Contents
Wiskott-Aldrich syndrome
Wiskott-Aldrich syndrome also known as “WAS” or “eczema-thrombocytopenia-immunodeficiency syndrome” is a rare X-linked disorder that primarily affects males with a characteristic Wiskott-Aldrich syndrome triad of abnormal immune system function (immune deficiency), thrombocytopenia (low platelets), and eczema (dermatitis) 1, 2, 3, 4, 5, 6, 7. Wiskott-Aldrich syndrome results from a genetic mutation in the WAS gene found on the X chromosome that codes for Wiskott-Aldrich syndrome protein (WASP) affecting the immune system and inducing a state of immunodeficiency, that results in increased risk of infections and to autoimmune diseases and inflammatory disorders and an increased risk for some types of cancer such as lymphoma and is also associated with microthrombocytopenia (low platelet count and abnormally reduced platelet size) and eczema (dermatitis) 8, 9, 10, 11. Different types of mutation within the WAS gene may vary between individuals resulting in the presence of the complete clinical spectrum in some cases, or only some of the features in others with severe phenotype known as classic Wiskott-Aldrich syndrome to milder ones called X-linked thrombocytopenia (XLT) and X-linked neutropenia (XLN) 11, 1.
Wiskott Aldrich Syndrome is named after two physicians who first recognized the disorder. It was first described in 1937 by Dr. Alfred Wiskott, a German pediatrician who noticed a bleeding disorder due to low platelets that ran only in boys in a family 12, 13. In 1954, Dr. Robert Aldrich, an American pediatrician, studied seven generations of an affected family and showed that this disease is passed on from mothers to their sons 14, 13.
Wiskott-Aldrich syndrome can be thought of as a spectrum known as “Wiskott-Aldrich syndrome-related disorders” are all caused by mutations in the WAS gene presenting with overlapping symptoms that fall between the severe features of classic Wiskott-Aldrich syndrome and the less severe form called X-linked thrombocytopenia (XLT), and X-linked neutropenia (XLN) 2, 1. Patients may shift in severity, depending on the progression of the disease over time, or the emergence of complications such as the development of lymphoma.
The WAS gene mutations result in deficiency of the Wiskott-Aldrich syndrome protein (WASP). The more deficient the WASP, the more severe the disease 15. The Wiskott-Aldrich syndrome protein (WASP) plays an essential role in relaying signals from the surface of the blood cell to the cell’s actin cytoskeleton, the network of fibers that make up the cell’s structural framework. Immune cells that lack the WASP protein have a decreased ability to respond to their
environment, fight invaders, and form functional platelets.
Approximately 160 different mutations or deletions in the WAS gene have been described. Because the WAS gene codes for WASP is X-linked, the majority of cases with Wiskott-Aldrich syndrome are males; rarely girls can be affected. Females with the abnormal WAS gene are usually unaffected carriers that pass the mutation on to the next generation 1. WAS gene carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk of Wiskott-Aldrich syndrome are possible if the pathogenic variant has been identified in the family.
Wiskott-Aldrich syndrome is estimated to occur in approximately 1 to 10 of every million boys 3. In a US study, Wiskott-Aldrich syndrome incidence was reported to be 3.3–5.2 per million live male births 16. National registry data on primary immunodeficiency diseases from Sweden and Switzerland estimated Wiskott-Aldrich syndrome incidence to be 3.7 and 4.1 per million live births, respectively 17, 18.
- Wiskott-Aldrich syndrome-related disorders usually present in infancy. Affected males have thrombocytopenia with intermittent mucosal bleeding, bloody diarrhea, and intermittent or chronic petechiae and purpura; eczema; and recurrent bacterial and viral infections, particularly of the ear. At least 40% of those who survive the early complications develop one or more autoimmune conditions including hemolytic anemia, immune thrombocytopenic purpura, immune-mediated neutropenia, rheumatoid arthritis, vasculitis, and immune-mediated damage to the kidneys and liver. Individuals with a Wiskott-Aldrich syndrome-related disorder, particularly those who have been exposed to Epstein-Barr virus (EBV), are at increased risk of developing lymphomas, which often occur in unusual, extranodal locations including the brain, lung, or gastrointestinal tract.
- Males with X-linked thrombocytopenia (XLT) have thrombocytopenia with small platelets; other complications of Wiskott-Aldrich syndrome, including eczema and immune dysfunction, are usually mild or absent.
- Males with X-linked neutropenia (XLN) have congenital neutropenia, myeloid dysplasia, and lymphoid cell abnormalities.
The estimated incidence of Wiskott-Aldrich syndrome is between 1 and 10 cases per million males worldwide; this condition is rarer in females.
Individuals with Wiskott-Aldrich syndrome have microthrombocytopenia, which is a decrease in the number and size of blood cell fragments involved in clotting called platelets. This platelet abnormality, which is typically present from birth, can lead to easy bruising, bloody diarrhea, or episodes of prolonged bleeding following minor trauma. Microthrombocytopenia can also lead to small areas of bleeding just under the surface of the skin, resulting in purplish spots called purpura or rashes of tiny red spots called petechiae. In some cases, the bleeding episodes can be life-threatening.
Wiskott-Aldrich syndrome is also characterized by abnormal or nonfunctional immune system cells known as white blood cells. Changes in white blood cells lead to an increased risk of several immune and inflammatory disorders in people with Wiskott-Aldrich syndrome. These immune problems vary in severity and include an increased susceptibility to infection and eczema (an inflammatory skin disorder characterized by abnormal patches of red, irritated skin). People with Wiskott-Aldrich syndrome are at greater risk of developing autoimmune disorders, such as rheumatoid arthritis or hemolytic anemia, which occur when the immune system malfunctions and attacks the body’s own tissues and organs. The chance of developing certain types of cancer, such as cancer of the immune system cells (lymphoma), is also increased in people with Wiskott-Aldrich syndrome.
Wiskott-Aldrich syndrome is often considered to be part of a disease spectrum with two other disorders: X-linked thrombocytopenia and severe congenital neutropenia. These conditions have overlapping signs and symptoms and the same genetic cause and is inherited in an X-linked manner.
Wiskott-Aldrich syndrome treatment may depends on the severity and symptoms in each person, but hematopoietic stem cell transplantation is the only known cure. Hematopoietic stem cells are the blood-forming stem cells that can be found mainly in the sponge-like material found inside bones (bone marrow), but also in the bloodstream called peripheral blood stem cells (PBSCs), and in the umbilical cord. Hematopoietic stem cells from any of these sources can be used in transplants. Hematopoietic stem cells divide to form more blood-forming stem cells, or they mature into one of three types of blood cells: white blood cells, which fight infection; red blood cells, which carry oxygen; and platelets, which help the blood to clot.
The other management of Wiskott-Aldrich syndrome mainly depends on supportive care which includes broad-spectrum antibiotics for bacterial infections, antivirals for viral infections or antifungals for fungal infections 2. Patients also require platelet transfusions to prevent bleeding. Topical steroids are used to treat eczema. Live vaccines such as BCG and MMR are contraindicated. Non-steroidal anti-inflammatory medications (ibuprofen, diclofenac, aspirin and others) should be avoided.
Wiskott-Aldrich syndrome prognosis have improved over time due to better management of the disease. People who have a successful and uncomplicated hematopoeitic cell transplantation, usually have normal immune function and, normal survival.
Wiskott-Aldrich syndrome causes
Mutations in the WAS gene located on short arm of the X-chromosome at Xp11.22-23 position (on the short arm of the X chromosome), cause Wiskott-Aldrich syndrome 19, 20, 21, 22, 23, 24, 25. The WAS gene provides instructions for making a protein called Wiskott-Aldrich protein (WASP). This protein is found in all blood cells. Wiskott-Aldrich protein (WASp) is a 502 amino acid protein expressed in the cytoplasm of non-erythroid hematopoietic cells, is involved in relaying signals from the surface of blood cells to the actin cytoskeleton, which is a network of fibers that make up the cell’s structural framework 26, 27, 28, 29, 30, 31, 32. WASP signaling triggers the cell to move and attach to other cells and tissues (adhesion). In white blood cells, this signaling allows the actin cytoskeleton to establish interactions between cells and the foreign invaders that they target (immune synapses) 26, 27, 28, 29, 30, 31, 32. Gain-of-function mutations WAS gene manifest as severe congenital X-linked neutropenia, which is characterized by recurrent bacterial infections, neutropenia, and monocytopenia without thrombocytopenia 33, 34.
WAS gene mutations that cause Wiskott-Aldrich syndrome lead to a lack of any functional Wiskott-Aldrich protein (WASp). Loss of Wiskott-Aldrich protein (WASp) signaling disrupts the function of the actin cytoskeleton in developing blood cells. White blood cells that lack Wiskott-Aldrich protein (WASp) have a decreased ability to respond to their environment and form immune synapses. As a result, white blood cells are less able to respond to foreign invaders, causing many of the immune problems related to Wiskott-Aldrich syndrome. Similarly, a lack of functional WASP in platelets impairs their development, leading to reduced size and early cell death.
More than 300 gene mutations have been identified leading to impaired protein configuration. The most common mutations are missense mutations followed by non-sense, splice site and short deletion mutations. Because of the wide range of genetic mutations, the disease itself has phenotypic variability ranging from severe (classic Wiskott-Aldrich syndrome) to mild disease X linked thrombocytopenia and X linked neutropenia.
As mentioned earlier, the Wiskott-Aldrich syndrome protein (WASP) expresses in non-erythroid hematopoietic cells where it functions as a bridge between signaling and movement of actin filaments in the cytoskeleton. This ultrastructural component of the cellular architecture is primarily responsible for intracellular and cell-substrate interactions and signaling because of its role in cell morphology and movements 35. The actin cytoskeleton is involved in various cellular functions such as growth, cytokinesis, endocytosis, and exocytosis. It also has involvement in the formation of an immunologic synapse, which is the site of interaction between T cells and antigen-presenting cells like dendritic cells. The interaction depends on the generation of lipid rafts, which provide a platform to recruit crucial molecules to ensure the stability of immunologic synapse 36. In Wiskott-Aldrich syndrome, there is abnormal cytoskeleton reorganization because of impaired gene expression leading to T cell dysfunction causing impaired migration, adhesion and insufficient interaction with other cells due to abnormal synapse formation; this affects B cells homeostasis resulting in selective depletion of circulating mature B cells splenic marginal zone precursors, and marginal zone B cells. This event of lymphocyte numbers declining over time is due to accelerated cell death 37. Circulating natural killer cells are normal or increased, but cytotoxicity of these WAS protein deficient cells is impeded as a result of impaired immunologic synapse formation. Interleukin-2 can help to restore cytotoxicity in natural killer (NK) cells by inducing the expression of a functionally related protein. Invariant natural killer T cells are completely absent in patients with Wiskott-Aldrich syndrome and X linked thrombocytopenia, which predisposes patients to increased risks for autoimmunity and cancer 38. Mechanisms of autoimmunity in Wiskott-Aldrich syndrome include inadequate Treg cell function, B cell-intrinsic loss of tolerance via positive selection of self-reactive transitional B cells, expansion of autoreactive B cells and production of autoantibodies, impaired Fas-mediated apoptosis of self-reactive lymphocytes, and defective phagocytosis of apoptotic cells resulting in chronic inflammation 39.
WASp-deficient myeloid lineage cells exhibit impaired phagocytosis and chemotaxis. Also, monocytes, macrophages, and dendritic cells from WASp-deficient patients demonstrate almost completely abrogated assembly of actin-rich structures responsible for cellular migration leading to impaired chemotaxis to specific chemoattractants 40. The explanation for thrombocytopenia is increased clearance, ineffective thrombocytopoiesis, reduced platelet survival due to intrinsic platelet abnormalities, and immune-mediated events.
Whereas “loss-of-function” mutations in the WAS gene cause either X-linked thrombocytopenia or Wiskott-Aldrich syndrome, unique “gain-of-function” missense mutations impair the autoinhibitory conformation of the molecule and lead to increased actin polymerization, resulting in congenital neutropenia.
Because they all have the same genetic cause, Wiskott-Aldrich syndrome, X-linked thrombocytopenia, and severe congenital neutropenia are sometimes collectively referred to as Wiskott-Aldrich syndrome-related disorders.
Figure 1. Wiskott Aldrich syndrome protein (WASP) structure and function
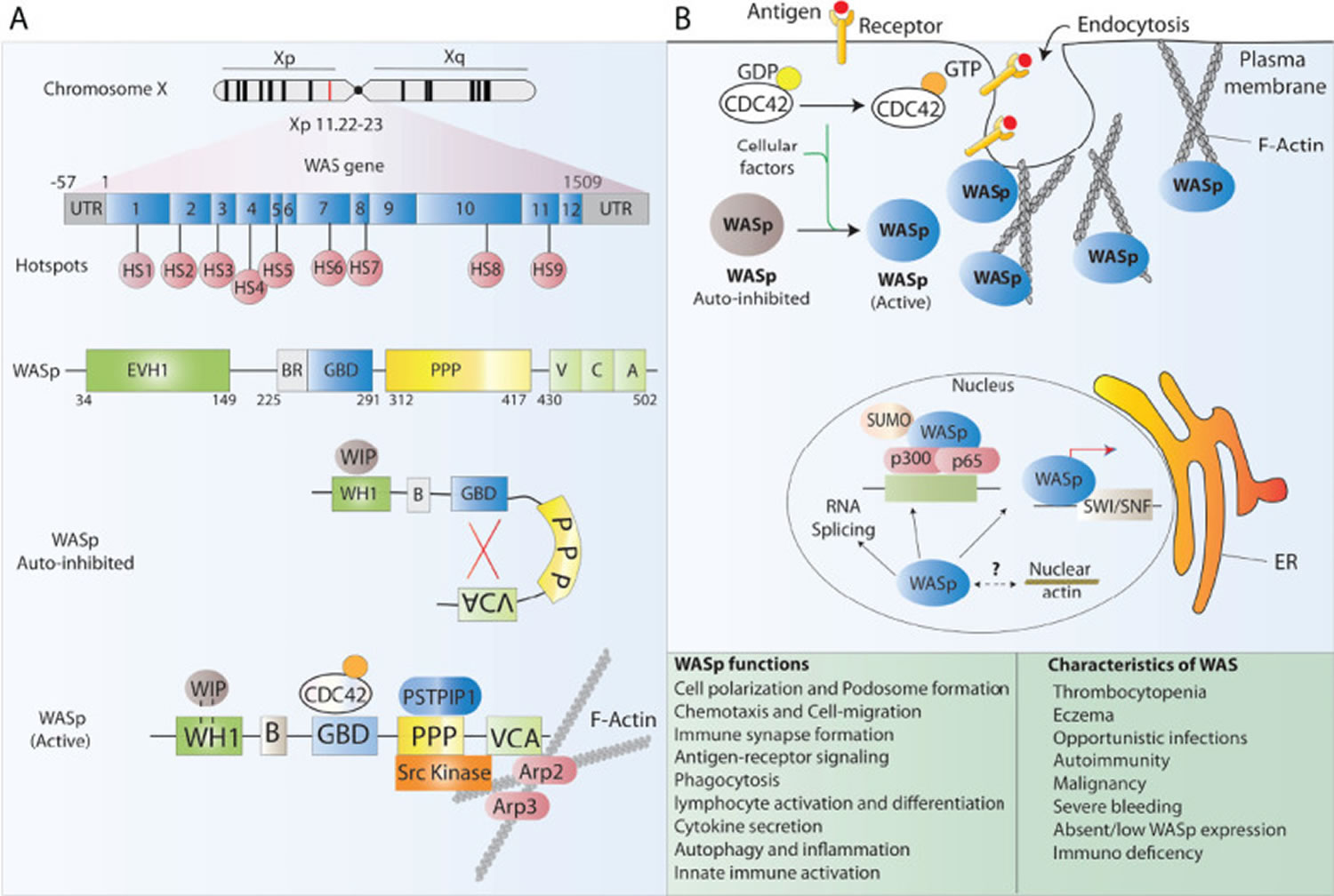
Footnotes: (A) Schematic representation of the WAS gene and its major functional protein domains. The WAS gene is located on the X chromosome and comprises 12 exons. Disease-causing mutations in WAS patients are scattered around the whole gene, however, 9 mutational hotspots (HS) have been detected in patients. WASp is made of 5 different domains and presents in the cell cytoplasm in an auto-inhibited conformation at resting state and leads to actin polymerization after receptor stimulation. (B) The many functions of WASp in lymphoid and myeloid immune cells relate to its role in regulating the polymerization of new branched actin filaments. Besides, WASp plays an important role as a scaffold protein in the regulation of some nuclear functions such as chromatin remodeling.
[Source 5 ]Wiskott-Aldrich syndrome inheritance pattern
Wiskott-Aldrich syndrome is inherited in an inherited in an X-linked recessive manner. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males, who have only one X chromosome, a mutation in the only copy of the gene in each cell is sufficient to cause the condition. In females, who have two copies of the X chromosome, one altered copy of the gene in each cell can lead to less severe features of the condition or may cause no signs or symptoms at all. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Although females have two X chromosomes, one of the X chromosomes in each cell is “turned off” and all of the genes on that chromosome are inactivated. Females who have a mutation in a gene on one of their X chromosomes are called carriers of the related condition. Carrier females usually do not have symptoms of the condition because usually the X chromosome with the mutated gene is turned off. Therefore, they have another X chromosome with a working copy of the gene. Sometimes, the X chromosome with the working copy of the gene is turned off, which may cause symptoms of the condition. However, females with symptoms are usually much more mildly affected than males. A male has only one X chromosome, so if he inherits a mutation on the X chromosome, he will have signs and symptoms (be affected).
Males with an X-linked recessive condition always pass the mutated gene to all of their daughters, who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome to male offspring 41.
Female carriers of an X-linked recessive condition have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have an affected son, and a 25% chance to have an unaffected son 41. This also means that each daughter of a carrier mother has a 50% chance of being a carrier, and each son has a 50% chance of having the condition.
Figure 2. Wiskott-Aldrich syndrome X-linked recessive inheritance pattern

People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Wiskott-Aldrich syndrome signs and symptoms
Wiskott-Aldrich Syndrome involves a spectrum of disorders with symptoms ranging from mild to severe. Common signs and symptoms of Wiskott-Aldrich syndrome include the following.
- Decreased numbers of platelets (thrombocytopenia), and very small platelets usually present at birth which can result in:
- Bleeding inside the brain, which can be very fatal
- Mucosal (such as inside the mouth) bleeding
- Bloody diarrhea
- Bruising (ecchymoses) or purplish areas on the skin or mucous membranes (purpura), caused by bleeding under the skin
- Pinpoint bleeding into the skin (petechiae).
- Life-threatening bleeding (occurs in 30% of males prior to diagnosis)
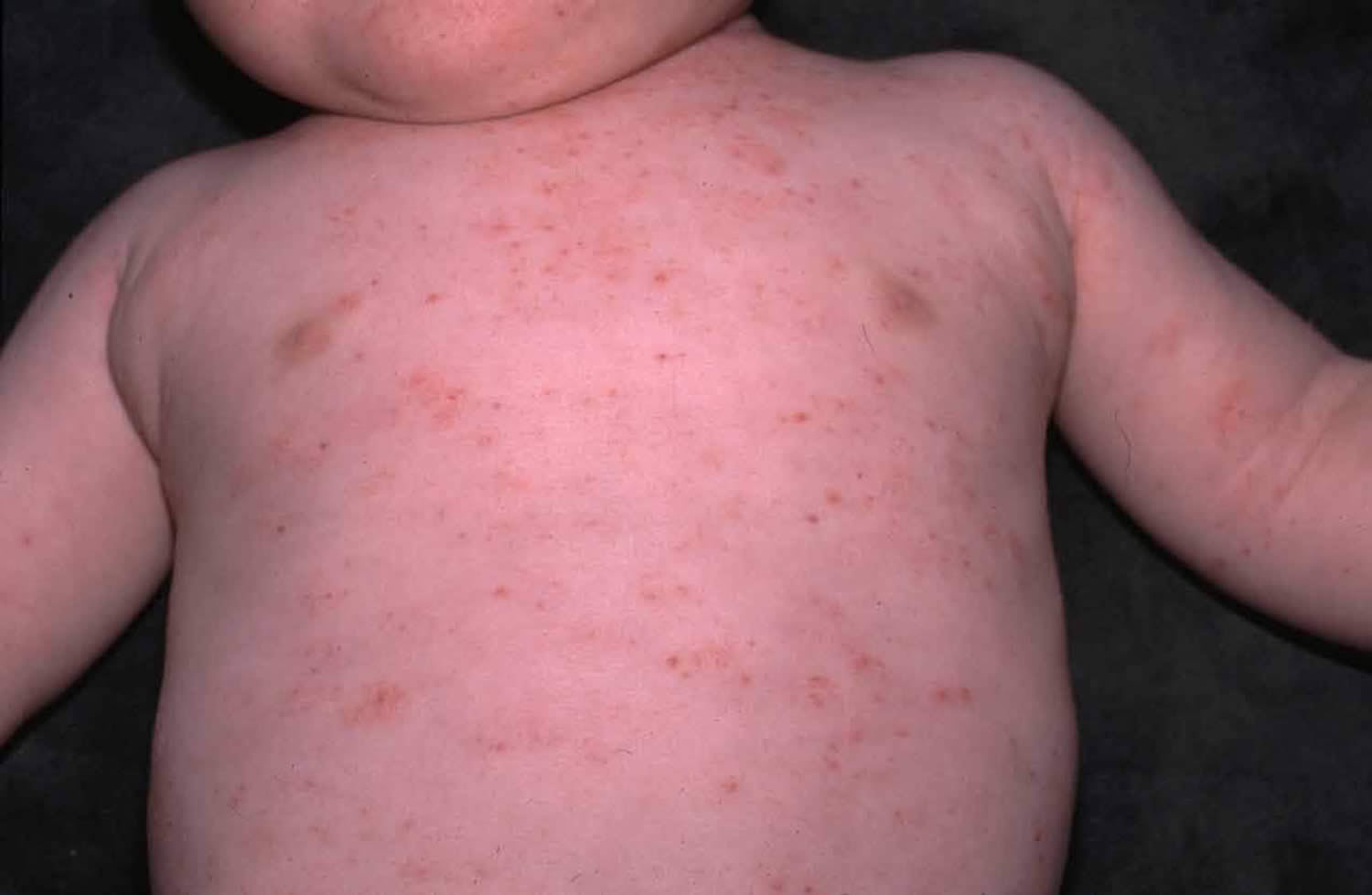
- Red patches of red and irritated skin (eczema), occurs in about 80% of the cases and can be mild to severe. Typically eczema (dermatitis) appears during infancy or early childhood. The features of the eczema are not distinguishable from atopic eczema. Wiskott-Aldrich syndrome patients often have elevated IgE levels and develop allergies.
- Opportunistic skin infections such as molluscum contagiosum, impetigo, cellulitis, herpes simplex, abscesses and bacterial sepsis may also be persistent or recurrent problems for Wiskott-Aldrich syndrome patients.
- Immunodeficiency may affect both T and B lymphocyte function. Immune deficiency increases the risk and frequency of a wide range of infections, especially to recurrent bacterial and viral infections, mostly recurrent ear infections with discharging ears, bacterial or viral pneumonia, bacterial skin infections and some viruses such as cytomegalovirus (CMV), herpes simplex virus (HSV, cold sore virus), Epstein-Barr virus (EBV). Opportunistic lung infection with Pneumocystis jiroveci can occur.
- Increased risk of developing autoimmune disorders (when the immune system mistakenly attacks the body’s own tissues and organs) specially when people get older, and that may include autoimmune hemolytic anemia (destruction of red blood cells), immune thrombocytopenic purpura, rheumatoid arthritis, vasculitis of small and large vessels, and immune-mediated damage to the kidneys and liver
- Increased risk of developing some types of cancer, such as leukemia and B-cell lymphoma, especially in people with Wiskott Aldrich syndrome who had an Epstein-Barr virus (EBV) infection or are older and have an autoimmune disease.
Wiskott-Aldrich syndrome manifests as follows:
- Bleeding: Thrombocytopenia is present at birth. It is the most common finding present at the time of diagnosis. Affected patients may present in the first days of life with petechiae and prolonged bleeding from the umbilical stump or after circumcision. Other manifestations may include purpura, hematemesis, melena, epistaxis, hematuria, and such life-threatening symptoms as oral, gastrointestinal, and intracranial bleeding. A subset of infants less than or equal to 2 years of age may present with “severe refractory thrombocytopenia,” possibly due to antiplatelet autoantibody, a complication that is associated with poor prognosis 42.
- Immunodeficiency: The severity of immunodeficiency depends largely on the type of mutations and resulting protein expression. Patients usually present with multiple recurrent infections and failure to thrive. Patients are susceptible to encapsulated organisms as Streptococcus pneumoniae, Neisseria meningitides, and Haemophilus influenzae. Manifestations include otitis media, sinusitis, pneumonia, meningitis, sepsis, and colitis. Splenectomy, which is occasionally performed to decrease the risk of bleeding, further increases the risk of severe infections and sepsis. This immunodeficiency also predisposes patients to opportunistic infections with Pneumocystis jirovecii, Molluscum contagiosum, as well as systemic varicella and cytomegalovirus infection. Fungal infections are relatively rare consisting primarily of mucocutaneous infection due to Candida albicans.
- Eczema: Eczema of varying severity develops in approximately one-half of Wiskott-Aldrich syndrome patients during the first year of life and resembles classical atopic dermatitis.
- Autoimmune manifestations: Reports exist of autoimmune diseases include hemolytic anemia, neutropenia, vasculitis involving both small and large vessels, inflammatory bowel disease, and renal diseases. A broad spectrum of autoantibodies has been observed both in classic Wiskott-Aldrich syndrome and in X-linked thrombocytopenia.
- Cancers: Cancers can occur during childhood but are most frequently present in adolescent and young adult males with the classic Wiskott-Aldrich syndrome phenotype. B cell lymphoma (often Epstein-Barr virus-positive) and leukemia are common in classic Wiskott-Aldrich syndrome but do occur in X-linked thrombocytopenia (XLT).
Wiskott-Aldrich syndrome has three main clinical phenotypic manifestations:
Classic (severe) Wiskott-Aldrich syndrome
Classic (severe) Wiskott-Aldrich syndrome is the severe phenotype of Wiskott-Aldrich syndrome. Affected boys present in early childhood with a bleeding disorder (hemorrhagic diathesis) due to thrombocytopenia (low number of platelets in the blood); recurrent bacterial, viral and fungal infections; and extensive eczema. Lymphadenopathy (enlarged lymph nodes) is frequently present, especially in those Wiskott-Aldrich syndrome patients with chronic eczema, and hepatosplenomegaly (enlarged liver and spleen) is common. Patients with classic Wiskott-Aldrich syndrome tend to develop autoimmune disorders and lymphoma or other malignancies, often leading to early death 43.
X-linked neutropenia (XLN)
X-linked neutropenia (XLN) presents mainly as congenital neutropenia (low number of neutrophils or white blood cells that are present at birth). Patients with X-linked neutropenia present with infections characteristic for neutropenia but may also develop infections associated with lymphocyte dysfunction. These patients also have an elevated risk for myelodysplasia (a group of blood cancers in which immature blood cells in the bone marrow do not mature or become healthy blood cells) 44.
X-linked thrombocytopenia (XLT)
X-linked thrombocytopenia (XLT) presents as congenital thrombocytopenia (low number of platelets in the blood present at birth) that is sometimes intermittent 45. Eczema is usually mild. These patients generally have a benign disease course and good long-term survival. They still carry an increased risk (lower than that for Wiskott-Aldrich syndrome) for severe events such as life-threatening infections (especially post-splenectomy), serious hemorrhage, autoimmune complications, and cancer 46. Any male with thrombocytopenia and small platelets should be evaluated for WASp expression and WAS gene mutations.
Wiskott-Aldrich syndrome diagnosis
A diagnosis of Wiskott-Aldrich syndrome or X-linked thrombocytopenia (XLT) is a consideration in any male patient who presents with petechiae, bruises, and congenital or early-onset thrombocytopenia associated with small platelet size. Identification of mutation in the WAS gene is essential for confirmation of diagnosis of Wiskott Aldrich syndrome. Presence of mild or severe eczema supports the diagnosis. Infections and immunologic abnormalities may be absent, mild, or severe. Autoimmune diseases and malignancies develop more often in patients with classic WAS than in those with X-linked thrombocytopenia. Screening for presence or absence of WAS protein (WASp) can be performed in lymphocytes by flow cytometry using an anti-WASp antibody 47. The diagnosis of X-linked neutropenia should be considered in any male patient presenting with severe congenital neutropenia.
- Immunology: Abnormal immunologic findings in patients with Wiskott-Aldrich syndrome include decrease number and function of T cells and regulatory T cells, abnormal immunoglobulin (Ig) isotypes, defective antigen-antibody response, impaired cytotoxicity of natural killer cells with normal to increased cell numbers, impaired chemotaxis of neutrophils and phagocytic cells. Absolute lymphocyte counts are usually normal during infancy, but T and B cell numbers decrease later in life in patients with classic Wiskott-Aldrich syndrome. Reported variations in the levels of Ig including normal levels of serum IgG, decreased levels of IgM, and elevated levels of IgA and IgE also exist.
- Histopathology: Abnormal findings in lymphoreticular tissue are commonly present, including varying degrees of T cell zone depletion in lymph nodes and spleen, decreased number of follicles and abnormal follicular formation devoid of marginal zone, and regressive or “burned out” germinal centers 48.
- Thrombocytopenia and platelet abnormalities: Thrombocytopenia (<70,000 platelets/mm3) associated with small platelet volume is a consistent finding in patients with WAS gene mutations, except for those presenting with an X-linked neutropenia phenotype. Platelet counts are generally 20,000 to 50,000 per mm³ but may drop below 10,000 per mm³.
Wiskott-Aldrich syndrome should be suspected in a male with:
- Profound thrombocytopenia (<70,000 platelets/mm³)
- Small platelet size (mean platelet volume >2 SD below the mean for the laboratory)
- Recurrent bacterial or viral infection or opportunistic infection in infancy or early childhood
- Eczema
- Autoimmune disorder
- Lymphoma
- Family history of one or more maternally related males with a Wiskott-Aldrich syndrome-related phenotype or disorder
- Absent or decreased intracellular Wiskott-Aldrich syndrome protein (WASp) detection in hematopoietic cells as determined by flow cytometry or western blotting
- Abnormal lymphocytes:
- Decreased T-cell subsets, especially proportion and absolute number of CD8+T cells
- Decreased NK cell function. Lymphocyte subsets, mitogen responses, and other tests of cell-mediated immunity can vary among individuals, and over time in the same individual.
- Note: (1) Some individuals, particularly children, have normal lymphocyte numbers and normal function. (2) Although the proportion of CD8+ cells is often decreased, it is occasionally increased.
- Abnormal immunoglobulin levels: decreased IgM, normal or decreased IgG, increased IgA, increased IgE
- Absent isohemagglutinins
- Note: Interpretation of the significance of isohemagglutinin titers is unreliable in children younger than age 18 years.
- Absent or greatly decreased antibody responses to polysaccharide vaccines (e.g., Pneumovax®)
X-linked thrombocytopenia (XLT) should be suspected in a male with:
- Congenital thrombocytopenia (5,000-50,000 platelets/mm³)
- Small platelet size (platelet volume <7.5 fL)
- Absence of other clinical findings of Wiskott-Aldrich syndrome
- Family history of one or more maternally related males with a WAS-related phenotype or disorder
- Decreased or absent WASp by flow cytometry or western blotting
- Note: Some affected individuals have near-normal amounts of WASp.
X-linked congenital neutropenia (XLN) should be suspected in a male with:
- Recurrent bacterial infections
- Persistent neutropenia
- Arrested development of the bone marrow in the absence of other clinical findings of Wiskott-Aldrich syndrome
- Normal WASp expression by flow cytometry or western blotting
Wiskott-Aldrich syndrome treatment
The management of Wiskott-Aldrich syndrome mainly depends supportive care which includes broad-spectrum antibiotics for bacterial infections, antivirals for viral infections or antifungals for fungal infections 1, 2. Patients also require platelet transfusions to prevent bleeding. Topical steroids are used to treat eczema. Live vaccines such as BCG and MMR are contraindicated. Non-steroidal anti-inflammatory medications (ibuprofen, diclofenac, aspirin and others) should be avoided.
- Trimethoprim-sulfamethoxazole prophylaxis daily to prevent Pneumocystis jiroveci infection
- Aciclovir prophylaxis to prevent herpes simplex infections
- Regular intravenous immunoglobulin infusions where there are abnormalities in B cell function
- Irradiated platelets and red blood cell transfusions to treat serious bleeding episodes.
- Hematopoietic stem cells transplantation also called bone marrow transplantation or stem cell transplantation: Hematopoietic stem cells transplantation is the only available curative treatment, with excellent results for patients with human leukocyte antigen (HLA)-matched family or unrelated donors or partially matched cord-blood donors. Affected males who receive hematopoietic cell transplantation from a matched healthy sibling or closely matched unrelated donor before their second birthday have a greater than 90% probability of being cured of the disorder 49, 50. Currently, males with a WAS pathogenic variant who meet the clinical diagnostic criteria for Wiskott-Aldrich syndrome (WAS score 3-5), have markedly decreased WASP expression, and have a suitably matched donor are candidates for hematopoietic stem cells transplantation. Myeloablative conditioning prior to transplantation is the most widely used approach, as reduced-intensity conditioning increases the risk of partial engraftment, which may not be curative 49. Some symptoms (e.g., autoimmune disease) may take months to resolve following complete engraftment. Since the phenotype of Wiskott-Aldrich syndrome may evolve over time, optimal timing of transplantation is challenging. Individuals with absent intracellular WASP detection in hematopoietic cells are likely to develop Wiskott-Aldrich syndrome, although a minority will exhibit the milder X-linked thrombocytopenia (XLT) phenotype throughout their life span. Hematopoietic cell transplantation before development of autoimmunity and malignancy is highly desirable, and younger age at transplant is associated with improved long-term outcome following hematopoietic cell transplantation; however, the use of multiple chemotherapeutic agents necessary to achieve myeloablation presents a risk in young infants. Individuals with Wiskott-Aldrich syndrome who do not have a suitably matched donor but who experience life-threatening complications are candidates for gene therapy.
- Intravenous immune globulin therapy: Intravenous immunoglobin (IVIG) therapy is indicated in Wiskott-Aldrich syndrome and X-linked thrombocytopenia (XLT) patients with significant antibody deficiency. The dose is usually higher than that used for other primary immunodeficiencies due to an increased catabolic rate observed in Wiskott-Aldrich syndrome patients. Immune globulin may also be given subcutaneously. This route of administration requires caution in this patient population because of the bleeding tendency 51.
- Eltrombopag: An oral thrombopoietin receptor agonist approved for the treatment of immune thrombocytopenia (ITP), may be useful in preventing bleeding in patients with Wiskott-Aldrich syndrome who are awaiting hematopoietic cell transplantation (HCT) 52.
- Immunosuppressive treatment: Immunosuppressive treatment may be necessary for autoimmune manifestations. Autoimmune cytopenias often respond to the monoclonal antibody rituximab which is relatively safe for those patients already receiving therapy with intravenous immunoglobin (IVIG).
- Splenectomy: Elective splenectomy has been advocated in selected patients to reverse the thrombocytopenia and arrest the bleeding tendency by increasing the number of circulating platelets. Patients who undergo splenectomy require lifelong antibiotic prophylaxis and are at increased risk of septicemia.
- Eczema. Topical steroids are the mainstay of therapy. When chronic infections of the skin worsen eczema, antibiotics may be useful.
- Infection. For those individuals with clinical signs or symptoms of infection, prompt evaluation and treatment is necessary. The initiation of empiric parenteral antibiotic treatment is necessary in the majority of individuals. The evaluation should be exhaustive until the source of the infection is uncovered. This may include invasive assessments, as cultures and isolation of the offending organism should be sought in order to guide therapy. If hematopoietic cell transplantation is being considered, attention to the prevention and treatment of infectious complications is necessary to limit pre-transplant morbidity.
- Autoimmune disease. Treatment usually consists of judicious use of immunosuppressants tailored to the individual’s diagnosis.
X-Linked Thrombocytopenia (XLT)
The primary management of individuals with X-linked thrombocytopenia (WAS score 1-2) remains controversial. Although long-term survival is excellent with conservative management of presenting symptoms, event-free survival is reduced by the substantial risk of severe, life-threatening or potentially debilitating complications 46. Serious bleeding episodes are generally restricted to the first 30 years of life. In contrast, the risk of developing autoimmune disease, malignancy, or a life-threatening infectious episode is rather constant throughout the individual’s lifetime. This persistent morbidity argues for hematopoietic cell transplantation as a treatment option for such individuals. Given the excellent success in young children with classic Wiskott-Aldrich syndrome, hematopoietic cell transplantation may be considered a viable option for individuals with X-linked thrombocytopenia if an HLA–identical donor can be identified. However, one needs to carefully weigh the advantage of a possible cure against the acute risks and long-term consequences of this procedure (e.g., risk of secondary malignancy, infertility). Thus, hematopoietic cell transplantation in X-linked thrombocytopenia needs to be decided on an individual basis.
X-Linked Neutropenia (XLN)
Treatment of X-linked neutropenia is with granulocyte colony-stimulating factor (G-CSF) and appropriate antibiotics.
Wiskott-Aldrich syndrome gene therapy
Gene therapy is an alternative, potentially curative therapy under investigation for Wiskott-Aldrich syndrome 53, 54, 55. However, initial attempts at gene therapy using a retroviral vector resulted in leukemic proliferation in several affected individuals.
More recent attempts using a lentiviral vector in two clinical trials are promising. In the first trial, Italian investigators showed improvement of platelet counts, immune function, and clinical manifestations of the disease in three individuals at one year or longer after gene therapy 56. In the second trial, six out of seven individuals treated in London and Paris also showed improvement of immune function and clinical manifestations six to 42 months after treatment, without evidence of clonal expansion 57. For reasons that are not yet clear, neither trial resulted in reconstitution of normal platelet numbers, although bleeding episodes significantly reduced in number and severity with individuals becoming independent from transfusion and need for thrombopoietin agonists. Thus far, no evidence of leukemic transformation has been reported with the use of the lentivirus vector 56. Based on these observations, it can be concluded that lentiviral-mediated gene therapy for Wiskott-Aldrich syndrome is feasible and can result in significant benefit for treated individuals. Clearly, however, long-term observation is warranted to confirm the superior safety of lentiviral gene transfer as an alternative treatment option for Wiskott-Aldrich syndrome.
Search ClinicalTrials.gov for access to information on clinical studies for a wide range of diseases and conditions.
Figure 3. Wiskott-Aldrich syndrome gene therapy
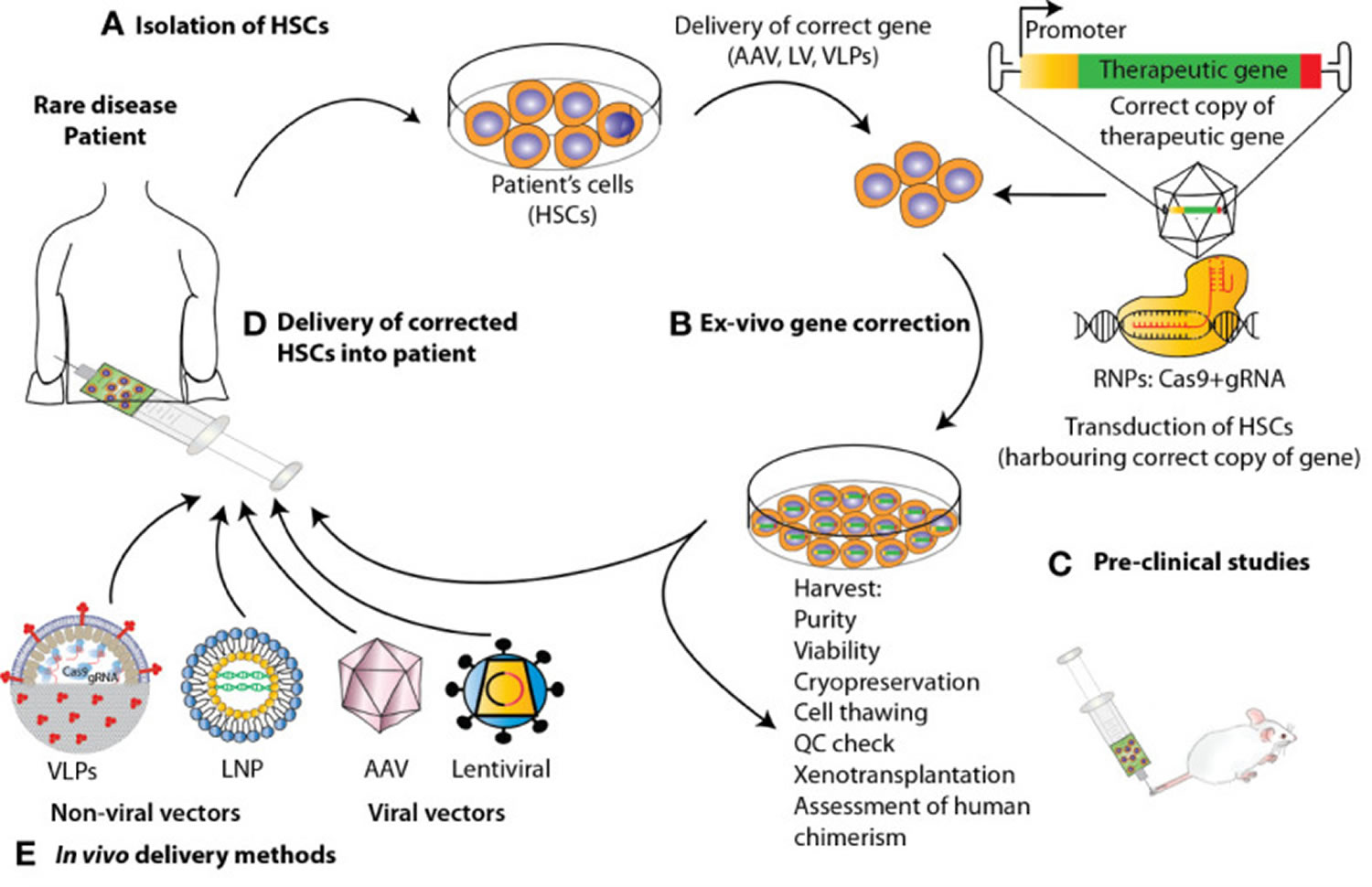
Footnotes: State-of-the-art ex vivo and in vivo gene therapy and gene editing strategies applied to hematopoietic stem and progenitor cells (HSPCs) to correct blood genetic disorders via delivery of a corrective gene. Schematic representation of the various steps involved in the development of a therapeutic product for the treatment of blood diseases via gene therapy. For the ex-vivo approach, (A) hematopoietic stem and progenitor cells (HSPCs) are isolated from mobilized peripheral blood (mPB) apheresis. (B) Using CRISPR/Cas9-based gene editing or lentivirus vector carrying the gene of interest, these cells are edited/transduced ex vivo and (C) after performing a thorough safety and efficacy quality control (QC) on the product (D) cells are infused back to the patient after pharmacokinetically adjusted myeloablation. (E) When performing gene therapy in vivo, the correct copy of the gene is directly infused into the patient via the use of viral vectors or non-viral methods such as Virus-like Particles (VLPs) and lipid nanoparticles (LNPs).
[Source 5 ]Prevention of primary manifestations
Infection
- Antibiotic prophylaxis. Prophylaxis for pneumonia secondary to Pneumocystis jiroveci, formerly known as Pneumocystis carinii (PCP) is indicated for infants with Wiskott-Aldrich syndrome as they are at risk of developing Pneumocystis carinii. Typical prophylaxis is Bactrim® (trimethoprim-sulfamethoxazole) orally or pentamidine by intravenous or inhalation therapy. Individuals with recurrent bacterial sinopulmonary infections may benefit from prophylactic antibiotic use.
- Intravenous immune globulin (IVIG). Replacement therapy with intravenous immune globulin (IVIG) by age six months is administered every three to four weeks or subcutaneously, usually on a weekly basis. Intravenous immune globulin (IVIG) is a highly purified blood derivative (a combination of many specific antimicrobial antibodies).
- Routine childhood immunizations. Live vaccines should be avoided. Other “non-live” vaccinations can be given safely to individuals with a Wiskott Aldrich syndrome-related disorder but may not generate protective levels of antibody.
Bleeding
- Splenectomy. Splenectomy is palliative, and while it may be life-saving in an individual with severe bleeding, it does not prevent any of the other possible complications of Wiskott Aldrich syndrome 58. In a survey of clinical immunologists performed by the European and Pan American Groups on Immunodeficiencies, respondents from centers treating the highest numbers of individuals with Wiskott-Aldrich syndrome did not recommend splenectomy 59. Because splenectomy significantly increases the risk of life-threatening infections in males with X-linked thrombocytopenia 60 as well as in males with Wiskott-Aldrich syndrome who subsequently undergo hematopoietic cell transplantation 61, it should be used with caution. Males who have had splenectomy must take antibiotics routinely for the rest of their lives because of the increased risk for overwhelming infection.
- Platelet transfusions. Platelet transfusions should be administered judiciously (e.g., for significant bleeding and surgical procedures).
Monitoring
Regular follow-up is indicated to monitor blood counts, adequacy of the IVIG replacement therapy, and other potential complications.
Wiskott-Aldrich syndrome prognosis
The prognosis of X-linked thrombocytopenia is good with the life expectancy as close to the normal population 46, but classic Wiskott-Aldrich syndrome has a poor prognosis with decreased life expectancy due to recurrent infections, autoimmune disease, and cancer. Bleeding is most frequently the cause of death in these patients 43.
The only curative treatment clinically available for Wiskott-Aldrich syndrome is allogeneic hematopoietic cell transplantation (when the stem cells come from another person, called a donor). Affected males who receive bone marrow transplantation or hematopoietic cell transplantation from a matched healthy sibling or closely matched unrelated donor before their second birthday have a greater than 90% probability of being cured of the disorder 49, 50.
Wiskott-Aldrich life expectancy
The average life expectancy for boys with Wiskott-Aldrich syndrome is about 15 – 20 years without hematopoietic stem cell transplantation. Affected males who receive bone marrow transplantation or hematopoietic cell transplantation from a matched healthy sibling or closely matched unrelated donor before their second birthday have a greater than 90% probability of being cured of the disorder 49, 50.
- Chandra S, Bronicki L, Nagaraj CB, et al. WAS-Related Disorders. 2004 Sep 30 [Updated 2016 Sep 22]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1178[↩][↩][↩][↩][↩]
- Malik MA, Masab M. Wiskott-Aldrich Syndrome. [Updated 2022 Dec 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539838[↩][↩][↩][↩]
- Stray-Pedersen A, Abrahamsen TG, Frøland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000 Nov;20(6):477-85. doi: 10.1023/a:1026416017763[↩][↩]
- Sudhakar M, Rikhi R, Loganathan SK, Suri D, Singh S. Autoimmunity in Wiskott-Aldrich Syndrome: Updated Perspectives. Appl Clin Genet. 2021 Aug 20;14:363-388. doi: 10.2147/TACG.S213920[↩]
- Naseem A, Steinberg Z, Cavazza A. Genome editing for primary immunodeficiencies: A therapeutic perspective on Wiskott-Aldrich syndrome. Front Immunol. 2022 Aug 18;13:966084. doi: 10.3389/fimmu.2022.966084[↩][↩][↩]
- Obydennyi SI, Artemenko EO, Sveshnikova AN, Ignatova AA, Varlamova TV, Gambaryan S, Lomakina GY, Ugarova NN, Kireev II, Ataullakhanov FI, Novichkova GA, Maschan AA, Shcherbina A, Panteleev M. Mechanisms of increased mitochondria-dependent necrosis in Wiskott-Aldrich syndrome platelets. Haematologica. 2020 Apr;105(4):1095-1106. doi: 10.3324/haematol.2018.214460[↩]
- Suri D, Rikhi R, Jindal AK, Rawat A, Sudhakar M, Vignesh P, Gupta A, Kaur A, Sharma J, Ahluwalia J, Bhatia P, Khadwal A, Raj R, Uppuluri R, Desai M, Taur P, Pandrowala AA, Gowri V, Madkaikar MR, Lashkari HP, Bhattad S, Kumar H, Verma S, Imai K, Nonoyama S, Ohara O, Chan KW, Lee PP, Lau YL, Singh S. Wiskott Aldrich Syndrome: A Multi-Institutional Experience From India. Front Immunol. 2021 Apr 16;12:627651. doi: 10.3389/fimmu.2021.627651[↩]
- Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117(4):725–738. doi: 10.1016/j.jaci.2006.02.005[↩]
- Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15(1):84–90. doi: 10.1016/j.bbmt.2008.10.007[↩]
- Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Curr Opin Hematol. 2008;15(1):30–36. doi: 10.1097/MOH.0b013e3282f30448[↩]
- Wiskott-Aldrich syndrome. https://rarediseases.info.nih.gov/diseases/7895/wiskott-aldrich-syndrome[↩][↩]
- Wiskott A. Familiarer, angeborener morbus werlhofii? Monatsschr Kinderheilkd. 1937;68:212–216.[↩]
- ABOUT WAS. https://www.wiskott.org/About-WAS[↩][↩]
- ALDRICH RA, STEINBERG AG, CAMPBELL DC. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics. 1954 Feb;13(2):133-9.[↩]
- Chandra S, Bronicki L, Nagaraj CB, et al. WAS-Related Disorders. 2004 Sep 30 [Updated 2016 Sep 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1178[↩]
- Perry GS 3rd, Spector BD, Schuman LM, et al. The Wiskott-Aldrich syndrome in the United States and Canada (1892–1979). J Pediatr. 1980;97(1):72–78. doi: 10.1016/s0022-3476(80)80133-8[↩]
- Fasth A. Primary immunodeficiency disorders in Sweden: cases among children, 1974–1979. J Clin Immunol. 1982;2(2):86–92. doi: 10.1007/BF00916891[↩]
- Ryser O, Morell A, Hitzig WH. Primary immunodeficiencies in Switzerland: first report of the national registry in adults and children. J Clin Immunol. 1988;8(6):479–485. doi: 10.1007/BF00916954[↩]
- Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78(4):635–644. doi: 10.1016/0092-8674(94)90528-2[↩]
- Kwan SP, Lehner T, Hagemann T, et al. Localization of the gene for the Wiskott-Aldrich syndrome between two flanking markers, TIMP and DXS255, on Xp11.22-Xp11.3. Genomics. 1991;10(1):29–33. doi: 10.1016/0888-7543(91)90480-3[↩]
- Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol. 1995;15(10):5725–5731. doi: 10.1128/mcb.15.10.5725[↩]
- Symons M, Derry JM, Karlak B, et al. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84(5):723–734. doi: 10.1016/s0092-8674(00)81050-8[↩]
- Miki H, Takenawa T. Direct binding of the verprolin-homology domain in N-WASP to actin is essential for cytoskeletal reorganization. Biochem Biophys Res Commun. 1998;243(1):73–78. doi: 10.1006/bbrc.1997.8064[↩]
- Miki H, Nonoyama S, Zhu Q, Aruffo A, Ochs HD, Takenawa T. Tyrosine kinase signaling regulates Wiskott-Aldrich syndrome protein function, which is essential for megakaryocyte differentiation. Cell Growth Differ. 1997 Feb;8(2):195-202.[↩]
- Malik MA, Masab M. Wiskott-Aldrich Syndrome. [Updated 2019 Apr 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539838[↩]
- Gallego MD, Santamaría M, Peña J, Molina IJ. Defective actin reorganization and polymerization of Wiskott-Aldrich T cells in response to CD3-mediated stimulation. Blood. 1997;90(8):3089–3097. doi: 10.1182/blood.V90.8.3089[↩][↩]
- Cory GO, MacCarthy-Morrogh L, Banin S, Gout I, Brickell PM, Levinsky RJ, Kinnon C, Lovering RC. Evidence that the Wiskott-Aldrich syndrome protein may be involved in lymphoid cell signaling pathways. J Immunol. 1996 Nov 1;157(9):3791-5.[↩][↩]
- Krawczyk C, Bachmaier K, Sasaki T, et al. Cbl-b is a negative regulator of receptor clustering and raft aggregation in T cells. Immunity. 2000;13(4):463–473. doi: 10.1016/s1074-7613(00)00046-7[↩][↩]
- Sasahara Y, Rachid R, Byrne MJ, et al. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol Cell. 2002;10(6):1269–1281. doi: 10.1016/s1097-2765(02)00728-1[↩][↩]
- Dupré L, Aiuti A, Trifari S, et al. Wiskott-Aldrich syndrome protein regulates lipid raft dynamics during immunological synapse formation. Immunity. 2002;17(2):157–166. doi: 10.1016/s1074-7613(02)00360-6[↩][↩]
- Linder S, Higgs H, Hüfner K, Schwarz K, Pannicke U, Aepfelbacher M. The polarization defect of Wiskott-Aldrich syndrome macrophages is linked to dislocalization of the Arp2/3 complex. J Immunol. 2000;165(1):221–225. doi: 10.4049/jimmunol.165.1.221[↩][↩]
- Jones GE, Zicha D, Dunn GA, Blundell M, Thrasher A. Restoration of podosomes and chemotaxis in Wiskott-Aldrich syndrome macrophages following induced expression of WASp. Int J Biochem Cell Biol. 2002;34(7):806–815. doi: 10.1016/s1357-2725(01)00162-5[↩][↩]
- Devriendt K, Kim AS, Mathijs G, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27(3):313–317. doi: 10.1038/85886[↩]
- Ancliff PJ, Blundell MP, Cory GO, et al. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108(7):2182–2189. doi: 10.1182/blood-2006-01-010249[↩]
- Blundell MP, Worth A, Bouma G, Thrasher AJ. The Wiskott-Aldrich syndrome: The actin cytoskeleton and immune cell function. Dis. Markers. 2010;29(3-4):157-75[↩]
- Malinova D, Fritzsche M, Nowosad CR, Armer H, Munro PM, Blundell MP, Charras G, Tolar P, Bouma G, Thrasher AJ. WASp-dependent actin cytoskeleton stability at the dendritic cell immunological synapse is required for extensive, functional T cell contacts. J. Leukoc. Biol. 2016 May;99(5):699-710.[↩]
- Meyer-Bahlburg A, Becker-Herman S, Humblet-Baron S, Khim S, Weber M, Bouma G, Thrasher AJ, Batista FD, Rawlings DJ. Wiskott-Aldrich syndrome protein deficiency in B cells results in impaired peripheral homeostasis. Blood. 2008 Nov 15;112(10):4158-69[↩]
- Gismondi A, Cifaldi L, Mazza C, Giliani S, Parolini S, Morrone S, Jacobelli J, Bandiera E, Notarangelo L, Santoni A. Impaired natural and CD16-mediated NK cell cytotoxicity in patients with WAS and XLT: ability of IL-2 to correct NK cell functional defect. Blood. 2004 Jul 15;104(2):436-43[↩]
- Maillard MH, Cotta-de-Almeida V, Takeshima F, Nguyen DD, Michetti P, Nagler C, Bhan AK, Snapper SB. The Wiskott-Aldrich syndrome protein is required for the function of CD4(+)CD25(+)Foxp3(+) regulatory T cells. J. Exp. Med. 2007 Feb 19;204(2):381-91[↩]
- Burns S, Thrasher AJ, Blundell MP, Machesky L, Jones GE. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood. 2001 Aug 15;98(4):1142-9[↩]
- WAS Related Disorders. https://rarediseases.org/rare-diseases/was-related-disorders[↩][↩]
- Mahlaoui N, Pellier I, Mignot C, Jais JP, Bilhou-Nabéra C, Moshous D, Neven B, Picard C, de Saint-Basile G, Cavazzana-Calvo M, Blanche S, Fischer A. Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome. Blood. 2013 Feb 28;121(9):1510-6.[↩]
- Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994 Dec;125(6 Pt 1):876-85. doi: 10.1016/s0022-3476(05)82002-5[↩][↩]
- Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, Verhoef GE, Boogaerts MA, Fryns JP, You D, Rosen MK, Vandenberghe P. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001 Mar;27(3):313-7. doi: 10.1038/85886[↩]
- Notarangelo LD, Mazza C, Giliani S, D’Aria C, Gandellini F, Ravelli C, Locatelli MG, Nelson DL, Ochs HD, Notarangelo LD. Missense mutations of the WASP gene cause intermittent X-linked thrombocytopenia. Blood. 2002 Mar 15;99(6):2268-9. doi: 10.1182/blood.v99.6.2268[↩]
- Albert MH, Bittner TC, Nonoyama S, Notarangelo LD, Burns S, Imai K, Espanol T, Fasth A, Pellier I, Strauss G, Morio T, Gathmann B, Noordzij JG, Fillat C, Hoenig M, Nathrath M, Meindl A, Pagel P, Wintergerst U, Fischer A, Thrasher AJ, Belohradsky BH, Ochs HD. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. 2010 Apr 22;115(16):3231-8. doi: 10.1182/blood-2009-09-239087[↩][↩][↩]
- Chiang SCC, Vergamini SM, Husami A, Neumeier L, Quinn K, Ellerhorst T, Sheppard L, Gifford C, Buchbinder D, Joshi A, Ifversen M, Kleiner GI, Bussel JB, Chandrakasan S, Pesek RD, Pozos TC, Rose MJ, Scurlock AM, Zhang K, Bryceson YT, Bleesing J, Marsh RA. Screening for Wiskott-Aldrich syndrome by flow cytometry. J. Allergy Clin. Immunol. 2018 Jul;142(1):333-335.e8[↩]
- Snover DC, Frizzera G, Spector BD, Perry GS, Kersey JH. Wiskott-Aldrich syndrome: histopathologic findings in the lymph nodes and spleens of 15 patients. Hum. Pathol. 1981 Sep;12(9):821-31[↩]
- Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, Cant AJ, Thrasher AJ, Cowan MJ, Albert MH, Small T, Pai SY, Haddad E, Lisa A, Hambleton S, Slatter M, Cavazzana-Calvo M, Mahlaoui N, Picard C, Torgerson TR, Burroughs L, Koliski A, Neto JZ, Porta F, Qasim W, Veys P, Kavanau K, Hönig M, Schulz A, Friedrich W, Notarangelo LD. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood. 2011 Aug 11;118(6):1675-84. doi: 10.1182/blood-2010-11-319376[↩][↩][↩][↩]
- Shin CR, Kim MO, Li D, Bleesing JJ, Harris R, Mehta P, Jodele S, Jordan MB, Marsh RA, Davies SM, Filipovich AH. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2012 Nov;47(11):1428-35. doi: 10.1038/bmt.2012.31[↩][↩][↩]
- Blaese RM, Strober W, Levy AL, Waldmann TA. Hypercatabolism of IgG, IgA, IgM, and albumin in the Wiskott-Aldrich syndrome. A unique disorder of serum protein metabolism. J Clin Invest. 1971 Nov;50(11):2331-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC292175/pdf/jcinvest00199-0105.pdf[↩]
- Gerrits AJ, Leven EA, Frelinger AL 3rd, Brigstocke SL, Berny-Lang MA, Mitchell WB, Revel-Vilk S, Tamary H, Carmichael SL, Barnard MR, Michelson AD, Bussel JB. Effects of eltrombopag on platelet count and platelet activation in Wiskott-Aldrich syndrome/X-linked thrombocytopenia. Blood. 2015 Sep 10;126(11):1367-78. doi: 10.1182/blood-2014-09-602573[↩]
- Klein C, Nguyen D, Liu CH, Mizoguchi A, Bhan AK, Miki H, Takenawa T, Rosen FS, Alt FW, Mulligan RC, Snapper SB. Gene therapy for Wiskott-Aldrich syndrome: rescue of T-cell signaling and amelioration of colitis upon transplantation of retrovirally transduced hematopoietic stem cells in mice. Blood. 2003 Mar 15;101(6):2159-66. doi: 10.1182/blood-2002-05-1423[↩]
- Qasim W, Gaspar HB, Thrasher AJ. Progress and prospects: gene therapy for inherited immunodeficiencies. Gene Ther. 2009 Nov;16(11):1285-91. doi: 10.1038/gt.2009.127[↩]
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, Böhm M, Nowrouzi A, Ball CR, Glimm H, Naundorf S, Kühlcke K, Blasczyk R, Kondratenko I, Maródi L, Orange JS, von Kalle C, Klein C. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010 Nov 11;363(20):1918-27. doi: 10.1056/NEJMoa1003548[↩]
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013 Aug 23;341(6148):1233151. doi: 10.1126/science.1233151[↩][↩]
- Hacein-Bey Abina S, Gaspar HB, Blondeau J, Caccavelli L, Charrier S, Buckland K, Picard C, Six E, Himoudi N, Gilmour K, McNicol AM, Hara H, Xu-Bayford J, Rivat C, Touzot F, Mavilio F, Lim A, Treluyer JM, Héritier S, Lefrère F, Magalon J, Pengue-Koyi I, Honnet G, Blanche S, Sherman EA, Male F, Berry C, Malani N, Bushman FD, Fischer A, Thrasher AJ, Galy A, Cavazzana M. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015 Apr 21;313(15):1550-63. doi: 10.1001/jama.2015.3253[↩]
- Mullen CA, Anderson KD, Blaese RM. Splenectomy and/or bone marrow transplantation in the management of the Wiskott-Aldrich syndrome: long-term follow-up of 62 cases. Blood. 1993 Nov 15;82(10):2961-6. https://doi.org/10.1182/blood.V82.10.2961.2961[↩]
- Conley ME, Saragoussi D, Notarangelo L, Etzioni A, Casanova JL; PAGID; ESID. An international study examining therapeutic options used in treatment of Wiskott-Aldrich syndrome. Clin Immunol. 2003 Dec;109(3):272-7. doi: 10.1016/j.clim.2003.08.005[↩]
- Albert MH, Notarangelo LD, Ochs HD. Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome. Curr Opin Hematol. 2011 Jan;18(1):42-8. doi: 10.1097/MOH.0b013e32834114bc[↩]
- Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ, Mazzolari E, Slatter MA, Le Deist F, Blanche S, Veys P, Fasth A, Bredius R, Sedlacek P, Wulffraat N, Ortega J, Heilmann C, O’Meara A, Wachowiak J, Kalwak K, Matthes-Martin S, Gungor T, Ikinciogullari A, Landais P, Cant AJ, Friedrich W, Fischer A. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008 Jan 1;111(1):439-45. doi: 10.1182/blood-2007-03-076679[↩]
{kind=link}