
Contents
What is hyperphosphatemia
Hyperphosphatemia is an abnormally high serum phosphate levels that is more than 4.7 mg/dL (greater than 1.45 mmol/L), that can result from increased phosphate intake, decreased phosphate excretion, or a disorder that shifts intracellular phosphate to extracellular space 1. Phosphorus is a mineral that combines with other substances to form organic and inorganic phosphate compounds. Phosphorus is a major structural component of bone in the form of a calcium phosphate salt called hydroxyapatite. Phospholipids (e.g., phosphatidylcholine) are major structural components of cell membranes. All energy production and storage are dependent on phosphorylated compounds, such as adenosine triphosphate (ATP) and creatine phosphate 2. Nucleic acids (DNA and RNA), which are responsible for the storage and transmission of genetic information, are long chains of phosphate-containing molecules 2. A number of enzymes, hormones, and cell-signaling molecules depend on phosphorylation for their activation. Phosphorus also helps maintain normal acid-base balance (pH) by acting as one of the body’s most important buffers. Additionally, the phosphorus-containing molecule 2,3-diphosphoglycerate (2,3-DPG) binds to hemoglobin in red blood cells and regulates oxygen delivery to the tissues of the body 3.
The terms phosphorus and phosphate are often used interchangeably when talking about testing, but it is the amount of inorganic phosphate in the blood that is measured with a serum phosphorus or phosphate test. Inorganic phosphorus is an intracellular anion. Phosphate (phosphorus) plays an essential role in cell functions, including structural component of cell membranes and nucleic acids, energy production, muscle and nerve function, membrane transport, cell signaling through phosphorylation reactions and bone growth (bone mineralization) 4. Phosphate (phosphorus) also play an important role as a buffer, helping to maintain the body’s acid-base balance. You get the phosphorus you need through the foods you eat. Phosphate (phosphorus) is found in many foods and is readily absorbed by the digestive tract. A wide variety of foods, such as beans, peas and nuts, cereals, dairy products, eggs, beef, chicken, and fish, contain significant amounts of phosphorus. Most of the body’s phosphates combine with calcium to help form bones in the form of hydroxyapatite and teeth. Smaller amounts are found in muscle and nerve tissue. The rest is found within cells throughout the body, where they are mainly used to store energy.
Normally, only about 1% of total body phosphates are present in the blood. Serum contains approximately 2.5 to 4.5 mg/dL of inorganic phosphate (the fraction measure in routine biochemical assays). Serum phosphate concentrations are dependent on meals and variation in the secretion of hormones such as parathyroid hormone (PTH) and may vary widely. The body maintains phosphorus or phosphate levels in the blood by regulating how much it absorbs from the intestines and how much it excretes via the kidneys. Phosphate levels are also affected by the interaction of parathyroid hormone (PTH), calcium, vitamin D and fibroblast growth factor-23 (FGF-23). The acute regulation of blood calcium and phosphorus concentrations is controlled through the actions of parathyroid hormone (PTH) and the active form of vitamin D (1,25-dihydroxyvitamin D; calcitriol). A slight drop in blood calcium levels (e.g., in the case of inadequate calcium intake) is sensed by the parathyroid glands, resulting in their increased secretion of parathyroid hormone (PTH), which rapidly decreases urinary excretion of calcium but increases urinary excretion of phosphorus and stimulates bone resorption. This results in the release of bone mineral (calcium and phosphate) — actions that restore serum calcium concentrations. Although the action is not immediate, parathyroid hormone (PTH) also stimulates conversion of vitamin D to its active form (1,25-dihydroxyvitamin D; calcitriol) in the kidneys. Increased circulating 1,25-dihydroxyvitamin D (calcitriol) in turn stimulates increased intestinal absorption of both calcium and phosphorus. A third hormone, Klotho fibroblast growth factor-23 (FGF-23), plays a central role in phosphorus homeostasis 5. Klotho works as a receptor for the phosphaturic hormone FGF-23. Fibroblast growth factor-23 (FGF-23) is secreted by bone-forming cells (osteoblasts/osteocytes) in response to increases in phosphorus intake. In a negative feedback loop, FGF-23 inhibits the production and stimulates the degradation of active form of vitamin D (1,25-dihydroxyvitamin D; calcitriol), as well as promotes an increase in urinary phosphorus excretion independently of parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D 6.
Increased secretion of parathyroid hormone (PTH) and fibroblast growth factor-23 (FGF-23) helps maintain phosphorus serum concentrations in the normal range (2.5-4.5 mg/dL) even in the setting of high phosphorus intake 7. This contributes to serum phosphorus being only weakly correlated to phosphorus consumption 8. Sustained increases in FGF-23 and PTH are commonly observed during chronic kidney disease (CKD) in order to maintain normal serum phosphorus concentrations despite a reduction in urinary phosphorus excretion 9. Elevated FGF-23, rather than serum phosphorus, appears to be an early marker of disordered phosphorus homeostasis and a predictor of adverse health outcomes in patients with early-stage chronic kidney disease (CKD) 9, 10. Therefore, it is reasonable to assume that measuring serum phosphorus in people with normal kidney function cannot adequately reflect early disturbances in phosphorus metabolism due to high phosphorus consumption.
Hyperphosphatemia is rare in the general population, but in patients with advanced chronic kidney disease, the rate of hyperphosphatemia is at least 70% 1. Almost all patients with dialysis-dependent kidney failure experience hyperphosphatemia at some time during the course of their disease. This is true for acute and chronic kidney disease (CKD).
Hyperphosphatemia within the normal range (2.5-4.5 mg/dL) has recently been associated with increased incidence of cardiovascular disease (CVD) in individuals with normal kidney function. Two studies conducted in the general population and in individuals with prior cardiovascular disease have linked high-normal serum phosphorus concentrations (≥3.5 mg/dL) to a greater cardiovascular risk 11, 12. Additional observational studies found that serum phosphorus concentrations equal to or above 4 mg/dL were associated with a doubling of the risk of developing incident chronic kidney disease (CKD) and end-stage renal disease in individuals free of renal disease at study inception 13. In a prospective cohort study, which followed 4,005 healthy young adults for more than 15 years, higher serum phosphorus within the normal range was also associated with left ventricular hypertrophy, a condition often linked to adverse cardiovascular outcomes 14. In another study of 3,088 middle-aged healthy participants followed for over 17 years, serum phosphorus concentrations in the top quartile of the normal range were associated with a two-fold higher risk of heart failure compared to the lowest quartile (≥3.5 mg/dL vs. <2.9 mg/dL) 11. It is thought that vascular calcification, which may explain the relationship between high phosphorus and cardiovascular disease risk in chronic kidney disease (CKD) patients (hyperphosphatemia in subjects with kidney disease), contributes to this association in individuals with normal kidney function, even when their serum phosphorus is within the normal range and their intakes are below the tolerable upper intake level (UL) 15, 16.
Decreased phosphate excretion represents a critical feature of pathophysiology of chronic kidney disease (CKD) 17. This pathophysiology contributes to the high rates of mortality observed in chronic kidney disease 18. Hyperphosphatemia is independently associated with high mortality risk in chronic kidney disease population, whether they are on dialysis, or not 19. Kestenbaum et al, 20 who noticed that serum phosphate levels of more than 3.5 mg/dL (1.13 mmol/L) were associated with significantly increased risk for death in chronic kidney disease patients who were not on dialysis (creatinine clearance >50.4 – 39.5 ml/min). Earlier, a national study in the USA showed a 27% increase in relative risk of death, associated with serum phosphorus >6.5 mg/dL among people who received chronic dialysis 19. Then, Block et al 19 again reviewed the database of >40,000 hemodialysis patients to find a strong association between higher levels of serum phosphorus (>5 mg/dL), and increased risk of death. More recently, Calo et al 21 in their study on sevelamer-resistant dialysis patients found a relationship between the reduction of serum phosphate induced by chitosan-loaded chewing gum and C-reactive protein reduction, which support a proinflammatory role of hyperphosphatemia “per se”.
In the community, hyperphosphatemia is associated with increased risk of cardiovascular disease in individuals without history of cardiovascular disease or chronic kidney disease as shown by Dhingra et al 22. Furthermore, several observational studies demonstrate that hyperphosphatemia is an independent cardiovascular risk factor in chronic kidney disease 23. Hyperphosphatemia has been linked to vascular calcification 24. Hyperphosphatemia in healthy normal individuals were associated with increased coronary artery calcium content with subsequent risk of cardiovascular disease 25.
The high mortality rate in chronic kidney disease patients is related to cardiovascular disease, which in fact, is responsible for more than 50% of deaths in patients with end stage renal disease (ESRD) 26. Sudden cardiac death, heart failure, ischemic heart disease, and peripheral arterial disease are the primary causes of cardiovascular mortality 27. These cardiovascular events are strongly correlated with vascular calcification; a pathology which is induced and promoted by hyperphosphatemia, elevated calcium x phosphorus (Ca x P) product, and increased calcium load (diet, dialysate) 28. Over the past 2 decades, several studies have shown the clinical significance of vascular calcification in chronic kidney disease. These studies demonstrated that arterial calcification is strongly associated with coronary ischemic disease in uremic patients 29. Intimal calcification is classically associated with advanced stages of atherosclerosis and subsequent ischemic heart disease, while calcification of media is most likely dependent upon the abnormalities of mineral metabolism typically found in chronic kidney disease patients with mineral and bone disorder. Medial arterial calcification leads to increased arterial wall stiffness, and increased pulse pressure resulting in the development of cardiomyopathy, arrhythmia, and sudden cardiac death 30. However, these 2 types of vascular calcification (intimal and medial) can co-exist in the same patient and the same vessel 31. In addition, calcification of cardiac valves and myocardium is also increased in dialysis patients leading to increased morbidity and mortality 32. Finally, vascular medial calcification of small arterioles is responsible for calciphylaxis, a syndrome of ischemic necrosis of the skin associated with extremely high mortality rates in uremic patients 26.
Approximately 11–15% of Americans have chronic kidney disease 33, and their risk of death due to a cardiovascular event related cause is higher than their risk of surviving and needing renal replacement therapy for end stage kidney disease (ESKD) 18. The mortality rates of patients surviving chronic kidney disease and receiving hemodialysis are extremely high such that a 30 year old patient with end stage kidney disease (ESKD) has a life expectancy similar to a ninety year old with normal renal function 34. The mechanisms of this excess risk of cardiovascular disease are not completely understood. The well characterized risks of cardiovascular disease in the general population do not explain the increased risk in chronic kidney disease 35. Observational studies suggest that the well known propensity of end stage kidney disease patients to develop heterotopic mineralization of soft tissues including the vasculature is an important component of the cardiovascular risks of end stage kidney disease 36.
Figure 1. Phosphate functions

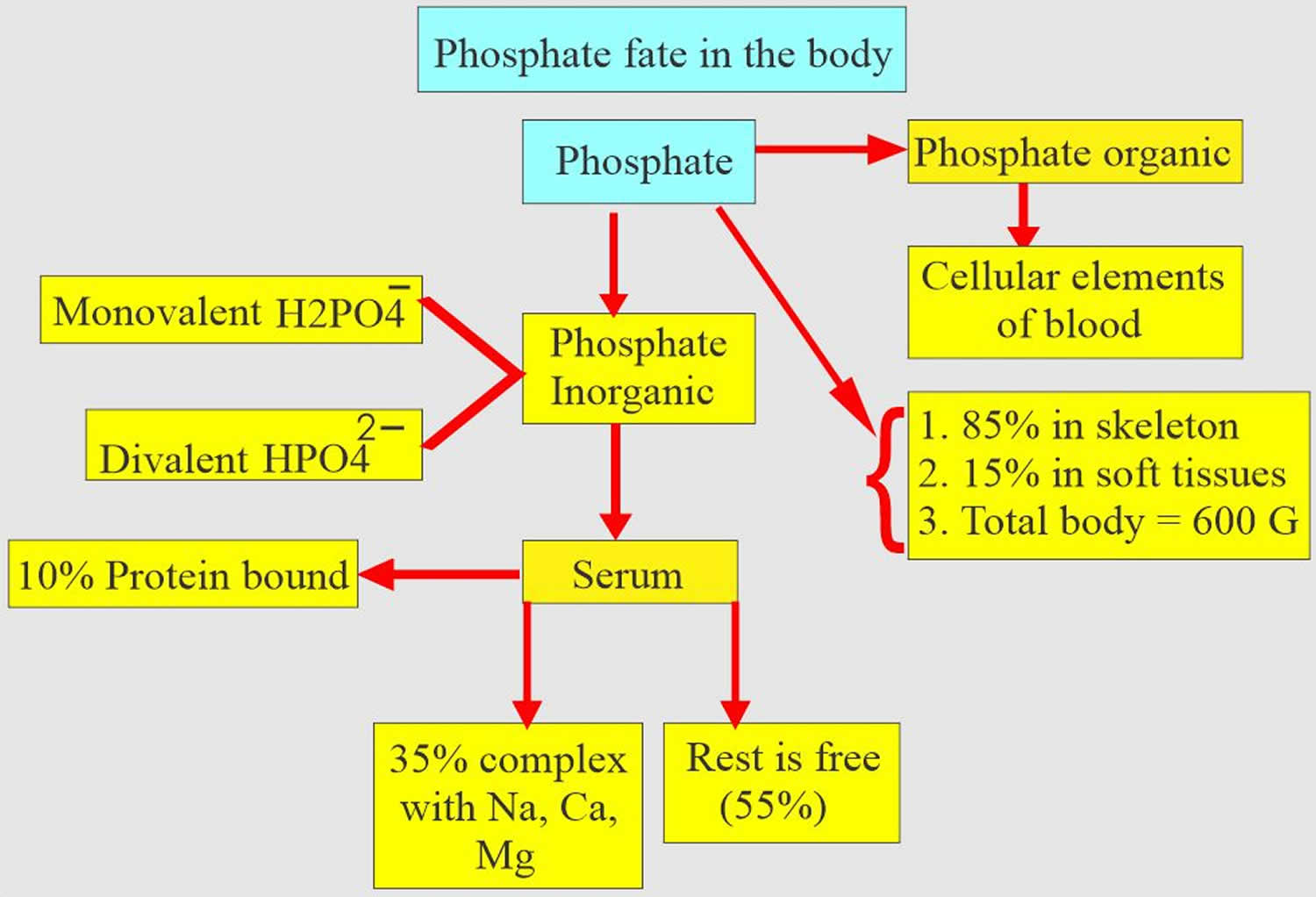
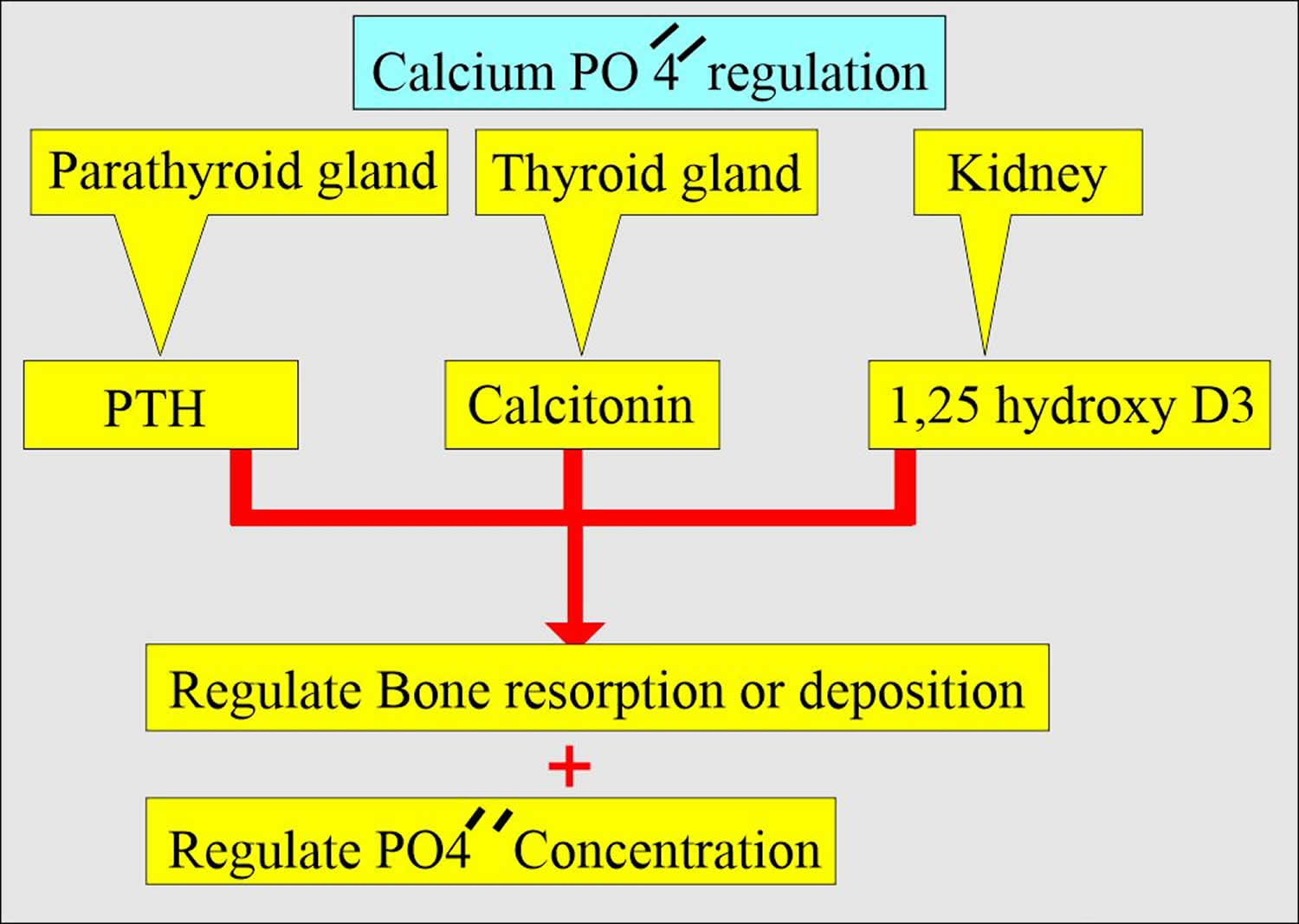
Figure 2. Phosphate regulation in the body
Phosphate homeostasis
The average American diet ingest approximately 1000–1200 mg of phosphorus per day (Figure 3) 37. Of this, a net of about 800 mg is absorbed into the exchangeable phosphorus pool. This pool consists of intracellular phosphorus (70%), the skeletal mineralization front (29%) and the serum phosphorus (<1%). Exit from the exchangeable pool is through skeletal deposition, renal excretion, and intestinal secretion (Figure 1). Exit from the exchangeable pool into the skeleton is matched by entry from the skeleton into the pool in adulthood, and therefore, the skeleton is a very important contributor to the level of phosphorus concentration in the pool, in chronic kidney disease. Regulation of phosphorus excretion by the kidney is the key mechanism of maintaining phosphate balance in normal day to day life. Kidney injury impairs the ability of mammals to maintain phosphorus balance, and in human chronic kidney disease, phosphorus homeostasis is lost and positive phosphate balance occurs in the later stages (4 and 5) of kidney diseases 38. Loss of phosphorus homeostasis due to excretion failure in chronic kidney disease results in hyperphosphatemia 39 due to positive balance increasing the concentration in the exchangeable phosphorus pool, often when the pool size is reduced as in the adynamic bone disorder (Figure 2). Surprisingly and not generally adequately considered, the skeleton contributes to hyperphosphatemia in chronic kidney disease and end stage kidney disease (ESKD) through the effects of disordered bone remodeling. There are multiple skeletal remodeling disorders discussed below in chronic kidney disease, but all of them are associated with excess bone resorption compared to bone formation. Thereby, they contribute to hyperphosphatemia and effectively block the skeleton from exerting its normal reservoir function when positive phosphate balance occurs.
Figure 3. Phosphate homeostasis
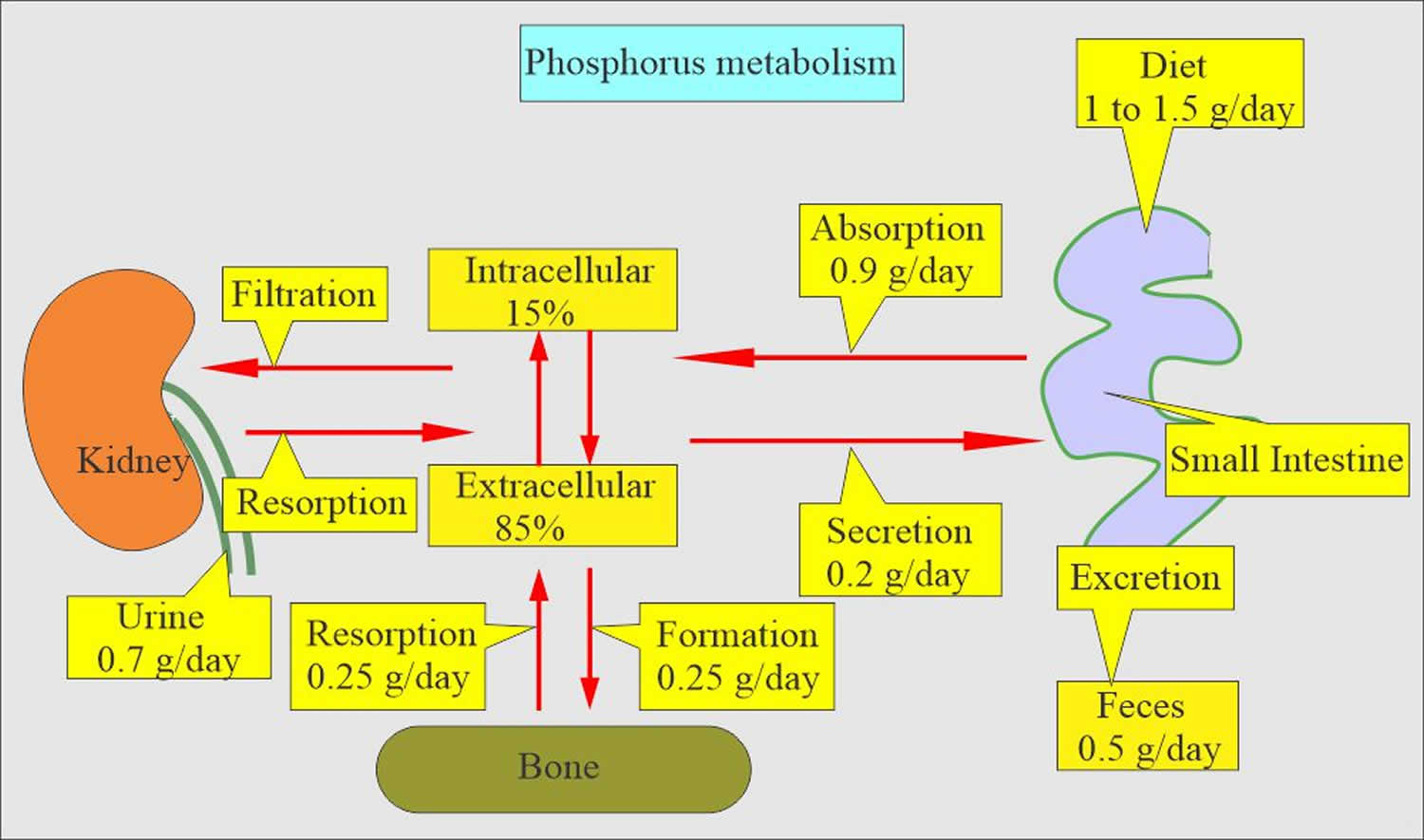
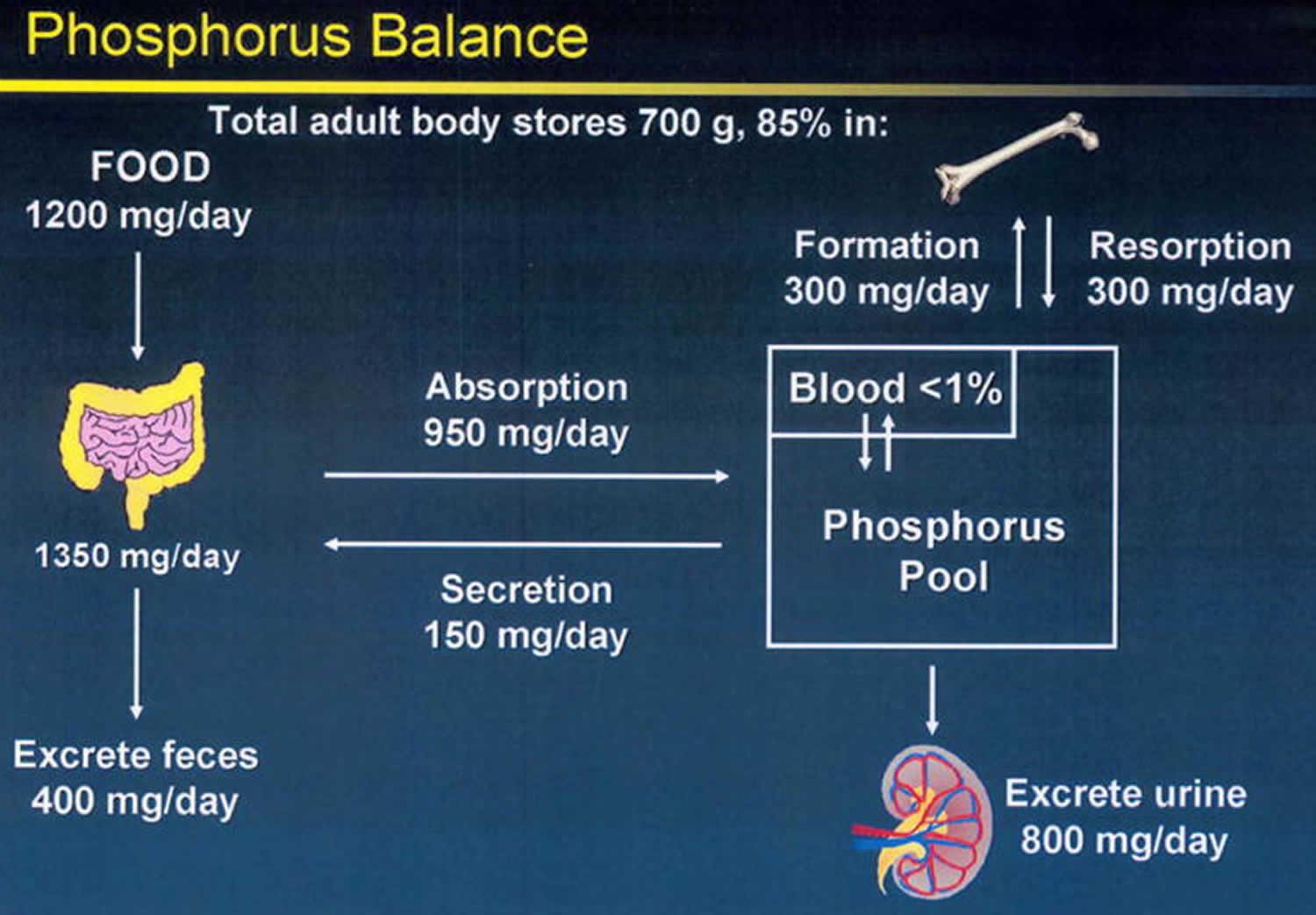
Footnote: Phosphorus balance in normal physiology. The kidney is the main regulator of human phosphate homeostasis. In adulthood, exit from the exchangeable phosphorus pool into the skeleton (bone formation) is roughly equal to entry into the exchangeable pool due to bone resorption. The skeleton is a storage depot for Pi and contains 85% of the total body phosphorus.
[Source 37 ]Figure 4. Hyperphosphatemia of chronic kidney disease
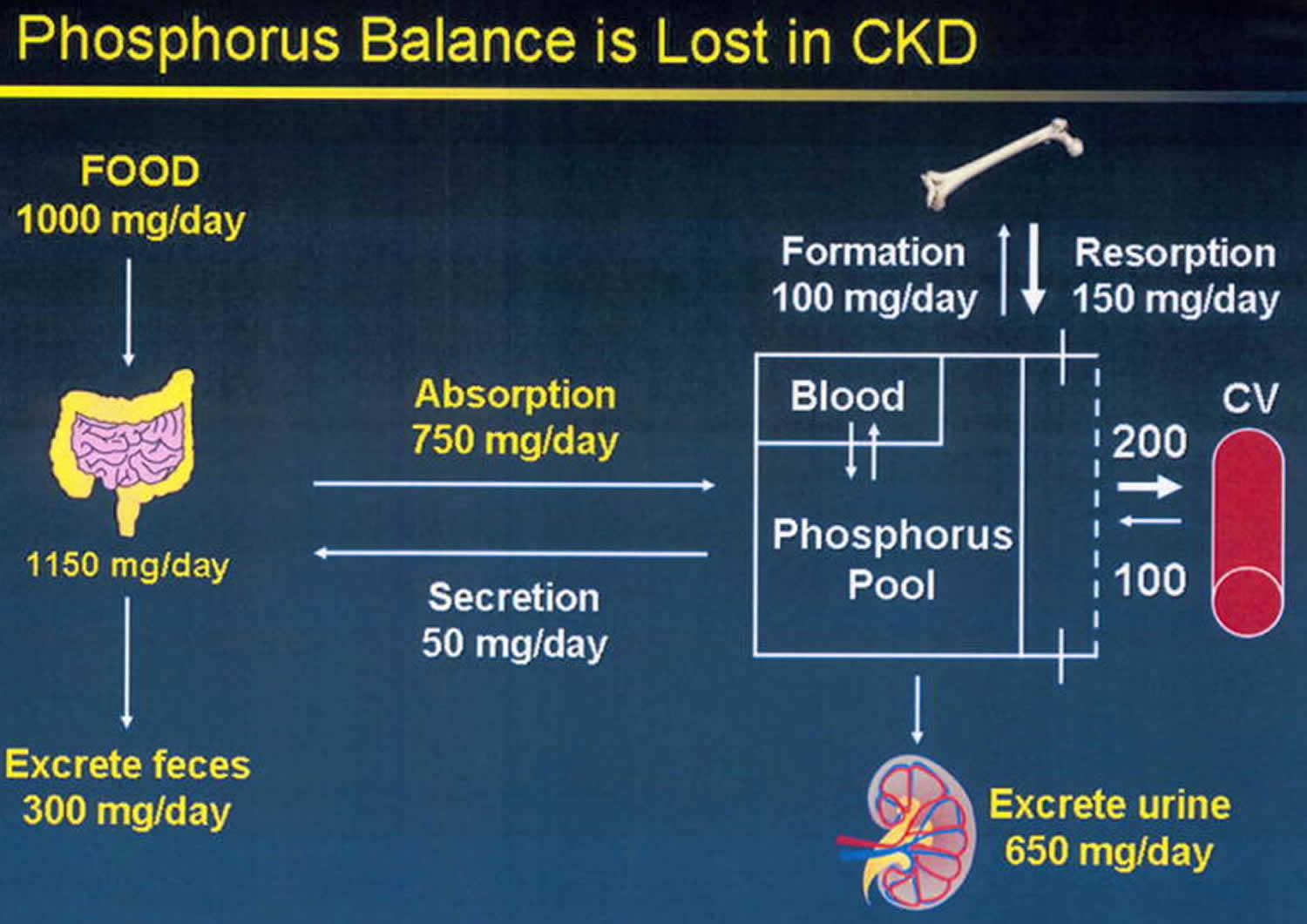
Footnote: Phosphorus homeostasis is lost in chronic kidney disease due to failure of excretion. Despite reductions in the fraction of filtered phosphorus that is reabsorbed, eventually the filtered load becomes insufficient to maintain homeostasis, and positive phosphorus balance ensues. Kidney disease decreases the exchangeable phosphorus pool size by inhibiting bone formation. The skeletal mineralization fronts at the sites of new bone formation are significant components of the exchangeable phosphorus pool. Positive phosphate balance is associated with establishment of heterotopic mineralization sites in soft tissue organs and the vasculature. Exit from the exchangeable phosphorus pool into the vasculature is portrayed as a bidirectional process because we have been able to demonstrate that stopping the exit into the vasculature results in diminishment of established vascular calcification levels.
[Source 37 ]Hyperphosphatemia causes
The most common cause of hyperphosphatemia is renal failure. Less common causes can be classified according to pathogenesis; ie, increased phosphate intake, decreased phosphate output, or a shift of phosphate from the intracellular to the extracellular space. Often, several mechanisms contribute. Impaired renal excretion is most frequently the major factor, with relatively increased intake or cell breakdown contributing to the problem.
Increased intake
This can result from the following:
- Excessive oral or rectal use of an oral phosphate-saline laxative (Phospho-soda)
- Excessive parenteral administration of phosphate
- Milk-alkali syndrome
- Vitamin D intoxication
Decreased excretion
This can result from the following:
- Renal failure, acute or chronic
- Hypoparathyroidism
- Pseudohypoparathyroidism
- Severe hypomagnesemia
- Tumoral calcinosis
- Bisphosphonate therapy
Shift of phosphate from intracellular to extracellular space
This can result from the following:
- Rhabdomyolysis
- Tumor lysis
- Acute hemolysis
- Acute metabolic or respiratory acidosis
Spurious
Results that falsely indicate the presence of hyperphosphatemia can result from the following:
- Blood sample taken from line containing heparin or alteplase 40
- High concentrations of paraproteins 41
- Hyperbilirubinemia 42
- In vitro hemolysis
- Hyperlipidemia 43
Therefore, information related to the causes of hyperphosphatemia, such as a history of diabetes mellitus or hypertension (causes of renal failure), a history of neck surgery or irradiation (causes of hypoparathyroidism), or a history of excessive vitamin D or milk ingestion, is important to obtain.
A search for the following historical clues may help to identify patients who are at risk for increased phosphorus levels:
- Kidney disease: Past or present hemodialysis; adherence to renal (low phosphorus) diet; use of oral phosphate binders
- Cancer: Leukemia, lymphoma, bone tumors, other cancers, chemotherapy treatment
- Endocrinopathies (hypoparathyroidism and pseudohypoparathyroidism)
- Trauma
- Burns or heat-related illnesses
- Prolonged immobilization
- Metabolic or hematologic disorders, including genetic predisposition
- Ischemic bowel (possible phosphorus elevations)
The patient’s medication history with regard to the following should also be obtained:
- Oral phosphate binders
- Potassium phosphate
- Antacids
- Bisphosphonates
- Laxatives (oral/rectal) and enemas 44
- Nutritional supplements or hyperalimentation
Hyperphosphatemia signs and symptoms
Typically, most patients with hyperphosphatemia are asymptomatic. Signs and symptoms of acute hyperphosphatemia result from the effects of hypocalcemia, with patients occasionally reporting symptoms such as muscle cramps, tetany, and perioral numbness or tingling. Other symptoms include bone and joint pain, pruritus (itch), and rash.
More commonly, patients report symptoms related to the underlying cause of the hyperphosphatemia. These generally are uremic symptoms, such as the following:
- Fatigue
- Shortness of breath
- Anorexia
- Nausea
- Vomiting
- Sleep disturbances
In acute hyperphosphatemia, especially that caused by parenteral phosphate administration, the patient may be hypotensive or exhibit signs of hypocalcemia, such as the following:
- Chvostek’s sign – is the manifestation of heightened neuromuscular excitability. It is the spasm of facial muscles in response to tapping the facial nerve near the angle of the jaw. Chvostek sign is an involuntary twitching of the facial muscles elicited by a light tapping of the facial nerve just anterior to the exterior auditory meatus. It is present in ≤ 10% of healthy people and in most people with acute hypocalcemia but is often absent in chronic hypocalcemia.
- Trousseau sign is the precipitation of carpal spasm by reduction of the blood supply to the hand with a tourniquet or blood pressure cuff inflated to 20 mm Hg above systolic blood pressure applied to the forearm for 3 min. Trousseau sign also occurs in alkalosis, hypomagnesemia, hypokalemia, and hyperkalemia and in about 6% of people with no identifiable electrolyte disturbance.
- Hyperreflexia
- Carpopedal spasm– Also referred to as Trousseau’s sign. It represents increased neuromuscular excitability which may be related to the gating function of calcium ion for ion channels at a cellular level (particularly in neurons). This manifests as a spasm of hand upon routine BP check.
- Seizures – Can be the sole manifestation or a part of the whole myriad of clinical presentation.
- Tetany – Generally induced by a rapid decline in serum ionized calcium. Tetany is usually most dangerous and most commonly seen in the presence of respiratory alkalosis causing hypocalcemia.
- Paresthesias – can be perioral or otherwise
- Psychiatric manifestations – Can be associated with anxiety/depression/emotional lability
- QTc prolongation – Can lead to Torsades de pointes although extremely rare, it can be fatal.
- Many other abnormalities may occur in patients with chronic hypocalcemia, such as dry and scaly skin, brittle nails, and coarse hair.
- Candida infections occasionally occur in hypocalcemia but most commonly occur in patients with idiopathic hypoparathyroidism.
- Cataracts occasionally occur with long-standing hypocalcemia and are not reversible by correction of serum calcium.
Hyperphosphatemia complications
Hyperphosphatemia, even of a quite severe degree, is largely a clinically asymptomatic condition. Associated morbidity most commonly results from an underlying condition than it does from the hyperphosphatemia itself.
The short-term complications of hyperphosphatemia include acute hypocalcemia with possible tetany and, more rarely, acute deposition of calcium/phosphate complexes into joints, as well as subcutaneous tissue and other areas of soft tissue. Acute hyperphosphatemia caused by excessive Phospho-soda ingestion may result in acute renal failure and, in some cases, chronic kidney disease 45. The inciting cause in acute hyperphosphatemia can usually be successfully treated.
In chronic hyperphosphatemia, however, the prognosis can be mixed, and the long-term complications can severely damage any organ system. The organs most commonly affected in chronic hyperphosphatemia include the vascular system, as well as the bones, skin, and heart. The joints are also commonly involved.
Prolonged hyperphosphatemia is an independent risk factor for cardiovascular disease in patients with chronic kidney disease. Patients with chronic phosphate levels above 6.5 mg/dL have an 18-39% higher mortality compared with patients with kidney failure who have near-normal serum phosphate levels.
Even in patients without chronic kidney disease, hyperphosphatemia may be a risk factor. In a Korean study of 92,756 individuals with normal kidney function, higher serum phosphorus levels were an independent predictor for all-cause mortality, particularly in men 46.
Hyperphosphatemia in kidney disease
Hyperphosphatemia is a risk factor for high rates of mortality in multiple populations, including kidney transplant recipients 47, patients with end-stage renal disease 48, and patients with chronic kidney disease (CKD) 49. A meta-analysis of 13 prospective cohort studies, conducted in over 90,000 chronic kidney disease (CKD) patients, found an 18% increase in all-cause mortality per 1 mg/dL increase in serum phosphorus concentration above 3.5 mg/dL. A 10% increased risk in cardiovascular disease (CVD)-related death was also calculated for each 1 mg/dL higher concentration in the meta-analysis of three studies 50.
Although no causality has been established between vitamin D deficiency and cardiovascular disease (CVD) risk, it has been suggested that failure to produce 1,25-dihydroxyvitamin D in hyperphosphatemic individuals may modify the risk of developing cardiovascular and renal disease, as well as worsen kidney insufficiency in chronic kidney disease (CKD) patients 51. Another plausible mechanism for hyperphosphatemia-induced cardiovascular dysfunction is the deposition of calcium phosphate in non-skeletal tissues, especially the blood vessels 52. High phosphorus concentrations may stimulate the expression of bone specific markers in blood vessel-forming cells, resulting in a shift in their functions; this process, called osteochondrogenic differentiation, transforms vascular smooth muscle cells into bone-like cells. The culture of human aortic vascular smooth muscle cells in hyperphosphatemic conditions was found to result in the mineralization of the extracellular media, mimicking in vivo vascular calcification 53. Blood vessel calcification has been associated with at least a three-fold increase in risk for cardiovascular events and mortality; the risk for cardiovascular events is twice as high (i.e., six-fold increased risk) in individuals with kidney insufficiency 54.
In patients with chronic kidney disease (CKD), disorders in bone remodeling may result in excess release of phosphorus and calcium into the blood, which worsens hyperphosphatemia and blood vessel calcification and accelerates the decline of kidney function. Currently, dietary phosphorus restriction is recommended to normalize serum concentrations in patients with chronic kidney disease (CKD), although the impact on cardiovascular disease (CVD) and mortality risks is not known.
If started early in the course of kidney failure, control of phosphate ingestion and absorption by appropriate changes in diet and the use of binders can successfully postpone the development of various complications. This has been demonstrated most convincingly in animal studies; however, and there is a paucity of human studies. On the other hand, if hyperphosphatemia is not adequately addressed early on, the changes that occur in bones, joints, and cardiovascular tissues can be very difficult, if not impossible, to eradicate.
Changes in baseline phosphorus values beyond the recommended targets of the Kidney Disease Outcome Quality Initiative (KDOQI) have been found to be robust predictors of higher death risk.
Interestingly, whether treatment to lower phosphate levels in patients with chronic or end-stage kidney disease results in lower morbidity and mortality has not really been definitively demonstrated. A study showed that patients treated with phosphate binders had a decrease in 1-year mortality, but the effect did not correlate with the degree of hyperphosphatemia 55. A more recent study demonstrated that when compared with placebo, treatment of patients with chronic kidney disease with one of a variety of phosphate binders resulted in lower serum phosphate but greater acceleration of vascular calcification, a condition associated with a higher mortality 56. This is an area that requires more intensive investigation.
Hyperphosphatemia diagnosis
Results from a full chemistry profile can be used as follows in determining the cause of hyperphosphatemia:
- Low serum calcium levels (hypocalcemia) along with high phosphate levels: Observed with renal failure, hypoparathyroidism, and pseudohypoparathyroidism
- Blood urea nitrogen (BUN) and creatinine values: Help to determine whether renal failure is the cause of hyperphosphatemia
- Elevated intact parathyroid hormone (PTH) levels: Higher likelihood in patients with renal failure or pseudohypoparathyroidism
- Relatively low levels of intact parathyroid hormone (PTH) and normal renal function: Found in patients with primary or acquired hypoparathyroidism
- High serum calcium and phosphate levels: Observed with vitamin D intoxication and milk-alkali syndrome.
- Relatively low levels of intact parathyroid hormone (PTH) and high 25 and 1,25 vitamin D: Also seen in vitamin D intoxication
- Low levels of parathyroid hormone (PTH) and vitamin D: Seen in milk-alkali syndrome
If renal function is normal, then more unusual disorders, such as the following, may be the cause:
- Vitamin D intoxication
- Laxative (Phospho-soda) abuse
- Tumor lysis
- Rhabdomyolysis
- Isolated hypoparathyroidism
- Pseudohypoparathyroidism
Rarely, if the cause of hyperphosphatemia is not clear, 24-hour measurement of urinary phosphate can be performed. Results indicate the following:
- Fractional renal excretion exceeding 15%: Suggests either massive phosphate ingestion (eg, laxative [Phospho-soda] abuse) or lysis of tissue and resulting release of intracellular phosphate
- Fractional renal excretion not exceeding 15%: Suggests that renal excretion is impaired because of either renal failure or hypoparathyroidism
Hyperphosphatemia treatment
Treatment of hyperphosphatemia consists of 3 main ways.
- Diagnose and treat the cause: Eg, hyperphosphatemia due to tumor lysis responds to forced saline diuresis to enhance urinary losses
- Limit phosphate intake: Renal failure is the clinical condition most often requiring curtailment of phosphate ingestion; patients with advanced renal insufficiency or complete renal failure also require phosphate binders, to inhibit gastrointestinal absorption of phosphate
- Enhance renal excretion: Used in patients with normal renal function and hyperphosphatemia; it can be accomplished most effectively by using volume repletion with saline coupled with forced diuresis with a loop diuretic such as furosemide or bumetanide
The clinical condition most often requiring curtailment of phosphate ingestion is renal failure. Because intestinal absorption of phosphate and phosphate content in a typical diet is high, maintenance of phosphate homeostasis is dependent on renal excretion of the ingested excess. Therefore, when renal failure develops and hyperphosphatemia ensues, the sole means of controlling it is limitation of intake.
Dietary phosphate restriction is the first step in the prevention and management of hyperphosphatemia 57. Protein restriction and avoidance of dairy products are the cornerstone of this dietary regimen. Furthermore, extra phosphate load from food, which contain phosphate additives (cooked ham, roast breast turkey/chicken, and so forth), and high phosphate content of beverage creates a special concern because this extra amount of phosphorous is almost completely absorbed by the intestinal tract 58. Effective dialysis plays an essential role in decreasing high serum phosphate levels.
Hyperphosphatemia diet
Patients with chronic kidney disease are advised to avoid foods that are especially high in phosphate; high-phosphate foods include dairy products; meats, nuts, and other high-protein foods; processed foods; and dark colas. Kidney Disease: Improving Global Outcomes (KDIGO) guidelines note that it is reasonable to consider phosphate source in making dietary recommendations, as approximately 40%-60% of animal-based phosphate is absorbed, compared with 20%-50% of plant-based phosphate, and that fresh and homemade foods are preferable to processed foods, which often contain inorganic phosphate additives 59.
Low phosphorus foods:
- Fresh fruits such as apples, apricots, blackberries, grapes, pear, peaches
- Popcorn and crackers
- Fresh vegetables such as cauliflower, carrot, cucumber, celery, green beans and broccoli
- Rice cereal, sherbet
- Coffee and tea without milk, light-colored sodas and fruit juices.
The foods that contain phosphorus are listed below:
- Meat products are good sources of phosphorus. Light meat products contain more phosphorus as compare to dark meat products.
- Chicken and turkey
- Beef contains 134-174mg of phosphorus per 75g of serving size
- Beef/chicken liver contains 345-373mg of phosphorus per 75g of serving size
- Beef/chicken kidneys contain 228mg of phosphorus per 75g of serving size.
- Lamb contains 144-180mg of phosphorus per 75g of serving size
- Deer contains 174-224mg of phosphorus per 75g of serving size
- Seafood: Many types of fishes are good sources of phosphorus
- Dairy products
- Milk contains 217-272mg of phosphorus per 250ml of serving size
- Cheese contains 232-302mg of phosphorus per 50g of serving size
- Yogurt contains 183-217mg of phosphorus per 175g of serving size
- Yogurt beverages contain 168mg of phosphorus per 200ml of serving size
- Buttermilk contains 212-230mg of phosphorus per 250ml of serving size
- Soy beverages contain 253mg of phosphorus per 250ml of serving size
- Egg contains 126-157mg of phosphorus per 2 large servings
- Grain products contain good levels of phosphorus. Some of the grain products are:
- Wheat bran contains 270mg of phosphorus per 30g of serving size
- Rice bran contains 335mg of phosphorus per 20g of serving size
- Quinoa contains 149mg of phosphorus per 125ml of serving size
- Oatmeal contains 138-177mg of phosphorus per 175ml of serving size
- Wheat germ cereals contain 344mg of phosphorus per 30g of serving size
- Nuts and seeds are good sources of phosphorus and contain 40% of the RDI. It includes:
- Sunflower seeds contain 375-393mg of phosphorus per 60ml of serving size
- Pumpkin seeds contain 676mg of phosphorus per 60ml of serving size.
- Almonds contain 174-208mg of phosphorus per 60ml of serving size
- Pistachios contain 146-153mg of phosphorus per 60ml of serving size
- Brazil nuts are high in phosphorus and contain 257mg of phosphorus per 60ml of serving size.
- Lentils are good sources of phosphorus and contain 264mg of phosphorus per 175ml of serving size
- Beans are good sources of phosphorus and contain 194-216mg of phosphorus per 175ml of serving size
- Tofu contains 146-204mg of phosphorus per 150mg of serving size
Phosphate binders
Oral phosphate binders are used to decrease the highly efficient gastrointestinal absorption of phosphorus. Calcium-containing phosphate binders are widely used but may produce hypercalcemia and may exacerbate metastatic calcification 60. There is already a clear-cut evidence indicating not to use calcium-based agents in patients with positive calcium balance to avoid vascular calcification 61. Aluminum salts are effective phosphate binders but may induce aluminum toxicity. Newer compounds containing iron or bile acid sequestrants are replacing calcium and aluminum binders. Iron salts are becoming a new phosphate binder still under investigation. Several studies suggest that ferric compounds can reduce intestinal phosphate uptake in uremic and non-uremic rats 62. So far, 2 compounds may enter the market in the near future; ferric citrate compound 63 and iron (III)-oxyhydroxide-based phosphate binder (PA21) 64. Efficacy and short-term safety have almost been proven for both compounds, however, side effects and cost-effectiveness needs to be evaluated.
The use of non-calcium-based agents like sevelamer has become a commonly used therapy. Experimental studies on phosphate binders showed a clear beneficial effect of these agents in suppressing ectopic calcification. For example, Katsumata et al 65 examined the effectiveness of sevelamer in preventing ectopic calcification of soft tissues in adenine induced renal failure rats. Uremic rats demonstrated an increase in ectopic calcification of blood vessels, kidney, and stomach, as well as hyperphosphatemia after 3 weeks of adenine diet. When serum phosphorus level had significantly increased, the rats were freely fed a normal diet that contained one or 2% sevelamer for another 5 weeks. The results showed that compared with controls (n=10), sevelamer-treated animals (n=16) had reduced serum phosphorus, and calcification was clearly suppressed in their aorta media. Block et al 66 assessed all-cause mortality in 127 patients new to hemodialysis assigned to calcium-containing binders, or sevelamer after a median follow-up of 44 months from randomization; a total of 34 deaths occurred during the period of follow-up: 23 deaths in the calcium group, and 11 deaths in the sevelamer group 66. The authors concluded that treatment with sevelamer was associated with a significant survival benefit, as compared with the use of calcium-containing phosphate binders. It seems that the effect of sevelamer on cardiovascular system is time-dependent. Suki et al 67, showed a clear improvement in survival among patients treated for more than 2 years with sevelamer, as compared with those treated with calcium salts. This time related effect of sevelamer was further supported by Savica et al 68, who observed the effect of sevelamer on cardiovascular morbidity (cardiovascular hospitalization events and duration) for 3 years in 16 patients on maintenance dialysis. At the end of the study, there was a significant reduction in serum calcium and phosphorous. Also, there was a decrease in hospitalization rate and hospitalization duration.
Lanthanum carbonate, another non-calcium-based phosphate binder was assessed by Hutchison et al 69 on patients who received either hemodialysis, or peritoneal dialysis treatment for end stage kidney disease. The results showed that lanthanum carbonate effectively reduced serum phosphate levels, and when used as a monotherapy, it provided effective phosphate control with a low tablet burden.
Takahashi et al 70 showed in their clinical study on 65 hemodialysis patients that Nicotinamide, a metabolite of nicotinic acid (Vitamin B3) decreased serum phosphorus levels significantly from 6.9±1.5 mg/dL to 5.4±1.3 mg/dL during the 12-weeks of Nicotinamide treatment. Nicotinic acid is known to be an inhibitor of sodium dependent phosphate cotransporter in renal tubule and small intestine of rats.
Increased excretion of salivary phosphate has been reported in patients with chronic kidney disease, and in hemodialysis patients’ independent from food intake 71. In an attempt to improve the treatment of hyperphosphatemia, Savica et al 72 assessed the phosphate-binding capacity of the natural cationic polymer-chitosan as chewing gum (the 20 mg chitosan loaded chewing gum was manufactured as cold compressed 3-layer tablets). Thirteen hemodialysis patients with serum phosphate levels >6.0 mg/dL despite treatment with sevelamer hydrochloride chewed 20 mg of chitosan-loaded chewing gum twice daily during fasting (that is, between meals) for 2 weeks in addition to their prescribed phosphate-binding regimen. Salivary phosphate and serum phosphate significantly decreased during the first week with chewing. By the end of the second week, salivary phosphate decreased by 55% from baseline, and serum phosphate decreased 31% from baseline. Parathyroid hormone (PTH) and serum calcium concentrations were not affected by the gum. These results suggest that adding salivary phosphate-binding to traditional phosphate binders could be a useful approach for improving treatment of hyperphosphatemia in dialysis patients 73.
Proximal diuretics are phosphuretic to the same extent that they are natriuretic. Acetazolamide is particularly efficient in promoting renal phosphate excretion.
- Hyperphosphatemia. https://emedicine.medscape.com/article/241185-overview[↩][↩]
- Phosphorus. https://lpi.oregonstate.edu/mic/minerals/phosphorus[↩][↩]
- Knochel JP. Phosphorus. In: Shils ME, Shike M, Ross AC, Caballero B, Cousins RJ, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lippincott Williams & Wilkins; 2006:211-222.[↩]
- Gilbert S, Weiner D, Gipson DS, Perazella MA, Tonelli M. 6th ed. Boston (MA): Elsevier; 2014. National Kidney Foundation’s Primer on Kidney Diseases.[↩]
- Kuro-o M. Klotho in health and disease. Curr Opin Nephrol Hypertens. 2012;21:362–368[↩]
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012 Jan;92(1):131-55. doi: 10.1152/physrev.00002.2011[↩]
- Calvo MS, Uribarri J. Public health impact of dietary phosphorus excess on bone and cardiovascular health in the general population. Am J Clin Nutr. 2013 Jul;98(1):6-15. doi: 10.3945/ajcn.112.053934[↩]
- de Boer IH, Rue TC, Kestenbaum B. Serum phosphorus concentrations in the third National Health and Nutrition Examination Survey (NHANES III). Am J Kidney Dis. 2009 Mar;53(3):399-407. doi: 10.1053/j.ajkd.2008.07.036[↩]
- Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011 Jun;79(12):1370-8. doi: 10.1038/ki.2011.47. Epub 2011 Mar 9. Erratum in: Kidney Int. 2012 Aug;82(4):498.[↩][↩]
- Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutiérrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M; Chronic Renal Insufficiency Cohort (CRIC) Study Group. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011 Jun 15;305(23):2432-9. doi: 10.1001/jama.2011.826[↩]
- Dhingra R, Sullivan LM, Fox CS, Wang TJ, D’Agostino RB Sr, Gaziano JM, Vasan RS. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007 May 14;167(9):879-85. doi: 10.1001/archinte.167.9.879[↩][↩]
- Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G; Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation. 2005 Oct 25;112(17):2627-33. doi: 10.1161/CIRCULATIONAHA.105.553198. Erratum in: Circulation. 2007 Dec 4;116(23):e556.[↩]
- O’Seaghdha CM, Hwang SJ, Muntner P, Melamed ML, Fox CS. Serum phosphorus predicts incident chronic kidney disease and end-stage renal disease. Nephrol Dial Transplant. 2011 Sep;26(9):2885-90. doi: 10.1093/ndt/gfq808[↩]
- Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphate and left ventricular hypertrophy in young adults: the coronary artery risk development in young adults study. Kidney Blood Press Res. 2009;32(1):37-44. doi: 10.1159/000203348[↩]
- Shuto E, Taketani Y, Tanaka R, Harada N, Isshiki M, Sato M, Nashiki K, Amo K, Yamamoto H, Higashi Y, Nakaya Y, Takeda E. Dietary phosphorus acutely impairs endothelial function. J Am Soc Nephrol. 2009 Jul;20(7):1504-12. doi: 10.1681/ASN.2008101106[↩]
- Tuttle KR, Short RA. Longitudinal relationships among coronary artery calcification, serum phosphorus, and kidney function. Clin J Am Soc Nephrol. 2009 Dec;4(12):1968-73. doi: 10.2215/CJN.01250209[↩]
- Askar AM. Hyperphosphatemia. The hidden killer in chronic kidney disease. Saudi Med J. 2015;36(1):13–19. doi:10.15537/smj.2015.1.9843 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4362181[↩]
- Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. New Engl J Med. 2004;351:1296–1305.[↩][↩]
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15:2208–2218.[↩][↩][↩]
- Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol. 2005;16:520–528.[↩]
- Calò LA, Savica V, Piccoli A, Fusaro M, D’Angelo A, Davis PA. Reduction of hyperphosphatemia is related with the reduction of C-reactive protein in dialysis patients. Study in sevelamer-resistant dialysis patients treated with chitosan chewing gum as salivary phosphate binder. Ren Fail. 2011;33:11–14.[↩]
- Dhingra R, Sullivan LM, Fox CS, Wang TJ, D’Agostino RB, Sr, Gaziano JM, et al. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch Intern Med. 2007;167:879–885[↩]
- Slinin Y, Foley RN, Collins AJ. Calcium, Phosphorus, Parathyroid Hormone, and Cardiovascular Disease in Hemodialysis Patients: The USRDS Waves 1, 3, and 4 Study. J Am Soc Nephrol. 2005;16:1788–1793.[↩]
- Block GA, Raggi P, Bellasi A, et al. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007;71:438–441.[↩]
- Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphorus levels associate with coronary atherosclerosis in young adults. J Am Soc Nephrol. 2009;20:397–404.[↩]
- Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol. 2004;15:2959–2964.[↩][↩]
- National Institute of Diseases and Digestive and Kidney Disease. US Renal Data System 2012 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Am J Kidney Dis. 2013;61:1–480.[↩]
- Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15:2208–2218[↩]
- Yang H, Curinga G, Giachelli CM. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 2004;66:2293–2299[↩]
- Glassock RJ, Pecoits-Filho R, Barberato SH. Left ventricular mass in chronic kidney disease and ESRD. Clin J Am Soc Nephrol. 2009;4(Suppl 1):S79–S91[↩]
- Nakamura S, Ishibashi-Ueda H, Niizuma S, Yoshihara F, Horio T, Kawano Y. Coronary calcification in patients with chronic kidney disease and coronary artery disease. Clin J Am Soc Nephrol. 2009;4:1892–1900[↩]
- Shamseddin MK, Parfrey PS. Sudden cardiac death in chronic kidney disease: epidemiology and prevention. Nat Rev Nephrol. 2011;7:145–154[↩]
- Coresh J, Selvin E, Stevens LA, et al. Prevalence of Chronic Kidney Disease in the United States. J Am Med Assoc. 2007;298:2038–2047[↩]
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am JKid Dis. 1998;32:S112–S119.[↩]
- Sarnak MJ, Levey AS, Schoolwerth AC, et al. Kidney Disease as a Risk Factor for Development of Cardiovascular Disease: A Statement From the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension. 2003;42:1050–1065[↩]
- London GM, Guerin AP, Marchais SJ, et al. Arterial media calcification in end-stage renal diseases: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731–1740.[↩]
- Hruska KA, Mathew S, Lund R, Qiu P, Pratt R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008;74(2):148–157. doi:10.1038/ki.2008.130 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2735026[↩][↩][↩]
- Slatopolsky E, Robson AM, Elkan I, et al. Control of phosphate excretion in uremic man. J Clin Invest. 1968;47:1865–1874[↩]
- Craver L, Marco MP, Martinez I, et al. Mineral metabolism parameters throughout chronic kidney disease stages 1–5–achievement of K/DOQI target ranges. Nephrol Dial Transplant. 2007;22:1171–1176[↩]
- Cachat F, Bardy D, Durussel C, Di Paolo E. Spurious hyperphosphatemia in a patient with alteplase-locked central venous catheter. Pediatr Nephrol. 2006 Feb. 21(2):301-2.[↩]
- Marcu CB, Hotchkiss M. Pseudohyperphosphatemia in a patient with multiple myeloma. Conn Med. 2004 Feb. 68(2):71-2[↩]
- Larner AJ. Pseudohyperphosphatemia. Clin Biochem. 1995 Aug. 28(4):391-3[↩]
- Leehey DJ, Daugirdas JT, Ing TS, Reid RW. Spurious hyperphosphatemia due to hyperlipidemia. Arch Intern Med. 1985 Apr. 145(4):743-4.[↩]
- Marraffa JM, Hui A, Stork CM. Severe hyperphosphatemia and hypocalcemia following the rectal administration of a phosphate-containing Fleet pediatric enema. Pediatr Emerg Care. 2004 Jul. 20(7):453-6.[↩]
- Markowitz GS, Stokes MB, Radhakrishnan J, D’Agati VD. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol. 2005. 16:3389-3396[↩]
- Yoo KD, Kang S, Choi Y, Yang SH, Heo NJ, Chin HJ, et al. Sex, Age, and the Association of Serum Phosphorus With All-Cause Mortality in Adults With Normal Kidney Function. Am J Kidney Dis. 2015 Sep 2[↩]
- Connolly GM, Cunningham R, McNamee PT, Young IS, Maxwell AP. Elevated serum phosphate predicts mortality in renal transplant recipients. Transplantation. 2009. 87:1041-1044[↩]
- Tentori F, Blayney MJ, Albert JM, Gillespie BW, Kerr PG, Bommer J, et al. Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2008. 52:519-530.[↩]
- Hruska KA, Mathew S, Lund R, Qiu P, Pratt R. Hyperphosphatemia of chronic kidney disease. Kidney International. 2008. 74:148-157.[↩]
- Palmer SC, Hayen A, Macaskill P, Pellegrini F, Craig JC, Elder GJ, Strippoli GF. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011 Mar 16;305(11):1119-27. doi: 10.1001/jama.2011.308[↩]
- Li YC. Vitamin D: roles in renal and cardiovascular protection. Curr Opin Nephrol Hypertens. 2012 Jan;21(1):72-9. doi: 10.1097/MNH.0b013e32834de4ee[↩]
- Hruska KA, Mathew S, Lund R, Qiu P, Pratt R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008 Jul;74(2):148-57. doi: 10.1038/ki.2008.130[↩]
- Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000 Sep 29;87(7):E10-7. doi: 10.1161/01.res.87.7.e10[↩]
- Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5(1):185-97. doi: 10.2147/vhrm.s4822[↩]
- Isakova T, Gutiérrez OM, Chang Y, et al. Phosphorus binders and survival on hemodialysis. J Am Soc Nephrol. 2009 Feb. 20(2):388-96.[↩]
- Block GA, Wheeler DC, Persky MS, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012 Aug. 23(8):1407-15.[↩]
- Askar AM. Hyperphosphatemia. The hidden killer in chronic kidney disease. Saudi Med J. 2015;36(1):13–19. doi:10.15537/smj.2015.1.9843 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4362181/[↩]
- Benini O, D’Alessandro C, Gianfaldoni D, Cupisti A. Extra-phosphate load from food additives in commonly eaten foods: a real and insidious danger for renal patients. J Ren Nutr. 2011;21:303–308.[↩]
- [Guideline] Ketteler M, Block GA, Evenepoel P, Fukagawa M, Herzog CA, McCann L, et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: what’s changed and why it matters. Kidney Int. 2017 Jul. 92 (1):26-36.[↩]
- Bellinghieri G, Santoro D, Savica V. Emerging drugs for hyperphosphatemia. Expert Opin Emerg Drugs. 2007;12:355–365[↩]
- Savica V, Bellinghieri G, Monardo P, Muraca U, Santoro D. An update on calcium metabolism alterations and cardiovascular risk in patients with chronic kidney disease: questions, myths and facts. J Nephrol. 2013;26:456–464.[↩]
- Iida A, Kemmochi Y, Kakimoto K, Tanimoto M, Mimura T, Shinozaki Y, et al. Ferric citrate hydrate, a new phosphate binder, prevents the complications of secondary hyperparathyroidism and vascular calcification. Am J Nephrol. 2013;37:346–358[↩]
- Dwyer JP, Sika M, Schulman G, Chang IJ, Anger M, Smith M, et al. Dose-response and efficacy of ferric citrate to treat hyperphosphatemia in hemodialysis patients: a short-term randomized trial. Am J Kidney Dis. 2013;61:759–766.[↩]
- Wüthrich RP, Chonchol M, Covic A, Gaillard S, Chong E, Tumlin JA. Randomized clinical trial of the iron-based phosphate binder PA21 in hemodialysis patients. Clin J Am Soc Nephrol. 2013;8:280–289[↩]
- Katsumata K, Kusano K, Hirata M, Tsunemi K, Nagano N, Burke SK, et al. Sevelamer hydrochloride prevents ectopic calcification and renal osteodystrophy in chronic renal failure rats. Kidney Int. 2003;64:441–450[↩]
- Block GA, Raggi P, Bellasi A, Kooienga L, Spiegel DM. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007;71:438–441.[↩][↩]
- Suki WN Dialysis Clinical Outcomes Revisited Investigators. Effects of sevelamer and calcium-based phosphate binders on mortality in hemodialysis patients: results of a randomized clinical trial. J Ren Nutr. 2008;18:91–98.[↩]
- Savica V, Monardo P, Santoro D, Mallamace A, Bellinghieri G. Time-dependent effect of sevelamerHCl on the cardiovascular system. Nephrol Dial Transplant. 2008;23:2102–2103[↩]
- Hutchison AJ, Laville M SPD405-313 Lanthanum Study Group. Switching to lanthanum carbonate monotherapy provides effective phosphate control with a low tablet burden. Nephrol Dial Transplant. 2008;23:3677–3684.[↩]
- Takahashi Y, Tanaka A, Nakamura T, Fukuwatari T, Shibata K, Shimada N, et al. Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int. 2004;65:1099–1104[↩]
- Savica V, Calò L, Santoro D, Monardo P, Granata A, Bellinghieri G. Salivary phosphate secretion in chronic kidney disease. J Ren Nutr. 2008;18:87–90.[↩]
- Savica V, Calò LA, Monardo P, Davis PA, Granata A, Santoro D, et al. Salivary phosphate-binding chewing gum reduces hyperphosphatemia in dialysis patients. J Am Soc Nephrol. 2009;20:639–644.[↩]
- Savica V, Calò LA, Santoro D, Monardo P, Santoro G, Muraca U, et al. Salivary glands: a new player in phosphorus metabolism. J Ren Nutr. 2011;21:39–42.[↩]




{kind=link}