Contents
What is Cornelia de Lange syndrome
Cornelia de Lange syndrome is a rare genetic disorder that is apparent at birth (congenital) causing delays in physical development before and after birth (prenatal and postnatal growth retardation) that affects many parts of the body 1. The severity of Cornelia de Lange syndrome and the associated signs and symptoms can vary widely among affected individuals, but may include distinctive facial characteristics, growth delays, intellectual disability and limb defects. Children with Cornelia de Lange syndrome are often short and below average in weight when compared to others their age. The average birth weight for children with Cornelia de Lange syndrome is 5 lbs., 1 oz., but birth weights have been reported ranging from 1 lb., 2 oz. to 10 lbs. Average birth length is approximately 18 inches. Small head size (microcephaly) is a feature commonly associated with Cornelia de Lange syndrome.
Cornelia de Lange syndrome is characterized by slow growth before and after birth leading to short stature; intellectual disability that is usually moderate to severe; and abnormalities of bones in the arms, hands, and fingers. The vast majority of children diagnosed with Cornelia de Lange syndrome are intellectually delayed, with the degree ranging from mild to severe. Learning disabilities and severe language delays are often present. Although intellectual delay is considered essential for diagnosis, there are cases of people with Cornelia de Lange syndrome who have borderline to normal intelligence.
Most people with Cornelia de Lange syndrome also have distinctive facial features, including arched eyebrows that often meet in the middle (synophrys), long eyelashes, low-set ears, small and widely spaced teeth, and a small and upturned nose. Many affected individuals also have behavior problems similar to autism, a developmental condition that affects communication and social interaction.
Additional signs and symptoms of Cornelia de Lange syndrome can include excessive body hair (hypertrichosis), an unusually small head (microcephaly), hearing loss, and problems with the digestive tract. Some people with this condition are born with an opening in the roof of the mouth called a cleft palate. Seizures, heart defects, and eye problems have also been reported in people with this condition.
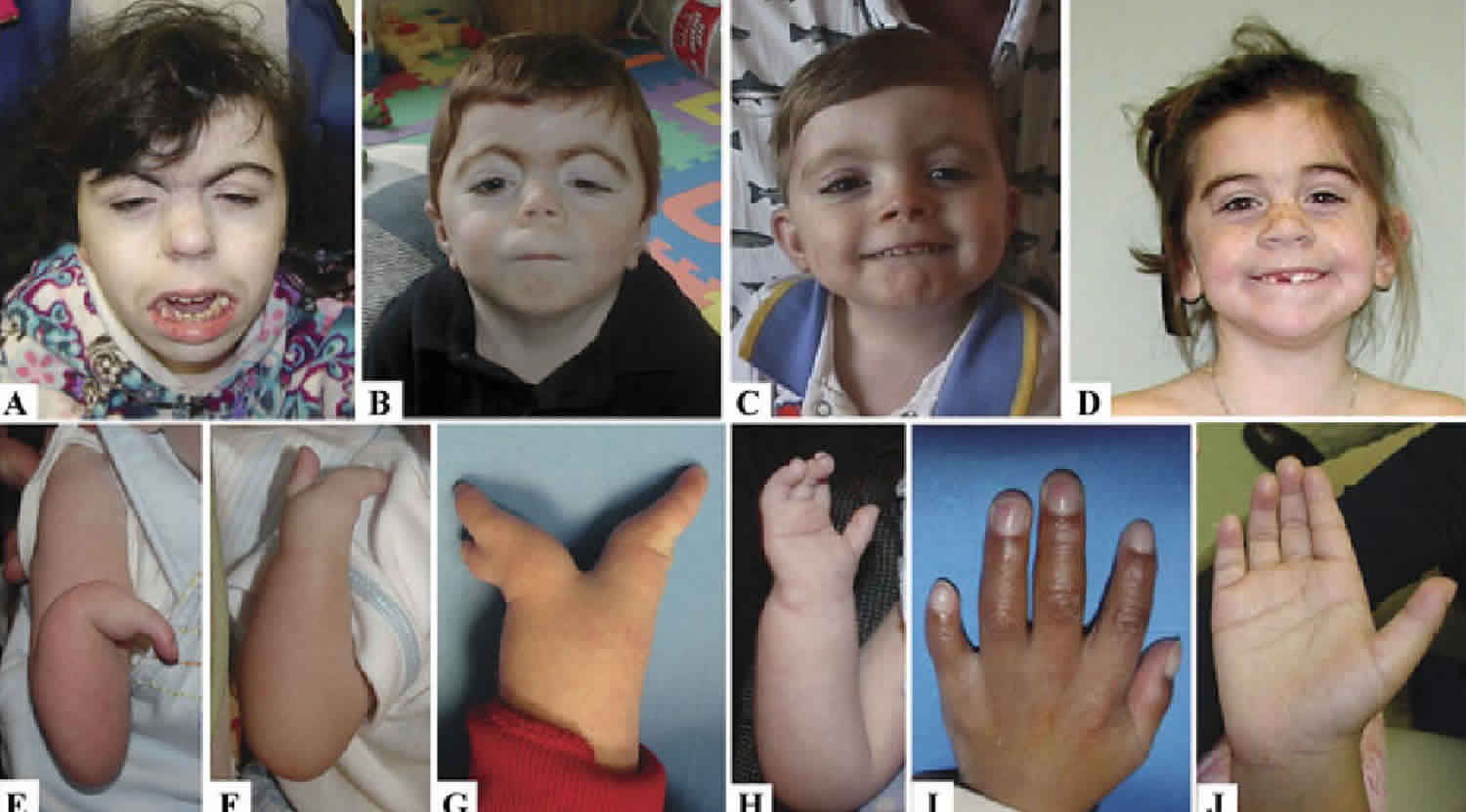
Figure 1. Cornelia de Lange syndrome characteristic features
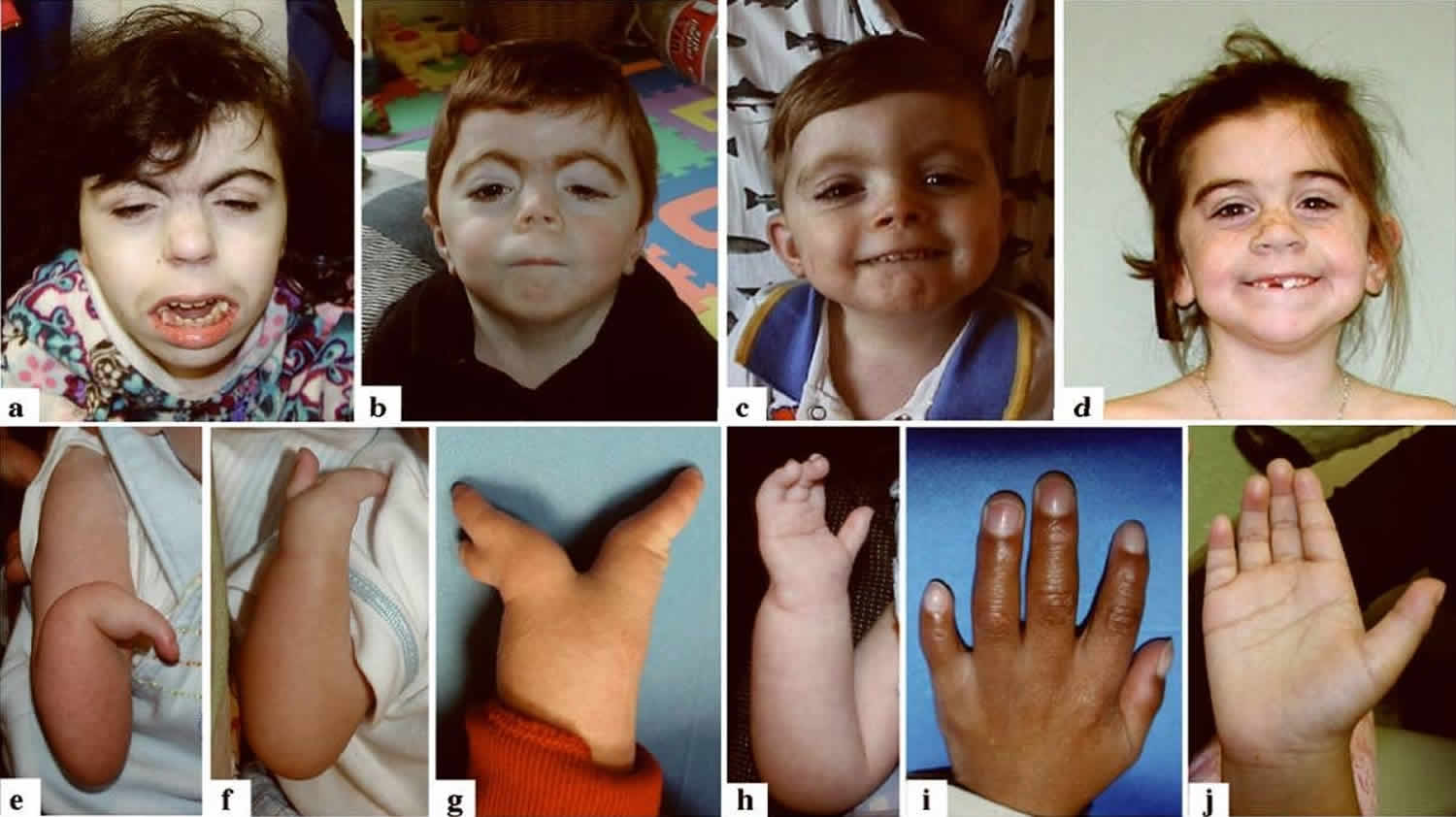
Footnote: Characteristic facial features in two individuals with Cornelia de Lange syndrome and NIPBL mutations (a and b) and in two children with SMC1A mutations (c and d). Note the arched eyebrows with synophrys, long eyelashes, ptosis (more noticeable in `a’ and `b’), the short upturned nose and long philtrum with thin upper lip. These features are still present in the SMC1A mutated individuals, but to a more subtle degree. The variability of the phenotype in Cornelia de Lange syndrome is highlighted by the range of involvement of the upper limbs as demonstrated in e-j. In `e’ the more severe end of the spectrum of upper limb involvement is depicted with severe ulnar hypoplasia of the forearm with only a single digit and underdevelopment of almost all boney structures. f-h depicts variable forms of oligodactyly and i-j demonstrates the milder end of the spectrum with small hands, 5th finger clinodactyly and proximally placed thumbs.
[Source 2 ]Approximately 60% of people affected by Cornelia de Lange syndrome have a disease-causing variation (mutation) in the NIPBL gene, and about 10% of cases are caused by mutations in one of four known genes: SMC1A, SMC3, HDAC8 and RAD21. In the remaining 30% of cases, the underlying genetic cause of the condition is unknown. Cornelia de Lange syndrome can be inherited in an autosomal dominant (NIPBL, SMC2, or RAD21) or X-linked (SMC1A or HDAC8) manner. However, most cases result from new (de novo) mutations and occur in people with no family history of the condition 3. Treatment is based on the signs and symptoms present in each person 3.
Although the exact incidence is unknown, Cornelia de Lange syndrome likely affects 1 in 10,000 to 30,000 newborns. Cornelia de Lange syndrome is probably underdiagnosed because affected individuals with mild or uncommon features may never be recognized as having Cornelia de Lange syndrome.
In 1933, Dutch pediatrician Dr. Cornelia de Lange described two children with similar features. The first child had pneumonia and feeding difficulties. She was very small for her age, with a proportionately smaller head circumference. Other unusual facial characteristics were noted by Dr. de Lange. Soon after, she saw a second little girl with common medical problems and physical characteristics. Nowhere was the puzzled physician able to find a similar patient described in the medical literature. Cornelia de Lange is now generally credited with describing the collection of symptoms comprising the syndrome that bears her name.
The syndrome is sometimes referred to as Brachmann-de Lange syndrome after Dr. W. Brachmann, who described a similar patient in 1916. Dr. de Lange may have overlooked his report because he concentrated on characteristics of the upper limbs and wrote on the facial symptoms less specifically.
Do all people with Cornelia de Lange syndrome have intellectual disability?
Intellectual disability is a common feature of Cornelia de Lange syndrome, occurring in more than 95 percent of people with this condition 3. The IQ test is one tool for measuring intelligence, and a score below 70 indicates that a person has intellectual disability. IQ scores in people with Cornelia de Lange syndrome range from 30 to 102, so there have been some people with IQ scores in the normal range. The average IQ score for people with Cornelia de Lange is 53 3.
- Classic Cornelia de Lange syndrome. Severe-to-profound pervasive developmental delay is characteristic.
- Mild Cornelia de Lange syndrome. Less affected individuals with higher functioning and higher IQs (some in the normal range) have been identified. The overall IQ in Cornelia de Lange syndrome ranges from below 30 to 102, with an average IQ of 53 4.
How can I find other families with Cornelia de Lange syndrome?
Here is a list of groups dedicated to Cornelia de Lange syndrome. You can contact these organizations to get in touch with other families.
- Cornelia de Lange Syndrome Foundation, Inc. (https://www.cdlsusa.org/)
- Cornelia de Lange Syndrome Foundation UK & Ireland (http://www.cdls.org.uk/)
- Cornelia de Lange Syndrome World federation (http://www.cdlsworld.org/xwiki/bin/view/Main/WebHome)
- Children’s Craniofacial Association (https://ccakids.org/)
Cornelia de Lange syndrome causes
To date Cornelia de Lange syndrome can result from mutations in at least five genes: NIPBL, SMC1A, HDAC8, RAD21, and SMC3. Mutations in the NIPBL gene have been identified in more than half of all people with Cornelia de Lange syndrome, fewer than 1% of individuals with NIPBL-related Cornelia de Lange syndrome have an affected parent; mutations in the other genes are much less common. There are still a small percentage of people with Cornelia de Lange syndrome in whom a change in one of these genes cannot be identified; it is likely that additional causative genes have yet to be identified. In the majority of cases, the mutation is not inherited, but occurs spontaneously as a new (de novo) change in an egg or sperm or very early in fetal development.
The proteins produced from all five genes contribute to the structure or function of the cohesin complex, a group of proteins with an important role in directing development before birth. Within cells, the cohesin complex helps regulate the structure and organization of chromosomes, stabilize cells’ genetic information, and repair damaged DNA. The cohesin complex also regulates the activity of certain genes that guide the development of limbs, face, and other parts of the body.
Mutations in the NIPBL, SMC1A, HDAC8, RAD21, and SMC3 genes cause Cornelia de Lange syndrome by impairing the function of the cohesin complex, which disrupts gene regulation during critical stages of early development.
The features of Cornelia de Lange syndrome vary widely, and the severity of the disorder can differ even in individuals with the same gene mutation. Researchers suspect that additional genetic or environmental factors may be important for determining the specific signs and symptoms in each individual. In general, SMC1A, RAD21, and SMC3 gene mutations cause milder signs and symptoms than NIPBL gene mutations. Mutations in the HDAC8 gene cause a somewhat different set of features, including delayed closure of the “soft spot” on the head (the anterior fontanelle) in infancy, widely spaced eyes, and dental abnormalities. Like affected individuals with NIPBL gene mutations, those with HDAC8 gene mutations may have significant intellectual disability.
In about 30 percent of cases, the cause of Cornelia de Lange syndrome is unknown. Researchers are looking for additional changes in the five known genes, as well as mutations in other genes, that may cause this condition.
Table 1. Molecular Genetic Testing Used in Cornelia de Lange Syndrome
| Gene | Proportion of Cornelia de Lange syndrome Attributed to Pathogenic Variants in This Gene | Test Method |
|---|---|---|
| NIPBL | ~60% 3 | Sequence analysis 4 |
| Gene-targeted deletion/duplication analysis 5 | ||
| SMC1A | ~5% 6 | Sequence analysis 4 |
| Gene-targeted deletion/duplication analysis 5 | ||
| HDAC8 | ~4% | Sequence analysis 4 |
| Gene-targeted deletion/duplication analysis 5 | ||
| SMC3 | 1%-2% | Sequence analysis 4 |
| Gene-targeted deletion/duplication analysis 5 | ||
| RAD21 | <1% | Sequence analysis 4 |
| Gene-targeted deletion/duplication analysis 5 | ||
| Unknown 8 | NA | NA |
Footnote: NA = not applicable
3. Gene-targeted deletion/duplication analysis of NIPBL detects ~3% of NIPBL-related Cornelia de Lange syndrome 5
4. Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
5. Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods that may be used include: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
8. Lack of identified NIPBL pathogenic variants in 50% of individuals with Cornelia de Lange syndrome suggest possible locus heterogeneity 6
Inheritance pattern
When Cornelia de Lange syndrome is caused by mutations in the NIPBL, RAD21, or SMC3 gene, the condition is considered to have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases result from new gene mutations and occur in people with no history of the condition in their family.
When Cornelia de Lange syndrome is caused by mutations in the HDAC8 or SMC1A gene, the condition has an X-linked dominant pattern of inheritance. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. Studies of X-linked Cornelia de Lange syndrome indicate that one copy of the altered gene in each cell may be sufficient to cause the condition. Unlike X-linked recessive conditions, in which males are more frequently affected or experience more severe symptoms than females, X-linked dominant Cornelia de Lange syndrome appears to affect males and females similarly. Most cases result from new mutations in the HDAC8 or SMC1A gene and occur in people with no history of the condition in their family.
Genetic counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
What does a negative genetic test result mean for my child?
In cases where the diagnosis is doubtful, it may contribute additional evidence against the diagnosis; however even in individuals with classic features of Cornelia de Lange syndrome, gene mutations are not always found. In about 30 percent of cases, the cause of Cornelia de Lange syndrome is unknown. Researchers are looking for additional changes in the five known genes, as well as mutations in other genes, that may cause this condition; it is likely that additional causative genes have yet to be identified.
Cornelia de Lange syndrome symptoms
Affected people typically have distinctive craniofacial features, as well, which may include microcephaly; arched eyebrows that often grow together in the middle (synophrys); long eyelashes; low-set ears; small, widely spaced teeth; and a small, upturned nose.
The signs and symptoms of Cornelia de Lange syndrome vary widely among affected people and can range from relatively mild to severe. Affected people may experience 3:
- Slowed growth before and after birth
- Intellectual disability
- Developmental delay
- Autistic and/or self-destructive behaviors
- Skeletal abnormalities of the arms and hands
- Gastrointestinal problems
- Hirsutism (excess hair growth)
- Hearing loss
- Myopia
- Congenital heart defects
- Genital abnormalities (i.e. cryptorchidism)
- Seizures. Approximately 25% of children have had seizures.
- Some parents have described temperature intolerance and decreased pain sensation.
Common physical characteristics of Cornelia de Lange syndrome include:
- Similar facial features (which may include an upturned nose, eyebrows that meet in the middle, long eyelashes and low-set ears)
- Gastroesophageal reflux disease (GERD), which can make eating painful and contribute to slow growth and other intestinal differences
- Upper limb differences ranging from small hands to missing fingers or forearms
- Cleft palate
- Diaphragmatic hernias, vision and hearing problems, excessive body hair (hirsutism), heart defects, seizures and dental issues are also common.
Cornelia de Lange syndrome is a very rare disorder characterized by growth delays; distinctive facial features; malformations of the hands, feet, arms, and/or legs (limb anomalies); other physical abnormalities; intellectual disability; and/or developmental delays. The range and severity of symptoms and physical characteristics may vary greatly from person to person.
Individuals with Cornelia de Lange syndrome exhibit abnormal growth delays that affect both weight and linear growth before and after birth (prenatal and postnatal growth retardation). Most affected infants may have a low birth weight and may fail to gain weight or grow at the expected rate (failure to thrive). Cornelia de Lange syndrome growth charts are available to compare growth to other affected individuals. Individuals may experience feeding, chewing, and swallowing difficulties during the first several months/years of life.
Many affected infants may frequently “spit up” food that has already been swallowed (regurgitation) and may experience episodes of severe, forceful vomiting (projectile vomiting). Infants with Cornelia de Lange syndrome may also demonstrate abnormally increased muscle tone (hypertonicity) and have an unusual, low-pitched, growling cry.
Individuals with Cornelia de Lange syndrome also have distinctive features of the head and facial (craniofacial) area including an abnormally small head (microcephaly) that may also be unusually short (brachycephaly); a short, thick neck; a low hairline; a small, broad, upturned nose with nostrils that tip upwards (anteverted nares); neat, arched eyebrows that grow together (synophrys); long, curly eyelashes; and/or excessive hair growth on various areas of the body (hypertrichosis). Additional features may include thin, downturned lips; an abnormally long vertical gap between the upper lip (philtrum) and the nose; an abnormally small, underdeveloped jaw (micrognathia); late-erupting, widely-spaced, unusually small teeth; and low-set ears. In some cases, affected infants may also exhibit incomplete closure of the roof of the mouth (cleft palate), a hidden incomplete closure (submucous cleft palate) and/or a highly arched palate.
In most infants with Cornelia de Lange syndrome, the hands and feet are small for their size. In addition, affected individuals may have short fingers that become smaller and thinner toward the ends (tapered fingers), fifth fingers that are permanently curved toward the ring finger (clinodactyly), and/or, in some people, absence of one or more fingers (oligodactyly). The thumbs may be abnormally positioned (i.e., proximally placed) and the arms may be permanently bent or flexed at the elbows due to bone fusions. In addition, in many cases, affected individuals may demonstrate underdevelopment (hypoplasia) of some of the bones of the fingers and toes, and the second and third toes are often abnormally fused or webbed (syndactyly). Some affected infants may also have, in rare cases, fingers, hands, and forearms which are missing. In individuals with Cornelia de Lange syndrome, upper limb abnormalities may involve one side (unilateral) or both sides (bilateral) of the body. If bilateral limb malformations are present, the abnormalities on one side of the body may be completely different from those on the other side (asymmetrical). Although the feet are small, only in extremely rare cases are there absent bones in the feet or lower legs.
Individuals with Cornelia de Lange syndrome also demonstrate delayed bone age (retarded osseous maturation). In addition, affected individuals may remain low in weight and exhibit abnormally short stature (prenatal and postnatal growth retardation), failure to thrive during infancy, delayed bone age, and/or other abnormalities. Many individuals with Cornelia de Lange syndrome also exhibit additional skeletal abnormalities. These may include a deformity of the hip (coxa valga), a short breastbone (sternum), and/or abnormally thin ribs.
Many infants and children with Cornelia de Lange syndrome may exhibit delays in the acquisition of skills requiring the coordination of mental and muscular activity (psychomotor retardation), have mild to severe intellectual disability, and/or demonstrate behavioral problems (e.g., episodes of biting, screaming, hitting themselves, etc.). In addition, although affected children have decreased facial expression based on emotion, they appear to respond positively to certain stimuli (e.g., fast movements). A Cornelia de Lange syndrome developmental chart is available to compare milestones.
Many children with Cornelia de Lange syndrome also have hearing impairment as well as abnormal speech development. Middle ear infections (otitis media), which sometimes occur chronically with an accumulation of sticky fluid (otitis media with effusion or glue ear), are common. Younger children may have difficulty speaking (dysphonia), while older children may have abnormally hoarse speech.
Many individuals with Cornelia de Lange syndrome also exhibit additional physical abnormalities. In many cases, the skin may appear “marbled” (cutis marmorata), and the skin above the eyes, mouth, and nose may have an unusual bluish tone. In addition, many affected individuals demonstrate irregularities in the skin ridge patterns on the palms of the hands (dermatoglyphics). As mentioned earlier, most affected individuals may exhibit excessive hair growth (hypertrichosis) on various areas of the body including the ears. Hair may also tend to appear on the lower back, limbs, and/or other areas of the body.
Many individuals with Cornelia de Lange syndrome also have various abnormalities of the gastrointestinal system including gastroesophageal reflux, a condition in which the acidic contents of the stomach flow upward into the lower esophagus; inflammation of the lining of the esophagus (esophagitis); and/or narrowing of the esophagus (esophageal stenosis). In addition, affected individuals may have abnormal twisting (malrotation) of the intestines, potentially causing intestinal obstruction (volvulus). In some children, the bands of muscle fibers (pyloric sphincter) at the junction between the stomach and small intestine (pyloric stenosis) may become abnormally narrowed (stenosis) in infancy, resulting in obstruction of the normal flow of stomach contents into the small intestine. In addition, some individuals with Cornelia de Lange syndrome may also have protrusion of portions of the large intestine through an abnormal opening in musculature lining the abdominal cavity in the area of the groin (inguinal hernia) and/or part of the stomach through an abnormal opening where the esophagus passes through the diaphragm (hiatal hernia). In some individuals with Cornelia de Lange syndrome, certain gastrointestinal abnormalities may lead to intestinal obstruction, potentially causing serious or life-threatening complications if left untreated.
Some individuals with Cornelia de Lange syndrome may also have malformations of the genitourinary tract. In affected males, such abnormalities may include underdevelopment (hypoplasia) of the genitals, failure of one or both of the testes to descend into the scrotum (cryptorchidism), and/or abnormal placement of the urinary opening (urinary meatus) on the underside of the penis (hypospadias). Affected females may have abnormal development of the uterus (e.g., bicornate or septate uterus), and menstruation may be irregular.
Many children with Cornelia de Lange syndrome have additional physical abnormalities including various heart (cardiac) abnormalities. Some affected individuals may also have an increased susceptibility to repeated respiratory infections, eye abnormalities such as nearsightedness (myopia), rapid, involuntary eye movements (nystagmus), and/or abnormal drooping of the upper eyelid(s) (ptosis). Some infants and children with Cornelia de Lange syndrome may also experience episodes of uncontrolled electrical disturbances in the brain (seizures).
Cornelia de Lange syndrome behavior
Many individuals demonstrate autistic behavior, including self-destructive tendencies, attention deficit hyperactivity disorder (ADHD) and they may avoid or reject social interactions and physical contact.
Behavior problems are often directly related to frustration from inability to communicate.
Cornelia de Lange syndrome life expectancy
Life expectancy is relatively normal for people with Cornelia de Lange syndrome and most affected children live well into adulthood 3. For example, one article mentioned a woman with Cornelia de Lange syndrome who lived to age 61 and an affected man who lived to age 54 7. However, certain features of Cornelia de Lange Syndrome, particularly severe congenital heart defects, recurrent pneumonia, intestinal issues or throat may decrease life expectancy in some affected people 3.
Beck & Fenger 7 and Jackson et al 8 reported a total of 24 deaths from aspiration pneumonia (9), recurrent apnea (3), congenital heart disease (3), volvulus and intestinal obstruction (2), post-surgical complications (thrombocytopenia and intracranial bleeding) (1), and cerebral edema and herniation after spine surgery (1). Severe bronchopulmonary dysplasia, mediastinitis, uremia, bronchial asthma, coronary artery occlusion, and pulmonary embolus were other causes of death.
Schrier et al 9 reported 295 affected individuals in whom a cause of death was known. Respiratory causes, including aspiration/reflux and pneumonias, were the most common primary causes (31%), followed by gastrointestinal disease, including obstruction/volvulus (19%). Congenital anomalies accounted for 15% of deaths and included congenital diaphragmatic hernia and congenital heart defects. Acquired cardiac disease accounted for 3% of deaths. Neurologic causes and accidents each accounted for 8%, sepsis for 4%, cancer for 2%, renal disease for 1.7%, and other causes 9% of deaths.
Cornelia de Lange syndrome diagnosis
The diagnosis is suspected when the following signs and symptoms are present 3:
- Head and face appearance (>95%): Very small and flat head (microbrachycephaly), unibrow (synophrys) and highly arched eyebrows (in 98% of the cases), long and thick eyelashes, low-set abnormally placed ears with a thick helix (curve of the outer ear), short nose with upturned tip with nares that are easily seeing from the front (anteverted nares), long space between the nose and the superior lip, thin downturned lips, high and arched palate with clefts (30% of the cases), very small jaw (micrognathia) in 80% of the cases, with spurs (42% of the cases), and short neck.
- Growth failure (>95%): Growth failure that starts while the baby is growing inside the womb, resulting in a very low height and weight throughout life, and failure to thrive secondary to gastroesophageal reflux and other issues with feeding.
- Intellectual disability (>95%): Severe-to-profound developmental delay
- Limb abnormalities (>95%): Small or absent forearms and missing fingers in about 30% of the cases. Some people do not have limb deficiencies but have micromelia (small hands), abnormal placed thumbs, and an abnormal curvature of the fifth finger (clinodactyly). A fusion of the bones of the forearm (radioulnar synostosis) is common and may result in a defect of the elbows. Small feet and joined toes (syndactyly) in more than 80% of the cases.
- Excess of hair in the face, back and arms (hirsutism) in more than 80% of the cases.
People with the milder Cornelia de Lange syndrome usually have many of the characteristic facial features but with less severe cognitive and upper extremities defects, and mild intellectual disability (intelligence is normal in some cases). A milder syndrome is more common in people with variants in the SMC3, RAD21, HDAC8 or SMC1A genes.
The diagnosis of Cornelia de Lange syndrome is established with the presence of the clinical features and/or by the genetic test showing a variation in any of the genes associated with the syndrome. However, about 30% of the people affected by Cornelia de Lange syndrome do not have any known cause.
Cornelia de Lange syndrome treatment
The treatment of Cornelia de Lange syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the efforts of a team of specialists working together to systematically and comprehensively plan an affected child’s treatment. Such specialists may include pediatricians; geneticists; surgeons; specialists who diagnose and treat skeletal disorders (orthopedists); plastic surgeons; orthopedic surgeons; specialists who diagnose and treat abnormalities of the digestive system (gastroenterologists), disorders of the urinary tract (urologists), and abnormalities of the ears, nose, and throat (otolaryngologists); pediatric heart specialists (cardiologists); dental specialists; speech pathologists; specialists who assess and treat hearing problems (audiologists); eye specialists; physical and occupational therapists; and/or other health care professionals.
Treatment for Cornelia de Lange syndrome varies based on the signs and symptoms present in each person. It may include 3:
- Supplemental formulas and/or gastrostomy tube placement to meet nutritional needs and improve the growth delay
- Ongoing physical, occupational, and speech therapies
- Surgery to treat skeletal abnormalities, gastrointestinal problems, congenital heart defects and other health problems
- Medications to prevent or control seizures.
Affected infants and children may be closely monitored for certain abnormalities potentially associated with Cornelia de Lange syndrome (e.g., potential intestinal obstruction due to gastrointestinal abnormalities, cardiac defects, gastroesophageal reflux, glue ear, and/or susceptibility to respiratory infections) to ensure early detection and prompt treatment.
Specific therapies for the treatment of Cornelia de Lange syndrome are symptomatic and supportive. In some children, surgery may be performed to help correct cleft palate, cardiac defects and/or diaphragmatic hernias. Plastic surgery may be helpful in reducing excessive hair. Some gastrointestinal, genitourinary, and/or cardiac malformations may be treated with certain medications, surgical intervention, and/or other techniques. The surgical procedures performed will depend upon the location and severity of the anatomical abnormalities and their associated symptoms. Respiratory infections may be treated with antibiotic drug therapy and/or other medications that may help fight infection.
Various orthopedic techniques may be used to help treat limb deformities. Hearing aids may be beneficial in some children. Treatment with anticonvulsant medications may help prevent, reduce, or control seizures in some affected children.
Early intervention is important in ensuring that children with Cornelia de Lange syndrome reach their highest potential. Services that may be beneficial include special remedial education, vocational training, speech therapy, and/or other medical and/or social services.
Genetic counseling is recommended for affected individuals and their families. Other treatment is symptomatic and supportive.
- Cornelia de Lange syndrome. https://rarediseases.org/rare-diseases/cornelia-de-lange-syndrome/[↩]
- Dorsett D, Krantz ID. On the molecular etiology of Cornelia de Lange syndrome. Ann N Y Acad Sci. 2009;1151:22-37. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2733214/[↩]
- Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange Syndrome. 2005 Sep 16 [Updated 2016 Jan 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1104[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Kline AD, Stanley C, Belevich J, Brodsky K, Barr M, Jackson LG. Developmental data on individuals with the Brachmann-de Lange syndrome. Am J Med Genet. 1993b;47:1053–8.[↩]
- Russo S, Masciadri M, Gervasini C, Azzollini J, Cereda A, Zampino G, Haas O, Scarano G, Di Rocco M, Finelli P, Tenconi R, Selicorni A, Larizza L. Intragenic and large NIPBL rearrangements revealed by MLPA in Cornelia de Lange patients. Eur J Hum Genet. 2012;20:734–41.[↩]
- Borck G, Redon R, Sanlaville D, Rio M, Prieur M, Lyonnet S, Vekemans M, Carter NP, Munnich A, Colleaux L, Cormier-Daire V. NIPBL mutations and genetic heterogeneity in Cornelia de Lange syndrome. J Med Genet. 2004;41:e128.[↩]
- Beck B, Fenger K. Mortality, pathological findings and causes of death in the de Lange syndrome. Acta Paediatrica Scandinavica. 1985; 74:765-769. https://www.ncbi.nlm.nih.gov/pubmed/4050424[↩][↩]
- Jackson L, Kline AD, Barr MA, Koch S. de Lange syndrome: a clinical review of 310 individuals. Am J Med Genet. 1993;47:940–6.[↩]
- Schrier SA, Sherer I, Deardorff MA, Clark D, Audette L, Gillis L, Kline AD, Ernst L, Loomes K, Krantz ID, Jackson LG. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am J Med Genet A. 2011;155A:3007–24.[↩]


{kind=link}