Contents
What is hypermobility syndrome
Hypermobility syndrome or joint hypermobility syndrome is commonly known as hypermobility type Ehlers-Danlos syndrome (hEDS), which is a group of inherited disorders that affect your connective tissues — primarily your skin, joints and blood vessel walls 1. Joint hypermobility syndrome is considered to be synonymous with hypermobility type Ehlers-Danlos syndrome 2. An updated classification of the diagnostic criteria for the various Ehlers-Danlos syndromes has been published 3, which includes hypermobile Ehlers-Danlos syndrome.
Those with joint hypermobility syndrome or hypermobility type Ehlers-Danlos syndrome report generalized joint hypermobility and symptoms like musculoskeletal complaints, recurrent joint dislocations/joint instability and chronic pain, sometimes starting in childhood 3. Physical therapy is considered to be the main mode of treatment 4, although there is little evidence relating to the success of these interventions 5; the reason for this is likely to be the complex individual nature of the condition.
Joint hypermobility syndrome or Ehlers-Danlos syndrome-hypermobility type is one of the heritable connective tissue disorders showing symptom overlap with Marfans Syndrome, Osteogeneis Imperfecta, Chronic Fatigue Syndrome and Fibromyalgia 6.
Hypermobile Ehlers-Danlos syndrome (hEDS) is generally considered the least severe type of Ehlers-Danlos syndrome, although significant complications, primarily musculoskeletal, can and do occur 7. The skin is often soft and may be mildly hyperextensible. Subluxations and dislocations are common; they may occur spontaneously or with minimal trauma and can be acutely painful. Degenerative joint disease is common. Chronic pain, distinct from that associated with acute dislocations, is a serious complication of the condition and can be both physically and psychologically disabling. Easy bruising, functional bowel disorders, and cardiovascular autonomic dysfunction are common. Aortic root dilation, when present, is typically of a mild degree with no increased risk of dissection in the absence of significant dilation. Psychological dysfunction, psychosocial impairment, and emotional problems are common.
The diagnosis of joint hypermobility syndrome (hypermobile Ehlers-Danlos syndrome) is based entirely on clinical evaluation and family history. The gene(s) in which mutation causes joint hypermobility syndrome (hypermobile Ehlers-Danlos syndrome) are unknown and unmapped 7.
Joint hypermobility syndrome causes
Mutations in at least 19 genes have been found to cause the Ehlers-Danlos syndromes. Mutations in the COL5A1 or COL5A2 gene, or rarely in the COL1A1 gene, can cause the classical type. Mutations in the TNXB gene cause the classical-like type and have been reported in a very small percentage of cases of the hypermobile type (although in most people with this type, the cause is unknown). The cardiac-valvular type and some cases of the arthrochalasia type are caused by COL1A2 gene mutations; mutations in the COL1A1 gene have also been found in people with the arthrochalasia type. Most cases of the vascular type result from mutations in the COL3A1 gene, although rarely this type is caused by certain COL1A1 gene mutations. The dermatosparaxis type is caused by mutations in the ADAMTS2 gene. PLOD1 or FKBP14 gene mutations result in the kyphoscoliotic type. Other rare forms of Ehlers-Danlos syndrome result from mutations in other genes.
Some of the genes associated with the Ehlers-Danlos syndromes, including COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2, provide instructions for making pieces of several different types of collagen. These pieces assemble to form mature collagen molecules that give structure and strength to connective tissues throughout the body. Other genes, including ADAMTS2, FKBP14, PLOD1, and TNXB, provide instructions for making proteins that process, fold, or interact with collagen. Mutations in any of these genes disrupt the production or processing of collagen, preventing these molecules from being assembled properly. These changes weaken connective tissues in the skin, bones, and other parts of the body, resulting in the characteristic features of the Ehlers-Danlos syndromes.
Some genes associated with recently described types of Ehlers-Danlos syndrome have functions that appear to be unrelated to collagen. For many of these genes, it is not clear how mutations lead to hypermobility, elastic skin, and other features of these conditions.
Joint hypermobility syndrome inheritance pattern
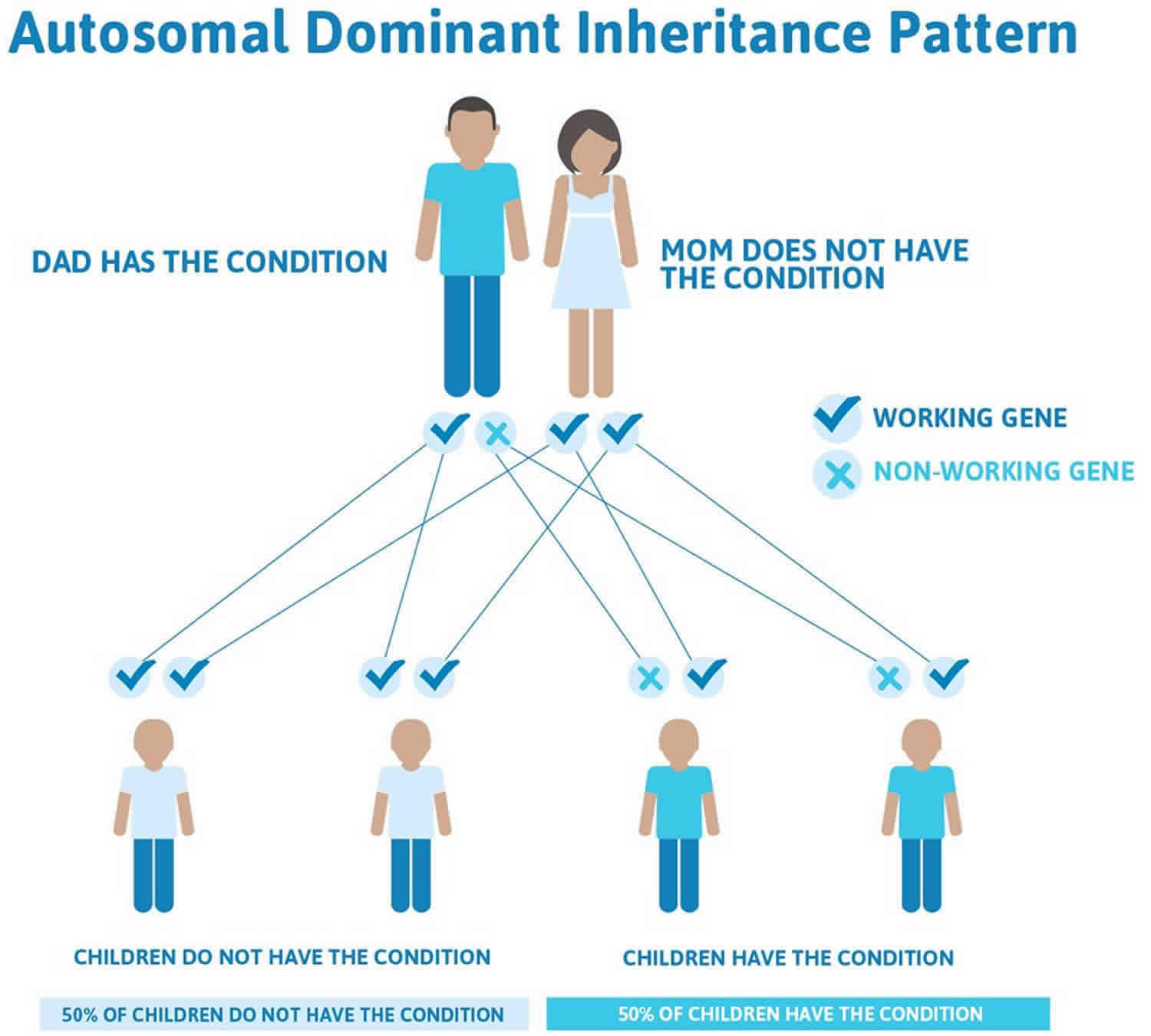
The inheritance pattern of the Ehlers-Danlos syndromes varies by type. The classical, vascular, arthrochalasia, and periodontal forms of the disorder, and likely the hypermobile type, have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new (de novo) gene mutations and occur in people with no history of the disorder in their family.
Figure 1. Joint hypermobility syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Hypermobility syndrome symptoms
Clinical distinction between the hypermobile and classic types of Ehlers-Danlos syndrome is sometimes very difficult. With the exception of skin and soft tissue complications, much of the information in this section is derived from publications that collectively analyzed individuals with hypermobile and classic Ehlers-Danlos syndrome, without specifying whether there was any difference in manifestations between the two types.
Hypermoblie Ehlers-Danlos syndrome (hEDS) is generally considered the least severe type of Ehlers-Danlos syndrome, although significant complications, primarily musculoskeletal, do occur. Clinical variability is substantial. Most individuals who seek medical care are female. Pain and major joint complications are much less common among affected males. This bias may result from differences between men and women with respect to pain perception and inherent joint stability, as well as the effects of sex hormones 8.
Skin
The skin is often soft and may be mildly hyperextensible. Piezogenic papules (small herniations of subcutaneous fat through the underlying dermis of the heel occurring only with weight bearing) are common but rarely painful.
Keratosis pilaris may be more common than in the general population 9.
Subcutaneous spheroids and molluscoid pseudotumors are not features of this Ehlers-Danlos syndrome type.
Clinically significant skin morbidity does not occur; its presence should prompt consideration of alternative diagnoses.
Musculoskeletal
Joint instability. Joint laxity and instability and excessive joint motion are frequently evident on routine activity, even in the absence of overt subluxation or dislocation.
- Subluxations and dislocations are common. They may occur spontaneously or with minimal trauma and can be acutely painful. Reduction often occurs spontaneously or can be accomplished by the affected individual or a bystander. For most affected individuals, medical intervention for an acute dislocation is not usually necessary, but pain can last for hours or days after an event.
- All sites can be involved, including the extremities, vertebral column, costo-vertebral and costo-sternal joints, clavicular articulations, and temporomandibular joints.
- Sprains or twisting of the ankles and buckling or “giving out” of the knees are common.
- Iliotibial band syndrome or “snapping hip” is a common symptom, often perceived by the affected individual as hip joint instability 10.
- Temporomandibular dysfunction (“TMJ syndrome”) is relatively common 11, and can be thought of as a specific example of joint degeneration and osteoarthritis.
- Females tend to have more substantial laxity than males.
- Younger individuals tend to have more substantial laxity than older individuals 9.
- Tendinitis and bursitis may occur 12, especially greater trochanteric bursitis in those with iliotibial band syndrome. Ehlers-Danlos syndrome does not directly cause inflammation, and these problems are likely secondary to joint instability.
Osteoarthritis
Degenerative joint disease occurs at a younger age than in the general population, possibly because of chronic joint instability resulting in increased mechanical stress.
Bone density
There is very limited and contradictory evidence regarding bone mineral density in hypermobile Ehlers-Danlos syndrome. Dolan et al 13 found bone density to be reduced by up to 0.9 SD in individuals with Ehlers-Danlos syndrome compared to healthy controls, but that study did not look specifically at individuals with hypermobile Ehlers-Danlos syndrome. Compared to age- and sex-matched controls, Gulbahar et al 14 reported bone density reduction of up to 0.5 SD among premenopausal women with joint hypermobility syndrome (now considered identical to hypermobile Ehlers-Danlos syndrome). However, Carbone et al 15 found no difference in bone density between women with hypermobile Ehlers-Danlos syndrome and normal controls after adjusting for height, weight, and physical activity.
Pain
Chronic pain, distinct from that associated with acute dislocations, is a serious complication of the condition and can be both physically and psychosocially disabling 12.
It is variable in age of onset (as early as adolescence or as late as the 5th-6th decade), number of sites, duration, quality, severity, and response to therapy.
Severity is typically greater than expected based on physical and radiologic examinations.
Severity sometimes correlates with degree of joint instability and with sleep impairment 16.
Fatigue and sleep disturbance are frequently associated 16. Affected individuals are often diagnosed with chronic fatigue syndrome, fibromyalgia, depression, hypochondriasis, and/or malingering prior to recognition of joint laxity and establishment of the correct underlying diagnosis.
Headaches, especially migraine, are common 12. Cervical muscle tension, temporomandibular dysfunction, and stress are some of the likely contributing factors.
Several recognizable pain syndromes are likely:
- Muscular or myofascial pain, localized around or between joints, often described as aching, throbbing, or stiff in quality, may be attributable to myofascial spasm, and palpable spasm with tender points (consistent with fibromyalgia) is often demonstrable, especially in the paravertebral musculature 12. Myofascial spasm possibly occurs in response to chronic joint instability, but this has not been systematically studied. Myofascial release often provides temporary relief.
- Neuropathic pain, variably described as electric, burning, shooting, numb, tingling, or hot or cold discomfort, may occur in a radicular or peripheral nerve distribution or may appear to localize to an area surrounding one or more joints 17. Nerve conduction studies are usually not diagnostic. Skin biopsy may reveal reduction or absence of small nerve fibers, but may be normal. One hypothesis is that neuropathic pain may result from direct nerve impingement (e.g., by subluxed vertebrae, herniated discs, vertebral osteoarthritis, or peripheral joint subluxations). In addition, there may be mild-to-moderate nerve compression within areas of myofascial spasm. Neither of these possibilities has been evaluated clinically.
- Osteoarthritic pain occurs later in life (but earlier than in the general population) and typically presents as aching pain in the joints, frequently associated with stiffness. It is often exacerbated by stasis and by resistance and/or highly repetitive activity.
Hematologic
Easy bruising is quite common, frequently without obvious trauma or injury 18. Mildly prolonged bleeding, epistaxis, bleeding from the gums (especially after dental extraction), and menometrorrhagia may also occur.
The underlying cause of the hematologic manifestations is unknown.
- Bleeding time may or may not be prolonged, but no consistent abnormalities of coagulation factors, von Willebrand factor or platelet number, release or aggregation have been reported 19.
- Capillary fragility and/or impaired soft tissue integrity may play a role 18.
Gastrointestinal
Functional bowel disorders are common and underrecognized, affecting 33%-67% of individuals with hypermobile Ehlers-Danlos syndrome 9.
Gastroesophageal reflux and gastritis may be symptomatic despite maximal doses of proton pump inhibitors with additional H2-blockers and acid-neutralizing medications.
Early satiety and delayed gastric emptying may occur and may be exacerbated by opioid (and other) medications.
Irritable bowel syndrome may manifest with diarrhea and/or constipation, associated with abdominal cramping and rectal mucus.
Cardiovascular
Autonomic dysfunction. Many affected individuals report atypical chest pain, palpitations at rest or on exertion, and/or orthostatic intolerance with syncope or near syncope 20. Holter monitoring usually shows normal sinus rhythm, but sometimes reveals premature atrial complexes or paroxysmal supraventricular tachycardia. Tilt table testing may reveal neurally mediated hypotension and/or postural orthostatic tachycardia syndrome (POTS).
Raynaud syndrome and acrocyanosis occur at an increased frequency, which may be another manifestation of autonomic dysfunction 9.
Aortic root dilation, usually of a mild degree, occurs in 11%-33% of individuals with hypermobile Ehlers-Danlos syndrome 21. The severity appears to be much less than occurs in Marfan syndrome, and there is no increased risk of aortic dissection in the absence of significant dilation. Dilation onset is in childhood and is usually stable over time. It is unlikely to progress or to develop later in life 21.
Mitral valve prolapse was previously considered a common feature of Ehlers-Danlos syndrome. Rigorous evaluations using modern diagnostic criteria have been inconsistent, with some studies showing no increase in the frequency of clinically significant mitral valve prolapse 21 and others showing an mitral valve prolapse frequency of 28%-67% 22. It is possible that mild mitral valve prolapse not meeting diagnostic criteria (and therefore not requiring special monitoring or treatment) may also explain some of the atypical chest pain and palpitations.
Oral/Dental
High, narrow palate and dental crowding are nonspecific features of most heritable disorders of connective tissue. Bifid uvula, submucous cleft palate, and overt cleft palate are not manifestations of hypermobile Ehlers-Danlos syndrome, and should prompt consideration of alternative diagnoses (see Differential Diagnosis).
The frequency of periodontal manifestations such as friability, gingivitis, and gum recession is probably increased but has not been adequately studied specifically in the hypermobile type 9. De Felice et al 23 reported an abnormally complex oral microvascular network in 12 individuals with classic or hypermobile Ehlers-Danlos syndrome; potential correlation of this with periodontal disease has not been reported.
Obstetric/Gynecologic
Pregnancy may be complicated by rapid labor and delivery (<4 hours), with small studies suggesting a frequency of 28%-36% 24.
Joint laxity and pain typically increase throughout gestation, especially in the third trimester, as normally occurs during pregnancy in unaffected women 24.
There is no clear advantage to vaginal vs cesarean delivery. Cesarean delivery may reduce the risk of hip dislocation 25, but carries the same risk for surgical complications as in the general population.
There is no increase in risk for cervical incompetence, and no evidence to support use of prophylactic cerclage 26.
No other pregnancy complications are associated with hypermobile Ehlers-Danlos syndrome.
Pelvic prolapse, dysmenorrhea, and dyspareunia occur at increased frequency in hypermobile Ehlers-Danlos syndrome 24.
Psychiatric
Psychological dysfunction, psychosocial impairment, and emotional problems are common 27.
Specific manifestations may include depression, anxiety, affective disorder, low self-confidence, negative thinking, hopelessness, and desperation 27.
Fatigue 28 and pain 27 exacerbate the psychological dysfunction.
Psychological distress exacerbates pain 29.
Fear of pain and/or joint instability may lead to avoidance behavior (kinesiophobia) and exacerbate dysfunction and disability 29.
Affected individuals may feel misunderstood, disbelieved, marginalized, and alone 29.
Resentment, distrust, and hostility may develop between the affected individual/family and the health care team (in both directions), adversely affecting the therapeutic relationship 10.
Ocular
Detailed and systematic evaluation of ocular findings in 44 eyes of 22 individuals with hypermobile Ehlers-Danlos syndrome was compared to age- and gender-matched controls 30. Findings included the following:
- Subjective and objective measures of xerophthalmia were rare, but more common in hypermobile Ehlers-Danlos syndrome than controls. It is unknown whether this represents an intrinsic feature of Ehlers-Danlos syndrome or possibly an indirect association (e.g., a side effect of medication).
- Clinically insignificant minor lens opacities were found in 13% of Ehlers-Danlos syndrome eyes, compared to none among controls.
- High myopia (more than -6.0 diopters) and vitreous degeneration were found in 16% of Ehlers-Danlos syndrome eyes and none of the controls. There was no difference in frequency of mild or moderate myopia between Ehlers-Danlos syndrome and control eyes.
- There was no significant difference in axial length of the globe between Ehlers-Danlos syndrome and control eyes.
- Ehlers-Danlos syndrome eyes averaged slightly increased corneal curvature compared to controls, but there was no overt keratoconus.
Neurologic & Neuromuscular
Delayed onset and/or resistance to local anesthesia is a frequent complaint 9
.
Dysautonomia may manifest as functional bowel disorders, cardiovascular autonomic dysfunction, and/or Raynaud syndrome/acrocyanosis.
Poor balance is common, with increased incidence of falls and occasionally fear of falling 31.
Diminished joint position sense has been reported at the knees, but not the shoulders. Vibration sensation is normal 32.
It is unclear whether weakness is an associated feature. Some studies suggest normal muscle strength 33, while others have demonstrated decreased ankle power 34, reduced passive muscle tension, and increased Achilles tendon compliance 33. One study reported lower-extremity weakness, but the findings could also be explained by reduced motor effort secondary to pain and/or fatigue 33.
Individuals with hypermobile Ehlers-Danlos syndrome tend to have a slower-than-normal gait with shorter gait length 34.
Kinematic studies are normal at the hips and knees 34, but the ankles demonstrate excess plantar flexion at ground contact and decreased dorsiflexion during motion 34.
In a series of 2,813 individuals with Chiari malformation type 1, 12.7% were felt to also have a hereditary disorder of connective tissue, including many with hypermobile Ehlers-Danlos syndrome 35. Among those with independently confirmed Ehlers-Danlos syndrome, Chiari malformation was found in only one (4.7%) of 21 individuals with hypermobile Ehlers-Danlos syndrome [9 and one (5.5%) of 18 individuals with headache and unspecified types of Ehlers-Danlos syndrome 36. The incidence of Chiari malformation among individuals with Ehlers-Danlos syndrome has not been systematically studied, and the clinical relevance of this potential association is uncertain.
Disability
Functional and psychosocial impairment are common, manifesting with decreased sport-related physical activity, diminished health-related quality of life, and significant impact on daily function 12.
Pain, fatigue, and sleep disturbance all may contribute to disability and functional impairment 12.
Other
Fragility of soft tissues with spontaneous ruptures or tears of internal organs is, by definition, not a feature of hypermobile Ehlers-Danlos syndrome. Such manifestations should prompt consideration of other hereditary connective tissue disorders (see Differential Diagnosis).
Joint hypermobility syndrome diagnosis
Joint hypermobility syndrome (hypermobile Ehlers-Danlos syndrome) should be suspected in individuals with joint laxity, soft skin, and easy bruising. Other organ systems (especially gastrointestinal and cardiovascular) are frequently involved. None of these features is specific to Ehlers-Danlos syndrome, and these features alone are insufficient to establish a diagnosis of any type of Ehlers-Danlos syndrome 37.
Establishing the diagnosis
The diagnostic criteria for hypermobility Ehlers-Danlos syndrome (and all other types of Ehlers-Danlos syndrome) were revised by the International Ehlers-Danlos syndrome Consortium in 2017 37. No underlying genetic cause has yet been identified in hypermobility Ehlers-Danlos syndrome, and thus the diagnosis is based entirely on clinical evaluation and family history.
Joint hypermobility is a feature of many heritable and acquired disorders (see Differential Diagnosis), and may also occur as an asymptomatic and/or nonsyndromic finding. In order to reduce heterogeneity and enhance efforts to identify the genetic etiology, a formal diagnosis of hypermobility Ehlers-Danlos syndrome should be made only when all of the diagnostic criteria are met. Individuals with signs and symptoms suggestive of a hereditary connective tissue disorder who fail to meet diagnostic criteria for hypermobility Ehlers-Danlos syndrome or any other described condition should be considered to have hypermobility spectrum disorder 38.
The clinical diagnosis of hypermobility Ehlers-Danlos syndrome requires the simultaneous presence of three criteria:
- Generalized joint hypermobility (Criterion 1)
- Evidence of syndromic features, musculoskeletal complications, and/or family history (Criterion 2)
- Exclusion of alternative diagnoses (Criterion 3)
Multiple other clinical features including (but not limited to) sleep disturbance, fatigue, postural orthostatic tachycardia, functional gastrointestinal disorders, dysautonomia, anxiety, and depression are associated with hypermobility Ehlers-Danlos syndrome. Some of these features were formerly included as minor diagnostic criteria for hypermobility Ehlers-Danlos syndrome 39. They were excluded from the 2017 hypermobility Ehlers-Danlos syndrome diagnostic criteria because they lack specificity for hypermobility Ehlers-Danlos syndrome 37.
Criterion 1
Generalized joint hypermobility. The Beighton score 40 remains the best-validated tool for assessing joint hypermobility 41. In order to reduce false-positive Beighton scores, the 2017 hypermobility Ehlers-Danlos syndrome diagnostic criteria recommend standardized performance of the Beighton test 37. One point is scored for each of the following:
- Passive dorsiflexion of each fifth finger greater than 90°. This should be assessed with the palm and forearm resting on a flat surface, and is considered positive only if the fifth metacarpal-phalangeal joint (MCP) can be extended more than 90°. Ability to extend the tip of the fifth finger to a position proximal to the MCP is insufficient to be called positive if the MCP does not extend more than 90°.
- Passive apposition of each thumb to the flexor surface of the forearm. This should be assessed with the elbow extended and hand pronated.
- Hyperextension of each elbow greater than 10°. This should be measured with a goniometer, with the hand supinated, elbow fully extended, and shoulder abducted to 90°.
- Hyperextension of each knee greater than 10°. This should be measured with a goniometer, with the patient standing and knees fully extended.
- Ability to place the palms flat on the floor with the knees fully extended. This should be assessed with the knees locked in extension and the feet together, and is considered positive only if the total palm of both hands lies flat on the floor just in front of the feet. Slight flexion of the knees, spreading of the feet, failure to get the heels of the palms to the floor, and positioning the hands more than a few inches in front of the feet are common causes for false positive scoring of this point.
The original Beighton publication defines a score of ≥5 as indicative of generalized joint hypermobility 40. However, joint range of motion typically decreases with age, leading to overdiagnosis of children and underdiagnosis of older adults with joint hypermobility. For the purposes of diagnosing hypermobility Ehlers-Danlos syndrome, generalized joint hypermobility is confirmed by a score of 37:
- ≥6 for prepubertal children
- ≥5 for pubertal children and adults up to age 50
- ≥4 for those age >50 years
Multiple other variables including (but not limited to) ethnicity, gender, trauma, surgery, arthritic change, conditioning, and stretching also affect the Beighton score. There is no validated method to account for this variation in assessing generalized joint hypermobility, but the 2017 hypermobility Ehlers-Danlos syndrome diagnostic criteria include an allowance for individuals with acquired limitation of joint mobility 37. Generalized joint hypermobility may be confirmed in an individual whose Beighton score is one point below the age-specific cutoff if there are two or more positive answers to the five-point questionnaire (5PQ) 42. Individuals with prior history of joint hypermobility, suggested by a positive 5PQ (≥2 positive answers) but scoring two or more points below the age-specific Beighton cutoff, should not be considered to have generalized joint hypermobility and should instead be evaluated for hypermobility spectrum disorder 38.
The Five-point questionnaire (5PQ):
- Can you now (or could you ever) place your hands flat on the floor without bending your knees?
- Can you now (or could you ever) bend your thumb to touch your forearm?
- As a child, did you amuse your friends by contorting your body into strange shapes or could you do the splits?
- As a child or teenager, did your shoulder or kneecap dislocate on more than one occasion?
- Do you consider yourself “double-jointed”?
Criterion 2
At least two of the following features (A, B, and/or C) must be present.
Feature A
Five or more of the following systemic manifestations of a more generalized connective tissue disorder (A Systemic Score calculator and a complete description of each component evaluation can be found at the National Marfan Foundation website (https://www.marfan.org/dx/score):
- Unusually soft or velvety skin. This is an inherently subjective feature. It should be assessed in the absence of recent application of moisturizer, and a high threshold is recommended.
- Mild skin hyperextensibility, assessed at a site lacking excess or loose skin and without evidence of prior trauma by gently pulling until resistance is met. An ideal location is the volar surface of the non-dominant forearm, where the upper limit of normal extensibility is 1.5 cm. Extensor surfaces of joints have excess skin and should not be used. More significant extensibility (e.g. >2.0 cm) should prompt consideration of other Ehlers-Danlos syndrome types.
- Unexplained* striae at the back, groins, thighs, breast, and/or abdomen in adolescents, men, or prepubertal females (* i.e., without a history of significant gain or loss of body fat or weight)
- Bilateral piezogenic papules* of the heel (* i.e., herniations of subcutaneous heel fat visible upon standing); must be present bilaterally to be considered positive
- Recurrent or multiple abdominal hernias, such as umbilical, curural, inguinal, or femoral. Hiatal hernia does not count toward this feature.
- Atrophic scarring involving at least two sites and without the formation of papyraceous and/or hemosideric scars as seen in classic Ehlers-Danlos syndrome. Atrophic scarring is defined as scars from linear traumatic lacerations or single surgery that are unusually shallow and/or wider than the original wound. Atrophic scars as the result of multiple incisions, wound infections, or inflammatory conditions do not count toward this feature and elliptical incisions (e.g., for removal of nevi) may be difficult to assess without knowing the size of the original wound.
- Pelvic floor, rectal, and/or uterine prolapse in children, men, or nulliparous women without a known predisposing medical condition
- Dental crowding (including a history of crowding corrected by orthodontia) and high or narrow palate. Both conditions must be positive to count toward this feature.
- Arachnodactyly, defined as either bilateral positive wrist sign or bilateral positive thumb sign
- Arm span to height ≥1.05
- Mitral valve prolapse, based on strict echocardiographic criteria
- Aortic root dilatation with a Z score >+2
Feature B
Positive family history, with one or more first-degree relatives independently meeting the current diagnostic criteria for hypermobility Ehlers-Danlos syndrome. Of note, a first-degree relative meeting prior diagnostic criteria for Ehlers-Danlos syndrome, hypermobility type or type III Ehlers-Danlos syndrome does not count toward this feature; the relative must meet current criteria for hypermobility Ehlers-Danlos syndrome.
Feature C
At least one of the following musculoskeletal complications (but see Criterion 3, Note):
- Musculoskeletal pain in two or more limbs, recurring daily for at least three months
- Chronic widespread pain for at least three months
- Recurrent joint dislocations or frank joint instability, in the absence of trauma:
- Three or more atraumatic dislocations in the same joint or two or more atraumatic dislocations in two different joints occurring at different times; OR
- Medical confirmation of joint instability at two or more sites not related to trauma
Criterion 3
ALL of the following prerequisites must be met:
- Absence of unusual skin fragility, which should prompt consideration of other types of Ehlers-Danlos syndrome
- Exclusion of other heritable and acquired connective tissue disorders including autoimmune rheumatologic conditions (see Note)
- Exclusion (based on history, physical exam, and/or molecular genetic testing) of alternative diagnoses that may also include joint hypermobility by means of hypotonia and/or connective tissue laxity such as neuromuscular disorders, other heritable connective tissue disorders and skeletal dysplasias (see Differential Diagnosis)
Note: In individuals with an acquired connective tissue disorder (e.g., lupus, rheumatoid arthritis), additional diagnosis of hypermobility Ehlers-Danlos syndrome requires meeting both Features A and B of Criterion 2. Feature C of Criterion 2 (chronic pain and/or instability) cannot be counted toward a diagnosis of hypermobility Ehlers-Danlos syndrome in this situation.
Joint hypermobility syndrome test
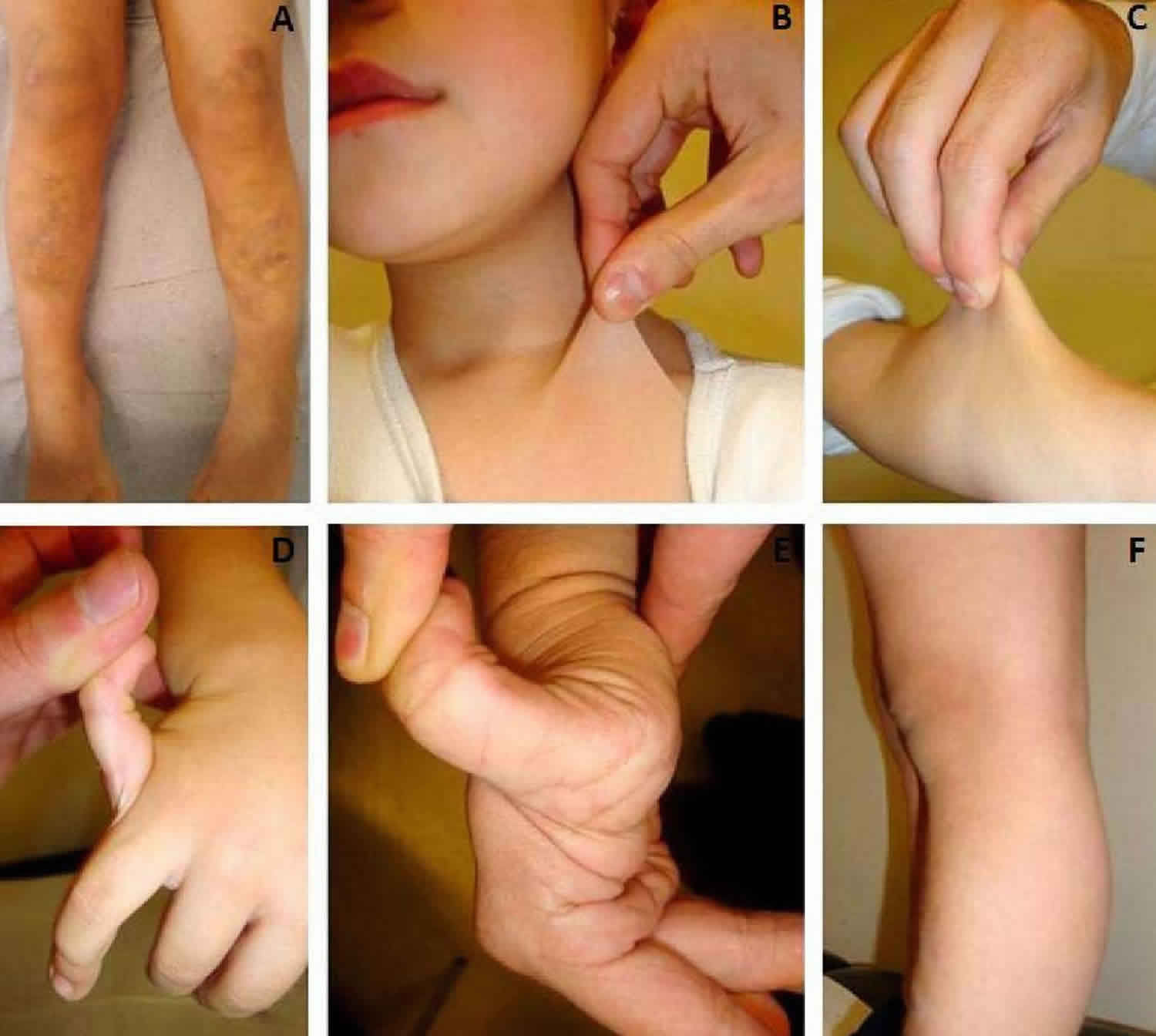
The Beighton Scoring System measures joint hypermobility on a 9-point scale. The joints assessed are:
- Knuckle of the little/fifth/pinky finger
- Base of the thumb
- Elbow
- Knee
- Spine
Where applicable, range of movement is measured using a goniometer, an instrument that measures the joint angle.
The movements that make up the Beighton score are:
A. 5th FINGER/PINKIES
Rest palm of the hand and forearm a flat surface with palm side down and fingers out straight. Can the 5th finger be bent/lifted upwards at the knuckle to go back beyond 90 degrees? If yes, add one point for each hand.
B. THUMBS
With the arm out straight, the palm facing down, and the wrist then fully bent downward, can the thumb be pushed back to touch the forearm? If yes, add one point for each thumb.
C. ELBOWS
With arms outstretched and palms facing upwards, does the elbow extend (bend too far) upwards more than an extra 10 degrees beyond a normal outstretched position? If yes, add one point for each side.
D. KNEES
While standing, with knees locked (bent backwards as far as possible), does the lower part of either leg extend more than 10 degrees forward? If yes, add one point for each side?
E. SPINE
Can you bend forward and place the palms of your hands flat on the floor in front of your feet without bending your knees? If yes, add one point.
NOTE:
- (A) With the palm of the hand and forearm resting on a flat surface with the elbow flexed at 90°, if the metacarpal-phalangeal joint of the fifth finger can be hyperextended more than 90° with respect to the dorsum of the hand, it is considered positive, scoring 1 point.
- (B) With arms outstretched forward but hand pronated, if the thumb can be passively moved to touch the ipsilateral forearm it is considered positive scoring 1 point.
- (C) With the arms outstretched to the side and hand supine, if the elbow extends more than 10°, it is considered positive scoring 1 point.
- (D) While standing, with knees locked in genu recurvatum, if the knee extends more than 10°, it is considered positive scoring 1 point.
- (E) With knees locked straight and feet together, if the patient can bend forward to place the total palm of both hands flat on the floor just in front of the feet, it is considered positive scoring 1 point.
Joint hypermobility syndrome differential diagnosis
All types of Ehlers-Danlos syndrome share some degree of joint laxity and skin / soft tissue manifestations.
The other forms of Ehlers-Danlos syndrome are distinguished by additional connective tissue manifestations 37:
- Classic Ehlers-Danlos syndrome (cEDS) includes skin and soft tissue fragility. Mild presentations of the classic type may be mistaken for the hypermobile type, including similar degrees of joint laxity, pain, pelvic prolapse, dyspareunia, and manifestations in the hematologic, gastrointestinal, cardiovascular, and dental systems. The diagnosis is sometimes revised from hypermobile to classic when the individual or a family member later develops more significant skin and soft tissue manifestations. Among individuals with all of the skin features of classic Ehlers-Danlos syndrome, including dystrophic scarring, more than 90% have an identifiable pathogenic variant in COL5A1 or COL5A2, the two genes encoding type V collagen 43. In individuals with milder skin manifestations (but still more than typically seen in the hypermobile type), no consistent pathogenic variants in any genes have been found. The diagnosis of classic Ehlers-Danlos syndrome is based on clinical findings and confirmed by molecular genetic testing. Classic Ehlers-Danlos syndrome is inherited in an autosomal dominant manner.
- In vascular Ehlers-Danlos syndrome (vEDS), the joint laxity is predominantly in small joints, as opposed to the generalized laxity typically observed in hypermobile Ehlers-Danlos syndrome. Vascular Ehlers-Danlos syndrome also usually manifests thin, translucent skin, fragility of skin and soft tissue, and atrophic scarring. Predisposition to spontaneous rupture of hollow organs (primarily arteries, intestines, and uterus) is a hallmark of the vascular type, but not all individuals with vEhlers-Danlos syndrome develop such complications. A family history of unexplained sudden death is potentially consistent with catastrophic internal organ rupture and could be sufficient to prompt diagnostic testing for the vascular type, especially if the event(s) occurred in a first-degree relative, multiple second-degree relatives, and/or at a young age (e.g., <50 years). Nonspecific venous and hematologic abnormalities including varicose veins, hemorrhoids, easy bruising, and bleeding diathesis are not suggestive of vascular Ehlers-Danlos syndrome, and should not prompt additional evaluation for this type. Dysfunction and/or deficiency of type III collagen, caused by pathogenic variants in COL3A1, is responsible for all cases of vascular Ehlers-Danlos syndrome. The diagnosis of vascular Ehlers-Danlos syndrome is based on clinical findings and confirmed by molecular genetic testing. Vascular Ehlers-Danlos syndrome is almost always inherited in an autosomal dominant manner, but rare examples of biallelic inheritance have been reported.
- Kyphoscoliotic Ehlers-Danlos syndrome (kEDS) and dermatosparaxis (dermatosparaxis Ehlers-Danlos syndrome, dEDS) are autosomal recessive, rare, and distinguished by more severe skin manifestations and other features. Kyphoscoliotic Ehlers-Danlos syndrome is caused by deficient activity of the enzyme procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1: lysyl hydroxylase 1); the diagnosis can be established by demonstration of an increased ratio of deoxypyridinoline to pyridinoline crosslinks in urine measured by HPLC, assay of lysyl hydroxylase enzyme activity in skin fibroblasts, or molecular genetic testing of PLOD1. Dermatosparaxis Ehlers-Danlos syndrome is caused by deficient activity of the enzyme procollagen I N-proteinase (ADAMTS2) and is diagnosed by molecular genetic testing of ADAMTS2 or biochemical analysis of type I procollagen extracted from dermis or in fibroblast cultures.
- Arthrochalasia Ehlers-Danlos syndrome (aEDS) is autosomal dominant, rare, and distinguished by congenital hip dislocation and more severe skin manifestations. Pathogenic variants occur in exon 6 of COL1A1 and COL1A2, causing impaired cleavage of the amino terminal propeptide of the corresponding type 1 procollagen molecules.
- Classic-like Ehlers-Danlos syndrome (clEDS) is caused by deficiency of tenascin-X, resulting from compound heterozygous or homozygous TNXB mutation/deletion. Classic-like Ehlers-Danlos syndrome is autosomal recessive and manifests joint laxity, hyperextensible skin, and easy bruising with normal wound healing and absence of atrophic scarring. Some, but not all, also have congenital adrenal hyperplasia as a result of contiguous gene deletion involving CYP21A2 44. Heterozygous mutation of TNXB has been reported in a small subset of individuals with joint laxity and soft skin typical of hypermobile Ehlers-Danlos syndrome, but skin hyperextensibility, easy bruising, and other hematologic manifestations are not part of this phenotype 45.
Joint laxity is a nonspecific manifestation of dozens of other disorders and syndromes. Functionally, joint hypermobility may be the result of ligamentous laxity (as in the heritable disorders of connective tissue and skeletal dysplasias) or hypotonia (as in mitochondrial diseases and other neuromuscular conditions). It can be difficult to distinguish between these mechanisms of pathology, especially in adults. When there is symptomatic joint hypermobility and no other specific diagnosis can be established, it is reasonable to diagnose hypermobility spectrum disorder 38.
Most of the following disorders are easily distinguished from Ehlers-Danlos syndrome by characteristic features and/or involvement of systems other than the joints and skin, but mild presentations can sometimes be misdiagnosed as hypermobile Ehlers-Danlos syndrome.
- Marfan syndrome results in additional skeletal, ocular, cardiovascular, pulmonary, and skin/integument manifestations beyond those seen in Ehlers-Danlos syndrome. Specific clinical criteria are available to establish a diagnosis of Marfan syndrome, and it can be confirmed by demonstration of a pathogenic variant in FBN1. Joint hypermobility is common in the MASS phenotype (myopia, mitral valve prolapse, mild aortic root dilatation, striae, and minor skeletal manifestations of Marfan syndrome), also caused by pathogenic variants in FBN1. Sometimes individuals with hypermobile Ehlers-Danlos syndrome can have a Marfanoid build and as such resemble individuals with Marfan syndrome or a Marfan-related disorder. However, application of the clinical diagnostic criteria for Marfan syndrome and FBN1 molecular analysis allow differentiation of these conditions. Marfan syndrome is inherited in an autosomal dominant manner.
- Loeys-Dietz syndrome (LDS) is characterized by vascular findings (cerebral, thoracic, and abdominal arterial aneurysms and/or dissections) and skeletal manifestations (pectus excavatum or pectus carinatum, scoliosis, joint laxity, arachnodactyly, talipes equinovarus). Approximately 75% of affected individuals have Loeys-Dietz syndrome type 1 (LDS-1) with craniofacial manifestations (widely spaced eyes, bifid uvula/cleft palate, craniosynostosis); approximately 25% have Loeys-Dietz syndrome type 2 (LDS-2) with systemic manifestations of LDS-1 but minimal or absent craniofacial features. Loeys-Dietz syndrome type 1 (LDS-1) and Loeys-Dietz syndrome type 2 (LDS-2) form a clinical continuum. The presentation often mimics Marfan syndrome or vascular Ehlers-Danlos syndrome, but prior to detection of the arterial abnormalities, individuals may be misdiagnosed with classic or hypermobile Ehlers-Danlos syndrome. The diagnosis is established by detection of a pathogenic variant in TGFBR1,TGFBR2, SMAD3, or TGFB2. Loeys-Dietz syndrome is inherited in an autosomal dominant manner.
- Arterial tortuosity syndrome, in addition to aneurysm, dissection, tortuosity, and stenosis of larger and medium-sized arteries, manifests craniofacial and skeletal features similar to Loeys-Dietz syndrome. In contrast to dominantly inherited connective tissue disorders predisposing to arterial fragility (vascular Ehlers-Danlos syndrome, Marfan syndrome, and Loeys-Dietz syndrome), Arterial tortuosity syndrome is inherited in an autosomal recessive manner. A diagnosis of arterial tortuosity syndrome is confirmed by identifying biallelic (homozygous or compound heterozygous) pathogenic variants in SLC2A10 in an individual with generalized arterial tortuosity and other typical features.
- Shprintzen-Goldberg syndrome is distinguished by cardinal features of craniosynostosis and mild-to-moderate intellectual disability. Cerebral abnormalities may include hydrocephalus, dilatation of the lateral ventricles, and Chiari 1 malformation. Aortic root dilation may occur, but less commonly than in Marfan syndrome or Loeys-Dietz syndrome. Some additional characteristic findings include C1/C2 spine malformation, reduced subcutaneous fat, abdominal wall defects, and cryptorchidism in males. Shprintzen-Goldberg syndrome is confirmed in individuals with typical findings by identifying a pathogenic variant in SKI. SKI-related Shprintzen-Goldberg syndrome is inherited in an autosomal dominant manner.
- Stickler syndrome. Distinguishing features include sensorineural hearing loss, vitreoretinal abnormalities, and cleft palate. Six genes have been associated with Stickler syndrome. However, a few families with features of Stickler syndrome are not linked to any of these loci; thus, pathogenic variants in other genes may also cause the disorder. Stickler syndrome is diagnosed based on clinical features. In many affected individuals and families the diagnosis can be confirmed by molecular genetic testing, although a negative test does not rule out the diagnosis. Stickler syndrome caused by mutation of COL2A1, COL11A1, or COL11A2 is inherited in an autosomal dominant manner; Stickler syndrome caused by mutation of COL9A1, COL9A2, or COL9A3 is inherited in an autosomal recessive manner.
- Williams syndrome is a contiguous gene deletion syndrome characterized by cardiovascular disease (elastin arteriopathy, peripheral pulmonary stenosis, supravalvular aortic stenosis, hypertension), distinctive facies, connective tissue abnormalities, intellectual disability (usually mild), a specific cognitive profile, unique personality characteristics, growth abnormalities, and endocrine abnormalities (hypercalcemia, hypercalciuria, hypothyroidism, and early puberty). The mainstay for diagnosis is detection of the contiguous gene deletion of the Williams-Beuren syndrome critical region (WBSCR) that encompasses the elastin gene (ELN). More than 99% of individuals with the clinical diagnosis of WS have this contiguous gene deletion, which can be detected using fluorescent in situ hybridization (FISH) or deletion/duplication analysis. Supravalvular aortic stenosis (SVAS) is caused by mutation (rather than deletion) of ELN. Individuals with either deletion or mutation of ELN have joint laxity, but the classic elastin arteriopathy is not seen in any type of Ehlers-Danlos syndrome. Williams syndrome is transmitted in an autosomal dominant manner.
- Aarskog-Scott syndrome (faciogenital dysplasia) is an X-linked condition resulting from mutation of FGD1. The most significant distinguishing feature is shawl scrotum, which may become less obvious in adulthood. Widow’s peak, short upturned nose, other dysmorphic features, and the inheritance pattern can be additional diagnostic clues. Intellectual disability, which is not associated with any of the types of Ehlers-Danlos syndrome, is sometimes present.
- Fragile X syndrome is not typically confused with hypermobile Ehlers-Danlos syndrome. When a full pathogenic variant of FMR1 is present, fragile X syndrome is characterized by moderate intellectual disability in affected males and mild intellectual disability in affected females. Males may have a characteristic appearance (large head, long face, prominent forehead and chin, protruding ears), joint laxity, and large testes (post-puberty). However, premutation carriers may have joint laxity and Ehlers-Danlos syndrome-like skin findings without other major manifestations. Family history of intellectual disability, premature ovarian failure, and/or fragile X tremor/ataxia syndrome is helpful when present. The frequency of FMR1 premutation among individuals diagnosed clinically with hypermobile Ehlers-Danlos syndrome has not been studied, but fragile X syndrome has not been reported among offspring of women with hypermobile Ehlers-Danlos syndrome. Fragile X syndrome is inherited in an X-linked manner.
- Achondroplasia and hypochondroplasia are distinguished by short stature with characteristic skeletal features (marked in achondroplasia, milder in hypochondroplasia). Achondroplasia can be diagnosed by characteristic clinical and radiographic findings in most affected individuals. Molecular genetic testing reveals a pathogenic variant in FGFR3 in 99% of individuals with achondroplasia and about 70% of individuals with hypochondroplasia. However, it is clear that locus heterogeneity exists for hypochondroplasia because pathogenic variants in other as-yet unidentified genes can result in similar, if not identical, phenotypes. Achondroplasia and hypochondroplasia are inherited in an autosomal dominant manner.
- Osteogenesis imperfecta is distinguished by the presence of fractures and, in some cases, dentinogenesis imperfecta (grey or brown teeth). Biochemical testing (i.e., analysis of the structure and quantity of type I collagen synthesized in vitro by cultured dermal fibroblasts) detects abnormalities in 98% of individuals with osteogenesis imperfecta type II, about 90% with osteogenesis imperfecta type I, about 84% with osteogenesis imperfecta type III, and about 84% with osteogenesis imperfecta type IV. About 90% of individuals with osteogenesis imperfecta types I, II, III, and IV (but none with osteogenesis imperfecta types V, VI, and VII) have an identifiable pathogenic variant in either COL1A1 or COL1A2. COL1A1/COL1A2-related OI is inherited in an autosomal dominant manner.
- Mitochondrial myopathies may cause joint hypermobility as a result of hypotonia, rather than the ligamentous laxity assumed to be the underlying mechanism in hereditary disorders of connective tissue. It can be difficult to distinguish between ligamentous laxity and hypotonia on physical exam, especially in adolescents and adults. Other features of mitochondrial disease that overlap with those seen in hypermobile Ehlers-Danlos syndrome include headache, neuropathy, myopathy, and autonomic dysfunction. Gastrointestinal dysmotility may also occur in mitochondrial disorders, especially Mitochondrial neurogastrointestinal encephalopathy disease.
- Many other neuromuscular disorders can also cause hypotonia, neuropathy, autonomic dysfunction, functional gastrointenstinal abnormalities, and other features overlapping with hypermobile Ehlers-Danlos syndrome. Since there are currently no laboratory tests available to definitively confirm or rule out hypermobile Ehlers-Danlos syndrome, it is important to thoroughly consider the possibility of a mitochondrial or other neuromuscular disorder in the differential diagnosis.
- Aneuploidies, including Down, Turner, and Klinefelter syndromes, are usually easily recognized based on dysmorphic features and/or intellectual disability. Small duplications or deletions may be less clinically obvious, but could be suggested by reduced fertility or recurrent pregnancy loss.
Chronic pain and fatigue are common in the general population, and the differential diagnosis of those symptoms is very long. Some conditions that may overlap with or complicate hypermobile Ehlers-Danlos syndrome:
- A subset of individuals with fibromyalgia and/or chronic fatigue syndrome may have Ehlers-Danlos syndrome as the underlying etiology, but not all individuals with chronic pain and/or fatigue have Ehlers-Danlos syndrome.
- Celiac disease may coexist with Ehlers-Danlos syndrome or be misdiagnosed as Ehlers-Danlos syndrome. Common features may include pain, fatigue, functional gastrointestinal disorders, and cardiovascular autonomic dysfunction.
- Vitamin D deficiency may also cause or exacerbate pain and fatigue.
- Familial Mediterranean fever most classically presents with episodic abdominal pain associated with fever, but low-grade fever may not be detected and the manifestations could be confused with irritable bowel syndrome. Joint pain may also occur in familial Mediterranean fever .
Clinically, the hematologic manifestations of Ehlers-Danlos syndrome mimic von Willebrand disease and respond to similar treatments, but von Willebrand factor and platelet number are almost always normal. Easy bruising and prolonged bleeding alone are insufficient to establish a diagnosis of Ehlers-Danlos syndrome, and the diagnostic evaluation of such problems should also consider von Willebrand disease, idiopathic thrombocytopenia purpura, and other hemorrhagic conditions.
Joint hypermobility syndrome treatment
Physical therapy
Myofascial release (any physical therapy modality that reduces spasm) provides short-term relief of pain, lasting hours to days. While the duration of benefit is short and it must be repeated frequently, this pain relief may be critical to facilitate participation in toning exercise for stabilization of the joints. Modalities must be tailored to the individual; a partial list includes heat, cold, massage, ultrasound, electrical stimulation, acupuncture, acupressure, biofeedback, and conscious relaxation.
Low-resistance muscle toning exercise can improve joint stability and reduce future subluxations, dislocations, and pain.
Transvaginal pelvic physical therapy and myofascial release (in which massage or ultrasound is applied to the pelvic musculature via a transvaginal approach) may improve dyspareunia, abdominal pain, back pain, and sometimes radicular lower-extremity pain. This should only be performed by a physical therapist with training and expertise in pelvic floor physical therapy and with patient consent.
Assistive devices
Braces are useful to improve joint stability. Orthopedists, rheumatologists, and physical therapists can assist in recommending appropriate devices for commonly problematic joints such as knees and ankles. Shoulders and hips present more of a challenge for external bracing. Occupational therapists may be consulted for ring splints (to stabilize interphalangeal joints) and wrist or wrist/thumb braces in affected individuals with small joint instability. A soft neck collar, if tolerated, may help with neck pain and headaches.
A wheelchair or scooter may be necessary to offload stress on lower-extremity joints. Special wheelchair customizations such as lightweight and/or motorized chairs, seat pads, and specialized wheels and wheel grasps may be necessary to accommodate pelvic and upper-extremity issues.
A waterbed, adjustable air mattress, or viscoelastic foam mattress (and/or pillow) may provide increased support with improved sleep quality and less pain.
Pain medication
Pain medication is frequently underprescribed, and should be tailored to the individual’s subjective symptoms and objective measures of pain, not to physical examination or radiologic findings. Individuals with mild to moderate pain may get sufficient relief from as-needed use. Those with more significant pain typically require higher doses and combinations of multiple medications. Prevention or control of pain with regularly scheduled dosing is often more successful than acute treatment with as-needed dosing. Many clinicians recruit a pain management specialist, but pain can be managed by the primary physician if desired.
Note: All of the following dose recommendations are for adults without hepatic or renal disease; adjustments may be necessary for other populations.
- Acetaminophen, 4,000 mg daily in three or four divided doses, will not completely alleviate pain but is a useful and well-tolerated adjunct in combination with other agents. Acetaminophen is often present in combination with other analgesic medications and cold/flu preparations, and careful attention should be paid to the total daily dose to avoid exceeding 4000 mg/day.
- NSAIDs (nonsteroidal anti-inflammatory drugs) (e.g., ibuprofen, naproxen, meloxicam, nabumetone) should be titrated to the maximum dose or as tolerated by upper gastrointestinal symptoms. NSAIDs are particularly useful for arthralgia, myalgia, and secondary inflammatory conditions (e.g. bursitis, tendinitis, costochodritis, or post-dislocation pain). Bruising is not a contraindication to NSAID therapy, but occasionally requires dose reduction or change to a Cox-2 inhibitor.
- Cox-2 inhibitors (e.g., celecoxib) in maximal doses are no stronger than dose-equivalent NSAIDS, but may be better tolerated and thus more effective.
- Topical lidocaine as a cream or patch is sometimes useful for localized areas of pain. Topical capsaicin is of questionable utility, but is safe.
- Skeletal muscle relaxants are useful in combination with all of the above to treat myofascial spasm. They are also sometimes helpful in treating neuropathic pain. Metaxalone may be the least sedating, but all are potentially limited by sedation. There is theoretic risk that skeletal muscle relaxants can increase joint instability by reducing muscle tone. This is not a contraindication to their use, but does justify monitoring for potential complications.
- Magnesium, either topically as Epsom salt baths or orally in any form, may also reduce muscle spasm and pain. No specific formulation or dosage has been established as superior to any other. The most common adverse effects – sedation, nausea, abdominal pain, and diarrhea – are more likely to occur with oral rather than topical supplementation.
- Tricyclic antidepressants are often effective for neuropathic pain, with additional benefits of mild sedation (for those with sleep disturbance) and a little mood elevation. Constipation, a common side effect, can be managed with fluids, fiber, stool softeners, and laxatives. For those with diarrhea-predominant irritable bowel syndrome, the constipating effect may be therapeutic. Typical doses are nortriptyline (25-150 mg) or trazadone (50-300 mg) every evening.
- Serotonin/norepinephrine receptor inhibitors (SNRIs), such as venlafaxine, desvenlafaxine, duloxetine, and milnacipran also offer combined benefit for depression and neuropathic pain. Venlaxafine may raise the blood pressure a few points, which potentially could be helpful for individuals with neurally mediated hypotension.
- Some anti-seizure medications are also effective for neuropathic pain and can be used in addition to tricyclic and/or SNRI antidepressants. All require gradual titration before reaching therapeutic levels. Gabapentin should be titrated as tolerated up to at least 1200 mg/3x/day before declaring failure, but is often limited by sedation and/or gastrointestinal side effects. Pregabalin can be dosed twice or three times daily up to a total daily dose of at least 300 mg, and tends to be better tolerated than gabapentin. Topiramate and lamotrigine have also been used successfully.
- Short courses of steroids can be very effective for controlling acute flares of pain associated with secondary inflammation. Ehlers-Danlos syndrome is not an intrinsically inflammatory condition, and there is no role for chronic steroid use.
- Glucosamine and chondroitin may help to prevent or treat osteoarthritis in the general population. They have not been studied specifically in Ehlers-Danlos syndrome, but are not contraindicated.
- Cannabinoids such as dronabinol and (where legal) marijuana may be helpful for several different types of pain. Their benefits should be weighed against the potential for dependency and/or psychoactive effects.
- Tramadol can be added to acetaminophen plus an NSAID or Cox-2 inhibitor before resorting to opioids. Nausea is the most common side effect.
- Opioids are effective for both myofascial pain and neuropathic pain, but should be reserved for use after failing the above medications. They should be administered in conjunction with all of the above, except tramadol, in order to minimize total opioid requirements. Since they are typically used chronically (or at least several months), the primary formulation should be long acting (e.g., sustained-release oxycodone or morphine or topical fentanyl patch) with short-acting forms of the same drug used as needed for breakthrough pain. Routine use of two or more daily doses of a short-acting form should prompt an increase in the long-acting dose or another adjustment to the pain regimen.
- Benzodiazepines may offer some short-term reduction in muscle spasm, but are poor choices for long-term muscle relaxation and carry high risk of tolerance, dependency, and addiction. Routine use of benzodiazepines is not recommended.
Medication precautions
- It is critically important to evaluate all potential sources of acetaminophen and assure that total daily use does not exceed 4,000 mg.
- Abrupt cessation of anti-seizure medications can precipitate a seizure. When discontinuing, they should be tapered gradually.
- Chronic opioid use can result in tolerance and dependency with escalating dose requirements and diminishing effect. Narcotic bowel syndrome can also develop, and may be confused with irritable bowel syndrome.
- Serotonin syndrome can occur when combining multiple serotonergic medications, such as tramadol, tricyclic antidepressants, SNRI antidepressants, anti-seizure medications, and opiods. Symptoms and signs may include agitation, restlessness, tremor, hyperreflexia, ataxia, confusion, irritability, nausea, diarrhea, hyperthermia, tachycardia, hyper/hypotension, and/or diaphoresis. Severity can range from mild to life threatening. Many of these manifestations overlap those associated with Ehlers-Danlos syndrome, making diagnosis difficult. Some individuals find mild-to-moderate serotonergic symptoms acceptable in order to achieve adequate pain control.
Surgery and other procedures
Many individuals will have undergone several orthopedic procedures prior to diagnosis. These often include joint debridement, tendon relocations, capsulorraphy, and arthroplasty. The degree of stabilization and pain reduction, overall patient satisfaction, and duration of improvement are variable, but usually less than in individuals without Ehlers-Danlos syndrome 12. In general, orthopedic surgery should be delayed in favor of physical therapy and bracing. When surgery is performed, the affected individual and physician should cautiously anticipate some improvement but expect less than optimal results. There is one report of long-term improvement in shoulder stability with Achilles tendon allograft reconstruction of the joint capsule in an individual with hypermobile Ehlers-Danlos syndrome 46. It is not yet known if this approach will be successful in other affected individuals. Unlike classic and vascular Ehlers-Danlos syndrome, hypermobile Ehlers-Danlos syndrome is not associated with increased risk for perioperative skin and soft tissue complications.
Prolotherapy, in which saline and/or other irritants are injected in tendons or around joints to induce scar formation and increase stability, has not been objectively studied. It is probably safe and probably subject to the same limitations as orthopedic surgery.
Anesthetic/corticosteroid injections for localized areas of pain and acute inflammation are often helpful, but cannot be repeated indefinitely. “Dry needling” without injection of any material sometimes provides similar benefit.
Anesthetic nerve blocks can provide temporary relief of neuropathic pain. These are sometimes followed by surgical nerve root destruction and/or implantable stimulators (sensory or motor), with variable results.
Constant intrathecal delivery of anesthetic and/or opioid medication may reduce the need for oral/systemic medications, but should only be considered as a last resort.
Bone density
Therapy is the same as for any other individual with low bone density, including supplementation of calcium and vitamin D. Simple weight-bearing exercise, such as walking or use of an elliptical trainer, should not be overlooked as a means to help maintain bone density as well as improve resting muscle tone.
Hematologic
Easy and spontaneous bruising does not require treatment, and does not require avoidance of NSAIDs.
For severe bleeding (e.g., epistaxis, menometrorrhagia) or operative prophylaxis, desmopressin acetate (ddAVP) is often beneficial 19.
Gastrointestinal
Gastritis and reflux symptoms may require intensive therapy, including proton pump inhibitor twice daily before meals, high-dose H2-blocker at bedtime (e.g., famotidine 20-40 mg or ranitidine 150-300 mg), sucralfate one gram four times daily, and over-the-counter acid-neutralizing agents. Other treatable causes, such as H. pylori infection, should be investigated. Upper endoscopy is indicated for resistant symptoms, but frequently is normal other than chronic gastritis.
Delayed gastric emptying should be identified if present and treated the same way as in individuals without Ehlers-Danlos syndrome. Assistance from a gastroenterologist experienced in managing GI (gastrointestinal) dysmotility can be helpful.
Irritable bowel syndrome is treated as usual with dietary modification, fiber, antispasmodics, antidiarrheals, and laxatives as needed. Lubiprostone is a motility enhancer that may be helpful for those with constipation only. Tricyclic antidepressants may be helpful for persons with both neuropathic pain and diarrhea.
Cardiovascular
Beta-blockade is rarely necessary, but should be considered for progressive aortic enlargement. Rarely, severe enlargement (>4.5-5.0 cm) requires surgical evaluation. When such severe enlargement is noted, other hereditary disorders of connective tissue should be considered (see Differential Diagnosis).
Neurally mediated hypotension and postural orthostatic tachycardia are treated as usual, with avoidance of sudden postural change, consideration of lower-extremity and/or abdominal compression garments, exercise to increase muscle tone, supplementation of sodium and water to expand the blood volume, and sometimes pharmacologic treatment with beta-blockers, midodrine, fludrocortisone, and/or other medications [Mathias et al 2011]. Commercially available electrolyte tablets can be added to water to facilitate oral expansion of blood volume.
Dental
Orthodontic and palatal corrections may tend to relapse, requiring prolonged use of a retainer.
Periodontal disease should be identified and treated.
Temporomandibular joint laxity and dysfunction are difficult to treat. There are no specific interventions of proven benefit. Intra-oral devices are sometimes helpful. Oral rest (minimization of chewing and talking), local myofascial release, and muscle relaxant medications may be beneficial for acute flares. Surgical intervention is often disappointing and should be considered only as a last resort.
Psychiatric
Validation of the affected individual’s symptoms can be immensely helpful, as many with hypermobile Ehlers-Danlos syndrome have been accused of malingering or diagnosed with primary psychiatric disorders by previous physicians.
Establishing trust, rapport, and a supportive relationship between patient and provider is important. Emphasis should be placed on chronic, rather than acute, pain management. Distraction and hypnosis are often helpful 10.
Depression is a common result of the chronic pain, disability, and other complications. Psychological and/or pain-oriented counseling can improve adaptation to and acceptance of these issues and the necessary physical limitations. Cognitive behavioral therapy can be particularly beneficial, but requires active patient participation 29. Antidepressants are also of great benefit. Many individuals initially resist a diagnosis of or therapy for depression because of concern that their problems are being written off as purely psychiatric.
Consumer support groups are available and can be very beneficial.
Prevention of Primary Manifestations
Improved joint stability may be achieved by low-resistance exercise to increase muscle tone (subconscious resting muscle contraction), as opposed to muscle strength (voluntary force exerted at will). Emphasis should be placed on both core and extremity muscle tone. Examples include walking, bicycling, low-impact aerobics, swimming or water exercise, and simple range-of-motion exercise without added resistance. Core toning activities, such as balance exercises and repetitive motions focusing on the abdominal, lumbar, and interscapular muscles, are also important. Progress should be made by gradually increasing repetitions, frequency, or duration, not resistance. It often takes months or years for significant progress to be recognized.
Wide-grip writing utensils can reduce strain on finger and hand joints. An unconventional grasp of a writing utensil, gently resting the shaft in the web between the thumb and index finger and securing the tip between the distal interphalangeal joints or middle phalanges of the index and third fingers (rather than using the tips of the fingers), results in substantially reduced axial stress to the interphalangeal, metacarpophalangeal, and carpometacarpal joints. These adjustments frequently result in marked reduction of pain in the index finger and at the base of the thumb.
Prevention of Secondary Complications
Calcium (500-600 mg/2x/day), vitamin D (400 or more units daily), and low-impact weight bearing exercise should be encouraged to maximize bone density.
Surveillance
DEXA should be repeated approximately every other year if bone loss is confirmed. Otherwise, routine population surveillance of bone density is all that is necessary.
Annual echocardiography is not necessary in those with a normal initial echocardiogram [Atzinger et al 2011]. In children and adolescents with a normal aortic root diameter, it is the author’s practice to repeat every two to three years until young adulthood (age ~25 years). If the aortic root diameter is increased or accelerating faster than body surface area, more frequent monitoring is appropriate. In adults with a normal aortic root diameter, no further monitoring is needed.
Agents/Circumstances to Avoid
Joint hyperextension may not need to be avoided. In a randomized controlled trial of physical therapy among 26 children and adolescents with joint hypermobility and knee pain, those allowed to exercise into hyperextension had similar improvement in pain score and better improvement in psychosocial score compared to those restricted to neutral joint position 47.
Resistance exercise can exacerbate joint instability and pain. In general, it is preferable to increase the number of repetitions of exercise rather than to increase the resistance.
High-impact activity increases the risk for acute subluxation/dislocation, chronic pain, and osteoarthritis. Some sports, such as tackle football, are therefore contraindicated. However, most sports and activities are acceptable with appropriate precautions.
Chiropractic adjustment is not strictly contraindicated, but must be performed cautiously to avoid iatrogenic subluxations or dislocations.
Joint hypermobility syndrome diet
Joint hypermobility syndrome or hypermobile Ehlers-Danlos syndrome is commonly associated with many gastrointestinal problems and symptoms, including acid reflux and irritable bowel syndrome, with many individuals reporting attempted symptom management through dietary adaptation. Despite this, there is minimal research looking into the effect of the diet in individuals with diagnosed hypermobile Ehlers-Danlos syndrome.
Good nutrition is essential for connective tissue repair and general healing. Certain nutrients are needed for the body to make collagen and support connective tissue function. Identifying specific nutrients needed for connective tissue function is important.
- Connelly E., Hakim A., Davenport H. S., & Simmonds J. V. (2015). A study exploring the prevalence of joint hypermobility syndrome in patients attending a musculoskeletal triage clinic. Physiotherapy Practice Research, 36(1), 43–53[↩]
- Tinkle B., Bird H. A., Grahame R., Lavallle M., Levy H. P., & Sillence D. (2009). The lack of clinical distinction between, the Ehlers-Danlos syndrome and the joint hypermobilty syndrome (a.k.a hypermobility syndrome). American Journal of Medical Genetics Part A, 149, 2368–2370. doi:10.1002/ajmg.a.33070[↩]
- Malfait F., Francomano C., Byers P., Belmont J., Berglund B., Black J., … Tinkle B. (2017). The 2017 international classification of the Ehlers–Danlos syndromes. American Journal of Medical Genetics Part C Seminars in Medical Genetics, 175C, 8–26. doi:10.1002/ajmg.c.31552[↩][↩]
- Simmonds J. V., & Keer R. J. (2007). Hypermobility and the hypermobility syndrome. Manual Therapy, 12, 298–309. doi:10.1016/j.math.2007.05.001[↩]
- Palmer S., Bailey S., Barker L., Barney L., & Elliott A. (2014). The effectiveness of therapeutic exercise for joint hypermobility syndrome: A systematic review. Physiotherapy, 100(3), 220–227. doi:10.1016/j.physio.2013.09.002[↩]
- Hakim A., Malfait F., De Paepe A., & Sahota A. (2010). The heritable disorders of connective tissue: Epidemiology, nosology and clinical features In Hakim A., Keer R., & Grahame R. (Eds.), Hypermobility, fibromyalgia and chronic pain. Philadelphia, PA: Elsevier[↩]
- Levy HP. Hypermobile Ehlers-Danlos Syndrome. 2004 Oct 22 [Updated 2018 Jun 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1279[↩][↩]
- Castori M, Camerota F, Celletti C, Grammatico P, Padua L. Ehlers-Danlos syndrome hypermobility type and the excess of affected females: possible mechanisms and perspectives. Am J Med Genet A. 2010b;152A:2406–8[↩]
- Castori M, Camerota F, Celletti C, Danese C, Santilli V, Saraceni VM, Grammatico P. Natural history and manifestations of the hypermobility type Ehlers-Danlos syndrome: a pilot study on 21 patients. Am J Med Genet A. 2010a;152A:556–64[↩][↩][↩][↩][↩][↩][↩]
- Branson JA, Kozlowska K, Kaczynski KJ, Roesler TA. Managing chronic pain in a young adolescent girl with Ehlers-Danlos syndrome. Harv Rev Psychiatry. 2011;19:259–70[↩][↩][↩]
- De Coster PJ, Martens LC, De Paepe A. Oral health in prevalent types of Ehlers-Danlos syndromes. J Oral Pathol Med. 2005;34:298–307[↩]
- Rombaut L, Malfait F, De Wandele I, Cools A, Thijs Y, De Paepe A, Calders P. Medication, surgery, and physiotherapy among patients with the hypermobility type of Ehlers-Danlos syndrome. Arch Phys Med Rehabil. 2011b;92:1106–12[↩][↩][↩][↩][↩][↩][↩]
- Dolan AL, Arden NK, Grahame R, Spector TD. Assessment of bone in Ehlers Danlos syndrome by ultrasound and densitometry. Ann Rheum Dis. 1998;57:630–3[↩]
- Gulbahar S, Sahin E, Baydar M, Bircan C, Kizil R, Manisali M, Akalin E, Peker O. Hypermobility syndrome increases the risk for low bone mass. Clin Rheumatol. 2006;25:511–4[↩]
- Carbone L, Tylavsky FA, Bush AJ, Koo W, Orwoll E, Cheng S. Bone density in Ehlers-Danlos syndrome. Osteoporos Int. 2000;11:388–92[↩]
- Voermans NC, Knoop H, Bleijenberg G, van Engelen BG. Pain in ehlers-danlos syndrome is common, severe, and associated with functional impairment. J Pain Symptom Manage. 2010a;40:370–8[↩][↩]
- Camerota F, Celletti C, Castori M, Grammatico P, Padua L. Neuropathic pain is a common feature in Ehlers-Danlos syndrome. J Pain Symptom Manage. 2011;41:e2–4[↩]
- De Paepe A, Malfait F. Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br J Haematol. 2004;127:491–500[↩][↩]
- Mast KJ, Nunes ME, Ruymann FB, Kerlin BA. Desmopressin responsiveness in children with Ehlers-Danlos syndrome associated bleeding symptoms. Br J Haematol. 2009;144:230–3[↩][↩]
- Mathias CJ, Low DA, Iodice V, Owens AP, Kirbis M, Grahame R. Postural tachycardia syndrome–current experience and concepts. Nat Rev Neurol. 2011;8:22–34[↩]
- Atzinger CL, Meyer RA, Khoury PR, Gao Z, Tinkle BT. Cross-sectional and longitudinal assessment of aortic root dilation and valvular anomalies in hypermobile and classic Ehlers-Danlos syndrome. J Pediatr. 2011;158:826–30.e1[↩][↩][↩]
- Kozanoglu E, Coskun Benlidayi I, Eker Akilli R, Tasal A. Is there any link between joint hypermobility and mitral valve prolapse in patients with fibromyalgia syndrome? Clin Rheumatol. 2016;35:1041–4[↩]
- De Felice C, Bianciardi G, Dileo L, Latini G, Parrini S. Abnormal oral vascular network geometric complexity in Ehlers-Danlos syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;98:429–34[↩]
- Castori M, Morlino S, Dordoni C, Celletti C, Camerota F, Ritelli M, Morrone A, Venturini M, Grammatico P, Colombi M. Gynecologic and obstetric implications of the joint hypermobility syndrome (a.k.a. Ehlers – Danlos syndrome hypermobility type) in 82 Italian patients. Am J Med Genet Part A. 2012;158A:2176–82[↩][↩][↩]
- Dutta I, Wilson H, Oteri O. Pregnancy and delivery in ehlers-danlos syndrome (hypermobility type): review of the literature. Obstet Gynecol Int. 2011;2011:306413[↩]
- Volkov N, Nisenblat V, Ohel G, Gonen R. Ehlers-Danlos syndrome: insights on obstetric aspects. Obstet Gynecol Surv. 2007;62:51–7[↩]
- Rombaut L, Malfait F, De Paepe A, Rimbaut S, Verbruggen G, De Wandele I, Calders P. Impairment and impact of pain in female patients with Ehlers-Danlos syndrome: a comparative study with fibromyalgia and rheumatoid arthritis. Arthritis Rheum. 2011a;63:1979–87[↩][↩][↩]
- Voermans NC, Knoop H, van de Kamp N, Hamel BC, Bleijenberg G, van Engelen BG. Fatigue is a frequent and clinically relevant problem in Ehlers-Danlos syndrome. Semin Arthritis Rheum. 2010b;40:267–74[↩]
- Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, Bravo JF. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31:1131–6[↩][↩][↩][↩]
- Gharbiya M, Moramarco A, Castori M, Parisi F, Celletti C, Marenco M, Mariani I, Grammatico P, Camerota F. Ocular features in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type: a clinical and in vivo confocal microscopy study. Am J Ophthalmol. 2012;154:593–600.e1[↩]
- Rombaut L, Malfait F, De Wandele I, Thijs Y, Palmans T, De Paepe A, Calders P. Balance, gait, falls, and fear of falling in women with the hypermobility type of Ehlers-Danlos syndrome. Arthritis Care Res (Hoboken). 2011c;63:1432–9[↩]
- Rombaut L, De Paepe A, Malfait F, Cools A, Calders P. Joint position sense and vibratory perception sense in patients with Ehlers-Danlos syndrome type III (hypermobility type). Clin Rheumatol. 2010a;29:289–95[↩]
- Rombaut L, Malfait F, Cools A, De Paepe A, Calders P. Musculoskeletal complaints, physical activity and health-related quality of life among patients with the Ehlers-Danlos syndrome hypermobility type. Disabil Rehabil. 2010b;32:1339–45[↩][↩][↩]
- Galli M, Cimolin V, Rigoldi C, Castori M, Celletti C, Albertini G, Camerota F. Gait strategy in patients with Ehlers-Danlos syndrome hypermobility type: a kinematic and kinetic evaluation using 3D gait analysis. Res Dev Disabil. 2011;32:1663–8[↩][↩][↩][↩]
- Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine. 2007;7:601–9[↩]
- Jacome DE. Headache in Ehlers-Danlos syndrome. Cephalalgia. 1999;19:791–6[↩]
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26[↩][↩][↩][↩][↩][↩][↩]
- Castori M, Tinkle B, Levy H, Grahame R, Malfait F, Hakim A. A framework for the classification of joint hypermobility and related conditions. Am J Med Genet Part C Semin Med Genet. 2017;175:148–57[↩][↩][↩]
- Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31–7[↩]
- Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis. 1973;32:413–8[↩][↩]
- Juul-Kristensen B, Schmedling K, Rombaut L, Lund H, Engelbert RHH. Measurement properties of clinical assessment methods for classifying generalized joint hypermobility—A systematic review. Am J Med Genet Part C Semin Med Genet. 2017;175:116–47[↩]
- Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: An adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57:163–166.[↩]
- Symoens S, Syx D, Malfait F, Callewaert B, De Backer J, Vanakker O, Coucke P, De Paepe A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat. 2012;33:1485–93[↩]
- Bristow J, Carey W, Egging D, Schalkwijk J. Tenascin-X, collagen, elastin, and the Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2005;139C:24–30[↩]
- Zweers MC, Dean WB, van Kuppevelt TH, Bristow J, Schalkwijk J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin Genet. 2005;67:330–4[↩]
- Chaudhury S, Gasinu S, Rodeo SA. Bilateral anterior and posterior glenohumeral stabilization using Achilles tendon allograft augmentation in a patient with Ehlers-Danlos syndrome. J Shoulder Elbow Surg. 2012;21:e1–5[↩]
- Pacey V, Tofts L, Adams RD, Munns CF, Nicholson LL. Exercise in children with joint hypermobility syndrome and knee pain: a randomised controlled trial comparing exercise into hypermobile versus neutral knee extension. Pediatr Rheumatol Online J. 2013;11:30–40.[↩]





{kind=link}