Contents
- What is Klippel Feil syndrome
- Is Klippel Feil syndrome inherited?
- Is it possible for only one identical twin to have Klippel Feil syndrome?
- Can Klippel Feil syndrome become life threatening?
- As a young adult I was diagnosed with a fusion of C4 and C5. My question is whether this condition is automatically considered Klippel Feil Syndrome?
- Is there any relationship between Klippel Feil syndrome and low birth weight?
- Could Chiari malfomation type 1 be connected to Klippel Feil syndrome?
- Klippel Feil syndrome causes
- Klippel Feil syndrome symptoms
- Klippel Feil syndrome diagnosis
- Klippel Feil syndrome life expectancy
- Klippel Feil syndrome treatment
What is Klippel Feil syndrome
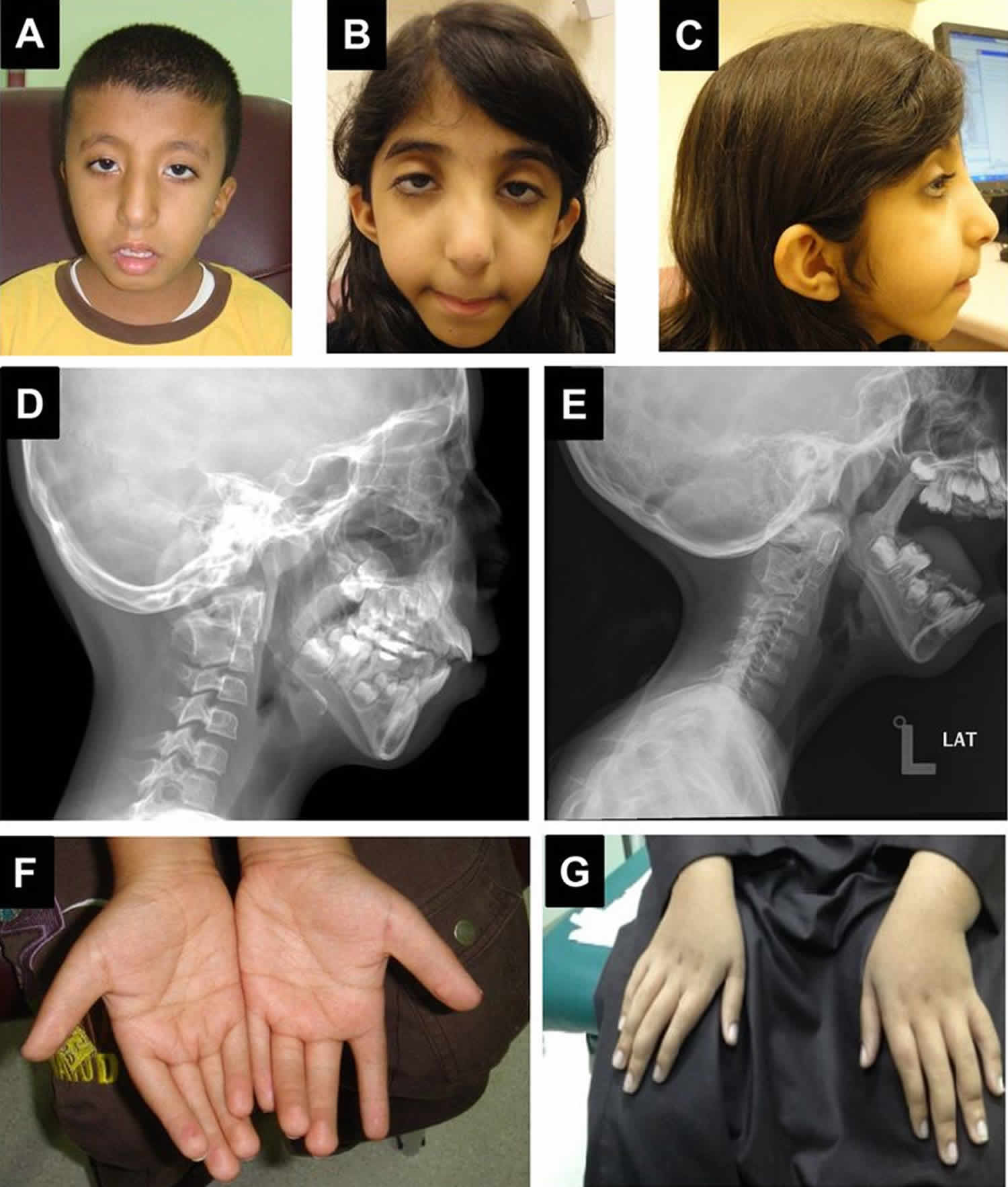
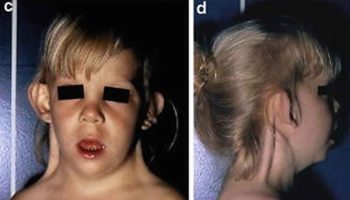
Klippel Feil syndrome is a rare congenital (present at birth) bone disorder characterized by the fusion of two or more spinal bones in the neck (cervical vertebrae). The vertebral fusion is present from birth. Klippel Feil syndrome is caused by a failure in the normal segmentation or division of the cervical vertebrae during the early weeks of fetal development. Three major features result from this vertebral fusion: a short neck, the resulting appearance of a low hairline at the back of the head, and a limited range of motion in the neck. Most affected people have one or two of these characteristic features. Less than half of all individuals with Klippel-Feil syndrome have all three classic features of this condition.
The most common symptoms of Klippel Feil syndrome are short neck, low hairline at the back of the head, and restricted mobility of the upper spine. The fused vertebrae can cause nerve damage and pain in the head, neck, or back. In people with Klippel-Feil syndrome, the fused vertebrae can limit the range of movement of the neck and back as well as lead to chronic headaches and muscle pain in the neck and back that range in severity. People with minimal bone involvement often have fewer problems compared to individuals with several vertebrae affected. The shortened neck can cause a slight difference in the size and shape of the right and left sides of the face (facial asymmetry). Trauma to the spine, such as a fall or car accident, can aggravate problems in the fused area. Fusion of the vertebrae can lead to nerve damage in the head, neck, or back. Over time, individuals with Klippel-Feil syndrome can develop a narrowing of the spinal canal (spinal stenosis) in the neck, which can compress and damage the spinal cord. Rarely, spinal nerve abnormalities may cause abnormal sensations or involuntary movements in people with Klippel-Feil syndrome. Affected individuals may develop a painful joint disorder called osteoarthritis around the areas of fused bone or experience painful involuntary tensing of the neck muscles (cervical dystonia). In addition to the fused cervical bones, people with this condition may have abnormalities in other vertebrae. Many people with Klippel-Feil syndrome have abnormal side-to-side curvature of the spine (scoliosis) due to malformation of the vertebrae; fusion of additional vertebrae below the neck may also occur.
People with Klippel-Feil syndrome may have a wide variety of other features in addition to their spine abnormalities. Some people with this condition have hearing difficulties, eye abnormalities, an opening in the roof of the mouth (cleft palate), genitourinary problems such as abnormal kidneys or reproductive organs, heart abnormalities, or lung defects that can cause breathing problems. Affected individuals may have other skeletal defects including arms or legs of unequal length (limb length discrepancy), which can result in misalignment of the hips or knees. Additionally, the shoulder blades may be underdeveloped so that they sit abnormally high on the back, a condition called Sprengel deformity. Rarely, structural brain abnormalities or a type of birth defect that occurs during the development of the brain and spinal cord (neural tube defect) can occur in people with Klippel-Feil syndrome.
In some cases, Klippel-Feil syndrome occurs as a feature of another disorder or syndrome, such as Wildervanck syndrome or hemifacial microsomia. In these instances, affected individuals have the signs and symptoms of both Klippel-Feil syndrome and the additional disorder.
Klippel-Feil syndrome is estimated to occur in 1 in 40,000 to 42,000 newborns worldwide. Females seem to be affected slightly more often than males.
Most cases of Klippel Feil syndrome are sporadic (happen on their own), but mutations in the GDF6 (growth differentiation factor 6) or GDF3 (growth differentiation factor 3) genes are inherited in an autosomal dominant manner; or, it may be caused by mutations in the MEOX1 (mesenchyme homeobox 1) gene and inherited in an autosomal recessive manner. These genes make proteins that are involved in bone development and segmentation of the vertebrae.
There is no cure for Klippel Feil syndrome. Treatment for Klippel-Feil Syndrome is generally symptomatic and supportive and may include surgery to relieve cervical or craniocervical instability and constriction of the spinal cord, and to correct scoliosis. Physical therapy may also be useful 1.
Is Klippel Feil syndrome inherited?
In some cases, Klippel Feil syndrome appears to occur randomly for unknown reasons (sporadically). In other cases, the condition appears to be genetic and may occur in more than one person in a family 2. Both autosomal dominant and autosomal recessive inheritance patterns have been reported, with different responsible genes 2.
When Klippel Feil syndrome is caused by mutations in the GDF6 or GDF3 genes, it is inherited in an autosomal dominant manner. This means that having a mutation in only one copy of the responsible gene is enough to cause features of the condition. When a person with an autosomal dominant condition has children, each child has a 50% (1 in 2) chance to inherit the mutated copy of the gene.
When Klippel Feil syndrome is caused by mutations in the MEOX1 gene, it is inherited in an autosomal recessive manner. This means that a person must have mutations in both copies of the responsible gene to be affected. The parents of a person with an autosomal recessive condition usually each carry one mutated copy of the gene and are referred to as carriers. Carriers are usually unaffected. When two carriers of the same autosomal recessive condition have children, each child has a 25% (1 in 4) risk to be affected, a 50% (1 in 2) chance to be an unaffected carrier like each parent, and a 25% risk to be unaffected and not be a carrier.
When Klippel Feil syndrome occurs as a feature of another condition, the inheritance pattern follows that of the other condition.
Is it possible for only one identical twin to have Klippel Feil syndrome?
It is theoretically possible for only one identical (monozygotic) twin to have Klippel Feil syndrome. While both autosomal and recessive inheritance patterns have been described, most cases of Klippel Feil syndrome are sporadic. There are various potential causes of Klippel Feil syndrome, many of which remain unknown.
An article published in 2006 described a pair of monozygotic twin girls, only one of which had Klippel Feil syndrome with no associated abnormalities. The authors suggested that Klippel Feil syndrome may result in part from a somatic mutation (a change in DNA occurring after conception), or from unidentified, environmental factors within the uterus during embryonic or fetal development 3.
Can Klippel Feil syndrome become life threatening?
The long-term outlook (prognosis) for people with Klippel Feil syndrome varies depending on the specific features and severity in each affected person. While all affected people have fusion of at least two vertebrae of the neck, additional signs and symptoms (if present) can vary greatly 4. In general, people with minimal involvement can lead normal, active lives and may have no significant restrictions or symptoms 4. People with additional abnormalities and/or severe forms of the condition may require careful and routine follow-up, but can have a good prognosis if symptoms and complications are treated early 1.
As a young adult I was diagnosed with a fusion of C4 and C5. My question is whether this condition is automatically considered Klippel Feil Syndrome?
Klippel Feil syndrome is typically diagnosed when X-rays or other imaging techniques show fusion of cervical vertebrae. X-rays of the entire spine should be performed to detect other spinal abnormalities, and additional imaging studies may be needed to assess the extent of the abnormality 5.
Klippel Feil syndrome can be associated with a wide range of other abnormalities involving many parts of the body. Therefore, other initial exams are needed to detect additional physical abnormalities or underlying conditions. These include 5:
- examination of the chest to rule out involvement of the heart and lungs
- examination of the chest wall to detect possible rib anomalies
- MRI for spinal stenosis or neurological deficits
- ultrasound of the kidneys for renal abnormalities
- hearing evaluation due to high incidence of hearing loss
- various lab tests to assess organ function
Additional tests or consultations with specialists may be recommended depending on the features present in each person with Klippel Feil syndrome.
Is there any relationship between Klippel Feil syndrome and low birth weight?
Klippel Feil syndrome may occur alone or in association with other birth defects or syndromes. Rarely Klippel Feil syndrome occurs in association with VACTERL association or Fetal Alcohol syndrome. VACTERL is an acronym with each letter representing the first letter of one of the more common findings seen in affected individuals: (V) = vertebral abnormalities; (A) = anal atresia; (C) = cardiac (heart) defects; (T) = tracheal anomalies including tracheoesophageal (TE) fistula; (E) = esophageal atresia; (R) = renal (kidney) and radial (thumb side of hand) abnormalities; and (L) = other limb abnormalities. Low birth weight and failure to thrive are commonly associated with these syndromes. Klippel Feil syndrome may also occur in association with other spine, kidney (and other genitourinary), hearing, heart, nerve, muscle, or skeletal defects. It has also been seen in people with Goldenhar syndrome and Mohr syndrome 6.
When Klippel Feil syndrome occurs alone, symptoms are more common in adults than in children and teens. Common symptoms in these cases include neck and arm pain, weakness, tingling, numbness, and/or loss of sensation 6.
Could Chiari malfomation type 1 be connected to Klippel Feil syndrome?
About 3-5% of people with Chiari malfomation type 1 (CM 1) also have a diagnosis of Klippel Feil syndrome. In fact medical researchers searching for the genes involved in CM1 found that their studies pointed to mutations or changes in some of the genes known to be linked to Klippel Feil syndrome 7.
Klippel Feil syndrome causes
The exact underlying causes and mechanisms of Klippel Feil syndrome are not well understood. In general, medical researchers believe Klippel Feil syndrome happens when the tissue of the embryo that normally develops into separate vertebrae does not divide correctly 8.
Isolated Klippel Feil syndrome (meaning not associated with another syndrome) can be sporadic or inherited. Although Klippel Feil syndrome may in some cases be caused by a combination of genetic and environmental factors, mutations in at least three genes have been linked to Klippel Feil syndrome: GDF6 (growth differentiation factor 6), GDF3 (growth differentiation factor 3), and MEOX1 (mesenchyme homeobox 1). These genes are involved in proper bone development. The protein produced from the GDF6 gene is necessary for the formation of bones and joints, including those in the spine. While the protein produced from the GDF3 gene is known to be involved in bone development, its exact role is unclear. The protein produced from the MEOX1 gene, called homeobox protein MOX-1, regulates the process that begins separating vertebrae from one another during early development.
GDF6 and GDF3 gene mutations that cause Klippel-Feil syndrome likely lead to reduced function of the respective proteins. MEOX1 gene mutations lead to a complete lack of homeobox protein MOX-1. Although the GDF6, GDF3, and homeobox protein MOX-1 proteins are involved in bone development, particularly formation of vertebrae, it is unclear how a shortage of one of these proteins leads to incomplete separation of the cervical vertebrae in people with Klippel-Feil syndrome.
When Klippel-Feil syndrome is a feature of another disorder, such as fetal alcohol syndrome, Goldenhar syndrome, Wildervanck syndrome or hemifacial microsomia, among others, it is caused by mutations in genes involved in the other disorder 9.
Klippel Feil syndrome inheritance pattern
In most cases, Klippel Feil syndrome appears to occur randomly for unknown reasons (sporadically). In other cases, the condition appears to be genetic and may occur in more than one person in a family. Both autosomal dominant and autosomal recessive inheritance patterns have been reported, with different responsible genes.
When Klippel Feil syndrome is caused by mutations in the GDF6 or GDF3 genes, it is inherited in an autosomal dominant manner. This means that having a mutation in only one copy of the responsible gene is enough to cause features of the condition. When a person with an autosomal dominant condition has children, each child has a 50% (1 in 2) chance to inherit the mutated copy of the gene.
When Klippel Feil syndrome is caused by mutations in the MEOX1 gene, it is inherited in an autosomal recessive manner. This means that a person must have mutations in both copies of the responsible gene to be affected. The parents of a person with an autosomal recessive condition usually each carry one mutated copy of the gene and are referred to as carriers. Carriers are usually unaffected. When two carriers of the same autosomal recessive condition have children, each child has a 25% (1 in 4) risk to be affected, a 50% (1 in 2) chance to be an unaffected carrier like each parent, and a 25% risk to be unaffected and not be a carrier.
When Klippel Feil syndrome occurs as a feature of another condition, the inheritance pattern follows that of the other condition.
Klippel Feil syndrome symptoms
Klippel Feil syndrome is characterized by the fusion of 2 or more spinal bones in the neck (cervical vertebrae). The condition is present from birth (congenital). The 3 most common features include a low posterior hairline (at the back of the head), a short neck, and limited neck range of motion. However, not all people with Klippel Feil syndrome have these features 10. Klippel Feil syndrome can also cause chronic headaches as well as pain in both the neck and the back 11.
Klippel Feil syndrome has been reported in people with a very wide variety of other conditions and abnormalities, including:
- scoliosis (curvature of the spine)
- cervical dystonia (painful, involuntary tensing of the neck muscles)
- genitourinary abnormalities (those of the reproductive organs and/or urinary system, including the kidneys)
- Sprengel deformity
- cardiac (heart) defects such as ventricular septal defect
- pulmonary abnormalities (relating to the lungs) and respiratory problems
- hearing deficits
- facial asymmetry, or other abnormalities of the head and face (such as cleft palate or hemifacial microsomia)
- torticollis
- central nervous system abnormalities (including Chiari malformation, spina bifida, or syringomyelia), and/or neurological symptoms
- other skeletal abnormalities (including those of the ribs, limbs and/or fingers)
- situs inversus
- short stature
- synkinesia (where movement in one hand involuntarily mimics the deliberate movement of the other hand)
- Wildervank syndrome
- Duane syndrome or other eye (ocular) abnormalities
In addition to the fusion of certain vertebrae, Klippel Feil syndrome can be associated with a wide variety of additional anomalies affecting many different organ systems of the body. The progression and severity of Klippel Feil syndrome can vary greatly depending upon the specific associated complications and the Class of Klippel Feil syndrome. Some cases may be mild; others may cause serious, life-long complications.
It is important to note that affected individuals will not have all of the symptoms discussed below. Affected individuals should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
Approximately 30 percent of affected individuals have additional skeletal abnormalities, such as fusion of certain ribs or other rib defects; abnormal sideways curvature of the spine (scoliosis); or a condition known as Sprengel’s deformity. This condition is characterized by elevation and/or underdevelopment of the shoulder blade (scapula), limited movement of the arm on the affected side, and the development of a lump at the base of the neck due to elevation of the shoulder blade. Also, in some individuals with Klippel Feil syndrome, a portion of the spinal cord may be exposed due to incomplete closure of certain vertebrae (spina bifida occulta). Associated findings may include the presence of a tuft of hair or dimple over the underlying abnormality and, in some cases, leg weakness, an inability to control urination (urinary incontinence), or other findings. As mentioned above, Klippel Feil syndrome type II may be associated with incomplete development of one half of certain vertebrae (hemivertebrae) and fusion of the first vertebra of the neck (atlas) with the bone at the back of the skull (occipital bone).
Approximately 25 to 50 percent of individuals with Klippel Feil syndrome also have hearing impairment. Such hearing loss may result from impaired transmission of sound from the outer or middle ear to the inner ear (conductive hearing loss); failed transmission of sound impulses from the inner ear to the brain (sensorineural hearing loss); or both (mixed hearing loss). Various eye (ocular) abnormalities may also be associated with Klippel Feil syndrome, such as deviation of one eye toward the other (cross-eye or convergent strabismus); involuntary, rapid eye movements (nystagmus); or absence or defects of ocular tissue (colobomas). In addition, some affected individuals may have other abnormalities of the head and facial (craniofacial) area including facial asymmetry, in which one side of the face appears dissimilar from other side, with one eye higher than the other. There may also be abnormal twisting of the neck (torticollis), causing the head to be rotated into an abnormal position. According to some reports, approximately 17 percent of individuals with Klippel Feil syndrome also have incomplete closure of the roof of the mouth (cleft palate).
Klippel Feil syndrome may sometimes be associated with additional physical abnormalities. These may include structural malformations of the heart (congenital heart defects), particularly ventricular septal defects (VSDs). Ventricular septal defects are characterized by the presence of an abnormal opening in the fibrous partition (septum) that separates the two lower chambers of the heart. Some individuals may also have kidney (renal) defects, such as underdevelopment (hypoplasia) or absence (agenesis) of one or both kidneys; abnormal renal rotation or placement (ectopia); or swelling of the kidneys with urine (hydronephrosis) due to blockage or narrowing of the tubes (ureters) that carry urine to the bladder.
Some individuals with the disorder may also develop neurological complications due to associated spinal cord injury. Such injury may result from instability of cervical vertebrae. For example, unfused vertebral segments adjacent to fused cervical vertebrae may be abnormally mobile (hypermobile), making them vulnerable to increased stress, which in turn may lead to vertebral instability or degenerative changes. Associated neurological complications tend to develop between the second and third decades of life and may occur spontaneously or following minor trauma. Such complications may include pain; abnormal sensations (paresthesia), such as tingling, prickling, or burning; or involuntary muscle movements accompanying certain voluntary actions (synkinesia or mirror movements). In addition, some individuals may develop increased reflex reactions (hyperreflexia); weakness or paralysis of one side of the body (hemiplegia) or of the legs and the lower part of the body (paraplegia); or impairment of certain nerves that emerge from the brain (cranial nerve palsies).
Klippel Feil syndrome diagnosis
Klippel Feil syndrome is typically diagnosed when X-rays or other imaging techniques show fusion of cervical vertebrae. X-rays of the entire spine should be performed to detect other spinal abnormalities, and additional imaging studies may be needed to assess the extent of the abnormality 8.
Klippel Feil syndrome can be associated with a wide range of other abnormalities involving many parts of the body. Therefore, other initial exams are needed to detect additional physical abnormalities or underlying conditions. These include 8:
- examination of the chest to rule out involvement of the heart and lungs
- examination of the chest wall to detect possible rib anomalies
- MRI for spinal stenosis or neurological deficits
- ultrasound of the kidneys for renal abnormalities
- hearing evaluation due to high incidence of hearing loss
- various lab tests to assess organ function
Additional tests or consultations with specialists may be recommended depending on the features present in each person with Klippel Feil syndrome.
Klippel Feil syndrome life expectancy
The long-term outlook (prognosis) for people with Klippel Feil syndrome varies depending on the specific features and severity in each affected person. While all affected people have fusion of at least two vertebrae of the neck, additional signs and symptoms (if present) can vary greatly 4. In general, people with minimal involvement can lead normal, active lives and may have no significant restrictions or symptoms 4. People with additional abnormalities and/or severe forms of the condition may require careful and routine follow-up, but can have a good prognosis if symptoms and complications are treated early. Activities that can injure the neck should be avoided.
Klippel Feil syndrome treatment
Treatment is generally symptomatic and supportive. Management depends on the features and severity in each person, and can be life-long. Careful evaluation, consistent follow-up, and coordination with various specialists are needed to improve outcome and make sure that no related diagnosis is missed 9.
There are various conservative therapies available, including the use of cervical collars, braces, traction, physical therapy, non-steroidal anti-inflammatory drugs (NSAIDs), and various pain medications 2. However, for many people with Klippel Feil syndrome, symptoms are progressive due to degenerative changes that occur in the spine 9.
Surgery may be indicated for a variety of reasons, including persistent pain; neurologic deficits; cervical or craniocervical instability; constriction of the spinal cord; or to correct severe scoliosis. Some people with Klippel Feil syndrome may need surgery to repair other skeletal abnormalities, or those related to the heart, kidneys, ears, eyes, or other parts of the body 2.
Those at an increased risk for neurological complications should be regularly monitored by their health care providers and may be advised to avoid activities that could lead to trauma or injury to cervical vertebrae 2.
Because some affected individuals may have an increased risk of neurological complications, they should be regularly monitored by physicians. In addition, they should avoid activities that could lead to trauma or injury to cervical vertebrae.
In some individuals with Klippel Feil syndrome, treatment may include surgical repair of certain skeletal, auditory, ocular, cardiac, renal, or other abnormalities potentially associated with the disorder. For example, in those with cervical spinal cord compression, surgery may be conducted to correct such compression or associated vertebral instability. The surgical procedures performed will depend upon the severity of the anatomical abnormalities, their associated symptoms, and other factors.
In addition, some individuals with hearing impairment may benefit from the use of specialized hearing aids. Genetic counseling may also be of benefit for individuals with Klippel Feil syndrome and their families.
- Klippel-Feil Syndrome Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Klippel-Feil-Syndrome-Information-Page[↩][↩]
- Klippel-Feil Syndrome. https://rarediseases.org/rare-diseases/klippel-feil-syndrome[↩][↩][↩][↩][↩]
- Toyoshima M, Maegaki Y, Yuasa I, Ohno K. Monozygotic twins discordant for Klippel-Feil syndrome. Pediatr Neurol. January, 2006; 34(1):76-78.[↩]
- Warner WC. Pediatric Cervical Spine. In: Canale & Beaty. Campbell’s Operative Orthopaedics, 11th ed. . Philadelphia, PA: Mosby; 2007[↩][↩][↩][↩]
- Klippel-Feil Syndrome. https://emedicine.medscape.com/article/1264848-overview#showall[↩][↩]
- Kliegman RM et al.,. The Neck. In: Kliegman RM et al.,. Nelson Textbook of Pediatrics. Saunders; 2011; Chapter 672:2377-2382.e1[↩][↩]
- Markunas CA, Soldano K, Dunlap K, Cope H, Asiimwe E, Stajich J, Enterline D, Grant G, Fuchs H, Gregor SG, and Ashley-Koch AE. Stratified Whole Genome Linkage Analysis of Chiari Type I Malformation Implicates Known Klippel-Feil Syndrome Genes as Putative Disease Candidates. PLoS ONE. April 19 2013; 8(4):e61521. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3631233[↩]
- Klippel-Feil Syndrome. https://rarediseases.org/rare-diseases/klippel-feil-syndrome/[↩][↩][↩]
- Klippel-Feil Syndrome. https://emedicine.medscape.com/article/1264848-overview[↩][↩][↩]
- Isolated Klippel-Feil syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=2345[↩]
- Klippel-Feil syndrome. https://ghr.nlm.nih.gov/condition/klippel-feil-syndrome[↩]




{kind=link}