Contents
Renal tubular acidosis
Renal tubular acidosis (RTA) is a general term for when your kidneys cannot properly remove acid from your body. Renal tubular acidosis (RTA) occurs when your kidneys are unable to adequately reclaim filtered bicarbonate [HCO3–] (a base) or excrete sufficient hydrogen ions (H+) because of defects in tubular transport 1, 2. The acid level in your blood then becomes too high causing your blood pH to fall below 7.35, a condition called acidosis. Some acid in the blood is normal, but too much acid can disturb many bodily functions. There are three main types of renal tubular acidosis that are characterized by: 1) a normal anion gap metabolic acidosis; 2) abnormalities in renal bicarbonate (HCO3-) absorption or new renal bicarbonate (HCO3-) generation; 3) changes in renal ammonium (NH4+), calcium (Ca2+), potassium (K+) and water (H2O) homeostasis; and 4) extrarenal manifestations that provide etiologic diagnostic clues 3, 4, 5.
The kidneys contain nephrons, which are hair-sized structures that are the basic filtering units of the kidneys. Each nephron consists of a glomerulus and a renal tubule. The renal tubule reabsorbs electrolytes such as sodium, chloride and potassium back into the blood so that not too much electrolyte is lost through the urine. The kidneys, through the tubules, reclaim bicarbonate, an electrolyte that helps to maintain the acid-base balance in the body, and then excrete acid through the urine. Acid is produced as a byproduct from a normal diet.
There are 3 main types of renal tubular acidosis.
- Type 1 renal tubular acidosis or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules in the kidneys not being able to rid the body of the daily acid load. This results in an inability to lower urine pH regardless of the degree of acidosis or acidemia (acid level in the blood). Distal refers to being “distant” from the point of origin. In the nephron, it means the defect occurs away from the point where fluid enters the tubule. Distal renal tubular acidosis occurs because the kidneys fail to secrete acids into the urine.
- Type 2 renal tubular acidosis or proximal renal tubular acidosis, occurs when there is a problem in the beginning or proximal part of the tubules in the kidneys.
- Type 3 renal tubular acidosis is rarely used as a classification now because it is thought to be a combination of type 1 and type 2 renal tubular acidosis with features of both distal and proximal renal tubular acidosis.
- Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood.
Table 1. Summary of renal tubular acidosis classification, diagnostic characteristics, and treatment options
| Type 1 RTA (Distal renal tubular acidosis) | Type 2 RTA (Proximal renal tubular acidosis) | Type 4 RTA (Hyperkalemic renal tubular acidosis) | |
|---|---|---|---|
| Primary defect | Decreased distal acid excretion or increased H+ membrane permeability | Decreased proximal reabsorption of bicarbonate (HCO3–) | Reduced excretion of acid and K+ in the collecting duct |
| Symptoms | Polydipsia, polyuria, muscle weakness, nephrolithiasis, nephrocalcinosis, growth retardation or failure to thrive, rickets | Muscle weakness or paralysis (if severely hypokalemic), growth retardation in early childhood | Often asymptomatic, occasional muscle weakness of cardiac arrhythmia |
| Urine pH | > 5.3 | < 5.5 | < 5.5 |
| Serum bicarbonate (HCO3-) | 10–20 mmol/L | 16–20 mmol/L | 16–22 mmol/L |
| Serum K+ | Low (< 3.5 mmol/L) | Low (< 3.5 mmol/L) | High (5.5–6.5 mmol/L) |
| Serum anion gap | Normal | Normal | Normal |
| Diagnostic tests | Positive urinary anion gap after ammonium (NH4+) loading test | Fractional excretion of bicarbonate (HCO3–) > 15% or urine pH > 7.5 after bicarbonate (HCO3–) loading test Glycosuria, hypophosphatemia, and hypouricemia indicates Fanconi syndrome | Urinary K+ < 40 mmol/L or fractional K+ excretion < 20%, abnormal serum aldosterone, with near-normal renal function |
| Treatment | |||
| Diet and lifestyle modifications | Increased citrus fruit and fluid intake, restricted intake of Na+, oxalate, fructose, and animal protein, normal Ca2+ intake | Limit acid-based foods (animal source protein), increase alkali-based foods (fruits and vegetables) | Dietary K+ restriction, increase alkali-based foods, limit acid-based foods |
| Pharmacotherapy | Sodium bicarbonate (NaHCO3) or potassium bicarbonate (KHCO3) (1–2 mmol/kg/day), potassium chloride (KCl) or potassium citrate (K-citrate) (in patients with severe hypokalemia) | Alkali therapy (usually K-citrate 10–15 mmol/kg/day), fluids, electrolytes, vitamin D, phosphate, hydrochlorothiazide | Low-dose fludrocortisone, loop diuretics (if fludrocortisone not tolerated), oral NaHCO3 if serum bicarbonate (HCO3–) < 22 mmol/L, K+ binders (patiromer or sodium zirconium cyclosilicate) |
Untreated renal tubular acidosis can affect a child’s growth, cause kidney stones, and other problems like bone or kidney disease. Fortunately, treatment is very helpful at preventing these things from happening. So it’s important to start treatment as soon as renal tubular acidosis (RTA) is diagnosed.
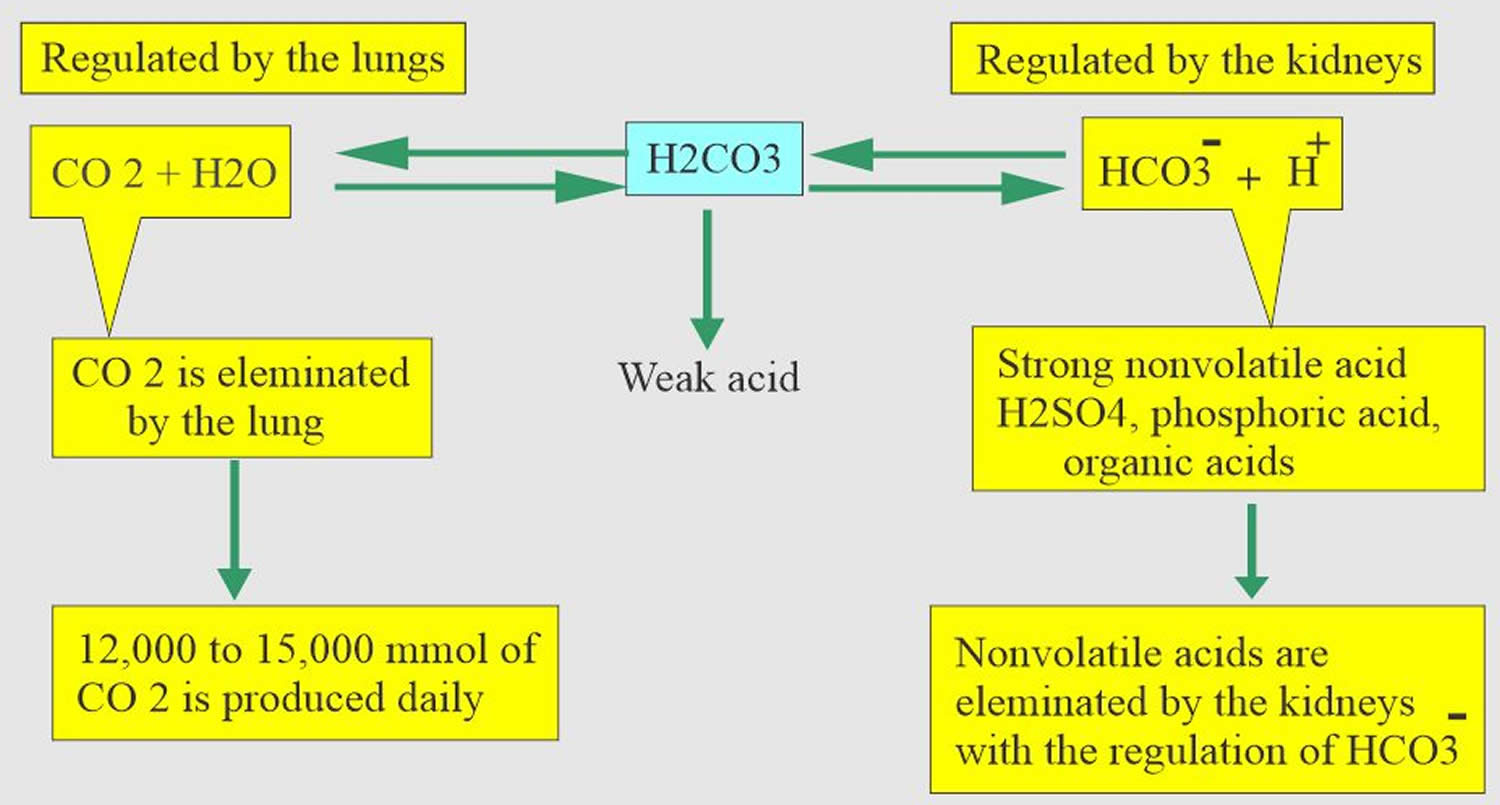
The pH is a number that shows how acidic or alkaline a substance is. A pH of less than 7 is acidic, and greater than 7 is alkaline. The pH of blood is about 7.4. A pH below 7.35 is called acidosis or acidemia. This is very unlikely to occur, as your body has multiple mechanisms for ensuring a very stable blood pH. Acidosis only becomes acidemia, where your blood pH is less than 7.35, when your body’s acid-base compensatory measures or buffering systems become overwhelmed. Your blood pH 7.4 is tightly regulated by your kidneys and respiratory system 6. The primary pH buffering system in the human body is the bicarbonate (HCO3–) and carbon dioxide (CO2). Bicarbonate (HCO3–) functions as an alkalotic substance. Carbon dioxide (CO2) functions as an acidic substance. Therefore, a decrease in serum bicarbonate (HCO3–) or an increase in CO2 (carbon dioxide) will make blood more acidic. The opposite is also true where an increase in bicarbonate (HCO3–) or a decrease in carbon dioxide (CO2) will make blood more alkaline. The carbon dioxide (CO2) levels are physiologically regulated by the pulmonary system through respiration, whereas the serum bicarbonate (HCO3–) levels are regulated through your kidneys by two mechanisms: bicarbonate [HCO3–] (a base) reclamation mainly in the proximal tubule and bicarbonate [HCO3–] (a base) generation predominantly in the distal nephron. Any excess acid is excreted in the urine. Your blood pH is not altered by your dietary intake.
For all types of RTA, drinking a solution of sodium bicarbonate or sodium citrate will lower the acid level in your blood. This alkali therapy can prevent kidney stones from forming and make your kidneys work more normally so kidney failure does not get worse.
Infants with type 1 renal tubular acidosis may need potassium supplements, but older children and adults rarely do because alkali therapy prevents the kidneys from excreting potassium into the urine.
Children with type 2 renal tubular acidosis will also drink an alkali solution (sodium bicarbonate or potassium citrate) to lower the acid level in their blood, prevent bone disorders and kidney stones, and grow normally. Some adults with type 2 RTA may need to take vitamin D supplements to help prevent bone problems.
People with type 4 renal tubular acidosis (hyperkalemic renal tubular acidosis) may need other medicines to lower the potassium levels in their blood.
If your RTA is caused by another condition, your health care professional will try to identify and treat it.
Figure 1. Acid-base disorders

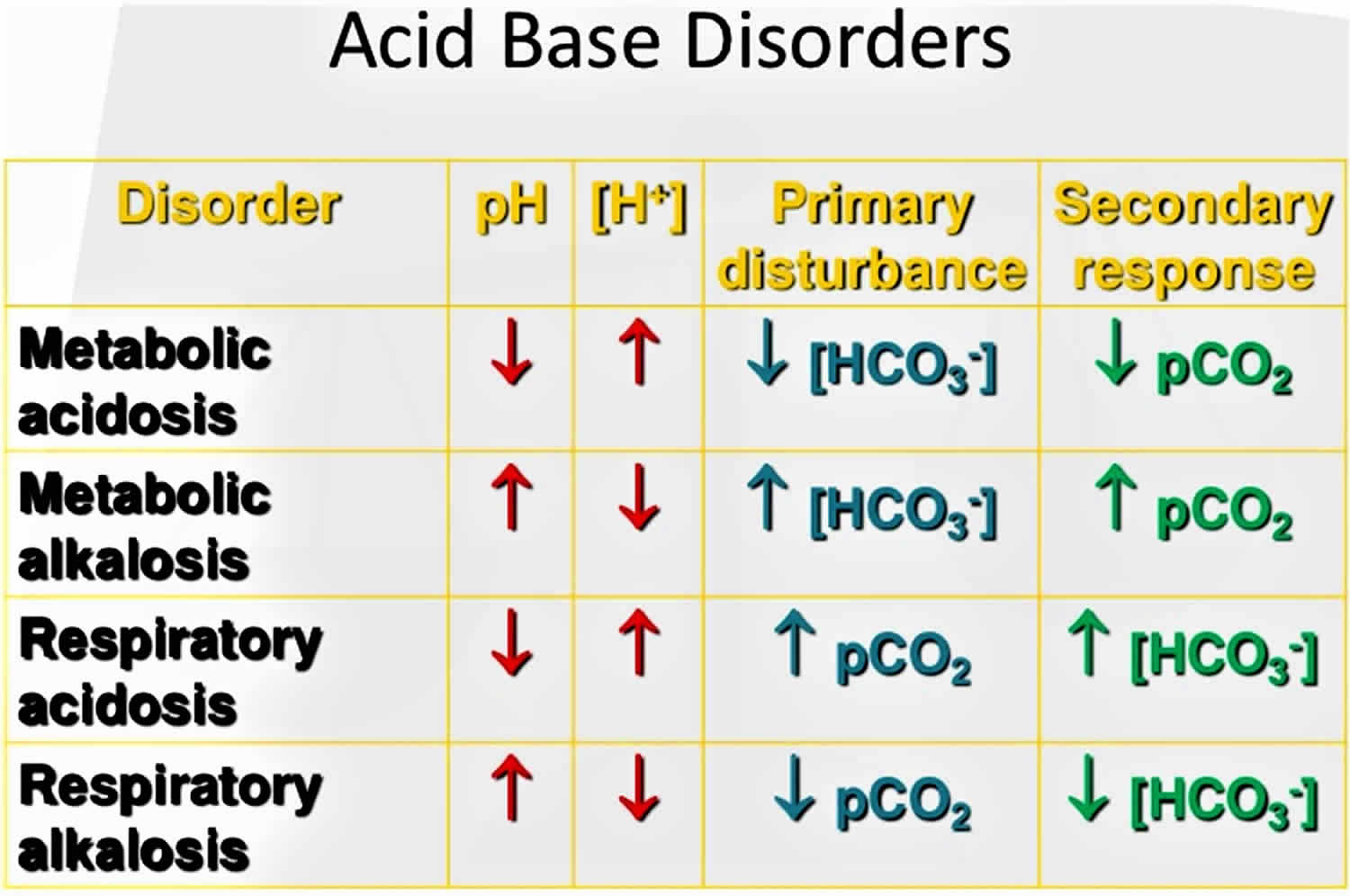
Figure 2. Normal anion gap levels
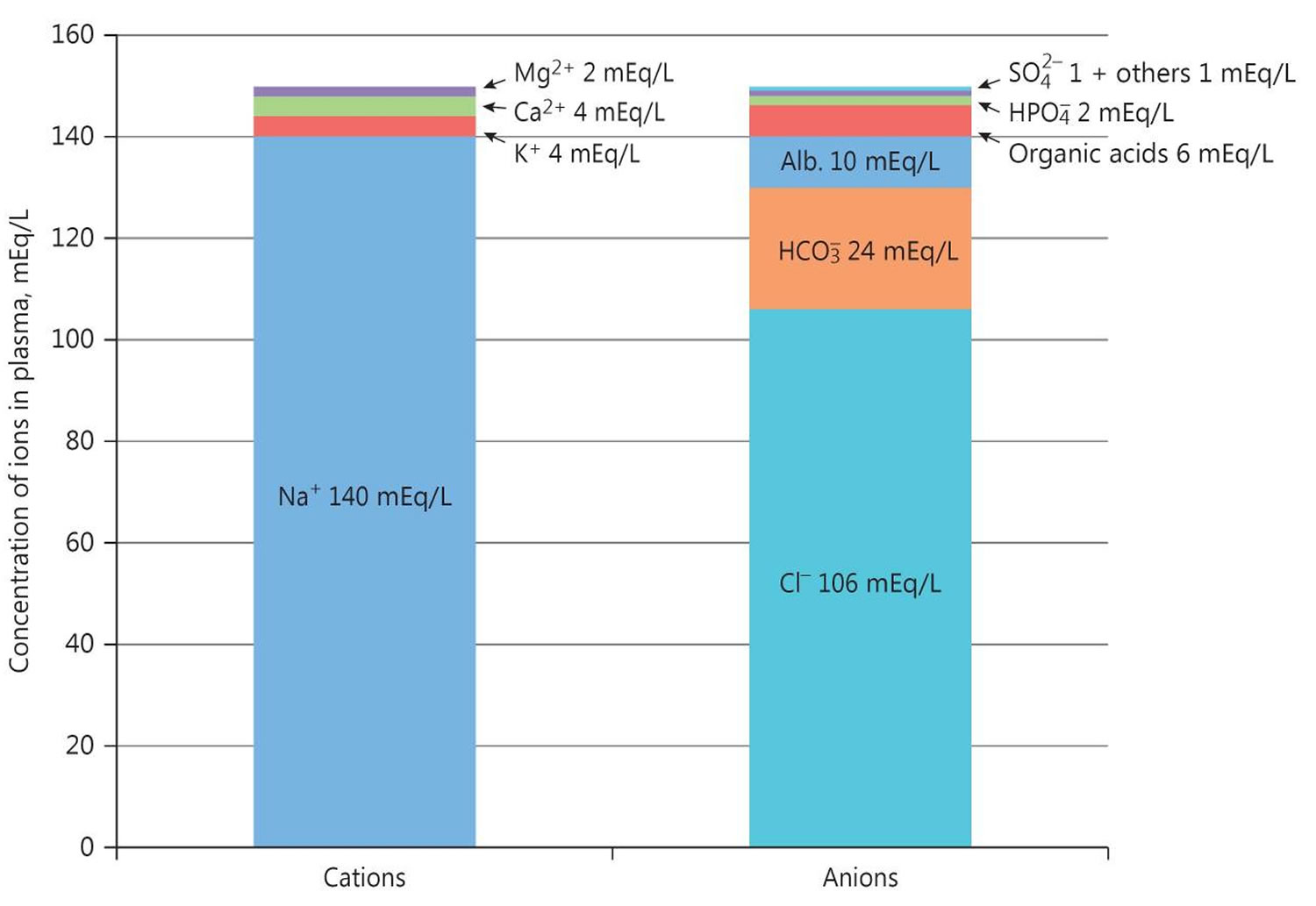
Figure 3. Henderson-Hasselbalch equation
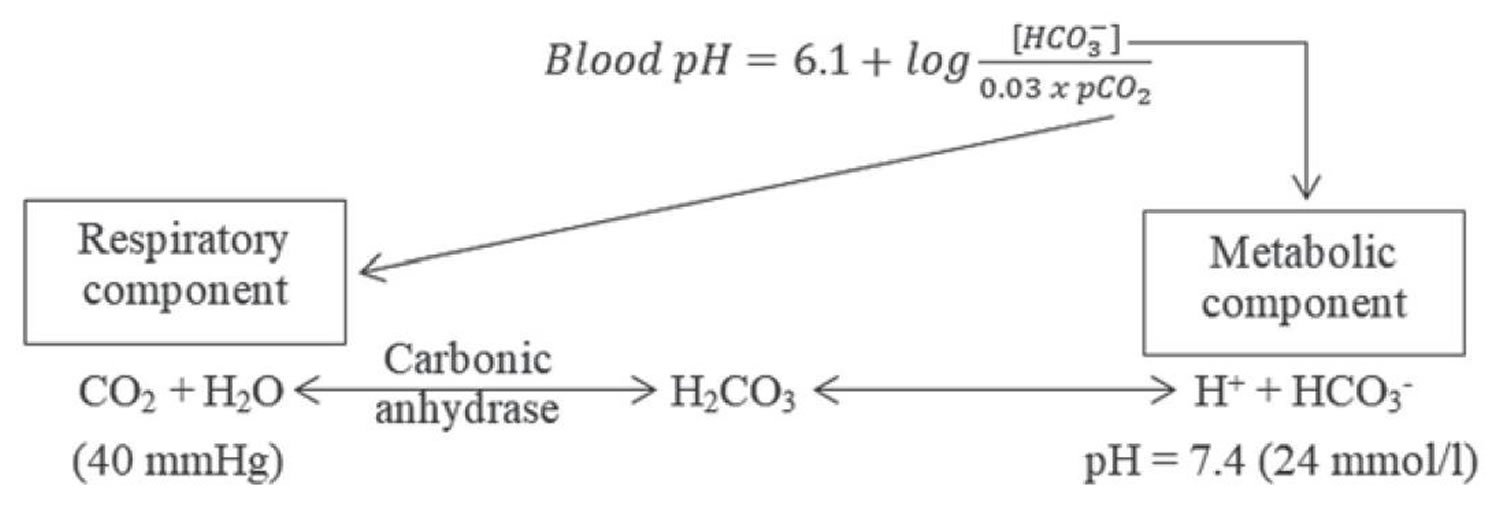
Figure 4. Acid-base buffering system
Who is more likely to have renal tubular acidosis?
You are more likely to have type 1 renal tubular acidosis (distal renal tubular acidosis) if you inherit specific genes from your parents or if you have certain autoimmune diseases such as Sjögren’s syndrome or lupus.
If you have Fanconi syndrome or are taking medicines to treat HIV or viral hepatitis, you are more likely to have type 2 RTA (proximal renal tubular acidosis). People who inherit genes for type 2 RTA from their parents may also have it. In adults, type 2 RTA (proximal renal tubular acidosis) can be a complication or side effect of multiple myeloma, exposure to toxins, or certain medications. In rare cases, type 2 RTA occurs in people who experience chronic rejection of a transplanted kidney.
If you have low levels of the hormone aldosterone, cannot urinate freely because of an obstruction, or had a kidney transplant, you are more likely to develop type 4 RTA ( (hyperkalemic renal tubular acidosis)). One in five people develop type 4 RTA if they experience rejection of a transplanted kidney or are taking immunosuppressive medications.
Kidneys
Your paired kidneys are reddish, kidney bean–shaped organs located just above the waist between the peritoneum and the posterior wall of the abdomen. Because their position is posterior to the peritoneum of the abdominal cavity, the organs are said to be retroperitoneal. The kidneys are located between the levels of the last thoracic vertebrae T12 and third lumbar (L3) vertebrae, a position where they are partially protected by ribs 11 and 12. If these lower ribs are fractured, they can puncture the kidneys and cause significant, even life-threatening damage. The right kidney is slightly lower than the left (see Figure 1) because the liver occupies considerable space on the right side superior to the kidney.
A typical adult kidney is 10–12 cm (4–5 in.) long, 5–7 cm (2–3 in.) wide, and 3 cm (1 in.) thick—about the size of a bar of bath soap—and weighs about 135–150 g (4.5–5 oz). The concave medial border of each kidney faces the vertebral column (see Figure 2). Near the center of the concave border is an indentation called the renal hilum, through which the ureter emerges from the kidney along with blood vessels, lymphatic vessels, and nerves.
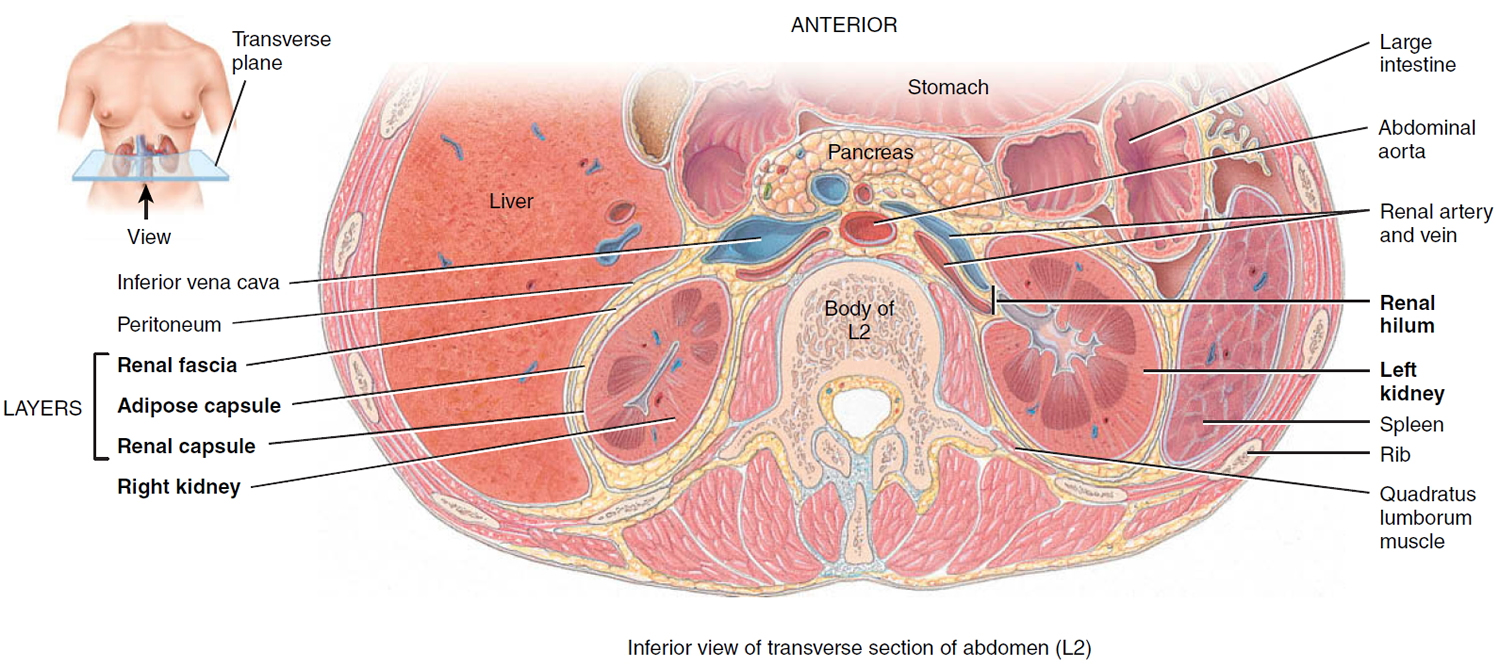
Three layers of tissue surround each kidney. The deep layer, the renal capsule, is a smooth, transparent sheet of dense irregular connective tissue that is continuous with the outer coat of the ureter. It serves as a barrier against trauma and helps maintain the shape of the kidney. The middle layer, the adipose capsule, is a mass of fatty tissue surrounding the renal capsule. It also protects the kidney from trauma and holds it firmly in place within the abdominal cavity. The superficial layer, the renal fascia, is another thin layer of dense irregular connective tissue that anchors the kidney to the surrounding structures and to the abdominal wall. On the anterior surface of the kidneys, the renal fascia is deep to the peritoneum.
Figure 5. Kidney location
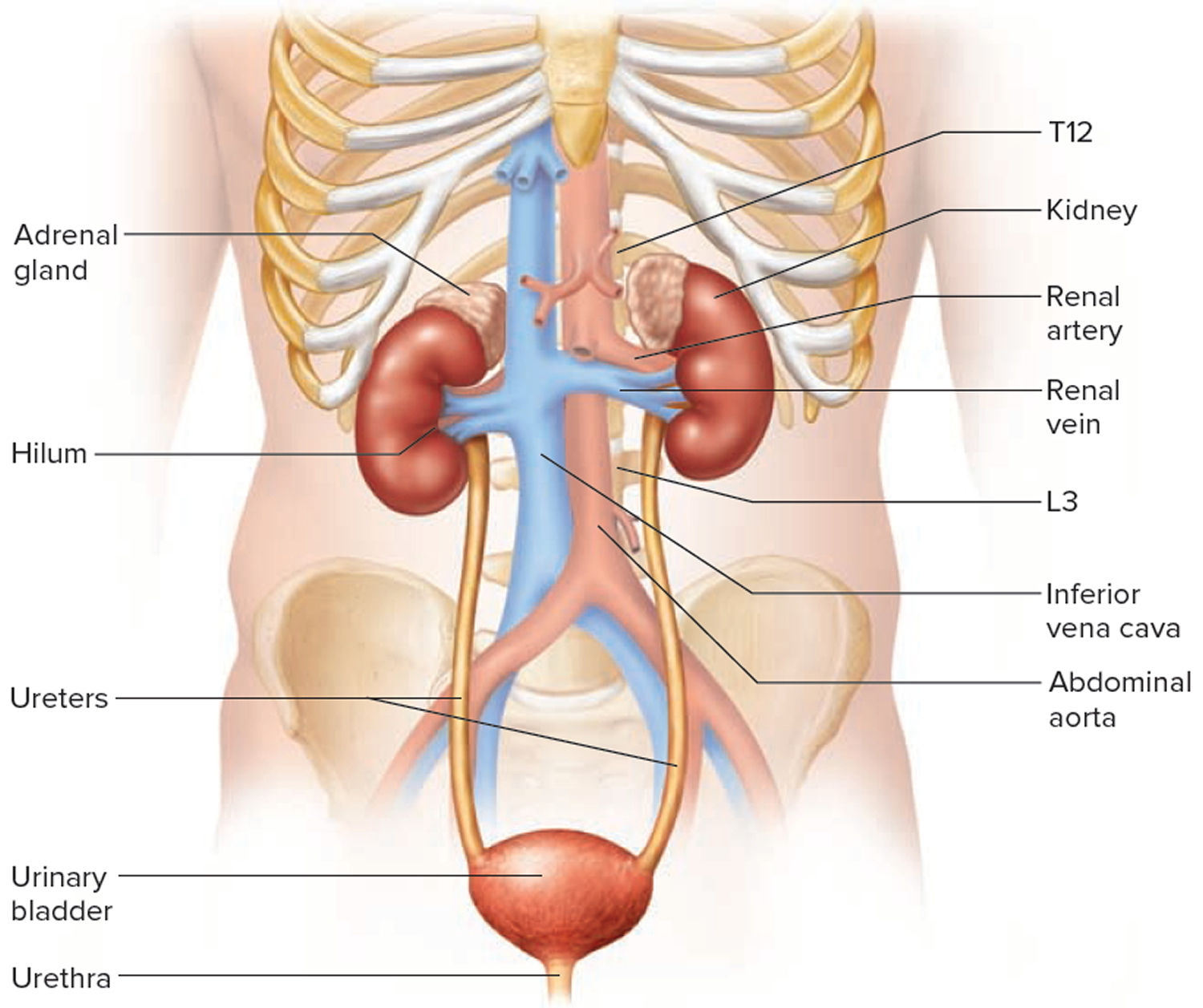
Figure 6. Kidney location (transverse section)
Kidney anatomy
A frontal section through the kidney reveals two distinct regions: a superficial, light red region called the renal cortex and a deep, darker reddish-brown inner region called the renal medulla (medulla = inner portion) (Figure 3). The renal medulla consists of several cone-shaped renal pyramids. The base (wider end) of each pyramid faces the renal cortex, and its apex (narrower end), called a renal papilla, points toward the renal hilum. The renal cortex is the smooth-textured area extending from the renal capsule to the bases of the renal pyramids and into the spaces between them. It is divided into an outer cortical zone and an inner juxtamedullary zone. Those portions of the renal cortex that extend between renal pyramids are called renal columns.
Together, the renal cortex and renal pyramids of the renal medulla constitute the parenchyma or functional portion of the kidney. Within the parenchyma are the functional units of the kidney—about 1 million microscopic structures called nephrons. Filtrate (filtered fluid) formed by the nephrons drains into large papillary ducts, which extend through the renal papillae of the pyramids. The papillary ducts drain into cuplike structures called minor and major calyces. Each kidney has 8 to 18 minor calyces and 2 or 3 major calyces. A minor calyx receives filtrate from the papillary ducts of one renal papilla and delivers it to a major calyx. Once the filtrate enters the calyces it becomes urine because no further reabsorption can occur. The reason for this is that the simple epithelium of the nephron and ducts becomes transitional epithelium in the calyces. From the major calyces, urine drains into a single large cavity called the renal pelvis and then out through the ureter to the urinary bladder.
The hilum expands into a cavity within the kidney called the renal sinus, which contains part of the renal pelvis, the calyces, and branches of the renal blood vessels and nerves. Adipose tissue helps stabilize the position of these structures in the renal sinus.
Figure 7. Kidney anatomy
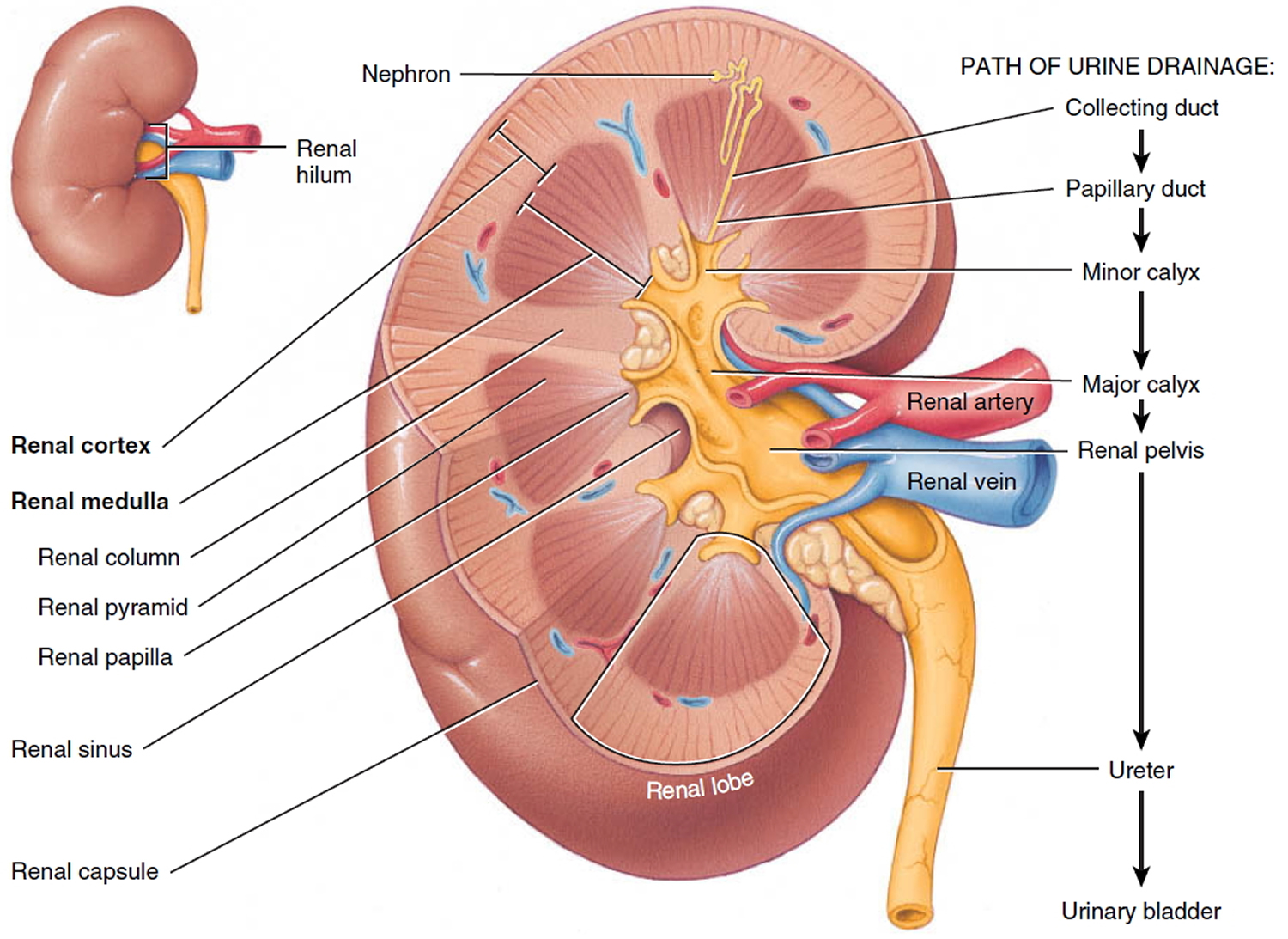
Figure 8. Kidney structure
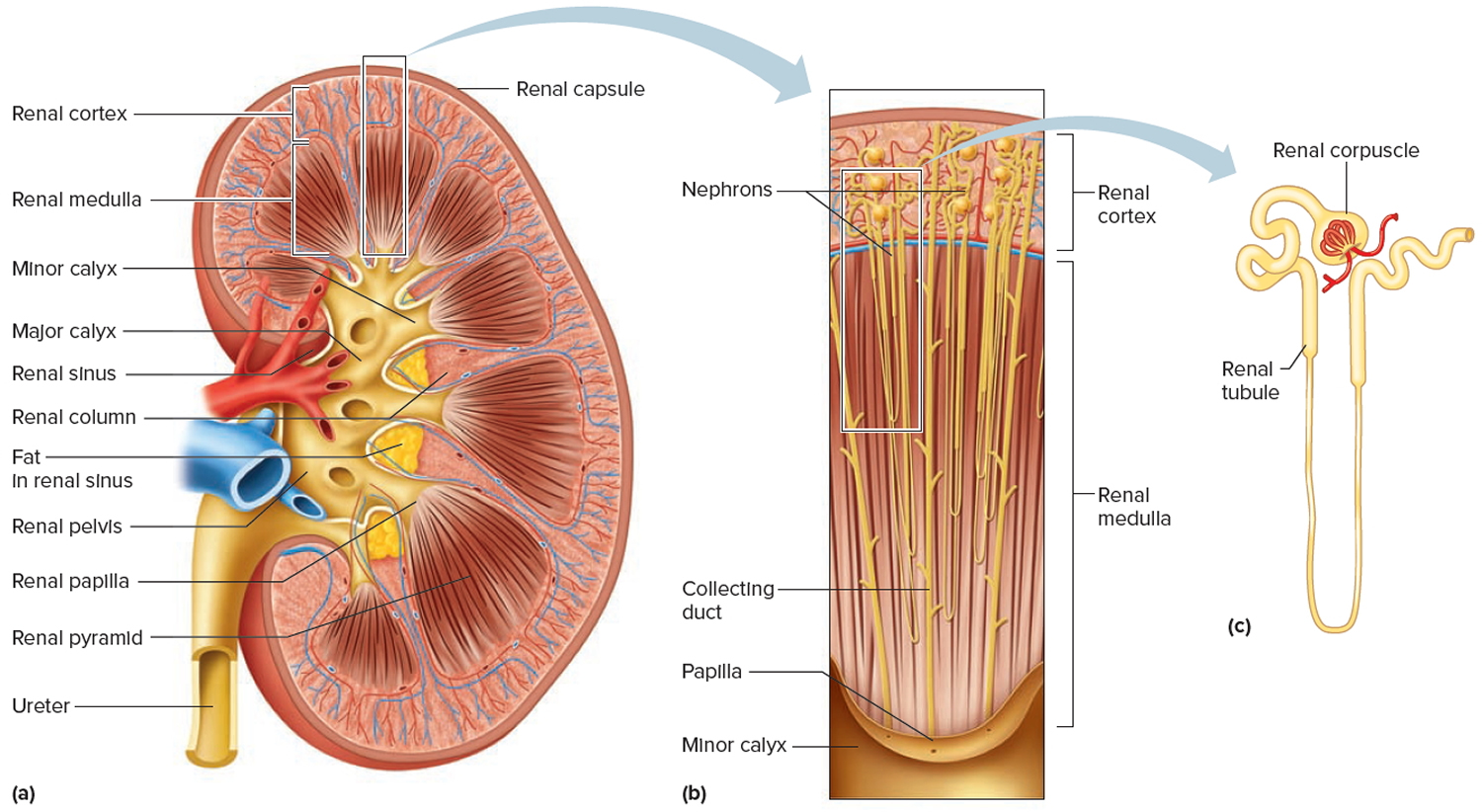
Figure 9. Microcirculation of the kidney
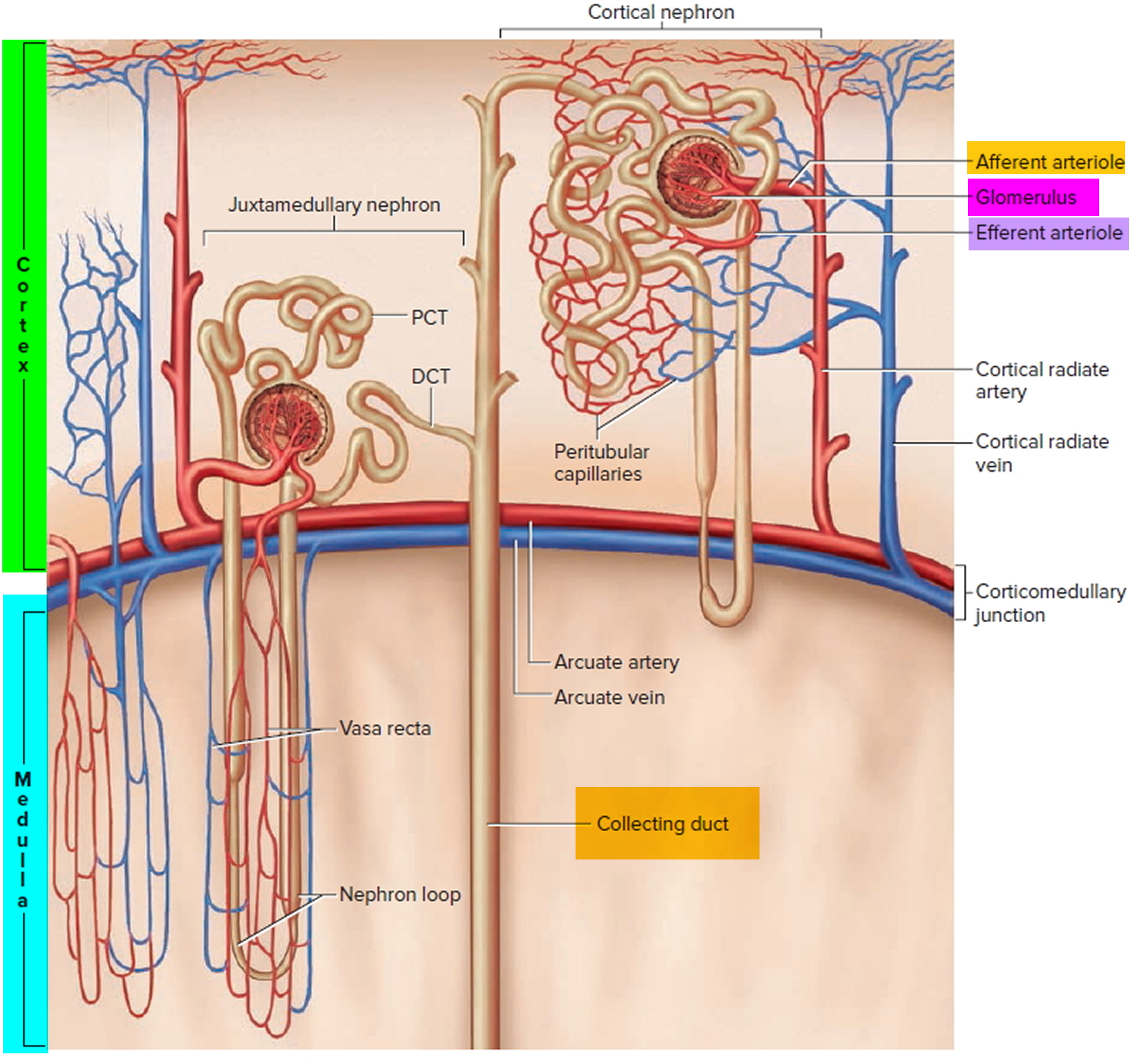
Note: DCT = distal convoluted tubule; PCT = proximal convoluted tubule
Kidney function
The primary function of the kidneys is to help maintain homeostasis by regulating the composition (including pH) and the volume of the extracellular fluid. The kidneys accomplish this by removing metabolic wastes from the blood and combining the wastes with excess water and electrolytes to form urine, which they then excrete.
Kidneys maintain homeostasis
- Regulatory function
- Control composition and volume of blood
- Maintain stable concentrations of inorganic anions such as sodium (Na), potassium (K), and calcium (Ca)
- Maintain acid-base balance
- Excretory function
- Produce urine
- Remove metabolic wastes including nitrogenous waste
Kidneys blood filtration and urine production
- Filtration: Glomeruli generate ultrafiltrate of the plasma.
- Reabsorption: Tubules selectively reabsorb substances from the ultrafiltrate.
- Secretion: Tubules secrete substances into the urine.
Examples:
- Potassium is reabsorbed from and secreted into the urine by the tubules.
- Sodium is generally reabsorbed by the tubules.
- Organic acids are secreted into the urine.
- Albumin is generally reabsorbed within the tubules.
Damaged kidneys allow albumin to cross the filtration barrier into the urine
- Increased glomerular permeability allows albumin (and other proteins) to cross the glomerulus into the urine.
- Higher levels of protein within the tubule may exacerbate kidney damage by exceeding tubules’ ability to reabsorb the proteins.
- An elevated urine albumin-to-creatinine ratio (UACR) is used to identify damaged kidneys. Urine albumin-to-creatinine ratio (UACR) results are used for screening, diagnosing, and treating chronic kidney disease. Forty percent of people are identified with chronic kidney disease on the basis of urine albumin alone.
The kidneys have several other important functions:
- Produce Erythropoietin which stimulates marrow production of red blood cells.
- Playing a role in the activation of vitamin D [activate 25(OH)D to 1,25 (OH)2D (active vitamin D)].
- Helping to maintain blood volume and blood pressure by secreting the enzyme Renin.
- Metabolize drugs and endogenous substances (e.g., insulin).
In patients with kidney failure:
- Kidneys cannot maintain homeostasis.
- Kidney failure is associated with fluid, electrolyte, and hormonal imbalances and metabolic abnormalities.
- End stage kidney failure means the patient is on dialysis or has a kidney transplant.
Acidosis compensation system
The primary ways the body deals with excessive acidity are through renal adaptations, respiration, and buffering with calcium from bone.
It is vital for life that pH does not waiver too far from normal, and the body will always attempt to return an abnormal pH towards normal when acid-base balance is disturbed. Compensation is the name given to this life-preserving process. To understand compensation, it is important to recall that pH is governed by the ratio bicarbonate [HCO3–] (a base)/arterial partial pressure of carbon dioxide (PaCO2) (an acid). So long as the ratio is normal, pH will be normal.
Normal body functions and metabolism generate large quantities of acids that must be neutralized and/or eliminated to maintain blood pH balance. In healthy individuals ingesting a typical Western diet, metabolism produces acid or alkali components 6, with a daily net acid production of approximately 1 mmol/kg of body weight per day 9. The amino acids lysine and arginine are metabolized to generate acids, while metabolism of glutamate and aspartate and organic anions, such as acetate and citrate, yield alkali 10. Metabolism of sulfur-containing amino acids (i.e., methionine and cysteine) yields sulfuric acid, while dietary phosphate yields phosphoric acid 10.
Most of the acid is carbonic acid, which is created from carbon dioxide (CO2) and water (H2O). Carbon dioxide (CO2) is produced as the body uses glucose (sugar) or fat for energy. In its normal state, the body maintains carbon dioxide (arterial partial pressure of carbon dioxide [PaCO2]) in a well-controlled range from 38 to 42 mm Hg by balancing its production and elimination. Lesser quantities of lactic acid, ketoacids, and other organic acids are also produced.
According to the Henderson-Hasselbalch equation (Figure 3), maintaining physiological pH depends on arterial partial pressure of carbon dioxide (PaCO2), which in turn depends on alveolar ventilation (hypoventilation causes acidosis and hyperventilation causes alkalosis). The kidneys participate in maintaining the stable pH by reabsorption of bicarbonate (3,600 mmol of bicarbonate is filtrated in glomeruli during 24 hour) and excretion of hydrogen ions from nonvolatile acids (including sulfur and phosphate) as titratable acidity (0.3 mmol hydrogen ions/kg/day) and in the form of ammonium ion (0.7 mmol hydrogen ions/kg/day) 11, 6.
The lungs and kidneys are the major organs involved in regulating blood pH. And to compensate for the metabolic acidosis, you increase your breathing rate (hyperventilation) to increase carbon dioxide (CO2) elimination 12, 13.
- The lungs flush acid out of your body by exhaling carbon dioxide (CO2). Raising and lowering the respiratory rate alters the amount of carbon dioxide (CO2) that is breathed out, and this can affect blood pH within minutes 14.
- The kidneys excrete acids in the urine, and they regulate the concentration of bicarbonate (HCO3–, a base) in blood. Acid-base changes due to increases or decreases in bicarbonate [HCO3–] concentration occur more slowly than changes in carbon dioxide (CO2), taking hours or days. Bicarbonate (HCO3–) reabsorption occurs in the kidneys in every part of the tubules. About 85–90% of the filtered bicarbonate is reabsorbed in the proximal tubules, 10% in the ascending arms of the Henle loop, 6% in the distal tubules, and 4% in the collecting tubules 11, 6.
Both of these processes are always at work, and they keep the blood pH in healthy people tightly controlled. The absolute quantities of acids or bases are less important than the balance between the two and its effect on blood pH.
Buffering systems that resist changes in pH also contribute to the regulation of acid and base concentrations. The main buffers in blood are hemoglobin (in red blood cells), plasma proteins, carbon dioxide (CO2), bicarbonate (HCO3–) and phosphates.
Carbon dioxide (CO2) plays a remarkable role in the human body mainly through pH regulation of the blood. The pH is the primary stimulus to initiate ventilation. In its normal state, the body maintains carbon dioxide (CO2) in a well-controlled range from 38 to 42 mm Hg by balancing its production and elimination. In a state of hypoventilation (breathing that is too shallow or too slow to meet the needs of the body), the body produces more carbon dioxide (CO2) than it can eliminate, causing a net retention of carbon dioxide (CO2). The increased carbon dioxide (CO2) is what leads to an increase in hydrogen ions (H+) and a slight increase in bicarbonate (HCO3–), as seen by a right shift in the following equilibrium reaction of carbon dioxide:
Carbon dioxide (CO2) + water (H2O) -> H2CO3 (carbonic acid) -> HCO3– + H+
The buffer system created by carbon dioxide consists of the following three molecules in equilibrium: carbon dioxide (CO2), H2CO3 (carbonic acid), and bicarbonate (HCO3–). When hydrogen ions (H+) is high, bicarbonate (HCO3–) buffers the low pH. When hydroxide (OH–) is high, H2CO3 (carbonic acid) buffers the high pH. In respiratory acidosis, the slight increase in bicarbonate (HCO3–) serves as a buffer for the increase in hydrogen ions (H+), which helps minimize the drop in pH. The increase in hydrogen ions inevitably causes the decrease in pH, which is the mechanism behind metabolic acidosis.
Respiration
The pulmonary system adjusts pH using carbon dioxide (CO2); upon expiration, carbon dioxide (CO2) is projected into the environment. Due to carbon dioxide (CO2) forming carbon dioxide (CO2) in the body when combining with water (H2O), the amount of carbon dioxide (CO2) expired can cause pH to increase or decrease. When the respiratory system is utilized to compensate for metabolic pH disturbances, the effect occurs in minutes to hours 15.
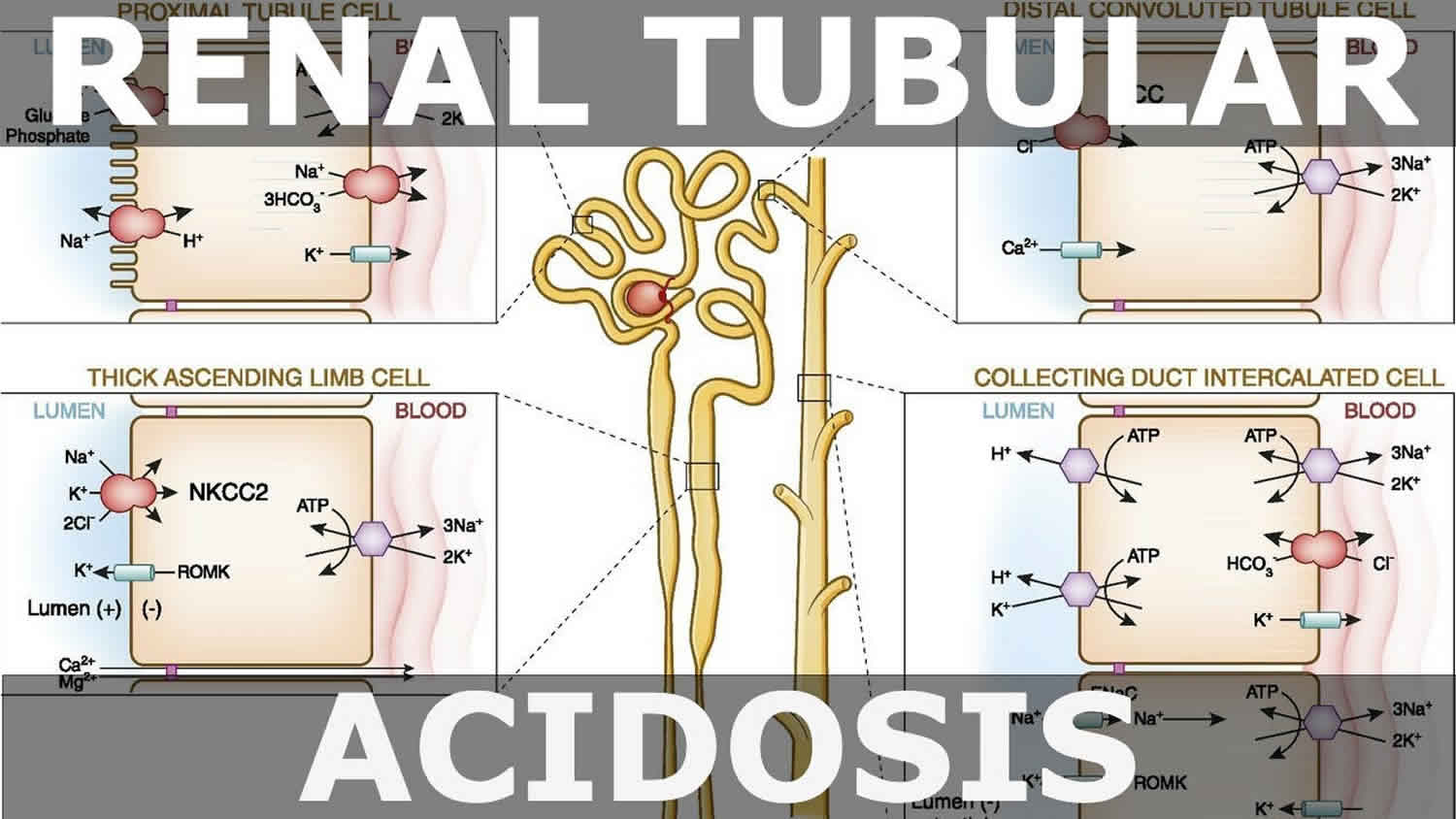
Renal adaptation
The kidneys contribute to the maintenance of acid–base homeostasis by reabsorbing bicarbonate (HCO3–) in the proximal tubule, as well as regenerating bicarbonate (HCO3–) in the cortical collecting duct 1, 6. Urine bicarbonate (HCO3–) reabsorption involves secretion of hydrogen ions (H+) into the proximal tubule through the Na+/H+ exchanger 3 (NHE3) and H+-adenosine triphosphatase (H+-ATPase) transporters, after which luminal bicarbonate (HCO3–) reacts with secreted hydrogen ion (H+) to form CO2 and water, a process that is catalyzed by membrane-bound carbonic anhydrase isoform 4 1, 6, 16. CO2 is then freely absorbed across the proximal tubular membrane, where cytosolic carbonic anhydrase II catalyzes its reaction with water to form carbonic acid that rapidly breaks down to HCO3–, which is reabsorbed into the blood through the Na+/HCO3– cotransporter (NBCe1), and H+, which is then secreted back into the tubular lumen [5]. HCO3– is also reabsorbed by a similar mechanism in the thick ascending limb of the loop of Henle (Fig. 1b) [6].
Most of the filtered bicarbonate (HCO3–) is reabsorbed in the proximal tubule. Virtually no bicarbonate (HCO3–) remains in the final urine 6.
Whether due to pathology or necessary compensation, the kidney excretes or reabsorbs these substances which affect pH. The nephron is the functional unit of the kidney. Blood vessels called glomeruli transport substances found in the blood to the renal tubules so that some can be filtered out while others are reabsorbed into the blood and recycled. This is true for hydrogen ions and bicarbonate. If bicarbonate (HCO3–) is reabsorbed and/or acid is secreted into the urine, the pH becomes more alkaline (pH increases). When bicarbonate (HCO3–) is not reabsorbed or acid is not excreted into the urine, pH becomes more acidic (pH decreases). The metabolic compensation from the renal system takes longer to occur, days rather than minutes or hours.
Figure 10. Kidneys control of plasma bicarbonate (HCO3–)
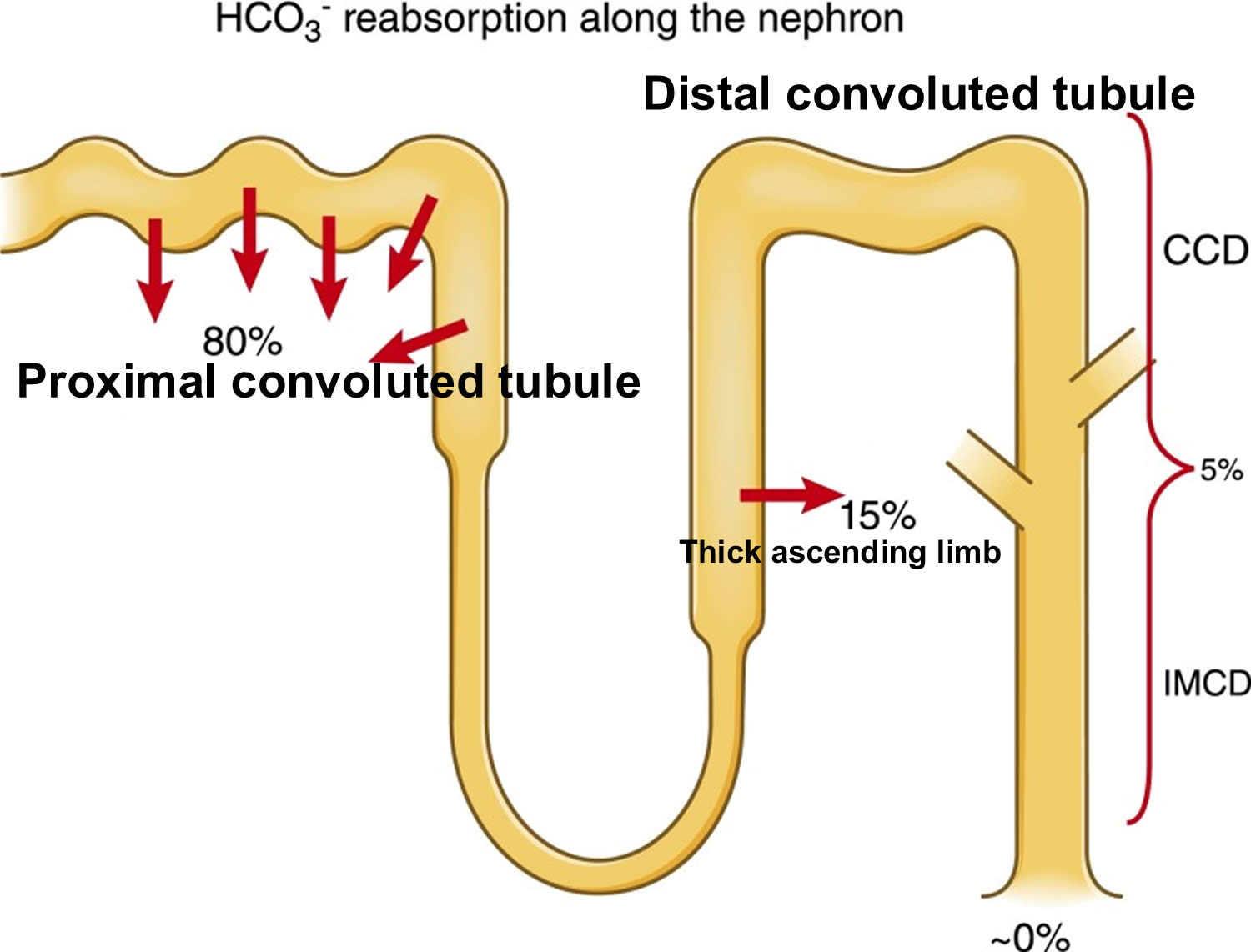
Abbreviations: CCD = cortical collecting duct; IMCD = inner medullary collecting duct
[Source 6 ]Figure 11. Schematic diagrams illustrating bicarbonate (HCO3–) reabsorption and regeneration in the kidney
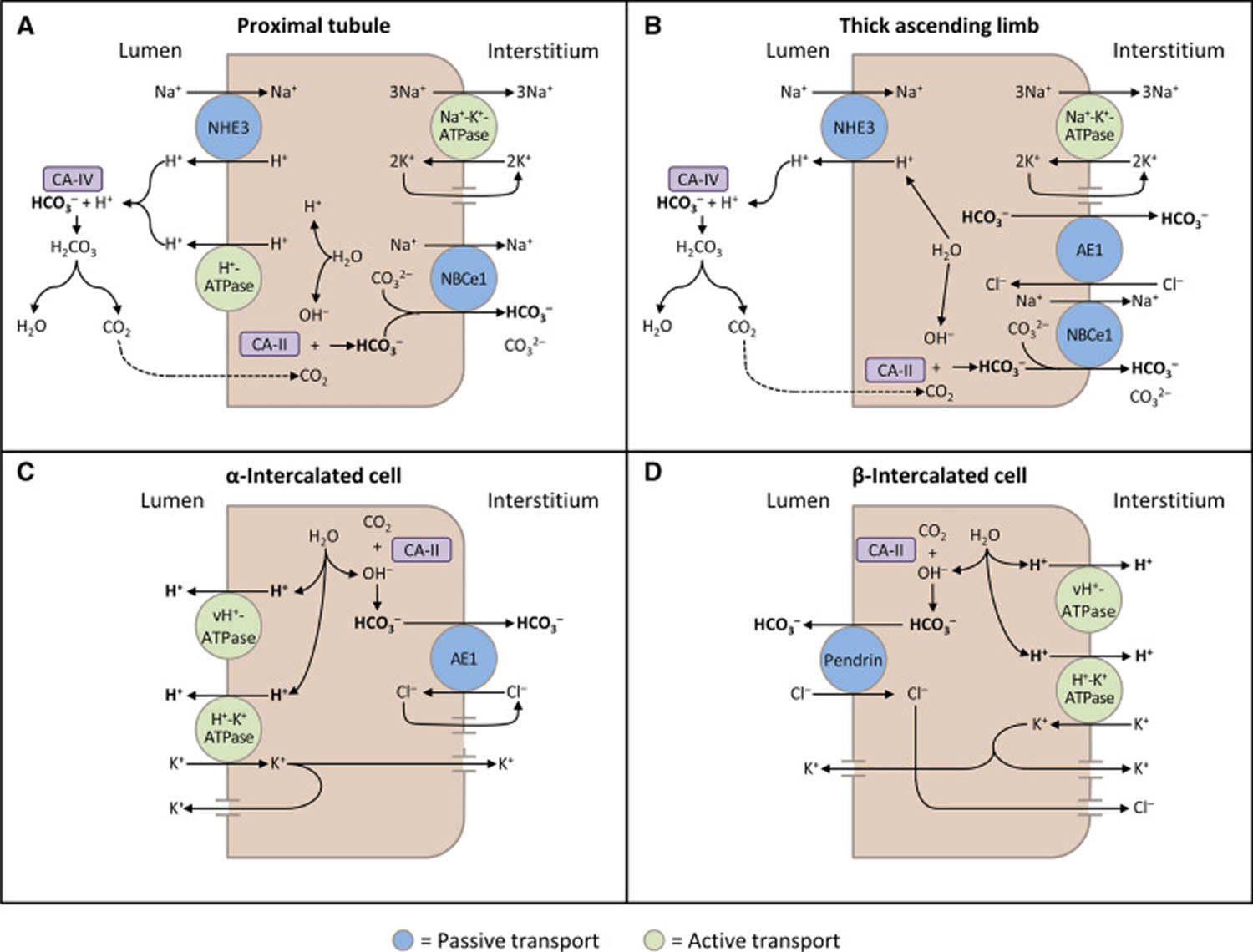
Footnotes: (A) Bicarbonate (HCO3–) reabsorption in the proximal tubule. Hydrogen ions (H+) are secreted into the lumen via apical sodium (Na+)/H+ exchanger 3 (NHE3) and H+-ATPase transporters. Apical carbonic anhydrase (CA) IV catalyzes the reaction between H+ and bicarbonate (HCO3–), which forms H2CO3 that rapidly dissociates to water and carbon dioxide (CO2). Carbon dioxide (CO2) diffuses back across the apical membrane, where CA-II catalyzes its reaction with intracellular hydroxide ions (OH–) to form H+ and bicarbonate (HCO3–). Bicarbonate (HCO3–) is transported across the basolateral membrane by the Na+/HCO3–/CCO32- cotransporter (NBCe1). (B) Bicarbonate (HCO3–) reabsorption in the thick ascending limb. As in the proximal tubule, H+ is secreted into the lumen via sodium (Na+)/H+ exchanger 3 (NHE3), where it reacts with bicarbonate (HCO3–) to release carbon dioxide (CO2) that diffuses back across the apical membrane. Bicarbonate (HCO3–) is transported across the basolateral membrane by the Na+/HCO3– cotransporter (NBCe1) and the kidney anion exchanger (AE1). (C) Hydrogen ions (H+) secretion by α-intercalated cells in cortical collecting duct (CCD). Hydrogen ion (H+) is secreted into the lumen by H+/K+-ATPase and vacuolar (v) H+-ATPase transporters on the apical membrane. Intracellular hydroxide ions (OH–) generated by hydrogen ion (H+) secretion reacts with carbon dioxide (CO2) via CA-II to form bicarbonate (HCO3–), which is removed by basolateral AE1. The resulting intracellular chloride (Cl–) exits via conductance channels in the basolateral membrane. Luminal K+ transported into the cell via H+/K+-ATPase can exit via channels in the apical or basolateral membrane, depending on K+ balance. (D) Bicarbonate (HCO3–) secretion by β-intercalated cells in cortical collecting duct (CCD). H+-ATPase transports H+ across the basolateral membrane. Intracellular hydroxide ion (OH–) generated by hydrogen ion (H+) secretion reacts with carbon dioxide (CO2) via CA-II to form bicarbonate (HCO3–), which is transported into the lumen by the apical chloride (Cl–)/bicarbonate (HCO3–) exchanger pendrin. Intracellular chloride (Cl–) exits via conductance channels in the basolateral membrane
[Source 16 ]The renal adaptations are extensive 17:
- Increased urinary excretion of sulfate, phosphate, urate, and chloride;
- Increased urinary excretion of calcium;
- Decreased urinary excretion of citrate;
- Increased urinary excretion of ammonium ions; and
- Kidney vasodilatation and increased glomerular filtration rate.
The kidneys mitigate but do not eliminate all the excess acidity. As the kidneys lose function with aging (when GFR is lower than 30 mL/min/1.73 m²), their ability to excrete acid becomes impaired, which may be another explanation for the loss of bone with aging 18. In fact, counteracting metabolic acidosis helps to preserve muscle mass and to improve bone metabolism 19, 20, 21.
Bone for acid buffering
The major reservoir of base is the skeleton (in the form of alkaline salts of calcium), which provides the buffer needed to maintain blood pH and plasma bicarbonate concentrations when renal and respiratory adaptations are inadequate. Acid-promoting diets are associated with increased urinary excretion of both calcium and bone matrix protein and decreased bone density 22. Neutralizing acid intake with diet or alkalinizing supplements decreases urine Calcium and bone matrix protein excretion. Also, to a much smaller degree, skeletal muscle can act as a buffer.
Other buffer systems
Other buffer systems in the human body include the phosphate buffer system, proteins, and hemoglobin. All of these contain bases which accept hydrogen ions which keep the pH from plummeting. The phosphate buffer system, while present globally, is important for the regulation of urine pH. Proteins assist with intracellular pH regulation. Red blood cells use the reaction above to help hemoglobin buffer; carbon dioxide can diffuse across red blood cells and combine with water. This alone would cause an increase in hydrogen ions; however, hemoglobin can bind hydrogen ions. Hemoglobin also can bind carbon dioxide without this reaction. This depends on the amount of oxygen that is bound to hemoglobin. This is called the Haldane effect and the Bohr effect. When hemoglobin is saturated with oxygen, it has a lower affinity for carbon dioxide (CO2) and hydrogen ions and is able to release it.
Renal tubular acidosis types
There are three main types of renal tubular acidosis that are characterized by: 1) a normal anion gap metabolic acidosis; 2) abnormalities in renal bicarbonate (HCO3-) absorption or new renal bicarbonate (HCO3-) generation; 3) changes in renal ammonium (NH4+), calcium (Ca2+), potassium (K+) and water (H2O) homeostasis; and 4) extrarenal manifestations that provide etiologic diagnostic clues 3, 4, 5.
- Type 1 renal tubular acidosis or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules.
- Type 2 renal tubular acidosis or proximal renal tubular acidosis, occurs when there is a problem in the beginning or proximal part of the tubules.
- Type 3 renal tubular acidosis is rarely used as a classification now because it is thought to be a combination of type 1 and type 2 renal tubular acidosis with features of both distal and proximal renal tubular acidosis.
- Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood.
Type 1 renal tubular acidosis
Type 1 renal tubular acidosis or distal renal tubular acidosis occurs when there is a problem at the distal part of the tubules. Distal renal tubular acidosis occurs because the kidneys fail to secrete acids into the urine and evidence of kidney K+ wasting with either normal or minimally reduced GFR and persistently alkaline urine pH > 5.3 23. Distal type 1 RTA or classic renal tubular acidosis usually referred to as distal renal tubular acidosis is characterized by a buildup of acids in the blood as a consequence of the distal tubules in the kidneys not being able to rid the body of the daily acid load. This results in an inability to lower urine pH regardless of the degree of acidemia (acid level in the blood). Distal refers to being “distant” from the point of origin. In the nephron, it means the defect occurs away from the point where fluid enters the tubule.
Untreated type 1 renal tubular acidosis (distal renal tubular acidosis) causes children to grow more slowly and adults to develop progressive kidney disease and bone diseases. Adults and children with untreated type 1 RTA may develop kidney stones because of abnormal calcium deposits that build up in the kidneys. These deposits prevent the kidneys from working properly.
Type 1 renal tubular acidosis causes
There are different forms of type 1 renal tubular acidosis (distal renal tubular acidosis). On the basis of the underlying defect, type 1 renal tubular acidosis (distal renal tubular acidosis) may be classified as hereditary (primary) or acquired (secondary) 1.
Type 1 RTA may be inherited, which is also known as primary distal renal tubular acidosis. Researchers have identified at least three different genes that may cause the inherited form of type 1 RTA 24. The inherited type 1 renal tubular acidosis (primary distal renal tubular acidosis) are caused by a variation (mutation) in one of at least three different genes; the SLC4A1 gene, the ATP6V0A4 gene, and the ATP6V1B1 gene. A variation in the SLC4A1 gene is usually inherited in an autosomal dominant pattern, and less often in an autosomal recessive pattern. Variations in the ATP6V0A4 and ATP6V1B1 genes are usually inherited in an autosomal recessive pattern 24. In some affected individuals (~20% cases), no variation in these three genes can be identified suggesting that other, as-yet-unidentified genes can play a role in primary distal renal tubular acidosis 25, 26.
The SLC4A1 gene contains instructions for producing (encoding) a protein called anion exchanger 1 or AE1. This protein helps negatively-charged atoms cross cell membranes; specifically, it helps exchange chlorine ions for bicarbonate ions. Bicarbonate is an electrolyte that helps maintain the acid-base balance in the body and is filtered by the kidneys; but, most of the bicarbonate is still retained in the blood and the urine contains very small amounts. Theanion exchanger 1 (AE1) protein is found in the membranes of kidney cells and red blood cells. The kidneys reclaim filtered bicarbonate and then release acid into the urine to be excreted from the body. Researchers have speculated that a variation in the SLC4A1 gene prevents enough functional anion exchanger 1 (AE1) protein from reaching the cell membranes of kidney and red blood cells. Ultimately, this prevents the kidneys from releasing acid into the urine. Acid then builds up in the blood and tissues of the body (metabolic acidosis). The reason why some people develop metabolic acidosis and others do not is not fully understood. In red blood cells, anion exchanger 1 (AE1) protein cannot reach the red cell membrane resulting in red blood cells that break down prematurely. Some altered AE1 protein can still reach the membranes of red blood cells because it is helped by another protein called glycophorin A. This is most likely why many people with a disease-causing variation in the SLC4A1 gene do not develop hemolytic anemia.
Variations in the SLCA41 gene are usually inherited in an autosomal dominant pattern, and, less often, in an autosomal recessive pattern. Disease-causing variations in the SLC4A1 gene can be inherited from a parent or it can occur as a new (sporadic or de novo) mutation, which means that the gene variation has occurred at the time of the formation of the egg or sperm for that child only, and no other family member will be affected. Affected individuals can then pass on the altered gene in an autosomal dominant pattern.
The ATP6V0A4 and the ATP6V1B1 genes encode specific proteins that are part of a protein complex called vacuolar H+-ATPase (V-ATPase). This protein complex acts as a proton pump that helps to move positively-charged atoms (protons) across cell membranes, and helps to regulate acid levels of cells and their surrounding areas. These proteins are commonly found in cells of the inner ear and within the nephron, which is the basic filtering unit of the kidneys. These proteins have a role in regulating the amount of acid removed from the blood to the urine, and in maintaining the proper acid balance within the ear. Variations in the ATP6V0A4 and the ATP6V1B1 genes are inherited in an autosomal recessive pattern.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
Disorders inherited in a recessive pattern occur when an individual inherits two variants in a gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Inherited type 1 RTA (primary distal renal tubular acidosis) affects females and males in equal numbers. The exact number of people who have this disorder is unknown. Rare disorders like primary distal renal tubular acidosis often go misdiagnosed or undiagnosed, making it difficult to determine their true frequency in the general population.
Acquired (secondary) forms of type 1 RTA are caused by autoimmune disorders including Sjögren’s syndrome or systemic lupus erythematosus. Autoimmune disorders are ones in which the body’s immune system mistakenly attacks healthy tissue. Sometimes, in certain autoimmune disorders the immune system attacks the distal portion of the renal tubules. Distal renal tubular acidosis can also occur in people with sickle cell anemia, chronic obstructive uropathy, Ehlers-Danlos syndrome, hypogammaglobulinemia, chronic liver disease, and following a kidney transplant. Acquired forms of type 1 renal tubular acidosis can also be caused by certain medications (e.g., lithium, amphotericin B, NSAIDs, lead, antivirals), including some used for pain and bipolar disorder, conditions causing high calcium in the urine, blocked urinary tract, or rejection of a transplanted kidney.
Diseases and conditions related to type 1 renal tubular acidosis:
- a hereditary form of deafness
- renal medullary cystic disease
- sickle cell disease
- sickle cell anemia
- Ehlers-Danlos syndrome
- urinary tract infections
- amyloidosis, a buildup of abnormal protein, called amyloid, in the tissues and organs
- Fabry disease, an abnormal buildup in the body of a certain type of fatty substance
- High level of calcium in the blood (hypercalcemia)
- Sjögren syndrome, an autoimmune disorder in which the glands that produce tears and saliva are destroyed
- Systemic lupus erythematosus, an autoimmune disease in which the body’s immune system mistakenly attacks healthy tissue
- Wilson disease, an inherited disorder in which there is too much copper in the body’s tissues
- Use of certain medicines, such as amphotericin B, lithium, and nonsteroidal anti-inflammatory drugs (NSAIDs)
Type 1 renal tubular acidosis signs and symptoms
Inherited type 1 RTA (primary distal renal tubular acidosis) is a highly variable disorder; this means that the disorder affects people differently. Some individuals may only have slightly elevated acid levels and no accompanying symptoms (asymptomatic). Some individuals living with type 1 renal tubular acidosis (primary distal renal tubular acidosis) may experience kidney stones and others may not. Generally, people with an autosomal dominant pattern of inheritance have milder symptoms and a later age of onset of symptoms than people with an autosomal recessive pattern of inheritance. However, this is not always true and sometimes more severe complications such as growth failure or rickets (bowing of the bones) can affect individuals with dominantly-inherited primary distal renal tubular acidosis.
Symptoms of type 1 renal tubular acidosis (distal renal tubular acidosis) include any of the following:
- Confusion or decreased alertness
- Fatigue
- Impaired growth in children
- Increased breathing rate
- Kidney stones
- Nephrocalcinosis (too much calcium deposited in the kidneys)
- Osteomalacia (softening of the bones)
- Muscle weakness
Other symptoms may include:
- Bone pain
- Decreased urine output
- Increased heart rate or irregular heartbeat
- Muscle cramps
- Pain in the back, flank, or abdomen
- Skeletal abnormalities
Inherited type 1 RTA (primary distal renal tubular acidosis) can cause severe complications in infants, especially if unrecognized and untreated. Affected infants can experience vomiting, dehydration, and poor growth that can result in being short for their age and gender (short stature). Additional symptoms can include excessive thirst (polydipsia), urinating frequently (polyuria), constipation, muscle weakness, and fatigue. Sometimes, affected individuals may have diminished reflexes. Many of these symptoms are related to metabolic acidosis, a serious and often life-threatening condition. Parents should seek prompt medical attention if a baby shows signs of metabolic acidosis. Some children develop rickets, which is a condition characterized by improper hardening (calcification) of the bones leading to softening and distortion/bowing of the bones and bone pain. If unrecognized and untreated, primary distal renal tubular acidosis usually causes too much calcium to build up in the kidneys (nephrocalcinosis), and the formation of kidney stones (nephrolithiasis). If untreated, nephrocalcinosis can progress to cause damage to the kidneys resulting in chronic kidney disease (CKD) and reduced kidney function.
In severe instances, if untreated, extreme muscle weakness (muscle paralysis), abnormal heartbeats (cardiac arrhythmia), and episodes of having difficulty breathing or stopping breathing (respiratory arrest) can develop. These symptoms are related to low levels of potassium in the blood (hypokalemia). Potassium is an important electrolyte for the health of nerve and muscles. The kidneys excrete excess potassium through the urine. However, in primary distal renal tubular acidosis the kidneys sometimes excrete too much potassium. Hypokalemia may also contribute to excessive urination (polyuria).
A subset of individuals with the autosomal recessive forms develop sensorineural hearing loss. Sensorineural hearing loss occurs when the nerves within the ear cannot properly send sensory input (sound) to the brain, and is not caused by problems with the ear itself. The degree and progression of sensorineural hearing loss can vary from one child to another, but often affects both ears (bilateral) and is usually severe.
Affected individuals with an autosomal dominant type 1 renal tubular acidosis (primary distal renal tubular acidosis) usually experience a milder form of the disorder with onset of symptoms in adolescence or adulthood. Affected adults may develop reduced bone mass (osteopenia) and abnormal softening of the bones (osteomalacia) and bone pain. Weakened bones may be prone to fracture. Some individuals may develop an abnormal increase in red blood cell mass (erythrocytosis). They may develop kidney stones or kidney issues as adolescents or adults if the disorder is unrecognized and untreated.
Occasionally, individuals with an inherited variation in the SLC4A1 gene have experienced the premature breakdown of red blood cells, which leads to low levels of circulating red blood cells (hemolytic anemia). The main function of red blood cells is to deliver oxygen throughout the body. People with hemolytic anemia can experience shortness of breath (dyspnea), lightheadedness, fatigue, weakness, pale skin color, and headaches.
Type 1 renal tubular acidosis treatment
The treatment of inherited type 1 RTA (primary distal renal tubular acidosis) may require the coordinated efforts of a team of specialists. A kidney doctor (nephrologist) who specialize in diagnosing and treating kidney disorders may be a critical member of the care team. A pediatric nephrologist specializes in kidney disorders in children. Physicians who specialize in diagnosing and treating skeletal disorders (orthopedists), an audiologist to monitor hearing and other healthcare professionals may need to systematically and comprehensively help guide treatment.
The goal is to restore normal acid level and electrolyte balance in the body. This will help correct bone disorders and reduce calcium buildup in the kidneys (nephrocalcinosis) and kidney stones.
The underlying cause of distal renal tubular acidosis should be corrected if it can be identified.
Individuals with inherited type 1 RTA (primary distal renal tubular acidosis) are treated with alkali therapy. Alkali are chemical compounds that neutralize acids. Medicines that may be prescribed include potassium citrate, sodium bicarbonate, and thiazide diuretics. A mixture of sodium and potassium salts in the form of sodium citrate or potassium citrate liquid solutions is usually recommended. These are alkaline medicines that help correct the acidic condition of the body. Sodium bicarbonate and potassium supplements may correct the loss of potassium and calcium. Liquid preparations, however, have poor palatability and acceptance among patients. In this case, sodium bicarbonate tablets are used instead or in addition to the drinking solution.
Alkali therapy usually leads to normal growth in children, and can improve other symptoms including lowering the tendency to develop calcium build up in the kidneys and calcium stones and reverse bone disease. Alkali therapy usually consists of drinking a solution of sodium bicarbonate (baking soda) or sodium citrate every day to counteract the acids produced from eating each day. The dose and specific type of alkali therapy depends upon the bicarbonate and potassium concentrations in the blood serum. Children generally require larger doses; these doses are adjusted as a child ages. Most individuals do not experience symptoms (asymptomatic) when properly treated, except for irreversible kidney or skeletal damage that has occurred before treatment was begun.
If low potassium levels persist (hypokalemia), then affected individuals may require treatment with alkylating potassium salt like potassium citrate. Potassium citrate (versus sodium citrate) may also be recommended when calcium stones are present because sodium can increase calcium stone formation. Citrate salts like potassium citrate correct low levels of citrate (hypocitraturia) and prevent calcium stone formation.
The focus of treatment in individuals with severe hypokalemia that causes paralysis or breathing problems (respiratory compromise) should be to correct the low potassium levels with an intravenous potassium chloride.
Children with autosomal recessive primary distal renal tubular acidosis should receive routine hearing assessments through childhood to detect hearing loss. Hearing loss does not usually respond to alkali therapy. Other treatments can include supplementation with vitamin D or oral calcium supplements to help reduce skeletal abnormalities such as rickets or osteomalacia.
Type 2 renal tubular acidosis
Type 2 renal tubular acidosis or proximal renal tubular acidosis occurs when there is a problem in the beginning or proximal part of the tubules. Isolated proximal type 2 RTA is characterized by defects in the reabsorption of filtered bicarbonate (HCO3–) in the proximal tubule without defects in the transport of other solutes (Table 1) 27, 28. The threshold serum concentration for bicarbonate (HCO3–) reabsorption (normally approximately 25 mmol/L) is reduced, leading to delivery of larger quantities of filtered bicarbonate (HCO3–) to the distal nephron (which has a low capacity for HCO3– reabsorption) and urinary bicarbonate (HCO3–) wastage 27, 28. Reductions in serum bicarbonate (HCO3–) cause acidosis; however, the urine pH remains alkaline because of the presence of urinary bicarbonate (HCO3–) 28. When serum bicarbonate (HCO3–) concentrations decrease below the lower threshold (16–20 mmol/L), a new steady state is reached, whereby all filtered bicarbonate (HCO3–) is reabsorbed. At this point, the urine contains no bicarbonate (HCO3–) and is maximally acidic 27.
A diagnosis of proximal renal tubular acidosis (type 2 renal tubular acidosis) may be suspected in patients who present with hypokalemia, normal anion gap metabolic acidosis, and acidic urine (pH < 5.5) 29. Other signs of proximal tubular dysfunction, including hypophosphatemia, hypouricemia, euglycemic glycosuria, and proteinuria, are also consistent with a proximal renal tubular acidosis (type 2 renal tubular acidosis) diagnosis and are reflective of generalized proximal tubular dysfunction 29. Fractional urinary bicarbonate (HCO3–) excretion, which is normally less than 5% of filtered bicarbonate (HCO3–), is usually approximately 15% in patients with proximal proximal renal tubular acidosis (type 2 renal tubular acidosis) 30. Isolated proximal RTA, that is decreased bicarbonate (HCO3–) reabsorption without abnormalities in the transport of other solutes, is rare 28.
The cause of isolated type 2 renal tubular acidosis or proximal renal tubular acidosis may be inherited (autosomal recessive mutations in SLC4A4, the gene encoding electrogenic NBCe1) or acquired (e.g., due to carbonic anhydrase inhibitors) 31, 28. Patients with isolated proximal renal tubular acidosis (type 2 RTA) typically present with growth retardation in early childhood 32.
In patients with proximal renal tubular acidosis (type 2 RTA), hypokalemia develops as a result of the loss of proximal bicarbonate (HCO3–) reabsorption 29. Increased urinary excretion of bicarbonate (HCO3–) causes a decrease in intravascular volume, which leads to renin-angiotensin-aldosterone system (RAAS) stimulation. Impaired proximal bicarbonate (HCO3–) reabsorption also causes increased distal Na+ delivery. The increase in renin-angiotensin-aldosterone system (RAAS) activity results in elevated aldosterone levels that, combined with elevated distal Na+ concentrations, cause an increase in urinary K+ excretion (i.e., K+ wasting) that leads to hypokalemia 29.
Proximal renal tubular acidosis (type 2 RTA) may occur as an isolated defect in bicarbonate (HCO3–) reabsorption, but more typically occurs in association with Fanconi syndrome, characterized by a widespread proximal tubular dysfunction resulting in the loss of phosphate, glucose, uric acid, amino acids, and low molecular weight proteins, as well as bicarbonate (HCO3–) 27, 28. Although proximal RTA is not associated with nephrolithiasis or nephrocalcinosis, patients with proximal renal tubular acidosis (type 2 RTA) and Fanconi syndrome may develop skeletal abnormalities, such as osteomalacia 33, 34, 35. Skeletal abnormalities result from impaired phosphate reabsorption, which causes chronic hypophosphatemia due to renal phosphate wasting 29, 33, 36. Active vitamin D deficiency may be present as a result of impaired conversion of 25(OH) vitamin D3 to 1,25 (OH)2 vitamin D3 in the proximal tubule 34. Osteopenia may also be evident as a result of acidosis-induced bone demineralization 37.
Proximal renal tubular acidosis (type 2 RTA) in association with Fanconi syndrome can also occur following exposure to some medications, including tenofovir 28, ifosfamide 38, sodium valproate 39 and topiramate 40. Topiramate is a carbonic anhydrase inhibitor that can cause simultaneous defects in both proximal and distal acidification mechanisms, presenting as type 3 RTA 41. Proximal renal tubular acidosis (type 2 RTA) can occur secondary to metabolic diseases, such as hereditary fructose intolerance and glycogen storage disease 42, 43.
The most common cause of acquired Fanconi syndrome in adults is multiple myeloma 44. The clinical manifestations of anemia, hypercalcemia, and bone pain suggest a diagnosis of multiple myeloma. The patient’s condition is also complicated by proximal renal tubular acidosis (type 2 RTA) , characterized by impaired proximal bicarbonate (HCO3–) reabsorption, hypokalemia, and variable urine pH. As the most common cause of proximal RTA in adults is multiple myeloma, this diagnosis should be excluded in all adults with proximal RTA unless another cause is found.
Untreated type 2 renal tubular acidosis may cause children to grow more slowly. In addition, type 2 RTA may cause rickets, a bone disease and dental disease in both children and adults 45, 46. A very low potassium level (hypokalemia) can develop during treatment of type 2 RTA with alkali 47.
Type 4 renal tubular acidosis
Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, is typically caused by selective aldosterone deficiency or intrinsic defects in the cortical collecting duct that lead to aldosterone resistance, which causes impaired distal H+ and K+ secretion 48, 29. Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood. Hyperkalemic renal tubular acidosis (type 4 RTA) commonly develops in patients with diabetes or interstitial nephritis and is characterized by a disturbance in distal nephron function, leading to a reduction in the excretion of H+ and K+ in the cortical collecting duct that results in hyperkalemic, hyperchloremic, normal anion gap acidosis (Table 1) 49, 50.
In type 4 renal tubular acidosis, the hypoaldosteronism causes reduced principal cell Na+ reabsorption and a decrease in transepithelial voltage in the cortical collecting duct, which results in diminished excretion of H+ and K+. Increased serum K+ concentrations inhibit ammonium (NH4) synthesis in the proximal tubule, further reducing the kidney’s capacity to excrete acid. Ammonium (NH4) availability is critical for normal distal H+ secretion. Lack of adequate ammonium (NH4) buffer results in a drop in urine pH creating a steep pH gradient, which impedes distal H+ secretion. Aldosterone deficiency can cause Na+ wasting, leading to decreased plasma volume that stimulates proximal Na+ reabsorption. The reduction in distal Na+ delivery secondarily inhibits secretion of K+ and H+ in the distal nephron 48, 29. Hyperkalemia reduces ammonium (NH4) production in the proximal tubule and inhibits ammonium (NH4) transport in the thick ascending limb as the high luminal K+ concentrations compete with NH4+ for Na+/K+/2Cl– cotransporters and apical K+ channels 51. Reductions in urine ammonium (NH4) can be detected by a positive urine anion gap and a failure to increase the urine osmolal gap.
Patients with hyperkalemic renal tubular acidosis or type 4 RTA are often asymptomatic and are typically diagnosed during routine laboratory analyses. When symptomatic, high levels of potassium in the blood (hyperkalemia) can lead to muscle weakness or heart problems, such as slow or irregular heartbeats and cardiac arrest 52, 48, 29. Hyperkalemic renal tubular acidosis may be diagnosed by the presence of hyperkalemia, normal anion gap metabolic acidosis, and abnormal serum aldosterone levels, although the glomerular filtration rate (GFR) may be near-normal or only moderately reduced (45 to less than 60 mL/min/1.73 m²) 29, 53.
Patients with hyperkalemic renal tubular acidosis or type 4 RTA usually have diabetes with mild-to-moderate decreases in glomerular filtration rate (GFR), a serum bicarbonate [HCO3–] concentration of 18–22 mmol/L, and a serum K+ concentration of 5.5–6.5 mmol/L 29. Other causes of hyperkalemia and normal anion gap acidosis include selective aldosterone deficiency, or defects in K+ or H+ secretion resulting from aldosterone resistance in the kidney (sickle cell nephropathy) 29, 53. Urinary obstruction can give rise to type 4 RTA [52, 53]. In patients with obstructive uropathy and hyperkalemic metabolic acidosis, those who are unable to acidify their urine pH to less than 5.5 are thought to have a voltage-dependent defect in Na+ transport in the distal nephron (i.e., voltage-dependent distal RTA) 48, 54.
Patients with chronic kidney disease (CKD) are also at risk of developing hyperkalemic renal tubular acidosis or type 4 RTA because of the progressive loss of functional kidney mass 29. Initially, there is an adaptive increase in NH4+ production and acid secretion [56]; however, as kidney impairment progresses (GFR 30–40 mL/min/1.73 m²), this adaptive increase is unable to maintain sufficient net H+ excretion to keep pace with endogenous acid production. Patients develop a hyperchloremic normal gap acidosis, often referred to as RTA of kidney insufficiency 29. In patients with more advanced chronic kidney disease (CKD) (GFR < 15–20 mL/min/1.73 m²), the ability to excrete phosphate and other anions is reduced and a high anion gap metabolic acidosis develops; the acidosis at this stage is termed uremic acidosis. During this transition, patients frequently manifest features of both a normal and increased anion gap metabolic acidosis 29. Furthermore, patients with stage 3 to 5 chronic kidney disease (CKD) and hyperkalemia commonly develop metabolic acidosis as progressive kidney impairment leads to compromised maintenance of electrolyte and acid–base balance 55.
Pseudohypoaldosteronism is a genetic condition associated with hyperkalemic, hyperchloremic metabolic acidosis with normal kidney function and either normal or high aldosterone levels 48. Type 1 pseudohypoaldosteronism may be caused by mutations in genes encoding the mineralocorticoid receptor or epithelial Na+ channels (ENaC) 48, whereas type 2 pseudohypoaldosteronism is caused by mutations in genes encoding the with-no-lysine (WNK) family of kinases 56. Mutations in epithelial Na+ channels (ENaC) cause Na+ wasting and are characterized by increased aldosterone levels, while WNK mutations give rise to Na+ retention and either normal or low circulating aldosterone levels 56, 48.
Hyperkalemic type 4 RTA may also be caused by medications, including K+-sparing diuretics (e.g., spironolactone, eplerenone, and amiloride), antibiotics (e.g., trimethoprim and pentamidine), nonsteroidal anti-inflammatory drugs (NSAIDs), including cyclooxygenase-2 (COX-2) inhibitors, ACE inhibitors, and heparin or low molecular weight heparin 1, 57. These agents cause hyperkalemic type 4 RTA by reducing aldosterone synthesis (ACE inhibitors), release (NSAIDs and heparin), or receptor binding (spironolactone, eplerenone), or through inhibition of ENaC (amiloride, trimethoprim, and pentamidine) 1. Hyperkalemic renal tubular acidosis or type 4 RTA may also be caused by immunosuppressant therapy with calcineurin inhibitors (e.g., tacrolimus and ciclosporin) 58, 59, 60. Calcineurin inhibitors block K+ and H+ secretion from the collecting duct through inhibition of basolateral Na+/K+-ATPase and Na+/K+/2Cl– cotransporter activity 60, 61. Calcineurin inhibitors also suppress expression of mineralocorticoid receptors, resulting in aldosterone resistance 62.
Renal tubular acidosis causes
Type 1 renal tubular acidosis causes
Type 1 renal tubular acidosis or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules in the kidneys not being able to rid the body of the daily acid load. This results in an inability to lower urine pH regardless of the degree of acidosis or acidemia (acid level in the blood). Distal refers to being “distant” from the point of origin. In the nephron, it means the defect occurs away from the point where fluid enters the tubule. Distal renal tubular acidosis occurs because the kidneys fail to secrete acids into the urine. On the basis of the underlying defect, type 1 renal tubular acidosis (distal renal tubular acidosis) may be classified as hereditary (primary) or acquired (secondary) 1.
Type 1 RTA may be inherited, which is also known as primary distal renal tubular acidosis. Researchers have identified at least three different genes that may cause the inherited form of type 1 RTA 24. The inherited type 1 renal tubular acidosis (primary distal renal tubular acidosis) are caused by a variation (mutation) in one of at least three different genes; the SLC4A1 gene, the ATP6V0A4 gene, and the ATP6V1B1 gene. A variation in the SLC4A1 gene is usually inherited in an autosomal dominant pattern, and less often in an autosomal recessive pattern. Variations in the ATP6V0A4 and ATP6V1B1 genes are usually inherited in an autosomal recessive pattern 24. In some affected individuals (~20% cases), no variation in these three genes can be identified suggesting that other, as-yet-unidentified genes can play a role in primary distal renal tubular acidosis 25, 26.
The SLC4A1 gene contains instructions for producing (encoding) a protein called anion exchanger 1 or AE1. This protein helps negatively-charged atoms cross cell membranes; specifically, it helps exchange chlorine ions for bicarbonate ions. Bicarbonate is an electrolyte that helps maintain the acid-base balance in the body and is filtered by the kidneys; but, most of the bicarbonate is still retained in the blood and the urine contains very small amounts. Theanion exchanger 1 (AE1) protein is found in the membranes of kidney cells and red blood cells. The kidneys reclaim filtered bicarbonate and then release acid into the urine to be excreted from the body. Researchers have speculated that a variation in the SLC4A1 gene prevents enough functional anion exchanger 1 (AE1) protein from reaching the cell membranes of kidney and red blood cells. Ultimately, this prevents the kidneys from releasing acid into the urine. Acid then builds up in the blood and tissues of the body (metabolic acidosis). The reason why some people develop metabolic acidosis and others do not is not fully understood. In red blood cells, anion exchanger 1 (AE1) protein cannot reach the red cell membrane resulting in red blood cells that break down prematurely. Some altered AE1 protein can still reach the membranes of red blood cells because it is helped by another protein called glycophorin A. This is most likely why many people with a disease-causing variation in the SLC4A1 gene do not develop hemolytic anemia.
Variations in the SLCA41 gene are usually inherited in an autosomal dominant pattern, and, less often, in an autosomal recessive pattern. Disease-causing variations in the SLC4A1 gene can be inherited from a parent or it can occur as a new (sporadic or de novo) mutation, which means that the gene variation has occurred at the time of the formation of the egg or sperm for that child only, and no other family member will be affected. Affected individuals can then pass on the altered gene in an autosomal dominant pattern.
The ATP6V0A4 and the ATP6V1B1 genes encode specific proteins that are part of a protein complex called vacuolar H+-ATPase (V-ATPase). This protein complex acts as a proton pump that helps to move positively-charged atoms (protons) across cell membranes, and helps to regulate acid levels of cells and their surrounding areas. These proteins are commonly found in cells of the inner ear and within the nephron, which is the basic filtering unit of the kidneys. These proteins have a role in regulating the amount of acid removed from the blood to the urine, and in maintaining the proper acid balance within the ear. Variations in the ATP6V0A4 and the ATP6V1B1 genes are inherited in an autosomal recessive pattern.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
Disorders inherited in a recessive pattern occur when an individual inherits two variants in a gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Inherited type 1 RTA (primary distal renal tubular acidosis) affects females and males in equal numbers. The exact number of people who have this disorder is unknown. Rare disorders like primary distal renal tubular acidosis often go misdiagnosed or undiagnosed, making it difficult to determine their true frequency in the general population.
Acquired (secondary) forms of type 1 RTA are caused by autoimmune disorders including Sjögren’s syndrome or systemic lupus erythematosus. Autoimmune disorders are ones in which the body’s immune system mistakenly attacks healthy tissue. Sometimes, in certain autoimmune disorders the immune system attacks the distal portion of the renal tubules. Distal renal tubular acidosis can also occur in people with sickle cell anemia, chronic obstructive uropathy, Ehlers-Danlos syndrome, hypogammaglobulinemia, chronic liver disease, and following a kidney transplant. Acquired forms of type 1 renal tubular acidosis can also be caused by certain medications (e.g., lithium, amphotericin B, NSAIDs, lead, antivirals), including some used for pain and bipolar disorder, conditions causing high calcium in the urine, blocked urinary tract, or rejection of a transplanted kidney.
Diseases and conditions related to type 1 renal tubular acidosis:
- a hereditary form of deafness
- renal medullary cystic disease
- sickle cell disease
- sickle cell anemia
- Ehlers-Danlos syndrome
- urinary tract infections
- amyloidosis, a buildup of abnormal protein, called amyloid, in the tissues and organs
- Fabry disease, an abnormal buildup in the body of a certain type of fatty substance
- High level of calcium in the blood (hypercalcemia)
- Sjögren syndrome, an autoimmune disorder in which the glands that produce tears and saliva are destroyed
- Systemic lupus erythematosus, an autoimmune disease in which the body’s immune system mistakenly attacks healthy tissue
- Wilson disease, an inherited disorder in which there is too much copper in the body’s tissues
- Use of certain medicines, such as amphotericin B, lithium, and nonsteroidal anti-inflammatory drugs (NSAIDs)
Type 2 renal tubular acidosis causes
Type 2 renal tubular acidosis or proximal renal tubular acidosis occurs when there is a problem in the beginning or proximal part of the tubules. Isolated proximal type 2 RTA is characterized by defects in the reabsorption of filtered bicarbonate (HCO3–) in the proximal tubule without defects in the transport of other solutes (Table 1) 27, 28.
Type 2 renal tubular acidosis (proximal renal tubular acidosis) may be inherited or caused by other inherited conditions such as:
- cystinosis, a rare disease in which cystine crystals are deposited in bones and other tissues
- hereditary fructose intolerance
- Wilson’s disease
Type 2 RTA can also be caused by acute lead poisoning or chronic exposure to cadmium. It can also occur in people treated with certain medications used in chemotherapy and to treat HIV, viral hepatitis, glaucoma, migraines, and seizures.
Type 2 renal tubular acidosis almost always occurs as part of Fanconi syndrome. The main features of Fanconi syndrome include:
- abnormal excretion of glucose, amino acids, citrate, bicarbonate, and phosphate into the urine
- low blood potassium levels
- low levels of vitamin D
The most common cause of acquired Fanconi syndrome in adults is multiple myeloma 44. The clinical manifestations of anemia, hypercalcemia, and bone pain suggest a diagnosis of multiple myeloma. As the most common cause of proximal RTA in adults is multiple myeloma, this diagnosis should be excluded in all adults with proximal RTA unless another cause is found.
Type 4 renal tubular acidosis causes
Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, is typically caused by selective aldosterone deficiency or when the kidneys do not respond to the aldosterone hormone (aldosterone resistance). Aldosterone directs the kidneys to regulate the level of sodium, which also affects the levels of chloride and potassium, in the blood. Hyperkalemic renal tubular acidosis (type 4 RTA) commonly develops in patients with diabetes or interstitial nephritis and is characterized by a disturbance in distal nephron function, leading to a reduction in the excretion of H+ and K+ in the cortical collecting duct that results in hyperkalemic, hyperchloremic, normal anion gap acidosis (Table 1) 49, 50. Hypoaldosteronism causes reduced principal cell Na+ reabsorption and a decrease in transepithelial voltage in the cortical collecting duct, which results in diminished excretion of H+ and K+. Increased serum K+ concentrations inhibit ammonium (NH4) synthesis in the proximal tubule, further reducing the kidney’s capacity to excrete acid. Ammonium (NH4) availability is critical for normal distal H+ secretion. Lack of adequate ammonium (NH4) buffer results in a drop in urine pH creating a steep pH gradient, which impedes distal H+ secretion. Aldosterone deficiency can cause Na+ wasting, leading to decreased plasma volume that stimulates proximal Na+ reabsorption. The reduction in distal Na+ delivery secondarily inhibits secretion of K+ and H+ in the distal nephron 48, 29. Hyperkalemia reduces ammonium (NH4) production in the proximal tubule and inhibits ammonium (NH4) transport in the thick ascending limb as the high luminal K+ concentrations compete with NH4+ for Na+/K+/2Cl– cotransporters and apical K+ channels 51. Reductions in urine ammonium (NH4) can be detected by a positive urine anion gap and a failure to increase the urine osmolal gap.
Certain medicines that interfere with the kidney’s task of moving electrolytes between your blood and urine may also cause hyperkalemic renal tubular acidosis or type 4 RTA. Some of these include:
- blood pressure medicines called angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs)
- certain diuretics used to treat congestive heart failure that do not decrease potassium in the blood
- certain medicines to prevent blood from clotting
- some immunosuppressive medicines that prevent the rejection of transplanted organs
- painkillers called nonsteroidal anti-inflammatory drugs (NSAIDs)
- antibiotics used to treat pneumonia, urinary tract infections, and traveler’s diarrhea
Type 4 RTA can also occur when diseases or an inherited disorder affect how the kidneys work, such as:
- Addison’s disease, due to disease or removal of the adrenal glands
- congenital adrenal insufficiency
- aldosterone synthase deficiency
- Gordon syndrome
- amyloidosis
- diabetic kidney disease
- HIV/AIDS
- kidney transplant rejection
- lupus
- sickle cell disease
- urinary tract obstruction
Renal tubular acidosis symptoms
The major signs of type 1 renal tubular acidosis (distal renal tubular acidosis) and type 2 renal tubular acidosis (proximal renal tubular acidosis) are low levels of potassium (K+) and bicarbonate (HCO3–), a waste product produced by your body in the blood. The potassium level drops if your kidneys send too much potassium into your urine instead of returning it to the blood.
Because potassium (K+) helps regulate your nerve and muscle health and heart rate, low potassium levels (hypokalemia) can cause:
- extreme weakness
- irregular heartbeat
- paralysis
- death
The major signs of type 4 renal tubular acidosis (hyperkalemic renal tubular acidosis) are high potassium and low bicarbonate levels in the blood. Symptoms of type 4 RTA include 63:
- abdominal pain
- fatigue that does not go away
- weak muscles
- not feeling hungry
- weight change
Renal tubular acidosis diagnosis
A diagnosis of distal renal tubular acidosis is based upon identification of characteristic symptoms, a detailed patient and family history, a thorough clinical evaluation and a variety of specialized tests. The disorder may be suspected in individuals with unexplained metabolic acidosis and an elevated plasma chloride (so called normal anion gap metabolic acidosis). In all cases of metabolic acidosis, plasma or serum anion gap should be the first laboratory assessment; hyperchloremic metabolic acidosis with a normal anion gap is present in all types of renal tubular acidosis (Table 1) 64, 53.
If your blood is more acidic than it should be and your urine is less acidic than it should be, RTA may be the reason, but a health care professional will need to rule out other causes.
Tests that may be ordered include:
- Arterial blood gas
- Basic metabolic panel, (a group of blood tests that measure your sodium, potassium, and chloride levels, kidney function, and other chemicals and functions)
- A complete blood count (CBC) to evaluate for an infectious cause with elevated white blood count and fluid body status with hemoglobin and hematocrit values is useful.
- Urine pH
- Urine ketones or blood ketones
- Acid-load test
- Bicarbonate infusion test
- Urinalysis
- Genetic testing. Molecular genetic testing can confirm a diagnosis. Genetic counseling may be of benefit for affected individuals and their families. Molecular genetic testing can detect disease-causing variants in the specific genes known to cause primary distal renal tubular acidosis, but is available only as a diagnostic service at specialized laboratories. Psychosocial support for the entire family can be essential as well.
Confirmatory testing of distal versus proximal renal tubular acidosis involves assessment of the markers of urinary acid and bicarbonate (HCO3–) secretion 53. An ammonium (NH4+) loading test is used to confirm distal renal tubular acidosis in patients with hypokalemic, hyperchloremic metabolic acidosis and urine pH > 5.5; at 6 hour after oral ingestion of NH4Cl 100 mg/kg, patients with distal renal tubular acidosis will develop a positive urine anion gap (Urine anion gap [UAG] = Urine Na + Urine K – Urine Cl) 53. As the NH4Cl test is associated with gastrointestinal adverse effects (e.g., nausea and vomiting), the furosemide–fludrocortisone test may be used as an alternative method of diagnosing distal renal tubular acidosis 65, 66.
In patients with distal renal tubular acidosis, measurement of the urine-to-blood (U-B) pCO2 gradient during an NaHCO3 infusion can be used to diagnose H+-ATPase secretory defect 67. Patients with an H+-ATPase defect have an abnormally low urine-to-blood pCO2 gradient (30 mmHg or lower), indicating impaired distal H+ secretion 67. In patients with a gradient defect, as occurs with amphotericin B-induced distal renal tubular acidosis 68, the urine-to-blood pCO2 gradient is normal (greater than 30 mmHg), suggesting an intact H+ secretory mechanism 69. In these patients, secreted H+ diffuses back into the cell as a result of a luminal permeability defect 68.
A diagnosis of proximal renal tubular acidosis may be confirmed using a NaHCO3 loading test; during an intravenous infusion of NaHCO3, the increase in serum bicarbonate (HCO3–) above the reabsorption threshold will lead to fractional excretion of bicarbonate (HCO3–) greater than 15% or urine pH > 7.5 in patients with proximal renal tubular acidosis 53. Patients with proximal RTA should also be evaluated for Fanconi syndrome by assessment of serum and urine samples for glycosuria, hypophosphatemia, and hypouricemia.
The differential diagnosis of type 4 hyperkalemic renal tubular acidosis may be divided into conditions associated with hypoaldosteronism or those caused by cortical collecting duct defects. A urine pH of less than 5.5 indicates defects caused by impaired aldosterone activity and a more severe reduction in NH3 availability than impairment of H+ secretion 29. In contrast, when the primary defect is caused by structural damage to the cortical collecting duct, the urine pH is more alkaline 29. Selective aldosterone deficiency may be confirmed after other causes of hyperkalemia are excluded, including transcellular shifts in K+ or the use of KCl, K+-sparing diuretics, or RAAS inhibitors [52]. After correction of serum K+, persistently low aldosterone levels indicate aldosterone deficiency; in most cases plasma renin levels are low. In patients with hyperkalemia, urinary K+ below 40 mmol/L or fractional K+ excretion below 20% are indicative of a defect in kidney K+ secretion 48. In patients with hypoaldosteronism, but normal renin levels, possible causes include adrenal gland damage, Addison disease, critical illness (i.e., direct renal suppression), RAAS inhibitor therapy, or heparin-induced suppression of aldosterone synthesis 49.
Extrarenal conditions may mimic RTA by causing normal gap metabolic acidosis through increased production of endogenous H+ and accelerated loss of extrarenal bicarbonate (HCO3–) 29. Extrarenal metabolic acidosis is associated with elevated levels of H+ and urinary NH4+ excretion 29. Severe or chronic diarrhea commonly causes hyperchloremic metabolic acidosis through the loss of large amounts of gastrointestinal bicarbonate (HCO3–), particularly as bicarbonate (HCO3–) concentrations are usually higher in diarrheal fluid than in plasma 7, 53. The reduction in plasma volume triggers an increase in kidney NaCl reabsorption which, in combination with bicarbonate (HCO3–) losses, leads to normal anion gap metabolic acidosis 29. Low serum pH, as well as hypokalemia caused by gastrointestinal losses, promotes ammoniagenesis in the proximal tubule; the increased NH3 concentrations allow for increased H+ excretion by the distal nephron. This can result in an increased urine pH in patients with chronic diarrhea because of increases in kidney NH3 metabolism29. Chronic laxative abuse has been reported to mimic distal RTA through intestinal losses of bicarbonate (HCO3–) and K+ 70. The normal increase in urinary NH3 excretion in these situations is reflected by a negative urinary anion gap and an increased urine osmolal gap.
Calcium deposits in the kidneys and kidney stones may be seen on:
- X-rays
- Ultrasound
- CT scan
Plain x-rays (radiographs) or specialized imaging techniques such as computerized tomography (CT) scanning or ultrasonography can help to further confirm a diagnosis, or help determine the extent of disease. These tests can show the accumulation or deposition of calcium in the kidneys and help to rule out other conditions. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of certain tissue structures. During ultrasonography, reflected sound waves are used to create an image of structures within the body including the kidneys.
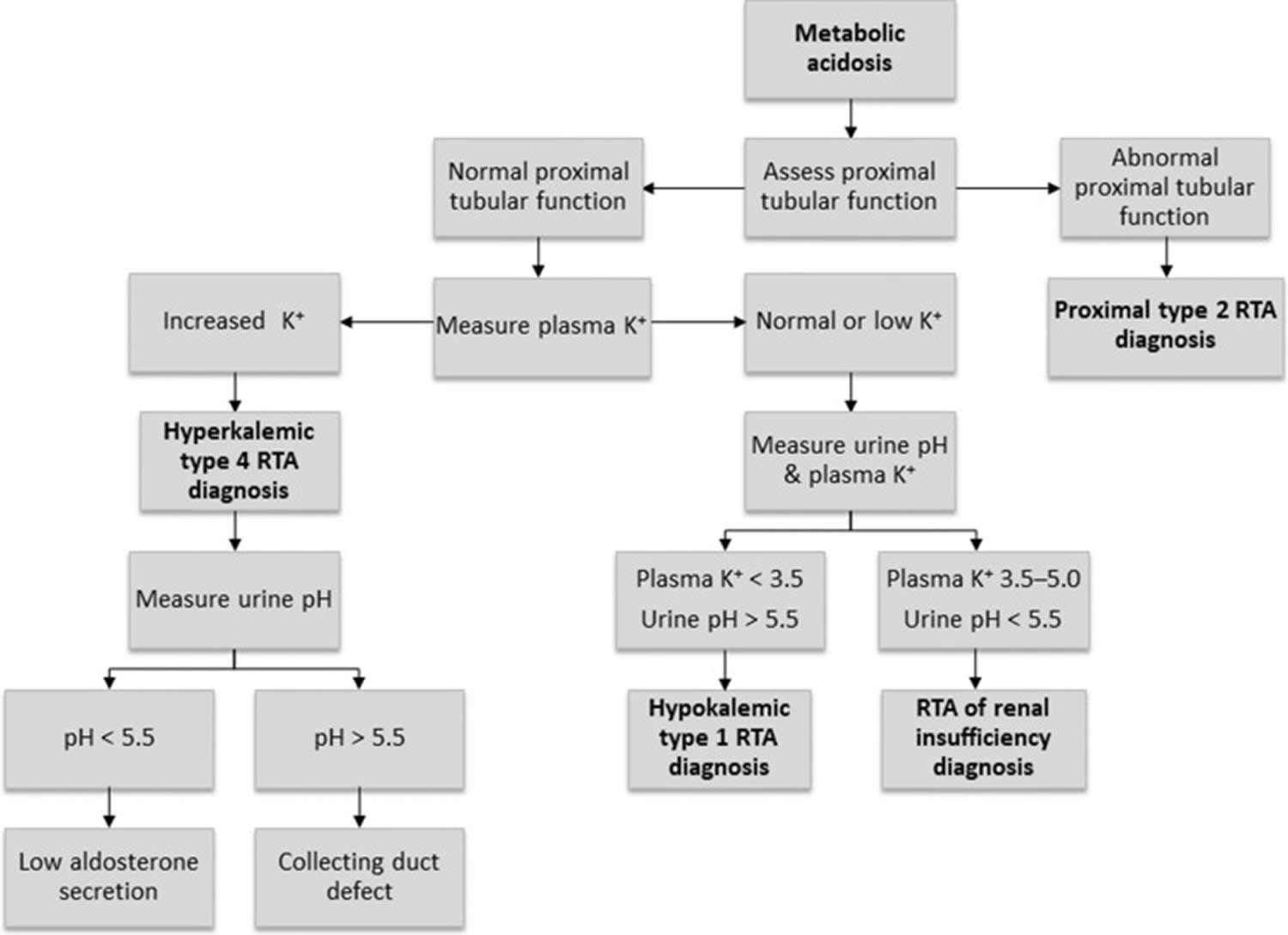
Figure 12. Renal tubular acidosis diagnostic algorithm
Arterial blood gas (ABG) analysis
Arterial blood gas (ABG) sampling, is a test often performed in an inpatient setting to assess the acid-base status of a patient. A needle is used to draw blood from an artery, often the radial artery, and the blood is analyzed to determine parameters such as the pH, arterial partial pressure of carbon dioxide (PaCO2), arterial partial pressure of oxygen (PaO2), bicarbonate (HCO3–), oxygen saturation (O2 Sat) and more. This allows the physician to understand the status of the patient better. ABGs are especially important in the critically ill. They are the main tool utilized in adjusting to the needs of a patient on a ventilator.
- Arterial partial pressure of carbon dioxide (PaCO2) as carbon dioxide tension, this measures the level of carbon dioxide in your blood.
- Arterial partial pressure of oxygen (PaO2) also known as oxygen tension, this measures how well oxygen is being transferred into your blood.
- Oxygen saturation (O2 Sat) is an assessment of the amount of oxygen in your blood that is based on measuring levels of hemoglobin. Hemoglobin is a protein found inside red blood cells that is responsible for carrying oxygen throughout the body.
- Bicarbonate (HCO3–) concentration: Bicarbonate (HCO3–) is an electrolyte, which is a type of mineral involved in managing your body’s acid-base balance. Most of the carbon dioxide (CO2) in your blood is stored in the form of bicarbonate, so this measurement helps reflect carbon dioxide (CO2) levels.
- Although not universal, some arterial blood gases tests include measurements of hemoglobin as well as altered forms of the hemoglobin protein. Examples of these potential additional measurements include:
- Methemoglobin: Methemoglobin is a form of hemoglobin that has been oxidized, changing its heme iron configuration from the ferrous (Fe2+) to the ferric (Fe3+) state. Unlike normal hemoglobin, methemoglobin does not bind oxygen and as a result cannot deliver oxygen to the tissues.
- Carboxyhemoglobin: Carboxyhemoglobin is a stable complex of carbon monoxide and hemoglobin that forms in red blood cells upon contact with carbon monoxide. This abnormal form of hemoglobin attaches to carbon monoxide and can interfere with oxygen’s ability to travel in the blood.
- Oxyhemoglobin: Oxyhemoglobin represents the fraction of oxygenated hemoglobin in relation to the total hemoglobin present, including non-oxygen-binding hemoglobins. In healthy individuals, oxyhemoglobin and oxygen saturation are approximately equal.
- Deoxyhemoglobin: This is the form of hemoglobin without oxygen in the blood.
The following are the most important Normal Values on an ABG:
- pH = 7.35 to 7.45
- Arterial partial pressure of carbon dioxide (PaCO2) = 35 to 45 mmHg
- Arterial partial pressure of oxygen (PaO2) = 75 to 100 mmHg
- Bicarbonate (HCO3–) = 22 to 26 mEq/L
- O2 Sat = greater than 95%
The ability to quickly and efficiently read an ABG is paramount to quality patient care.
- Look at the pH. Decide whether it is acidotic, alkalotic, or within the physiological range
- Arterial partial pressure of carbon dioxide (PaCO2) level determines respiratory contribution; a high level means the respiratory system is lowering the pH and vice versa.
- Bicarbonate (HCO3–) level denotes metabolic/kidney effect. An elevated bicarbonate (HCO3–) is raising the pH and vice versa.
- If the pH is acidotic, look for the number that corresponds with a lower pH. If it is a respiratory acidosis, the carbon dioxide (CO2) should be high. If the patient is compensating metabolically, the bicarbonate (HCO3–) should be high as well. A metabolic acidosis will be depicted with an bicarbonate (HCO3–) that is low.
- If the pH is alkalotic, again, determine which value is causing this. A respiratory alkalosis will mean the carbon dioxide (CO2) is low; a metabolic alkalosis should lend an bicarbonate (HCO3–) that is high. Compensation with either system will be reflected oppositely; for a respiratory alkalosis the metabolic response should be a low bicarbonate (HCO3–) and for metabolic alkalosis, the respiratory response should be a high carbon dioxide (CO2).
- If the pH level is in the physiological range but the arterial partial pressure of carbon dioxide (PaCO2) and/or bicarbonate (HCO3–) are not within normal limits, there is likely a mixed disorder. Also, compensation does not always occur; this is when clinical information becomes paramount.
- Sometimes it is difficult to ascertain whether a patient has a mixed disorder.
Other tests that are important to perform when analyzing the acid-base status of a patient include those that measure electrolyte levels and renal function. This helps the clinician gather information that can be used to determine the exact mechanism of the acid-base imbalance as well as the factors contributing to the disorders 71, 72.
Urinalysis
Urine pH is normally acidic, at less than 5.0. In acidemia, the urine normally becomes more acidic. If the urine pH is above 5.5 in the face of acidemia, this finding is consistent with a type 1 renal tubular acidosis (RTA). Alkaline urine is typical in salicylate toxicity.
Patients with ethylene glycol toxicity may present with calcium oxalate crystals, which appear needle shaped, in the urine.
Serum Anion Gap
The formula for anion gap is:
- Serum anion gap = (Na+) – [(HCO3– + Cl–)]
Where Na+ is plasma sodium concentration, HCO3– is plasma bicarbonate concentration, and Cl– is plasma chloride concentration. The anions are negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]. The anion gap is the difference between measured cations (positively charged ions like sodium [Na+] and potassium [K+]) and measured anions (negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]) 73. The most common application of the anion gap is classifying cases of metabolic acidosis, states of lower than normal blood pH. Specifically, classifying into either those that do and those that do not have unmeasured anions in the plasma. The human body is electrically neutral; therefore, in reality, does not have a true anion gap 73. A normal serum anion gap is measured to be 5 to 16 mEq/L, with autoanalyzers using an ion-selective electrode. However, the anion gap value is dependent on the type of instrument used to measure its components 7. Therefore, you should know the reference range of the analyzer used and, if known, the patient’s baseline anion gap, too.
The most common application of the anion gap is classifying cases of metabolic acidosis, states of lower than normal blood pH. Specifically, classifying into either those that do and those that do not have unmeasured anions in the plasma. The human body is electrically neutral; therefore, in reality, does not have a true anion gap 73. A normal serum anion gap is measured to be 5 to 16 mEq/L, with autoanalyzers using an ion-selective electrode. However, the anion gap value is dependent on the type of instrument used to measure its components 7. Therefore, you should know the reference range of the analyzer used and, if known, the patient’s baseline anion gap, too.
The anion gap is a calculation to determine the quantity of ionically active components within your blood that are not routinely measured. Serum anion gap is affected by the concentrations of all anions and cations which are not included in its calculations: i.e., albumin, globulin, potassium, calcium, magnesium, and organic and inorganic acids (see Figure 1). Because of the narrow extracellular concentration, most ions are omitted from the anion gap calculation. Since there are always components not directly measured, we expect this value to not equal 0. Most of this number is due to albumin (Alb); this anion is not accounted for in the anion gap formula, which is a large reason why the anion gap is not closer to zero. According to James Gamble 74, electrical neutrality in solution demands that the sum of the cations is equal to the sum of the anions (Figure 1). Sodium, chloride, bicarbonate, and albumin are quantitatively the major ions in the extracellular fluid compartment and are therefore used to calculate the anion gap 7. A true “ion gap,” however, does not exist in vivo which makes the anion gap a fundamental tool to evaluate acid-base disorders 75. Albumin is normally 4 mg/dL. Because of the large effect of albumin on anion gap, if a patient’s albumin level is abnormal, their expected anion gap will not be accurate 76. This can be corrected using simple math. The correction factor for albumin is 2.3–2.5 × [albumin], in g/dL 7. Therefore, each g/dL albumin decline will decrease the anion gap with about 2.5 mEq/L. To appreciate these facts, the anion gap formula should be: [Na+] − [Cl−] − [HCO3−] − 2.5 [albumin, in g/dL]. This equation is about zero in health, to stress the balance of ions, and also shows the relevance of albumin as a negative ion 7. As opposed to high anion gap acidosis which involves increased organic acid production, normal anion gap acidosis involves either increased production of chloride (hyperchloremic acidosis) or increased excretion of bicarbonate (HCO3–).
If a patient has an anion gap over 12, these mnemonics are helpful to remember the possible causes of the disorder 77, 78. The mnemonic MUDPILES has classically been used by clinicians to summarize the causes of high anion gap metabolic acidosis.
MUDPILES stands for:
- Methanol,
- Uremia,
- Diabetic ketoacidosis,
- Paraldehyde,
- Infection,
- Lactic acidosis,
- Ethylene glycol, and
- Salicylates.
A new mnemonic, GOLDMARK, has been suggested to be an improvement 76.
GOLDMARK is an anagram for:
- Glycols (ethylene and propylene),
- Oxoproline,
- Lactate,
- Methanol,
- Aspirin,
- Renal failure, or chronic kidney disease (CKD) and
- Ketones.
Urine Anion Gap
Calculating the urine anion gap is helpful in evaluating some cases of non-anion gap metabolic acidosis. The major measured urinary cations are Na+ and K+, and the major measured urinary anion is Cl-:
- Urine anion gap = Urine Na + Urine K – Urine Cl
In the face of metabolic acidosis, the kidneys increase the amount of NH3 synthesized to buffer the excess H+ and NH4 Cl excretion increases. The increased unmeasured ammonium (NH4+) thus increases the measured anion Cl- in the urine, and the net effect is a negative anion gap, representing a normal response to systemic acidification. The finding of a positive urine anion gap in the face of non-anion gap metabolic acidosis points toward a renal acidification defect (eg, renal tubular acidosis) 79.
Salicylate levels and Iron levels
Therapeutic salicylate levels range up to 20-35 mg/dL. Plasma levels exceeding 40-50 mg/dL are in the toxic range.
Plasma levels provide some information as to the severity of intoxication: 40-60 mg/dL is considered mild; 60-100 mg/dL is moderate; and greater than 100 mg/dL is considered severe.
Iron toxicity is associated with lactic acidosis. Iron levels greater than 300 mg/dL are considered toxic.
Special tests
Measuring the transtubular potassium gradient (TTKG) is useful in determining the cause of hyperkalemia or hypokalemia associated with metabolic acidosis.
- Transtubular Potassium Gradient (TTKG) = urine K+ × serum osmolality/serum K+ × urine osmolality
A transtubular potassium gradient (TTKG) of greater than 8 indicates that aldosterone is present and that the collecting duct is responsive to it. A transtubular potassium gradient (TTKG) of less than 5 in the presence of hyperkalemia indicates aldosterone deficiency or resistance. For the test to be interpretable, the urine Na+ level should be greater than 10 mEq/L and the urine osmolality should be greater than or equal to serum osmolality.
Plasma renin activity and plasma aldosterone levels are useful in determining the cause of the hyperkalemia and hypokalemia that accompany metabolic acidosis.
Calculation of fractional excretion of bicarbonate (FEHCO3–) is useful in the diagnosis of proximal renal tubular acidosis (RTA).
The ammonium chloride (NH4Cl) loading test is useful in patients with nephrocalcinosis and/or nephrolithiasis, who may have an incomplete form of distal renal tubular acidosis. These patients may not have a pH less than 7.35 or a drop in serum bicarbonate (HCO3–); metabolic acidosis can be induced by administration of NH4Cl (0.1 g/kg for 3 days). Under these circumstances of induced acidemia, a urine pH greater than 5.3 indicates distal renal tubular acidosis (RTA).
An alternative to the ammonium chloride (NH4Cl) loading test involves the simultaneous oral administration of furosemide to increase distal Na+ delivery and fludrocortisone to increase collecting duct Na+ absorption and proton secretion 80. Under these circumstances, a urine pH greater than 5.3 indicates distal renal tubular acidosis (RTA).
Measuring the urine-blood arterial partial pressure of carbon dioxide (PaCO2) gradient following an bicarbonate (HCO3–) load is useful in some patients with classic distal renal tubular acidosis to differentiate a permeability defect from other defects. This test is useful in patients with nephrocalcinosis in whom distal renal tubular acidosis (RTA) is suspected but urine is acidified appropriately in the face of metabolic acidosis. Some of these patients have a rate-dependent defect in proton secretion, revealed by a low urine-blood PaCO2 gradient following bicarbonate (HCO3–) loading.
Abdominal radiographs (eg, kidneys, ureters, bladder), CT scans, and/or renal ultrasound images may show renal stones or nephrocalcinosis in patients with distal renal tubular acidosis.
Renal tubular acidosis treatment
For all types of RTA, drinking a solution of sodium bicarbonate (NaHCO3) or sodium citrate will lower the acid level in your blood. This alkali therapy can prevent kidney stones from forming and make your kidneys work more normally so kidney failure does not get worse.
Infants with type 1 renal tubular acidosis may need potassium supplements, but older children and adults rarely do because alkali therapy prevents the kidneys from excreting potassium into the urine.
Children with type 2 renal tubular acidosis will also drink an alkali solution (sodium bicarbonate or potassium citrate) to lower the acid level in their blood, prevent bone disorders and kidney stones, and grow normally. Some adults with type 2 RTA may need to take vitamin D supplements to help prevent bone problems.
People with type 4 renal tubular acidosis (hyperkalemic renal tubular acidosis) may need other medicines to lower the potassium levels in their blood.
If your RTA is caused by another condition, your health care professional will try to identify and treat it.
Medications to treat renal tubular acidosis
Alkali therapy may be used to correct acidosis in patients with distal or proximal RTA 29. In patients with distal renal tubular acidosis (type 1 RTA), alkali therapy also corrects for hypokalemia. Alkali therapy with 1–2 mmol/kg/day sodium bicarbonate (NaHCO3) or potassium bicarbonate (KHCO3) is normally sufficient to equal daily acid production 1; however, in patients with kidney stones or nephrocalcinosis, the elevated Na+ load with sodium bicarbonate (NaHCO3) therapy may cause increased urine calcium excretion, which can precipitate kidney stone formation. In these patients, potassium citrate (K-citrate) administration is preferable; this will also increase urine citrate excretion and prevent recurrence of kidney stones 81. Patients with severe hypokalemia should also receive K+ replacement (i.e., with KCl or K-citrate) to prevent further lowering of serum K+ concentrations and symptomatic hypokalemia 29. Long-term treatment of distal renal tubular acidosis (type 1 RTA) generally requires a combination of sodium bicarbonate (NaHCO3) and potassium bicarbonate (KHCO3) 29, 82. Children with distal renal tubular acidosis (type 1 RTA) require sufficient sodium bicarbonate (NaHCO3) or potassium bicarbonate (KHCO3) (usually 4–8 mmol/kg/day) to maintain normal serum bicarbonate (HCO3–) concentrations and prevent growth retardation 1.
In contrast, treatment of proximal renal tubular acidosis (type 2 RTA) is often challenging and patients require larger quantities of alkali therapy (10–15 mmol/kg/day), which are usually administered as a K+ salt (e.g., K-citrate) to avoid worsening hypokalemia 1, 29. However, as exogenous alkali is rapidly excreted in the urine, correction of acidosis is often impossible despite administration of large amounts of alkali therapy 29. In addition to alkali therapy, patients with Fanconi syndrome are treated with fluid and electrolyte replacement to prevent volume depletion, as well as supplementation with vitamin D and phosphate (1–3 g/day) to prevent bone disease 83. Impaired proximal bicarbonate (HCO3–) reabsorption leads to most of the administered bicarbonate (HCO3–) being lost in the urine with minimal increases in serum concentrations 84, while increased distal Na+ delivery and elevated aldosterone levels cause increased renal K+ wasting 29. Hydrochlorothiazide may be beneficial in increasing bicarbonate (HCO3–) reabsorption capacity, preventing volume expansion, and increasing the effectiveness of alkali therapy; however, supplemental K+ or K+-sparing diuretics are needed to prevent hypokalemia 83. After initiation of therapy, patients should be closely monitored for severe electrolyte abnormalities. Children with proximal renal tubular acidosis (type 2 RTA) often require aggressive alkali therapy (5–15 mmol/kg/day) to mitigate bone disease and growth retardation 85, whereas alkali therapy is typically administered in adults with serum bicarbonate (HCO3–) concentrations of less than 18 mmol/L to prevent severe acidosis 29.
Sodium bicarbonate (NaHCO3) therapy is often associated with gastrointestinal adverse effects, most commonly bloating and belching, and should be taken on an empty stomach 86. If these adverse effects limit patient adherence, Na-citrate or enteric-coated sodium bicarbonate (NaHCO3) (where available) may be considered 86.
In patients with hyperkalemic type 4 renal tubular acidosis, lowering of serum K+ concentrations often leads to correction of metabolic acidosis 29. The reduction in serum K+ leads to increased NH3 production in the proximal tubule and medullary transfer, thereby increasing the availability of buffer supply for distal acidification 29. Any non-essential medications affecting renal K+ excretion or aldosterone synthesis or activity should be discontinued 29. Discontinuation of the COX-2 inhibitor celecoxib would be beneficial in reducing serum K+ concentrations. ACE inhibitors and ARBs reduce urinary K+ excretion through inhibition of aldosterone secretion in the adrenal gland and may cause hyperkalemia in patients with pre-existing conditions that cause impaired K+ excretion 87. For this reason, RAAS inhibitor therapy is avoided; however, ACE inhibitors and ARBs are normally continued in patients with CKD because of their cardiovascular and renoprotective benefits 29. Low-dose fludrocortisone therapy may also be effective in managing hyperkalemia and hyponatremia in patients with type 4 RTA who do not have heart failure or hypertension 48, 88. Since thiazide diuretics are largely ineffective in patients with an estimated GFR of less than 30 mL/min/1.73 m², loop diuretics and NaHCO3 therapy may be beneficial in patients with type 4 RTA, particularly when fludrocortisone is not tolerated. Loop diuretics may reduce serum K+ and help control volume overload by increasing Na+ delivery and flow rates to the cortical collecting duct while lowering blood pressure 48. The Kidney Disease-Improving Global Outcomes guidelines suggest administration of oral HCO3– therapy to maintain serum bicarbonate (HCO3–) in the normal range in patients with CKD and serum bicarbonate (HCO3–) less than 22 mmol/L 89. Sodium bicarbonate (NaHCO3) administration will correct metabolic acidosis and further minimize the risk of hyperkalemia, and the addition of Na+ would not be problematic in the setting of effective diuretic therapy. However, patients on sodium bicarbonate (NaHCO3) therapy should be closely monitored for volume overload and hypertension 29.
There are newer K+-binding agents available (i.e., patiromer and sodium zirconium cyclosilicate [sodium zirconium cyclosilicate]) for patients with hyperkalemic type 4 RTA, which can be used to treat hyperkalemia and improve acidosis. Patiromer is a polymeric cation exchange resin that binds K+ ions in exchange for calcium ions in the colon 90, whereas sodium zirconium cyclosilicate is non-polymeric, selective K+-binder, which entraps K+ and NH4+ ions in exchange for H+ and Na+ ions throughout the gastrointestinal tract 91. Both patiromer and sodium zirconium cyclosilicate are effective in patients receiving RAAS inhibitor therapy, which allows continuation of these therapeutic agents in the presence of hyperkalemia 92. sodium zirconium cyclosilicate has also been associated with significant increases in serum bicarbonate (HCO3–) concentrations by approximately 2 mmol/L versus placebo during the initial 48-h treatment period and 2–3 mmol/L during maintenance therapy for 1 month, regardless of chronic kidney disease (CKD) stage 92. This increase in serum bicarbonate (HCO3–) with sodium zirconium cyclosilicate is most likely as a result of gastrointestinal NH4+ binding ions and associated decreases in serum urea concentrations 93. Reducing serum K+ concentrations with secondary increases in NH3 synthesis may be an additional mechanism by which acidosis is improved with sodium zirconium cyclosilicate. Whether patiromer produces similar effects on serum bicarbonate (HCO3–) remains to be determined; however, treatment with sodium zirconium cyclosilicate may provide therapeutic benefits in patients with hyperkalemic renal tubular acidosis (type 4 RTA). Studies are needed to confirm the efficacy and safety of sodium zirconium cyclosilicate or patiromer in hyperkalemic renal tubular acidosis (type 4 RTA).
Renal tubular acidosis diet
In general, plant-based diets contain a lower net acid load and less bioavailable phosphate than animal-based diets 94. In patients with metabolic acidosis, the dietary acid load can be decreased by limiting acid-producing foods (e.g., animal protein) and increasing alkali-producing foods (e.g., fruits and vegetables) (Table 1) 95, 86.. Reduction in the consumption of dietary protein obtained from animal sources also results in increases in serum total carbon dioxide (CO2) concentrations 86.
Changes in dietary consumption of citrus fruit and juices, as well as restricted Na+, oxalate, fructose, and animal protein intake, in combination with normal calcium intake may also benefit patients with distal renal tubular acidosis and nephrolithiasis 81. Increased fruit and vegetable intake is associated with increased urinary citrate excretion in patients with hypocitraturia and nephrolithiasis 96, 81. In patients with hyperkalemic renal tubular acidosis, dietary restriction of K+ has previously been the standard of care 1; however, new data suggest that increased intake of alkali-producing fruits and vegetables (which are often high in K+) and limiting intake of acid-producing foods may correct acidosis 97.
- Soleimani M, Rastegar A. Pathophysiology of renal tubular acidosis: core curriculum 2016. Am J Kidney Dis. 2016;68:488–498. doi: 10.1053/j.ajkd.2016.03.422[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Palmer BF, Kelepouris E, Clegg DJ. Renal Tubular Acidosis and Management Strategies: A Narrative Review. Adv Ther. 2021 Feb;38(2):949-968. doi: 10.1007/s12325-020-01587-5[↩][↩]
- Kurtz I. Renal Tubular Acidosis: H+/Base and Ammonia Transport Abnormalities and Clinical Syndromes. Adv Chronic Kidney Dis. 2018 Jul;25(4):334-350. doi: 10.1053/j.ackd.2018.05.005[↩][↩]
- Renal Tubular Acidosis. https://www.niddk.nih.gov/health-information/kidney-disease/renal-tubular-acidosis[↩][↩]
- Roth KS, Chan JC. Renal tubular acidosis: a new look at an old problem. Clin Pediatr (Phila). 2001 Oct;40(10):533-43. doi: 10.1177/000992280104001001[↩][↩]
- Hamm LL, Nakhoul N, Hering-Smith KS. Acid-Base Homeostasis. Clin J Am Soc Nephrol. 2015 Dec 7;10(12):2232-42. doi: 10.2215/CJN.07400715[↩][↩][↩][↩][↩][↩][↩][↩]
- Berend K. Review of the Diagnostic Evaluation of Normal Anion Gap Metabolic Acidosis. Kidney Dis (Basel). 2017 Dec;3(4):149-159. doi: 10.1159/000479279[↩][↩][↩][↩][↩][↩][↩]
- Adamczak M, Surma S. Metabolic Acidosis in Patients with CKD: Epidemiology, Pathogenesis, and Treatment. Kidney Dis (Basel). 2021 Jun 4;7(6):452-467. doi: 10.1159/000516371[↩]
- Scialla JJ, Asplin J, Dobre M, et al. Higher net acid excretion is associated with a lower risk of kidney disease progression in patients with diabetes. Kidney Int. 2017;91:204–215. doi: 10.1016/j.kint.2016.09.012[↩]
- Halperin ML, Jungas RL. Metabolic production and renal disposal of hydrogen ions. Kidney Int. 1983;24:709–713. doi: 10.1038/ki.1983.217[↩][↩]
- Koeppen BM. The kidney and acid-base regulation. Adv Physiol Educ. 2009 Dec;33(4):275-81. doi: 10.1152/advan.00054.2009[↩][↩]
- Kisaka T, Cox TA, Dumitrescu D, Wasserman K. CO2 pulse and acid-base status during increasing work rate exercise in health and disease. Respir Physiol Neurobiol. 2015 Nov;218:46-56. doi: 10.1016/j.resp.2015.07.005[↩]
- Brinkman JE, Toro F, Sharma S. Physiology, Respiratory Drive. [Updated 2022 Jun 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482414[↩]
- Brinkman JE, Sharma S. Respiratory Alkalosis. [Updated 2022 Jul 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482117[↩]
- Hopkins E, Sharma S. Physiology, Acid Base Balance. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507807[↩]
- Palmer BF. Normal acid-base balance. In: Johnson RJ, Feehally J, Floege J, editors. Comprehensive clinical nephrology. 5. Philadelphia: Elsevier; 2014. pp. 142–148.[↩][↩]
- Acidosis: An Old Idea Validated by New Research. Integr Med (Encinitas). 2015;14(1):8-12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4566456/[↩]
- Frassetto LA, Morris RC, Jr, Sebastian A. Effect of age on blood acid-base composition in adult humans: role of age-related renal functional decline. Am J Physiol. 1996;271(6 Pt 2):F1114–F1122[↩]
- Kopple J.D., Kalantar-Zadeh K., Mehrotra R. Risks of chronic metabolic acidosis in patients with chronic kidney disease. Kidney Int. 2005;67:S21–S27. doi: 10.1111/j.1523-1755.2005.09503.x[↩]
- Dubey A.K., Sahoo J., Vairappan B., Haridasan S., Parameswaran S., Priyamvada P.S. Correction of metabolic acidosis improves muscle mass and renal function in chronic kidney disease stages 3 and 4: A randomized controlled trial. Nephrol. Dial. Transplant. 2020;35:121–129. doi: 10.1093/ndt/gfy214[↩]
- Noce A., Marrone G., Ottaviani E., Guerriero C., Di Daniele F., Pietroboni Zaitseva A., Di Daniele N. Uremic sarcopenia and its possible nutritional approach. Nutrients. 2021;13:147. doi: 10.3390/nu13010147[↩]
- Diet acids and alkalis influence calcium retention in bone. Buclin T, Cosma M, Appenzeller M, Jacquet AF, Décosterd LA, Biollaz J, Burckhardt P. Osteoporos Int. 2001; 12(6):493-9.[↩]
- Trepiccione F, Prosperi F, de la Motte LR, et al. New findings on the pathogenesis of distal renal tubular acidosis. Kidney Dis (Basel) 2017;3:98–105. doi: 10.1159/000478781[↩]
- Primary Distal Renal Tubular Acidosis. https://rarediseases.org/rare-diseases/primary-distal-renal-tubular-acidosis[↩][↩][↩][↩]
- Mohebbi N, Wagner CA. Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol. 2018 Aug;31(4):511-522. doi: 10.1007/s40620-017-0447-1[↩][↩]
- Gil-Peña H, Mejía N, Santos F. Renal tubular acidosis. J Pediatr. 2014 Apr;164(4):691-698.e1. doi: 10.1016/j.jpeds.2013.10.085[↩][↩]
- Kashoor I, Batlle D. Proximal renal tubular acidosis with and without Fanconi syndrome. Kidney Res Clin Pract. 2019;38:267–281. doi: 10.23876/j.krcp.19.056[↩][↩][↩][↩][↩]
- Haque SK, Ariceta G, Batlle D. Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol Dial Transplant. 2012;27:4273–4287. doi: 10.1093/ndt/gfs493[↩][↩][↩][↩][↩][↩][↩][↩]
- Palmer BF, Clegg DJ. Hyperchloremic normal gap metabolic acidosis. Minerva Endocrinol. 2019 Dec;44(4):363-377. doi: 10.23736/S0391-1977.19.03059-1[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Unwin RJ, Capasso G. The renal tubular acidoses. J R Soc Med. 2001;94:221–225. doi: 10.1177/014107680109400506[↩]
- Igarashi T, Inatomi J, Sekine T, et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet. 1999;23:264–266. doi: 10.1038/15440[↩]
- Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2171–2177. doi: 10.1097/01.ASN.0000025281.70901.30[↩]
- Gutierrez JO, Zurita MF, Zurita LA. Sjögren’s Syndrome Associated with Fanconi’s Syndrome and Osteomalacia. Am J Case Rep. 2018 Apr 3;19:392-396. doi: 10.12659/ajcr.907503[↩][↩]
- Yamaguchi S, Maruyama T, Wakino S, Tokuyama H, Hashiguchi A, Tada S, Homma K, Monkawa T, Thomas J, Miyashita K, Kurihara I, Yoshida T, Konishi K, Hayashi K, Hayashi M, Itoh H. A case of severe osteomalacia caused by Tubulointerstitial nephritis with Fanconi syndrome in asymptomotic primary biliary cirrhosis. BMC Nephrol. 2015 Nov 11;16:187. doi: 10.1186/s12882-015-0184-4[↩][↩]
- Lemann J Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int. 2000 Sep;58(3):1267-77. doi: 10.1046/j.1523-1755.2000.00282.x[↩]
- Brenner RJ, Spring DB, Sebastian A, et al. Incidence of radiographically evident bone disease, nephrocalcinosis, and nephrolithiasis in various types of renal tubular acidosis. N Engl J Med. 1982;307:217–221. doi: 10.1056/NEJM198207223070403[↩]
- Lee YS, Kim BK, Lee HJ, Dan J. Pathologic femoral neck fracture due to Fanconi syndrome induced by adefovir dipivoxil therapy for hepatitis B. Clin Orthop Surg. 2016;8:232–236. doi: 10.4055/cios.2016.8.2.232[↩]
- Negro A, Regolisti G, Perazzoli F, Davoli S, Sani C, Rossi E. Ifosfamide-induced renal Fanconi syndrome with associated nephrogenic diabetes insipidus in an adult patient. Nephrol Dial Transplant. 1998;13:1547–1549. doi: 10.1093/ndt/13.6.1547[↩]
- Knights M, Thekkekkara T, Morris A, Finlay E. Sodium valproate-induced Fanconi type proximal renal tubular acidosis. BMJ Case Rep. 2016;2016:bcr2015213418[↩]
- Izzedine H, Launay-Vacher V, Deray G. Topiramate-induced renal tubular acidosis. Am J Med. 2004;116:281–282. doi: 10.1016/j.amjmed.2003.08.021[↩]
- Sacré A, Jouret F, Manicourt D, Devuyst O. Topiramate induces type 3 renal tubular acidosis by inhibiting renal carbonic anhydrase. Nephrol Dial Transplant. 2006;21:2995–2996. doi: 10.1093/ndt/gfl251[↩]
- Nagai T, Matsuo N, Tsuchiya Y, Cho H, Hasegawa Y, Igarashi Y. Proximal renal tubular acidosis associated with glycogen storage disease, type 9. Acta Paediatr Scand. 1988;77:460–463. doi: 10.1111/j.1651-2227.1988.tb10681.x[↩]
- Richardson RM, Little JA, Patten RL, Goldstein MB, Halperin ML. Pathogenesis of acidosis in hereditary fructose intolerance. Metabolism. 1979;28:1133–1138. doi: 10.1016/0026-0495(79)90152-5[↩]
- Mathur M, Chacko B, Vankalakunti M, Patil C. Fanconi syndrome due to light chain proximal tubulopathy in a patient with multiple myeloma. Saudi J Kidney Dis Transpl. 2016;27:805–807. doi: 10.4103/1319-2442.185268[↩][↩]
- Lacruz RS, Habelitz S, Wright JT, Paine ML. DENTAL ENAMEL FORMATION AND IMPLICATIONS FOR ORAL HEALTH AND DISEASE. Physiol Rev. 2017 Jul 1;97(3):939-993. doi: 10.1152/physrev.00030.2016[↩]
- Chanchlani R, Nemer P, Sinha R, Nemer L, Krishnappa V, Sochett E, Safadi F, Raina R. An Overview of Rickets in Children. Kidney Int Rep. 2020 Apr 11;5(7):980-990. doi: 10.1016/j.ekir.2020.03.025[↩]
- Menegussi J, Tatagiba LS, Vianna JGP, Seguro AC, Luchi WM. A physiology-based approach to a patient with hyperkalemic renal tubular acidosis. J Bras Nefrol. 2018 Oct-Dec;40(4):410-417. doi: 10.1590/2175-8239-JBN-3821[↩]
- Batlle D, Arruda J. Hyperkalemic forms of renal tubular acidosis: clinical and pathophysiological aspects. Adv Chronic Kidney Dis. 2018;25:321–333. doi: 10.1053/j.ackd.2018.05.004[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Karet FE. Mechanisms in hyperkalemic renal tubular acidosis. J Am Soc Nephrol. 2009;20:251–254. doi: 10.1681/ASN.2008020166[↩][↩][↩]
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med. 2015;373:548–559. doi: 10.1056/NEJMra1503102[↩][↩]
- Mount DB. Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol. 2014;9:1974–1986. doi: 10.2215/CJN.04480413[↩][↩]
- Montford JR, Linas S. How Dangerous Is Hyperkalemia? J Am Soc Nephrol. 2017 Nov;28(11):3155-3165. doi: 10.1681/ASN.2016121344[↩]
- Yaxley J, Pirrone C. Review of the Diagnostic Evaluation of Renal Tubular Acidosis. Ochsner J. 2016 Winter;16(4):525-530. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5158160[↩][↩][↩][↩][↩][↩][↩]
- Batlle DC, Arruda JA, Kurtzman NA. Hyperkalemic distal renal tubular acidosis associated with obstructive uropathy. N Engl J Med. 1981;304:373–380. doi: 10.1056/NEJM198102123040701[↩]
- Cook E, Davis J, Israni R, et al. Prevalence of metabolic acidosis among patients with CKD and hyperkalemia [abstract 89] Am J Kidney Dis. 2020;75:561–562.[↩]
- Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844[↩][↩]
- Harris RC, Breyer MD. Update on cyclooxygenase-2 inhibitors. Clin J Am Soc Nephrol. 2006;1:236–245. doi: 10.2215/CJN.00890805[↩]
- Lin W, Mou L, Tu H, et al. Clinical analysis of hyperkalemic renal tubular acidosis caused by calcineurin inhibitors in solid organ transplant recipients. J Clin Pharm Ther. 2017;42:122–124. doi: 10.1111/jcpt.12485[↩]
- Riveiro-Barciela M, Campos-Varela I, Tovar JL, et al. Hyperkalemic distal renal tubular acidosis caused by immunosuppressant treatment with tacrolimus in a liver transplant patient: case report. Transplant Proc. 2011;43:4016–4018. doi: 10.1016/j.transproceed.2011.09.064[↩]
- Schmoyer C, Mishra S, Fulco F. Tacrolimus-induced type IV renal tubular acidosis following liver transplantation. Case Rep Hepatol. 2017;2017:9312481[↩][↩]
- Tumlin JA, Sands JM. Nephron segment-specific inhibition of Na+/K+-ATPase activity by cyclosporin A. Kidney Int. 1993;43:246–251. doi: 10.1038/ki.1993.38[↩]
- Heering PJ, Kurschat C, Vo DT, Klein-Vehne N, Fehsel K, Ivens K. Aldosterone resistance in kidney transplantation is in part induced by a down-regulation of mineralocorticoid receptor expression. Clin Transplant. 2004;18:186–192. doi: 10.1046/j.1399-0012.2003.00154.x[↩]
- Adrenal Insufficiency & Addison’s Disease. https://www.niddk.nih.gov/health-information/endocrine-diseases/adrenal-insufficiency-addisons-disease[↩]
- Santos F, Ordonez FA, Claramunt-Taberner D, Gil-Pena H. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol. 2015;30:2099–2107. doi: 10.1007/s00467-015-3083-9[↩]
- Kyono Y, Nozu K, Nakagawa T, et al. Combination of furosemide and fludrocortisone as a loading test for diagnosis of distal renal tubular acidosis in a pediatric case. CEN Case Rep. 2020;9:81–86. doi: 10.1007/s13730-019-00432-1[↩]
- Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007;71:1310–1316. doi: 10.1038/sj.ki.5002220[↩]
- Kim S, Lee JW, Park J, et al. The urine-blood PCO2 gradient as a diagnostic index of H+-ATPase defect distal renal tubular acidosis. Kidney Int. 2004;66:761–767. doi: 10.1111/j.1523-1755.2004.00801.x[↩][↩]
- Overview and pathophysiology of renal tubular acidosis and the effect on potassium balance. https://www.uptodate.com/contents/overview-and-pathophysiology-of-renal-tubular-acidosis-and-the-effect-on-potassium-balance[↩][↩]
- Stinebaugh BJ, Schloeder FX, Tam SC, Goldstein MB, Halperin ML. Pathogenesis of distal renal tubular acidosis. Kidney Int. 1981;19:1–7. doi: 10.1038/ki.1981.1[↩]
- Sidler M, Mohebbi N, Hoorn EJ, Wagner CA. Gut it out: laxative abuse mimicking distal renal tubular acidosis. Kidney Blood Press Res. 2019;44:1294–1299. doi: 10.1159/000501855[↩]
- Patel S, Sharma S. Respiratory Acidosis. [Updated 2022 Jun 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482430[↩]
- Rajkumar P, Pluznick JL. Acid-base regulation in the renal proximal tubules: using novel pH sensors to maintain homeostasis. Am J Physiol Renal Physiol. 2018 Nov 1;315(5):F1187-F1190. doi: 10.1152/ajprenal.00185.2018[↩]
- Pandey DG, Sharma S. Biochemistry, Anion Gap. [Updated 2019 Apr 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539757[↩][↩][↩]
- Gamble JL. Extracellular fluid and its maintenance. N Engl J Med. 1936;250:1150–1152.[↩]
- Berend K, de Vries AP, Gans RO. Physiological approach to assessment of acid-base disturbances. N Engl J Med. 2015 Jan 8;372(2):195. doi: 10.1056/NEJMc1413880[↩]
- Hopkins E, Sanvictores T, Sharma S. Physiology, Acid Base Balance. [Updated 2022 Sep 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507807[↩][↩]
- Burger MK, Schaller DJ. Metabolic Acidosis. [Updated 2022 Jul 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482146[↩]
- Marano M. Sobre el uso de la fórmula de Winters en la acidosis metabólica crónica [On the use of Winters’ formula in chronic metabolic acidosis]. Rev Psiquiatr Salud Ment. 2015 Jan-Mar;8(1):45-6. Spanish. doi: 10.1016/j.rpsm.2014.07.006[↩]
- Pereira PC, Miranda DM, Oliveira EA, Silva AC. Molecular pathophysiology of renal tubular acidosis. Curr Genomics. 2009 Mar;10(1):51-9. doi: 10.2174/138920209787581262[↩]
- Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007 Jun;71(12):1310-6. doi: 10.1038/sj.ki.5002220[↩]
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol. 2009 Summer;11(3):134-44. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2777061[↩][↩][↩]
- Mohebbi N, Wagner CA. Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol. 2018;31:511–522. doi: 10.1007/s40620-017-0447-1[↩]
- Karatzas A, Paridis D, Kozyrakis D, Tzortzis V, Samarinas M, Dailiana Z, Karachalios T. Fanconi syndrome in the adulthood. The role of early diagnosis and treatment. J Musculoskelet Neuronal Interact. 2017 Dec 1;17(4):303-306. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5749037[↩][↩]
- Sebastian A, McSherry E, Morris RC., Jr On the mechanism of renal potassium wasting in renal tubular acidosis associated with the Fanconi syndrome (type 2 RTA) J Clin Invest. 1971;50:231–243. doi: 10.1172/JCI106479[↩]
- Nash MA, Torrado AD, Greifer I, Spitzer A, Edelmann CM Jr. Renal tubular acidosis in infants and children. Clinical course, response to treatment, and prognosis. J Pediatr. 1972 May;80(5):738-48. doi: 10.1016/s0022-3476(72)80124-0[↩]
- Raphael KL. Metabolic acidosis in CKD: core curriculum 2019. Am J Kidney Dis. 2019;74:263–275. doi: 10.1053/j.ajkd.2019.01.036[↩][↩][↩][↩]
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004;351:585–592. doi: 10.1056/NEJMra035279[↩]
- Dobbin SJH, Petrie JR, Lean MEJ, McKay GA. Fludrocortisone therapy for persistent hyperkalaemia. Diabet Med. 2017;34:1005–1008. doi: 10.1111/dme.13359[↩]
- Kidney Disease Improving Global Outcomes KDIGO clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150. doi: 10.1038/kisup.2012.73[↩]
- Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ. Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double-blind, placebo-controlled study in patients with chronic heart failure (the PEARL-HF) trial. Eur Heart J. 2011;32:820–828. doi: 10.1093/eurheartj/ehq502[↩]
- Stavros F, Yang A, Leon A, Nuttall M, Rasmussen HS. Characterization of structure and function of ZS-9, a K+ selective ion trap. PLoS One. 2014;9:e114686. doi: 10.1371/journal.pone.0114686[↩]
- Roger SD, Lavin PT, Lerma EV, et al. Long-term safety and efficacy of sodium zirconium cyclosilicate for hyperkalaemia in patients with mild/moderate versus severe/end-stage chronic kidney disease: comparative results from an open-label, phase 3 study. Nephrol Dial Transplant. 2020 doi: 10.1093/ndt/gfz285[↩][↩]
- Roger SD, Spinowitz BS, Lerma EV, et al. Sodium zirconium cyclosilicate increases serum bicarbonate concentrations among patients with hyperkalaemia: exploratory analyses from three randomized, multi-dose, placebo-controlled trials. Nephrol Dial Transplant. 2020 doi: 10.1093/ndt/gfaa158[↩]
- Carrero JJ, Gonzalez-Ortiz A, Avesani CM, et al. Plant-based diets to manage the risks and complications of chronic kidney disease. Nat Rev Nephrol. 2020;16:525–542. doi: 10.1038/s41581-020-0297-2[↩]
- Navaneethan SD, Shao J, Buysse J, Bushinsky DA. Effects of treatment of metabolic acidosis in CKD: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2019;14:1011–1020. doi: 10.2215/CJN.13091118[↩]
- Meschi T, Maggiore U, Fiaccadori E, et al. The effect of fruits and vegetables on urinary stone risk factors. Kidney Int. 2004;66:2402–2410. doi: 10.1111/j.1523-1755.2004.66029.x[↩]
- Goraya N, Wesson DE. Management of the metabolic acidosis of chronic kidney disease. Adv Chronic Kidney Dis. 2017;24:298–304. doi: 10.1053/j.ackd.2017.06.006[↩]




{kind=link}