
Contents
- What is dysautonomia
- Autonomic Nervous System
- Dysautonomia life expectancy
- Dysautonomia types
- Dysautonomia symptoms
- Dysautonomia causes
- Amyloidosis
- Antiphospholipid Syndrome
- Celiac Disease
- Charcot-Marie-Tooth Disease
- Chiari Malformation
- Chronic Inflammatory Demyelinating Polyneuropathy
- Crohn’s Disease and Ulcerative Colitis
- Deconditioning
- Delta Storage Pool Deficiency
- Diabetes and Pre-Diabetes
- Ehlers-Danlos Syndrome
- Mast Cell Disorders
- Mitchochondrial Diseases
- Paraneoplastic Syndrome
- Parkinson’s Disease
- Sarcoidosis
- Sjogren’s Syndrome
- Toxicity
- Physical Trauma, Surgery and Pregnancy
- Vitamin Deficiencies
- Dysautonomia treatment
What is dysautonomia
Dysautonomia is an umbrella term that refers to a disorder of autonomic nervous system function that generally involves failure of the sympathetic or parasympathetic components of the autonomic nervous system, but dysautonomia involving excessive or overactive autonomic nervous system actions also can occur 1. The autonomic nervous system controls the “automatic” functions of the body that you do not consciously think about, such as heart rate, blood pressure, digestion, dilation and constriction of the pupils of the eye, kidney function, and temperature control. People living with various forms of dysautonomia have trouble regulating these systems, which can result in lightheadedness, fainting, unstable blood pressure, abnormal heart rates, erectile dysfunction in men, trouble with breathing and swallowing, malnutrition, and in severe cases, death. Dysautonomia can be local, as in reflex sympathetic dystrophy, or generalized, as in pure autonomic failure. Dysautonomia can be acute and reversible, as in Guillain-Barre syndrome, or chronic and progressive. Several common conditions such as diabetes and alcoholism can include dysautonomia. Dysautonomia also can occur as a primary condition or in association with degenerative neurological diseases such as Parkinson’s disease and multiple sclerosis. Dysautonomia can also occur secondary to other medical conditions, such as diabetes, rheumatoid arthritis, celiac, Sjogren’s syndrome and lupus. Other diseases with generalized, primary dysautonomia include multiple system atrophy and familial dysautonomia. Hallmarks of generalized dysautonomia due to sympathetic failure are impotence (in men) and a fall in blood pressure during standing (orthostatic hypotension). Excessive sympathetic activity can present as hypertension or a rapid pulse rate.
Dysautonomia is not rare. Over 70 million people worldwide live with various forms of dysautonomia 2. People of any age, gender or race can be impacted. There is no cure for any form of dysautonomia at this time, but Dysautonomia International is funding research to develop better treatments, and hopefully someday a cure for each form of dysautonomia. Despite the high prevalence of dysautonomia, most patients take years to get diagnosed due to a lack of awareness amongst the public and within the medical profession.
Some of the different forms of dysautonomia include:
Postural Orthostatic Tachycardia Syndrome (POTS) – estimated to impact 1 out of 100 teenagers and, including adult patients, a total of 1,000,000 to 3,000,000 Americans. Postural Orthostatic Tachycardia Syndrome can cause lightheadness, fainting, tachycardia, chest pains, shortness of breath, GI upset, shaking, exercise intolerance, temperature sensitivity and more. While postural orthostatic tachycardia syndrome predominantly impacts young women who look healthy on the outside, researchers compare the disability seen in postural orthostatic tachycardia syndrome to the disability seen in conditions like COPD and congestive heart failure.
Neurocardiogenic Syncope – neurocardiogenic syncope is the most common form of dysautonomia, neurocardiogenic syncope impacts tens of millions of individuals worldwide. Many individuals with neurocardiogenic syncope have a mild case, with fainting spells once or twice in their lifetime. However, some individuals have severe neurocardiogenic syncope which results in fainting several times per day, which can lead to falls, broken bones and sometimes traumatic brain injury. Individuals with moderate to severe neurocardiogenic syncope have difficulty engaging in work, school and social activities due to the frequent fainting attacks.
Multiple System Atrophy – multiple system atrophy is a fatal form of dysautonomia that occurs in adult ages 40 and up. It is a neurodegenertive disorder with some similarities to Parkinson’s disease, but unlike Parkinson’s patients, multiple system atrophy patients usually become fully bedridden within a 2 years of diagnosis and die within 5-10 years. Multiple System Atrophy is considered a rare disease, with an estimated 350,000 patients worldwide.
There is currently no cure for dysautonomia, but secondary forms may improve with treatment of the underlying disease 1. There are some treatments available to improve quality of life, both with medications and lifestyle changes/adaptations, but even using all treatments available, many dysautonomia patients experience disabling symptoms that significantly reduce their quality of life.
If you have questions about the other causes of dysautonomia, it’s best to speak with your doctor. If you need help finding a doctor with expertise in autonomic disorders, you can look for one using Dysautonomia International Interactive Global Dysautonomia Map (http://dysautonomiainternational.org/map.php).
Autonomic Nervous System
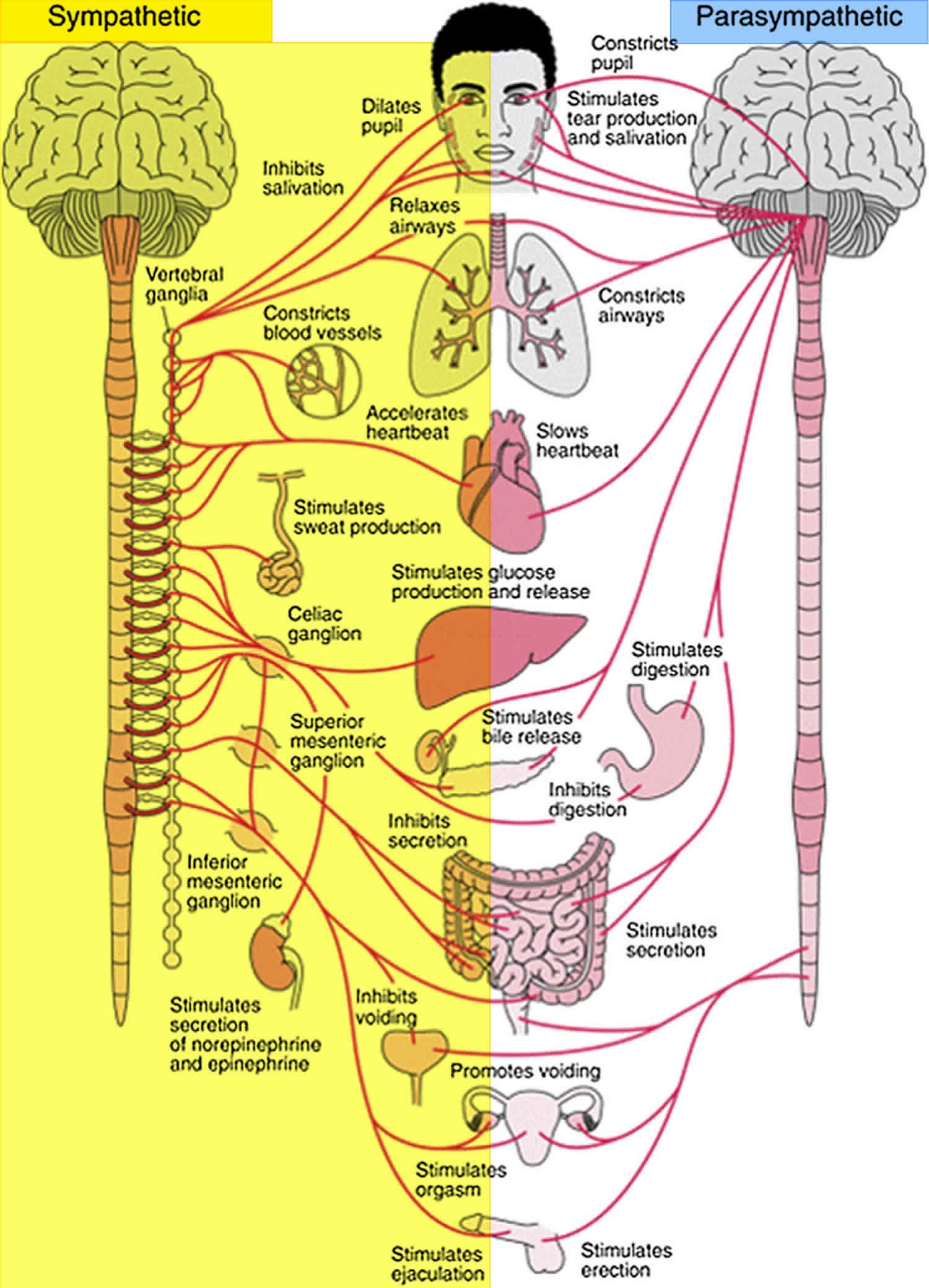
In order to understand the various forms of dysautonomia, it is a good idea to learn a little bit about the basics of the autonomic nervous system. The autonomic nervous system is a very complex system of nerves in the brain, spinal cord, and peripheral nerves that reach out to the limbs and organs. The autonomic nervous system can be divided into three main areas. The central (brain) portions of the autonomic nervous system are found in the medulla oblogata in the lower brain stem, and also in the hypothalmus. The other two portions of the autonomic nervous system are found in the peripheral nerves, including the Sympathetic Nervous System branch, and the Parasympathetic Nervous System branch.
The medulla oblongata is a part of the brain that regulates cardiac, respiratory, vasomotor control, as well as reflexes like coughing, sneezing, vommitting and swallowing. The hypothalmus, another part of the brain, performs a supporting role by linking the nervous system to the endocrine system. The hypothalmus regulates body temperature, thirst, hunger, sleep and circadian rythyms in the body. Through endocrine control, the hypothamlus also plays a role in regulating blood volume and blood pressure.
The Sympathetic Nervous System is commonly associated with the “fight or flight” responses – those bodily reactions that you need to respond quickly in an emergency. When faced with a life threatening situation, your human instinct takes over and you either fight the danger you are facing, or you take flight and run away from the danger. Your Sympathetic Nervous System allows your body to do this rapidly. For example, in the face of danger, your Sympathetic Nervous system will cause bronchial dilation – this allows you to breathe better while you are fighting or running away from the dangerous situation. Likewise, your heart will beat stronger and faster, also prepping the body to fight or take flight.
The Parasympathetic Nervous System is commonly associated with the “rest and digest” responses – those bodily actions needed to restore energy and rest the body. For example, chewing food triggers the Parasympathetic Nervous System to increase production of saliva and to increase digestion in the gut. The Parasympathetic Nervous System also increases gallbladder function, which assists in the digestive process.
Figure 1. Autonomic nervous system
Dysautonomia life expectancy
The outlook for individuals with dysautonomia depends on the particular diagnostic category. People with chronic, progressive, generalized dysautonomia in the setting of central nervous system degeneration have a generally poor long-term prognosis. Death can occur from pneumonia, acute respiratory failure, or sudden cardiopulmonary arrest.
Dysautonomia types
There are many forms of dysautonomia. Some are quite rare, such as Familial Dysautonomia, while others are fairly common, such as Diabetic Autonomic Neuropathy. The following is not an all-inclusive list.
- Neurocardiogenic Syncope/Vasovagal Syncope: syncope is the formal medical term for fainting, describing a temporary loss of consciousness due to a sudden decline in blood flow to the brain.
- Inappropriate Sinus Tachycardia: the syndrome of inappropriate sinus tachycardia is defined as a sinus heart rate over 100 beats per minute (bpm) at rest, with a mean 24-hour heart rate of over 90 bpm not due to identifiable causes, and is associated with distressing symptoms of palpitations 3. Inappropriate sinus tachycardia, a form of dysautonomia that is estimated to impact around 1.2% of the population 3. Inappropriate sinus tachycardia is characterized by unexpectedly fast heart rates at rest, with minimal physical activity, or both 4.
- Pure Autonomic Failure: pure autonomic failure is a peripheral degenerative disorder of the autonomic nervous system 5. Pure Autonomic Failure was formerly known as Bradbury-Eggleston Syndrome, after the two researchers who first described it in 1925. Pure Autonomic Failure is also referred to as idiopathic orthostatic hypotension by some physicians. Pure Autonomic Failure is one of three diseases classified as a primary autonomic disorder, the other two being Multiple System Atrophy and Parkinson’s Disease.
- Autoimmune Autonomic Ganglionopathy: autoimmune autonomic ganglionopathy is a very rare form of dysautonomia in which the bodies own immune system damages a receptor in the autonomic ganglia (part of the peripheral autonomic nerve fiber). Autoimmune autonomic ganglionopathy is often associated with high titers of ganglionic acetylcholine receptor antibody (g-AChR antibody). Autoimmune Autonomic Ganglionopathy can impact people of all ages and both sexes. Approximately 100 Americans are diagnosed with autoimmune autonomic ganglionopathy per year. Autoimmune Autonomic Ganglionopathy is a treatable antibody-mediated disorder of autonomic ganglionic synaptic transmission. Prior names for autoimmune autonomic ganglionopathy include acute pandysautonomia, autoimmune autonomic neuropathy and idiopathic subacute autonomic neuropathy.
- Multiple System Atrophy: multiple system atrophy also known a Shy-Drager Syndrome is a rare neurological condition that causes Parkinson’s-like symptoms, however Multiple System Atrophy patients have more widespread autonomic nerve damage than typical Parkinson’s patients. Since multiple system atrophy can cause widespread nerve damage, it may cause diverse symptoms throughout the body. Physicians often classify multiple system atrophy as either multiple system atrophy-P ormultiple system atrophy-C. Multiple System Atrophy-P patients have predominantly Parkinsonian-like symptoms: tremors, muscle rigidity and slowness of voluntary movements. Multiple System Atrophy-C patients predominantly show signs of cerebellar dysfunction: gait and limb ataxia (ataxia being a lack of control of muscle movements) 6.
- Autonomic Dysreflexia: autonomic dysreflexia is a form of autonomic dysfunction associated with spinal cord injuries. Usually, when an autonomic dysreflexia attack occurs, there is an irritant impacting the person below the level of their spinal cord injury, and because their autonomic nervous system cannot process messages properly, this results in a severe spike in blood pressure, flushing above the spinal injury, nasal stuffiness and other symptoms. Autonomic dysreflexia is serious and can lead to a stroke if not treated properly during an attack.
- Baroreflex Failure: the baroreceptor reflex, or baroreflex, is one of the body’s homeostatic mechanisms for maintaining blood pressure. If the baroreceptor itself or part of its messaging system fails, this is referred to as Baroreflex Failure. Patients often deal with normal or low resting blood pressure and very high or volatile blood pressure during periods of stress.
- Cerebral Salt Wasting Syndrome: cerebral salt-wasting syndrome is a rare condition featuring hyponatremia and dehydration in response to a physical injury or the presence of tumors in or surrounding the brain. The hyponatraemia is due to excessive sodium excretion from the kidney resulting from a centrally mediated process.
- Diabetic Autonomic Neuropathy: diabetic autonomic neuropathy is a secondary form of autonomic dysfunction, but it is likely the most common form of autonomic dysfunction in the world. An estimated 20% of all diabetics suffer from diabetic autonomic neuropathy, which equates to approximately 69 million people worldwide. Diabetic autonomic neuropathy is a serious complication of diabetes. Diabetic autonomic neuropathy is associated with an increased risk of cardiovascular mortality.
- Panayiotopoulos Syndrome: panayiotopoulos syndrome is also known as autonomic epilepsy or benign childhood autonomic epilepsy. It is a fairly common childhood seizure disorder characterized by seizures, often prolonged, with predominantly autonomic symptoms, and by an EEG [electroencephalogram] that shows shifting and/or multiple foci, often with occipital predominance. The typical patient does not “convulse” as seen in grand mal seizures. The child is awake, able to speak and may vomit or feel like he is going to vomit. Sometimes the child is arisen from sleep during the seizure.
- Reflex Sympathetic Dystrophy (Complex Regional Pain Syndrome): reflex sympathetic dystrophy is an extremely painful neurological condition. Some doctors consider it a form of dysautonomia, while others don’t.
Familial dysautonomia
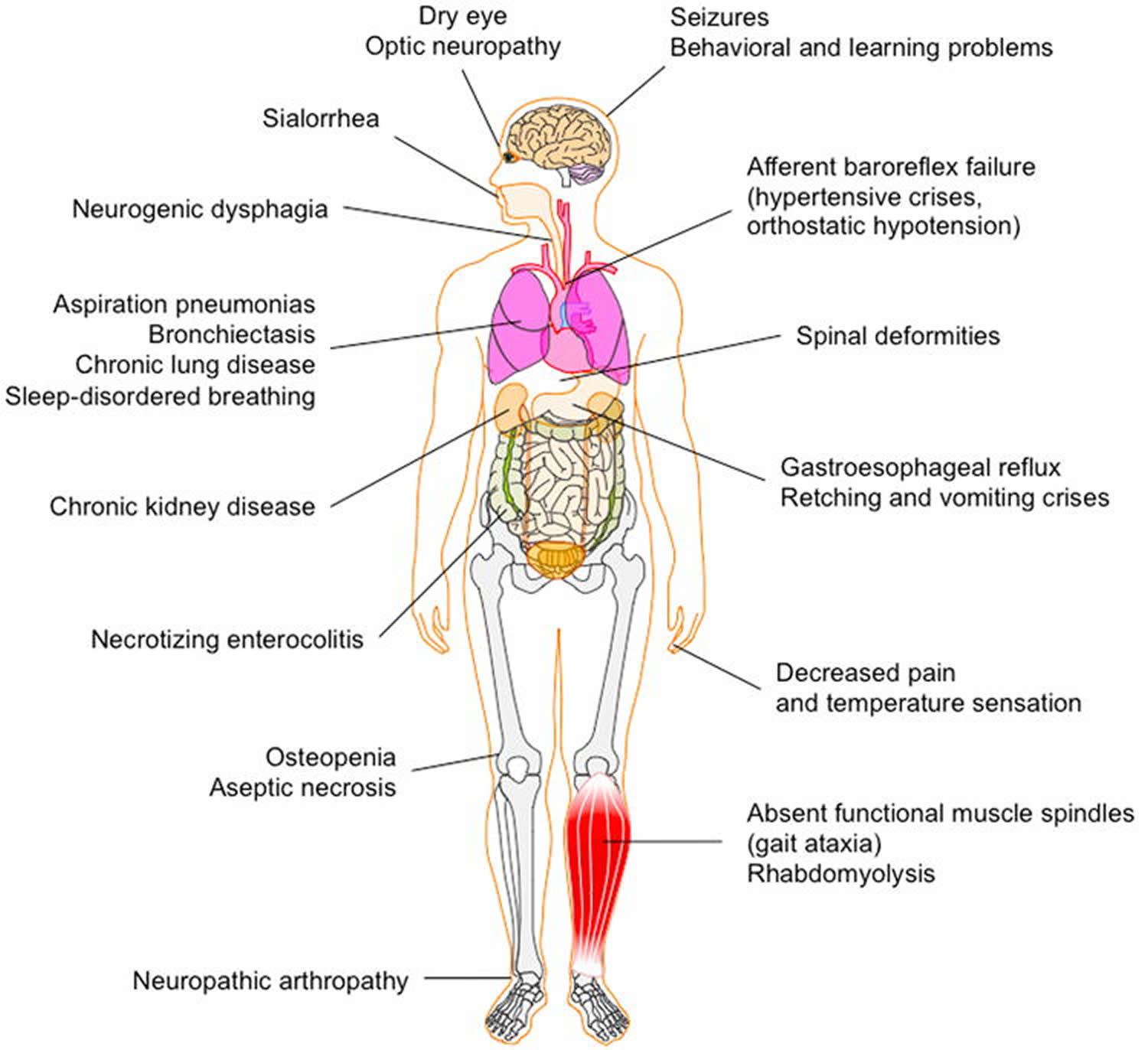
Familial dysautonomia, also known as Riley-Day Syndrome and HSAN type III, is a rare genetic disease that affects the autonomic and sensory nervous systems of children from birth 7. Familial dysautonomia primarily affects people of Eastern European Jewish heritage. The most striking symptoms of familial dysautonomia are reduced sensitivity to pain and fluctuations of body temperature, the inability to produce tears, unstable blood pressure and a decrease in the number of knob-like projections that cover the tongue (fungiform papillae) 8. But familial dysautonomia is much more than “no pain and no tears,” it affects every major system of the body, causing severe respiratory, cardiovascular, orthopedic, digestive, renal and vision problems.
Children with familial dysautonomia lack the most basic reflexes and instincts that we take for granted. As a result, their bodies cannot function normally. They cannot control their blood pressure or heart rate, and they lack the ability to suck at birth and the ability to swallow properly. Because they often swallow into their lungs rather than their stomachs, they are prone to pneumonia. As a result, most familial dysautonomia patients have a feeding tube, so they can be fed directly into their stomachs and reduce the risk of pneumonia.
Familial dysautonomia was once thought of as a fatal childhood disease, with most children expected to live, on average, only to five years of age. Advances in treatment have dramatically extended life expectancy, but children with familial dysautonomia still suffer from chronic and often debilitating symptoms that prevent them from leading normal lives. familial dysautonomia causes a mysterious syndrome called “autonomic crisis” in which patients experience extreme swings in blood pressure and heart rate, along with dramatic personality changes, and a complete shut down of the digestive system. Once familial dysautonomia patients go into crisis, they cannot engage in any normal activity until hours or days later, and they may require hospitalization for observation, sedation and hydration until the crisis abates.
Figure 2. Familial dysautonomia
Familial dysautonomia life expectancy
Currently, the mean age of the familial dysautonomia population is approximately 15 years 7. By statistical projection, babies born with familial dysautonomia in 2006 have a 50% chance of surviving to 40 years of age. The major causes of death are the result of pulmonary complications or sudden death due to autonomic instability.
Familial dysautonomia symptoms
Familial dysautonomia causes dysfunction of the autonomic and sensory nervous systems. Familial dysautonomia is a progressive neurogenetic disease. Symptoms and severity vary in each patient. Symptoms include:
- Absence of overflow tears / corneal drying
- Poor suck at birth
- Drooling
- Swallowing & feeding problems
- Hypotonia / poor muscle tone
- Short stature
- Delayed developmental milestones: motor, language, social
- Inappropriate temperature controls
- Wide swings in blood pressure
- Gastro-esophageal reflux
- Frequent lung infections or pneumonias
- Decreased or no reaction to pain and temperature
- Episodic vomiting
- Excessive sweating
- Blotchy reddening of skin with excitement and/or feeding
- Smooth tongue / lack of taste buds
- Spinal curvature
- Poor weight gain and growth
- Impaired renal function
- Osteoporosis and osteopenia
- Fainting and cardiac arrhythmias
- Sleep apnea
- Restrictive lung disease
An infant born with familial dysautonomia typically has poor sucking ability, impaired swallowing reflexes, poor muscle tone (hypotonia), and/or abnormally low body temperature (hypothermia). Infants with this disorder may have cold hands and feet and experience unstable body temperature (from 94 to 108 degrees) during the course of infectious diseases. Profuse sweating and drooling may also occur. Crying without tears is one of the most striking symptoms of familial dysautonomia. Sometimes a lack of tears and insensitivity of the eyes to pain from foreign objects (corneal anesthesia) can lead to inflammation of the corneas and ulcerations in the eyes.
Children with familial dysautonomia have a decreased perception of pain and lack of sensitivity to hot and/or cold temperatures; this can result in unnoticed injuries to the skin. Unstable blood pressure is usually present in infants with familial dysautonomia. Blood pressure readings may vary greatly and may be abnormally high or low.
Other symptoms of familial dysautonomia may include the absence of the sense of taste, impaired speech, and/or red blotches on the skin that appear with emotional excitement. Approximately 40 percent of children with this disorder experience episodes of vomiting. Occasionally there may be skeletal defects, absence of tendon reflexes, stunted height, and/or repeated episodes of pneumonia due to the inhalation of food (aspiration).
By adolescence, 95 percent of individuals with familial dysautonomia have evidence of side-to side spinal curvature (scoliosis). In addition, they may experience increased sweating and an accelerated heart rate. A decreased awareness of pain makes it difficult for children with this disorder to be aware of injuries; bone fractures may go unrecognized. Other symptoms that may appear during adolescence include weakness, leg cramps, and/or difficulty concentrating. Personality changes may also occur including depression, irritability, inability to sleep (insomnia), and/or negativism.
Approximately 20 percent of adults with familial dysautonomia over 20 years of age develop kidney insufficiency. Neurological deterioration also appears and unsteadiness in walking may become more apparent at this age.
A medical test is available that can determine if an infant has familial dysautonomia. Histamine is injected under the skin and response is measured along nerve cell fibers (axon flare). A lack of response confirms the diagnosis of familial dysautonomia.
Familial dysautonomia causes
Familial dysautonomia is a rare genetic disorder that affects males and females in equal numbers. Familial dysautonomia primarily affects infants of Ashkenazi Jewish or Eastern European ancestry; approximately 1 in 30 people of East European Jewish ancestry are thought to be carriers of the defective gene that causes this disorder.
Familial dysautonomia is inherited as a recessive genetic trait. Human traits, including the classic genetic diseases, are the product of the interaction of two genes, one received from the father and one from the mother. In recessive disorders, the condition does not appear unless a person inherits the same defective gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk of transmitting the disease to the children of a couple, both of whom are carriers for a recessive disorder, is 25 percent. Fifty percent of their children risk being carriers of the disease, but generally will not show symptoms of the disorder. Twenty-five percent of their children may receive both normal genes, one from each parent, and will be genetically normal (for that particular trait). The risk is the same for each pregnancy.
Researchers have identified the gene that causes familial dysautonomia. Two mutations of the gene known as IKBKAP can cause familial dysautonomia. A carrier test is now available for all Ashkenazi Jews. Consult your local physician for details.
Familial dysautonomia diagnosis
To diagnose familial dysautonomia, physicians use health and family history in addition to physical examination. A definitive diagnosis can be made with a blood test. Over 99% of affected individuals will have two copies of the most common gene mutation.
Carrier testing
Carrier testing is available for the two most common mutations. All patients have one or two copies of a single splicing mutation; over 99% of cases of familial dysautonomia in the Ashkenazi Jewish population have two copies of this splicing mutation. A second mutation paired with the Ashkenazi Jewish splicing mutation accounts for all other cases in the Ashkenazi Jewish population. A third mutation from a non-Jewish parent, again paired with the Ashkenazi Jewish splicing mutation, is responsible for a single case of familial dysautonomia. Carrier screening is based on DNA analysis. Screening is 99% accurate for the Ashkenazi Jewish population.
Familial dysautonomia treatment
There is no cure for familial dysautonomia. Treatments are supportive and preventative. Supportive therapies include medications to maintain and regulate cardiovascular, respiratory, and gastrointestinal function. Surgical interventions include fundoplication, gastrostomy, spinal fusion, and tear duct cautery. Various therapies are used to promote strength and speech development.
Drugs used to relieve the symptoms of familial dysautonomia include diazepam, metoclopramide, and chloral hydrate. Artificial tears may be needed to lubricate the eyes.
Physical therapy, chest physiotherapy, occupational therapy, feeding facilitation, and/or speech therapy may also be useful to alleviate the symptoms of familial dysautonomia.
People with familial dysautonomia may also benefit from a variety of other orthopedic and ocular (vision) aids.
Genetic counseling will be of benefit for patients with familial dysautonomia and their families.
Postural orthostatic tachycardia syndrome (POTS)
Postural orthostatic tachycardia syndrome (POTS) is a form of dysautonomia that have orthostatic intolerance as their primary symptom. Orthostatic intolerance describes a condition in which an excessively reduced volume of blood returns to the heart after an individual stands up from a lying down position. The primary symptom of orthostatic intolerance is lightheadedness or fainting. In POTS (postural orthostatic tachycardia syndrome), the lightheadedness or fainting is also accompanied by a rapid increase in heartbeat of more than 30 beats per minute, or a heart rate that exceeds 120 beats per minute, within 10 minutes of rising. The faintness or lightheadedness of postural orthostatic tachycardia syndrome are relieved by lying down again. Anyone at any age can develop postural orthostatic tachycardia syndrome (POTS), but the majority of individuals affected (between 75 and 80 percent) are women between the ages of 15 to 50 years of age. Some women report an increase in episodes of postural orthostatic tachycardia syndrome right before their menstrual periods. POTS often begins after a pregnancy, major surgery, trauma, or a viral illness. POTS may make individuals unable to exercise because the activity brings on fainting spells or dizziness.
Some patients have fairly mild symptoms and can continue with normal work, school, social and recreational activities. For others, symptoms may be so severe that normal life activities, such as bathing, housework, eating, sitting upright, walking or standing can be significantly limited 10. Physicians with expertise in treating POTS have compared the functional impairment seen in POTS patients to the impairment seen in chronic obstructive pulmonary disease (COPD) or congestive heart failure 11. Approximately 25% of POTS patients are disabled and unable to work 11. Researchers found that quality-of-life in POTS patients is comparable to patients on dialysis for kidney failure 12.
Doctors aren’t sure yet what causes the reduced return of blood to the heart that occurs in orthostatic intolerance, or why the heart begins to beat so rapidly in postural orthostatic tachycardia syndrome. Current thinking is that there are a number of mechanisms. Some individuals have peripheral denervation (neuropathic postural orthostatic tachycardia syndrome); some have symptoms that are due to sustained or parosyxmal overactivity of the sympathetic nervous system (hyperadrenergic postural orthostatic tachycardia syndrome); and many individuals with postural orthostatic tachycardia syndrome have significant deconditioning.
Some researchers have described POTS based on some of its more prominent characteristics: hypovolemic POTS, which is associated with low blood volume; partial dysautonomic or neuropathic POTS which is associated with a partial autonomic neuropathy; and hyperandrenergic POTS which is associated with elevated levels of norepinephrine 11. These are not distinct medical conditions and many POTS patients have two or three of the different characteristics present. For example, one patient can have neuropathy, low blood volume and elevated norepineprhine.
Postural orthostatic tachycardia syndrome (POTS) is estimated to impact between 1,000,000 and 3,000,000 Americans, and millions more around the world.
POTS may follow a relapsing-remitting course, in which symptoms come and go, for years. In most cases (approximately 80 percent), an individual with POTS improves to some degree and becomes functional, although some residual symptoms are common.
POTS prognosis
Currently, there is no cure for POTS, however researchers believe that some patients will see an improvement in symptoms over time. Detailed long term follow up studies on the course of POTS are sparse. With proper lifestyle adjustments, exercise, diet and medical treatments, many patients see an improvement in their quality of life.1 If an underlying cause can be identified, and if that cause is treatable, the POTS symptoms may subside. While the prognosis is good for most patients, researchers have noted that some patients will not improve and may actually worsen over time 11.
The longest follow-up study done to date comes from Mayo Clinic 13. Mayo Clinic did a survey of their pediatric POTS patients seen between 2003 and 2010. Of those who responded to the survey, 18.2% reported a complete resolution of their POTS symptoms, while 52.8% reported persistent but improved symptoms. Male patients were twice as likely to report recovery. The average survey respondent had been diagnosed for about 5 years. Both patients who fully recovered and those who did not had mental health scores similar to the national norm.
POTS signs and symptoms
While the diagnostic criteria focus on the abnormal heart rate increase upon standing, POTS usually presents with symptoms much more complex than a simple increase in heart rate. It is fairly common for POTS patients to have a drop in blood pressure upon standing, but some POTS patients have no change or even an increase in blood pressure upon standing. POTS patients often have hypovolemia (low blood volume) and high levels of plasma norepinephrine while standing, reflecting increased sympathetic nervous system activation.3 Approxiamtely 50% of POTS patients have a small fiber neuropathy that impacts their sudomotor nerves. Many POTS patients also experience fatigue, headaches, lightheadedness, heart palpitations, exercise intolerance, nausea, diminished concentration, tremulousness (shaking), syncope (fainting), coldness or pain in the extremities, chest pain and shortness of breath. Patients can develop a reddish purple color in the legs upon standing, believed to be caused by blood pooling or poor circulation. The color change subsides upon returning to a reclined position.
POTS Diagnostic Criteria
The current diagnostic criteria for postural orthostatic tachycardia syndrome (POTS) is a heart rate increase of 30 beats per minute (bpm) or more, or over 120 bpm, within the first 10 minutes of standing, in the absence of orthostatic hypotension 14. In children and adolescents, a revised standard of a 40 bpm or more increase has recently been adopted 15. POTS is often diagnosed by a Tilt Table Test, but if such testing is not available, POTS can be diagnosed with bedside measurements of heart rate and blood pressure taken in the supine (laying down) and standing up position at 2, 5 and 10 minute intervals. Doctors may perform more detailed tests to evaluate the autonomic nervous system in POTS patients, such as Quantitative Sudomotor Axon Reflex Test (QSART, sometimes called Q-Sweat), Thermoregulatory Sweat Test (TST), skin biopsies looking at the small fiber nerves, gastric motility studies and more.
What causes POTS?
POTS is a heterogeneous (meaning it has many causes) group of disorders with similar clinical manifestations 11. POTS itself is not a disease; it is simply a cluster of symptoms that are frequently seen together. This is why the ‘S’ in POTS stands for “Syndrome.” Since POTS is not a disease, it is fair to say that POTS is caused by something else. However, figuring out what is causing the symptoms of POTS in each patient can be very difficult, and in many cases, patients and their doctors will not be able to determine the precise underlying cause. When doctors cannot pinpoint the underlying cause of a patient’s POTS, it may be called Primary or Idiopathic POTS 11. Idiopathic simply means “of an unknown origin.”
While researchers are still working to identify the root causes and pathology of POTS, there are several underlying diseases and conditions that are known to cause or be associated with POTS or POTS like symptoms in some patients. This is a partial list:
- Amyloidosis;
- Autoimmune Diseases such as Autoimmune Autonomic Ganglionopathy, Sjogren’s Syndrome, Lupus, Sarcoidosis, Antiphospholipid Syndrome;
- Chiari Malformation;
- Deconditioning;
- Delta Storage Pool Deficiency;
- Diabetes and prediabetes;
- Ehlers Danlos Syndrome a collagen protein disorder than can lead to joint hypermobility and “stretchy” veins;
- Genetic Disorders/Abnormalities;
- Infections such as Mononucleosis, Epstein Barr Virus, Lyme Disease, extrapulmonary Mycoplasma pneumonia and Hepatitis C;
- Multiple Sclerosis;
- Mitochondrial Diseases;
- Mast Cell Activation Disorders;
- Paraneoplastic Syndrome rare small tumors of the lung, ovary, breast and pancreas that produce antibodies;
- Toxicity from alcoholism, chemotherapy and heavy metal poisoning;
- Traumas, pregnancy or surgery;
- Vaccinations;
- Vitamin Deficiencies/Anemia.
While some of the physical symptoms of POTS overlap with the symptoms of anxiety, such as tachycardia and palpitations, POTS is not caused by anxiety. POTS patients are often misdiagnosed as having anxiety or panic disorder, but their symptoms are real and can severely limit a person’s ability to function 11. Research has shown that POTS patients are similarly or even less likely to suffer from anxiety or panic disorder than the general public. 16. Research surveys that evaluate mental health show similar results between POTS patients and national norms 13.
Postural orthostatic tachycardia syndrome treatment
Therapies for POTS are targeted at relieving low blood volume or regulating circulatory problems that could be causing the disorder. No single treatment has been found to be effective for all. A number of drugs such as Fludrocortisone, Beta Blockers, Midodrine, Clonidine, Pyridostigmine, Benzodiazepines, SSRIs, SNRIs, Erythropoietin and Octreotide seem to be effective in the short term 11. Whether they help in long term is uncertain. Simple interventions such as adding extra salt to the diet and attention to adequate fluid intake are often effective. The drugs fludrocortisone (for those on a high salt diet) and midodrine in low doses are often used to increase blood volume and narrow blood vessels. Drinking 16 ounces of water (2 glassfuls) before getting up can also help raise blood pressure. Some individuals are helped by beta receptor blocking agents. There is some evidence that an exercise program can gradually improve orthostatic tolerance.
The most common treatments for POTS include increasing fluid intake to 2-3 liters per day; increasing salt consumption to 3,000 mg to 10,000 mg per day; wearing compression stockings; raising the head of the bed (to conserve blood volume); reclined exercises such as rowing, recumbent bicycling and swimming; a healthy diet; avoiding substances and situations that worsen orthostatic symptoms; and finally, the addition of medications meant to improve symptoms 10. Many different medications are used to treat POTS, If an underlying cause of the POTS symptoms can be identified, treating the underlying cause is very important as well.
Dysautonomia symptoms
People living with various forms of dysautonomia have trouble regulating these systems, which can result in lightheadedness, fainting, unstable blood pressure, abnormal heart rates, malnutrition, and in severe cases, death.
Dysautonomia causes
Dysautonomia is not a specific medical diagnosis. Dysautonomia is an umbrella term used to describe any malfunction of the autonomic nervous system. There are many underlying diseases and conditions that can lead to dysfunction of the autonomic nervous system. This is not an all inclusive list, so check with your doctor if you believe you might have an underlying cause for your Postural Orthostatic Tachycardia Syndrome (POTS) or other form of dysautonomia. Remember that not every dysautonomia patient can find a specific underlying cause, and some of these conditions are very rare.
Amyloidosis
Amyloidosis is a group of rare disorders caused by the accumulation of harmful protein in various tissues of the body. Amyloidosis can be an inherited condition or an acquired condition. Symptoms depend on which organs and tissues are impacted. If amyloid proteins are deposited on the heart or the peripheral nerves, it can cause symptoms of dysautonomia.
Antiphospholipid Syndrome
Antiphospholipid syndrome, also known as “Hughes syndrome” for the British rheumatologist who first described the syndrome in 1983, is an autoimmune blood clotting disorder. It is also known as “sticky blood.” Antiphospholipid syndrome may cause clotting of arteries (most commonly causing stroke or heart attack) as well as veins (most commonly causing deep vein thrombosis of the legs and pulmonary embolus of the lungs). Antiphospholipid syndrome may also cause recurrent miscarriage due to clotting within the placenta. Less well known to physicians is that antiphospholipid syndrome also causes many non-thrombotic manifestations due to “sludging” of the blood. These include a number of neurological manifestations such as headache, memory loss, word finding difficulty, trouble with balance, multiple sclerosis-like syndrome, neuropathy and disorders of the autonomic nervous system (most commonly postural tachycardia syndrome and neurocardiogenic syncope). Antiphospholipid syndrome may occur in association with another autoimmune disorder (most commonly lupus, but also Sjogren’s syndrome and rheumatoid arthritis); this is known as secondary antiphospholipid syndrome. Antiphospholipid syndrome may also occur on its own (primary antiphospholipid syndrome). The clotting manifestations are treated with anticoagulation (warfarin or heparin). Less well known is that the non-thrombotic manifestations may also be treated with anti-platelet agents (aspirin or plavix) or anticoagulants with significant improvement or even resolution of the symptoms. Antiphospholipid syndrome is diagnosed when there is at least one clinical manifestation and at least one of the following antibodies: anticardiolipin IgG or IgM, beta 2 glycoprotein I IgG or IgM, or the lupus anticoagulant. Research is currently underway to determine if intravenous immunoglobulin (IVIG) may benefit those with autonomic dysfunction caused by antiphospholipid syndrome.
Celiac Disease
Celiac disease or coeliac disease is a genetic, multisystem autoimmune disease in which the small intestine is the major site of injury. When a person with celiac disease eats gluten [the protein portion in wheat (gliadin), rye (secalin) and barley (hordein), and for some with celiac disease, oats (avenin)], the immune system responds by damaging or destroying the small intestine’s villi, which are the structures that enable the intestine to absorb the nutrients needed to survive. From the small intestine, the disease can go on to impact other parts of the body as it progresses. Celiac disease is not rare. The prevalence of celiac disease in the United States is approximately one in every 133, which translates to approximately three million Americans. Of those three million, recent research shows that 83% remain undiagnosed. The average length of time to diagnosis in the U.S. for those experiencing symptoms is four years. This delay in diagnosis increases a person’s chance of developing neurological disorders, additional autoimmune diseases, osteoporosis and cancer. By some estimates, neurological disorders are thought to occur in 6-10% of people with celiac disease. Autonomic neuropathy, peripheral neuropathy and ataxia are types of neurological disorders that can appear in people with celiac disease.
Charcot-Marie-Tooth Disease
Charcot-Marie-Tooth Disease is one of the most common inherited neurological disorders. It affects approximately 1 in 2,500 people. Charcot-Marie-Tooth Disease includes a group of disorders caused by genetic mutations that impact the normal function of the peripheral nerves (nerves outside the brain and spinal cord). A typical feature of Charcot-Marie-Tooth Disease is weakness of the foot and lower leg muscles, often resulting in a foot drop or a high-step gait. Charcot-Marie-Tooth Disease can also impact the autonomic nervous system. Although currently no cure or standard treatment for Charcot-Marie-Tooth Disease exists, patients find that physical therapy, orthopedic devices and orthopedic surgery can be of benefit.
Chiari Malformation
Arnold-Chairi malformation or Chiari is a brain condition present from the time of birth in which the tonsils of the cerebellum herniate at least 5 mm past the foramen magnum or opening of the skull. Some cases of Chiari are “acquired,” meaning they developed later in life and were not present at birth. A Chiari malformation can impair the flow of cerebral spinal fluid causing a syringomyelia or fluid build-up in the spinal cord. Chiari can produce a variety of symptoms relating to autonomic and vestibular impairment including dizziness, numbness in the hands or feet, difficulty swallowing, impaired motor coordination, and chronic headaches. Chairi malformation is diagnosed by a brain CT or MRI and a neurological exam.
Chronic Inflammatory Demyelinating Polyneuropathy
Chronic inflammatory demyelinating polyneuropathy is an autoimmune disease that involves inflammation of nerve roots and peripheral nerves (nerves outside the brain and spinal cord), along with corresponding destruction of myelin sheath (the fatty protective covering over the nerves). The loss of myelin can occur in motor or sensory nerves. Some patients also develop autonomic dysfunction, experiencing fluctuations in their blood pressure and cardiac arrhythmias. Treatment for chronic inflammatory demyelinating polyneuropathy includes corticosteroids, intravenous immunoglobulin (IVIG) and plasmapheresis (plasma exchange). Chronic inflammatory demyelinating polyneuropathy is often considered the chronic counterpart of the acute Guillan-Barre Syndrome.
Crohn’s Disease and Ulcerative Colitis
Crohn’s disease is a chronic inflammatory condition of the gastrointestinal tract. Ulcerative colitis is a disease of the large intestine, in which the lining of the large intestine becomes inflamed and develops ulcers that can produce pus and mucous. The combined inflammation and ulceration can cause abdominal discomfort and frequent emptying of the colon. Both Crohn’s and Colitis are forms of Inflammatory Bowel Disease (IBD), and both conditions can be associated with autonomic neuropathy and symptoms of autonomic dysfunction. Some studies have documented that about 50% of Crohn’s disease and ulcerative colitis patient have autonomic nervous system complications.
Deconditioning
Deconditioning is a physical change in the way the body functions due to a decrease in activity. Deconditioning can be caused by many different health conditions. One of the most common causes of deconditioning is bed rest, from intentional bed rest after surgery, to unintentional bed rest during an acute viral illness. Due to the debilitating nature of their symptoms, which can make exercise, standing and sometimes even sitting upright difficult, many patients with Postural Orthostatic Tachycardia Syndrome (POTS) or other forms of dysautonomia become deconditioned over time. Deconditioning may exacerbate symptoms, so it is important to take steps to prevent deconditioning from occurring, or to reverse it if the patient has already become deconditioned.
Delta Storage Pool Deficiency
Delta storage pool deficiency occurs when platelets are unable to secrete granules into the blood to promote clotting which can result in severe bleeding. It is diagnosed by microscopic examination of blood cells by a hematologist. Severe blood loss due to storage pool deficiency can result in hypovolemia, which can lead to autonomic dysfunction. Hypovolemia is the medical term for low blood volume.
Diabetes and Pre-Diabetes
Diabetes is the most common identifable cause of neuropathy. Approximately 50% of individuals with Type 1 and Type 2 diabetes will develop neuropathy. This can occur in pre-diabetes as well. When only the peripheral small fiber autonomic nerves are impacted, this may present with symptoms similar to Postural Orthostatic Tachycardia Syndrome (POTS). However, approximately 20% of diabetics develop a more serious Cardiac Autonomic Neuropathy, and this is associated with an increased risk of mortality. The likelihood of developing diabetic neuropathy increases with the patient’s age and duration of having diabetes. New research indicates that, not only can excessive blood sugar cause autonomic neuropathy, so can too little blood sugar, hypoglycemia, caused by overuse of blood sugar control drugs.
Ehlers-Danlos Syndrome
Ehlers-Danlos Syndome is a hereditary connective tissue disorder that typically presents with stretchy skin and joint hypermobility, frequent dislocations, and pain. There are some genetic tests but the diagnosis is usually based on a clinical evaluation by a geneticist or rheumatologist. There are many variants of Ehlers-Danlos Syndome yet the most common variants are hypermobile Ehlers-Danlos Syndome (formerly type III) and classical Ehlers-Danlos Syndome (formerly types I and II). Ehlers-Danlos Syndome, especially the hypermobile type, appears to be common in patients with autonomic dysfunction. Observed vascular abnormalities in patients with Ehlers-Danlos Syndome may link Ehlers-Danlos Syndome to orthostatic intolerance. Cranio-cervical instability and Chiari malformation are also seen in patients with Ehlers-Danlos Syndome and may contribute to autonomic symptoms. Further research is needed to find the etiology of autonomic dysfunction in patients with Ehlers-Danlos Syndome. It is important to note that not everyone who has Ehlers-Danlos Syndome develops autonomic dysfunction.
Mast Cell Disorders
Mast cell activation disorders can be divided generally into two categories. Mastocytosis, in which there is an actual problem with the number of mast cells, and mast cell activation disorder, in which the mast cells are normal in quantity, but behave adbnormally. Patients with mast cell activation disorder usually have episodes of flushing and sometimes rashes followed by autonomic symptoms. It is hypothesized that histamine release from degranulated mast cells triggers can cause excessive vasodilation and trigger a hyperadrenergic response. Mast cell activation disorder can be difficult to diagnose because some patients have symptoms with normal blood and urine tests.
Mitchochondrial Diseases
Mitochondria are membrane encapsulated structures found in every cell in the human body. They are considered the “power plant” of energy production, but they are also responsible for other important tasks in the body. There are many different forms of mitochondrial disease, but most mitochondrial diseases present with neurological symptoms including neuromuscular, respiratory, gastrointestinal, and autonomic dysfunction. Mitochondrial diseases are difficult to diagnose and seeing a specialist is key as the process usually involves a combination of clinical evaluation, blood tests, brain imaging, and muscle biopsies.
Paraneoplastic Syndrome
Sometimes when there is a tumor in the body, whether it is cancerous or not, the body tries to get rid of the tumor by producing antibodies meant to attack and remove the tumor. Unfortunately, sometimes these antibodies can also attack part of the nervous system. When this occurs, it is referred to as paraneoplastic syndrome. The antibodies involved are called paraneoplastic antibodies. This is considered quite rare, however, some studies have shown that up to 1% of patients with solid tumors may have paraneoplastic antibodies. If paraneopastic antibodies attack the autonomic nervous system, the patient can develop symptoms of dysautonomia. Frequently, the neurological symptoms present before the cancer is diagnosed. In some cases, the paraneoplastic syndrome improves once the tumor is removed. In other cases, intravenous immunoglobulin or other immune modulating treatments are used to try to reduce the harmful antibody levels.
Parkinson’s Disease
Autonomic dysfunction impacts up to 80% of people with Parkinson’s disease. Sometimes this is a side effect of the medication used to treat Parkinson’s, but sometimes it can be part of the Parkinson’s neurodegenerative disease process. All Parkinson’s patients should see a neurologist to discuss symptoms that may involve the autonomic nervous system, such as low blood pressure, digestive problems, bladder problems, and problems with sexual function. Parkinson’s disease is typically seen in older adults, and it impacts about 1 million Americans.
Sarcoidosis
Sarcoidosis is an inflammatory disorder in which the body’s immune system causes too much inflammation and then the build up of immune cells in organs such as the lungs, eyes and liver. Sarcoidosis can also impact the nervous system, and in some cases this can result in symptoms of dysautonomia. Approximately 60-70% of sarcoidosis patients go into remission without treatment, but in rare cases sarcoidosis can be fatal.
Sjogren’s Syndrome
Sjogren’s syndrome is one of the most common autoimmune disease in the United States, and possibly worldwide. One million people in the U.S. are living with Sjogren’s syndrome. Experts believe there may be another three million people in the U.S. who have Sjogren’s syndrome, but remain undiagnosed. Due to a lack of awareness within the medical profession and the complex and diverse symptoms Sjogren’s can present with, the average patient can take five years to get diagnosed. Typical symptoms can include dry eyes, dry mouth, fatigue and joint pain. However, Sjogren’s can attack any tissue or organ in the body, and not every patient has the classic dryness symptoms. Sjogren’s syndrome can initially present as Postural Orthostatic Tachycardia Syndrome (POTS). There is some evidence that younger patients, or those earlier in the disease process, may present initially with neurological symptoms and may be less likely to have the traditional symptoms of dryness. In the past, autonomic neuroapthy has been considered to be a rare manifestation of Sjogren’s, but newer recent research indicates that approximately half of all Sjogren’s patients experiences symptoms of autonomic dysfunction. Many physicians rely only on blood tests (SS-A, SS-B and ANA) to rule out Sjogren’s as a possible diagnosis. However, about 50% of Sjogren’s patients with neurological manifestations do not test positive for any of the antibody tests. A minor salivary gland lip biopsy is currently considered the gold standard test to diagnose or rule out Sjogren’s. The diagnostic criteria for Sjogren’s continues to be hotly debated by experts. The American-European Consensus Criteria for Sjogren’s Syndrome is the most widely accepted diagnostic criteria at this time. Approximately 50% of Sjogren’s patients have “primary” Sjogren’s, meaning just Sjogren’s and no other autoimmune disease, and the other 50% have Secondary Sjogren’s. Secondary Sjogren’s is Sjogren’s in association with another autoimmune disease, most commonly Lupus, Rheumatoid Arthritis or Hashimotos Thyroiditis. In some cases, the immune system becomes so overactive that Sjogren’s patients can develop three or more autoimmune conditions at once.
Toxicity
Alcoholism, chemotherapy drugs and heavy metal poisoning can cause damage to the autonomic nerves due to the toxicity of these substances. In addition to the toxicity of alcohol, chronic alcohol consumptions can also lead to nutritional deficiencies that contribute to autonomic nerve damage. “Heavy” metal poisoning doesn’t just include heavy metals, it can include any metal on the periodic table that is potentially harmful to human health. Some metals have no know benefit or need in human health such as lead, mercury and cadmium. Other metals such as iron and copper are essential for human health, but can be toxic at higher doses.
Physical Trauma, Surgery and Pregnancy
While physical traumas, surgeries and pregnancy might not seem to have much in common at first, all three can cause a rapid and significant change in structure and function of the body. The onset of autonomic dysfunction, particularly Postural Orthostatic Tachycardia Syndrome (POTS), has been well documented after car accidents, serious injuries, surgeries and pregnancies.
Vitamin Deficiencies
Vitamins are organic substances made by plants or animals that are required for human health. Many of these vitamins, including Vitamins E, B1 (thiamine), B3 (niacin) B6 (pyridoxine), and B12 are essential to healthy nerve function. Thiamine deficiency, in particular, is common among people with alcoholism. People who have digestive problems, which are very common in people who have autonomic disorders, are often deficient in B12. Vitamin deficiencies can usually be corrected with a proper diet, and if that is not sufficient, supplementation with oral, intravenous, or injectable vitamins may be necessary.
Dysautonomia treatment
There is usually no cure for dysautonomia. Secondary forms may improve with treatment of the underlying disease. In many cases treatment of primary dysautonomia is symptomatic and supportive. Measures to combat orthostatic hypotension include elevation of the head of the bed, water bolus (rapid infusion of water given intravenously), a high-salt diet, and drugs such as fludrocortisone and midodrine.
- Dysautonomia Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Dysautonomia-Information-Page[↩][↩]
- What is dysautonomia? http://dysautonomiainternational.org/page.php?ID=34[↩]
- 2015 heart rhythm society expert consensus statement on the diagnosis and treatment of postural tachycardia syndrome, inappropriate sinus tachycardia, and vasovagal syncope. Sheldon RS, Grubb BP, Olshansky B, Shen WK, Calkins H, Brignole M, Raj SR, Krahn AD, Morillo CA, Stewart JM, Sutton R, Sandroni P, Friday KJ, Hachul DT, Cohen MI, Lau DH, Mayuga KA, Moak JP, Sandhu RK, Kanjwal K. Heart Rhythm. 2015 Jun;12(6):e41-63.[↩][↩]
- Inappropriate sinus tachycardia. Olshansky B, Sullivan RM. J Am Coll Cardiol 2013;61:793-801.[↩]
- What is Pure Autonomic Failure (PAF)? https://www.rarediseasesnetwork.org/cms/autonomic/Learn-More/Disorder-Definitions[↩]
- Consensus statement on the diagnosis of multiple system atrophy. Journal of the Neurological Sciences. S. Gilman, P.A. Low, N. Quinn, A. Albanese, Y. Ben-Shlomo, C.J. Fowler, H. Kaufmann, T. Klockgether, A.E. Lang, P.L. Lantos, I. Litvan, C.J. Mathias, E. Oliver, D. Robertson, I. Schatz, G.K. Wenning; Volume 163, Issue 1; Pages 94-98; 1 February 1999[↩]
- About FD. http://www.familialdysautonomia.org/about-fd.htm[↩][↩]
- Familial Dysautonomia. https://rarediseases.org/rare-diseases/dysautonomia-familial/[↩]
- Palma J-A, Kaufmann L, Fuente C, Percival L, Mendoza C, Kaufmann H. Current Treatments in Familial Dysautonomia. Expert opinion on pharmacotherapy. 2014;15(18):2653-2671. doi:10.1517/14656566.2014.970530. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4236240/[↩]
- The Postural Tachycardia Syndrome (POTS): Pathophysiology, Diagnosis & Management. Satish R Raj, MD MSCI, Indian Pacing Electrophysiol J. 2006 April-Jun; 6(2): 84-99.[↩][↩]
- Postural Tachycardia Syndrome. Blair P. Grubb, Circulation. 2008; 117: 2814-2817. https://www.ahajournals.org/doi/pdf/10.1161/CIRCULATIONAHA.107.761643[↩][↩][↩][↩][↩][↩][↩][↩]
- Bagai K, Wakwe CI, Malow B, et al. Estimation of Sleep Disturbances Using Wrist Actigraphy in Patients with Postural Tachycardia Syndrome. Autonomic neuroscience : basic & clinical. 2013;177(2):260-265. doi:10.1016/j.autneu.2013.02.021. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3700681/[↩]
- Long-term outcomes of adolescent-onset postural orthostatic tachycardia syndrome. S.J. Kizilbash, S.P. Ahrens, R. Bhatia, J.M. Killian, S. A. Kimmes, E.E. Knoebel, P. Muppa, A.L. Weaver, P.R. Fischer. Clin. Auton. Res. October 2013. Abstract presented at the 24th International Symposium on the Autonomic Nervous System.[↩][↩]
- Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Auton Neurosci. 2011 Apr 26;161(1-2):46-8. doi: 10.1016/j.autneu.2011.02.004. Epub 2011 Mar 9. https://www.ncbi.nlm.nih.gov/pubmed/21393070[↩]
- Postural tachycardia in children and adolescents: what is abnormal? Singer W, Sletten DM, Opfer-Gehrking TL, Brands CK, Fischer PR, Low PA, J Pediatr. 2012 Feb;160(2):222-6. Epub 2011 Oct 11. https://www.jpeds.com/article/S0022-3476(11)00884-5/fulltext[↩]
- Excessive heart rate response to orthostatic stress in postural tachycardia syndrome is not caused by anxiety; Masuki S, Eisenach JH, Johnson C et al. Journal of Applied Physiology 2006; 102: 1134-42.[↩]
{kind=link}