Contents
What is Alagille syndrome
Alagille syndrome is a rare inherited multisystem disorder that can affect the liver, heart, skeleton, eyes, kidneys and other parts of the body 1. One of the major features of Alagille syndrome is liver damage caused by abnormalities in the bile ducts. These ducts carry bile (which helps to digest fats) from the liver to the gallbladder and small intestine. In Alagille syndrome, the bile ducts may be narrow, malformed, and fewer in number (bile duct paucity). As a result, bile builds up in the liver and causes scarring (cirrhosis) that prevents the liver from working properly to eliminate wastes from the bloodstream. The specific symptoms and severity of Alagille syndrome can vary greatly from one person to another, even within the same family. Symptoms can range from very mild as to go unnoticed to severe heart and/or liver disease requiring transplantation. The liver problems may be the first symptoms of Alagille syndrome, and may include yellow color of the skin and whites of the eyes (jaundice); itchy skin; bumps on the skin caused by deposits of cholesterol and fats (xanthomas); pale, loose bowel movements; and poor growth. Alagille syndrome can also affect other parts of the body including the heart, brain, kidneys, blood vessels, eyes, face, and skeleton.
Alagille syndrome signs and symptoms are generally noticed in infancy or early childhood due to liver damage may include a yellowish tinge in the skin and the whites of the eyes (jaundice), itchy skin, and deposits of cholesterol in the skin (xanthomas).
Alagille syndrome is also associated with several heart problems, including impaired blood flow from the heart into the lungs (pulmonic stenosis). Pulmonic stenosis may occur along with a hole between the two lower chambers of the heart (ventricular septal defect) and other heart abnormalities. This combination of heart defects is called tetralogy of Fallot.
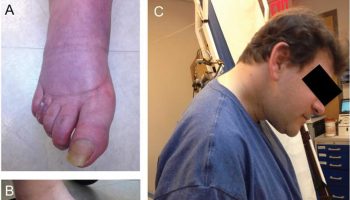
People with Alagille syndrome may have distinctive facial features including a broad, prominent forehead; deep-set eyes; and a small, pointed chin (see Figure 2). Alagille syndrome may also affect the blood vessels within the brain and spinal cord (central nervous system) and the kidneys. Affected individuals may have an unusual butterfly shape of the bones of the spinal column (vertebrae) that can be seen in an x-ray.
Some people with Alagille syndrome may have isolated signs of the disorder, such as a heart defect like tetralogy of Fallot, or a characteristic facial appearance. These individuals do not have liver disease or other features typical of the disorder.
The estimated prevalence of Alagille syndrome is 1 in 70,000 newborns 1. This figure is based on diagnoses of liver disease in infants, and may be an underestimation because some people with Alagille syndrome do not develop liver disease during infancy.
Alagille syndrome is caused by changes or mutations in the JAG1 and NOTCH2 genes. Inheritance is autosomal dominant. However, in about half of cases the mutation occurs as a new change (“de novo”) without being inherited from either parents. While there is no known cure for Alagille syndrome, there are treatments that can help control symptoms. Possible treatments may include medication that increases the flow of bile and careful management of diet to minimize nutrition and vitamin related problems. In severe cases, a liver transplant may be necessary.
Treatment may include:
- Medication that increases bile flow (ursodeoxycholic acid) and that reduces the itching, such as cholestyramine, rifampin, and naltrexone.
- Biliary diversion procedures (partial internal biliary diversion and ileal exclusion) that interrupt the normal bile circulation between the intestines and liver resulting in the bile being eliminated and therefore lowering the blood bile levels. These procedures relieve symptoms of liver disease, such as itching, and improve the quality of life, but do not prevent the progression of liver disease.
- Vitamin and special nutrition supplementation to support proper growth and development. Infants with Alagille syndrome are given a special formula that helps the small intestine absorb the fats. The diet usually is a high-calorie diet, calcium, and with vitamins A, D, E, and K, and sometimes zinc. Sometimes a child may receive additional calories through a tiny tube that is passed through the nose into the stomach. If extra calories are needed for a long time, a doctor may place a tube, called a gastrostomy tube, directly into the stomach through a small opening made in the abdomen. Nutrition management may need to continue throughout a person’s lifetime, but especially during childhood, with careful monitoring and balancing of nutrients, fat, calories, fat-soluble vitamins, calcium, and zinc. Each person with Alagille syndrome may have different dietary needs and the needs may change throughout their lifetime, so regular consultation with a nutrition specialists is recommended.
- Liver Transplantation to increase life span and improve liver function for those with end stage liver disease. Although there is some catch-up growth after a liver transplant in children with Alagille syndrome, it is not as much as seen in children with other liver diseases.
- Heart surgery or a catheter based procedure to correct a heart defect. Heart problems are treated the same as similar heart problems in children who do not have Alagille syndrome (standard medical care) although may be complicated if other organs are affected by the syndrome.
- Kidney surgery or treatment to correct any kidney abnormalities. Kidney problems are treated the same as kidney problems in children who do not have Alagille syndrome (standard medical care), although may be complicated if other organs are affected by the syndrome.
Heart problems may complicate liver transplantataion, especially, pulmonary artery stenosis with right ventricular pressure overload. In these cases a procedure known as congenital heart transcatheter intervention may be done before the liver transplant.
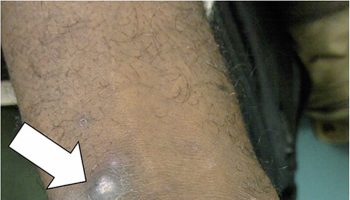
Figure 1. Alagille syndrome xanthomas on the extensor surface of the buttocks and thighs
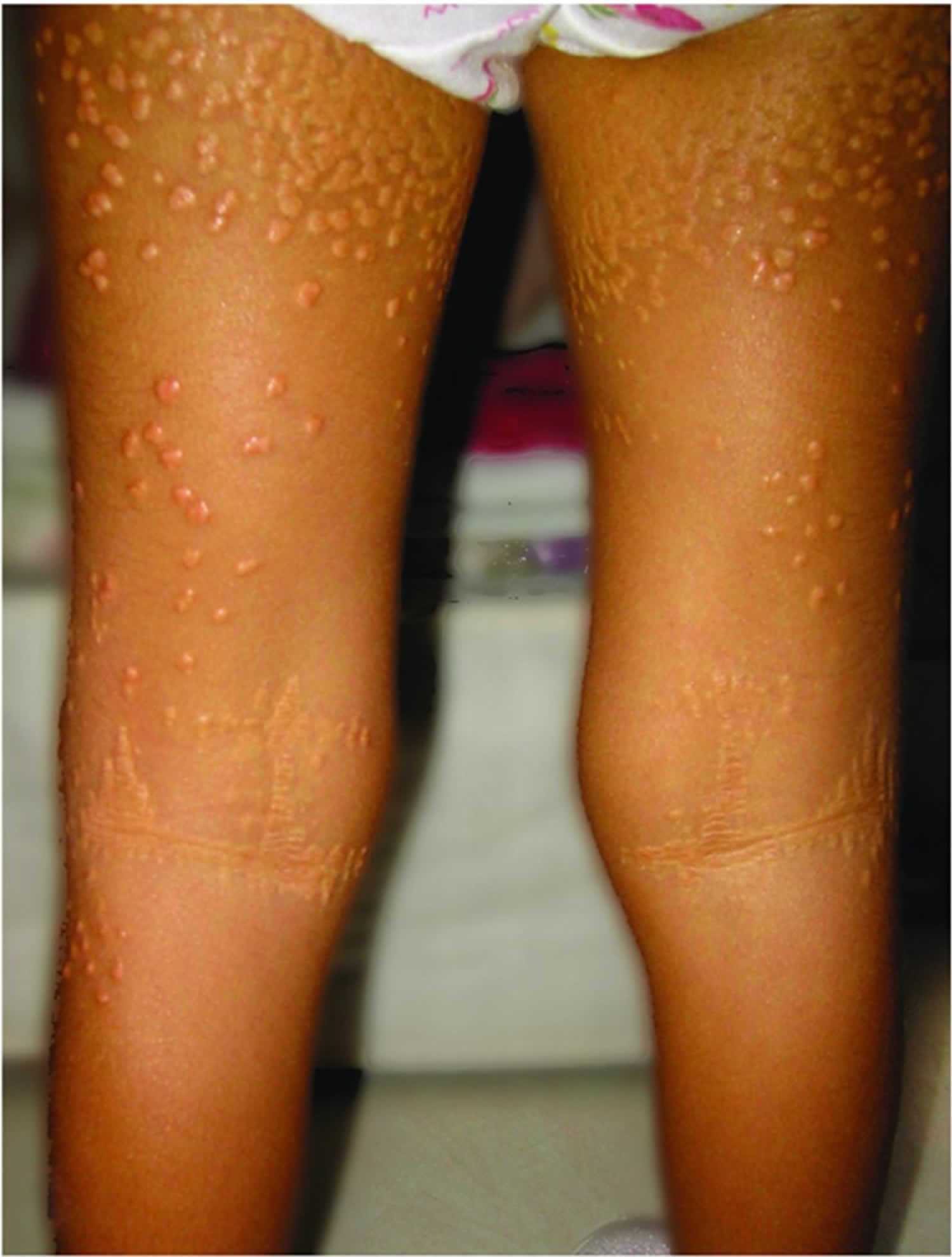
Figure 2. Alagille syndrome characteristic facial features
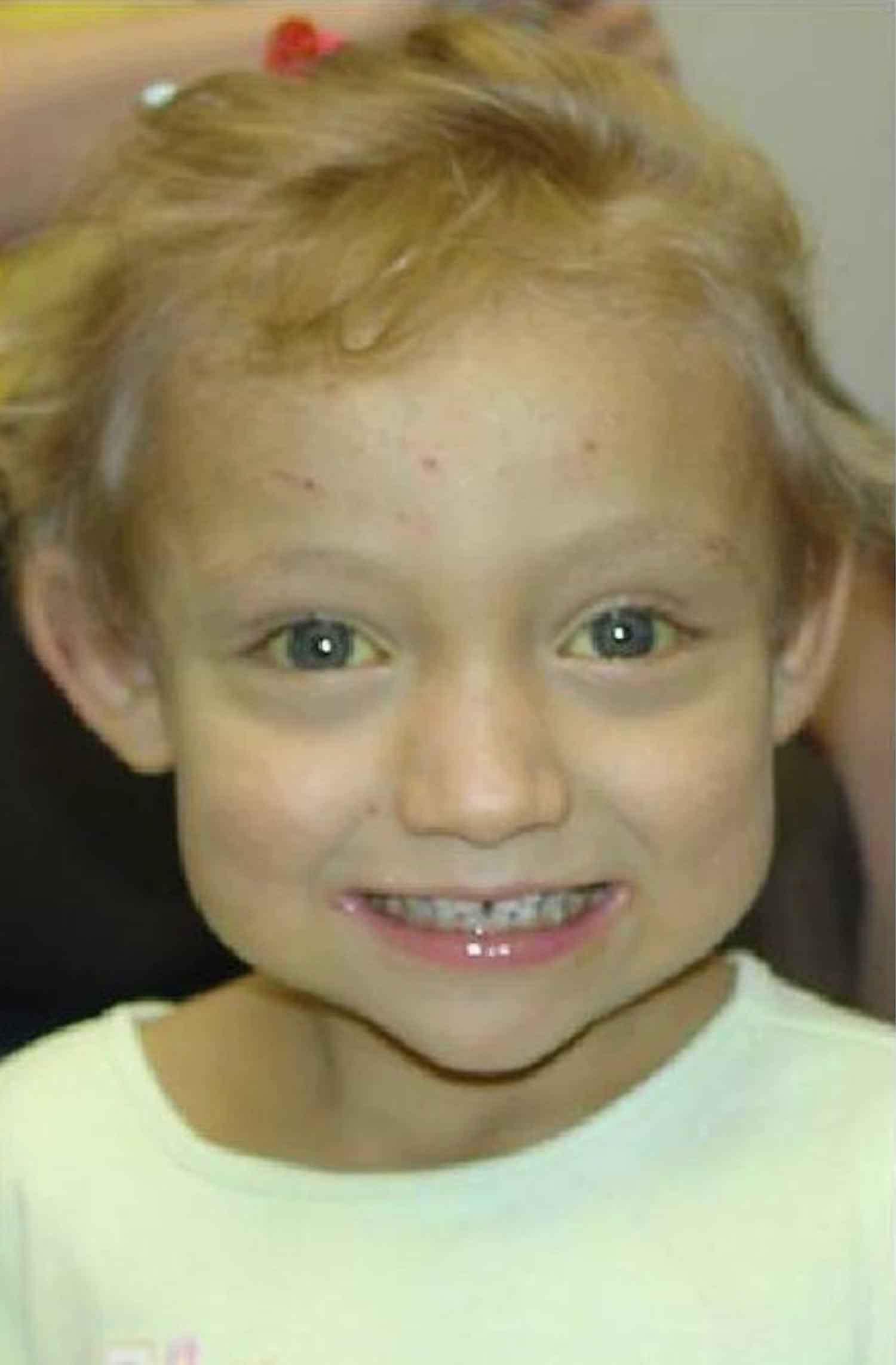
Footnote: Triangular appearance of the face with the high forehead, deep-set eyes with moderate hypertelorism, pointed chin, and saddle or straight nose with a bulbous tip.
[Source 2 ]Complications of Alagille syndrome
The complications of Alagille syndrome include liver failure, portal hypertension, and growth problems. People with Alagille syndrome usually have a combination of complications, and may not have every complication listed below.
Liver failure
Over time, the decreased number of bile ducts may lead to chronic liver failure, also called end-stage liver disease. This condition progresses over months, years, or even decades. The liver can no longer perform important functions or effectively replace damaged cells. A person may need a liver transplant. A liver transplant is surgery to remove a diseased or an injured liver and replace it with a healthy whole liver or a segment of a liver from another person, called a donor.
Portal hypertension
The spleen is the organ that cleans blood and makes white blood cells. White blood cells attack bacteria and other foreign cells. Blood flow from the spleen drains directly into the liver. When a person with Alagille syndrome has advanced liver disease, the blood flow backs up into the spleen and other blood vessels. This condition is called portal hypertension. The spleen may become larger in the later stages of liver disease. A person with an enlarged spleen should avoid contact sports to protect the organ from injury. Advanced portal hypertension can lead to serious bleeding problems.
Growth problems
Alagille syndrome can lead to poor growth in infants and children, as well as delayed puberty in older children. Liver disease can cause malabsorption, which can result in growth problems. Malabsorption is the inability of the small intestine to absorb nutrients from foods, which results in protein, calorie, and vitamin deficiencies. Serious heart problems, if present in Alagille syndrome, can also affect growth.
Malabsorption
People with Alagille syndrome may have diarrhea—loose, watery stools—due to malabsorption. The condition occurs because bile is necessary for the digestion of food. Malabsorption can lead to bone fractures, eye problems, blood-clotting problems, and learning delays.
Long-term Outlook
The long-term outlook for people with Alagille syndrome depends on several factors, including the severity of liver damage and heart problems. Predicting who will experience improved bile flow and who will progress to chronic liver failure is difficult. Ten to 30 percent of people with Alagille syndrome will eventually need a liver transplant 3.
Many adults with Alagille syndrome whose symptoms improve with treatment lead normal, productive lives. Deaths in people with Alagille syndrome are most often caused by chronic liver failure, heart problems, and blood vessel problems.
Alagille syndrome life expectancy
The prognosis is usually favorable, but complications such as cirrhosis, variceal hemorrhage, refractory ascites, and spontaneous bacterial peritonitis may occur. Alagille syndrome usually stabilizes between ages 4 and 10 years. When hepatic failure and/or cardiac lesions are present, mortality risk is increased.
Some people with Alagille syndrome only have one or two minor findings associated with the syndrome, such as one of the harmless eye findings, butterfly vertebrae, innocent heart murmurs, or characteristic facial features. In fact, before genetic testing, a person with only these findings would not have been diagnosed with Alagille syndrome. People with only minor findings will not need treatment and can lead normal, productive lives unaffected by the complications of Alagille syndrome.
For those with more serious symptoms of Alagille syndrome, the outlook depends on several factors, including the severity of liver damage and heart problems, and the early and continued correction of nutrition problems. Predicting who will experience improved bile flow and who will progress to end-stage liver failure is difficult since for some liver function improves over time. About 10% to 30% or more of the people with Alagille syndrome will eventually require a liver transplant 3.
Children born with severe heart problems or who have serious liver problems may have a shortened lifespan 4. However, due to improvements in liver and heart therapies, people are living longer, and those who respond well to the treatments and management of the syndrome, may lead normal, productive lives as adults. Deaths associated with Alagille syndrome are most often caused by liver failure, serious heart problems, or blood vessel abnormalities that cause bleeding in the brain or skull or strokes 5.
Alagille syndrome symptoms
The symptoms and severity of Alagille syndrome can vary greatly from one person to another, even among members of the same family. Some individuals may have mild forms of Alagille syndrome while others may have more serious forms. Common symptoms, which often develop during the first three months of life, include blockage of the flow of bile from the liver (cholestasis), yellowing of the skin and mucous membranes (jaundice) due to conjugated hyperbilirubinemia, poor weight gain and growth, severe itching (pruritis) and pale, loose stools. Additional symptoms include heart murmurs, congenital heart defects, vertebral (back bone) differences, thickening of the ring that normally lines the cornea in the eye (posterior embryotoxon) and distinctive facial features.
It is important to note that affected individuals may not have all of the symptoms discussed here. Affected individuals should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
- Liver problems: One of the major features of Alagille syndrome is liver damage caused by abnormalities in the bile ducts. These ducts carry bile, which helps to digest fats, from the liver to the gallbladder and small intestine. In Alagille syndrome, the bile ducts may be narrow, malformed, and reduced in number. This results in a build-up of bile causing scarring that prevents the liver from working properly. This may lead to jaundice, itchy skin, and deposits of cholesterol in the skin (xanthomas).
- Nutrition problems: Since bile is important for the digestion and absorption of fat and fat-soluble vitamins, people with Alagille syndrome may have a hard time getting enough calories and may develop Vitamin A, D, E and/or K deficiencies. Calcium and zinc deficiencies may also be associated with Alagille syndrome. Many people with Alagille syndrome continue to have nutrition problems even after a liver transplant, suggesting that other factors of the syndrome may play a role. Malnutrition can lead to slow growth, delayed puberty and other health concerns.
- Heart problems: Alagille syndrome is also associated with several heart problems, including impaired blood flow from the heart into the lungs (pulmonic stenosis). Other associated heart-related problems include a hole between the two lower chambers of the heart (ventricular septal defect) and a combination of heart defects called tetralogy of Fallot.
- Distinctive facial features: People with Alagille syndrome may have distinctive facial features, such as a broad, prominent forehead; deep-set eyes; and a small, pointed chin.
- Neurologic problems: Some people with Alagille syndrome may have intellectual disabilities and developmental delay.
- Vascular problems: Alagille syndrome can be associated with abnormalities of the blood vessels in the head and neck, which can lead to spontaneous or injury-induced bleeding in the skull or brain, or a stroke. Alagille syndrome can also cause narrowing (stenosis) or bulging (aneurysm) of other blood vessels in the body.
- Kidney problems: Some children may have impaired kidney function such as kidney artery stenosis, lipoid nephrosis, or glomerulosclerosis.
- Bone problems: People may have an unusual butterfly shape of the bones of the spinal column (vertebrae) but this shape almost never causes any problems with the function of the nerves in the spinal cord. However, Vitamin D deficiency can lead to rickets or weakened bones and poor growth. Calcium deficiency and other features of Alagille syndrome can also lead to poor growth, abnormally shaped bones, and increased risk of broken bones.
- Eye problems: Eye abnormalities are common in Alagille syndrome, but for the most part do not require treatment and do not cause vision loss. The most common eye finding is an extra, circular line on the surface of the eye, called a posterior embryotoxon, that can be detected during a specialized eye examination. Although about 90% of people with Alagille syndrome have this eye finding, so do up to 15% of people who do not have this syndrome. Other eye findings may include Axenfeld anomaly, in which strands of the colored part of the eye (iris) are abnormally attached to the cornea, and color changes of the retina (retinal pigmentary changes).
- Pancreatic insufficiency: Alagille syndrome can cause pancreatic insufficiency. People whose pancreas does not produce enough enzymes to help digest food do not properly absorb fat from food (fat malabsorption) and excrete fat in their bowel movements. Pancreatic insufficiency can cause poor nutrition and growth and therefore is believed to play a larger role than first suspected in the nutritional problems for people with Alagille syndrome.
A main finding of Alagille syndrome is liver disease that often becomes apparent within the first three months of life. However, in individuals with mild liver involvement, a diagnosis may not be made into later in life. Liver disease in Alagille syndrome, if present, may range in severity from jaundice or mild cholestasis to severe, progressive liver disease that can potentially result in liver failure.
Approximately 90 percent of individuals with Alagille syndrome have a reduced number of bile ducts (bile duct paucity) within the liver. Bile ducts are small tube-like structures that carry bile from the liver to the small intestines. The formation of bile is one of the functions of the liver. Bile is a fluid that contains water, certain minerals that carry an electric charge (electrolytes), and other materials including bile salts, phospholipids, cholesterol, and an orange-yellow pigment (bilirubin) that is a byproduct of the natural breakdown of the hemoglobin of red blood cells. Bile flow accomplishes two important tasks within the body: it aids in digestion and absorption of dietary fats, vitamins, and other nutrients and helps eliminate excess cholesterol, bilirubin, waste, and toxins from the body. Therefore, a problem with bile flow often results in malabsorption of vital nutrients and the accumulation of toxic materials in the body.
Because of the reduced number of bile ducts, individuals with Alagille syndrome can develop jaundice and cholestasis usually during the first four months of life. Cholestasis refers to reduced or obstructed flow of bile from the liver. Cholestasis can cause yellowing of the skin and whites of the eyes (jaundice), itching (pruritus) that may be intense, pale-colored stools, dark urine, fatty sores or bumps (xanthomas) just under the surface of the skin, and an abnormally enlarged liver (hepatomegaly) and/or enlarged spleen (splenomegaly). Because the body cannot properly absorb fats and fat-soluble vitamins (vitamins A, D, E, and K), affected children may also experience growth deficiencies and failure to thrive. Malabsorption of vital nutrients can also lead to rickets, a condition marked by softened, weakened bones (vitamin D deficiency), vision problems (vitamin A deficiency), poor coordination and developmental delays (vitamin E deficiency) and blood clotting problems (vitamin K deficiency).
In approximately 15 percent of patients, progressive liver disease results in scarring of the liver (cirrhosis) and liver failure. There is no way to tell which children are at risk for serious, progressive liver disease in Alagille syndrome.
Many individuals with Alagille syndrome have heart (cardiac) abnormalities that can range from benign heart murmurs to serious structural defects. A heart murmur is an extra sound that is heard during a heartbeat. Heart murmurs in children with Alagille syndrome are usually caused by narrowing of the blood vessels of the lungs (pulmonary artery stenosis). Some children with Alagille syndrome may have complex heart defects, the most common of which is tetralogy of Fallot. Tetralogy of Fallot is a rare form of cyanotic heart disease. Cyanosis is abnormal bluish discoloration of the skin and mucous membranes that occurs due to low levels of circulating oxygen in the blood. Tetralogy of Fallot consists of a combination of four different heart defects: ventricular septal defect, obstructed outflow of blood from the right ventricle to the lungs due to an abnormal narrowing of the opening between the pulmonary artery and the right ventricle of the heart (pulmonary stenosis), displaced aorta that causes blood to flow into the aorta from both the right and left ventricles, and abnormal enlargement of the right ventricle.
Additional heart defects that can occur in Alagille syndrome include ventricular septal defects, atrial septal defects, patent ductus arteriosus, and coarctation of the aorta. Some studies have shown that in rare cases there is an association with Wolff-Parkinson-White syndrome, a condition characterized by electrical disturbances in the heart. (For more information on these disorders, choose the specific disorder name as your search term in the Rare Disease Database.)
Some individuals with Alagille syndrome may have eye (ocular) abnormalities, especially posterior embryotoxon, a condition marked by thickening of the ring that normally lines the cornea in the eye. The cornea is the thin, transparent membrane that covers the eyeballs. In most cases, posterior embryotoxon is a benign finding that primarily helps to establish a clinical diagnosis and vision is usually unaffected, although mild decreases in the clarity of vision may occur. Less commonly, other eye abnormalities may occur such as Axenfeld anomaly, a condition in which strands of the iris are abnormally attached to the cornea, or progressive degeneration of the retina (pigmentary retinopathy). The retina is the thin layers of nerve cells that lines that inner surface of the back of the eyes and senses light and converts it to nerve signals, which are then relayed to the brain through the optic nerve.
Individuals with Alagille syndrome usually have distinctive facial features including deeply-set and widely spaced (hypertelorism) eyes, a pointed chin, broad forehead, and low-set, malformed eyes. In older individuals and adults the chin may appear larger and more prominent (prognathia).
Skeletal abnormalities may occur in some individuals with Alagille syndrome including butterfly vertebrae, a condition in which certain bones of the spinal column are irregularly-shaped. This condition is often noted on an x-ray, but usually does not cause any symptoms or problems (asymptomatic).
Additional symptoms may occur in some individuals with Alagille syndrome including kidney (renal) abnormalities, pancreatic insufficiency, vascular anomalies, mild developmental delays and cognitive impairment. Kidney abnormalities may be more prevalent in individuals with Alagille syndrome caused by mutations in the NOTCH2 gene and include abnormally small kidneys, the presence of cysts on the kidneys and decreased or impaired kidney function. The pancreas is a small organ located behind the stomach that secretes enzymes that travel to the intestines and aid in digestion. The pancreas also secretes other hormones such as insulin, which helps to break down sugar. Pancreatic insufficiency is when the pancreas cannot produce or transport enough enzymes to the intestines to aid in the breakdown and absorption of food and nutrients.
Individuals with Alagille syndrome can also develop abnormalities of certain blood vessels (vascular anomalies) including those in the brain, liver, lungs, heart, and kidneys. Vascular anomalies in the brain can lead to bleeding inside the brain (intracranial bleeding) and stroke. Some individuals with Alagille syndrome have developed a condition known as Moyamoya syndrome. Moyamoya syndrome is a progressive disorder that is characterized by narrowing (stenosis) and/or closing (occlusion) inside the skull of the carotid artery, the major artery that delivers blood to the brain. Intracranial bleeding and other vascular anomalies are potentially life-threatening complications and account for a significant percentage of mortality and morbidity in Alagille syndrome.
Alagille syndrome causes
In more than 90 percent of cases, mutations in the JAG1 gene cause Alagille syndrome. Another 7 percent of individuals with Alagille syndrome have small deletions of genetic material on chromosome 20 that include the JAG1 gene. A few people with Alagille syndrome have mutations in a different gene, called NOTCH2. The JAG1 and NOTCH2 genes provide instructions for making proteins that fit together to trigger interactions called Notch signaling between neighboring cells during embryonic development. This signaling influences how the cells are used to build body structures in the developing embryo. Changes in either the JAG1 gene or NOTCH2 gene probably disrupt the Notch signaling pathway. As a result, errors may occur during development, especially affecting the bile ducts, heart, spinal column, and certain facial features.
Inheritance pattern
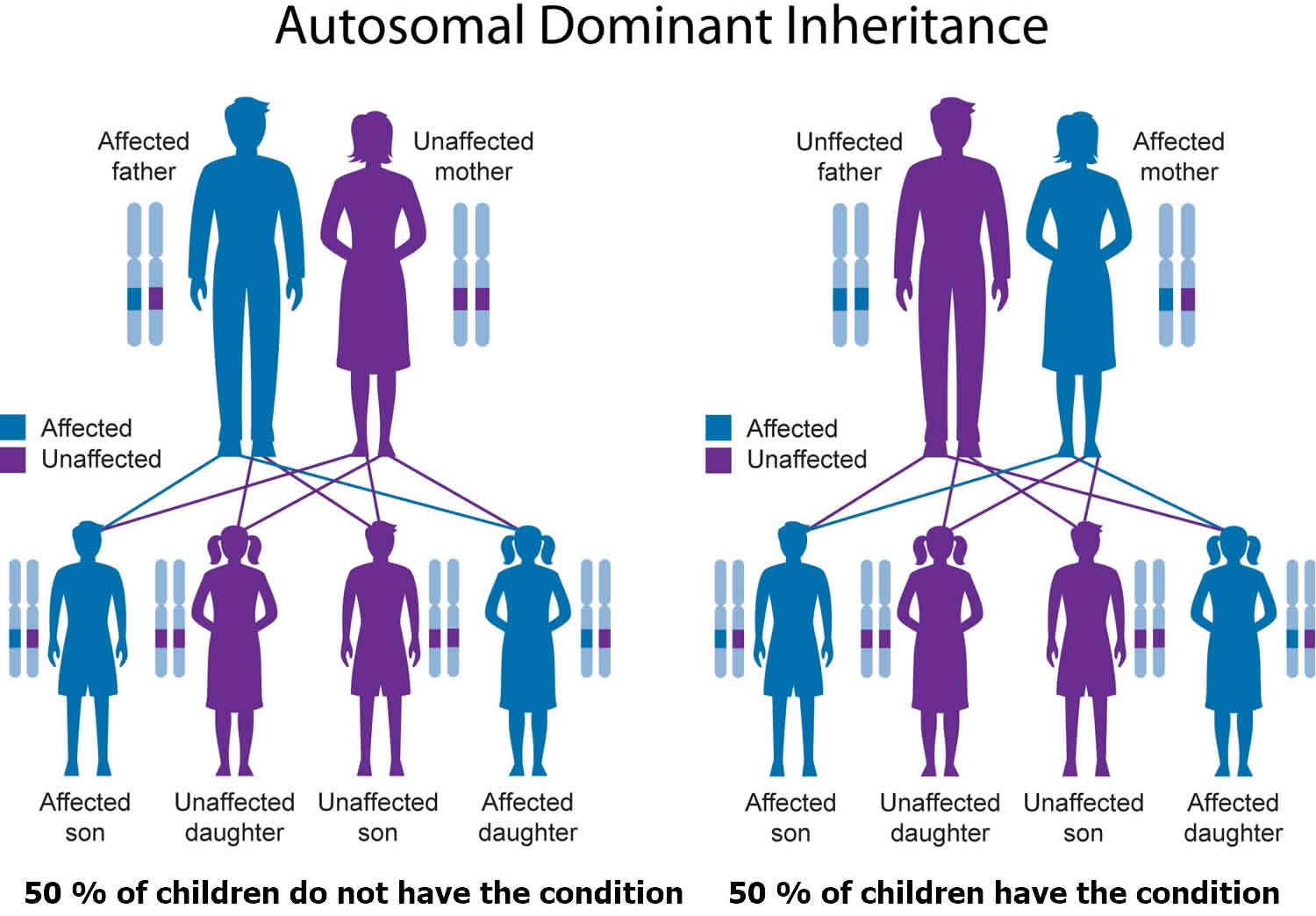
Alagille syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered or deleted gene in each cell is sufficient to cause the disorder. A parent with Alagille syndrome has a 50% chance of passing the genetic change causing Alagille syndrome onto each of their children, and it is not possible to predict how severely affected any of their children who inherit Alagille syndrome will be 5.
In approximately 30 to 50 percent of cases, an affected person inherits the mutation or deletion from one affected parent. Other cases result from new mutations in the gene or new deletions of genetic material on chromosome 20 that occur as random events during the formation of reproductive cells (eggs or sperm) or in early fetal development. These cases occur in people with no history of the disorder in their family.
Figure 3. Alagille syndrome autosomal dominant inheritance pattern
When to consider genetic counseling
People who are considering genetic testing may want to consult a genetics counselor. Genetic counseling can help family members understand how test results may affect them individually and as a family. Genetic counseling is provided by genetics professionals—health care professionals with specialized degrees and experience in medical genetics and counseling. Genetics professionals include geneticists, genetics counselors, and genetics nurses.
Genetics professionals work as members of health care teams, providing information and support to individuals or families who have genetic disorders or a higher chance of having an inherited condition.
Genetic counseling may be useful when a family member is deciding whether to have genetic testing and again later when test results are available.
The following online resources can also help you find a genetics professional in your community:
- The National Society of Genetic Counselors (https://www.nsgc.org/) provides a searchable directory of US and international genetic counseling services.
- The American College of Medical Genetics (https://www.acmg.net/) has a searchable database of US genetics clinics.
- The University of Kansas Medical Center (http://www.kumc.edu/gec/prof/genecntr.html) provides a list of US and international genetic centers, clinics, and departments.
- The American Society of Human Genetics (http://www.ashg.org/membership/member_search.shtml) maintains a database of its members, which includes individuals who live outside the United States. Visit the link to obtain a list of the geneticists in your country, some of whom may be researchers that do not provide medical care.
Pregnancy and Alagille syndrome
A few cases of successful pregnancy in Alagille syndrome have been described by different groups and stemming from these cases, several issues should be considered before attempting embarking on a pregnancy 6:
- the severity of liver disease and the degree of portal hypertension, which could further worsen during pregnancy;
- the degree of cardiac dysfunction and, in particular, the severity of pulmonary hypertension; and
- the 50% chance for the fetus to inherit the maternal or paternal Alagille syndrome mutation, although the severity of the clinical manifestations cannot be predicted in view of the observed intrafamilial variability.
Thus, genetic counseling is crucial before conception 6. This includes the discussion of preimplantation and prenatal diagnosis as well as the use of surrogate mother if the pregnancy can be associated with maternal risk for deterioration.
Alagille syndrome diagnosis
A diagnosis of Alagille syndrome is made based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. Because the symptoms of Alagille syndrome are highly variable, obtaining a diagnosis can be difficult. Surgical removal and microscopic study of liver tissue (liver biopsy) can reveal bile duct paucity. Although bile duct paucity is considered a key characteristic of Alagille syndrome, this finding is not always present in infants with the disorder.
A physician may suspect Alagille syndrome if an individual has three of the following five clinical findings in addition to bile duct paucity: symptoms of liver disease or cholestasis, heart defect, skeletal abnormality, eye (ophthalmologic) abnormality, and/or distinctive facial features.
In addition to a liver biopsy, physicians may conduct other tests to aid in the diagnosis of Alagille syndrome. Such tests may include blood tests to determine liver function and detect fat-soluble vitamin deficiencies, an eye examination, x-rays of the spine to detect characteristic changes such as butterfly vertebrae, an abdominal ultrasound of the hepatobiliary tree (e.g., liver, pancreas, gall bladder and spleen) to detect abnormalities or rule out other conditions, and an examination of heart structure and function to detect potential heart abnormalities.
The diagnosis of Alagille syndrome can be confirmed in many cases by molecular genetic testing, which reveals the presence of a JAG1 or NOTCH2 mutation. However, in some people with Alagille syndrome, genetic testing may not reveal a JAG1 or NOTCH2 mutation.
Alagille syndrome treatment
The treatment of Alagille syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, gastroenterologists, cardiologists, ophthalmologists, and other healthcare professionals may need to systematically and comprehensively plan the child’s treatment. Individuals with Alagille syndrome should have a baseline echocardiogram (ultrasound of the heart) to screen for heart involvement, ultrasound of the abdomen to screen for liver and kidney anomalies, and a screening eye (ophthalmology) exam, In addition, if not previously obtained for specific symptoms, a screening imaging study of the blood vessels of the head (MRI [magnetic resonance imaging] or MRA [magnetic resonance angiogram]) is recommended for children who are old enough to sit through the study without need for anesthesia or sedation. Supplemental treatment with vitamins and nutrients is essential for individuals with malabsorption. Such treatment may include restoring vitamins A, D, E and K. Young children may be given formula with medium chain triglycerides because this form of fat is better absorbed by individuals with Alagille syndrome who have cholestasis. In some cases, affected children may need to receive extra calories through a tube that runs from the nose to the stomach (nasogastric tube) or through a tube placed directly into the stomach through a small incision in the abdominal wall and stomach (gastrostomy tube).
Specific treatment may be indicated for individuals with cholestatic liver disease. The drug ursodeoxycholic acid is given to help improve bile flow, which can lead to a reduction in some symptoms such as itching (pruritus) or fatty deposits (xanthomas). However, pruritus associated with Alagille syndrome often is resistant to therapy. Additional drugs that have been used to treat pruritus include antihistamines, rifampin, cholestyramine, and naltrexone. Keeping the skin properly hydrated with moisturizers is also recommended. Cholestyramine may also be indicated for individuals with elevated cholesterol levels or xanthomas.
Some affected infants and children with Alagille syndrome who do not respond to pharmacologic and dietary therapies may be treated by a surgical procedure known as partial biliary diversion. This surgical procedure is used to disrupt or divert recirculation of bile acids between the liver and the gastrointestinal tract. This therapy has demonstrated that, in some cases, it can improve certain symptoms such as reducing itchiness or xanthoma formation.
In severe cases of Alagille syndrome (i.e., cases that have progressed to cirrhosis or liver failure or in which other therapies were unsuccessful), liver transplantation may be required.
Additional complications that can be associated with Alagille syndrome including heart, blood vessel and kidney abnormalities are treated in the standard manner. In some cases, this may include surgery.
Genetic counseling may be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Eating, Diet, and Nutrition
Researchers have not found that eating, diet, and nutrition play a role in causing or preventing Alagille syndrome. However, these factors are important for people with Alagille syndrome, particularly children, who are malnourished, growing poorly, or have delayed puberty. Caregivers and parents of children with Alagille syndrome should try to maximize their children’s potential for growth through good eating, diet, and nutrition.
A nutritionist or a dietitian—a person with training in nutrition and diet—can work with someone with Alagille syndrome and his or her health care team to build an appropriate healthy eating plan. A person with Alagille syndrome may need to take dietary supplements or vitamins in addition to eating a set number of calories, based on the type of complications the person has. Researchers consider good nutrition to be one of the most important aspects of managing the disorder.
If potential liver problems are present, a person with Alagille syndrome should not drink alcoholic beverages without talking with his or her health care provider first.
Additionally, eating, diet, and nutrition play a part in overall health and preventing further health problems.
- Alagille syndrome. https://ghr.nlm.nih.gov/condition/alagille-syndrome[↩][↩]
- Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet. 2016;9:75-82. Published 2016 Jun 30. doi:10.2147/TACG.S86420 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4935120/[↩][↩]
- Kamath BM, Loomes KM, Piccoli DA. Medical management of Alagille syndrome. Journal of Pediatric Gastroenterology and Nutrition. 2010;50(6):580–586.[↩][↩]
- Alagille Syndrome. https://www.niddk.nih.gov/health-information/liver-disease/alagille-syndrome[↩]
- Alagille syndrome. https://www.chop.edu/conditions-diseases/alagille-syndrome[↩][↩]
- Successful pregnancy in Alagille Syndrome. Ferrarese A, Senzolo M, Burra P. Dig Liver Dis. 2015 Jan; 47(1):86-7.[↩][↩]

{kind=link}