the phenotype varies from case to case 4.
5 alpha reductase deficiency symptoms
The presentation of patients with deficiency of 5 alpha reductase deficiency can be varying. Two individuals with the same gene defects in 5 alpha reductase deficiency can present with completely different phenotype, which clearly shows that there are other additional genes which probably controls the phenotype along with the gene under discussion 6.
Diagnosis of 5 alpha reductase deficiency is usually made in the newborn period when the infant presents with ambiguous genitalia 7. The newborns might have genitalia resembling labia majora which in reality would be an unfused labioscrotal folds. The phallus in these children may more look like a clitoris than a penis 8. At the same time, the internal genitalia in these children will include seminal vesicles, epididymis, vas deferens, and ejaculatory duct and one may not see any Mullerian structures. The testes in these children might be present in the inguinal sac, and very rarely they can also be found within the abdomen. These children tend to be raised as females till puberty, by which time they start exhibiting virilization 4. At puberty the phallus may grossly enlarge to form a penis, the testes may descend into the unfused labioscrotal folds, the voice deepens, and the beard starts growing. Development of all these secondary sexual characteristics during puberty does not need the presence of DHT, but only the presence of testosterone 8.
Neonatal presentation
Phenotypic findings in a newborn are limited to the genitalia 9. The spectrum of findings ranges from minimal undervirilization presenting with normal male anatomy, except for isolated micropenis or hypospadias, to severe undervirilization presenting as normal female external genitalia with mild clitoral enlargement as the only physical finding. In a study published in 2011, a cohort of 55 patients with 5 alpha reductase deficiency displayed widely varied phenotypes in the neonatal period. In this cohort, clitoromegaly was the most common phenotype in 49% of patients, followed by microphallus with varying degrees of hypospadias in 33% of patients 10.
Most commonly, the external genitalia exhibit labial appearance to the labioscrotal folds with some mild rugation or pigmentation present in some patients.
The phallus is often indeterminate in size and morphology. Its length falls between 1 cm (ie. usual maximum for a clitoris) and 2 cm (ie. lower limit of normal for a penis).
The urethra may empty anywhere from the tip of the phallus to the perineum, with the latter observed more frequently.
The testes are usually in the inguinal canals bilaterally; however, in some individuals with 5 alpha reductase deficiency, the testes can be found in the labioscrotal folds or retained in the abdomen.
A pseudovaginal blind-ending introitus may be present with a normal hymen.
The uterus and cervix are absent on imaging and by examination.
The rest of the examination findings are within normal limits.
Presentation of 46XY males at puberty
Clear signs of virilization predominate at puberty. The escutcheon is male in distribution. The phallus exhibits definite enlargement. The shoulders are relatively broad and the hips are narrow.
Muscularity and body hair may increase. Generally, no breast development is present. A prominentia laryngea (Adam’s apple) may start to develop. In addition, facial hair develops and the child’s voice may begin to deepen.
The mucosa of the vaginal introitus remains atrophic in appearance (remaining red) rather than the thickened pink of estrogen-stimulated mucosa.
Presentation of 46XX females
Females homozygous for 5 alpha reductase deficiency have very subtle manifestations, such as delayed menarche, minimal acne, and minimal body hair 11. Fertility is normal in females.
Gender identity problems in children with 5 alpha reductase deficiency
Although some children with 5 alpha reductase deficiency are raised as girls, many would change their gender to males at puberty after virilization. There are multiple factors like the culture and environmental pressures which can influence the gender changes in these children, but the most significant factor responsible for the gender change is the exposure of the child’s brain to androgen, including testosterone and not to the female sex hormone. A brain exposed to androgens will develop more masculine behavior and influences the child to identify itself as a male child rather than a female 12.
5 alpha reductase deficiency diagnosis
Initial evaluation should begin with a careful history 13. Assess prenatal and maternal past medical and family history. Specific questions should be asked regarding relatives with disorders of sexual development, neonatal deaths, amenorrhea, or infertility and consanguinity. Parental consanguinity increases the child’s risk for autosomal recessive disorders including 5-alpha reductase type 2 deficiency.
No risk factors or clinical markers in pregnancy are known. Genital ambiguity is occasionally diagnosed prenatally when an infant who is demonstrated by amniocentesis or chorionic villus sampling to have XY karyotype fails to have a demonstrable penis on ultrasonography.
The evaluation of 5 alpha reductase deficiency includes biochemical assays and gene analysis.
Biochemical assay
Traditionally the biochemical test of choice for the diagnosis of 5 alpha reductase deficiency has been the estimation of the ratio between testosterone to dihydrotestosterone (DHT), after stimulation with human chorionic gonadotropin (hCG). With this disorder, the children will show an increase in the ratio of testosterone to dihydrotestosterone (DHT) after hCG administration 14. It merits recall that 5 alpha reductase deficiency cannot be ruled out entirely if the testosterone to dihydrotestosterone (DHT) ratio does not rise with hCG stimulation, making it more difficult to diagnose this condition 15. Furthermore, this test is not very much reliable as the ratio of testosterone to dihydrotestosterone (DHT) may vary based on various factors like severity of enzyme deficiency and age of the children. This biochemical test is also not useful in case of partial enzyme deficiencies.
Gene analysis
Human gene SRD5A2 gene encodes for a protein which makes up enzyme 5 alpha-reductase 2 containing 254 amino acids. This variant has more affinity to testosterone than 5 alpha-reductase 1. At least 54 different types of mutations have been seen involving this gene, among which the primary type is missense mutations. The severity of the disease depends on the degree of loss of enzymatic activity due to a gene mutation. About half of these missense mutations result in an enzyme with no functional biological activity and rest half of the times; the gene may produce an enzyme with a very little measurable biological activity 4.
If 5-alpha-reductase deficiency is considered in a newborn, a broad approach to ambiguous genitalia should be taken 13. The utmost care should be taken not to assign a gender to the child before evaluation and consultation with an experienced, multidisciplinary team. The first words spoken to the parents are likely to be remembered and should focus on the overall health of the infant. A sensitive discussion about the uncertainty of the child’s gender and explanation of the child’s genital anatomy can be aided by online developmental website, such as the Disorders of Sexual Development Guidelines for clinicians (http://www.dsdguidelines.org/files/clinical.pdf) and for parents (http://www.dsdguidelines.org/files/parents.pdf).
5 alpha reductase deficiency treatment
The treatment of a child with 5 alpha reductase deficiency depends on many factors, the most important being the phenotypic findings and gender of the child at the time the physician diagnoses the problem. If there is a critical defect in the formation of external male genitalia, then it will be better if the child is raised as a female. Further, if the child wants to be raised as a female, then the testes have to be removed, and the corrective surgery will be necessary. The removal of testes has to occur before the child attains puberty and virilization of the child 16. The surgical correction if the child opts to be a female would be external genitalia reconstruction, creating a vaginal opening in perineum with a clear separation of urethra and vagina. When the child becomes a teenager, then vaginoplasty would be a potential treatment option 17.
If the child is raised as a male, then corrective surgery should be done depending on the phenotype of the child. The developmental size of the penis at the time of diagnosis and its ability to develop into a functional penis is the main criteria of consideration before raising the child as a male 18. The corrective procedures in males include hypospadias correction, correction of chordee and reconstruction of the urethra. These surgeries are normally performed during the first or second year of life 17.
The parents of the child with ambiguous genitalia have legal rights to seek assistance, support and any information on the child’s problem. They can take adequate time on deciding the treating options and deciding the gender of the child 19.
- Nascimento RLP, de Andrade Mesquita IM, Gondim R, Dos Apóstolos RAAC, Toralles MB, de Oliveira LB, Canguçu-Campinho AK, Barroso U. Gender identity in patients with 5-alpha reductase deficiency raised as females. J Pediatr Urol. 2018 Oct;14(5):419.e1-419.e6.[↩]
- Kumar G, Barboza-Meca JJ. 5 Alpha Reductase Deficiency. [Updated 2019 Mar 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539904[↩]
- Baetens D, Mladenov W, Delle Chiaie B, Menten B, Desloovere A, Iotova V, Callewaert B, Van Laecke E, Hoebeke P, De Baere E, Cools M. Extensive clinical, hormonal and genetic screening in a large consecutive series of 46,XY neonates and infants with atypical sexual development. Orphanet J Rare Dis. 2014 Dec 14;9:209.[↩]
- Cheon CK. Practical approach to steroid 5alpha-reductase type 2 deficiency. Eur. J. Pediatr. 2011 Jan;170(1):1-8.[↩][↩][↩][↩]
- Evaluation of the newborn with developmental anomalies of the external genitalia. American Academy of Pediatrics. Committee on Genetics. Pediatrics. 2000 Jul;106(1 Pt 1):138-42.[↩]
- Avendaño A, Paradisi I, Cammarata-Scalisi F, Callea M. 5-α-Reductase type 2 deficiency: is there a genotype-phenotype correlation? A review. Hormones (Athens). 2018 Jun;17(2):197-204.[↩]
- Bertelloni S, Scaramuzzo RT, Parrini D, Baldinotti F, Tumini S, Ghirri P. Early diagnosis of 5alpha-reductase deficiency in newborns. Sex Dev. 2007. 1(3):147-51.
[↩] - Byne W. Developmental endocrine influences on gender identity: implications for management of disorders of sex development. Mt. Sinai J. Med. 2006 Nov;73(7):950-9.[↩][↩]
- Praveen EP, Desai AK, Khurana ML, et al. Gender identity of children and young adults with 5alpha-reductase deficiency. J Pediatr Endocrinol Metab. 2008 Feb. 21(2):173-9.[↩]
- Maimoun L, Philibert P, Cammas B, Audran F, Bouchard P, Fenichel P. Phenotypical, biological, and molecular heterogeneity of 5a-reductase deficiency: an extensive international experience of 55 patients. J Clin Endocrinol Metab. 2011 Feb. 96(2):296-307.[↩]
- Katz MD, Cai LQ, Zhu YS, et al. The biochemical and phenotypic characterization of females homozygous for 5 alpha-reductase-2 deficiency. J Clin Endocrinol Metab. 1995 Nov. 80(11):3160-7.[↩]
- Cohen-Kettenis PT. Gender change in 46,XY persons with 5alpha-reductase-2 deficiency and 17beta-hydroxysteroid dehydrogenase-3 deficiency. Arch Sex Behav. 2005 Aug;34(4):399-410.[↩]
- [Guideline] Houk CP, Lee PA. Consensus statement on terminology and management: disorders of sex development. Sex Dev. 2008. 2(4-5):172-80.[↩][↩]
- Hiort O, Willenbring H, Albers N, Hecker W, Engert J, Dibbelt L, Sinnecker GH. Molecular genetic analysis and human chorionic gonadotropin stimulation tests in the diagnosis of prepubertal patients with partial 5 alpha-reductase deficiency. Eur. J. Pediatr. 1996 Jun;155(6):445-51.[↩]
- Kim SH, Kim KS, Kim GH, Kang BM, Yoo HW. A novel frameshift mutation in the 5alpha-reductase type 2 gene in Korean sisters with male pseudohermaphroditism. Fertil. Steril. 2006 Mar;85(3):750.e9-750.e12.[↩]
- Sultan C, Paris F, Terouanne B, Balaguer P, Georget V, Poujol N, Jeandel C, Lumbroso S, Nicolas JC. Disorders linked to insufficient androgen action in male children. Hum. Reprod. Update. 2001 May-Jun;7(3):314-22.[↩]
- Houk CP, Hughes IA, Ahmed SF, Lee PA., Writing Committee for the International Intersex Consensus Conference Participants. Summary of consensus statement on intersex disorders and their management. International Intersex Consensus Conference. Pediatrics. 2006 Aug;118(2):753-7.[↩][↩]
- Kojima Y, Mizuno K, Nakane A, Kato T, Kohri K, Hayashi Y. Long-term physical, hormonal, and sexual outcome of males with disorders of sex development. J. Pediatr. Surg. 2009 Aug;44(8):1491-6.[↩]
- Guerra-Júnior G, Maciel-Guerra AT. The role of the pediatrician in the management of children with genital ambiguities. J Pediatr (Rio J). 2007 Nov;83(5 Suppl):S184-91.[↩]




{kind=link}