Contents
- What is X chromosome
What is X chromosome
The X chromosome is one of the two sex chromosomes in humans (the other is the Y chromosome). The X chromosome is a sex chromosome that determines your sex. In females, these are two Xs (XX). In males they are an X and a Y (XY). The X chromosome spans about 155 million DNA building blocks (base pairs) and represents approximately 5 percent of the total DNA in cells. The X chromosome has hundreds of genes on it and many of these are controlling characters related to organs other than the reproductive system. Genes are sections of DNA that are passed from generation to generation and perform one function. For example, gene for colorblindness present on X, is responsible for maintenance of photoreceptors in healthy cone cells of your retina. Then there are genes on X chromosome which allow hepatic cells to produce clotting factors; hence mutation in such X linked genes cause hemophilia.
The sex chromosomes form one of the 23 pairs of human chromosomes in each cell. Each person normally has one pair of sex chromosomes in each cell. Women have two ‘X’ chromosomes (XX) for their 23rd pair, and men have one ‘X’ chromosome and one ‘Y’ chromosome (XY). Early in embryonic development in females, one of the two X chromosomes is randomly and permanently inactivated in cells other than egg cells. This phenomenon is called X-inactivation or lyonization. X-inactivation ensures that females, like males, have one functional copy of the X chromosome in each body cell. Because X-inactivation is random, in normal females the X chromosome inherited from the mother is active in some cells, and the X chromosome inherited from the father is active in other cells.
Some genes on the X chromosome escape X-inactivation. Many of these genes are located at the ends of each arm of the X chromosome in areas known as the pseudoautosomal regions. Although many genes are unique to the X chromosome, genes in the pseudoautosomal regions are present on both sex chromosomes. As a result, men and women each have two functional copies of these genes. Many genes in the pseudoautosomal regions are essential for normal development.
Identifying genes on each chromosome is an active area of genetic research. Because researchers use different approaches to predict the number of genes on each chromosome, the estimated number of genes varies. The X chromosome likely contains 800 to 900 genes that provide instructions for making proteins. These proteins perform a variety of different roles in the body.
Figure 1. Human karyotype (male/female chromosomes, also called a karyotype)
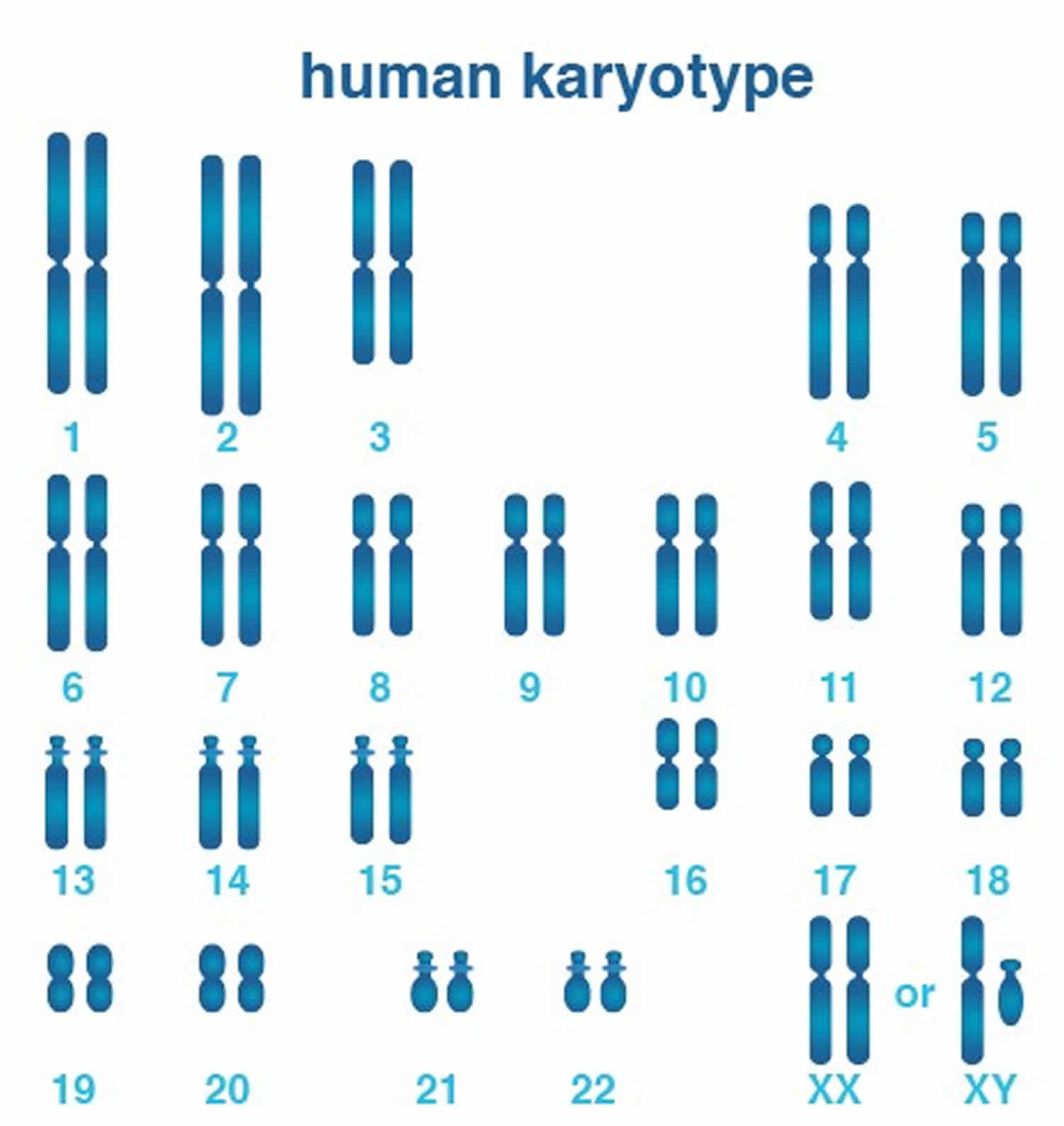
Figure 2. Sex chromosomes
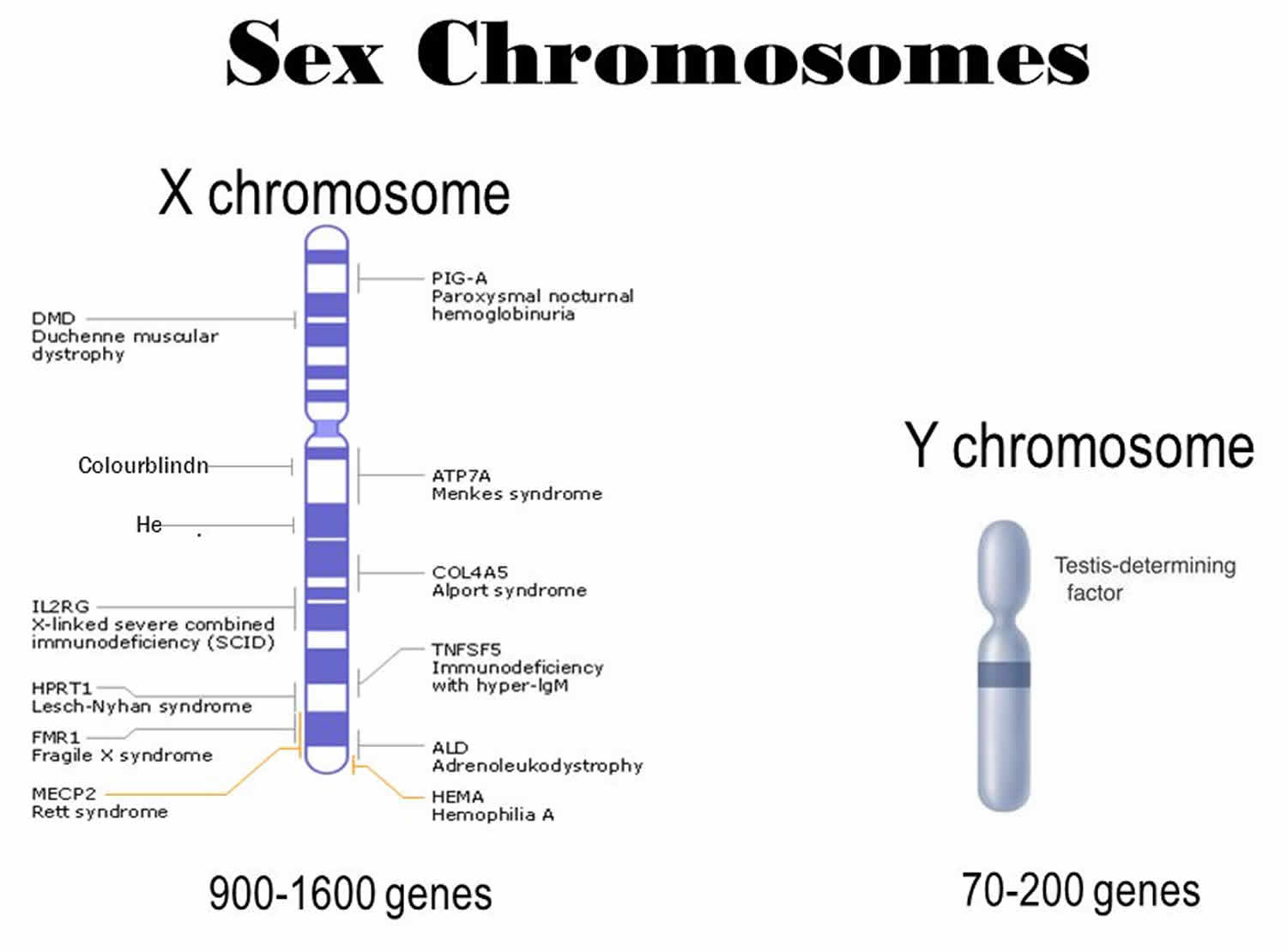
X chromosome disorders
The following chromosomal conditions are associated with changes in the structure or number of copies of X chromosome.
- 46,XX testicular disorder of sex development
- 48,XXXY syndrome
- 48,XXYY syndrome
- 49,XXXXY syndrome
- Intestinal pseudo-obstruction
- Klinefelter syndrome
- Microphthalmia with linear skin defects syndrome
- Triple X syndrome
- Turner syndrome
- X-linked acrogigantism
- Other chromosomal conditions
46,XX testicular disorder of sex development
46,XX testicular disorder of sex development is a condition in which individuals with two X chromosomes in each cell, the pattern normally found in females, have a male appearance. In most individuals with 46,XX testicular disorder of sex development, the condition results from an abnormal exchange of genetic material between chromosomes (translocation). This exchange occurs as a random event during the formation of sperm cells in the affected person’s father. The translocation affects the gene responsible for development of a fetus into a male (the SRY gene). The SRY gene, which is normally found on the Y chromosome, is misplaced in this disorder, almost always onto an X chromosome. A fetus with an X chromosome that carries the SRY gene will develop as a male despite not having a Y chromosome.
Approximately 1 in 20,000 individuals with a male appearance have 46,XX testicular disorder.
People with 46,XX testicular disorder of sex development have male external genitalia. They generally have small testes and may also have abnormalities such as undescended testes (cryptorchidism) or the urethra opening on the underside of the penis (hypospadias). A small number of affected people have external genitalia that do not look clearly male or clearly female (ambiguous genitalia). Affected children are typically raised as males and have a male gender identity.
At puberty, most affected individuals require treatment with the male sex hormone testosterone to induce development of male secondary sex characteristics such as facial hair and deepening of the voice (masculinization). Hormone treatment can also help prevent breast enlargement (gynecomastia). Adults with this disorder are usually shorter than average for males and are unable to have children (infertile).
46,XX testicular disorder of sex development causes
People normally have 46 chromosomes in each cell. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male or female sex characteristics. Females typically have two X chromosomes (46,XX), and males usually have one X chromosome and one Y chromosome (46,XY).
The SRY gene, normally located on the Y chromosome, provides instructions for making the sex-determining region Y protein. The sex-determining region Y protein causes a fetus to develop as a male.
In about 80 percent of individuals with 46,XX testicular disorder of sex development, the condition results from an abnormal exchange of genetic material between chromosomes (translocation). This exchange occurs as a random event during the formation of sperm cells in the affected person’s father. The translocation causes the SRY gene to be misplaced, almost always onto an X chromosome. If a fetus is conceived from a sperm cell with an X chromosome bearing the SRY gene, it will develop as a male despite not having a Y chromosome. This form of the condition is called SRY-positive 46,XX testicular disorder of sex development.
About 20 percent of people with 46,XX testicular disorder of sex development do not have the SRY gene. This form of the condition is called SRY-negative 46,XX testicular disorder of sex development. The cause of the disorder in these individuals is often unknown, although changes affecting other genes have been identified. Individuals with SRY-negative 46,XX testicular disorder of sex development are more likely to have ambiguous genitalia than are people with the SRY-positive form.
46,XX testicular disorder of sex development inheritance pattern
SRY-positive 46,XX testicular disorder of sex development is almost never inherited. This condition results from the translocation of a Y chromosome segment containing the SRY gene during the formation of sperm (spermatogenesis). Affected people typically have no history of the disorder in their family and cannot pass on the disorder because they are infertile.
In rare cases, the SRY gene may be misplaced onto a chromosome other than the X chromosome. This translocation may be carried by an unaffected father and passed on to a child with two X chromosomes, resulting in 46,XX testicular disorder of sex development. In another very rare situation, a man may carry the SRY gene on both the X and Y chromosome; a child who inherits his X chromosome will develop male sex characteristics despite having no Y chromosome.
The inheritance pattern of SRY-negative 46,XX testicular disorder of sex development is unknown. A few families with unaffected parents have had more than one child with the condition, suggesting the possibility of autosomal recessive inheritance. Autosomal recessive means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
46,XX testicular disorder of sex development symptoms
The symptoms associated with 46,XX testicular disorder of sex development will depend on the underlying cause. They may include:
- Ambiguous genitalia at birth
- Decreased testicular size
- Male hypogonadism
- Polycystic ovaries
- Ovotestis
- Azoospermia
- Bifid scrotum
- Decreased serum testosterone level
- Hypoplasia of the vagina
- Micropenis
- Scrotal hypoplasia
- Perineal hypospadias
- Sex reversal
- True hermaphroditism
48,XXXY syndrome
48,XXXY syndrome is a chromosomal condition in boys and men that causes intellectual disability, developmental delays, physical differences, and an inability to father biological children (infertility). This condition results from having two extra X chromosomes in each cell. Boys and men with 48,XXXY syndrome have the usual single Y chromosome plus three copies of the X chromosome, for a total of 48 chromosomes in each cell.
Having extra copies of multiple genes on the X chromosome affects many aspects of development, including sexual development before birth and at puberty. Researchers are working to determine which genes contribute to the specific developmental and physical differences that occur with 48,XXXY syndrome.
48,XXXY syndrome is sometimes described as a variant of Klinefelter syndrome (described below). However, the features of 48,XXXY syndrome tend to be more severe than those of Klinefelter syndrome and affect more parts of the body. As doctors and researchers have learned more about the differences between these sex chromosome disorders, they have started to consider them as separate conditions.
48,XXXY syndrome affects between 1 in 17,000 and 1 in 50,000 newborn boys. It is a relatively uncommon sex chromosome disorder, which is a group of conditions caused by changes in the number of sex chromosomes (the X chromosome and the Y chromosome).
48,XXXY syndrome signs and symptoms vary among affected individuals. Most boys and men with 48,XXXY syndrome have mild intellectual disability with learning difficulties. Speech and language development is particularly affected. Most affected boys and men can understand what other people say more easily than they themselves can speak. The problems with speech and communication can contribute to behavioral issues, including irritability and outbursts or temper tantrums. Boys and men with 48,XXXY syndrome tend to have anxiety, a short attention span, and impaired social skills.
48,XXXY syndrome is also associated with weak muscle tone (hypotonia) and problems with coordination that delay the development of motor skills, such as sitting, standing, and walking. Affected boys and men tend to be taller than their peers, with an average adult height of over 6 feet.
Other physical differences associated with 48,XXXY syndrome include abnormal fusion of certain bones in the forearm (radioulnar synostosis), an unusually large range of joint movement (hyperextensibility), elbow abnormalities, curved pinky fingers (fifth finger clinodactyly), and flat feet (pes planus). Affected individuals may have distinctive facial features, including widely spaced eyes (ocular hypertelorism), outside corners of the eyes that point upward (upslanting palpebral fissures), and skin folds covering the inner corner of the eyes (epicanthal folds). However, some boys and men with 48,XXXY syndrome do not have these differences in their facial features.
48,XXXY syndrome disrupts male sexual development. The penis is shorter than usual, and the testes may be undescended, which means they are abnormally located inside the pelvis or abdomen. The testes are small and do not produce enough testosterone, which is the hormone that directs male sexual development. The shortage of testosterone often leads to incomplete puberty. Starting in adolescence, affected boys and men may have sparse body hair, and some experience breast enlargement (gynecomastia). Their testes typically do not produce sperm, so most men with this condition are infertile.
48,XXXY syndrome is not inherited; it occurs as a random event during the formation of reproductive cells (eggs or sperm) in one of an affected person’s parents. During cell division, an error called nondisjunction prevents X chromosomes from being distributed normally among reproductive cells as they form. Typically, as cells divide, each egg cell gets a single X chromosome, and each sperm cell gets either an X chromosome or a Y chromosome. However, because of nondisjunction, an egg cell or a sperm cell can also end up with two extra copies of the X chromosome.
If an egg cell with two extra X chromosomes (XXX) is fertilized by a sperm cell with one Y chromosome, the resulting child will have 48,XXXY syndrome. Similarly, if a sperm cell with a Y chromosome and two extra X chromosomes (XXY) fertilizes an egg cell with a single X chromosome, the resulting child will have 48,XXXY syndrome.
48,XXXY syndrome causes
48,XXXY syndrome is a sex chromosome disorder in boys and men that results from having two extra X chromosomes in each cell. People typically have 46 chromosomes in each cell, two of which are the sex chromosomes. Females have two X chromosomes (46,XX), and males have one X and one Y chromosome (46,XY). Boys and men with 48,XXXY syndrome have the usual single Y chromosome, but they have three copies of the X chromosome, for a total of 48 chromosomes in each cell.
Boys and men with 48,XXXY syndrome have extra copies of multiple genes on the X chromosome. The activity of these extra genes affects many aspects of development, including sexual development before birth and at puberty. Researchers are working to determine which genes contribute to the specific developmental and physical differences that occur with 48,XXXY syndrome.
48,XXXY syndrome is sometimes described as a variant of another sex chromosome disorder called Klinefelter syndrome. Boys and men with Klinefelter syndrome have one extra copy of the X chromosome, for a total of 47 chromosomes in each cell (47,XXY). Like 48,XXXY syndrome, Klinefelter syndrome affects male sexual development and can be associated with learning disabilities and problems with speech and language development. However, the features of 48,XXXY syndrome tend to be more severe than those of Klinefelter syndrome and affect more parts of the body. As doctors and researchers have learned more about the differences between these sex chromosome disorders, they have started to consider them as separate conditions.
48,XXYY syndrome
48,XXYY syndrome is a chromosomal condition that causes infertility, developmental and behavioral disorders, and other health problems in affected boys and men. 48,XXYY syndrome is caused by the presence of an extra X chromosome and an extra Y chromosome in a male’s cells. Extra genetic material from the X chromosome interferes with male sexual development, preventing the testes from functioning normally and reducing the levels of testosterone (a hormone that directs male sexual development) in adolescent and adult males. Extra copies of genes from the pseudoautosomal regions of the extra X and Y chromosomes contribute to the signs and symptoms of 48,XXYY syndrome; however, the specific genes have not been identified.
48,XXYY disrupts male sexual development. Adolescent and adult males with this condition typically have small testes that do not produce enough testosterone, which is the hormone that directs male sexual development. A shortage of testosterone during puberty can lead to reduced facial and body hair, poor muscle development, low energy levels, and an increased risk for breast enlargement (gynecomastia). Because their testes do not function normally, males with 48, XXYY syndrome have an inability to father children (infertility).
48,XXYY syndrome can affect other parts of the body as well. Males with 48,XXYY syndrome are often taller than other males their age with an average adult height of 6 feet 4 inches (193 cm). They tend to develop a tremor that typically starts in adolescence and increases with age. Dental problems are frequently seen with this condition; they include delayed appearance of the primary (baby) or secondary (adult) teeth, thin tooth enamel, crowded and/or misaligned teeth, and multiple cavities. As affected males get older, they may develop a narrowing of the blood vessels in the legs, called peripheral vascular disease. Peripheral vascular disease can cause skin ulcers to form. Affected males are also at risk for developing a type of clot called a deep vein thrombosis (DVT) that occurs in the deep veins of the legs. Additionally, males with 48,XXYY syndrome may have flat feet (pes planus), elbow abnormalities, abnormal fusion of certain bones in the forearm (radioulnar synostosis), allergies, asthma, type 2 diabetes, seizures, and congenital heart defects.
Most males with 48,XXYY syndrome have an IQ that ranges from 70-80 with some degree of difficulty with speech and language development. Learning disabilities, especially those that are language-based, are very common in males with this disorder. Affected males seem to perform better at tasks focused on math, visual-spatial skills such as puzzles, and memorization of locations or directions. Some boys with 48,XXYY syndrome have delayed development of motor skills such as sitting, standing, and walking that can lead to poor coordination. Affected males have higher than average rates of behavioral disorders, such as attention deficit hyperactivity disorder (ADHD); mood disorders, including anxiety and bipolar disorder; and autism spectrum disorder, which affects communication and social interaction.
48,XXYY syndrome is estimated to affect 1 in 18,000 to 40,000 males.
48,XXYY syndrome is not inherited; it usually occurs as a random event during the formation of reproductive cells (eggs and sperm). An error in cell division called nondisjunction results in a reproductive cell with an abnormal number of chromosomes. In 48,XXYY syndrome, the extra sex chromosomes almost always come from a sperm cell. Nondisjunction may cause a sperm cell to gain two extra sex chromosomes, resulting in a sperm cell with three sex chromosomes (one X and two Y chromosomes). If that sperm cell fertilizes a normal egg cell with one X chromosome, the resulting child will have two X chromosomes and two Y chromosomes in each of the body’s cells.
In a small percentage of cases, 48,XXYY syndrome results from nondisjunction of the sex chromosomes in a 46,XY embryo very soon after fertilization has occurred. This means that a normal sperm cell with one Y chromosome fertilized a normal egg cell with one X chromosome, but right after fertilization nondisjunction of the sex chromosomes caused the embryo to gain two extra sex chromosomes, resulting in a 48,XXYY embryo.
48,XXYY syndrome causes
48,XXYY syndrome is a condition related to the X and Y chromosomes (the sex chromosomes). People normally have 46 chromosomes in each cell. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male or female sex characteristics. Females typically have two X chromosomes (46,XX), and males have one X chromosome and one Y chromosome (46,XY). 48,XXYY syndrome results from the presence of an extra copy of both sex chromosomes in each of a male’s cells (48,XXYY). Extra copies of genes on the X chromosome interfere with male sexual development, preventing the testes from functioning normally and reducing the levels of testosterone. Many genes are found only on the X or Y chromosome, but genes in areas known as the pseudoautosomal regions are present on both sex chromosomes. Extra copies of genes from the pseudoautosomal regions of the extra X and Y chromosome contribute to the signs and symptoms of 48,XXYY syndrome; however, the specific genes have not been identified.
49,XXXXY syndrome
49,XXXXY syndrome is a chromosomal condition in boys and men that causes intellectual disability, developmental delays (especially in speech and language), physical differences, and an inability to father biological children (infertility). This condition results from having three extra X chromosomes in each cell. Boys and men with 49,XXXXY syndrome have the usual single Y chromosome plus four copies of the X chromosome, for a total of 49 chromosomes in each cell.
Having extra copies of multiple genes on the X chromosome affects many aspects of development, including sexual development before birth and at puberty. Researchers are working to determine which genes contribute to the specific developmental and physical differences that occur with 49,XXXXY syndrome.
49,XXXXY syndrome is sometimes described as a variant of Klinefelter syndrome (described below). However, the features of 49,XXXXY syndrome tend to be more severe than those of Klinefelter syndrome and affect more parts of the body. As doctors and researchers have learned more about the differences between these sex chromosome disorders, they have started to consider them as separate conditions.
49,XXXXY syndrome signs and symptoms vary among affected individuals.
Boys and men with 49,XXXXY syndrome have mild or moderate intellectual disability with learning difficulties. Speech and language development is particularly affected. Most affected boys and men can understand what other people say more easily than they themselves can speak. People with 49,XXXXY syndrome tend to be shy and friendly, but problems with speech and communication can contribute to behavioral issues, including irritability, difficulty tolerating frustration, defiant behavior, and outbursts or temper tantrums.
49,XXXXY syndrome is also associated with weak muscle tone (hypotonia) and problems with coordination that delay the development of motor skills, such as sitting, standing, and walking. Affected infants and young boys are often shorter than their peers, but some catch up in height later in childhood or adolescence.
Other physical differences associated with 49,XXXXY syndrome include abnormal fusion of certain bones in the forearm (radioulnar synostosis), an unusually large range of joint movement (hyperextensibility), elbow abnormalities, curved pinky fingers (fifth finger clinodactyly), and flat feet (pes planus). Affected individuals have distinctive facial features that can include widely spaced eyes (ocular hypertelorism), outside corners of the eyes that point upward (upslanting palpebral fissures), skin folds covering the inner corner of the eyes (epicanthal folds), and a flat bridge of the nose. Dental abnormalities are also common in this disorder.
49,XXXXY syndrome disrupts male sexual development. The penis is often short and underdeveloped, and the testes may be undescended, which means they are abnormally located inside the pelvis or abdomen. The testes are small and do not produce enough testosterone, which is the hormone that directs male sexual development. The shortage of testosterone often leads to incomplete puberty. Starting in adolescence, affected boys and men may have sparse body hair, and some experience breast enlargement (gynecomastia). Their testes do not produce sperm, so all men with 49,XXXXY syndrome are infertile.
49,XXXXY syndrome affects an estimated 1 in 85,000 to 100,000 newborn boys. It is among the rarest of the sex chromosome disorders, which are conditions caused by changes in the number of sex chromosomes (the X chromosome and the Y chromosome).
49,XXXXY syndrome is not inherited; it occurs as a random event during the formation of reproductive cells (eggs) in an affected person’s mother. During cell division, an error called nondisjunction prevents X chromosomes from being distributed among egg cells as they form. Typically, as cells divide, each egg cell gets a single X chromosome. However, because of nondisjunction, a single egg cell can end up with four X chromosomes that would usually have been distributed among four separate egg cells. If a sperm cell containing a single Y chromosome fertilizes this egg cell, the resulting child will have four X chromosomes and one Y chromosome (49,XXXXY) in each of the body’s cells.
49,XXXXY syndrome causes
49,XXXXY syndrome is a sex chromosome disorder in boys and men that results from having three extra X chromosomes in each cell. People typically have 46 chromosomes in each cell, two of which are the sex chromosomes. Females have two X chromosomes (46,XX), and males have one X and one Y chromosome (46,XY). Boys and men with 49,XXXXY syndrome have the usual single Y chromosome, but they have four copies of the X chromosome, for a total of 49 chromosomes in each cell.
Boys and men with 49,XXXXY syndrome have extra copies of multiple genes on the X chromosome. The activity of these extra genes affects many aspects of development, including sexual development before birth and at puberty. Researchers are working to determine which genes contribute to the specific developmental and physical differences that occur with 49,XXXXY syndrome.
49,XXXXY syndrome is sometimes described as a variant of another sex chromosome disorder called Klinefelter syndrome. Boys and men with Klinefelter syndrome have one extra copy of the X chromosome, for a total of 47 chromosomes in each cell (47,XXY). Like 49,XXXXY syndrome, Klinefelter syndrome affects male sexual development and can be associated with learning disabilities and problems with speech and language development. However, the features of 49,XXXXY syndrome tend to be more severe than those of Klinefelter syndrome and affect more parts of the body. As doctors and researchers have learned more about the differences between these sex chromosome disorders, they have started to consider them as separate conditions.
Fragile X chromosome
Fragile X syndrome is a genetic condition, which results from mutations in a FMR1 gene on the X chromosome, is the most commonly inherited form of developmental and intellectual disability. Fragile X syndrome causes a range of developmental problems including learning disabilities and cognitive impairment. Usually, males are more severely affected by this disorder than females.
Affected individuals usually have delayed development of speech and language by age 2. Most males with fragile X syndrome have mild to moderate intellectual disability, while about one-third of affected females are intellectually disabled. Children with fragile X syndrome may also have anxiety and hyperactive behavior such as fidgeting or impulsive actions. They may have attention deficit disorder (ADD), which includes an impaired ability to maintain attention and difficulty focusing on specific tasks. About one-third of individuals with fragile X syndrome have features of autism spectrum disorder that affect communication and social interaction. Seizures occur in about 15 percent of males and about 5 percent of females with fragile X syndrome.
Most males and about half of females with fragile X syndrome have characteristic physical features that become more apparent with age. These features include a long and narrow face, large ears, a prominent jaw and forehead, unusually flexible fingers, flat feet, and in males, enlarged testicles (macroorchidism) after puberty.
Fragile X syndrome occurs in approximately 1 in 4,000 males and 1 in 8,000 females.
Fragile X chromosome causes
Mutations in the FMR1 gene cause fragile X syndrome. The FMR1 gene provides instructions for making a protein called FMRP. This protein helps regulate the production of other proteins and plays a role in the development of synapses, which are specialized connections between nerve cells. Synapses are critical for relaying nerve impulses.
Nearly all cases of fragile X syndrome are caused by a mutation in which a DNA segment, known as the CGG triplet repeat, is expanded within the FMR1 gene. Normally, this DNA segment is repeated from 5 to about 40 times. In people with fragile X syndrome, however, the CGG segment is repeated more than 200 times. The abnormally expanded CGG segment turns off (silences) the FMR1 gene, which prevents the gene from producing FMRP. Loss or a shortage (deficiency) of this protein disrupts nervous system functions and leads to the signs and symptoms of fragile X syndrome.
Males and females with 55 to 200 repeats of the CGG segment are said to have an FMR1 gene premutation. Most people with a premutation are intellectually normal. In some cases, however, individuals with a premutation have lower than normal amounts of FMRP. As a result, they may have mild versions of the physical features seen in fragile X syndrome (such as prominent ears) and may experience emotional problems such as anxiety or depression. Some children with a premutation may have learning disabilities or autistic-like behavior. The premutation is also associated with an increased risk of disorders called fragile X-associated primary ovarian insufficiency (FXPOI) and fragile X-associated tremor/ataxia syndrome (FXTAS).
Fragile X chromosome inheritance pattern
Fragile X syndrome is inherited in an X-linked dominant pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. (The Y chromosome is the other sex chromosome.) The inheritance is dominant if one copy of the altered gene in each cell is sufficient to cause the condition. X-linked dominant means that in females (who have two X chromosomes), a mutation in one of the two copies of a gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of a gene in each cell causes the disorder. In most cases, males experience more severe symptoms of the disorder than females.
In women, the FMR1 gene premutation on the X chromosome can expand to more than 200 CGG repeats in cells that develop into eggs. This means that women with the premutation have an increased risk of having a child with fragile X syndrome. By contrast, the premutation in men does not expand to more than 200 repeats as it is passed to the next generation. Men pass the premutation only to their daughters. Their sons receive a Y chromosome, which does not include the FMR1 gene.
The gene for Fragile X syndrome is carried on the X chromosome. Because both males (XY) and females (XX) have at least one X chromosome, both can pass on the mutated gene to their children.
- A father with the altered gene for Fragile X on his X chromosome will pass that gene on only to his daughters. To his sons he will pass on a Y chromosome, which doesn’t transmit Fragile X syndrome. Therefore, a father with the altered gene on his X chromosome and a mother with normal X chromosomes would have daughters with the altered gene for Fragile X, while none of their sons would have the mutated gene.
- A father can pass on the premutation form of the FMR1 gene to his daughters but not the full mutation. Even if the father himself has a full mutation of this gene, it appears that sperm can carry only the premutation. Scientists don’t understand how or why fathers can pass on only the milder form of Fragile X to their daughters. This remains an area of focused research.
- Mothers pass on only X chromosomes to their children, so if a mother has the altered gene for Fragile X, she can pass that gene to either her sons or her daughters. If a mother has the mutated gene on one X chromosome and has one normal X chromosome, and the father has no mutations, all the children have a 50-50 chance of inheriting the mutated gene.
- These 50-50 odds apply for each child the parents have. Having one child with the FMR1 mutation does not increase or decrease the chances of having another child with the mutated FMR1 gene. This is also true for the severity of the symptoms. Having one child with mild symptoms does not mean that the other children will have severe symptoms, and having a child with severe symptoms does not mean that the other children will have mild symptoms.
Fragile X chromosome symptoms
People with fragile X syndrome do not all have the same signs and symptoms, but they do have some things in common. Symptoms are often milder in females than in males.
- Intelligence and learning. Many people with Fragile X have problems with intellectual functioning.
- These problems can range from the mild, such as learning disorders or problems with mathematics, to the severe, such as an intellectual or developmental disability.
- The fragile X syndrome may affect the ability to think, reason, and learn.
- Because many people with fragile X syndrome also have attention disorders, hyperactivity, anxiety, and language-processing problems, a person with Fragile X may have more capabilities than his or her IQ (intelligence quotient) score suggests.
- Physical. Most infants and younger children with Fragile X don’t have any specific physical features of this syndrome. When these children start to go through puberty, however, many will begin to develop certain features that are typical of those with Fragile X.
- These features include a narrow face, large head, large ears, flexible joints, flat feet, and a prominent forehead.
- These physical signs become more obvious with age.
- Behavioral, social, and emotional. Most children with Fragile X have some behavioral challenges.
- They may be afraid or anxious in new situations.
- They may have trouble making eye contact with other people.
- Boys, especially, may have trouble paying attention or be aggressive.
- Girls may be shy around new people. They may also have attention disorders and problems with hyperactivity.
- Speech and language. Most boys with Fragile X have some problems with speech and language.
- They may have trouble speaking clearly, may stutter, or may leave out parts of words. They may also have problems understanding other people’s social cues, such as tone of voice or specific types of body language.
- Girls usually do not have severe problems with speech or language.
- Some children with Fragile X begin talking later than typically developing children. Most will talk eventually, but a few might stay nonverbal throughout their lives.
- Sensory. Many children with Fragile X are bothered by certain sensations, such as bright light, loud noises, or the way certain clothing feels on their bodies.
These sensory issues might cause them to act out or display behavior problems.
Fragile X chromosome diagnosis
Health care providers often use a blood sample to diagnose Fragile X syndrome. The health care provider will take a sample of blood and will send it to a laboratory, which will determine what form of the FMR1 gene is present 1.
Prenatal Testing (During Pregnancy)
Pregnant women who have an FMR1 premutation or full mutation may pass that mutated gene on to their children. A prenatal test allows health care providers to detect the mutated gene in the developing fetus. This important information helps families and providers to prepare for Fragile X syndrome and to intervene as early as possible.
Possible types of prenatal tests include:
- Amniocentesis. A health care provider takes a sample of amniotic fluid, which is then tested for the FMR1 mutation.
- Chorionic villus sampling. A health care provider takes a sample of cells from the placenta, which is then tested for the FMR1 mutation 1.
Because prenatal testing involves some risk to the mother and fetus, if you or a family member is considering prenatal testing for Fragile X, discuss all the risks and benefits with your health care provider.
Prenatal testing is not very common, and many parents do not know they carry the mutation. Therefore, parents usually start to notice symptoms in their children when they are infants or toddlers. The average age at diagnosis is 36 months for boys and 42 months for girls 2.
Diagnosis of Children
Many parents first notice symptoms of delayed development in their infants or toddlers. These symptoms may include delays in speech and language skills, social and emotional difficulties, and being sensitive to certain sensations. Children may also be delayed in or have problems with motor skills such as learning to walk.
A health care provider can perform developmental screening to determine the nature of delays in a child. If a health care provider suspects the child has Fragile X syndrome, he/she can refer parents to a clinical geneticist, who can perform a genetic test for Fragile X syndrome 2.
Fragile X chromosome treatment
There is no single treatment for Fragile X syndrome, but there are treatments that help minimize the symptoms of the condition. Individuals with Fragile X who receive appropriate education, therapy services, and medications have the best chance of using all of their individual capabilities and skills. Even those with an intellectual or developmental disability can learn to master many self-help skills.
Early intervention is important. Because a young child’s brain is still forming, early intervention gives children the best start possible and the greatest chance of developing a full range of skills. The sooner a child with Fragile X syndrome gets treatment, the more opportunity there is for learning.
Klinefelter syndrome
Klinefelter syndrome is a chromosomal condition in boys and men that can affect physical and intellectual development. It is caused by an extra copy of the X chromosome. Boys and men with Klinefelter syndrome have the usual single Y chromosome plus two copies of the X chromosome, for a total of 47 chromosomes in each cell (47,XXY). Having an extra copy of genes on the X chromosome affects many aspects of development, including sexual development before birth and at puberty. Researchers are working to determine which genes contribute to the specific developmental and physical differences that can occur with Klinefelter syndrome.
Most commonly, affected individuals are taller than average are unable to father biological children (infertile); however the signs and symptoms of Klinefelter syndrome vary among boys and men with this condition. In some cases, the features of the condition are so mild that the condition is not diagnosed until puberty or adulthood, and researchers believe that up to 75 percent of affected men and boys are never diagnosed.
Some people with features of Klinefelter syndrome have an extra X chromosome in only some of their cells; other cells have one X and one Y chromosome. In these individuals, the condition is described as mosaic Klinefelter syndrome (46,XY/47,XXY). Boys and men with mosaic Klinefelter syndrome may have milder signs and symptoms than those with the extra X chromosome in all of their cells, depending on what proportion of cells have the additional chromosome.
Several conditions resulting from the presence of more than one extra sex chromosome in each cell are sometimes described as variants of Klinefelter syndrome. These conditions include 48,XXXY syndrome and 49,XXXXY syndrome (both described above). The features of these disorders tend to be more severe than those of Klinefelter syndrome and affect more parts of the body. As doctors and researchers have learned more about the differences between these sex chromosome disorders, they have started to consider them as separate conditions.
Boys and men with Klinefelter syndrome typically have small testes that produce a reduced amount of testosterone (primary testicular insufficiency). Testosterone is the hormone that directs male sexual development before birth and during puberty. Without treatment, the shortage of testosterone can lead to delayed or incomplete puberty, breast enlargement (gynecomastia), decreased muscle mass, decreased bone density, and a reduced amount of facial and body hair. As a result of the small testes and decreased hormone production, affected males are infertile but may benefit from assisted reproductive technologies. Some affected individuals also have differences in their genitalia, including undescended testes (cryptorchidism), the opening of the urethra on the underside of the penis (hypospadias), or an unusually small penis (micropenis).
Other physical changes associated with Klinefelter syndrome are usually subtle. Older children and adults with the condition tend to be somewhat taller than their peers. Other differences can include abnormal fusion of certain bones in the forearm (radioulnar synostosis), curved pinky fingers (fifth finger clinodactyly), and flat feet (pes planus).
Children with Klinefelter syndrome may have low muscle tone (hypotonia) and problems with coordination that may delay the development of motor skills, such as sitting, standing, and walking. Affected boys often have learning disabilities, resulting in mild delays in speech and language development and problems with reading. Boys and men with Klinefelter syndrome tend to have better receptive language skills (the ability to understand speech) than expressive language skills (vocabulary and the production of speech) and may have difficulty communicating and expressing themselves.
Individuals with Klinefelter syndrome tend to have anxiety, depression, impaired social skills, behavioral problems such as emotional immaturity and impulsivity, attention deficit hyperactivity disorder (ADHD), and limited problem-solving skills (executive functioning). About 10 percent of boys and men with Klinefelter syndrome have autism spectrum disorder.
Nearly half of all men with Klinefelter syndrome develop metabolic syndrome, which is a group of conditions that include type 2 diabetes, high blood pressure (hypertension), increased belly fat, high levels of fats (lipids) such as cholesterol and triglycerides in the blood. Compared with unaffected men, adults with Klinefelter syndrome also have an increased risk of developing involuntary trembling (tremors), breast cancer (if gynecomastia develops), thinning and weakening of the bones (osteoporosis), and autoimmune disorders such as systemic lupus erythematosus and rheumatoid arthritis. (Autoimmune disorders are a large group of conditions that occur when the immune system attacks the body’s own tissues and organs.)
Klinefelter syndrome affects about 1 in 650 newborn boys. It is among the most common sex chromosome disorders, which are conditions caused by changes in the number of sex chromosomes (the X chromosome and the Y chromosome).
Klinefelter syndrome is not inherited; the addition of an extra X chromosome occurs during the formation of reproductive cells (eggs or sperm) in one of an affected person’s parents. During cell division, an error called nondisjunction prevents X chromosomes from being distributed normally among reproductive cells as they form. Typically, as cells divide, each egg cell gets a single X chromosome, and each sperm cell gets either an X chromosome or a Y chromosome. However, because of nondisjunction, an egg cell or a sperm cell can also end up with an extra copy of the X chromosome.
If an egg cell with an extra X chromosome (XX) is fertilized by a sperm cell with one Y chromosome, the resulting child will have Klinefelter syndrome. Similarly, if a sperm cell with both an X chromosome and a Y chromosome (XY) fertilizes an egg cell with a single X chromosome, the resulting child will have Klinefelter syndrome.
Mosaic Klinefelter syndrome (46,XY/47,XXY) is also not inherited. It occurs as a random error during cell division early in fetal development. As a result, some of the body’s cells have the usual one X chromosome and one Y chromosome (46,XY), and other cells have an extra copy of the X chromosome (47,XXY).
Klinefelter syndrome causes
Klinefelter syndrome is a sex chromosome disorder in boys and men that results from the presence of an extra X chromosome in cells. People typically have 46 chromosomes in each cell, two of which are the sex chromosomes. Females have two X chromosomes (46,XX), and males have one X and one Y chromosome (46,XY). Most often, boys and men with Klinefelter syndrome have the usual X and Y chromosomes, plus one extra X chromosome, for a total of 47 chromosomes (47,XXY).
Boys and men with Klinefelter syndrome have an extra copy of multiple genes on the X chromosome. The activity of these extra genes may disrupt many aspects of development, including sexual development before birth and at puberty, and are responsible for the common signs and symptoms of Klinefelter syndrome. Researchers are working to determine which genes contribute to the specific developmental and physical differences that can occur with Klinefelter syndrome.
Some people with features of Klinefelter syndrome have an extra X chromosome in only some of their cells; other cells typically have one X and one Y chromosome. (Rarely, other cells may have additional chromosome abnormalities.) In these individuals, the condition is described as mosaic Klinefelter syndrome (46,XY/47,XXY). It is thought that less than 10 percent of individuals with Klinefelter syndrome have the mosaic form. Boys and men with mosaic Klinefelter syndrome may have milder signs and symptoms than those with the extra X chromosome in all of their cells, depending on what proportion of cells have the additional chromosome.
Several conditions resulting from the presence of more than one extra sex chromosome in each cell are sometimes described as variants of Klinefelter syndrome. These conditions include 48,XXXY syndrome, 48,XXYY syndrome, and 49,XXXXY syndrome. Like Klinefelter syndrome, these conditions affect male sexual development and can be associated with learning disabilities and problems with speech and language development. However, the features of these disorders tend to be more severe than those of Klinefelter syndrome and affect more parts of the body. As doctors and researchers have learned more about the differences between these sex chromosome disorders, they have started to consider them as separate conditions.
Triple X syndrome
Triple X syndrome also called 47,XXX or trisomy X, results from an extra copy of the X chromosome in each of a female’s cells. Females with triple X syndrome have three X chromosomes, for a total of 47 chromosomes per cell. An extra copy of the X chromosome can be associated with tall stature, developmental delays, learning problems, and other features in some girls and women.
Although females with this condition may be taller than average, this chromosomal change typically causes no unusual physical features. Most females with triple X syndrome have normal sexual development and are able to conceive children.
Triple X syndrome is associated with an increased risk of learning disabilities and delayed development of speech and language skills. Delayed development of motor skills (such as sitting and walking), weak muscle tone (hypotonia), and behavioral and emotional difficulties are also possible, but these characteristics vary widely among affected girls and women. Seizures or kidney abnormalities occur in about 10 percent of affected females.
Some females with triple X syndrome have an extra X chromosome in only some of their cells. This phenomenon is called 46,XX/47,XXX mosaicism.
Females with more than one extra copy of the X chromosome (48,XXXX or 49,XXXXX) have been identified, but these chromosomal changes are rare. As the number of extra sex chromosomes increases, so does the risk of learning problems, intellectual disability, birth defects, and other health issues.
Triple X syndrome occurs in about 1 in 1,000 newborn girls. Five to 10 girls with triple X syndrome are born in the United States each day.
Most cases of triple X syndrome are not inherited. The chromosomal change usually occurs as a random event during the formation of reproductive cells (eggs and sperm). An error in cell division called nondisjunction can result in reproductive cells with an abnormal number of chromosomes. For example, an egg or sperm cell may gain an extra copy of the X chromosome as a result of nondisjunction. If one of these atypical reproductive cells contributes to the genetic makeup of a child, the child will have an extra X chromosome in each of the body’s cells.
46,XX/47,XXX mosaicism is also not inherited. It occurs as a random event during cell division in early embryonic development. As a result, some of an affected person’s cells have two X chromosomes (46,XX), and other cells have three X chromosomes (47,XXX).
Triple X syndrome causes
People normally have 46 chromosomes in each cell. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male or female sex characteristics. Females typically have two X chromosomes (46,XX), and males have one X chromosome and one Y chromosome (46,XY).
Triple X syndrome results from an extra copy of the X chromosome in each of a female’s cells. As a result of the extra X chromosome, each cell has a total of 47 chromosomes (47,XXX) instead of the usual 46. An extra copy of the X chromosome is associated with tall stature, learning problems, and other features in some girls and women.
Some females with triple X syndrome have an extra X chromosome in only some of their cells. This phenomenon is called 46,XX/47,XXX mosaicism.
Turner syndrome
Turner syndrome (45,X) is a genetic disorder that affects a girl’s development. Girls who have Turner syndrome (45,X) are short, and their ovaries don’t work properly. Turner syndrome results when one normal X chromosome is present in a female’s cells and the other sex chromosome is missing or structurally altered. The missing genetic material affects development before and after birth, leading to short stature, ovarian malfunction, and other features of Turner syndrome.
Turner syndrome occurs in about 1 in 2,500 newborn girls worldwide, but it is much more common among pregnancies that do not survive to term (miscarriages and stillbirths).
About half of individuals with Turner syndrome have monosomy X (45,X), which means each cell in an individual’s body has only one copy of the X chromosome instead of the usual two sex chromosomes. Turner syndrome can also occur if one of the sex chromosomes is partially missing or rearranged rather than completely absent.
Some women with Turner syndrome have a chromosomal change in only some of their cells, which is known as mosaicism. Some cells have the usual two sex chromosomes (either two X chromosomes or one X chromosome and one Y chromosome), and other cells have only one copy of the X chromosome. Women with Turner syndrome caused by X chromosome mosaicism (45,X/46,XX or 45,X/46,XY) are said to have mosaic Turner syndrome.
Researchers have not determined which genes on the X chromosome are responsible for most of the features of Turner syndrome. They have, however, identified one gene called SHOX that is important for bone development and growth. The SHOX gene is located in the pseudoautosomal regions of the sex chromosomes. Missing one copy of this gene likely causes short stature and skeletal abnormalities in women with Turner syndrome.
The most common feature of Turner syndrome is short stature, which becomes evident by about age 5. An early loss of ovarian function (ovarian hypofunction or premature ovarian failure) is also very common. The ovaries develop normally at first, but egg cells (oocytes) usually die prematurely and most ovarian tissue degenerates before birth. Many affected girls do not undergo puberty unless they receive hormone therapy, and most are unable to conceive (infertile). A small percentage of females with Turner syndrome retain normal ovarian function through young adulthood.
About 30 percent of females with Turner syndrome have extra folds of skin on the neck (webbed neck), a low hairline at the back of the neck, puffiness or swelling (lymphedema) of the hands and feet, skeletal abnormalities, or kidney problems. One third to one half of individuals with Turner syndrome are born with a heart defect, such as a narrowing of the large artery leaving the heart (coarctation of the aorta) or abnormalities of the valve that connects the aorta with the heart (the aortic valve). Complications associated with these heart defects can be life-threatening.
Most girls and women with Turner syndrome have normal intelligence. Developmental delays, nonverbal learning disabilities, and behavioral problems are possible, although these characteristics vary among affected individuals.
Other physical features typical of Turner syndrome are:
- Short, “webbed” neck with folds of skin from tops of shoulders to sides of neck
- Low hairline in the back
- Low-set ears
- Swollen hands and feet
Most women with Turner syndrome are infertile. They are at risk for health difficulties such as high blood pressure, kidney problems, diabetes, cataracts, osteoporosis, and thyroid problems.
Most cases of Turner syndrome are not inherited. When this condition results from monosomy X, the chromosomal abnormality occurs as a random event during the formation of reproductive cells (eggs and sperm) in the affected person’s parent. An error in cell division called nondisjunction can result in reproductive cells with an abnormal number of chromosomes. For example, an egg or sperm cell may lose a sex chromosome as a result of nondisjunction. If one of these atypical reproductive cells contributes to the genetic makeup of a child, the child will have a single X chromosome in each cell and will be missing the other sex chromosome.
Mosaic Turner syndrome is also not inherited. In an affected individual, it occurs as a random event during cell division in early fetal development. As a result, some of an affected person’s cells have the usual two sex chromosomes, and other cells have only one copy of the X chromosome. Other sex chromosome abnormalities are also possible in females with X chromosome mosaicism.
Rarely, Turner syndrome caused by a partial deletion of the X chromosome can be passed from one generation to the next.
Doctors diagnose Turner syndrome based on symptoms and a genetic test. Sometimes it is found in prenatal testing. There is no cure for Turner syndrome, but there are some treatments for the symptoms. Growth hormone often helps girls reach heights that are close to average. Hormone replacement can help start sexual development. Assisted reproduction techniques can help some women with Turner syndrome get pregnant.
Turner syndrome causes
Turner syndrome is related to the X chromosome, which is one of the two sex chromosomes. People typically have two sex chromosomes in each cell: females have two X chromosomes, while males have one X chromosome and one Y chromosome. Turner syndrome results when one normal X chromosome is present in a female’s cells and the other sex chromosome is missing or structurally altered. The missing genetic material affects development before and after birth.
About half of individuals with Turner syndrome have monosomy X, which means each cell in the individual’s body has only one copy of the X chromosome instead of the usual two sex chromosomes. Turner syndrome can also occur if one of the sex chromosomes is partially missing or rearranged rather than completely absent. Some women with Turner syndrome have a chromosomal change in only some of their cells, which is known as mosaicism. Women with Turner syndrome caused by X chromosome mosaicism are said to have mosaic Turner syndrome.
Researchers have not determined which genes on the X chromosome are associated with most of the features of Turner syndrome. They have, however, identified one gene called SHOX that is important for bone development and growth. The loss of one copy of this gene likely causes short stature and skeletal abnormalities in women with Turner syndrome.
X-linked acrogigantism
Duplication of a small amount of genetic material on the X chromosome causes X-linked acrogigantism (X-LAG), which is characterized by abnormally fast growth beginning in infancy or early childhood. Babies with this condition are a normal size at birth but begin to grow rapidly in infancy or early childhood, and affected children are taller than their peers.
This rapid growth is caused by an abnormality of the pituitary gland. The pituitary gland, which is found at the base of the brain, produces hormones that control many important body functions, including growth. Individuals with X-LAG may have the condition as a result of enlargement (hyperplasia) of the gland or development of a noncancerous tumor in the gland (called a pituitary adenoma). Rarely, an affected individual has both pituitary hyperplasia and an adenoma. The abnormal gland releases excess amounts of growth hormone, a hormone that normally helps direct growth of the body’s bones and tissues. The abnormal gland releases more growth hormone than normal, causing rapid growth in individuals with X-LAG. Some people with X-LAG also have excess amounts of a hormone called growth hormone releasing hormone (GHRH), which is produced by a part of the brain called the hypothalamus. This hormone stimulates the release of growth hormone from the pituitary gland.
Some people with X-LAG have additional signs and symptoms such as facial features that are described as coarse; disproportionately large hands or feet (acral enlargement); an increased appetite; and a skin condition called acanthosis nigricans, in which the skin in body folds and creases becomes thick, dark, and velvety.
The duplication, often referred to as an Xq26.3 microduplication, occurs on the long (q) arm of the chromosome at a location designated q26.3. It can include several genes, but only duplication of the GPR101 gene is necessary to cause X-LAG. The GPR101 gene provides instructions for making a protein whose function is unknown, although it is thought to be involved in the growth of cells in the pituitary gland or in the release of growth hormone from the gland.
Duplication of the GPR101 gene leads to an excess of GPR101 protein. It is unclear how extra GPR101 protein results in the development of a pituitary adenoma or hyperplasia or in the release of excess growth hormone.
X-linked acrogigantism (X-LAG) is thought to be a rare condition, although the prevalence is not known. It occurs more frequently in females than in males. X-LAG accounts for one in ten cases of abnormally fast growth in children that is caused by pituitary gland abnormalities (pituitary gigantism).
X-linked acrogigantism causes
X-LAG is caused by a genetic change in which a small amount of genetic material on the X chromosome is abnormally copied (duplicated). The duplication, often referred to as an Xq26.3 microduplication, occurs on the long (q) arm of the chromosome at a location designated q26.3. It can include several genes, but only duplication of the GPR101 gene is necessary to cause X-LAG.
The GPR101 gene provides instructions for making a protein whose function is unknown. Studies suggest that the GPR101 protein is involved in the growth of cells in the pituitary gland or in the release of growth hormone from the gland.
Duplication of the GPR101 gene leads to an excess of GPR101 protein. It is unclear how extra GPR101 protein results in the development of a pituitary adenoma or hyperplasia or in the release of excess growth hormone or GHRH.
X-linked acrogigantism inheritance pattern
X-LAG follows an X-linked dominant inheritance pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a duplication of one of the two copies of the GPR101 gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a duplication of the only copy of the gene in each cell causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In females, the condition results from new (de novo) duplications involving the GPR101 gene that occur during the formation of a parent’s reproductive cells (eggs or sperm). The duplication is found in all of the cells in the affected person’s body.
In males, the condition often results from somatic mosaicism, in which some of an affected person’s cells have the duplication and others do not. The genetic changes, which are called somatic mutations, arise randomly in one cell during embryonic development. As cells continue to divide, only cells arising from the first abnormal cell will have the mutation. Other affected males inherit the duplication from their affected mother, and it is found in all the body’s cells.
Intestinal pseudo-obstruction
Intestinal pseudo-obstruction, a condition characterized by impairment of the coordinated waves of muscle contractions that move food through the digestive tract (peristalsis), can be caused by genetic changes involving the X chromosome.
Some individuals with intestinal pseudo-obstruction have mutations, duplications, or deletions of genetic material on the X chromosome that affect the FLNA gene. The protein produced from this gene, filamin A, helps form the branching network of filaments called the cytoskeleton, which gives structure to cells and allows them to change shape and move.
Researchers believe that the changes in the X chromosome that affect the FLNA gene impair the function of the filamin A protein. Studies suggest that impaired filamin A function affects the shape of cells in the smooth muscles of the gastrointestinal tract during development before birth, causing abnormalities in the layering of these muscles. Smooth muscles line the internal organs; they contract and relax without being consciously controlled. In the digestive tract, abnormal layering of these muscles may interfere with peristalsis.
Deletions or duplications of genetic material that affect the FLNA gene can also include adjacent genes on the X chromosome. Changes in adjacent genes may account for some of the other signs and symptoms, such as neurological abnormalities and unusual facial features, that occur in some affected individuals.
The overall prevalence of intestinal pseudo-obstruction is unknown. Researchers in Japan have estimated the prevalence of chronic intestinal pseudo-obstruction in that country as 9 cases per million people.
Intestinal pseudo-obstruction is often not inherited, and most affected individuals do not have a family history of the disorder. When it does run in families, it can have different inheritance patterns.
Intestinal pseudo-obstruction caused by FLNA gene mutations is inherited in an X-linked recessive pattern. The FLNA gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Intestinal pseudo-obstruction caused by ACTG2 gene mutations is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Some other cases of intestinal pseudo-obstruction are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Intestinal pseudo-obstruction causes
In some individuals with primary intestinal pseudo-obstruction, the condition is caused by genetic changes affecting the FLNA or ACTG2 gene.
The protein produced from the FLNA gene, filamin A, attaches (binds) to proteins called actins and helps them form the branching network of filaments that make up the cytoskeleton, which gives structure to cells and allows them to change shape and move. FLNA gene mutations that cause intestinal pseudo-obstruction are thought to reduce levels of the filamin A protein or impair its function. Research suggests that decreased filamin A function may affect the shape of cells in the smooth muscles of the gastrointestinal tract during development before birth, causing abnormalities in the layering of these muscles. Smooth muscles line the internal organs; they contract and relax without being consciously controlled. In the gastrointestinal tract, abnormal layering of these muscles interferes with the ability to produce the coordinated waves of contractions (peristalsis) that move food along during digestion.
Deletions or duplications of genetic material can affect all or part of the FLNA gene, and may also include adjacent genes on the X chromosome. Changes in adjacent genes may account for some of the other signs and symptoms that can occur with intestinal pseudo-obstruction.
The ACTG2 gene provides instructions for making a member of the actin family called gamma (γ)-2 actin. The γ-2 actin protein is found in smooth muscle cells of the intestinal and urinary tracts. It is necessary for contraction of the smooth muscles in the intestines and bladder. These contractions move food through the intestines as part of the digestive process and empty urine from the bladder. ACTG2 gene mutations hinder the formation of actin filaments in the cytoskeleton and reduce the ability of smooth muscles in the intestines and bladder to contract, leading to the signs and symptoms of intestinal pseudo-obstruction.
Secondary intestinal pseudo-obstruction occurs as a complication of other disorders that damage muscles or nerves in the intestinal tract, such as Parkinson disease, type 2 diabetes, various types of muscular dystrophy, or Kawasaki disease. Additionally, the condition is a characteristic feature of certain inherited syndromes such as megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS) or mitochondrial neurogastrointestinal encephalopathy disease (MNGIE disease). Infections, surgery, or certain drugs can also cause secondary intestinal pseudo-obstruction.
Mutations in other genes involved in smooth muscle contraction can also cause intestinal pseudo-obstruction. Studies suggest that mutations in additional genes that have not been identified can also result in this condition. In some affected individuals, the cause of intestinal pseudo-obstruction is unknown.
Microphthalmia with linear skin defects syndrome
A deletion of genetic material in a region of the X chromosome called Xp22 causes microphthalmia with linear skin defects syndrome. This condition is characterized by small or poorly developed eyes (microphthalmia) and unusual linear skin markings on the head and neck.
The Xp22 region includes a gene called HCCS, which carries instructions for producing an enzyme called holocytochrome c-type synthase. This enzyme helps produce a molecule called cytochrome c. Cytochrome c is involved in a process called oxidative phosphorylation, by which mitochondria generate adenosine triphosphate (ATP), the cell’s main energy source. It also plays a role in the self-destruction of cells (apoptosis).
A deletion of genetic material that includes the HCCS gene prevents the production of the holocytochrome c-type synthase enzyme. In females (who have two X chromosomes), some cells produce a normal amount of the enzyme and other cells produce none. The resulting overall reduction in the amount of this enzyme leads to the signs and symptoms of microphthalmia with linear skin defects syndrome.
In males (who have only one X chromosome), a deletion that includes the HCCS gene results in a total loss of the holocytochrome c-type synthase enzyme. A lack of this enzyme appears to be lethal very early in development, so almost no males are born with microphthalmia with linear skin defects syndrome. A few affected individuals with male appearance who have two X chromosomes have been identified.
A reduced amount of the holocytochrome c-type synthase enzyme can damage cells by impairing their ability to generate energy. In addition, without the holocytochrome c-type synthase enzyme, the damaged cells may not be able to undergo apoptosis. These cells may instead die in a process called necrosis that causes inflammation and damages neighboring cells. During early development this spreading cell damage may lead to the eye and skin abnormalities characteristic of microphthalmia with linear skin defects syndrome.
The prevalence of microphthalmia with linear skin defects syndrome is unknown. More than 50 affected individuals have been identified.
Microphthalmia with linear skin defects syndrome causes
Mutations in the HCCS gene or a deletion of genetic material that includes the HCCS gene cause microphthalmia with linear skin defects syndrome. The HCCS gene carries instructions for producing an enzyme called holocytochrome c-type synthase. This enzyme is active in many tissues of the body and is found in the mitochondria, the energy-producing centers within cells.
Within the mitochondria, the holocytochrome c-type synthase enzyme helps produce a molecule called cytochrome c. Cytochrome c is involved in a process called oxidative phosphorylation, by which mitochondria generate adenosine triphosphate (ATP), the cell’s main energy source. It also plays a role in the self-destruction of cells (apoptosis).
HCCS gene mutations result in a holocytochrome c-type synthase enzyme that cannot perform its function. A deletion of genetic material that includes the HCCS gene prevents the production of the enzyme. A lack of functional holocytochrome c-type synthase enzyme can damage cells by impairing their ability to generate energy. In addition, without the holocytochrome c-type synthase enzyme, the damaged cells may not be able to undergo apoptosis. These cells may instead die in a process called necrosis that causes inflammation and damages neighboring cells. During early development this spreading cell damage may lead to the eye abnormalities and other signs and symptoms of microphthalmia with linear skin defects syndrome.
Microphthalmia with linear skin defects syndrome inheritance pattern
This condition is inherited in an X-linked dominant pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. Some cells produce a normal amount of the holocytochrome c-type synthase enzyme and other cells produce none. The resulting overall reduction in the amount of this enzyme leads to the signs and symptoms of microphthalmia with linear skin defects syndrome.
In males (who have only one X chromosome), mutations result in a total loss of the holocytochrome c-type synthase enzyme. A lack of this enzyme appears to be lethal very early in development, so almost no males are born with microphthalmia with linear skin defects syndrome. A few affected individuals with male appearance but who have two X chromosomes have been identified.
Most cases of microphthalmia with linear skin defects syndrome occur in people with no history of the disorder in their family. These cases usually result from the deletion of a segment of the X chromosome during the formation of reproductive cells (eggs and sperm) or in early fetal development. They may also result from a new mutation in the HCCS gene.
- Fragile X Syndrome Testing & Diagnosis. https://fragilex.org/understanding-fragile-x/fragile-x-101/testing-diagnosis/[↩][↩]
- Bailey, D. B., Raspa, M., Bishop, E., & Holiday, D. (2009). No change in the age of diagnosis for fragile x syndrome: findings from a national parent survey. Pediatrics, 124, 527–533.[↩][↩]





{kind=link}