Contents
Biotinidase deficiency
Biotinidase deficiency is a rare autosomal recessive inherited disorder in which the body is unable to recycle biotin (a water-soluble B vitamin sometimes also called vitamin B7 or vitamin H), leading to biotin deficiency despite normal biotin intake from foods such as liver, egg yolks, and milk 1. Biotin is an important vitamin that helps the body break down protein, fats, and carbohydrates. Biotinidase deficiency is caused by genetic changes (mutations) in the BTD gene. The BTD gene provides instructions for making an enzyme called biotinidase 2. The biotinidase enzyme recycles biotin. Biotinidase removes biotin that is bound to proteins in food, leaving the vitamin in its free (unbound) state. Free biotin is needed by enzymes called biotin-dependent carboxylases to break down fats, proteins, and carbohydrates. Because several of these enzymes are impaired in biotinidase deficiency, the condition is considered a form of multiple carboxylase deficiency 1. If biotinidase deficiency is not recognized and treated, its signs and symptoms typically appear within the first few months of life, although it can also become apparent later in childhood.
Biotinidase deficiency is sometimes categorized into groups depending on how much of the biotinidase enzyme is working. These two categories are profound biotinidase deficiency (less than 10% of enzyme activity) and partial biotinidase deficiency (10 to 30% of enzyme activity).
Profound biotinidase deficiency, the more severe form of biotinidase deficiency, can cause seizures, weak muscle tone (hypotonia), breathing problems, hearing and vision loss, problems with movement and balance (ataxia), skin rashes, hair loss (alopecia), and a fungal infection called candidiasis. Affected children also have delayed development. Lifelong treatment can prevent these complications from occurring or improve them if they have already developed. Profound biotinidase deficiency can lead to coma or death if treatment is not initiated rapidly.
Partial biotinidase deficiency is a milder form of this condition. Without treatment, affected children may experience hypotonia, skin rashes, and hair loss, but these problems may appear only during illness, infection, or other times of stress.
Profound or partial biotinidase deficiency occurs in approximately 1 in 60,000 newborns 3. The early-onset form (profound biotinidase deficiency) usually begins during the newborn (neonatal) period. The juvenile form (partial biotinidase deficiency) usually begins at about three months of age. Both males and females are affected in equal numbers.
- One in 140,000 people have profound biotinidase deficiency.
- One in 110,000 people have partial biotinidase deficiency.
- One in 60,000 people have either profound or partial biotinidase deficiency.
According to the worldwide neonatal screening survey, the incidence of profound biotinidase deficiency is one in 112,271, and the incidence of partial biotinidase deficiency is one in 129,282 4. The combined incidence of profound and partial biotinidase deficiency is one in 60,089 live births 4. In 2006, the incidence of profound biotinidase deficiency was 1 in 80,000, and the incidence of partial biotinidase deficiency cases was from 1 per 31,000 to 1 per 40,000 in the US. Biotinidase deficiency has been diagnosed more commonly in children of the White race. Research has observed a higher incidence of biotin deficiency in Brazil, Turkey, and Saudi Arabia 5, 6.
Approximately 1 in 123 people are carriers of one gene for biotinidase deficiency, but this number may be higher in the Hispanic population and lower in the African American population. The rates vary from country to country 7, 8, 9.
Early diagnosis and treatment of biotinidase deficiency can prevent symptoms from happening. Nearly all infants with either profound or partial biotinidase deficiency can be detected in the US by newborn screening. However, not every country has added biotinidase deficiency to its newborn screening program.
Biotinidase deficiency can be treated by giving people with the condition extra biotin for their body to use. If treated early, people with biotinidase deficiency can avoid all symptoms of the condition and lead a normal, healthy life.
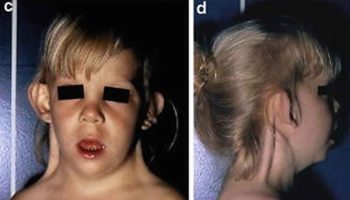
Figure 1. Biotinidase deficiency
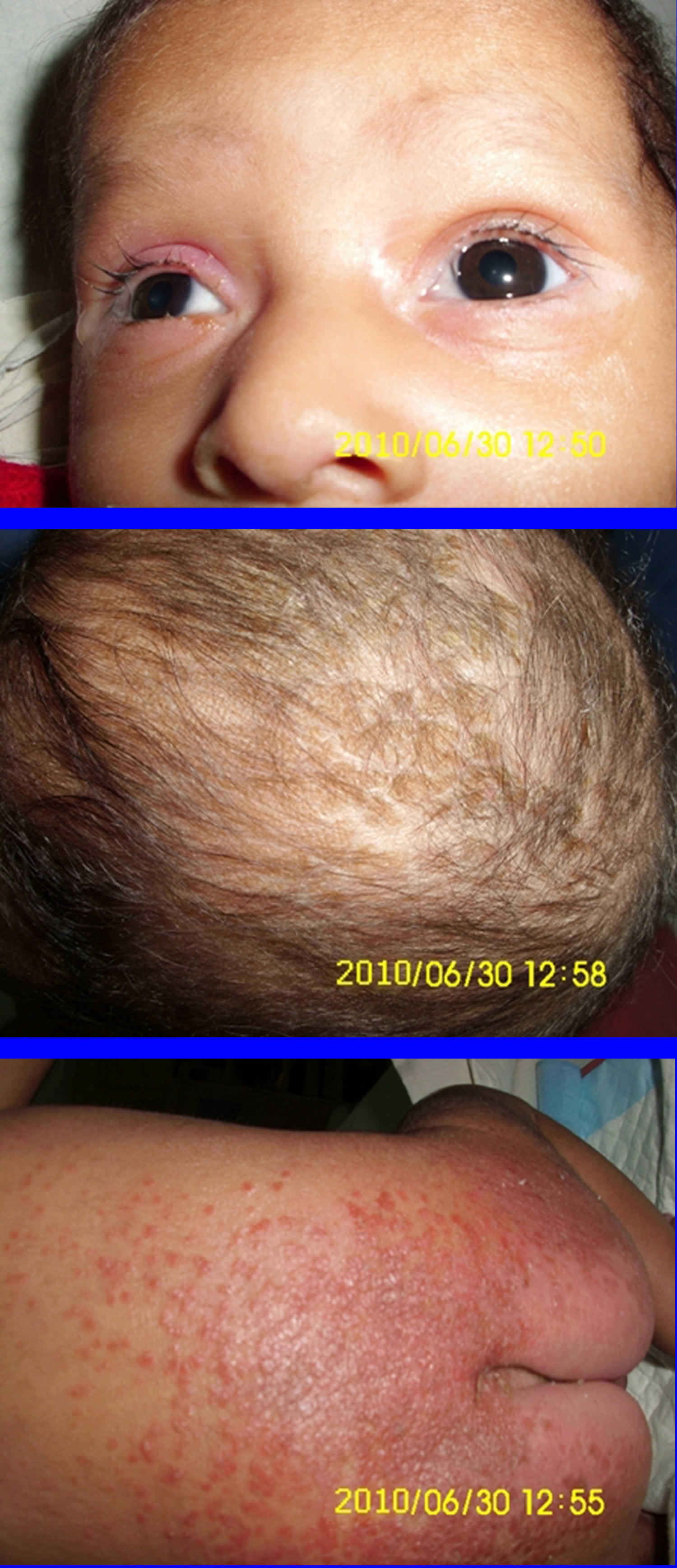
Footnotes: Biotinidase deficiency in a 58-day-old male infant with seizures for 2 weeks prior to presentation. At admission, the child had multiple episodes of generalized tonic and myoclonic seizures not associated with fever. He also had respiratory distress and stridor. The skin showed erythematous maculopapular rash in the perioral and perianal regions. He had alopecia (hair loss), scanty eyebrows, blepharitis, conjunctivitis, balanitis and seborrheic dermatitis involving the scalp. Hair over the scalp was hypopigmented. On neurological examination, the child was convulsing intermittently. Hypertonia was noted in all the four limbs with exaggerated deep tendon reflexes. Fundus examination revealed papilloedema. Rest of the systemic examination was normal. Within 24 hour of hospitalization, the possibility of biotinidase deficiency was entertained and oral biotin was started empirically at a dose of 10 mg twice a day, after obtaining a blood sample for biotinidase assay. There was total cessation of seizures and considerable improvement in the rash by 48 hour. The hypertonia in the upper limbs resolved within 72 hours though it persisted in the lower limbs, even at discharge, albeit much less than before. The balanitis and blepharitis also resolved by 72 hours. Serum biotinidase levels showed a profound deficiency. The child was then started on regular biotin supplementation and was discharged after 5 days. Anticonvulsants were successfully stopped without recurrence of seizures. The child is currently 10 months old, healthy, with adequate weight gain and normal developmental milestones. At follow-up, there are no sequelae except for a mild persisting lower limb hypertonia.
[Source 10 ]What is Biotin?
Biotin also known as vitamin B7 or vitamin H, is a water soluble vitamin and is naturally present in liver, soy, beans and egg yolks. Raw egg whites, however, contain the protein avidin that binds to biotin and reduces its availability. Eating 2 or more uncooked egg whites daily for several months has caused biotin deficiency that is serious enough to produce symptoms. Biotin acts as a carrier of carbon dioxide and plays a role in carboxylase enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) that catalyze critical steps fatty acid metabolism, gluconeogenesis (the formation of glucose from sources other than carbohydrates, such as pyruvate, lactate, glycerol, and the glucogenic amino acids), and amino acids 11. Biotin also plays key roles in histone modifications, gene regulation (by modifying the activity of transcription factors), and cell signaling 12.
The recommended daily dietary allowance for biotin has not been formally established, but the amounts needed are small and biotin is found in many foods and is produced by intestinal bacteria. An adequate intake for biotin has been estimated as 30 micrograms (mcg) daily. Thus, most diets provide adequate amounts of biotin and its deficiency is rare. Although there are no nationally representative estimates of biotin intakes in the United States, the average biotin intake from foods in other western populations is about 35–70 mcg/day, indicating that most people in these countries consume adequate amounts of biotin and biotin deficiency is rare 11, 13.
Most biotin in foods is bound to protein, although some dietary biotin is in the free form 14. Gastrointestinal proteases and peptidases break down the protein-bound forms of ingested biotin into biocytin and biotin-oligopeptides, which undergo further processing by biotinidase, an enzyme, in the intestinal lumen to release free biotin 14. The free biotin is then absorbed in the small intestine, and most biotin is stored in the liver 13.
Biotin is available generically in many over-the-counter forms in doses of 5 to 10 mg and is included in most multivitamin preparations, usually in concentrations of 30 to 300 mcg. Biotin is typically added to parenteral nutrition and the doses for deficiencies is in the range of 10 mg daily.
Biotin deficiency has occurred in humans on parenteral nutrition (intravenous administration of nutrition). However, biotin deficiency is very rare in the United States. The signs and symptoms of biotin deficiency typically appear gradually and can include thinning hair with progression to loss of all hair on the body (alopecia); a scaly red rash around the eyes, nose, mouth, and anal area (seborrheic dermatitis); pinkeye (conjunctivitis); lactic acidosis (which occurs when lactate production exceeds lactate clearance) and aciduria (abnormal amounts of acid in urine); skin infection; brittle nails; nervous system disorders (e.g., depression, lethargy, seizures, hallucinations, ataxia and numbness and tingling [paresthesias] of the extremities) in adults; and hypotonia (weak muscle tone), lethargy, sluggishness and developmental delay in infants 11. The characteristic facial rash, together with unusual facial fat distribution in people with biotin deficiency is known as “biotin deficiency facies” 15. Individuals with hereditary disorders of biotin metabolism (inborn metabolic disorders) resulting in functional biotin deficiency often have similar physical findings, as well as seizures and evidence of impaired immune system function and increased susceptibility to bacterial and fungal infections 16, 17.
A limited number of reliable indicators of biotin status is available 18. In healthy adults, the concentration of biotin is 133–329 pmol/L in serum and 18–127 nmol/24 hours in urine 11. Abnormally low urinary excretion of biotin is an indicator of biotin deficiency, as is abnormally high excretion of 3-hydroxyisovaleric acid (higher than 3.3 mmol/mol creatinine) or 3-hydroxyisovalerylcarnitine (higher than 0.06 mmol/mol creatinine) resulting from reduced activity of methylcrotonyl-CoA carboxylase 18. The most reliable individual markers of biotin status, including deficiency and sufficiency, are biotinylated methylcrotonyl-CoA carboxylase and propionyl-CoA carboxylase in white blood cells 18. Oral administration of large doses of biotin increases serum concentrations of biotin and its metabolites 19. However, serum concentrations of biotin and its catabolites are not good indicators of marginal biotin deficiency because they do not decrease sufficiently in people with marginal biotin deficiency for these changes to be detectable with existing tests 13.

What does biotin do?
Biotin helps turn the carbohydrates, fats, and proteins in the food you eat into the energy you need. Biotin functions as a coenzyme; involved in carboxylation, transcarboxylation, and decarboxylation reactions of gluconeogenesis, lipogenesis, fatty acid synthesis, propionate metabolism, and the catabolism of leucine. Biotin acts as a carrier of carbon dioxide and functions as a covalently bound cofactor required for the biological activity of the five known mammalian biotin-dependent carboxylases enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) that catalyze critical steps fatty acid metabolism, gluconeogenesis, and amino acids 11. For acetyl-CoA carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2), biotin serves as a cofactor responsible for transfer of bicarbonate to acetyl-CoA, converting it to malonyl-CoA for fatty acid synthesis. Pyruvate carboxylase (PC) participates in gluconeogenesis. Methylcrotonyl-CoA carboxylase (MCC) catalyzes a step in leucine metabolism. Propionyl-CoA carboxylase (PCC) catalyzes a step in the metabolism of propionyl-CoA 20, 21. Metabolic degradation of the biotinylated carboxylases leads to the formation of biocytin. This compound is further degraded by biotinidase to release biotin, which is then reutilized by holocarboxylase synthetase 21.
Biotin also plays key roles in histone modifications, gene regulation (by modifying the activity of transcription factors), and cell signaling 12.
Biotin is an essential cofactor to five known mammalian biotin-dependent carboxylases enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) in intermediary metabolism and a key regulator of gene expression 22.
Five mammalian carboxylases catalyze essential metabolic reactions 22:
- Both acetyl-Coenzyme A (CoA) carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2) catalyze the conversion of acetyl-CoA to malonyl-CoA using bicarbonate and ATP; however, the two enzymes have different roles in metabolism and different intracellular locations. ACC1 is located in the cytosol, and the malonyl CoA generated by ACC1 is a rate-limiting substrate for the synthesis of fatty acids (Figures 2 and 3). Acetyl-Coenzyme A (CoA) carboxylase 1 (ACC1) is found in all tissues and is particularly active in lipogenic tissues (i.e., liver, white adipose tissue, and mammary gland), heart, and pancreatic islets. Acetyl-CoA carboxylase 2 (ACC2) is located on the outer mitochondrial membrane, and the malonyl CoA generated via ACC2 inhibits CPT1, an enzyme that regulates malonyl-CoA entry into the inner mitochondria, thereby regulating fatty acid oxidation (Figure 3). ACC2 is especially abundant in skeletal muscle and heart 23.
- Pyruvate carboxylase (PC) is a critical enzyme in gluconeogenesis (the formation of glucose from sources other than carbohydrates, such as pyruvate, lactate, glycerol, and the glucogenic amino acids). Pyruvate carboxylase (PC) catalyzes the ATP-dependent incorporation of bicarbonate into pyruvate, producing oxaloacetate; hence, pyruvate carboxylase is anaplerotic for the citric acid cycle (Figure 4). Oxaloacetate can then be converted to phosphoenolpyruvate and eventually to glucose.
- Methylcrotonyl-CoA carboxylase (MCC) catalyzes an essential step in the catabolism of leucine, an essential branched-chain amino acid (BCAA). Methylcrotonyl-CoA carboxylase (MCC) enzyme catalyzes the production of 3-methylglutaconyl-CoA from methylcrotonyl-CoA (Figure 5).
- Propionyl-CoA carboxylase (PCC) produces D-malonylmalonyl-CoA from propionyl-CoA, a by-product in the beta-oxidation of fatty acids with an odd number of carbon atoms (Figure 5). The conversion of propionyl-CoA to D-malonylmalonyl-CoA is also required in the catabolic pathways of two branched-chain amino acids (isoleucine and valine) and the side chain of cholesterol (Figure 5) and of the amino acids methionine and threonine (Figure 6).
Figure 2. Roles of the 5 biotin-dependent carboxylases
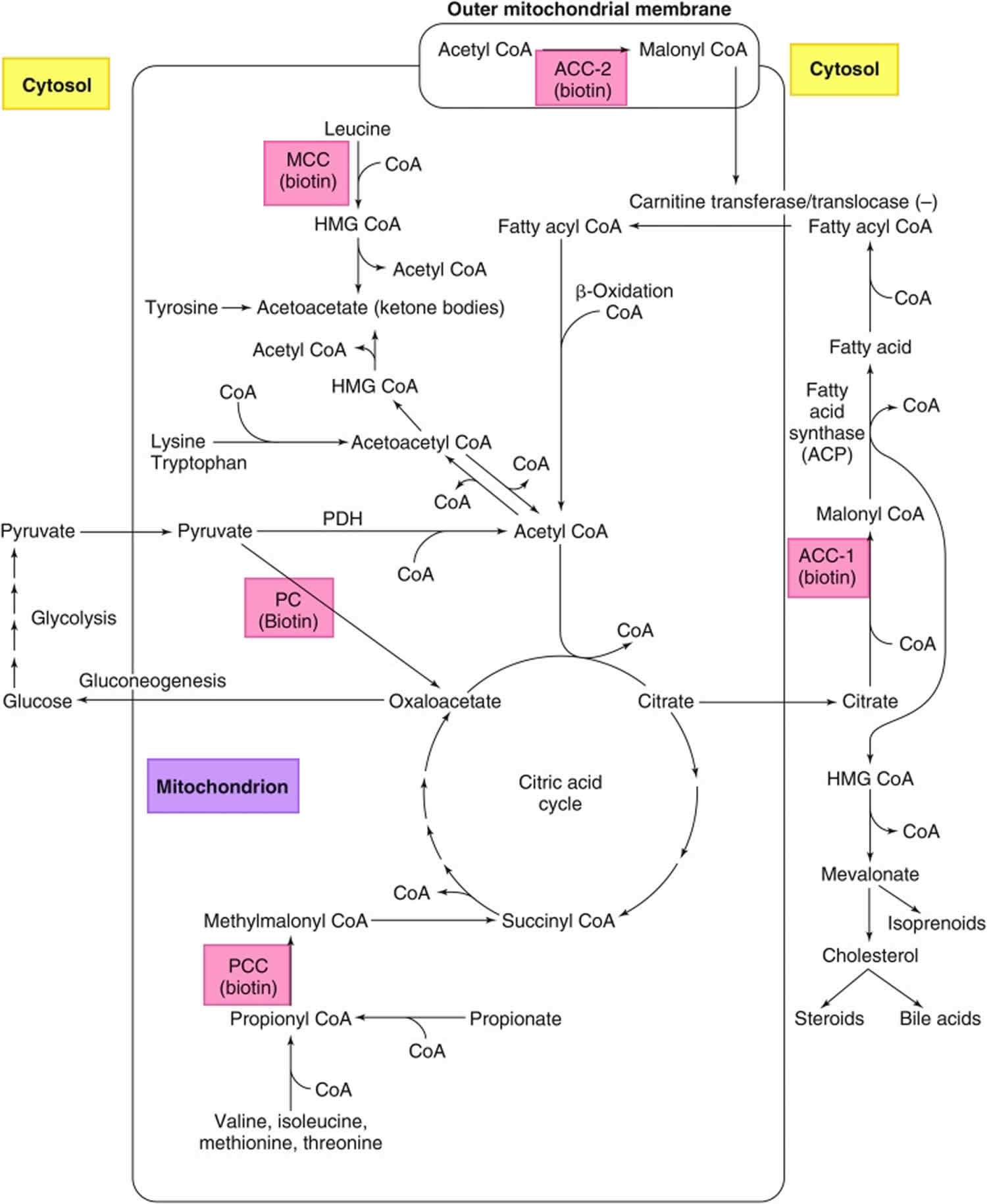
Footnotes: Roles of the 5 biotin-dependent carboxylases of coenzyme A (CoA) and acyl carrier protein (ACP) within the cell. Shown is an overview of the metabolic pathways of acetyl-CoA carboxylase 1 (ACC1) (cytosolic) and acetyl-CoA carboxylase 2 (ACC2) (outer mitochondrial membrane) and the 3 mitochondrial carboxylases propionyl-CoA carboxylase (PCC), methylcrotonyl-CoA carboxylase (MCC) and pyruvate carboxylase (PC).
Abbreviations: ACC1 = acetyl-CoA carboxylase 1; ACC2 = acetyl-CoA carboxylase 2; ACP = acyl carrier protein; HMG = 3-hydroxy-3-methylglutaryl; MCC = methylcrotonyl-CoA carboxylase; PC = pyruvate carboxylase; PCC = propionyl-CoA carboxylase; PDH = pyruvate dehydrogenase.
[Source 24 ]Figure 3. Biotin function as enzyme cofactor
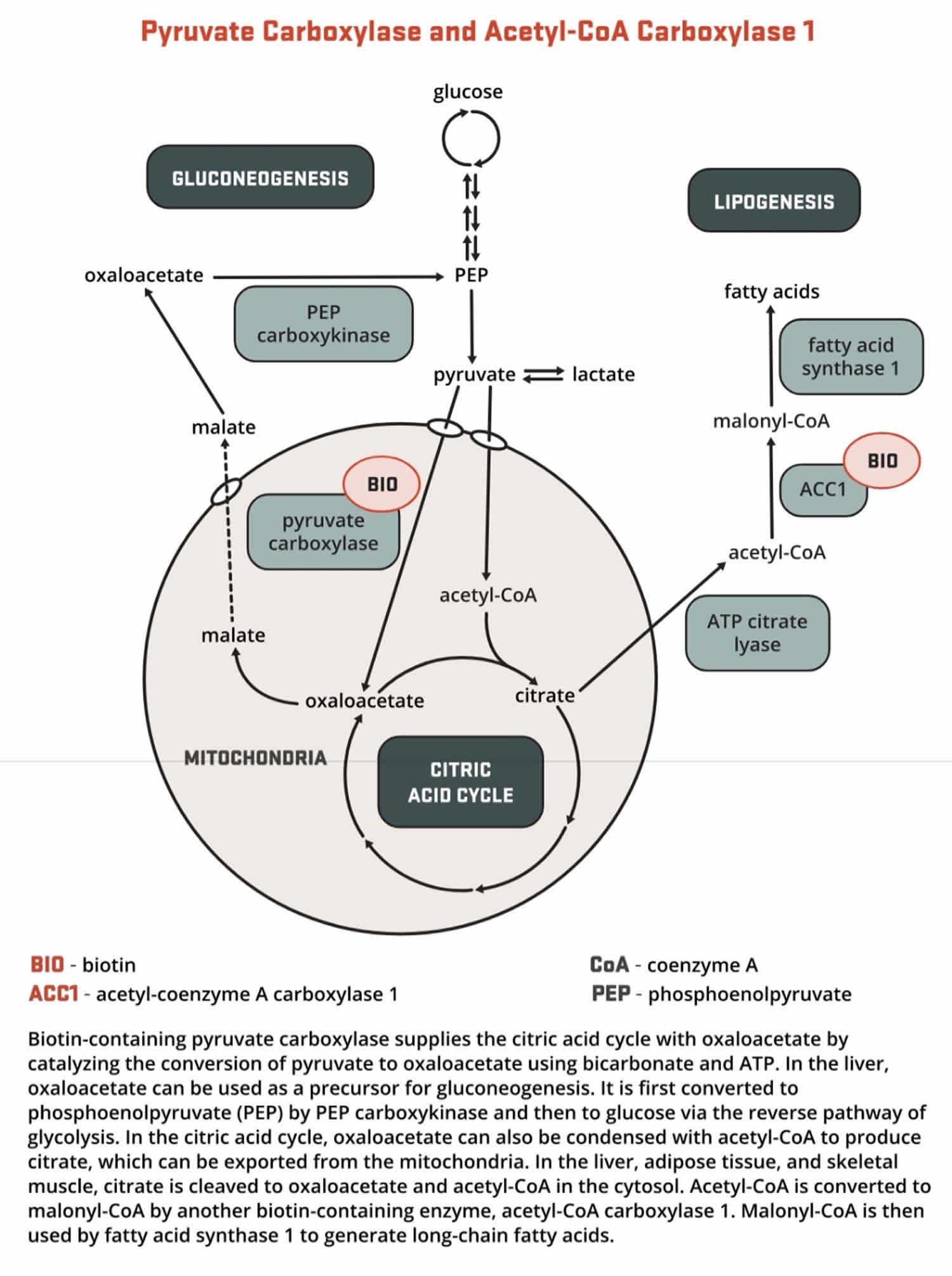
Figure 4. Biotin as enzyme cofactor in gluconeogenesis
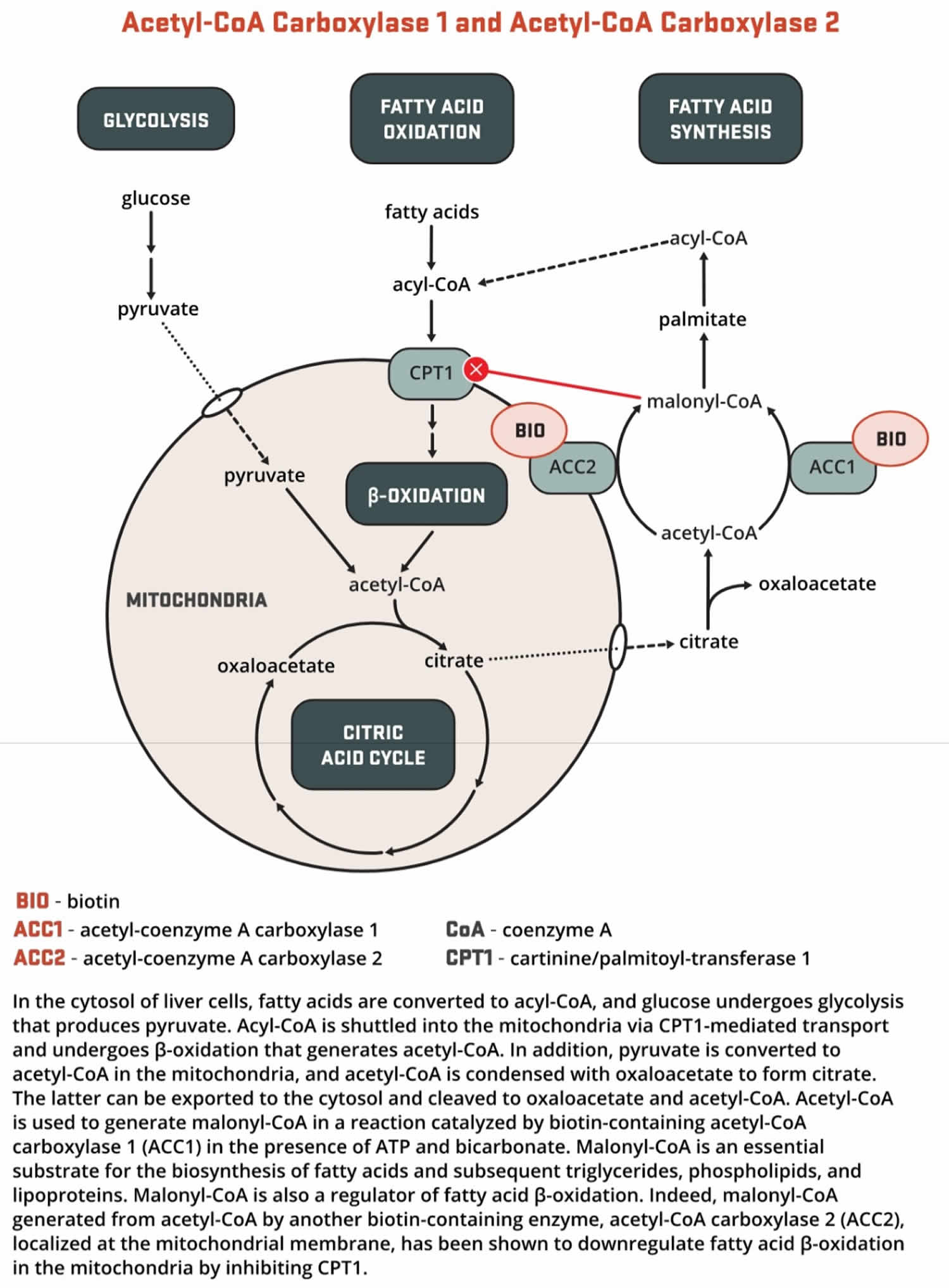
Figure 5. Biotin as enzyme cofactor in fatty acids, amino acids and cholesterol metabolism
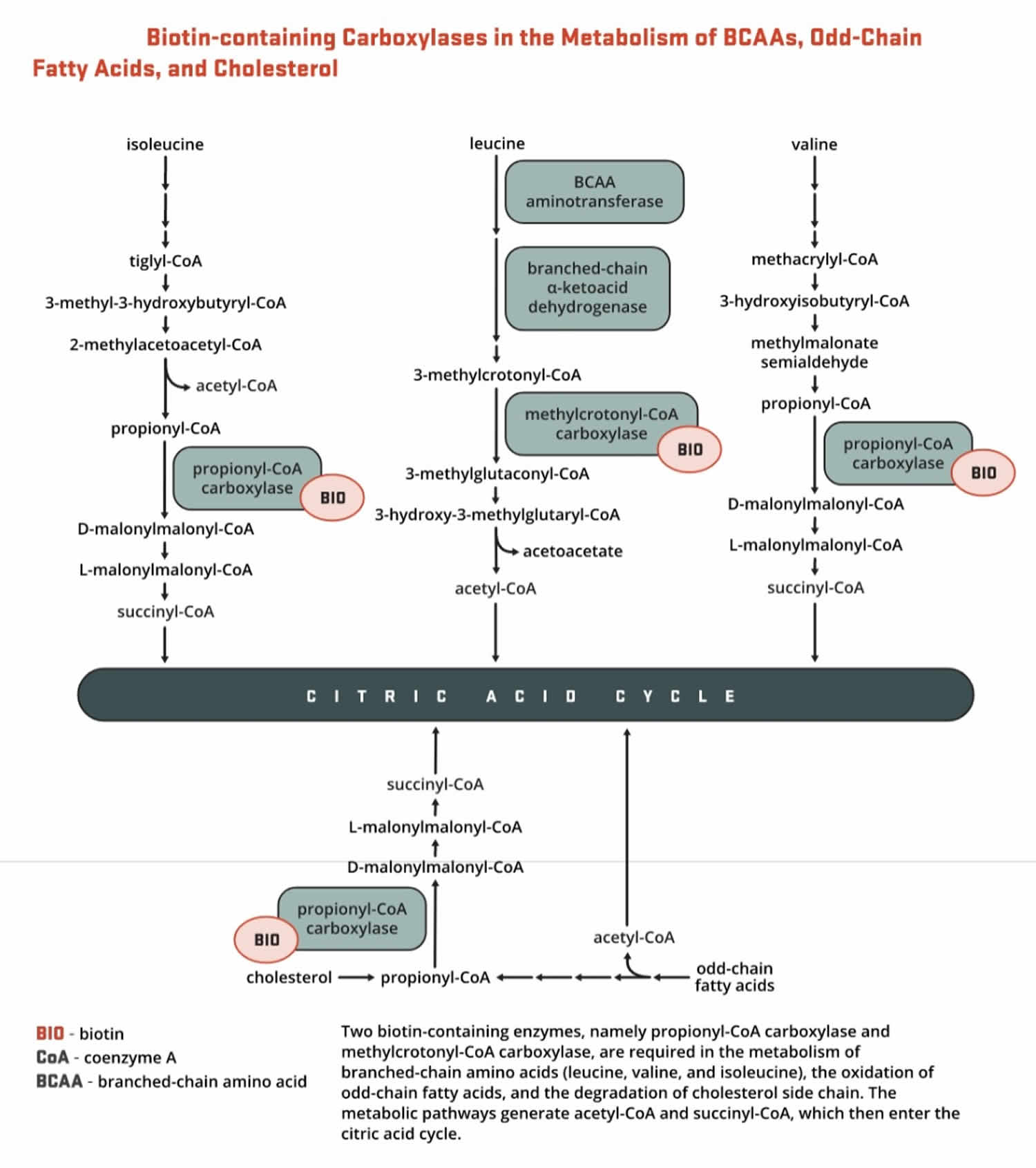
Figure 6. Biotin as enzyme cofactor in amino acids methionine and threonine metabolism
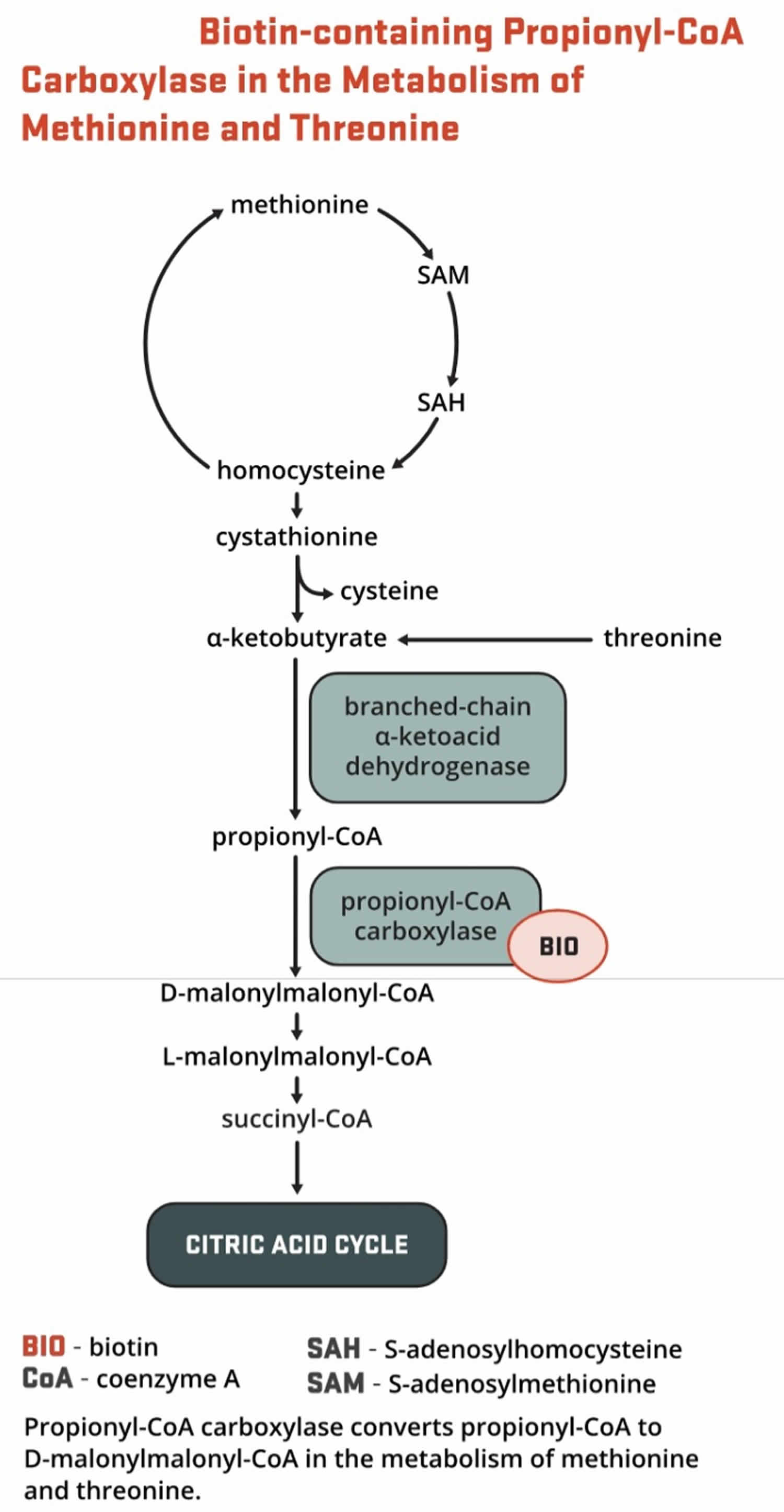
What is known about biotinidase deficiency and pregnancy?
There is limited information available in the medical literature about biotinidase deficiency during pregnancy. Biotinidase deficiency was first described as a distinct disorder in 1983, so there have not been many years of experience with females being of childbearing age. A 2005 case report 25 described a successful pregnancy in a woman being treated with biotin for biotinidase deficiency throughout her pregnancy. Her laboratory studies (plasma acylcarnitine profile and urine organic acids) were normal when analyzed during the pregnancy. The pregnancy was uneventful and her infant was healthy after birth and was developmentally normal at 3 months of age. Animal studies have raised concern that a deficiency of biotin could cause birth defects 26. However, this remains to be well-studied in humans.
Is biotin known to cause birth defects?
Biotin is a vitamin (a B vitamin) that is found in numerous foods, and is an important component of enzymes in the body that break down certain substances like fats and carbohydrates 26.
Current evidence in the medical literature does not show that taking biotin during pregnancy causes birth defects. On the contrary, animal studies have raised concern that a deficiency (too little) of biotin could cause birth defects. Maternal biotin deficiency in some animals has been shown to cause cleft palate and other malformations. However, whether biotin deficiency causes birth defects in humans has not been well-studied 27. It is known that a number of birth defects are potentially related to nutrient deficiencies, or preventable by nutrient supplementation 26.
Current research indicates that at least one-third of women develop marginal biotin deficiency during pregnancy 28. Small observational studies in pregnant women have reported an abnormally high urinary excretion of 3-hydroxyisovaleric acid in both early and late pregnancy, suggesting decreased activity of biotin-dependent methylcrotonyl-CoA carboxylase (MCC) 29, 30. In a randomized, single-blinded intervention study in 26 pregnant women, supplementation with 300 mcg/day of biotin for two weeks limited the excretion of 3-hydroxyisovaleric acid compared to placebo, confirming that increased 3-hydroxyisovaleric acid excretion indeed reflected marginal biotin deficiency in pregnancy 31. A small cross-sectional study in 22 pregnant women reported an incidence of low lymphocyte propionyl-CoA carboxylase (PCC) activity greater than 80% 32. Although these levels of biotin deficiency are not associated with overt signs of deficiency in pregnant women, such observations are sources of concern because subclinical biotin deficiency has been shown to cause cleft palate and limb hypoplasia in several animal species 32. In addition, biotin depletion has been found to suppress the expression of biotin-dependent carboxylases, remove biotin marks from histones, and decrease the proliferation in human embryonic palatal mesenchymal cells in culture 33. Impaired carboxylase activity may result in alterations in lipid metabolism, which have been linked to cleft palate and skeletal abnormalities in animals. Furthermore, biotin deficiency leading to reduced histone biotinylation at specific genomic loci may increase genomic instability and result in chromosome anomalies and fetal malformations 32.
Analogous to pregnant women who are advised to consume supplemental folic acid (vitamin B9) prior to and during pregnancy to prevent neural tube defects, it would also be prudent to ensure adequate biotin intake throughout pregnancy. The current Adequate Intake (intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an Recommended Dietary Allowance [RDA], the average daily level of intake sufficient to meet the nutrient requirements of nearly all (97%–98%) healthy individuals; often used to plan nutritionally adequate diets for individuals) for pregnant women is 30 mcg/day of biotin, and no toxicity has ever been reported at this level of intake.
Women with biotinidase deficiency who are planning a pregnancy, or are pregnant, should consult with their health care provider about their treatment plan and discuss the amount of biotin that is recommended. The amount of biotin recommended when trying to become pregnant, or during pregnancy, may be different from during other stages of adulthood.
Biotinidase deficiency causes
Mutations in the BTD gene cause biotinidase deficiency. The BTD gene provides instructions for making an enzyme called biotinidase. The biotinidase enzyme recycles biotin, a B vitamin found in foods such as liver, egg yolks, and milk. Biotinidase removes biotin that is bound to proteins in food, leaving the vitamin in its free (unbound) state. Free biotin is needed by enzymes called biotin-dependent carboxylases to break down fats, proteins, and carbohydrates. Because several of these enzymes are impaired in biotinidase deficiency, the condition is considered a form of multiple carboxylase deficiency.
Mutations in the BTD gene reduce or eliminate the activity of biotinidase. Profound biotinidase deficiency results when the activity of biotinidase is reduced to less than 10 percent of normal. Partial biotinidase deficiency occurs when biotinidase activity is reduced to between 10 percent and 30 percent of normal. Without enough of this enzyme, biotin cannot be recycled. The resulting shortage of free biotin impairs the activity of biotin-dependent carboxylases, leading to a buildup of potentially toxic compounds in the body. If the condition is not treated promptly, this buildup damages various cells and tissues, causing the signs and symptoms of biotinidase deficiency.
Figure 7. Biotin recycling
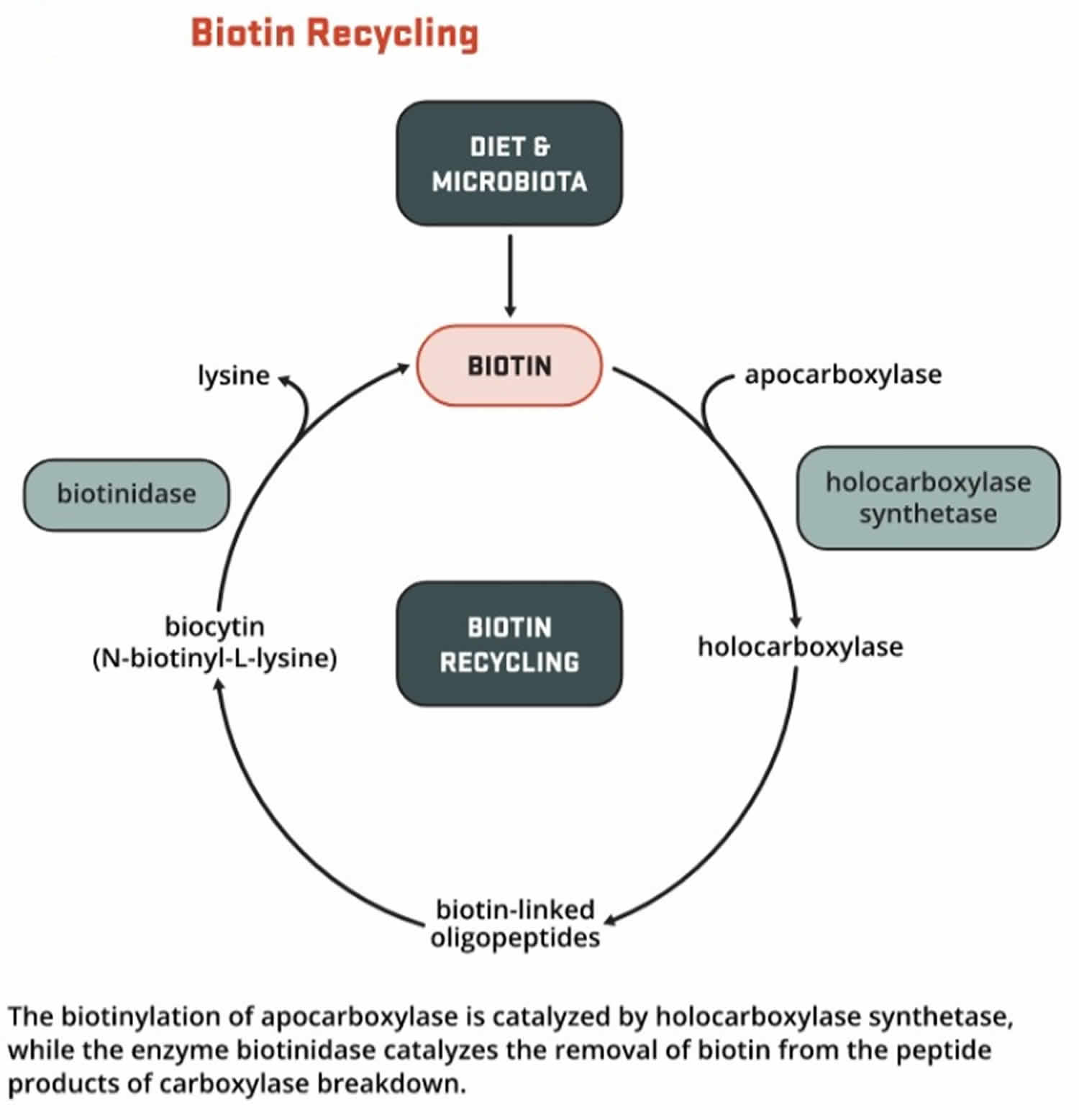
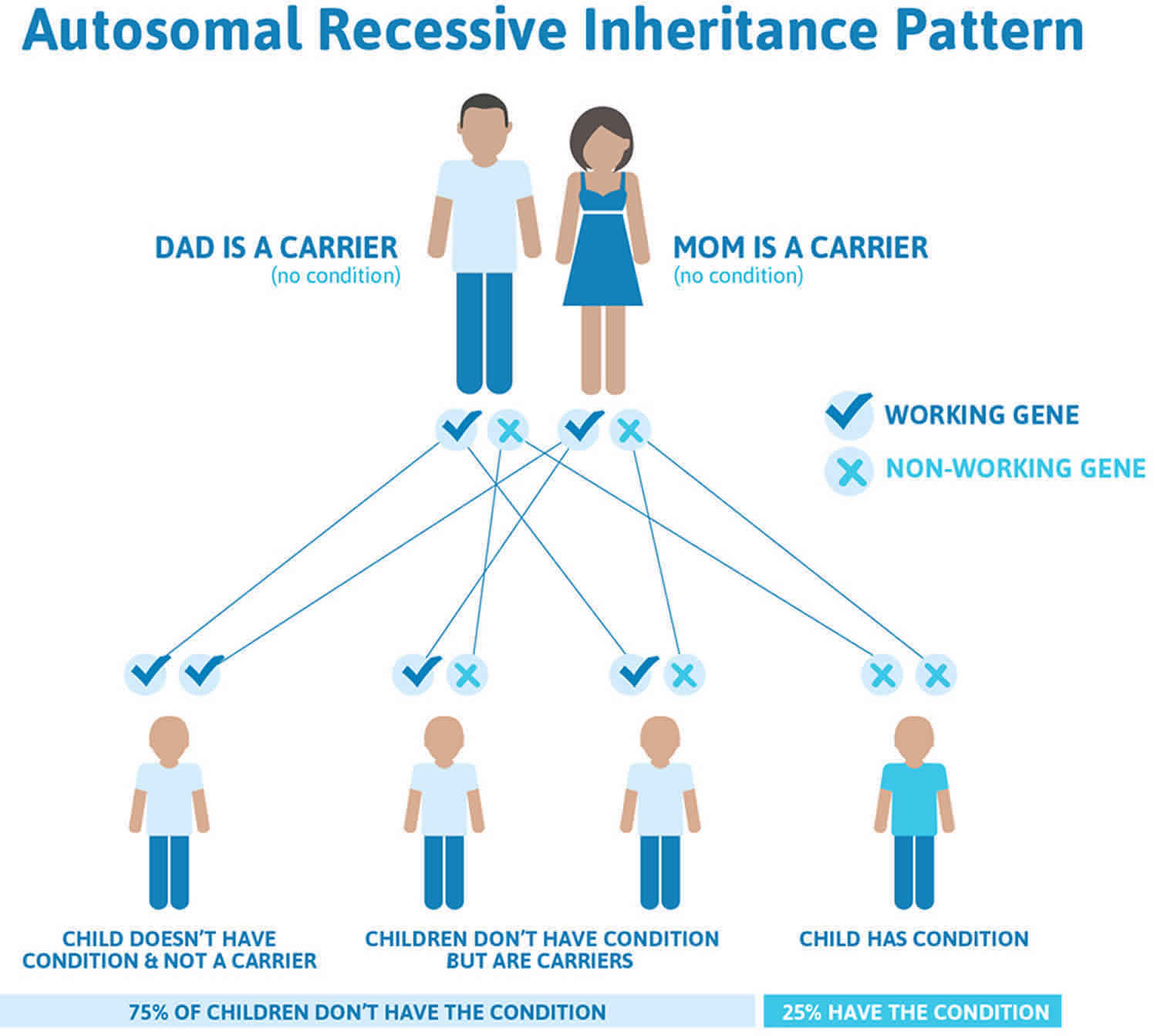
Biotinidase deficiency inheritance pattern
Biotinidase deficiency is inherited in an autosomal recessive pattern, which means both copies of the BTD gene in each cell have mutations. The parents of an individual with biotinidase deficiency each carry one copy of the mutated gene, but they typically do not have any health problems associated with the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 8 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 8. Biotinidase deficiency autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Biotinidase deficiency symptoms
Infants with biotinidase deficiency may be born without signs of the condition. The signs and symptoms of biotinidase deficiency typically appear within the first few months of life, but the age of onset varies. Below is a list of symptoms that infants and children with untreated biotinidase deficiency may have. It is important to know that not every person with biotinidase deficiency will show all of these symptoms.
Children with profound biotinidase deficiency, the more severe form of the condition, may have seizures, weak muscle tone (hypotonia), breathing problems, and delayed development. Infants with hypotonia may look abnormally “floppy.” Hypotonia can affect feeding and motor skills such sitting up without assistance. Biotinidase deficient infants and children may experience delays in reaching developmental milestones, including holding one’s head up or pulling up to stand. If left untreated, biotinidase deficiency can lead to hearing loss, eye abnormalities like eye infections (pink eye or conjunctivitis) and loss of vision, developmental delay, problems with body movement and balance (ataxia), skin rashes, hair loss (alopecia), and a fungal infection called candidiasis. Immediate treatment and lifelong management with biotin supplements can prevent many of these complications.
About 70% of infants with biotinidase deficiency will experience seizures if they are not treated 2. This is often the first symptom of biotinidase deficiency. Seizures in infants may look different than seizures in adults. Some signs of seizures in infants include:
- Staring spells
- Jerking arm or leg movements
- Stiffening of the body
- Flickering of the eyelids
Because the seizures are caused by the body being unable to recycle biotin, they may not stop with seizure medications (anticonvulsants) 2. However, the biotinidase deficiency seizures do respond to biotin therapy and often should stop within minutes to hours of receiving biotin treatment 2.
Infants with biotinidase deficiency may also have breathing problems, hearing and vision loss, problems with movement and balance (ataxia) 1. These issues can be prevented if biotin therapy is started early. Some other common features of biotinidase deficiency include eye infections, like pink eye (conjunctivitis), hair loss (alopecia), certain type of skin rash called eczema and a fungal infection called candidiasis. Infants with biotinidase deficiency may have specific molecules in their urine, such as lactic acid (lactic aciduria) or low but noticeable amounts of ammonia.
Some infants with biotinidase deficiency may have other symptoms like:
- Trouble controlling their body’s movements (ataxia)
- Breathing problems
- Drowsiness (lethargy)
- Enlarged liver (hepatomegaly)
- Enlarged spleen (splenomegaly)
- Speech problems.
Without treatment with biotin, infants with biotinidase deficiency can develop coma and may even die.
Partial biotinidase deficiency is a milder form of this condition. Affected children and adults may experience muscle weakness (hypotonia), skin rashes (eczema) and hair loss (alopecia), but these problems may appear only during illness, infection, or other times of stress on the body.
Treating biotinidase deficiency with biotin supplements before symptoms show up can prevent them from happening.
Biotinidase deficiency diagnosis
Biotinidase deficiency can be diagnosed in newborns through newborn screening. Newborn screening is a special type of screening test that newborns receive to see if they have certain diseases. Because the newborn screen is a screening test, a positive result does not mean that an infant definitely has the disease. Often, a repeat test must be done to confirm the diagnosis. A clinical diagnosis is possible after birth by testing for biotinidase activity in the blood. Usually, this is performed when signs and symptoms of biotinidase deficiency become clearer. In some infants, a genetic test may be ordered to identify the specific gene changes (mutation) that are causing biotinidase deficiency. Prenatal testing of sample fluid from the womb for biotinidase activity is available as early as 12 weeks of pregnancy (this includes chorionic villi sampling and amniocentesis).
Biotinidase activity can be measured in serum, plasma, and also in fibroblasts and leukocytes and other tissue extracts by radioassay 34. The measurement of biotinidase activity in plasma or serum by colorimetric assay is the most frequently used method for the diagnosis of biotinidase deficiency 35. Normal serum biotinidase activity in humans ranges from 4.4 to 10 nmol/min/mL with a mean activity and standard deviation of 7.1 ± 1.2 nmol/min/mL 36.
Serum biotinidase activity of carriers may be similar to those with partial biotinidase deficiency, confounding diagnosis based on enzyme analysis 37. Wolf 38 suggested that evaluation of parental biotinidase activity may be helpful. Mutation in the biotinidase deficiency gene results in deficient levels of enzyme activity 39. In some cases the enzyme activity does not differentiate partial deficiency from heterozygosity for profound deficiency, and genetic analysis is necessary 40.
Biochemically, in untreated patients, metabolic ketoacidosis, lactic acidosis, and/or hyperammonemia can occur 7. Elevation of 3-hydroxyisovaleric, 3-hydroxypropionic, lactic acid, and 3-methylcrotonylglycine can be detected in urine organic acid analysis 41, 42. In previous reports, it showed that urinary excretion of 3-hydroxyisovaleric acid was an indicator of biotin status 43.
Biotinidase deficiency treatment
Biotinidase deficiency is treated with oral biotin, typically prescribed at a starting dose of 5 to 20 milligrams (mg) of biotin daily. Treatment is with free biotin 7, 44. Bound biotin contained in oral multivitamin supplements does not treat the body deficiency. Treatment should begin as soon as the diagnosis is made. With biotin treatment, symptoms of the disorder may disappear. However, it takes a few hours to days for seizures and movement disorders to improve and some weeks for skin manifestations to improve.
Some patients require higher daily doses of free biotin. If the enzymatic defect is present but a healthy clinical response does not occur with lower doses, a high-dose therapy is considered (up to 40 mg/d) 44.
Treatment with free biotin is lifelong in patients with profound deficiency. Patients with a partial deficiency (10%-25% activity remaining) may not require lifelong treatment. Both patient populations, however, require knowledgeable continuity of care to address ongoing medical care and treatment approach.
Children with residual neurologic disease may require medical interventions for developmental delay, spasticity, and bulbar dysfunction, in addition to oral biotin treatment. Spasticity and dystonia associated with inborn errors of metabolism have been treated with intrathecal baclofen and neurotoxins 45.
Genetic counseling is recommended for families of a child with biotinidase deficiency. Genetic counselors are healthcare providers that help families understand genetic conditions and make genetic testing decisions.
Biotinidase deficiency prognosis
With immediate treatment and proper continuance and compliance of care, patients with biotinidase deficiency have an excellent prognosis and potential for a normal lifestyle and lifespan 46, 16. The prognosis for asymptomatic biotinidase deficiency cases is good if they receive treatment before the symptoms appear. For symptomatic biotinidase deficiency patients, pharmacological biotin therapy improves most clinical features but cannot reverse neurologic damage that has already occurred 47.
- Biotinidase deficiency. https://medlineplus.gov/genetics/condition/biotinidase-deficiency[↩][↩][↩]
- Biotinidase Deficiency. https://rarediseases.org/rare-diseases/biotinidase-deficiency[↩][↩][↩][↩]
- Biotinidase deficiency. https://ghr.nlm.nih.gov/condition/biotinidase-deficiency[↩]
- Saleem F, Soos MP. Biotin Deficiency. [Updated 2023 Feb 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547751[↩][↩]
- Zempleni J, Hassan YI, Wijeratne SS. Biotin and biotinidase deficiency. Expert Rev Endocrinol Metab. 2008 Nov 1;3(6):715-724. doi: 10.1586/17446651.3.6.715[↩]
- Hsu RH, Chien YH, Hwu WL, Chang IF, Ho HC, Chou SP, Huang TM, Lee NC. Genotypic and phenotypic correlations of biotinidase deficiency in the Chinese population. Orphanet J Rare Dis. 2019 Jan 7;14(1):6. doi: 10.1186/s13023-018-0992-2[↩]
- Canda E, Kalkan Uçar S, Çoker M. Biotinidase Deficiency: Prevalence, Impact And Management Strategies. Pediatric Health Med Ther. 2020 May 4;11:127-133. doi: 10.2147/PHMT.S198656[↩][↩][↩]
- Pindolia K, Chen J, Cardwell C, Cui X, Chopp M, Wolf B. Neurological deficits in mice with profound biotinidase deficiency are associated with demylination and axonal degeneration. Neurobiol Dis. 2012 Sep;47(3):428-35. doi: 10.1016/j.nbd.2012.04.016[↩]
- Wolf B. Biotinidase Deficiency. 2000 Mar 24 [Updated 2023 May 25]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1322[↩]
- Rajendiran A, Sampath S. Biotinidase deficiency–clinching the diagnosis rapidly can make all the difference! BMJ Case Rep. 2011 Sep 28;2011:bcr0720114494. doi: 10.1136/bcr.07.2011.4494[↩]
- Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:390-8.[↩][↩][↩][↩][↩]
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:359-74[↩][↩]
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:359-74.[↩][↩][↩]
- Said HM. Biotin: biochemical, physiological and clinical aspects. Subcell Biochem 2012;56:1-19.[↩][↩]
- Mock DM. Biotin. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:43-51[↩]
- Elrefai S, Wolf B. Disorders of biotin metabolism. In: Rosenberg RN, Pascual JM, eds. Rosenberg’s Molecular and Genetic basis of Neurological and Psychiatric Disease. 5th ed. United States of America: Elsevier; 2015:531-539.[↩][↩]
- Regula Baumgartner, E. and Suormala, T. (1999), Inherited defects of biotin metabolism. BioFactors, 10: 287-290. https://doi.org/10.1002/biof.5520100229[↩]
- Eng WK, Giraud D, Schlegel VL, Wang D, Lee BH, Zempleni J. Identification and assessment of markers of biotin status in healthy adults. Br J Nutr 2013;110:321-9[↩][↩][↩]
- Li D, Radulescu A, Shrestha RT, Root M, Karger AB, Killeen AA, et al. Association of biotin ingestion with performance of hormone and nonhormone assays in healthy adults. JAMA 2017;318:1150-60[↩]
- Zempleni J, Mock DM. Biotin biochemistry and human requirements. J Nutr Biochem. 1999 Mar;10(3):128-38. doi: 10.1016/s0955-2863(98)00095-3[↩]
- Present Knowledge in Nutrition. Basic Nutrition and Metabolism 11th Edition pp. 289–304, July 17, 2020. ISBN: 9780323661621[↩][↩]
- Biotin. https://lpi.oregonstate.edu/mic/vitamins/biotin[↩][↩][↩][↩][↩][↩][↩]
- Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253-72. doi: 10.1146/annurev.nutr.28.061807.155434[↩]
- Mock D, Matthews N. Biotin and pantothenic acid In: Stipanuk MH, Caudill MA, editors. Biochemical, physiological and molecular aspects of human nutrition. 3rd ed Amsterdam (Netherlands): Elsevier; 2012. p. 610–25.[↩]
- Hendriksz CJ, Preece MA, Chakrapani A. Successful pregnancy in a treated patient with biotinidase deficiency. J Inherit Metab Dis. 2005; 28(5):791-792.[↩]
- Donald M. Mock. Marginal Biotin Deficiency is Common in Normal Human Pregnancy and Is Highly Teratogenic in Mice. J Nutr.. January, 2009; 139(1):154-157.[↩][↩][↩]
- Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr.. April, 2008; 138(4):680-684.[↩]
- Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:390-398.[↩]
- Mock DM, Stadler DD. Conflicting indicators of biotin status from a cross-sectional study of normal pregnancy. J Am Coll Nutr. 1997 Jun;16(3):252-7. doi: 10.1080/07315724.1997.10718682[↩]
- Mock DM, Stadler DD, Stratton SL, Mock NI. Biotin status assessed longitudinally in pregnant women. J Nutr. 1997 May;127(5):710-6. doi: 10.1093/jn/127.5.710[↩]
- Mock DM, Quirk JG, Mock NI. Marginal biotin deficiency during normal pregnancy. Am J Clin Nutr. 2002 Feb;75(2):295-9. doi: 10.1093/ajcn/75.2.295[↩]
- Mock DM. Marginal biotin deficiency is common in normal human pregnancy and is highly teratogenic in mice. J Nutr. 2009 Jan;139(1):154-7. doi: 10.3945/jn.108.095273[↩][↩][↩]
- Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr. 2008 Apr;138(4):680-4. doi: 10.1093/jn/138.4.680[↩]
- Wolf B, Secor McVoy J. A sensitive radioassay for biotinidase activity: deficient activity in tissues of serum biotinidase-deficient individuals. Clin Chim Acta. 1983;135(3):275–281. doi: 10.1016/0009-8981(83)90286-3[↩]
- Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131(3):273–281. doi: 10.1016/0009-8981(83)90096-7[↩]
- Wolf B, Grier RE, Secor McVoy JR, Heard GS. Biotinidase deficiency: a novel vitamin recycling defect. J Inherit Metab Dis. 1985;8(Suppl 1):53–58. doi: 10.1007/BF01800660[↩]
- Gannavarapu S, Prasad C, DiRaimo J, et al. Biotinidase deficiency: spectrum of molecular, enzymatic and clinical information from newborn screening Ontario, Canada (2007–2014). Mol Genet Metab. 2015;116(3):146–151. doi: 10.1016/j.ymgme.2015.08.010[↩]
- Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med. 2012;14(6):565–575. doi: 10.1038/gim.2011.6[↩]
- Hymes J, Stanley CM, Wolf B. Mutations in BTD causing biotinidase deficiency. Hum Mutat. 2001;18(5):375–381. doi: 10.1002/(ISSN)1098-1004[↩]
- Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100(1):6–13. doi: 10.1016/j.ymgme.2010.01.003[↩]
- Strovel ET, Cowan TM, Scott AI, Wolf B. ERRATUM: Laboratory diagnosis of biotinidase deficiency, 2017 update: a technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med. 2018 Feb;20(2):282. https://doi.org/10.1038/gim.2017.201 Erratum Genet Med online publication 6 July 2017; doi:https://doi.org/10.1038/gim.2017.84[↩]
- Schubiger G, Caflisch U, Baumgartner R, Suormala T, Bachmann C. Biotinidase deficiency: clinical course and biochemical findings. J Inherit Metab Dis. 1984;7(3):129-30. doi: 10.1007/BF01801771[↩]
- Horvath TD, Matthews NI, Stratton SL, Mock DM, Boysen G. Measurement of 3-hydroxyisovaleric acid in urine from marginally biotin-deficient humans by UPLC-MS/MS. Anal Bioanal Chem. 2011;401(9):2805–2810. doi: 10.1007/s00216-011-5356-x[↩]
- Saleem H, Simpson B. Biotinidase Deficiency. [Updated 2023 Feb 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560607[↩][↩]
- Armstrong RW, Steinbok P, Cochrane DD, Kube SD, Fife SE, Farrell K. Intrathecally administered baclofen for treatment of children with spasticity of cerebral origin. J Neurosurg. 1997 Sep;87(3):409-14. doi: 10.3171/jns.1997.87.3.0409[↩]
- Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med. 2012 Jun;14(6):565-75. doi: 10.1038/gim.2011.6[↩]
- Szymańska E, Średzińska M, Ługowska A, Pajdowska M, Rokicki D, Tylki-Szymańska A. Outcomes of oral biotin treatment in patients with biotinidase deficiency – Twenty years follow-up. Mol Genet Metab Rep. 2015 Oct 6;5:33-35. doi: 10.1016/j.ymgmr.2015.09.004[↩]




{kind=link}