
Contents
What is aneuploidy
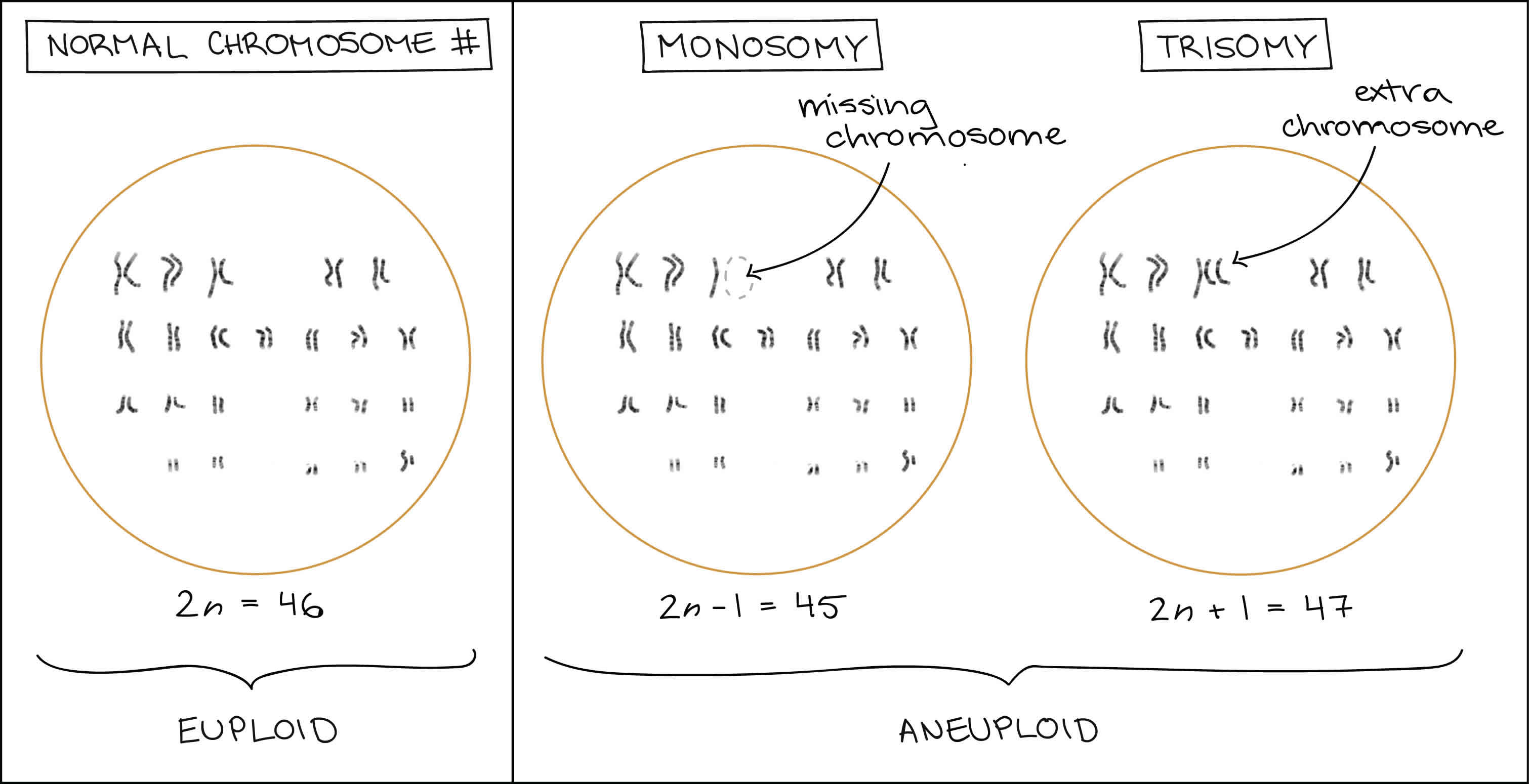
Aneuploidy is a genetic condition due to missing chromosome or having extra chromosomes. Aneuploidy is defined as change in chromosome number that is not the exact multiple of the haploid karyotype 1. Aneuploidy interferes with growth and development of an organism and frequently causes lethality and has been associated with disease, sterility, and solid tumors formation 1.
Each species has a characteristic chromosome number, such as 46 chromosomes for a typical human body cell. In organisms with two full chromosomes sets, such as humans, this number is given the name 2n. When an organism or cell contains 2n chromosomes (or some other multiple of n), it is said to be euploid (eu- = good), meaning that it contains chromosomes correctly organized into complete sets.
If a cell is missing one or more chromosomes, it is said to be aneuploid (an- = not, “not good”). For instance, human somatic cells with chromosome numbers of (2n−1) = 45 or (2n+1) = 47 are aneuploid.
Aneuploidy also includes cases where a cell has larger numbers of extra or missing chromosomes, as in (2n−2), (2n+3), etc. However, if there is an entire extra or missing chromosome set (e.g., 3n), this is not formally considered to be aneuploidy, even though it may still be bad for the cell or organism. Organisms with more than two complete sets of chromosomes are said to be polyploid. Polyploidy is defined as having a chromosome number that is a multiple greater than two of the monoploid number 1. Polyploidy is frequently found in nature; can be part of the normal physiology of plants and animals, including a few types of human cells; and generally does not lead to gross defects in the development of an organism or its physiology 2. In addition, duplications of an entire genome have taken place in the evolution of several groups such as plants and yeasts and may be a natural event necessary for this process (Wolfe and Shields 1997; Kellis et al. 2004; Adams and Wendel 2005). In contrast, aneuploidy frequently causes lethality and has been associated with disease, sterility, and tumor formation.
Similarly, a normal human egg or sperm has just one set of chromosomes (n=23). An egg or sperm with (n−1)=22 or (n+1)=24 chromosomes is considered to be aneuploid.
Two common types of aneuploidy have their own special names:
- Monosomy is when an organism has only one copy of a chromosome that should be present in two copies (2n−1).
- Trisomy is when an organism has a third copy of a chromosome that should be present in two copies (2n+1).
The risk of having a child with an aneuploidy increases as a woman ages. It is estimated that about 20–80% of human embryos are chromosomally aneuploid 3. This phenomenon is the result of meiotic and mitotic developmental errors and appears to occur at a much higher frequency in humans than in experimental animals 4.
Figure 1. Aneuploidy
Aneuploidy disorders
Trisomy is the most common aneuploidy. In trisomy, there is an extra chromosome (three chromosomes), rather than the usual pair of chromosomes. A common trisomy is Trisomy 21 (Down syndrome). Other trisomies include trisomy 13 (Patau syndrome) and trisomy 18 (Edwards syndrome) 5. Because many babies born with trisomy 18 (Edwards syndrome) and trisomy 13 (Patau syndrome) may not live beyond the first few days or weeks of life, it is possible that parents may have to face the fact that the baby may never go home from the hospital. Many times, parents are frightened and overwhelmed by all of the information associated with trisomy 18 and 13. Decisions surrounding the care of an infant with trisomy 18 and 13 are difficult and personal. There are many resources available to parents to help them during this time, including early intervention services, hospice care, social workers, the hospital chaplain or clergyman, and genetic counselors. Families who have or have had a baby with trisomy 18 or trisomy 13 are particularly helpful and supportive since they have experienced many of the same questions and emotions.
Monosomy is another type of aneuploidy in which one member of a chromosome pair is missing. So there are too few chromosomes rather than too many. A baby with a missing autosome has little chance of survival. However, a baby with a missing sex chromosome can survive in certain cases. For example, girls with Turner syndrome — who are born with missing (just one X chromosome) or damaged X chromosome — can live normal, productive lives as long as they receive medical care for any health problems associated with their condition.
Fetal aneuploidy
Down syndrome (Trisomy 21)
Trisomy 21 (Down syndrome) is the most common chromosomal anomaly in humans, affecting about 5,000 babies born each year and more than 350,000 people in the United States.
Also known as Down syndrome, trisomy 21 is a genetic condition caused by an extra chromosome. Most babies inherit 23 chromosomes from each parent, for a total of 46 chromosomes. Babies with Down syndrome however, end up with three chromosomes at position 21, instead of the usual pair.
Other examples of trisomies occur at position 13 and 18. Trisomy 21 is the most common of the three, occurring in 1 out of every 691 births. The disorder was first identified in 1866 by John Langdon Down, a British physician, and later named after him.
As your child with Down syndrome grows, he is at greater risk for certain medical problems and may develop:
- Congenital heart disease
- Gastrointestinal abnormalities
- Musculoskeletal and movement problems
- Spine disorders such as scoliosis, kyphosis or lordosis
- Endocrinologic disorders
- Epilepsy
- Hearing loss
- Speech apraxia (difficulty making speech sounds)
- Sleep disorders
- Feeding disorders
- Developmental disabilities (learning disabilities, intellectual disabilities and autism)
Problems in any of these areas can affect your child’s development and behavior.
What causes Trisomy 21
Down syndrome occurs because of the extra copy of chromosome 21, which can cause the body and brain to develop differently than a child without the syndrome.
The risk of having a baby with Down syndrome increases as a woman ages — women older than 35 are often encouraged to have prenatal genetic testing done of their unborn babies — but, because younger women have more babies, they give birth to 80 percent of babies with Down syndrome.
Trisomy 21 signs and symptoms
Symptoms of Down syndrome may include:
- Distinctive facial features
- Mild to moderate intellectual disabilities
- Heart, kidney and thyroid issues
- Numerous respiratory infections, from colds to bronchitis and pneumonia
- Skeletal abnormalities, including spine, hip, foot and hand disorders
- Flexible joints and weak, floppy muscles
- Overly quiet baby
- Less responsive to stimuli
- Vision and hearing impairment
- Inwardly curved little finger
- Abnormal space between the great and second toe
- Unusual creases on the soles of the feet and one or both hands
Trisomy 21 testing and diagnosis
Tests to confirm Down syndrome are often done before a baby is born through amniocentesis or chorionic villus sampling (CVS). For amniocentesis, a needle is inserted through the mother’s abdominal wall into the amniotic sac and a small sample of amniotic fluid is drawn out and tested in a laboratory.
If your child was not prenatally diagnosed with Down syndrome, diagnosis usually begins at birth based on your child’s physical appearance. Doctors will ask you for a thorough family medical history, do a physical exam of your child, and analyze your child’s chromosomes. Once a diagnosis is made, additional testing may be ordered to help clinicians better understand how Down syndrome may affect your child and help spot any early complications from the disorder.
Tests may include:
- Genetic testing, in which a sample of your child’s saliva is used to identify your child’s DNA.
- Blood tests, which can help determine drug usage and effectiveness, biochemical diseases and organ function.
- X-rays, which produce images of bones.
- Magnetic resonance imaging (MRI), which uses a combination of large magnets, radiofrequencies and a computer to produce detailed images of organs and structures within the body.
- Computed tomography (CT) scan, which uses a combination of X-rays and computer technology to produce cross-sectional images (“slices”) of the body.
- EOS imaging, an imaging technology that creates 3-dimensional models from two planar images. Unlike a CT scan, EOS images are taken while the child is in an upright or standing position, enabling improved diagnosis — for some conditions — due to weight-bearing positioning.
Trisomy 21 treatment
There is no cure for Down syndrome. Treatment is ordered when certain issues — such as heart problems, muscle weaknesses or spinal curvatures — occur and need to be treated.
Many children with Down syndrome are also diagnosed with a variety of secondary conditions that are present at birth and can be treated when your child is young. This is often true for conditions such as heart issues, hand and foot anomalies.
In other cases, the complications from Down syndrome may only become evident — or problematic — as your child grows. This is often true for spinal deformities such as scoliosis and hip conditions that might require surgical correction.
Every child’s condition is different, so treatment is determined on a case-by-case basis. Depending on your child’s needs, specialists from cardiology, orthopaedics, endocrinology and others will treat your child. Treatment may include therapy, surgery or additional support services for your child and family.
Patau syndrome (trisomy 13)
Trisomy 13 is called “Patau syndrome,” in honor of the physician who first described it. Trisomy 13 affects 1 out of every 15,000 to 25,000 births. Children with Patau syndrome often have cleft lip and palate, extra fingers or toes, foot abnormalities, and many different structural abnormalities of the skull and face. Trisomy 13 also can cause birth defects of the ribs, heart, abdominal organs, and sex organs. Long-term survival is unlikely but possible. More than 90% of children with trisomy 13 die in the first year.
Most infants with trisomy 13 have congenital heart disease.
Complications begin almost immediately and may include:
- Breathing difficulty or lack of breathing (apnea)
- Deafness
- Feeding problems
- Heart failure
- Seizures
- Vision problems
What causes Trisomy 13
Trisomy 13 occurs when extra DNA from chromosome 13 appears in some or all of the body’s cells.
- Trisomy 13: the presence of an extra (third) chromosome 13 in all of the cells.
- Mosaic trisomy: the presence of an extra chromosome 13 in some of the cells.
- Partial trisomy: the presence of a part of an extra chromosome 13 in the cells.
The extra material interferes with normal development.
Most cases are not passed down through families (inherited). Instead, the events that lead to trisomy 13 occur in either the sperm or the egg that forms the fetus.
Usually, each egg and sperm cell contains 23 chromosomes (half the normal number in body cells). The union of these cells creates 23 matched pairs, or 46 total chromosomes at the time of fertilization. In this manner, a person receives exactly half of their genetic material from each biological parent. Sometimes, an error occurs when an egg or sperm cell is forming, causing it to have an extra chromosome 13 inside. If this egg or sperm cell contributes that extra chromosome to the embryo, then trisomy results. If this egg or sperm cell contributes that extra chromosome 13 to the embryo, then trisomy 13 results. The extra chromosome 13 can come from either the mother or father. The features of trisomy 13 result from having this extra copy of chromosome 13 in each of the body’s cells.
Occasionally, the extra chromosome 13 is attached to another chromosome in the egg or sperm; this is called a translocation. This is the only form of trisomy 13 that can be inherited from a parent. Sometimes, a parent can carry a “balanced” rearrangement, where chromosome 13 is attached to another chromosome. However, since the parent does not have any extra or missing chromosome material, they are said to have a “balanced translocation” and they are usually normal and healthy. Rarely, mosaic trisomy 13 may occur when the error in cell division occurs after fertilization. These affected persons have some cells with an extra chromosome 13 and others with the normal number.
What is the risk for parents of a child with trisomy 13 having another child with trisomy 13?
In general, for women under 35 years of age, the chance of having another baby with trisomy 13 depends on several factors. The risk to having a baby with trisomy 13 does increase slightly with each added year of maternal age.
After birth, the physician usually takes a blood sample from a baby suspected of having trisomy 13, to perform a chromosomal analysis (called a karyotype). This confirms the physical findings of trisomy 13 and determines the underlying chromosomal abnormality. This information is important in determining the risk in future pregnancies. Translocation and mosaic trisomy 13 have different recurrence risks for future pregnancies. The physician may refer parents to a genetic physician or genetic counselor who can explain the results of chromosomal tests in detail. This includes what the recurrence risks may be in another pregnancy and what tests are available to diagnose chromosome problems before a baby is born.
Trisomy 13 prevention
Trisomy 13 can be diagnosed before birth by amniocentesis with chromosome studies of the amniotic cells.
Parents of infants with trisomy 13 that is caused by a translocation should have genetic testing and counseling. This may help them avoid having another child with the condition.
Trisomy 13 symptoms
Patau syndrome symptoms include:
- Cleft lip or palate
- Clenched hands (with outer fingers on top of the inner fingers)
- Close-set eyes — eyes may actually fuse together into one
- Decreased muscle tone
- Extra fingers or toes (polydactyly)
- Hernias: umbilical hernia, inguinal hernia
- Hole, split, or cleft in the iris (coloboma)
- Low-set ears
- Intellectual disability, severe
- Scalp defects (missing skin)
- Seizures
- Single palmar crease
- Skeletal (limb) abnormalities
- Small eyes
- Small head (microcephaly)
- Small lower jaw (micrognathia)
- Undescended testicle (cryptorchidism)
Babies with trisomy 13 often have a low birthweight, even when born full-term. They have a small head, with a sloping forehead. Usually, there are major structural problems with the brain that are diagnosed shortly after birth. Often, the front of the brain does not divide properly, resulting in a condition called holoprosencephaly. This can cause changes in the development of the baby’s face, where the eyes are close set, or the nose or nostrils are underdeveloped. Cleft lip and cleft palate are common in babies with trisomy 13.
Eye problems are common and the ears are low-set and unusual in shape. Sometimes, babies with trisomy 13 can have scalp abnormalities (cutis aplasia) which resemble ulcers. They can also have birthmarks that are purplish-red in color; the color is due to tiny blood vessels close to the skin (hemangiomas).
Many babies with trisomy 13 have extra fingers and toes (polydactyly). The feet may have prominent heels. In many cases, there are other health problems present at birth. These include heart defects, kidney problems, and/or an omphalocele (a condition in which some of the abdominal organs protrude through an opening in the abdominal muscles in the area of the umbilical cord). In males, the testes sometimes fail to descend into the scrotum. Females may have an abnormally shaped uterus, called a bicornuate uterus.
Trisomy 13 diagnosis
The infant with trisomy 13 may have a single umbilical artery at birth. There are often signs of congenital heart disease, such as:
- Abnormal placement of the heart toward the right side of the chest instead of the left
- Atrial septal defect (ASD)
- Patent ductus arteriosus (PDA)
- Ventricular septal defect (VSD)
Gastrointestinal x-rays or ultrasound may show rotation of the internal organs.
MRI or CT scans of the head may reveal a problem with the structure of the brain. The problem is called holoprosencephaly. It is the joining together of the 2 sides of the brain.
Chromosome studies show trisomy 13, trisomy 13 mosaicism, or partial trisomy.
Trisomy 13 treatment
There is no specific treatment for trisomy 13. Treatment varies from child to child and depends on the specific symptoms.
Edwards syndrome (trisomy 18)
Trisomy 18 is also called “Edwards syndrome,” named after the physician who first described the disorder. Trisomy 18 affects 1 out of every 7,500 births. Children with Edwards syndrome have a low birth weight and a small head, mouth, and jaw. Their hands typically form clenched fists with fingers that overlap. They also might have birth defects involving the hips and feet, heart and kidney problems, and intellectual disability (also called mental retardation).
Half of infants with this condition do not survive beyond the first week of life. Only about 5% of these children are expected to live longer than 1 year. Some children have survived to the teenage years, but with serious medical and developmental problems.
Trisomy 18 causes
Usually, each egg and sperm cell contains 23 chromosomes (half the normal number in body cells). The union of these cells creates 23 matched pairs, or 46 total chromosomes at the time of fertilization. In this manner, a person receives exactly half of their genetic material from each biological parent. Sometimes, an error occurs when an egg or sperm cell is forming, causing it to have an extra chromosome 18 inside. If this egg or sperm cell contributes that extra chromosome to the embryo, then trisomy results. If this egg or sperm cell contributes that extra chromosome 18 to the embryo, then trisomy 18 results. The extra chromosome 18 can come from either the mother or father. The features of trisomy 18 result from having this extra copy of chromosome 18 in each of the body’s cells.
Occasionally, the extra chromosome 18 is attached to another chromosome in the egg or sperm; this is called a translocation. This is the only form of trisomy 18 that can be inherited from a parent. Sometimes, a parent can carry a “balanced” rearrangement, where chromosome 18 is attached to another chromosome. However, since the parent does not have any extra or missing chromosome material, they are said to have a “balanced translocation” and they are usually normal and healthy. Rarely, mosaic trisomy 18 may occur when the error in cell division occurs after fertilization. These affected persons have some cells with an extra chromosome 18 and others with the normal number.
What is the risk for parents of a child with trisomy 18 having another child with trisomy 18?
In general, for women under 35 years of age, the chance of having another baby with trisomy 18 depends on several factors. The risk to having a baby with trisomy 18 does increase slightly with each added year of maternal age.
After birth, the physician usually takes a blood sample from a baby suspected of having trisomy 18, to perform a chromosomal analysis (called a karyotype). This confirms the physical findings of trisomy 18 and determines the underlying chromosomal abnormality. This information is important in determining the risk in future pregnancies. Translocation and mosaic trisomy 18 have different recurrence risks for future pregnancies. The physician may refer parents to a genetic physician or genetic counselor who can explain the results of chromosomal tests in detail. This includes what the recurrence risks may be in another pregnancy and what tests are available to diagnose chromosome problems before a baby is born.
Trisomy 18 symptoms
Edwards syndrome symptoms may include:
- Clenched hands
- Crossed legs
- Feet with a rounded bottom (rocker-bottom feet)
- Low birth weight
- Low-set ears
- Mental delay
- Poorly developed fingernails
- Small head (microcephaly)
- Small jaw (micrognathia)
- Undescended testicle
- Unusual shaped chest (pectus carinatum)
Babies with trisomy 18 appear thin and frail. They fail to thrive and have problems feeding. Trisomy 18 causes a small head size, with the back of the head (occiput) prominent. Ears are usually low-set on the head. The mouth and jaw are unusually small, and there is a shortened sternum (breastbone).
At birth, these babies are small for their age, even when delivered full-term, and have a weak cry. Their response to sound is decreased and there is often a history of infrequent fetal activity during the pregnancy. Most babies with trisomy 18 have heart defects. They clench their fists in a characteristic manner and fully extending their fingers is difficult. Joint contractures?where the arms and legs are in a bent position, rather than relaxed?are usually present. The feet may be referred to as “rocker bottom,” due to their curved shape.
Babies with trisomy 18 may also have spina bifida, eye problems, cleft lip and palate, and hearing loss. It is also common to see feeding problems, slow growth, seizures, high blood pressure, kidney problems, and scoliosis (curvature of the spine). In males, the testes sometimes fail to descend into the scrotum.
Most babies with trisomy 18 have problems that affect all parts of the body in some way. The majority of children with trisomy 18 will have most, but not all, of the health problems mentioned here. Heart problems, feeding difficulties, and an increased susceptibility to infection are factors which, most often, contribute to the death of these children.
Trisomy 18 diagnosis
An exam during pregnancy may show an unusually large uterus and extra amniotic fluid. There may be an unusually small placenta when the baby is born. A physical exam of the infant may show unusual fingerprint patterns. X-rays may show a short breast bone.
Chromosome studies will show trisomy 18. The chromosome abnormality may be present in every cell or present in only a certain percentage of the cells (called mosaicism). Studies may also show part of the chromosome in some cells. Rarely, part of the chromosome 18 becomes attached to another chromosome. This is called translocation.
Other signs include:
- Hole, split, or cleft in the iris of the eye (coloboma)
- Separation between the left and right side of the abdominal muscle (diastasis recti)
- Umbilical hernia or inguinal hernia
There are often signs of congenital heart disease, such as:
- Atrial septal defect (ASD)
- Patent ductus arteriosus (PDA)
- Ventricular septal defect (VSD)
Tests may also show kidney problems, including:
- Horseshoe kidney
- Hydronephrosis
- Polycystic kidney
Trisomy 18 treatment
There are no specific treatments for trisomy 18. Which treatments are used depend on the person’s individual condition.
Turner syndrome
Turner syndrome is a genetic disorder resulting in short stature and lack of puberty, along with several other medical issues. The severity of these problems varies among affected individuals. Typically, a female has two X chromosomes. Turner syndrome results from missing all or part of one of the X chromosomes. Turner syndrome occurs in 1 in 2,000 to 2,500 females.
The name “Turner syndrome” comes from Dr. Henry Turner, the physician who first described the collection of findings in 1938. It was not until 1959 that the cause of Turner syndrome was identified.
Turner syndrome causes
Normally during reproduction, a sperm with 23 chromosomes fertilizes an egg with 23 chromosomes resulting in a complete set of 46 chromosomes, half from the father and half from the mother. Sometimes, an error occurs when an egg or sperm cell is forming, causing it to have a missing or abnormal sex chromosome. Turner syndrome occurs when either the egg or sperm fails to contribute a normal sex chromosome to the embryo, resulting in all or part of one of the X chromosomes being missing.
In some cases, the embryo may initially contain 46 chromosomes, however shortly after conception, the sex chromosomes do not divide properly, leaving a set of cells with only one X chromosome.
- About 50 percent of Turner syndrome results from missing an entire X chromosome.
- About one-third of girls with Turner syndrome have the normal number of chromosomes (46 total), but they are missing a portion of the second X chromosome. In some cases, this is because the ends of the chromosome join together forming a ring; in other cases, the chromosome may be made up of two copies of the long (q) arm, and be missing the short, or p, arm. This is called an isochromosome.
When only part of an X chromosome is missing (called a deletion) the features of Turner syndrome that are present may vary depending upon which part of the X chromosome is missing.
Often, girls with Turner syndrome have a mosaic pattern (two or more chromosome patterns in the cells).
What is the risk for parents of a child with Turner syndrome having another child with Turner syndrome?
Although Turner syndrome is a genetic condition, in most cases it is not something that is inherited and not something that runs in families. Therefore, for most families, the chance of having another child with Turner syndrome is low.
Every pregnant woman has a risk for having a child with a chromosome abnormality that is associated with her age; as the woman gets older, this risk increases. The chance for having another baby with Turner syndrome is generally not increased above this population risk.
Your healthcare provider may refer you to a geneticist or genetic counselor who can explain what tests are available to diagnose chromosome problems before a baby is born.
Turner syndrome signs and symptoms
Short stature
Short stature is the most common feature of girls with Turner syndrome. The average adult height of a woman with Turner syndrome is 4 feet 8 inches, although some women reach 5 feet. Growth hormone therapy can be used in girls with Turner syndrome to increase their height.
Absent puberty
Most girls with Turner syndrome have either poorly formed or absent ovaries. Ovaries produce estrogen and without estrogen, normal pubertal development does not occur. Signs of puberty, such as breast development and menstruation (periods), often do not occur without the help of hormone therapy.
Other medical issues
Other medical issues that can occur in girls with Turner syndrome include cardiac abnormalities, renal (kidney) abnormalities, hearing problems, skin disorders, dental/orthodontic issues and learning issues.
Turner syndrome diagnosis
The diagnosis of Turner syndrome can be made in utero (during pregnancy), shortly after birth, during childhood or in the late teens.
Testing during pregnancy
In many cases, chromosomal abnormalities like those causing Turner syndrome can be diagnosed before a baby is born. This is done by analyzing cells in the amniotic fluid or from the placenta. A chromosome analysis, whether performed on a blood sample, cells from the amniotic fluid or the placenta, is over 99.9 percent accurate.
Fetal ultrasound during pregnancy can also give information about the possibility of Turner syndrome. However, ultrasounds are not 100 percent accurate, and many babies with Turner syndrome may look the same on ultrasound as those without Turner syndrome.
At birth
Sometimes features of Turner syndrome are noted at birth which lead to the diagnosis of Turner syndrome. When a girl is born with features suggestive of Turner syndrome, the healthcare provider usually takes a blood sample for chromosomal analysis (called a karyotype).
This confirms the diagnosis of Turner syndrome and determines the underlying chromosomal abnormality. Your healthcare provider may explain the results of the test to you or refer you to a geneticist or genetic counselor who can explain the results of chromosomal tests.
During childhood
Sometimes a healthcare provider may recommend a chromosomal analysis as part of the workup for short stature if a girl’s height is noted to be less than what is expected for her age and family, and she is growing at a slower than expected rate.
During adolescence
Sometimes girls with Turner syndrome do not exhibit any signs of the condition as infants or children. It is only when they fail to go through puberty that a healthcare provider begins to suspect the diagnosis of Turner syndrome.
Turner syndrome treatment
There is no cure for Turner syndrome; however, many of the more serious problems can be treated. For example, growth hormone can be given to improve final height, and hormone replacement therapy can be given so that girls will develop signs of puberty.
Aneuploidy screening
Aneuploidy screening is commonly known as preimplantation genetic testing for aneuploidy (PGT-A), preimplantation genetic screening (PGS) or preimplantation genetic testing (PGT), is a technique used to identify genetic defects in embryos created through in vitro fertilization (IVF) before pregnancy 6. Preimplantation genetic testing for aneuploidy (PGT-A) assesses the chromosome content of the embryos. Current technologies involve day 5 trophectoderm biopsy with testing for the entire chromosome set. As with any other intervention, detailed counseling is important. Couples need to know what to expect from preimplantation genetic testing for aneuploidy (PGT-A) for example the potential to increase time to pregnancy, lower miscarriage rates and who can benefit the most (e.g, women with recurrent pregnancy loss, recurrent implantation failure, severe male factor infertility. They need to know the added costs and the consequences of testing. These include having no euploid embryos for transfer.
Since it has been reported that in humans 90% of embryos are aneuploid as a result of malsegregation mechanisms in maternal meiosis I, the detection of abnormal oocytes in IVF treatments has become of considerable importance 7. Preimplantation genetic diagnosis (PGD) refers specifically to when one or both genetic parents has a known genetic abnormality and testing is performed on an embryo to determine if it also carries a genetic abnormality. In contrast, preimplantation genetic screening (PGS) refers to techniques where embryos from presumed chromosomally normal genetic parents are screened for aneuploidy 6. Because only unaffected embryos are transferred to the uterus for implantation, preimplantation genetic testing provides an alternative to current post conception diagnostic procedures (i.e., amniocentesis or chorionic villus sampling [CVS]), which are frequently followed by the difficult decision of pregnancy termination if results are unfavorable. Preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS) are presently the only options available for avoiding a high risk of having a child affected with a genetic disease prior to implantation. It is an attractive means of preventing heritable genetic disease, thereby eliminating the dilemma of pregnancy termination following unfavorable prenatal diagnosis.
Embryo implantation is the step that most limits the success of in vitro fertilization (IVF) 8. Successful implantation requires a euploid embryo and a receptive endometrium. It is well known that some embryos created in vitro are aneuploid and do not implant, or if they do, will probably be miscarried 9. The proportion of unhealthy embryos increases as a woman ages and IVF success rates decline 10.
Most IVF laboratories select embryos for transfer on the basis of morphology. Technical developments in the past two decades, however, have enabled us to assess the embryos beyond their morphology. Time-lapse monitoring of morphokinetic changes and the different “omics” technologies have allowed us to assess the genetic, metabolic, or functional capacities of developing embryos 11.
Several genetic tests can be used to screen embryos for aneuploidy using a few cells removed at various developmental stages.
Established means of pre-implantation embryo genetic profiling entail the collection of embryo-derived material for genetic testing 12. Many studies on day-3 embryo biopsies showed reassuring long-term neonatal outcomes 13, and no significant differences in developmental morbidities and major congenital abnormalities among children born from biopsied embryos compared to children normally conceived 14. The safety profile of conventional pre-implantation genetic testing (PGT), however, has been contested by other investigators in view of reports suggesting that the invasive removal of cells from pre-implantation embryos may interfere with embryonic development. Besides from invasive disruptive handling, embryo biopsies also require dedicated equipment and highly trained personnel, which amount to a measurable commitment in time and cost. Taken together, while embryo biopsy remains the cornerstone for preimplantation genetic testing (PGT), interest in investing non-invasive alternatives seems very timely, reasonable and could improve the cost-efficiency and safety of the procedure.
Preimplantation genetic screening (PGS) has been shown to significantly improved the ongoing pregnancy and live birth rate of young women carrying monogenic diseases in the first frozen/thawed embryo transfer cycles during in vitro fertilization (IVF). Preimplantation genetic screening (PGS) has evolved from using Fluorescent In Situ Hybridization (FISH) techniques to detect meiotic aneuploidies in polar body biopsies to less invasive comprehensive chromosome screening by array comparative genomic hybridization (aCGH) or next generation sequencing in trophectoderm biopsies. The next generation of screening is moving towards non-invasive screening of early embryos—blastocysts–via analysis of cell-free embryonic DNA obtained from culture medium surrounding embryos.
The Single Embryo Transfer of Euploid Embryo (STAR) Trial was a randomized controlled trial evaluating the effectiveness of preimplantation genetic screening using next generation sequencing in trophectoderm biopsies to optimize euploid embryo selection for elective single embryo transfer. The primary outcome of this trial was ongoing pregnancy at 20 weeks post embryo transfer by comparing preimplantation genetic screening/next generation sequencing in the experimental arm versus standard embryo selection through morphological analysis in controls. 650 subjects were randomized in both arms of the trial. The only significant difference in ongoing pregnancy rates between the two arms was seen in 35-40-year old women, and found an improvement in pregnancy rates from 37% to 51% in the control and preimplantation genetic screening arms, respectively. The Single Embryo Transfer of Euploid Embryo (STAR) Trial miscarriage rates were surprisingly low. Therefore, preimplantation genetic screening in trophectoderm biopsies has the potential to significantly improve successful pregnancies in 35-40-year old women who undergo in vitro fertilization (IVF).
In vitro fertilization (IVF) is a complex series of procedures used to treat fertility or genetic problems and assist with the conception of a child. During IVF, mature eggs are collected (retrieved) from your ovaries and fertilized by sperm in a lab. Then the fertilized egg (embryo) or eggs are implanted in your uterus. One cycle of IVF takes about two weeks. IVF is the most effective form of assisted reproductive technology. The procedure can be done using your own eggs and your partner’s sperm. Or IVF may involve eggs, sperm or embryos from a known or anonymous donor. In some cases, a gestational carrier — a woman who has an embryo implanted in her uterus — might be used.
Your chances of having a healthy baby using IVF depend on many factors, such as your age and the cause of infertility. In addition, IVF can be time-consuming, expensive and invasive. If more than one embryo is implanted in your uterus, IVF can result in a pregnancy with more than one fetus (multiple pregnancy).
Your doctor can help you understand how IVF works, the potential risks and whether this method of treating infertility is right for you.
In summary, preimplantation genetic screening (PGS) was previously thought to provide a binary output of either euploid or aneuploid, but with increased sensitivity associated with next generation sequencing, preimplantation genetic screening now detects more subtle abnormalities and mosaicism, including copy number variants, that fall within a continuum that may indicate risk factors for the fetus. More studies are needed to understand the fate and impact of embryonic mosaicism on fetal and neonatal development. The future development of non-invasive preimplantation genetic screening of DNA fragments from embryonic culture medium is an exciting concept, especially as questions still remain about the safety of embryo biopsy. There are still many questions to answer about this approach: how are cell fragments getting through the zona, will this approach exacerbate mosaicism questions, and will it prove to be a more sensitive but less specific test?
- Aneuploidy: Cells Losing Their Balance. Genetics June 1, 2008 vol. 179 no. 2 737-746; https://doi.org/10.1534/genetics.108.090878[↩][↩][↩]
- Otto, S. P., and J. Whitton, 2000 Polyploid incidence and evolution. Annu. Rev. Genet. 34: 401–437.[↩]
- Vera-Rodriguez M, Chavez SL, Rubio C, Pera RAR, Simon C. Prediction model for aneuploidy in early human embryo development revealed by single-cell analysis. Nat Commun 2015;6:7601.[↩]
- Bond D, Chandley A. The origins and causes of aneuploidy in experimental organisms. In: Bond D, Chandlev A (eds). Aneuploidy. Oxford and New York: Oxford University Press, 1983:27–54.[↩]
- Genetic Disorders. https://www.acog.org/Patients/FAQs/Genetic-Disorders[↩]
- Preimplantation Genetic Diagnosis. https://emedicine.medscape.com/article/273415-overview[↩][↩]
- Navarro J, Gutiérrez-Mateo C, Pujol A, et al. Preimplantation Genetic Diagnosis (PGD): Screening for Aneuploidy in Human Oocytes and Polar Bodies. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK6460[↩]
- Preimplantation Genetic Testing for Aneuploidy: A Boon for Infertility? https://www.medscape.com/viewarticle/897633[↩]
- Peter Kovacs. Preimplantation Genetic Testing for Aneuploidy: A Boon for Infertility? – Medscape – May 30, 2018 https://www.medscape.com/viewarticle/897633[↩]
- Ata B, Kaplan B, Danzer H, et al. Array CGH analysis shows that aneuploidy is not related to the number of embryos generated. Reprod Biomed Online. 2012;24:614-20.[↩]
- Montag M, Toth B, Strowitzki T. New approaches to embryo selection. Reprod Biomed Online. 2013;27:539-546[↩]
- Non-invasive Pre-implantation Genetic Testing of Human Embryos. Hum Reprod. 2018;33(12):2162-2167. https://www.medscape.com/viewarticle/905641[↩]
- Liebaers I, Desmyttere S, Verpoest W, De Rycke M, Staessen C, Sermon K, Devroey P, Haentjens P, Bonduelle M. Report on a consecutive series of 581 children born after blastomere biopsy for preimplantation genetic diagnosis. Hum Reprod 2009;25:275–282.[↩]
- Harper JC, Wilton L, Traeger-Synodinos J, Goossens V, Moutou C, SenGupta S, Pehlivan Budak T, Renwick P, De Rycke M, Geraedts J et al. The ESHRE PGD Consortium: 10 years of data collection. Hum Reprod Update 2012;18:234–247.[↩]




{kind=link}