Contents
- Rosai Dorfman disease
- Rosai-Dorfman disease types
- Rosai-Dorfman disease cause
- Rosai-Dorfman disease pathology outlines
- Clinical features
- Classic (nodal) Rosai-Dorfman disease
- Extranodal Rosai-Dorfman disease
- Cutaneous Rosai-Dorfman disease
- Central nervous system Rosai-Dorfman disease
- Ophthalmic Rosai-Dorfman disease
- Head and neck Rosai-Dorfman disease
- Intrathoracic Rosai-Dorfman disease
- Retroperitoneal and genitourinary Rosai-Dorfman disease
- Gastrointestinal Rosai-Dorfman disease
- Bone Rosai-Dorfman disease
- Hematologic Rosai-Dorfman disease
- Clinical features
- Rosai-Dorfman disease symptoms
- Rosai-Dorfman disease diagnosis
- Rosai Dorfman disease treatment
- Rosai Dorfman disease prognosis
Rosai Dorfman disease
Rosai-Dorfman disease (RDD) also called Rosai-Dorfman-Destombes disease or sinus histiocytosis with massive lymphadenopathy (SHML), is a rare benign non-Langerhans cell histiocytosis which is characterized by over-production and accumulation of a specific type of white blood cell called histiocyte in the lymph nodes of your body (lymphadenopathy), most often those in your neck (cervical lymphadenopathy) 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 11. Other lymph node groups may also be involved and in some cases, abnormal accumulation of histiocytes may occur in other areas of your body (extranodal disease is present in approximately 43% of cases) usually occurring in the skin, nasal cavity, paranasal sinus, upper respiratory tract, salivary gland, head, and neck, central nervous system (CNS), soft tissues, bone, kidneys, testis, digestive system, eyelid, orbit, and salivary gland is present with or without lymphadenopathy 6, 12, 13, 14, 15. These areas include your skin, central nervous system (brain and spinal cord), kidney and digestive tract. Histiocytes are large phagocytic cells (macrophages) that normally play a role in responding to infection and injury. A phagocytic cell is any “scavenger cell” that engulfs and destroys invading microorganisms or cellular debris. Histiocytes normally function to destroy foreign substances and protect the body from infection. In Rosai-Dorfman disease, the overproduction and accumulation of histiocytes (histiocytosis) in the channels (sinuses) that allow for the passage of lymph (sinus histiocytosis) and less commonly the interfollicular area of the lymph nodes are infiltrated with distinctive histiocytes with round or oval vesicular nuclei with well-defined, delicate nuclear membranes and a single prominent nucleolus 16, 17, 12.
Symptoms of Rosai-Dorfman disease is more frequently seen in children or young adults, but most often affects young adults under the age of 20 with peak incidence in the second or third decade (mean age, 20.6 years), although it has been reported up to age 74 years 15, 14. Rosai-Dorfman disease is more common in males and people of African descent, with the cutaneous form more common in female Asians 1, 17, 18. Some studies suggest that Rosai-Dorfman disease affects males more often than females. Skin Rosai-Dorfman disease occurs more often in females in their 20s or 30s.
The symptoms and physical findings associated with Rosai-Dorfman disease vary depending upon the specific areas of the body that are affected. In most Rosai-Dorfman disease cases, individuals exhibit painless swelling or enlargement of affected lymph nodes (lymphadenopathy), most often those of the neck (cervical lymphadenopathy). Many individuals with Rosai-Dorfman disease do not develop any additional symptoms of the disorder (asymptomatic). In some Rosai-Dorfman disease cases, affected individual may experience nonspecific symptoms that are common to many different conditions including fever, paleness of the skin (pallor), unintended weight loss, a general feeling of ill health (malaise) and a chronically runny nose (rhinitis). In extremely rare cases, affected individuals may experience abnormal enlargement of the liver and/or spleen (hepatosplenomegaly).
Rosai-Dorfman disease is a rare disease with a prevalence of 1 per 200,000 and an estimated 100 new cases diagnosed per year in the United States 1, 19. More than 1000 cases have been reported in the medical literature since the disorder’s first described by the French pathologist Destombes in 1965 20, then recognized as a distinct clinicopathological entity by Juan Rosai and Ronald F. Dorfman in 1969 16. In 1969 and 1972 Rosai and Dorfman described a total of 34 cases of patients with painless, bilateral, massive cervical lymphadenopathy, fever, and distinctive abnormalities in laboratory tests: normochromic anaemia, leukocytosis with neutrophilia, increased erythrocyte sedimentation rate (ESR), and often hypergammaglobulinaemia. Subsequently, it was established as being a distinct clinical entity 17, 16.
The cause of Rosai-Dorfman disease remains unknown, although altered immune responses and infectious agents may play a role 17, 21. Recent studies showed that approximately 1/3 of Rosai-Dorfman disease these patients harbor gene mutations involving the MAPK/ERK pathway, e.g., NRAS, KRAS, MAP2K1, and, rarely, BRAF, indicating a neoplastic process rather than a reactive disorder 5. Genetic predisposition or hereditary forms of Rosai-Dorfman disease has been hypothesized, as cases have been described in twins or family members, which supports this hypothesis 22. Germline mutations in SLC29A3 at 10q23, which is associated with histiocytosis-lymphadenopathy plus syndrome (H syndrome), Faisalabad histiocytosis, and pigmented hypertrichotic dermatosis with insulin-dependent diabetes, have been found in cases of familial Rosai-Dorfman disease 5. Another germline mutation (TNFRSF) that is found in autoimmune lymphoproliferative syndrome (ALPS) type 1 is also found in Rosai-Dorfman disease 5. The emergence of novel molecular data indicates that Rosai-Dorfman disease is a neoplastic process. Rosai-Dorfman disease was recently listed in the 5th Edition of the World Health Organization (WHO) Classification of Myeloid and Histiocytic/Dendritic Neoplasms 23.
The diagnosis of Rosai-Dorfman disease may be confirmed by a thorough clinical evaluation, a detailed patient history and a variety of specialized tests, such as surgical removal and microscopic examination of affected tissue (biopsy). Histopathological examination remains the mainstay of diagnosis – lymph nodes have massive sinusoidal dilation, containing histiocytes positive for S-100 and CD68, and negative for CD1a. The hallmark of Rosai-Dorman disease is lymphophagocytosis or emperipolesis, wherein the viable lymphocytes are located in well-defined cytoplasmic vacuoles of intact histiocytes. Emperipolesis is derived from the Greek (em – inside; peri – around; polemai – wander about) and it is an uncommon biological process, in which a cell penetrates another living cell 24. Unlike in phagocytosis where the engulfed cell is killed by lysosomal enzymes of the macrophage, the cell exists as viable cell within another in emperipolesis and can exit at any time without any structural or functional abnormalities for either of them 24. Plasma cells, neutrophils and red blood cells may also occupy this unique intracytoplasmic niche. The involved histiocytes are activated macrophages with features of phagocytic cells as well as immune accessory cells and thus express S-100 protein, HAM 56, α1 antitrypisn, α1 chymotrypsin, lysozyme, Mac 387, Ki-1 (CD 30, Ber-H2), but are negative for CD1a (leu 6) 25. Rosai-Dorfman disease involving extranodal sites shows similar morphologic features to its nodal counterpart with more fibrosis and fewer histiocytes with emperipolesis 13. The presence of benign histiocytes with emperipolesis, absence of cellular atypia, immunohistochemical profile, and associated clinical features distinguish Rosai-Dorfman disease from other simulating disorders. In laboratory tests the most common abnormalities are increased erythrocyte sedimentation rate (ESR), leukocytosis with neutrophilia, normocytic anemia, and hypergammaglobulinemia 11.
Because Rosai-Dorfman disease is so rare, no large research studies have been performed, and there is no established, widely accepted treatment.
In many cases, the symptoms of Rosai-Dorfman disease may disappear without treatment (spontaneous remission) within in months or a few years. The “watch and wait” approach is used without treatment is preferred for individuals with Rosai-Dorfman disease whenever possible 18. In many cases, no therapy will be necessary.
In some cases, various treatment options may become necessary. In these cases, the treatment of Rosai-Dorfman disease is directed toward the specific symptoms that are apparent in each individual. Several different treatment options have been used to treat individuals with Rosai-Dorfman disease including surgical removal of histiocytic lesions. In more serious cases, additional treatment options have included therapy with certain drugs including steroids (e.g., prednisone), alfa-interferon, and a regimen of certain anticancer drugs (chemotherapy). In some cases, affected individuals have shown improvement of symptoms with these treatments. In other cases, drug therapies have been ineffective.
Other treatment is symptomatic and supportive.
Figure 1. Rosai Dorfman disease with enlarged lymph node in the neck (cervical lymphadenopathy)
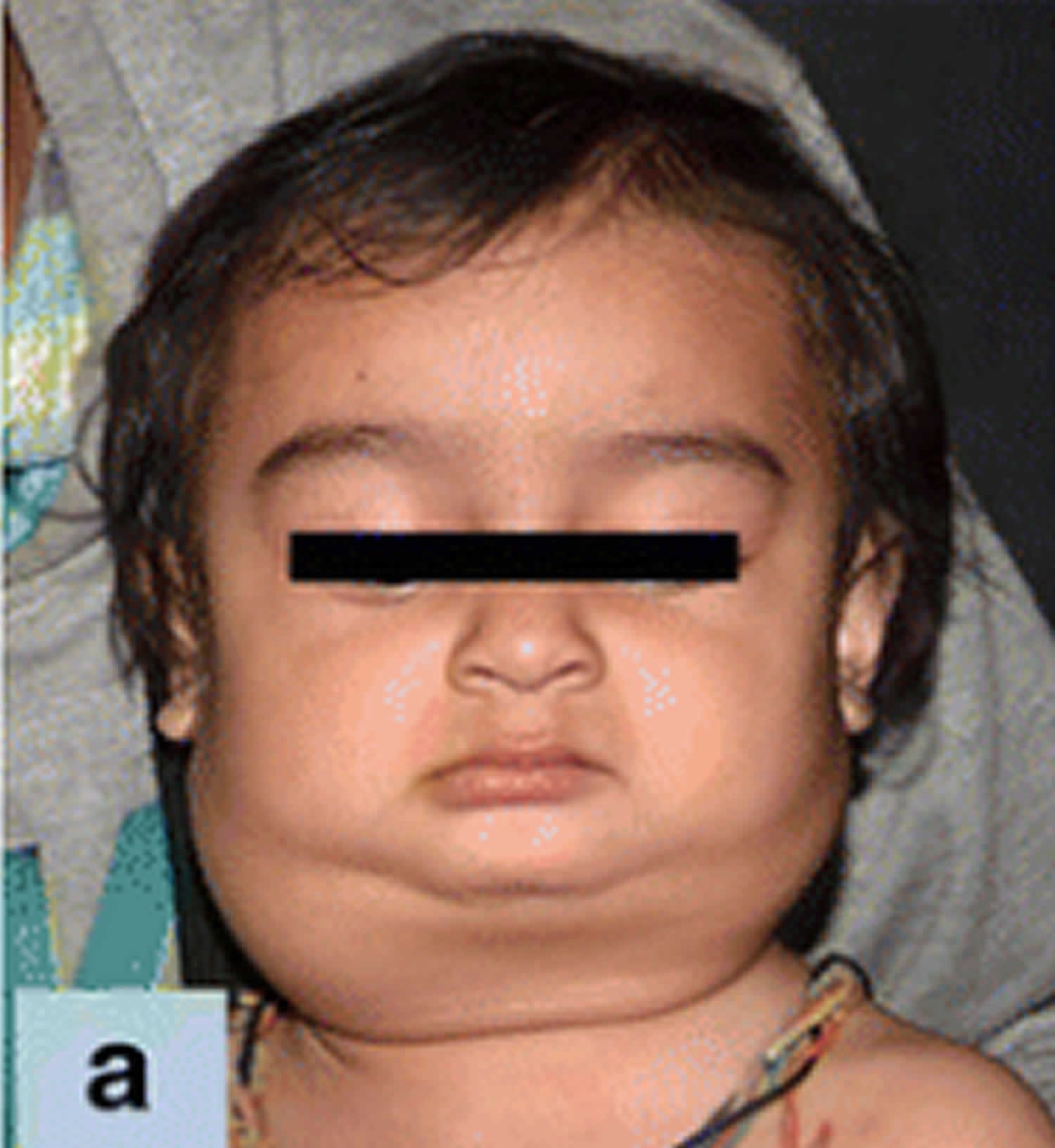
Figure 2. Rosai dorfman disease of the orbit
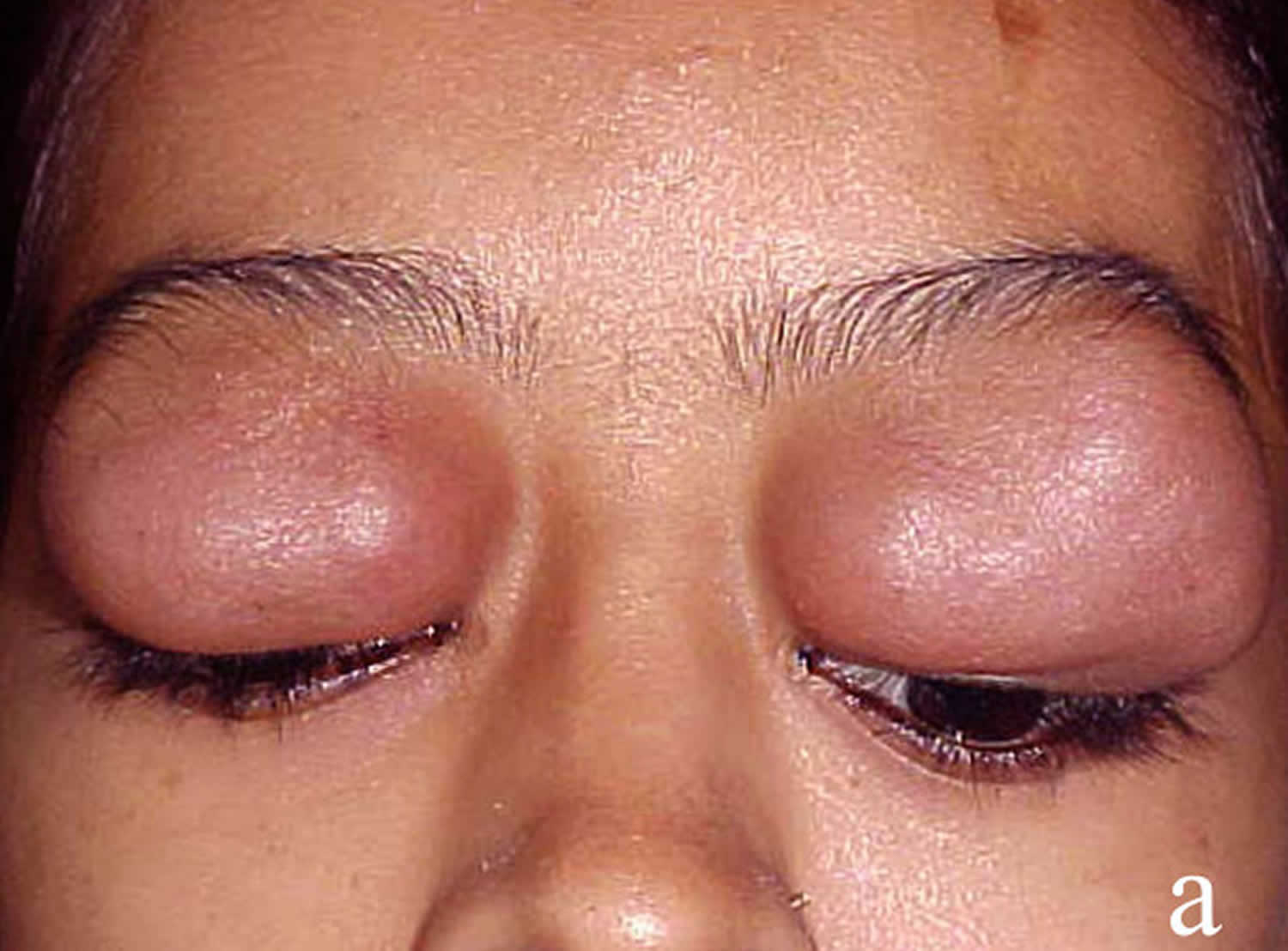
Footnote: Bilateral upper eyelid and preseptal orbital mass in a 16-year-old female with complete mechanical ptosis.
[Source 13 ]Figure 3. Cutaneous Rosai Dorfman disease
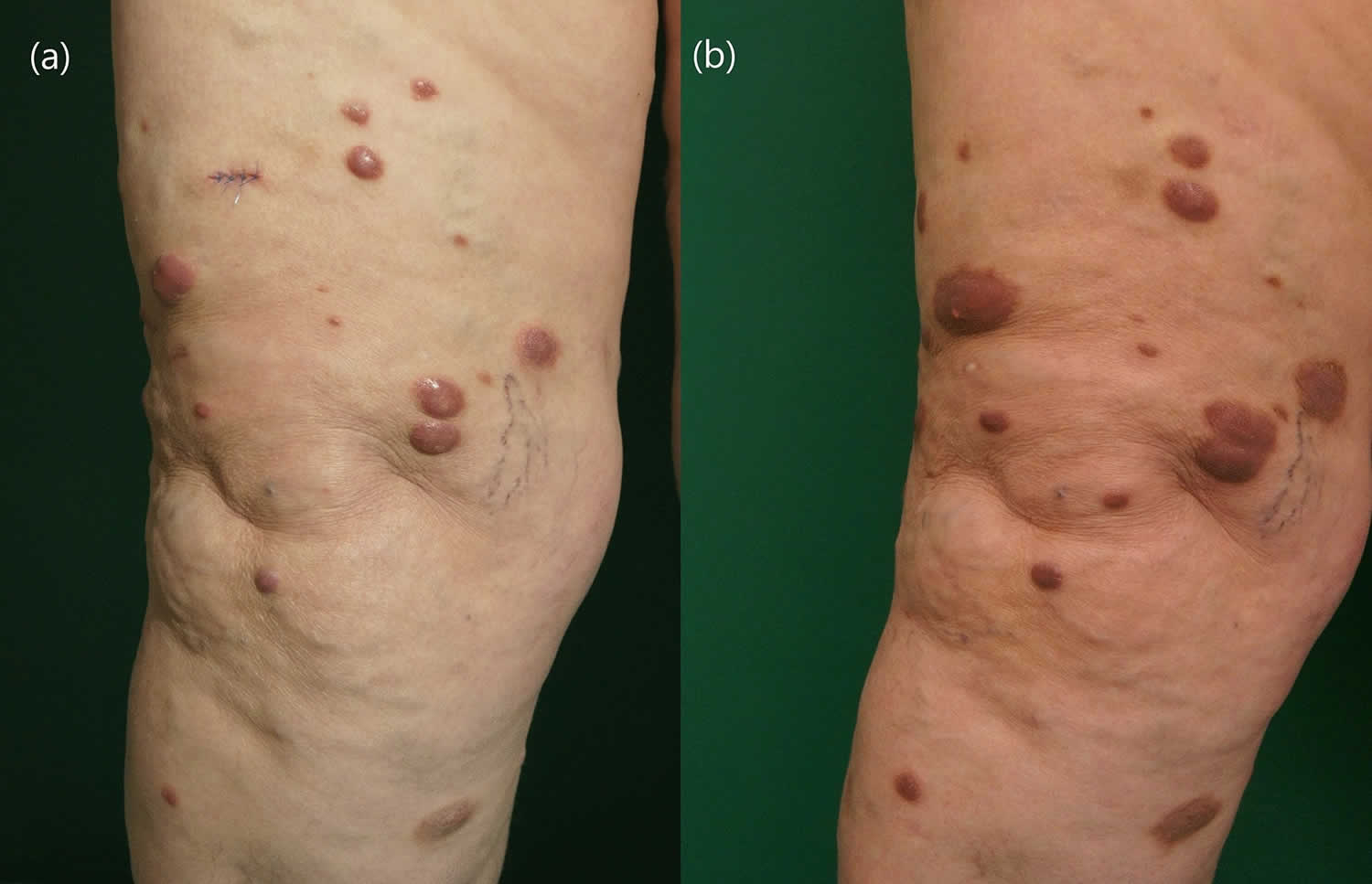
Footnote: A 68-year-old Caucasian woman presented to the dermatology department with multiple erythematous nodules localized on the right thigh that had been slowly progressing for 9 months. The lesions were slightly tender on palpation, but no other general symptoms were reported. (A) Multiple violaceous nodules disseminated on the right thigh before the treatment and (B) after 55 weeks on 15 mg methotrexate subcutaneously once weekly with additional 10 mg of prednisone once daily for 12 weeks.
[Source 27 ]Figure 4. Cutaneous Rosai-Dorfman disease
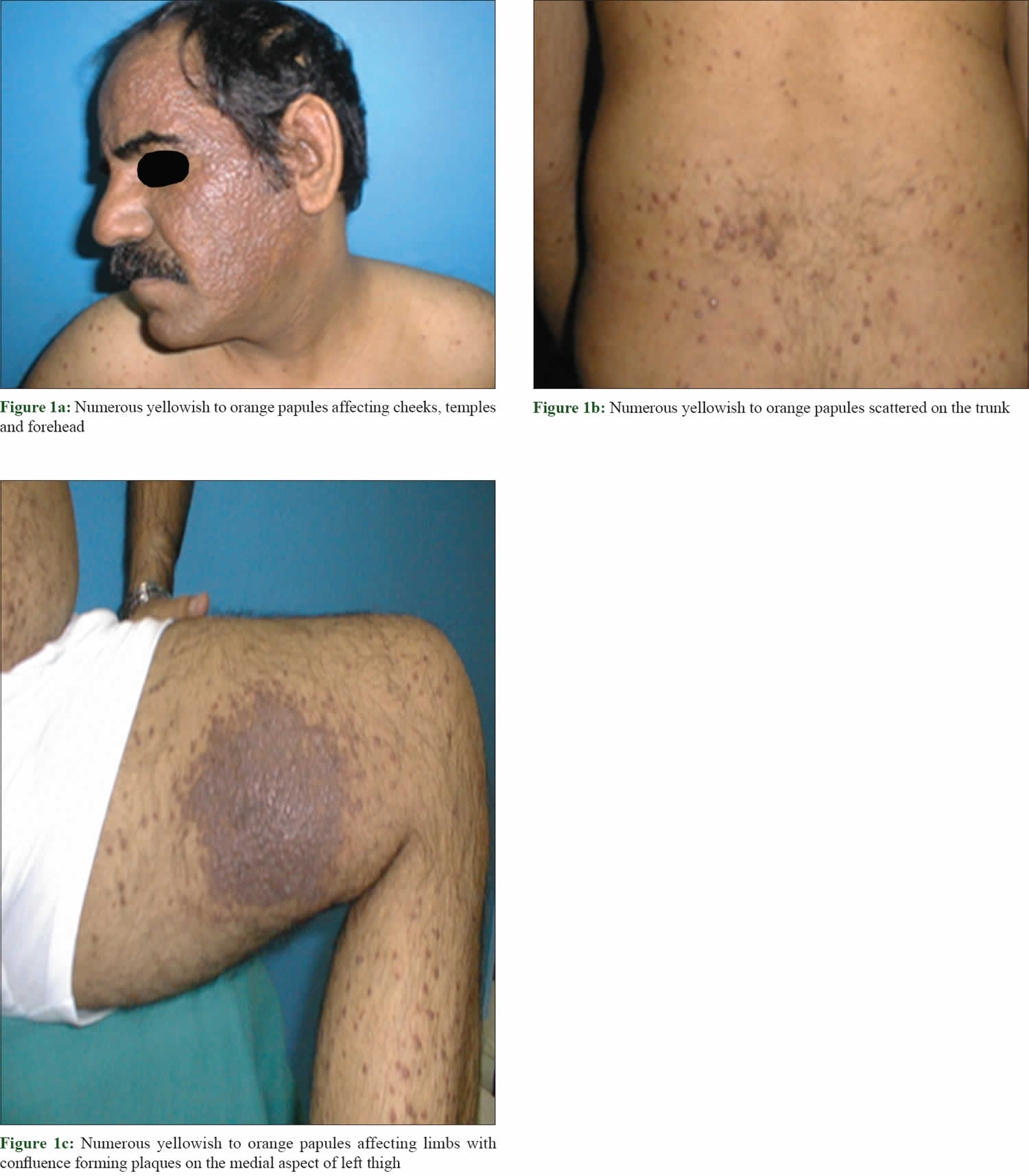
Footnote: A 46-year-old male reported appearance of asymptomatic widespread skin lesions 4 months ago. The lesions grew rapidly on the trunk and left thigh and later spread to involve other extremities and face within a span of 2 months. The patient had no related medical history and was otherwise healthy. The patient reported no systemic symptoms such as fever, malaise or weight loss. There was no organomegaly or lymph node enlargement. Cutaneous examination revealed innumerable generalized papules mainly affecting the face and limbs. The lesions were yellowish to orange papules with confluence forming non infiltrated plaques mainly on the thighs, cheeks, temples and peri-auricular area. There was no mucosal or nail involvement.
[Source 28 ]Figure 5. Rosai-Dorfman disease in the brain
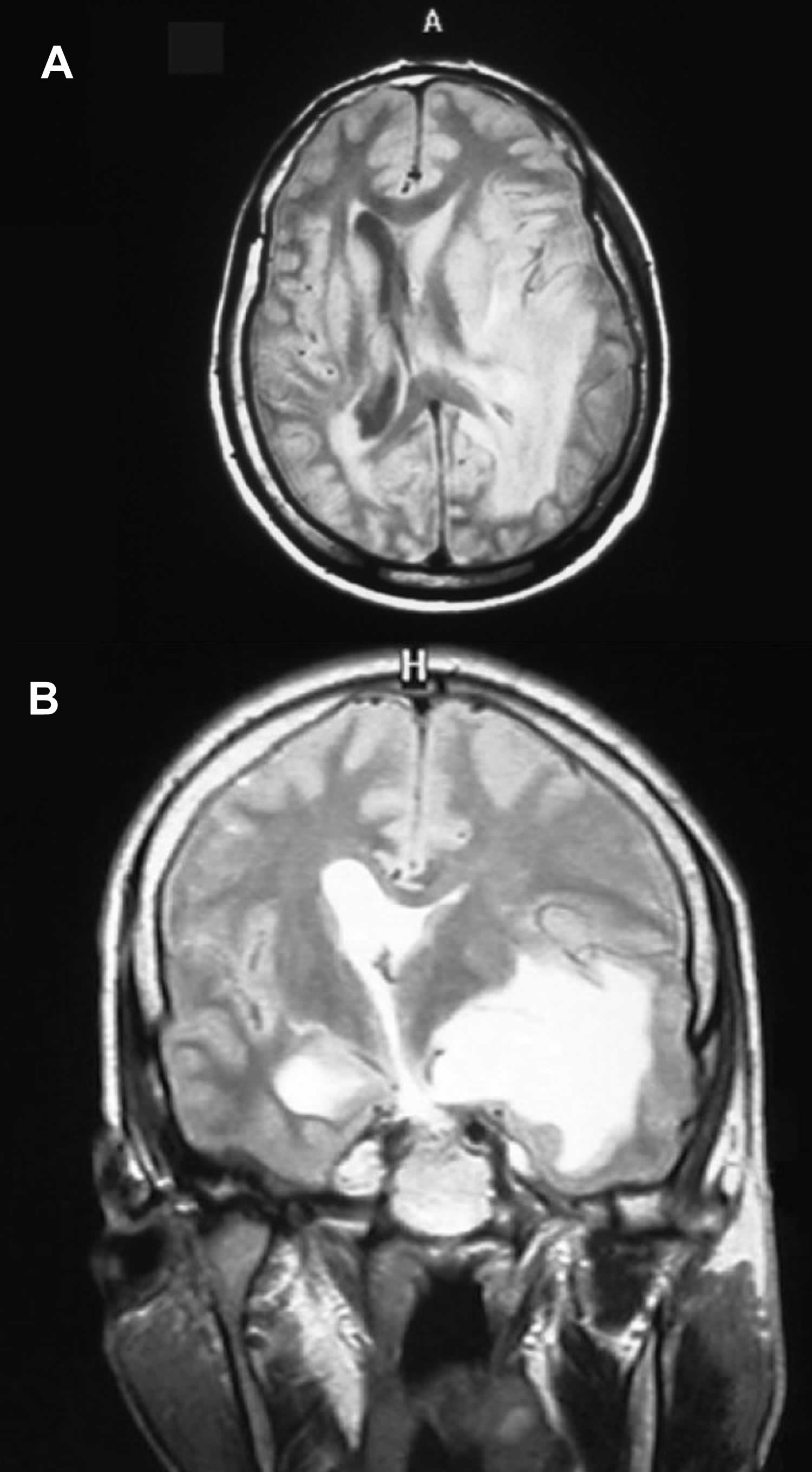
Footnotes: A 33-years-old man was suffering from fainting, frontal headache, ataxia and dizziness with no sensory or motor defect. MRI showed infiltrating mass adjacent to lateral ventricle with effacement of anatomical sign of the left parietal region. (A) Left periventricular mass that occupy temporal white matter and extend to adjacent gray matter. Left frontal horn was collapsed and left temporal horn was dilated. (B) There is enhancing mass that shows subfalcine hernia to right side. No definite lesion was found at the dura. Laboratory tests were unremarkable.
[Source 19 ]Figure 6. Rosai-Dorfman disease pathology outlines
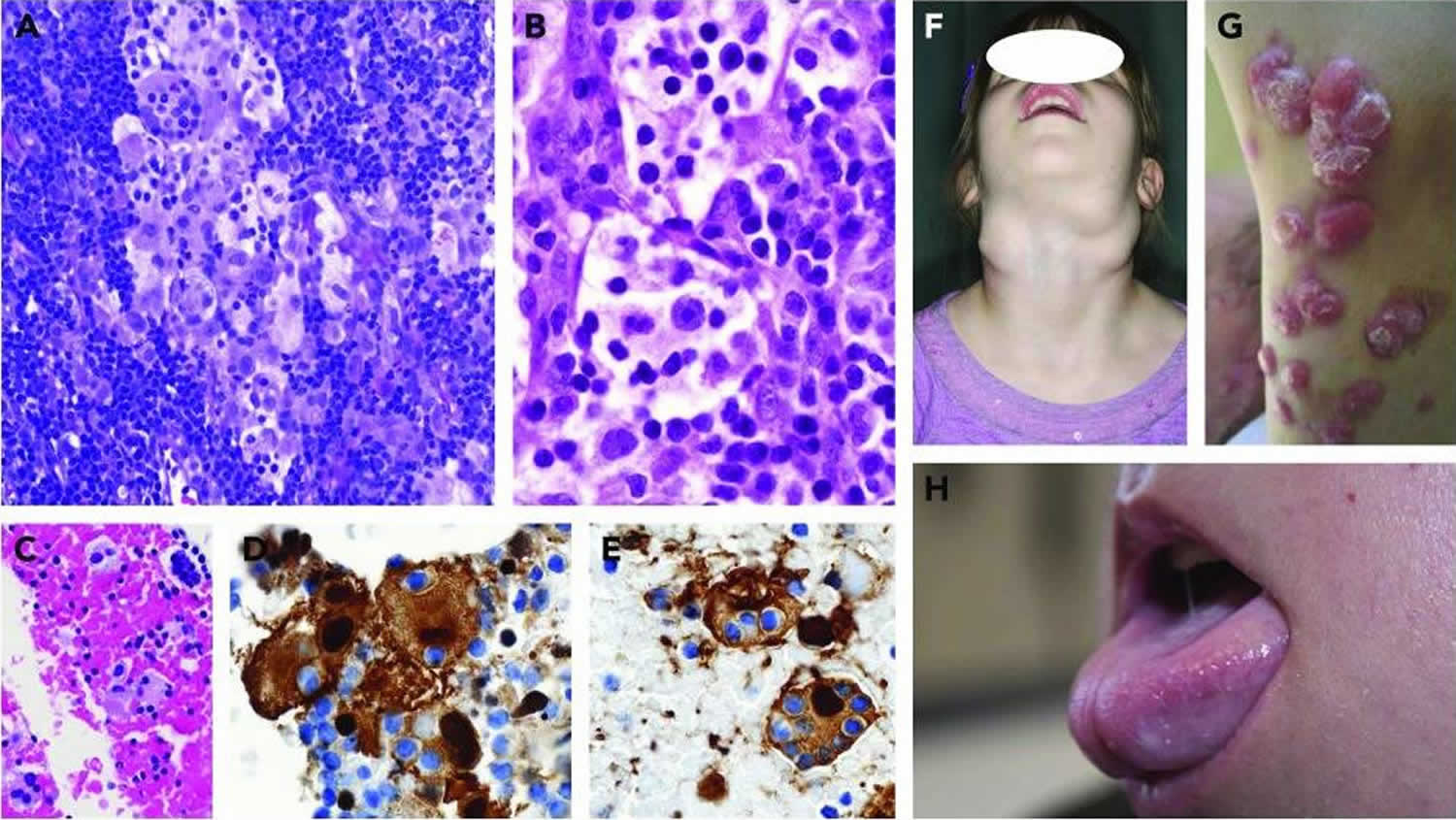
Footnotes: Pathologic and clinical features of Rosai-Dorfman disease. (A-E) Images of Rosai-Dorfman nodal disease and lymph node tissue biopsies (A-B) and fine-needle aspiration (C-E). (A) Mixed Rosai-Dorfman disease/Langerhans cell histiocytosis (LCH) case with sinus expansion. The large Rosai-Dorfman disease histiocytes display conspicuous emperipolesis with pale cytoplasm, as compared with the intermixed LCH cells with dense eosinophilic cytoplasm and convoluted nuclei (magnification ×400; hematoxylin and eosin [H&E] stain). (B) The Rosai-Dorfman disease histiocytes show pale watery-clear cytoplasm, a central round nucleus with a conspicuous nucleolus, and emperipolesis (magnification ×1000; H&E stain). (C) Cell block preparation shows clusters of Rosai-Dorfman disease histiocytes (magnification ×400; H&E stain), (D) with nuclear and cytoplasmic staining for S100 (magnification ×1000) and (E) fascin (magnification ×1000); the trafficking intact leukocytes are negative. (F) A child with immunodeficiency and Rosai-Dorfman disease with massive cervical lymphadenopathy. (G) Rosai-Dorfman disease of the skin showing red nodular lesions. (H) Tongue enlargement resulting from oral Rosai-Dorfman disease.
[Source 1 ]What is histiocytosis?
Histiocytosis is a general name for a group of disorders or “syndromes” that involve an abnormal increase in the number of specialized white blood cells that are called histiocytes 29. The term histiocyte refers to a bone marrow derived progenitor cell that differentiates, depending on the cytokine and growth factor milieu, into a dendritic antigen presenting cell (Langerhans cell, dermal dendrocyte) or a phagocytic cell (tissue macrophage) 30.
Recently, new knowledge about this family of diseases has led experts to develop a new classification. Five categories have been proposed 31, 32:
- L group — includes Langerhans cell histiocytosis (LCH), Erdheim-Chester disease, Indeterminate-cell histiocytosis and mixed Langerhans cell histiocytosis/Erdheim-Chester disease
- C group — includes non-Langerhans cell histiocytosis that involves the skin
- Cutaneous non-Langerhans cell histiocytosis
- Xanthomatous granuloma (XG) family: juvenile xanthogranuloma, adult xanthogranuloma, solitary reticulohistiocytoma, benign cephalic histiocytosis, generalized eruptive histiocytosis, progressive nodular histiocytosis
- Non-Xanthomatous granuloma (non-XG) family: Cutaneous Rosai-Dorfman disease, necrobiotic xanthogranuloma, other
- Cutaneous non-Langerhans cell histiocytosis with a major systemic component
- Xanthomatous granuloma (XG) family: xanthoma disseminatum
- Non-Xanthomatous granuloma (non-XG) family: multicentric reticulohistiocytosis
- Cutaneous non-Langerhans cell histiocytosis
- M group — includes Primary malignant histiocytoses and Secondary malignant histiocytoses
- R group — includes Rosai-Dorfman disease
- Familial Rosai-Dorfman disease
- Sporadic Rosai-Dorfman disease
- Classical Rosai-Dorfman disease
- Extranodal Rosai-Dorfman disease
- Rosai-Dorfman disease with neoplasia or immune disease
- Unclassified
- H Group — includes hemophagocytic lymphohistiocytosis
- Primary hemophagocytic lymphohistiocytosis : Monogenic inherited conditions leading to hemophagocytic lymphohistiocytosis
- Secondary hemophagocytic lymphohistiocytosis (non-Mendelian hemophagocytic lymphohistiocytosis)
- Hemophagocytic lymphohistiocytosis of unknown/uncertain origin.
Rosai-Dorfman disease types
Under the R group of non-Langerhans cell histiocytosis, Rosai-Dorfman disease can be further classified into five subgroups: classical (nodal), familial, extranodal, neoplasia associated, and immune disease-associated Rosai-Dorfman disease 32.
- Familial Rosai-Dorfman disease
- Sporadic Rosai-Dorfman disease
- Classical (nodal) Rosai-Dorfman disease
- The classical (nodal) Rosai-Dorfman disease subgroup includes IgG4 lymphoproliferative disorder or those without IgG4 syndrome.
- Extranodal Rosai-Dorfman disease
- Bone Rosai-Dorfman disease,
- Central nervous system (CNS) Rosai-Dorfman disease with or without IgG4 syndrome,
- Single-organ: The single-organ Rosai-Dorfman disease does not include lymph nodes or central nervous system (CNS) Rosai-Dorfman disease and is further divided into two subtypes: with or without IgG4 syndrome.
- Disseminated Rosai-Dorfman disease
- Rosai-Dorfman disease with neoplasia or immune disease
- Unclassified
- Classical (nodal) Rosai-Dorfman disease
- Of note, cutaneous Rosai-Dorfman disease has been reclassified to be included in the C group under the Non-Xanthomatous granuloma (non-XG) family 32.
Familial Rosai-Dorfman disease
Germ line mutations in SLC29A3 have been reported in patients with familial Rosai-Dorfman disease 33, 1. Two mutations have been reported to date: p.G427S and p.G437R 5. The spectrum of diseases associated with a mutation in the SLC29A3 gene mainly include the following three diseases:
- Familial or Faisalabad histiocytosis: Children present with sensorineural deafness and joint contractures. It is autosomal recessive disorder 33, 34. Histologically, it resembles Rosai-Dorfman disease; therefore, obtaining a clinical history along with a genetic consultation is an important step;
- H syndrome: Children present with hypogonadism, indurated, hyperpigmented, hypertrichotic skin plaques, hepatomegaly, cardiac abnormalities, and hearing loss 35, 36. Skin lesions share histologic features with Rosai-Dorfman disease;
- Pigmented hypertrichotic dermatosis with insulin-dependent diabetes syndrome: Children present with insulin-dependent diabetes mellitus and pigmented hypertrichosis 35.
All these diseases are described as histiocytosis-lymphadenopathy plus syndrome. A recent study confirmed that, despite sharing a missense mutation c.1088 G > A [p.Arg363Gln] of the SLC29A3 gene, the clinical phenotype of the intrafamilial SLC29A3 disorders could be heterogeneous 37.
Another cause of familial Rosai-Dorfman disease is heterozygous germline mutations in the FAS and TNFRSF genes, which also cause autoimmune lymphoproliferative syndrome (ALPS) type 1 38, 39. Patients with autoimmune lymphoproliferative syndrome (ALPS) tend to have a male predominance, more aggressive disease behavior, and an earlier age of disease onset, but the Rosai-Dorfman disease-like changes are usually self-limited 40.
Sporadic Rosai-Dorfman disease
Neoplasia-associated Rosai-Dorfman disease
Histologic features of Rosai-Dorfman disease have been observed in patients with Hodgkin lymphoma and non-Hodgkin lymphoma (NHL), where lymphoma and Rosai-Dorfman disease can either precede or follow each other or occur in the same lymph node 41. Rosai-Dorfman disease was also reported after myelodysplastic syndrome 42 or bone marrow transplantation for acute leukemia 43, concurrent with cutaneous clear-cell sarcoma 44 and concurrent with or following L-group histiocytoses or malignant histiocytoses 45. Small foci of Rosai-Dorfman disease-like histopathology (ie, histiocytes with emperipolesis) are nonspecific; therefore, >10% of a specimen should demonstrate Rosai-Dorfman disease morphology to constitute a neoplasia-associated Rosai-Dorfman disease as a distinct entity rather than a reactive process 1.
Rosai-Dorfman disease coexists with an immunologic disease in 10% of cases 12. Rosai-Dorfman disease has been associated with systemic lupus erythematous (SLE), idiopathic juvenile arthritis, autoimmune hemolytic anemia 46 and 1 case of RAS-associated autoimmune leukoproliferative disease, which is caused by gain-of-function mutations in the RAS-family (NRAS and KRAS) 47.
Some forms of extranodal Rosai-Dorfman disease, such as those involving the liver, lungs, or colon, have been associated with an increased number of immunoglobulin G4-positive (IgG4+) plasma cells 48, although other studies have shown a low number of IgG4+ plasma cells and low IgG4/IgG ratios (<40%) as compared with IgG4-related disease samples 49. No clear evidence suggests that the 2 disorders share the same pathogenesis; however, the most recent classification of histiocytoses recommends evaluating the IgG4/IgG ratio in all patients with Rosai-Dorfman disease 32.
Rosai-Dorfman disease cause
The exact cause of Rosai-Dorfman disease is unknown (idiopathic), but it does not seem to be of neoplastic nature 50. Over the years there have been many theories attempting to explain Rosai-Dorfman disease’s pathogenesis, but each of them has led to conflicting results. Researchers have suggested Rosai-Dorfman disease may be caused by an infectious agent, immunodeficiency, or autoimmunity. Studies have associated Rosai-Dorfman-Destombes disease with viral infections such as human herpesvirus 6 (HHV-6), human herpesvirus-8 (HHV-8), parvovirus B19, Epstein-Barr virus (EBV), cytomegalovirus (CMV), HIV, varicella zoster virus (also known as human herpesvirus 3), Brucella, and Klebsiella infection 51, 52, 53, 54, 55, although a clear link has not been proven 1. In some cases, the human herpesvirus 6 (HHV-6) antigen has been identified in the pathological histiocytes, while Epstein-Barr virus (EBV) and parvovirus B19 have been present in lymphocytes, which may eventually be phagocytosed by histiocytes 18. In one of the articles, it was observed that primarily infected lymphocytes induce a secondary histiocytic reaction that eventually causes the characteristic microscopic image of Rosai-Dorfman disease 56.
Other potential causes may be broadly-defined immunological disorders (e.g. immunodeficiency, autoimmune diseases). In view of the fact that some patients have hypergammaglobulinemia, scientists have been looking for a relationship between Rosai-Dorfman disease and IgG4-related diseases, but detailed analyses have put this theory into question 18, 57, 49.
One of the more recent studies considers the coexistence of Langerhans cell histiocytosis and Rosai-Dorfman disease 58, 59, 60. Although under the World Health Organization (WHO) classification Langerhans cell histiocytosis and Rosai-Dorfman disease are separate, it has been emphasized that both diseases share the same bone marrow precursor cell, which later undergoes further differentiation 59. Common features of histiocytes in these two disorders are, i.a., positive reaction to S-100 staining, and expression of cathepsin D and E 59. There are reports of patients treated for Langerhans cell histiocytosis, in whom after chemotherapy Rosai-Dorfman disease is revealed. This may be caused by immunological abnormalities connected with one disease, eventually leading to the disclosure of another one, or, alternatively, exposure to systemic chemotherapy causes disturbed differentiation in cells. In one of the reported cases, the authors observed histiocytes with immunohistochemical phenotype typical for Langerhans cell histiocytosis (S100+, CD1a+, presence of Birbeck granules), which underwent changes during chemotherapy (S100+, CD1a+, no Birbeck granules), finally presenting a profile typical for Rosai-Dorfman disease (S100+, CD1a–) 59, 61. There are also patients diagnosed with Rosai-Dorfman disease during or after treatment for leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma 62, 41.
In light of the finding of recurrent BRAF-V600E mutations in Erdheim-Chester disease (ECD), twenty three Rosai-Dorfman-Destombes disease samples were analyzed and found to be BRAF-V600E wild type 63. Similarly, Chakraborty et al 64 did not identify any somatic alterations by whole-exome sequencing of 4 Rosai-Dorfman-Destombes disease cases. However, recent studies have found NRAS, KRAS, MAP2K1, and ARAF mutations in Rosai-Dorfman disease tissues (see Figure 7), raising the possibility of a clonal origin in some forms of Rosai Dorfman disease 1. Characteristic lesional histiocytes are positive for S100, CD68, and negative for CD1a. Further research is needed to investigate the cell of origin of neoplastic forms of Rosai-Dorfman-Destombes disease 65.
Scientists are also looking for genetic disorders that could be responsible for Rosai-Dorfman disease development. The literature refers to, i.a., SLC29A3 mutations and familial Rosai-Dorfman disease 18, 66, 67.
Table 1. Summary of kinase mutations discovered in Rosai-Dorfman disease
| Gene | Genetic alteration | Amino acid variant | Protein domain |
|---|---|---|---|
| ARAF | Missense | p.N217K | CR2 domain |
| MAP2K1 | Missense | p.F53V | N-terminal negative regulatory |
| MAP2K1 | Missense | p.L115V | N-terminal catalytic core of kinase |
| MAP2K1 | Missense | p.P124R | N-terminal catalytic core of kinase |
| MAP2K1 | Missense | p.G128D | N-terminal catalytic core of kinase |
| NRAS | Missense | p.G13D | P-loop and GTP-binding 1 |
| KRAS | Missense | p.G12D | P-loop and GTP-binding 1 |
| KRAS | Missense | p.G12R | P-loop and GTP-binding 1 |
| KRAS | Missense | p.Q22K | GTP-binding 1 |
| KRAS | Missense | p.K117N | GTP-binding 3 |
| KRAS | Missense | p.A146T | GTP-binding 4 |
| KRAS | Missense | p.A146V | GTP-binding 4 |
Figure 7. Rosai-Dorfman disease kinase mutations
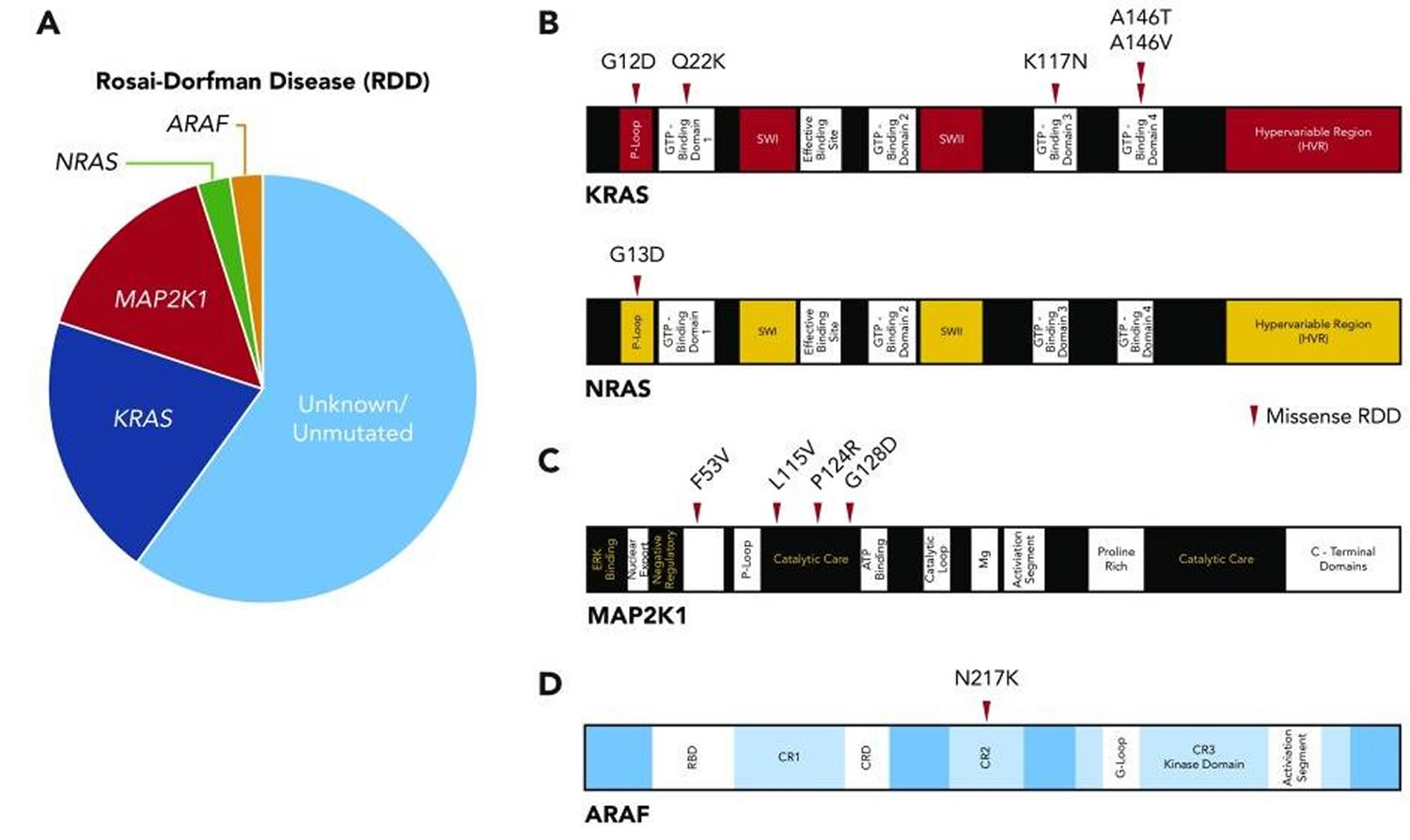
Footnotes: (A) Pie chart illustrating the known activating kinase mutations in Rosai-Dorfman disease (N = 34). (B) Diagrams of somatic mutations described in KRAS and NRAS. (C) Diagram of somatic mutations uncovered in MAP2K1. (D) Diagram of somatic mutation discovered in ARAF.
[Source 1 ]Rosai-Dorfman disease pathology outlines
The diagnostic pathologic features of nodal Rosai-Dorfman disease include the sinus expansion of large histiocytes, described by Destombes as possessing ample pale or “watery-clear” cytoplasm with a large hypochromatic nucleus and prominent nucleolus 1. Nodal Rosai-Dorfman disease is often accompanied by numerous plasma cells in the medullary cords and around the venules, with varying proportions of IgG4/IgG plasma cells 1, 68. Consistent features, regardless of the site, include the cytomorphology of the large pale histiocytes and their immunophenotype. Emperipolesis, the trafficking of intact leukocytes through the cytoplasm, is a helpful finding but is not required for diagnosis, because it can be focal, especially at extranodal sites, and may be seen focally in other histiocytoses such as Erdheim-Chester disease 69, juvenile xanthogranuloma, and malignant histiocytoses. Extranodal lesions are usually associated with more fibrosis, fewer Rosai-Dorfman disease histiocytes and less emperipolesis 1. In such cases, immunostains are needed to highlight the residual Rosai-Dorfman disease histiocytes in a rich lymphoplasmacytic background with stromal fibrosis and a variable xanthomatous histiocytic reaction 1. These extranodal cases can be difficult to differentiate from Erdheim-Chester disease and require clinical correlation.
The immunophenotype of the large Rosai-Dorfman disease histiocytes is characterized by cytoplasmic and nuclear S100 (Figure 6D) and fascin (Figure 6E) positivity, with CD68 and variable CD163 and CD14 positivity. The cells are CD1a negative/CD207 negative in contrast to Langerhans cell histiocytosis (LCH). An important aspect of making the diagnosis of nodal Rosai-Dorfman disease is evaluation for superimposed pathology, either within the node itself or other associated conditions 68. The presence of Rosai-Dorfman disease histology is required, but not sufficient, for the diagnosis of Rosai-Dorfman disease, which depends on the appropriate clinical and radiologic context and exclusion of primary malignant disorders in relation to which Rosai-Dorfman disease histology may represent a minor (<10%) reactive process.
Because the pathologic features of Rosai-Dorfman disease are variable, compounded by its heterogeneous clinical manifestations, in many instances the primary hurdle to establishing an Rosai-Dorfman disease diagnosis is suspecting the disorder in the first place, particularly for extranodal disease 1. The presence of slowly progressive or subacutely clinical symptomatology; biopsy specimens demonstrating ostensibly nonspecific inflammation, including lymphoid aggregates and plasma cells with a large histiocytic presence; and clinical findings compatible with those described in “clinical features” should raise concern for an Rosai-Dorfman disease diagnosis and, in difficult cases, prompt clinical and histopathologic evaluation by experts familiar with the disease.
Clinical features
Classic (nodal) Rosai-Dorfman disease
Most patients with Rosai-Dorfman disease present with bilateral, massive, and painless cervical lymphadenopathy (Figure 6F) with or without intermittent fevers, night sweats, and weight loss 70. Mediastinal, armpit, and inguinal lymph nodes may also be involved, but retroperitoneal lymphadenopathy is uncommon 71. Classic (nodal) Rosai-Dorfman disease prognosis has been found to correlate with the number of nodal groups involved by Rosai-Dorfman disease 12.
Extranodal Rosai-Dorfman disease
Extranodal involvement has been reported in 43% of Rosai-Dorfman disease cases 12. Extranodal Rosai-Dorfman disease usually occurring in the skin, nasal cavity, paranasal sinus, upper respiratory tract, salivary gland, head, and neck, central nervous system (CNS), soft tissues, bone, kidneys, testis, digestive system, eyelid, orbit, and salivary gland is present with or without lymphadenopathy 6, 12, 13, 14, 15. Multisystem involvement occurs in 19% of cases, and prognosis is correlated with the number of extranodal systems involved 12.
Cutaneous Rosai-Dorfman disease
Cutaneous Rosai-Dorfman disease was established as a separated clinical entity, in which only skin is involved – patients do not present lymphadenopathy. The skin is involved in 10% of extranodal Rosai-Dorfman disease cases, and isolated cutaneous disease is rare 70. Cutaneous Rosai-Dorfman disease usually affects individuals around the age of 50 years, predominantly women, especially from the Caucasian population. Lesions are typically slow growing, painless, nonpruritic nodules, plaques, or papules with coloration varying from yellow to red to brown (Figure 3 and 4). Any skin site can be affected. The differential diagnosis includes acne vulgaris, varicella-zoster virus, sarcoidosis, cutaneous lymphoma, and metastasis 70. Cutaneous Rosai-Dorfman disease is associated with an excellent prognosis 72, 18, 73.
Central nervous system Rosai-Dorfman disease
Central nervous system (CNS) involvement occurs in <5% of Rosai-Dorfman disease cases, with 75% occurring as intracranial and 25% as spinal lesions. Neurologic Rosai-Dorfman disease has been reported in >300 cases 74 and usually occurs in older patients and without lymphadenopathy 75. Symptoms include headaches, seizures, gait difficulty, motor or sensory abnormalities, and cranial nerve deficits, usually evolving over weeks or months 74. Familial cases are associated with damage to the auditory nerve pathway and deafness 67, 76. The most common radiographic appearance of intracranial Rosai-Dorfman disease is a solitary extraaxial, homogeneously enhancing dural mass mimicking a meningioma 77, 78, although Rosai-Dorfman disease can cause diffuse pachymeningitis. Parenchymal lesions are frequently infratentorial (brainstem and pons) 79, whereas supratentorial, intraventricular, and multifocal lesions are rare 80, 81. Cerebrospinal fluid (CSF) is often unremarkable although may show lymphocytic pleocytosis, elevated protein, low glucose, and emperipolesis 78.
Spinal dural or epidural lesions are most common in the cervical and thoracic regions 82 and present with myelopathy or symptoms of spinal cord compression. Although central nervous system (CNS) Rosai-Dorfman disease can have a rapidly progressive and even fatal course, many patients will have a favorable outcome after surgical resection when this is feasible 75.
Ophthalmic Rosai-Dorfman disease
Ophthalmic manifestations occur in 11% of Rosai-Dorfman disease cases 12, manifesting as a mass in the orbital soft tissues (Figure 2), eyelids, lacrimal glands, conjunctiva, or cornea and as uveitis or compressive optic neuropathy 83.
Head and neck Rosai-Dorfman disease
Involvement of the nasal cavity and paranasal sinuses occurs in 11% of Rosai-Dorfman disease cases 12 and is more common in patients of Asian descent. Symptoms of sinonasal Rosai-Dorfman disease include nasal obstruction, epistaxis, nasal dorsum deformity, facial asymmetry, and aural fullness 84. Oral cavity involvement can present as soft and hard palate nodules, gingival and oral mucosa swelling, tongue enlargement (Figure 6H), thickened mucosa of the oropharynx, enlarged tonsils, or frequent tonsillitis 12. Other less frequently involved sites include the salivary and parotid glands, larynx, pharynx, thymus, and thyroid gland, which can cause symptoms related to mass effect 85, 86.
Intrathoracic Rosai-Dorfman disease
Intrathoracic Rosai-Dorfman disease is described in 2% of patients, usually with concurrent lymphadenopathy 87. Manifestations include interstitial lung disease, pulmonary nodules, tracheobronchial disease, and pleural effusions with an obstructive pattern on pulmonary function tests 88. Symptoms include chronic dry cough, progressive dyspnea, or acute respiratory failure. Pulmonary Rosai-Dorfman disease can mimic primary lung cancers, interstitial lung disease or organizing pneumonias, sarcoidosis, granulomatous polyangiitis, rheumatoid arthritis–related lung disease, and mycobacterial and fungal infections 87. Rosai-Dorfman disease affecting the lower respiratory tract can have an aggressive phenotype, with a mortality rate of almost 45% 12. Cardiac involvement with Rosai-Dorfman disease is rare, occurring in 0.1% to 0.2% of cases 89.
Retroperitoneal and genitourinary Rosai-Dorfman disease
The kidneys are affected in 4% of Rosai-Dorfman disease cases, with a discrete mass or diffuse infiltration 90, 91. Symptoms include blood in urine (hematuria), flank pain, abdominal fullness, renal failure, hypercalcemia, or nephrotic syndrome caused by amyloidosis or renal vein thrombosis 92, 93, 94, 95. Hydronephrosis and ureteral obstruction can occur 96. Patients with renal involvement have a poor prognosis, with 40% mortality rate 12.
Testicular involvement is rare and manifests as a painful testicular or epididymal mass mimicking tumor or epididymitis 12, 97. Anecdotally, testicular Rosai-Dorfman disease can also present as diffuse enlargement and hardening of the testes, with or without pain.
Adrenal gland involvement is also possible but rare 98.
Gastrointestinal Rosai-Dorfman disease
Gastrointestinal (GI) involvement occurs in less than 1% of Rosai-Dorfman disease cases, most commonly in middle-age women with concurrent lymphadenopathy or other extranodal disease 12. Gastrointestinal Rosai-Dorfman disease can be solitary or segmental and has a predilection for the ileocecal area, appendix, and distal colon, with most cases being located beyond the pylorus 53, 99. Symptoms include passage of fresh blood per anus (hematochezia), constipation, abdominal pain, abdominal mass, and intestinal occlusion, although asymptomatic cases have been identified after colonoscopy or appendectomy 99, 100. Almost 20% of the reported patients in 1 series died as a result of disease 53. Pancreas or liver involvement is reported but is extremely rare 12, 101, 102.
Bone Rosai-Dorfman disease
Bone involvement occurs in 5% to 10% of Rosai-Dorfman disease cases, typically in association with nodal disease 103. Bone pain is common, whereas pathologic fractures are rare 92. Bone lesions typically occur in the metaphysis or diaphysis, are osteolytic or mixed lytic/sclerotic, and have a narrow zone of transition. Soft tissue extension can occur. The clinical differential diagnosis includes chronic osteomyelitis, fibrous dysplasia, lymphoma, and Ewing sarcoma. Lesions in the femurs and tibia should raise concern for Erdheim-Chester disease. The prognosis of bony Rosai-Dorfman disease is generally good 104.
Hematologic Rosai-Dorfman disease
Normochromic normocytic anemia (in 67% of cases), leukocytosis (in 60%, typically neutrophilia), thrombocytopenia, eosinophilia, hypergammaglobulinemia, and elevated erythrocyte sedimentation rate (ESR) are common, although bone marrow infiltration is rare 38, 105.
Rosai-Dorfman disease symptoms
The symptoms and physical findings associated with Rosai-Dorfman disease vary greatly from one person to another depending upon the extent of the disorder and the specific organ systems affected 72. Some Rosai-Dorfman disease cases may only affect the lymph nodes and may not cause any serious complications. Less often, some Rosai-Dorfman disease cases may affect various organ systems of the body and may potentially cause serious complications. Any organ system of the body may become affected.
In most Rosai-Dorfman disease cases, affected individuals exhibit painless swelling or enlargement of affected lymph nodes (lymphadenopathy), most often those of the neck (cervical lymphadenopathy) 12. Other locations such as inguinal (26%), axillary (24%) and mediastinal lymph nodes (15%) are also reported to be involved 12. Many individuals with Rosai-Dorfman disease do not develop any additional symptoms of the disorder (asymptomatic).
In some cases, affected individual may experience nonspecific symptoms that are common to many different conditions including fever, paleness of the skin (pallor), unintended weight loss, a general feeling of ill health (malaise) and a chronically runny nose (rhinitis). In extremely rare cases, affected individuals may experience abnormal enlargement of the liver and/or spleen (hepatosplenomegaly).
In approximately 43 percent of patients, other areas of the body besides the lymph nodes may also be affected (extranodal disease). Some individuals may have extranodal disease without the presence of lymphadenopathy 12. The skin is the most common extranodal site. Skin lesions associated with Rosai-Dorfman disease are usually yellow or purple. A reddish rash-like inflammation of the skin (erythema), small solid elevations on the skin (papules), or knots visible under the skin (nodules) may be present. Skin lesions may occur anywhere on the body, but most often affect the head and neck. In some cases, skin abnormalities precede the development of lymphadenopathy.
The salivary glands, nasal cavity, upper respiratory tract, various bones, and the eyes and eye sockets (orbits) may also be affected. In rare cases, the central nervous system (CNS), digestive system, or the kidneys may be affected. Hepatosplenomegaly is uncommon 12. Central nervous system (brain and spinal cord) can be involved less than 5% of Rosai-dorfman disease 106, 107. In 70% of central nervous system-Rosai-Dorfman disease, the presentation is limited to the brain or to the spinal cord and is not associated with lymphadenopathy 108. Approximately, 75% of central nervous system (CNS) afflictions are intracranial, whereas 25% involve the spine 108. Over 90% of central nervous system Rosai-Dorfman disease leptomeninges are only involved 109. Involvement of these areas may result in additional (secondary) symptoms. For example, loss of vision may occur secondary to the involvement of the eyes, and seizures, headache, ataxia, motor or sensory abnormalities, dizziness and cranial nerve deficits may occur secondary to involvement of the central nervous system (CNS) 74. In some cases of Rosai-Dorfman disease, the accumulation of histiocytes into masses may cause compression of vital organs potentially resulting in serious complications 106, 110, 111, 112, 113, 114, 21.
In some cases, the lymph nodes may not be affected. Instead a specific area of the body such as the skin, a solitary bone, or the central nervous system may be affected. These cases may be known as isolated Rosai-Dorfman disease. A distinct type of isolated Rosai-Dorfman disease has been identified called cutaneous Rosai-Dorfman disease, in which only the skin is affected. Initial reports indicate that cutaneous Rosai-Dorfman disease is more common in female adults.
Table 2. Rosai-Dorfman disease signs and symptoms
| Site | Incidence | Symptoms/Signs | Radiologic Findings |
|---|---|---|---|
| Nodal | |||
| Lymph node | 57% of cases | Bilateral cervical lymphadenopathy or other lymph node sites, manifested with palpable masses | Enlarged lymph nodes |
| Extranodal | |||
| Skin | 10% of cases | Painless, macular, slow-growing papules, subcutaneous nodules. Any skin site can be affected. | Nodule(s) or mass(es) |
| Central nervous system (CNS) | <5% of cases (75% intracranial and 25% spinal lesions), more than 300 cases have been reported. Cervical and thoracic regions are the most common areas affected in spinal-dural or epidural lesion | Headaches, seizures, gait difficulty. In familial cases, there is an association with damage to the auditory nerve pathway and deafness. | Dural lesion, extra-axial, homogeneously enhancing, mimicking nodular meningioma; or parenchymal (infratentorial) involvement |
| Orbit | 11% of cases | Presents as a mass in different part of the orbit, e.g., conjunctiva, lacrimal glands, and cornea. It can also present as uveitis. | Orbital mass |
| Head and neck | 11% of cases involving nasal cavity, more common among Asians | Nasal obstruction, epistaxis, and nasal dorsum deformity | Nodules, swelling, mass(es) |
| Intrathoracic | 2% of patients, with pulmonary disease concurrent lymphadenopathy or systemic disease. Cardiac involvement is extremely rare ~0.1–0.2% of cases. | Chronic dry cough, progressive dyspnea, or acute respiratory failure. Rosai-Dorfman disease affecting lower respiratory tract have a high mortality rate, reaching 45%. | Pulmonary nodular consolidation in all lobes of the lungs; pleural effusion with fibrosis or nodules |
| Retroperitoneal /genitourinary tract | Kidneys are affected in approximately 4% of cases. | Abdominal or flank pain, fullness, hematuria, renal failure, hypercalcemia, and/or nephrotic syndrome | Mass(es), hydronephrosis, urethral obstruction |
| Gastrointestinal tract | <1% of cases, commonly in middle-aged women with concurrent nodal or extranodal affection | Abdominal pain, constipation, hematochezia, and intestinal obstruction | Mass(es) |
| Bone | 5% to 10% of cases, usually with concurrent nodal affection | Bone pain and, rarely, pathologic fractures | Cortex-based osteolytic lesion, commonly long bones, vertebrae, and sacrum |
Rosai-Dorfman disease diagnosis
The diagnostic and staging evaluation of patients with newly diagnosed Rosai-Dorfman disease should include an assessment of disease extent, as well as evaluation for conditions either known to be associated with Rosai-Dorfman disease, particularly autoimmune disorders, or known to contain an Rosai-Dorfman disease-like reactive component secondary to malignancies (Table 3).
According to the international expert consensus at the 32nd Histiocyte Society Meeting in 2018 and National Comprehensive Cancer Network (NCCN) Guidelines for Histiocytic Neoplasms 2020, A comprehensive medical history and physical and neurologic examinations should be performed, including head and neck, intrathoracic/pulmonary/cardiovascular, gastrointestinal (GI), renal, genitourinary (GU) system, neuroendocrine, central nervous system (CNS), and cutaneous symptoms 116.
The following recommendations are used for baseline evaluation of new/suspected cases of Rosai-Dorfman disease 116, 1:
- Medical history
- Constitutional symptoms;
- Organ affection (head, eyes, ears, nose, and throat; cardiovascular; pulmonary; gastrointestinal (GI); genitourinary (GU); skin; central nervous system (CNS); and endocrine);
- History of autoimmune disease, Langerhans cell histiocytosis (LCH), or other histiocytic lesions, as well as hematologic malignancies;
- Family history for children.
- Physical examination
- Lymphadenopathy;
- Organomegaly;
- Cutaneous and extranodal lesions;
- Neurologic changes.
- Radiological evaluation
- All patients should have whole-body positron emission tomography (PET)/computed tomography (CT);
- Selected patients should have CT sinuses with contrast, high-resolution CT chest, magnetic resonance imaging of orbit/brain with contrast and magnetic resonance imaging of spine with contrast;
- Selected patients for organ-specific ultrasound.
- Laboratory evaluation
- Complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate;
- Serum immunoglobulins;
- Coagulation studies, C-reactive protein, uric acid, and lactate dehydrogenase (LDH);
- Hemolysis panel (Coombs, haptoglobin, reticulocytes, and blood smear);
- Panel for autoimmune diseases (autoimmune lymphoproliferative syndrome (ALPS) panel, antinuclear antibody, rheumatoid factor, HLA B27);
- Next-generation sequencing (NGS) targeted gene mutations in RAF-RAS-MEK-ERK pathway;
- If familial form is suspected, then NGS test for SLC29A3 (germline mutations);
- Bone-marrow biopsy (if cytopenia or abnormal peripheral blood smear are present);
- Lumbar puncture for CNS involvement.
- Subspecialty consultation as needed
- Dermatology and ophthalmology evaluation before initiating MEK-inhibitor therapy.
Rosai-Dorfman disease diagnosis is made by a biopsy. Grossly, the excised lymph node is usually very large and is often matted to overlying soft tissues or adjacent lymph nodes. The lymph node architecture is dominated by a striking sinus expansion, filled with characteristic histiocyte-like cells. The cytoplasm of these histiocyte-like cells often contains phagocytosed intact small lymphocytes (lymphophagocytosis) or a variety of cells, including red blood cells, neutrophils, or plasma cells (emperipolesis). Typically, there are multiple ingested cells forming a partial or complete ring around the nuclei. Late lesions may show extensive fibrosis. Immunohistochemical studies show that the proliferating cells express the histiocytic markers CD163 and CD68 as well as S-100 protein. They are negative for the Langerhans cell marker CD1a as well as the dendritic markers CD21 and CD35. They lack almost all lymphoid markers, although they may express CD30 in up to 50% of cases and show a germline configuration of their antigen receptor genes. They lack Birbeck granules on ultrastructural examination.
Although Rosai-Dorfman disease has many prominent features, which can help to distinguish this entity from other histiocytic disorders, certain features are nonspecific, e.g., emperipolesis can be identified in Erdheim-Chester disease, juvenile xanogranuloma, and malignant histiocytosis 1. In addition to S100, histiocytic markers (Fascin, CD68, CD163, CD4, CD14) positive for Rosai-Dorfman disease can be seen with numerous histiocytic disorders, especially when Rosai-Dorfman disease concurs with other histiocytic lesions. Differentiation between Rosai-Dorfman disease and other histiocytic disorders is sometimes challenging. For nodal Rosai-Dorfman disease, differential diagnoses mainly include Langerhans cell histiocytosis, Erdheim-Chester disease, and ALK1-positive histiocytosis and infection or other malignancies associated with reactive histiocytosis.
Table 3. Baseline clinical evaluation for patients with Rosai-Dorfman disease
| Recommendation |
|---|
| Medical history |
|
|
|
|
|
|
|
|
|
|
autoimmune lymphoproliferative syndrome (ALPS), malignancy, Langerhans cell histiocytosis (LCH), or another histiocytic disorder |
|
| Physical examination |
|
|
|
|
|
|
|
|
| Radiological evaluation |
| All patients |
|
|
| Selected patients based on symptoms or organ involvement |
|
|
|
|
|
|
|
| Laboratory evaluation |
|
|
|
|
|
|
|
|
|
|
|
Abbreviations: CSF = cerebrospinal fluid; LDH = lactate dehydrogenase
[Source 1 ]Imaging studies
In children, a chest X-ray with neck and abdominal ultrasounds are routinely performed initially. For older patients, CT of the neck/chest/abdomen and pelvis is recommended. Rosai-Dorfman disease lesions are known to be FDG-avid, including extranodal areas 117 and FDG-PET/CT is used by some investigators for initial staging when possible, similarly to patients with Erdheim-Chester disease 69. Of note, Rosai-Dorfman disease lesions can have appearance and avidity on FDG-PET similar to those of intermediate- and high-grade lymphomas, and these should be diagnostic considerations. Rather than skull-to-thigh scanning, full-body PET including the distal extremities should be performed for comprehensive osseous evaluation. However, there is no consensus among the authors about the benefits of PET/CT in patients with Rosai-Dorfman disease as compared with anatomic imaging 1. In children, efforts must be made to minimize radiation exposure and need for anesthesia. Whole-body MRI is usually recommended instead of CT scans, and PET scans should be used judiciously 1. Patients with orbital or neurologic symptoms should have a gadolinium-enhanced MRI of the brain, orbits, or total spine depending on the localizing symptoms; screening MRI of the brain and spine with contrast may be appropriate to identify asymptomatic neurologic involvement 1. Dedicated organ-specific imaging (ie, MRI of the heart or abdomen) may be necessary to evaluate structural lesions identified but poorly characterized by CT or PET 1.
Laboratory tests
Laboratory tests should include comprehensive metabolic panel, a complete blood count with differential, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and quantitative immunoglobulin levels. Serologies for HIV and hepatitis B and C are suggested to exclude these as associated diagnoses. Testing for antinuclear antibodies (ANA) and rheumatoid factor (RF) is suggested, because screening for associated systemic lupus erythematous (SLE) or idiopathic juvenile arthritis and further evaluation for evidence of autoimmunity should be carried out if other diagnoses are considered on the basis of medical history and physical examination. Bone marrow aspirate and biopsy are required only for patients with unexplained cytopenias or abnormal peripheral blood cells.
Laboratory evaluation of Rosai-Dorfman disease patients shows approximately 2/3 of patients present with normochromic normocytic anemia, leukocytosis (neutrophilia most commonly) 5. Poly-clonal hypergammaglobulinemia was reported in approximately 90% of patients; increased erythrocyte sedimentation rate in approximately 90%; and hypoalbuminemia in approximately 60%. The CD4:CD8 ratio was found to be decreased. Other laboratory findings are present, such as elevated ferritin level and autoimmune hemolytic anemia 12.
Tissue biopsy
It is important that sufficient tissue be acquired to establish a Rosai-Dorfman disease diagnosis and that biopsies be reviewed by a pathologist familiar with Rosai-Dorfman disease 1. Flow cytometry and cytogenetic testing may be required to rule out a lymphoproliferative disorder. Immunohistochemistry for IgG4 should be performed when pathology samples show an enriched plasma-cell presence 1. In the event of severe or refractory Rosai-Dorfman disease, lesional tissue should be analyzed to detect gain-of-function mutations of genes of the MAPK pathway amenable to targeted therapies (including at least KRAS, NRAS, HRAS, ARAF, BRAF, and MAP2K1) 1.
Histopathology
Histologic examination shows enlarged, matted, grossly involved lymph nodes and capsular fibrosis. Under microscopic examination, lymph nodes have a thickened capsule, abnormal architecture, and massive sinus expansion. Sinuses are obstructed by a mixed population of cells, including histiocytes, lymphocytes, plasma cells, and Rosai-Dorfman disease histiocytes. Rosai-Dorfman disease histiocytes are usually large with round-to-oval nuclei, dispersed chromatin, prominent nucleoli, and abundant clear-to-foamy or vacuolated cytoplasm. The hallmark of Rosai-Dorfman disease histiocytes is lymphophagocytosis or emperipolesis – the phagocytosis of a variety of intact cells, including red blood cells, neutrophils, or plasma cells 58. During the process of emperipolesis, Rosai-Dorfman disease histiocytes engulf intact cells and sometimes nuclear debris and lipids. The engulfed cells remain viable and can exit histiocytes in contrast to the process of phagocytosis (Figure 7). These findings are observed in both nodal and extranodal sites; however, there is a greater degree of fibrosis and Rosai-Dorfman disease histiocytes with less frequent or absent emperipolesis on extranodal sites. Extranodal Rosai-Dorfman disease also appears more frequently to be fibrosis and less frequently to be histiocytosis. Rosai-Dorfman disease is often associated with abundant plasma cells in the medullary cords and around the venules 22.
Fine-needle aspiration–smears and touch imprints are typically highly cellular with many histiocytes and engulfed lymphocytes (emperipolesis) against a background of mixed inflammatory cells, including plasma cells and lymphocytes 5. Histiocytes tend to be large with abundant cytoplasm and a round, vesicular nucleus with a small central nucleolus. In smear and imprint preparations, there tends to be a diagnostic dilemma; overlapping lymphocytes can be mistaken for emperipolesis as engulfed lymphocytes do not appear surrounded by a halo, as seen in tissue sections. In later stages of Rosai-Dorfman disease, there tends to be increased plasma cells and cytoplasmic immunoglobulin inclusion (Russell bodies) 5.
Immunohistochemical stains are crucial in confirming a diagnosis. The typical Rosai-Dorfman disease histiocytes express S100+, CD4+, CD11c+, CD14+, CD68+, CD163+, α-1-antitripsin+, α-1-antichymotripsin+, and CD1a negative 118, 119, 25, 58, 18. Uniquely, in Rosai-Dorfman disease, the histiocytes express S100, which is a useful feature for visualizing the emperipolesis, as first described in a single case by Aoyama et al. 120 and confirmed in a larger series by Miettinen et al. 121 (see Figure 8). Rosai-Dorfman disease is usually negative for pan B- or T-cell antigens, markers for Langerhans cells (CD1a and langerin/CD207), and follicular dendritic cell markers (CD21, CD23, CD35, and clusterin) 5. Rosai-Dorfman disease histiocytes are also reactive to α1-antichymotrypsin and α1-antitrypsin, which might suggest lysosomal activity 5.
Recent studies have shown that 1/3 of Rosai-Dorfman disease cases harboring mutations in the MAPK/ERK pathway that were found to be gain-of-function mutations leading to the upregulation of p-ERK and cyclin D1/BCL-1 in the histiocytes 122, 123, 124. Cyclin D1/BCL-1, a key cell-cycle regulator, represents a major downstream target of the MAPK/ERK pathway. Expression that is positive for Cyclin D1/BCL-1 can be associated with phosphorylated -ERK (p-ERK), reflecting the constitutive activation of MAPK pathway 123. However, some cases show cyclin D1 upregulation and absence of p-ERK expression; hence, cyclin D1 might be regulated by other oncogenic mechanisms bypassing the ERK pathway 124. Positive cyclin D1/BCL-1 staining in Rosai-Dorfman disease is not associated with an underlying translocation of the CCND1 gene. Be cautious: some reactive histiocytes also express cyclin D1/BCL-1; however, this is usually dimly expressed. The marker becomes less specific for Rosai-Dorfman disease if it is weakly expressed on histiocytes 125, 126. Besides cyclin D1/bcl-1, the Mayo group study showed a subset of Rosai-Dorfman disease cases also expressed p16 (64%), Factor XIIIa (30%) and phosphorylated extracellular signal-related kinase (45%) 127. The latter two parameters appeared to be associated with multifocality of Rosai-Dorfman disease 127. As Factor XIIIa is also seen with Erdheim-Chester disease, this should be interpreted with caution.
Another marker that was frequently expressed in many Rosai-Dorfman disease cases is BCL-2. Since Rosai-Dorfman disease cases usually have low Ki-67 proliferation index, the expression of BCL-2 might be caused by the activation of the anti-apoptotic process 124, 127. OCT2, a unique monocyte-macrophage marker, has also been found to be expressed in most cases of Rosai-Dorfman disease, in contrast to other histiocytic disorders, such as Langerhans cell histiocytosis and Erdheim-Chester disease 127. Plasma cell markers (IgG, IgG4, kappa and lambda) are used to identify concurrent IgG4 disease (Figure 10).
BRAF-V600E mutation and Birbeck granules, both found in Langerhans cell histiocytosis (LCH), are negative in Rosai-Dorfman disease 18. BRAF-V600E is more common in Rosai-Dorfman disease overlapped with Langerhans cell histiocytosis or Erdheim-Chester disease 128.
Figure 8. Rosai-Dorfman disease histopathology
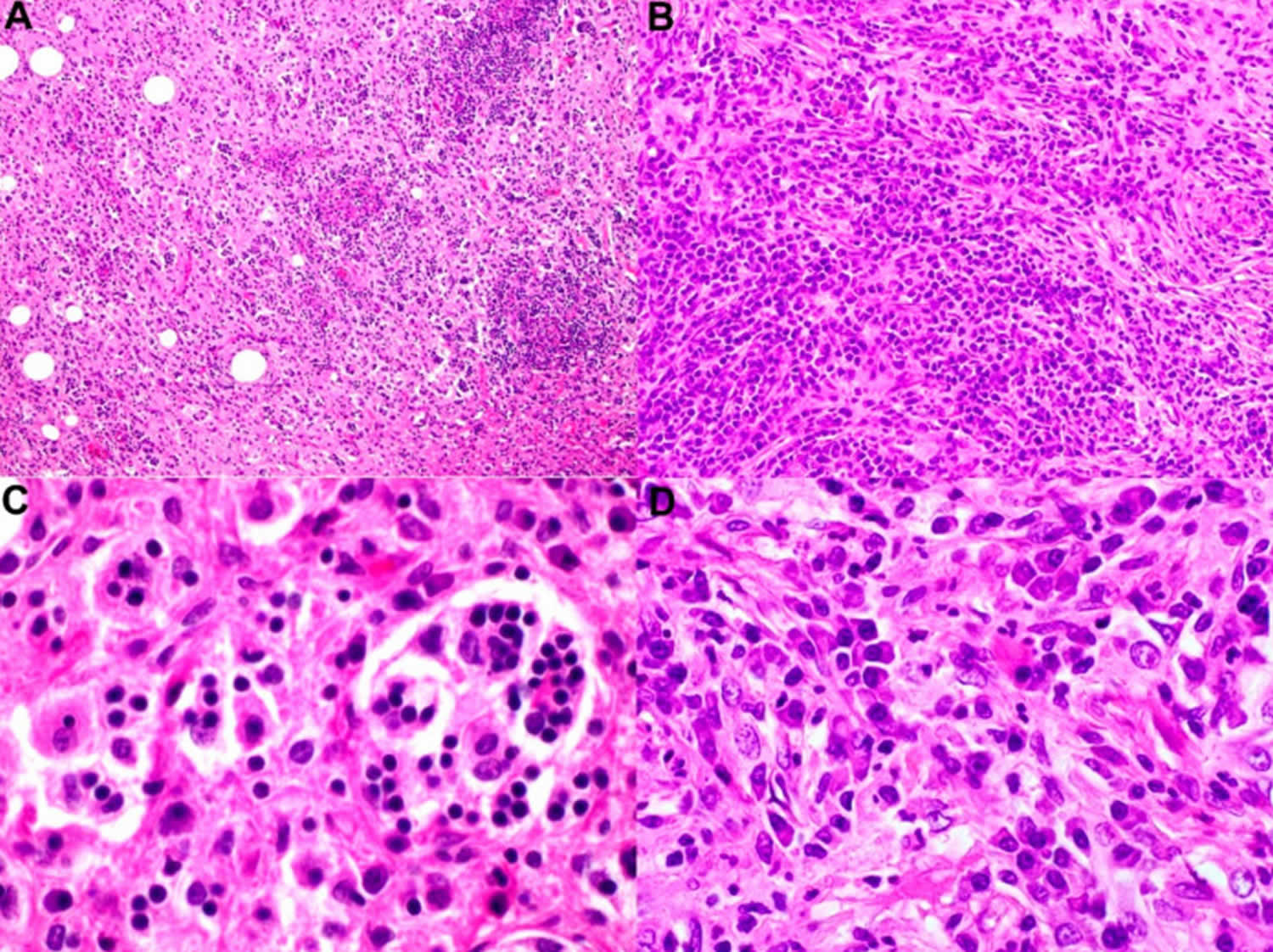
Footnotes: The selected images show a case of nodal Rosai–Dorfman disease with concurrent IgG4-related disease. (A) Low-power magnification shows effacement of normal lymph-node architecture by histiocytes (H & E, magnification 40×). (B) Higher-power magnification demonstrates increased plasma cells arranged in nests and singly associated with background fibrosis and histiocytes (immunoperoxidase, magnification 200×). (C) Loaded histiocytes with emperipolesis and (D) an increased number of plasma cells (immunoperoxidase, magnification 600× and 600×, respectively).
[Source 5 ]Figure 9. Rosai–Dorfman disease immunohistochemical stains
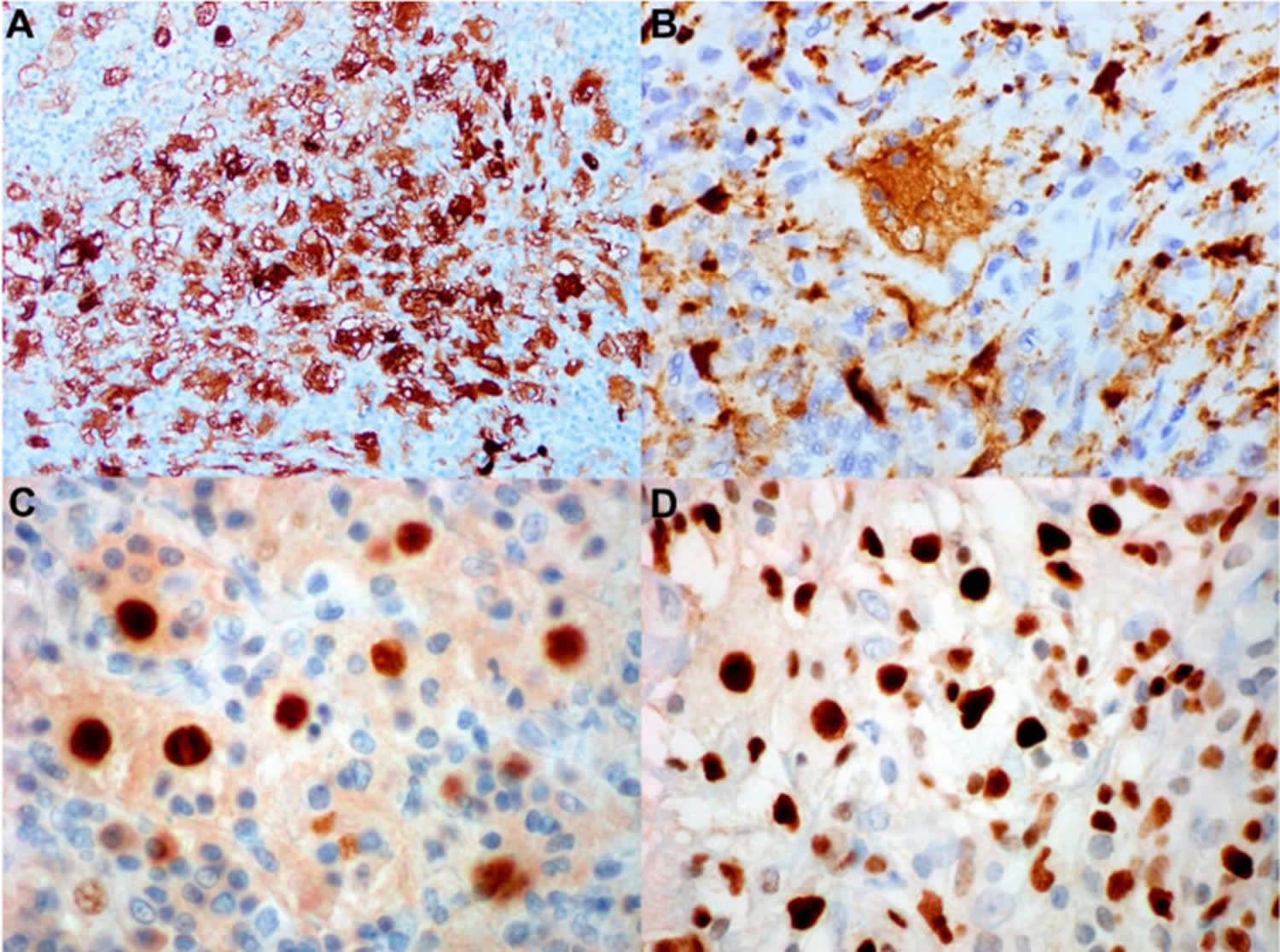
Footnotes: (A) S100 immunohistochemical stain highlighting the histiocytes, which also demonstrates emperipolesis (immunoperoxidase, magnification 600×). (B) CD68 immunohistochemical stain highlighting the histiocytes (immunoperoxidase, magnification 600×). (C,D) BCL-1 and Oct2 immunohistochemical stains are positive for histiocytes of Rosai–Dorfman disease (immunoperoxidase, magnification 600×).
[Source 5 ]Figure 10. Rosai–Dorfman disease with IgG4 disease
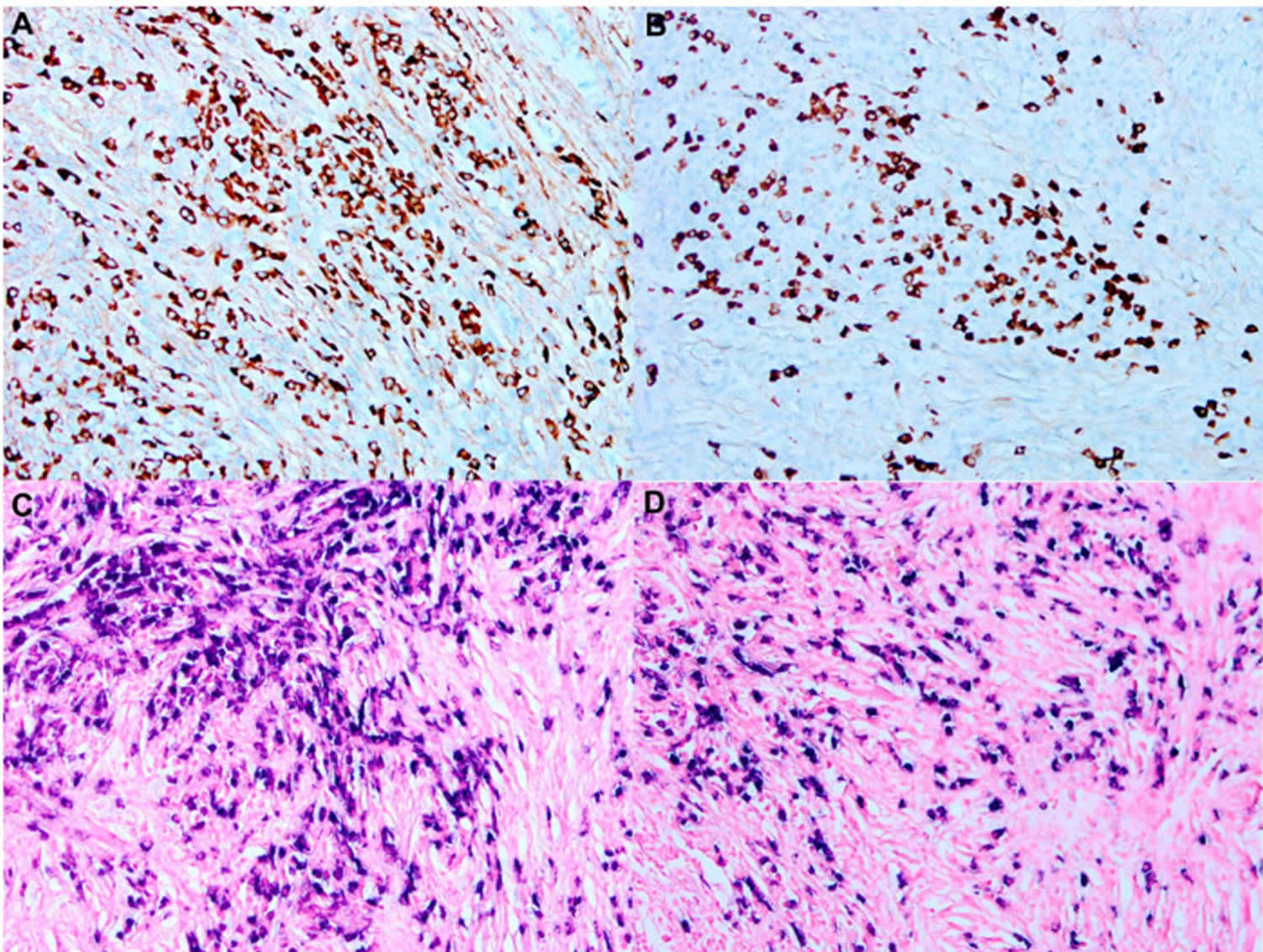
Footnotes: IgG4 disease in a patient with Rosai–Dorfman disease. Increased plasma cells in a background of fibrosis. (A,B) Immunohistochemical stains demonstrate plasma cells positive for IgG and proportionally positive forIgG4 (A,B). immunoperoxidase 100×, respectively). (C,D) In situ hybridization using kappa and lambda light chain probes reveal the plasma cells to be polyclonal (in situ hybridization 100×, respectively).
[Source 5 ]Gene mutations
Approximately 50% of cases of Rosai-Dorfman disease do not have a known distinct genetic mutation profile 5. From 30% to 50% of patients with Rosai-Dorfman disease-harboring somatic mutations are frequently involved in ARAF, NRAS, KRAS, MAP2K1, CSF1, and CBL genes, of which MA2P2K1 and KRAS were the most frequent, making up 14% and 12.5% of all Rosai-Dorfman disease, respectively (see Table 4) 129, 130, 131, 132, 133. Garces et al. 122 showed mutually excluded KRAS and MAP21 gene mutations in Rosai-Dorfman disease, together making up 33% of cases. NRAS mutation was detected in numerous cases of purely cutaneous Rosai-Dorfman disease. A study of Wu et al. 134 showed NRAS (A146T) mutation was the most common among point mutants, followed by NRAS G13S, suggesting that the mutations may play a role in pathogenesis of cutaneous Rosai-Dorfman disease. PTPN11, NF1 mutations have also occasionally been reported 123. BRAF mutations can occasionally occur in Rosai-Dorfman disease, and not just V600E mutations; other mutations were reported (BRAF Y472C and BRAF R188G, deletion in exon 12 of BRAF) 135, 136, 137. BRAF-V600E is more common in Rosai-Dorfman disease overlapped with Langerhans cell histiocytosis or Erdheim-Chester disease 128. Given the low frequency of BRAF mutations in cases of Rosai-Dorfman disease, one should pay an attention to Rosai-Dorfman disease-overlapping diseases.
There are many other genes that are mutated in Rosai-Dorfman disease, including SNX24, and are involved in intracellular trafficking. INTS2, CIC, SFR1, BRD4, and PHOX2B genes play roles in the transcriptional regulation as well as cell-cycle regulation genes (PDS5A, MUC4); ubiquitin-proteasome pathway (USP35); and DNA-mismatch repair genes (BRCA1, LATS2, ATM) 132, 138.
Some gene mutations frequently identified in myeloid or lymphoid neoplasms have also been observed in Rosai-Dorfman disease, e.g., ASXL1, TET2, and DNMT3A 123. A large cohort of 28 patients reported many mutations, which are implicated in other myeloid malignancies, e.g., TET2, MLL4, NF1, ALK, and ASXL1; many of these genes are driver mutations in different myeloid and lymphoid neoplasms 137. Whether they might play a role in the pathogenesis of Rosai-Dorfman disease or if they might cause transformation or transdifferentiation to these neoplasms has yet to be determined. Many other mutations have been reported: SEC62, FCGBP, PIK3R2, PIK3CA, VCL, EGFR, ERBB2 and TLR8 (minimum variable allele frequency in these mutations is 2%) 137. The exact roles of these genes are of further exploration.
Cytogenetic testing usually does not have a role in Rosai-Dorfman disease; however, testing can be conducted to rule out a neoplastic process. One case was reported to have a normal karyotype with a minor clone lacking chromosome 20 139.
Table 4. Certain gene mutations identified in Rosai-Dorfman disease
| Gene | Molecular Alteration |
|---|---|
| ARAF | N217K |
| MAP2K1 | F53V, L115V, P124R, G128D, V50M, D65M |
| KRAS | A146T, A146V, K117N, G12D, G12R, G13S, Q22K, |
| NRAS | G13D |
| CBL | C384Y, GNAQ Q209H |
| KDM5A | amplification |
| FBXW7 | E113D |
| BRAF | V600E, deletion (p. 486–491), Y472C and R188G |
| SMAD4 | T521I (variant of unknown significance, 1 case) |
Rosai Dorfman disease treatment
In many cases, Rosai-Dorfman disease is a self-limited and seldom life-threatening disease which commonly does not require therapy. The “watch and wait” approach is used without treatment is preferred for individuals with Rosai-Dorfman disease whenever possible 18, 140.
In some cases, various treatment options may become necessary. In these cases, the treatment of Rosai-Dorfman disease is directed toward the specific symptoms that are apparent in each individual. Several different treatment options have been used to treat individuals with Rosai-Dorfman disease including surgical removal of histiocytic lesions. In more serious cases, additional treatment options have included therapy with certain drugs including steroids (e.g., prednisone), interferon-alpha (IFN-α) 141, rituximab 142, 143, imatinib 144 and retinoids 145 and a regimen of certain anticancer drugs (chemotherapy). Numerous chemotherapeutic agents were used, i.e. vinca alkaloids, alkylating agents, anthracyclines, cladribine 146, clofarabine 147, methotrexate 148, mercaptopurine 149, azathioprine 150 and chlorodeoxyadenosine 151. In some cases, affected individuals have shown improvement of symptoms with these treatments. In other cases, drug therapies have been ineffective.
Other treatment is symptomatic and supportive. Radiotherapy was considered a palliative method in patients with symptomatic Rosai-Dorfman disease 18, but according to the latest findings it can give better results than chemotherapy in some cases 58. Radiotherapy appears to be an alternative in steroid-resistant patients 152, 153.
There are reports describing Rosai-Dorfman disease patients with a high level of HHV-6/VZV antibody titres, in which there was significant improvement after the application of acyclovir 53, 154. In other cases, complete remission was observed after using thalidomide 58.
Figure 11. Rosai-Dorfman disease treatment algorithm
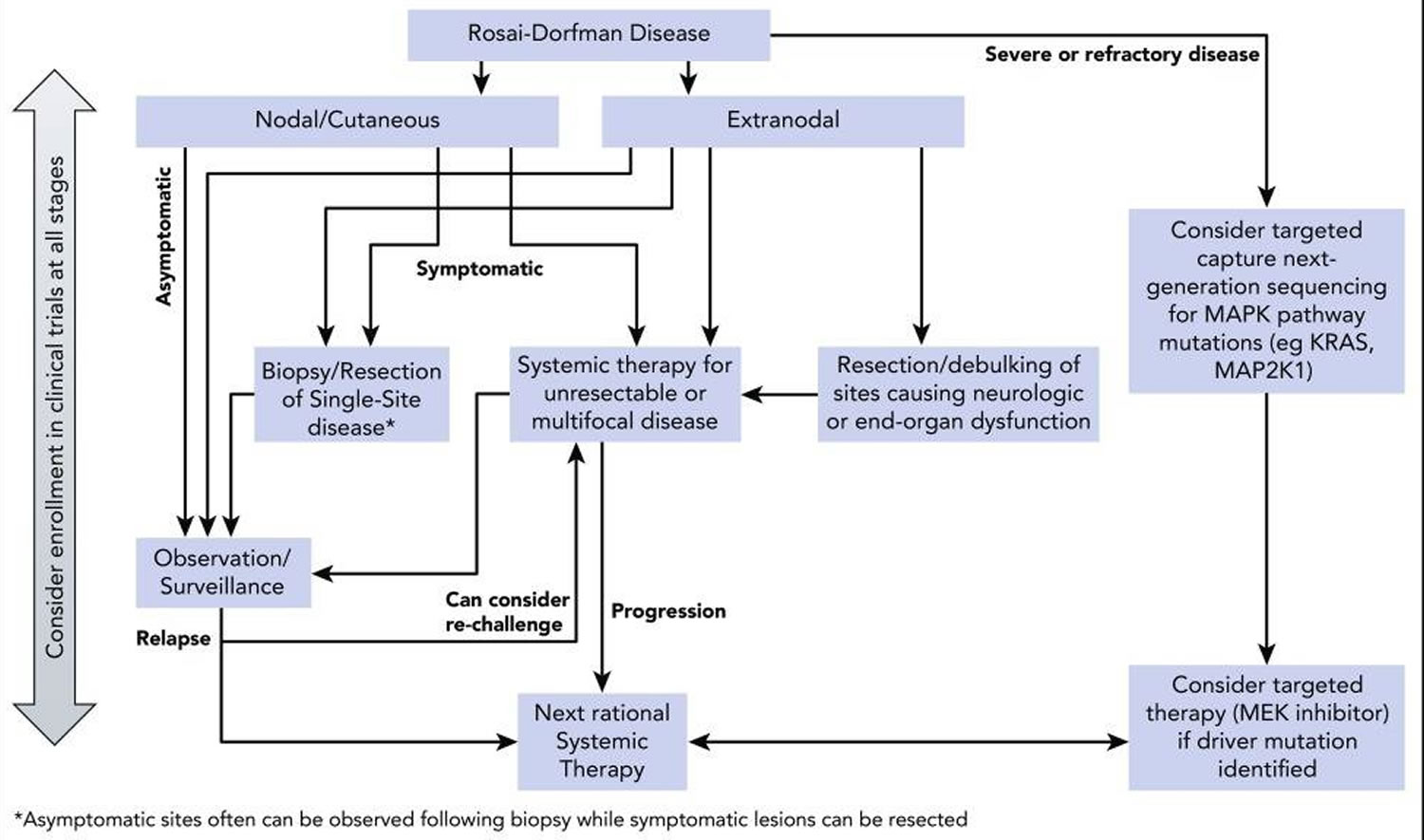
Figure 12. Rosai-Dorfman disease with nervous system disease treatment algorithm
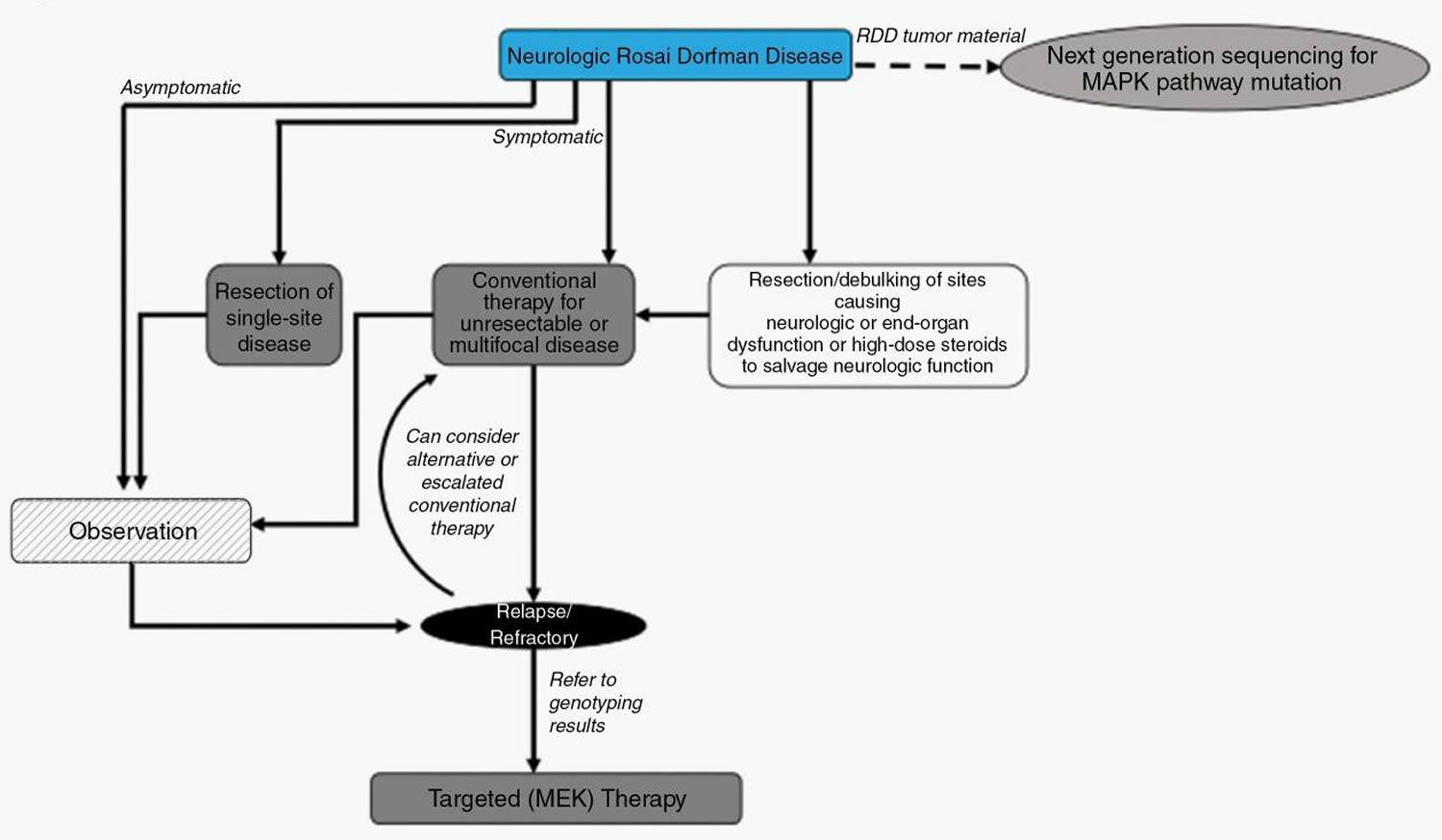
Table 5. Treatment options for Rosai-Dorfman disease
| Treatment | Dose/schedule | Indication | Supportive evidence | Comments |
|---|---|---|---|---|
| Observation | NA | Uncomplicated adenopathy | Case series of 80 patients showed 50% spontaneous remission 155 | Can consider observation for near-complete resection of unifocal lesions with minimal residual disease after surgery |
| Asymptomatic cutaneous Rosai-Dorfman disease | ||||
| Postoperatively for resected unifocal disease | ||||
| Corticosteroids | Prednisone: 40-70 mg or 1 mg/kg per day followed by taper | Symptomatic nodal or cutaneous disease | Several case reports and case series with prednisone showing responses in orbital, central nervous system (CNS), and bone Rosai-Dorfman disease and autoimmune hemolytic anemia-associated disease 156, 157 | Responses, when favorable, are unpredictable in their durability |
| Dexamethasone: 8-20 mg per day followed by taper | Nonresectable or multifocal extranodal disease requiring systemic treatment | Dexamethasone was effective in central nervous system (CNS) and nodal Rosai-Dorfman disease in case reports 158, 159 | Optimal duration of treatment is not known because early relapse can occur | |
| Case report of intralesional steroids in orbital Rosai-Dorfman disease 160 | One reasonable approach is to treat to optimal response followed by slow taper | |||
| No response to steroids in other case reports of orbital, tracheal, renal, or soft tissue Rosai-Dorfman disease 161, 162 | After successful steroid treatment: consider second-line agents to maintain response | |||
| Surgical resection | NA | Unifocal extranodal disease | Case series with long-term remission after resection of isolated cutaneous and intracranial disease 70, 156 | Local recurrences can occur 70, 84, for which case systemic treatment should be considered |
| Symptomatic cranial, spinal, sinus, or airway disease | ||||
| Sirolimus | 2.5 mg/m2 per day for 18 months, then taper off over 6 months | Prolonged complete response in Rosai-Dorfman disease with autoimmune cytopenia 163 | Reasonable first choice in Autoimmune lymphoproliferative syndrome (ALPS)-associated Rosai-Dorfman disease | |
| Radiotherapy | 30-50 Gy, lymphoma-like schedule 164 | Refractory or symptomatic disease not amenable to resection, recurrent after resection, or with a contraindication to systemic therapy | Palliative benefit in case reports, including refractory soft tissue and orbital Rosai-Dorfman disease with visual compromise 152 and for mass effect causing airway obstruction 155, 153 | No established fractionation schedule |
| Can also be considered as adjuvant treatment after resection of cranial or spinal lesions with residual but not bulky disease | ||||
| Chemotherapy | ||||
| Cladribine | 5 mg/m² per day for 5 days, every 28 days for up to 6 cycles | Severe, disseminated, or refractory disease | 11 patients reported: 7 with complete response, 1 with partial response, and 3 with progressive disease 165, 92, 151, 146, 166 | Can cause myelosuppression with associated infections |
| CNS involvement | ||||
| methotrexate or 6-mercaptopurine/methotrexate | 20 mg/m² per week of methotrexate, alone or with 6-mercaptopurine (50 mg/m² per day) or steroids | Multifocal, skin, or CNS Rosai-Dorfman disease | Partial response or complete response in refractory cases 167, 148, 168, 169, 170, 171, 172 | Reasonable as maintenance therapy after surgery or steroids |
| Optimal duration unknown | ||||
| Vinca alkaloids | Standard doses; vinblastine usually combined with prednisone | Vincristine effective in 1 report of skin Rosai-Dorfman disease 173 | Variable responses | |
| Several reports showing prolonged complete response when combined with other agents 169, 170, 171 | Optimal duration unknown | |||
| Immunomodulatory | ||||
| Thalidomide | 50-300 mg per day; variable duration | Refractory cutaneous Rosai-Dorfman disease | Recent review: several reports with prolonged complete response 174 | Notable toxicities include skin rash and neuropathy |
| Variable responses | Optimal dose and duration unknown | |||
| Lenalidomide | Not known | Refractory disease | Sustained complete response in an adult with multiply relapsed Rosai-Dorfman disease 175 | Myelosuppressive but less neuropathy and skin rash than thalidomide |
| Rituximab | 500 mg/m² per dose every 1 or 2 week for 2-6 cycles | For refractory nodal and immune-related Rosai-Dorfman disease | Efficacy described in single case reports 176 | Mechanism of efficacy is not understood |
| – alone or with chemotherapy | There are reports of refractoriness and relapses 167, 166 and therefore, reports of success are interpreted with caution | |||
| Imatinib mesylate | 400-600 mg per day for 7 months | Refractory/relapsed Rosai-Dorfman disease | Anecdotal activity in 1 adult with refractory Rosai-Dorfman disease 177 | Variable responses |
| 1 case of skin Rosai-Dorfman disease was refractory 144 | May work only in progressive disease GFR α/β+ cases | |||
| Clinical trial (experimental) | ||||
| Cobimetinib (NCT02649972) | Per trial guidelines | Refractory Rosai-Dorfman disease | Substantial regression of abdominal masses in a single patient with KRAS p.G12R–mutated Rosai-Dorfman disease 178 | Several case reports of successful treatment of Erdheim-Chester disease with cobimetinib 179, 180 |
| Clofarabine (NCT02425904) | 25 mg/m² per day for 5 days, every 28 days for 6 cycles | Severe, disseminated, or refractory disease | 3 patients: 2 with complete response, 1 had partial response 147 | Myelosuppressive and expensive |
| CNS involvement | Prospective studies ongoing to determine optimal dosing, long-term efficacy, and toxicity |
Footnote: NA = not applicable
[Source 1 ]Watch and wait
After the diagnosis of Rosai-Dorfman disease is established, observation is reasonable in many cases, because 20% to 50% of patients with nodal or cutaneous Rosai-Dorfman disease will have spontaneous remissions 155, 181. Watch and wait strategy is suitable for patients with uncomplicated lymphadenopathy or asymptomatic cutaneous Rosai-Dorfman disease and potentially for those with asymptomatic disease in other sites 1.
Surgery
Surgery for Rosai-Dorfman disease is usually limited to biopsy, but complete surgical resection can be curative for unifocal disease, and debulking may be warranted for upper airway obstruction, spinal cord compression, or large lesions causing end-organ damage 5. Long-term remissions with resection alone have been reported in isolated intracranial disease 156. The most effective treatment of cutaneous Rosai-Dorfman disease is surgical excision 70. Endoscopic resection of sinonasal Rosai-Dorfman disease can achieve symptomatic control and restoration of function 84. In cases of multifocal disease, surgical resection of single foci should be reserved for bulky disease with neurologic or end-organ dysfunction 1.
Corticosteroids
Corticosteroids are usually helpful in reducing nodal size and symptoms, although responses have been variable 1. The optimal corticosteroid (prednisone or dexamethasone) dose and duration are not clearly defined. Prednisone (40-70 mg per day) has produced complete or partial responses in cases of orbital, central nervous system (CNS), bone, and autoimmune hemolytic anemia–associated disease 157, 158. Compared with other immune diseases (eg, sarcoidosis), therapeutic prednisone doses are usually higher (>0.5 mg/kg per day). Similarly, dexamethasone (8-20 mg per day) was effective in cases of central nervous system (CNS) Rosai-Dorfman disease and hilar lymphadenopathy 159, 182. One viable approach is to treat to the best observed response, followed by slow taper 1. An anecdotal response to intralesional steroids has been reported in an adult with orbital Rosai-Dorfman disease and optic nerve compression 160. Nevertheless, other reports of orbital, tracheal, renal, or soft tissue Rosai-Dorfman disease have shown a failure to respond to steroids 161, 162. Furthermore, relapses of Rosai-Dorfman disease lesions can sometimes occur after a short period of interruption 1. Anecdotally, the experience of the authors is that patients with extranodal disease do not generally demonstrate a durable response to steroids alone 1. A case series of 57 patients reported by Mayo Clinic group showed that corticosteroid treatment was associated with 56% overall response rate (ORR) in treatment-naïve patients 183. Relapses occurred in 53% of patients. When steroids were used as a second-line therapy, the overall response rate (ORR) was 67% 183.
Sirolimus
Mammalian target of rapamycin is a critical pathway for the control of proliferation and cytokine production from immune cells and has been found to be dysregulated in Rosai-Dorfman disease 163. Sirolimus and prednisone induced objective responses and disease stabilization in 80% of patients with Erdheim-Chester disease in 1 report 184 and sirolimus was found to be beneficial in a child with resistant Rosai-Dorfman disease and recurrent autoimmune cytopenias 163. Sirolimus is frequently used for treating autoimmune lymphoproliferative syndrome (ALPS)- or autoimmune-associated Rosai-Dorfman disease, but further study are needed.
Chemotherapy
Treatment of Rosai-Dorfman disease with chemotherapeutic agents has shown mixed results 1. Although chemotherapy is generally reserved for refractory or relapsed cases, sometimes it is used as initial therapy in disseminated or life-threatening disease. Anthracyclines and alkylating agents have little efficacy, whereas vinca alkaloids have shown variable responses 155. Low-dose methotrexate and 6-mercaptopurine administered in combination were effective in few patients 155, 148. Sustainable remissions after regimens containing vinblastine/methotrexate/6-mercaptopurine and 6-thioguanine 168, vinblastine/prednisone/methotrexate/6-mercaptopurine 169 or vinorelbine/methotrexate 170 have been reported. Single-agent 6-mercaptopurine was effective in halting disease in an adult with orbital and intracranial Rosai-Dorfman disease 171. Furthermore, long-term remission of intracranial Rosai-Dorfman disease has been reported after postsurgical maintenance with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)–like regimens 172. A patient with multiply-relapsed nodal Rosai-Dorfman disease responded to cytarabine/prednisone/vincristine followed by methotrexate/6-mercaptopurine maintenance 167. Successful treatment of refractory cutaneous Rosai-Dorfman disease was reported with single-agent vincristine 173 and low-dose methotrexate 185. In addition, azathioprine and interferon-alpha (IFN-α) induced long-term remissions in patients with Rosai-Dorfman disease 150, 186. However, interferon-alpha (IFN-α) in combination with chemotherapy failed to induce any response in another report 155. Therefore, these agents could be considered for steroid-refractory disease or early recurrence after interruption of steroids or when steroids are contraindicated 1.
Nucleoside analogs cladribine and clofarabine have induced responses in Rosai-Dorfman disease 92, 151, 146, 166, 147. They impair the function of monocytes through inhibition of interleukin-6 (IL-6), IL-1β, and tumor necrosis factor alpha (TNF-α) production. Cladribine (2.1-5 mg/m² per day for 5 days every 28 days for 6 months) induced prolonged remissions in cases of recurrent or refractory systemic Rosai-Dorfman disease 92, 151, 146, 166 and clofarabine (25 mg/m² per day for 5 days every 28 days for 6 months) was effective as salvage therapy in a series of patients with refractory or relapsed Rosai-Dorfman disease 147. These agents should be considered in severe or refractory Rosai-Dorfman disease cases when the potential benefit justifies their myelosuppressive toxicity 1. A multicenter prospective study (NCT02425904) is being conducted by the North American Consortium for Histiocytosis to study the efficacy and safety of clofarabine salvage in patients with histiocytoses, including Rosai-Dorfman disease.
Immunomodulatory therapy
Tumor necrosis factor alpha (TNF-α) inhibitors thalidomide and lenalidomide have shown promising results in Rosai-Dorfman disease because the identification of high levels of TNF-α and IL-6 provides a rational basis for their effectiveness 1. A recent review showed that low-dose thalidomide (100 mg per day) was effective in refractory cutaneous Rosai-Dorfman disease 174. However, responses to thalidomide have not been universal, and optimal dosing and duration of this drug remain unknown. Lenalidomide recently showed an excellent response in an adult with multiply-refractory nodal and bone Rosai-Dorfman disease and may be more tolerable than thalidomide (fewer skin rashes and less neuropathy), although more myelosuppressive 175.
The efficacy of rituximab has been described, especially in autoimmune-related Rosai-Dorfman disease cases 176, although refractoriness 185 and recurrences 152 have also been described.
Targeted therapies
Imatinib mesylate, a tyrosine kinase inhibitor, showed some activity in 1 patient with refractory Rosai-Dorfman disease. Lesional histiocytes were positive for the imatinib target proteins PDGFRB and KIT by immunohistochemistry, but no concurrent mutation was found 177. In another case, cutaneous Rosai-Dorfman disease was refractory to imatinib 144.
Unlike Erdheim-Chester disease and Langerhans cell histiocytosis, BRAF-V600E mutations have not been observed in Rosai-Dorfman disease 63, 187; therefore, the use of BRAF inhibitors is not relevant 1. MEK inhibition has shown preliminary activity in BRAF–wild-type Erdheim-Chester disease and in an adult with KRAS-mutated Rosai-Dorfman disease 179, 178. A phase 2 trial of cobimetinib for patients with BRAF–wild-type histiocytoses, including Rosai-Dorfman disease, is ongoing (NCT02649972), with promising early results 164. The robust activity of targeted therapies in other histiocytoses raises interest in their potential for Rosai-Dorfman disease, especially in cases with demonstrated somatic mutations; however, currently, the broad applicability of tumor sequencing and targeted treatments has not yet been established 1.
Radiotherapy
External-beam radiation therapy has modest efficacy in Rosai-Dorfman disease, although it can be beneficial in localized unresectable symptomatic steroid-refractory soft tissue masses and orbital bone disease with visual compromise 152 or resistant airway obstruction or to palliate local symptoms 155, 153. Radiotherapy has also been implemented in patients whose symptoms persist or recur after resection of isolated disease and in those who are not suitable candidates for surgery and/or when other treatments are contraindicated 74, 104. No standard doses of radiotherapy have been established, but doses between 20 and 50 Gy with 2 Gy per fraction have been employed 153. Overall response rate (ORR) was approximately 40% in patients treated with doses ranging from 30 to 49 Gy and 27% with doses less than 30 Gy 5.
Treatment course and disease surveillance
The optimal duration of steroids or other systemic therapies for Rosai-Dorfman disease is not known 1. Six to 12 months of systemic therapy followed by observation, assuming tolerance and a favorable response to treatment, is a reasonable approach. When initiating treatment, a first response assessment should be carried out within 4 months; if disease stabilizes or is in remission, interval of surveillance can be extended to 6 to 12 months 1.
Rosai Dorfman disease prognosis
To date, there has not been a large case series monitoring the prognosis of Rosai-Dorfman disease patients 5. Prognosis is usually favorable for cutaneous, nodal, and bone Rosai-Dorfman disease. However, multiorgan involvement or dysfunction and association with immune dysfunction are poor prognostic indicators and may lead to death 11, 12. In extranodal Rosai-Dorfman disease forms, prognosis also correlated with the number of organ systems that were involved. CNS involvement with Rosai-Dorfman disease has variable outcomes 188, 189, 190. In most cases, Rosai-Dorfman disease has a good prognosis; however, some patients have a progressive and fatal course. Regarding gastrointestinal involvement by Rosai-Dorfman disease, a case series reported approximately 20% mortality 5. Genitourinary-system involvement has a worse prognosis of approximately 40% mortality in Rosai-Dorfman disease involving the kidney 5. The largest series of Rosai-Dorfman disease was reported by Foucar et al. in 1990 12; it showed that 17 (7%) of 238 patients died because of direct complications of their disease, infections, or amyloidosis. The most common causes of reported deaths were identified immunological abnormalities, severe infections, surgical complications, complications after radiotherapy, and the compression of airways by enlarged lymph nodes 17, 191. There are reports of Rosai-Dorfman disease leading to lymphoma, amyloidosis, and consequently death resulting from these diseases 191.
In 2011 the Histiocyte Society divided Rosai-Dorfman disease patients into three major groups 1:
- Patients with sudden enlargement of the lymph nodes, in which spontaneous regression is observed, without any further recurrences – consistent with the best prognosis;
- Patients with immunological abnormalities, in which lymphadenopathy is more generalized – the prognosis being worse; and
- Patients with extranodal site involvement and/or multinodal disease, with repeated relapses and remissions over a period of years – the prognosis depends on the type and number of extranodal sites.
In a review by Pulsoni et al in 2002 155, 10 (12%) of 80 patients died as a result of Rosai-Dorfman disease. Patients with multifocal and extranodal Rosai-Dorfman disease, particularly those with kidney, liver, or lower respiratory tract disease, seem to have an unfavorable prognosis. Therefore, intensive systemic chemotherapy, targeted therapies, and investigational agents may be justified in this context. Further research of the role of these therapies in refractory Rosai-Dorfman disease may mitigate the poor prognosis of some of these cases 1.
Classic symptoms, such as cervical lymphadenopathy, fever, and good general condition, may persist from several weeks to even a few years (the average time is 3-9 months) 17. Twenty percent of Rosai-Dorfman disease patients have spontaneous resolution or improvement within 3–9 months. Other patients experience an unpredictable clinical course, with alternating periods of remission and reactivation that may last years 1. The majority of Rosai-Dorfman disease patients have stable and persistent disease lasting up to several years. Without treatment, relapsing-remitting Rosai-Dorfman disease will occur in 70% of cases, spontaneous regression in 20%, and in 10% there will be a progression of the disease 18. Seven percent of patients have a fatal outcome, especially if immunologic abnormalities and extranodal involvement are present. It is recommended that a clinical examination and laboratory tests be performed every 3-6 months during the first two years following diagnosis, then every year 18.
- Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile JF, Durham BH, Braier J, Charlotte F, Donadieu J, Cohen-Aubart F, Rodriguez-Galindo C, Allen C, Whitlock JA, Weitzman S, McClain KL, Haroche J, Diamond EL. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018 Jun 28;131(26):2877-2890. doi: 10.1182/blood-2018-03-839753[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Tatit RT, Raffa PEAZ, de Almeida Motta GC, Bocchi AA, Guimaraes JL, Franceschini PR, de Aguiar PHP. Rosai-Dorfman disease mimicking images of meningiomas: Two case reports and literature review. Surg Neurol Int. 2021 Jun 21;12:292. doi: 10.25259/SNI_918_2020[↩]
- Hu PP, Wei F, Liu XG, Liu ZJ. Diagnosis and treatment of Rosai-Dorfman disease of the spine: a systematic literature review. Syst Rev. 2021 Jan 18;10(1):31. doi: 10.1186/s13643-021-01581-0[↩]
- Doglioni C. Rosai-Dorfman disease. A legacy of Professor Rosai that is still not exploited completely. Pathologica. 2021 Oct;113(5):388-395. doi: 10.32074/1591-951X-548[↩]
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman Disease between Proliferation and Neoplasia. Cancers (Basel). 2022 Oct 27;14(21):5271. doi: 10.3390/cancers14215271[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Cohen Aubart F, Idbaih A, Emile JF, Amoura Z, Abdel-Wahab O, Durham BH, Haroche J, Diamond EL. Histiocytosis and the nervous system: from diagnosis to targeted therapies. Neuro Oncol. 2021 Sep 1;23(9):1433-1446. doi: 10.1093/neuonc/noab107[↩][↩][↩][↩]
- Adam R, Harsovescu T, Tudorache S, Moldovan C, Pogarasteanu M, Dumitru A, Orban C. Primary Bone Lesions in Rosai-Dorfman Disease, a Rare Case and Diagnostic Challenge-Case Report and Literature Review. Diagnostics (Basel). 2022 Mar 23;12(4):783. doi: 10.3390/diagnostics12040783[↩]
- Azari-Yaam A, Abdolsalehi MR, Vasei M, Safavi M, Mehdizadeh M. Rosai-Dorfman Disease: A Rare Clinicopathological Presentation and Review of the Literature. Head Neck Pathol. 2021 Mar;15(1):352-360. doi: 10.1007/s12105-020-01183-7[↩]
- Weng X, Yang Y, Zhang M, Cai C, Sun Y, Wu X, Zhang R, Gui H, Li W, Xu Q, Liu X. Primary intraosseous Rosai-Dorfman disease: An analysis of clinicopathologic characteristics, molecular genetics, and prognostic features. Front Oncol. 2022 Sep 15;12:950114. doi: 10.3389/fonc.2022.950114[↩]
- Iancu G, Gica N, Mustata LM, Panaitescu AM, Vasile D, Peltecu G. Rosai-Dorfman Disease: Breast Involvement-Case Report and Literature Review. Medicina (Kaunas). 2021 Oct 27;57(11):1167. doi: 10.3390/medicina57111167[↩]
- Miękus A, Stefanowicz J, Kobierska-Gulida G, Adamkiewicz-Drożyńska E. Rosai-Dorfman disease as a rare cause of cervical lymphadenopathy – case report and literature review. Cent Eur J Immunol. 2018;43(3):341-345. doi: 10.5114/ceji.2018.80055[↩][↩][↩][↩]
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990 Feb;7(1):19-73.[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Vemuganti GK, Naik MN, Honavar SG. Rosai dorfman disease of the orbit. J Hematol Oncol. 2008 Jun 28;1:7. doi: 10.1186/1756-8722-1-7[↩][↩][↩][↩]
- Fukushima T, Yachi K, Ogino A, Ohta T, Watanabe T, Yoshino A, Katayama Y. Isolated intracranial Rosai-Dorfman disease without dural attachment–case report. Neurol Med Chir (Tokyo). 2011;51(2):136-40. doi: 10.2176/nmc.51.136[↩][↩][↩]
- Walid MS, Grigorian AA. Ethmo-spheno-intracranial Rosai-Dorfman disease. Indian J Cancer. 2010 Jan-Mar;47(1):80-1. doi: 10.4103/0019-509X.58870[↩][↩][↩]
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969 Jan;87(1):63-70.[↩][↩][↩]
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a pseudolymphomatous benign disorder. Analysis of 34 cases. Cancer. 1972 Nov;30(5):1174-88. doi: 10.1002/1097-0142(197211)30:5<1174::aid-cncr2820300507>3.0.co;2-s[↩][↩][↩][↩][↩][↩]
- Dalia S, Sagatys E, Sokol L, Kubal T. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014 Oct;21(4):322-7. doi: 10.1177/107327481402100408[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Mahzoni P, Zavareh MH, Bagheri M, Hani N, Moqtader B. Intracranial ROSAI-DORFMAN Disease. J Res Med Sci. 2012 Mar;17(3):304-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3527051[↩][↩]
- Destombes P. Adénites avec surcharge lipidique, de l’enfant ou de l’adulte jeune, observées aux Antilles et au Mali. (Quatre observations) [Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases)]. Bull Soc Pathol Exot Filiales. 1965 Nov-Dec;58(6):1169-75. French.[↩]
- McClain KL, Natkunam Y, Swerdlow SH. Atypical cellular disorders. Hematology Am Soc Hematol Educ Program. 2004:283-96. doi: 10.1182/asheducation-2004.1.283[↩][↩]
- Kismet E., Koseoglu V., Atay A.A., Deveci S., Demirkaya E., Tuncer K. Sinus histiocytosis with massive lymphadenopathy in three brothers. Pediatr. Int. 2005;47:473–476. doi: 10.1111/j.1442-200x.2005.02096.x[↩][↩]
- Khoury J.D., Solary E., Abla O., Akkari Y., Alaggio R., Apperley J.F., Bejar R., Berti E., Busque L., Chan J.K.C., et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–1719. doi: 10.1038/s41375-022-01613-1[↩]
- Rastogi V, Sharma R, Misra SR, Yadav L, Sharma V. Emperipolesis – a review. J Clin Diagn Res. 2014 Dec;8(12):ZM01-2. doi: 10.7860/JCDR/2014/10361.5299[↩][↩]
- Eisen RN, Buckley PJ, Rosai J. Immunophenotypic characterization of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Semin Diagn Pathol. 1990 Feb;7(1):74-82.[↩][↩]
- Gupta, A., Jindal, A., Dey, P. et al. Rosai Dorfman Disease in an Infant: An Unusual Cause of Cervical Lymphadenopathy. Indian J Pediatr 84, 481–482 (2017). https://doi.org/10.1007/s12098-017-2299-z[↩]
- Gawdzik, A., Ziarkiewicz-Wróblewska, B., Chlebicka, I. et al. Cutaneous Rosai-Dorfman Disease: A Treatment Challenge. Dermatol Ther (Heidelb) 11, 1443–1448 (2021). https://doi.org/10.1007/s13555-021-00557-1[↩]
- El-Kamel MF, Selim MK, Gawad MMA. A new presentation of isolated cutaneous Rosai-Dorfman disease: Eruptive xanthoma-like lesions. Indian J Dermatol Venereol Leprol. 2020 Mar-Apr;86(2):158-161. https://ijdvl.com/a-new-presentation-of-isolated-cutaneous-rosai-dorfman-disease-eruptive-xanthoma-like-lesions[↩]
- Histiocytosis. https://medlineplus.gov/ency/article/000068.htm[↩]
- 15 – Histiocytic Proliferative Disorders. Dermatopathology Foundations in Diagnostic Pathology 2010, Pages 619-630. https://doi.org/10.1016/B978-0-443-06654-2.00015-9[↩]
- Rodriguez-Galindo C, Allen CE. Langerhans cell histiocytosis. Blood. 2020 Apr 16;135(16):1319-1331. https://doi.org/10.1182/blood.2019000934[↩]
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, Charlotte F, Diamond EL, Egeler RM, Fischer A, Herrera JG, Henter JI, Janku F, Merad M, Picarsic J, Rodriguez-Galindo C, Rollins BJ, Tazi A, Vassallo R, Weiss LM; Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016 Jun 2;127(22):2672-81. doi: 10.1182/blood-2016-01-690636[↩][↩][↩][↩]
- Morgan N.V., Morris M.R., Cangul H., Gleeson D., Straatman-Iwanowska A., Davies N., Keenan S., Pasha S., Rahman F., Gentle D., et al. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010;6:e1000833. doi: 10.1371/journal.pgen.1000833[↩][↩]
- Moynihan L.M., Bundey S.E., Heath D., Jones E.L., McHale D.P., Mueller R.F., Markham A.F., Lench N.J. Autozygosity mapping, to chromosome 11q25, of a rare autosomal recessive syndrome causing histiocytosis, joint contractures, and sensorineural deafness. Am. J. Hum. Genet. 1998;62:1123–1128. doi: 10.1086/301824[↩]
- Cliffe S.T., Kramer J.M., Hussain K., Robben J.H., de Jong E.K., de Brouwer A.P., Nibbeling E., Kamsteeg E.J., Wong M., Prendiville J., et al. SLC29A3 gene is mutated in pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum. Mol. Genet. 2009;18:2257–2265. doi: 10.1093/hmg/ddp161[↩][↩]
- Bolze A., Abhyankar A., Grant A.V., Patel B., Yadav R., Byun M., Caillez D., Emile J.F., Pastor-Anglada M., Abel L., et al. A mild form of SLC29A3 disorder: A frameshift deletion leads to the paradoxical translation of an otherwise noncoding mRNA splice variant. PLoS ONE. 2012;7:e29708. doi: 10.1371/journal.pone.0029708[↩]
- Chouk H., Ben Rejeb M., Boussofara L., Elmabrouk H., Ghariani N., Sriha B., Saad A., H’Mida D., Denguezli M. Phenotypic intrafamilial variability including H syndrome and Rosai-Dorfman disease associated with the same c.1088G > A mutation in the SLC29A3 gene. Hum. Genom. 2021;15:63. doi: 10.1186/s40246-021-00362-z[↩]
- Maric I, Pittaluga S, Dale JK, Niemela JE, Delsol G, Diment J, Rosai J, Raffeld M, Puck JM, Straus SE, Jaffe ES. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am J Surg Pathol. 2005 Jul;29(7):903-11. doi: 10.1097/01.pas.0000157997.61177.08[↩][↩]
- Xie Y, Pittaluga S, Price S, Raffeld M, Hahn J, Jaffe ES, Rao VK, Maric I. Bone marrow findings in autoimmune lymphoproliferative syndrome with germline FAS mutation. Haematologica. 2017 Feb;102(2):364-372. doi: 10.3324/haematol.2015.138081[↩]
- Maric I., Pittaluga S., Dale J.K., Niemela J.E., Delsol G., Diment J., Rosai J., Raffeld M., Puck J.M., Straus S.E., et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am. J. Surg. Pathol. 2005;29:903–911. doi: 10.1097/01.pas.0000157997.61177.08[↩]
- Lu D, Estalilla OC, Manning JT Jr, Medeiros LJ. Sinus histiocytosis with massive lymphadenopathy and malignant lymphoma involving the same lymph node: a report of four cases and review of the literature. Mod Pathol. 2000 Apr;13(4):414-9. doi: 10.1038/modpathol.3880071[↩][↩]
- Long E, Lassalle S, Cheikh-Rouhou R, Hofman V, Lacour JP, Hofman P. Intestinal occlusion caused by Rosai-Dorfman disease mimicking colonic diverticulitis. Pathol Res Pract. 2007;203(4):233-7. doi: 10.1016/j.prp.2007.01.008[↩]
- Ambati S, Chamyan G, Restrepo R, Escalon E, Fort J, Pefkarou A, Khatib ZA, Dehner LP. Rosai-Dorfman disease following bone marrow transplantation for pre-B cell acute lymphoblastic leukemia. Pediatr Blood Cancer. 2008 Sep;51(3):433-5. doi: 10.1002/pbc.21606[↩]
- Hassani J, Porubsky C, Berman C, Zager J, Messina J, Henderson-Jackson E. Intraperitoneal Rosai-Dorfman disease associated with clear cell sarcoma: first case report. Pathology. 2016 Dec;48(7):742-744. doi: 10.1016/j.pathol.2016.07.015[↩]
- O’Malley DP, Duong A, Barry TS, Chen S, Hibbard MK, Ferry JA, Hasserjian RP, Thompson MA, Richardson MS, Jaffe R, Sidhu JS, Banks PM. Co-occurrence of Langerhans cell histiocytosis and Rosai-Dorfman disease: possible relationship of two histiocytic disorders in rare cases. Mod Pathol. 2010 Dec;23(12):1616-23. doi: 10.1038/modpathol.2010.157[↩]
- Vaiselbuh SR, Bryceson YT, Allen CE, Whitlock JA, Abla O. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014 Jul;61(7):1329-35. doi: 10.1002/pbc.25017[↩]
- Ragotte RJ, Dhanrajani A, Pleydell-Pearce J, Del Bel KL, Tarailo-Graovac M, van Karnebeek C, Terry J, Senger C, McKinnon ML, Seear M, Prendiville JS, Tucker LB, Houghton K, Cabral DA, Guzman J, Petty RE, Brown KL, Tekano J, Wu J, Morishita KA, Turvey SE. The importance of considering monogenic causes of autoimmunity: A somatic mutation in KRAS causing pediatric Rosai-Dorfman syndrome and systemic lupus erythematosus. Clin Immunol. 2017 Feb;175:143-146. doi: 10.1016/j.clim.2016.12.006[↩]
- Menon MP, Evbuomwan MO, Rosai J, Jaffe ES, Pittaluga S. A subset of Rosai-Dorfman disease cases show increased IgG4-positive plasma cells: another red herring or a true association with IgG4-related disease? Histopathology. 2014 Feb;64(3):455-9. doi: 10.1111/his.12274[↩]
- Liu L, Perry AM, Cao W, Smith LM, Hsi ED, Liu X, Mo JQ, Dotlic S, Mosunjac M, Talmon G, Weisenburger DD, Fu K. Relationship between Rosai-Dorfman disease and IgG4-related disease: study of 32 cases. Am J Clin Pathol. 2013 Sep;140(3):395-402. doi: 10.1309/AJCPFH0SJ6YILXJU[↩][↩]
- Paulli M, Bergamaschi G, Tonon L, Viglio A, Rosso R, Facchetti F, Geerts ML, Magrini U, Cazzola M. Evidence for a polyclonal nature of the cell infiltrate in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Br J Haematol. 1995 Oct;91(2):415-8. doi: 10.1111/j.1365-2141.1995.tb05313.x[↩]
- Levine PH, Jahan N, Murari P, Manak M, Jaffe ES. Detection of human herpesvirus 6 in tissues involved by sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). J Infect Dis. 1992 Aug;166(2):291-5. doi: 10.1093/infdis/166.2.291[↩]
- Luppi M, Barozzi P, Garber R, Maiorana A, Bonacorsi G, Artusi T, Trovato R, Marasca R, Torelli G. Expression of human herpesvirus-6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998 Sep;153(3):815-23. doi: 10.1016/S0002-9440(10)65623-4[↩]
- Lauwers GY, Perez-Atayde A, Dorfman RF, Rosai J. The digestive system manifestations of Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy): review of 11 cases. Hum Pathol. 2000 Mar;31(3):380-5. doi: 10.1016/s0046-8177(00)80254-3[↩][↩][↩][↩]
- Delacrétaz F, Meugé-Moraw C, Anwar D, Borisch B, Chave JP. Sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease) in an HIV-positive patient. Virchows Arch A Pathol Anat Histopathol. 1991;419(3):251-4. doi: 10.1007/BF01626356[↩]
- Tsang W.Y., Yip T.T., Chan J.K. The Rosai-Dorfman disease histiocytes are not infected by Epstein-Barr virus. Histopathology. 1994;25:88–90. doi: 10.1111/j.1365-2559.1994.tb00604.x[↩]
- Mehraein Y, Wagner M, Remberger K, Füzesi L, Middel P, Kaptur S, Schmitt K, Meese E. Parvovirus B19 detected in Rosai-Dorfman disease in nodal and extranodal manifestations. J Clin Pathol. 2006 Dec;59(12):1320-6. doi: 10.1136/jcp.2005.029850[↩]
- Kuo TT, Chen TC, Lee LY, Lu PH. IgG4-positive plasma cells in cutaneous Rosai-Dorfman disease: an additional immunohistochemical feature and possible relationship to IgG4-related sclerosing disease. J Cutan Pathol. 2009 Oct;36(10):1069-73. doi: 10.1111/j.1600-0560.2008.01222.x[↩]
- Maia RC, de Meis E, Romano S, Dobbin JA, Klumb CE. Rosai-Dorfman disease: a report of eight cases in a tertiary care center and a review of the literature. Braz J Med Biol Res. 2015 Jan;48(1):6-12. doi: 10.1590/1414-431X20144110[↩][↩][↩][↩][↩]
- Cohen-Barak E, Rozenman D, Schafer J, Krausz J, Dodiuk-Gad R, Gabriel H, Shani-Adir A. An unusual co-occurrence of Langerhans cell histiocytosis and Rosai-Dorfman disease: report of a case and review of the literature. Int J Dermatol. 2014 May;53(5):558-63. doi: 10.1111/ijd.12051[↩][↩][↩][↩]
- Wang KH, Cheng CJ, Hu CH, Lee WR. Coexistence of localized Langerhans cell histiocytosis and cutaneous Rosai-Dorfman disease. Br J Dermatol. 2002 Oct;147(4):770-4. doi: 10.1046/j.1365-2133.2002.04879.x[↩]
- Patrizi A, Neri I, Bianchi F, Guerrini V, Misciali C, Paone G, Burnelli R. Langerhans cell histiocytosis and juvenile xanthogranuloma. Two case reports. Dermatology. 2004;209(1):57-61. doi: 10.1159/000078589[↩]
- Moore JC, Zhao X, Nelson EL. Concomitant sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman Disease) and diffuse large B-cell lymphoma: a case report. J Med Case Rep. 2008 Mar 5;2:70. doi: 10.1186/1752-1947-2-70[↩]
- Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, Cohen-Aubart F, Launay D, Lesot A, Mokhtari K, Canioni D, Galmiche L, Rose C, Schmalzing M, Croockewit S, Kambouchner M, Copin MC, Fraitag S, Sahm F, Brousse N, Amoura Z, Donadieu J, Emile JF. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012 Sep 27;120(13):2700-3. doi: 10.1182/blood-2012-05-430140[↩][↩]
- Chakraborty R, Hampton OA, Shen X, Simko SJ, Shih A, Abhyankar H, Lim KP, Covington KR, Trevino L, Dewal N, Muzny DM, Doddapaneni H, Hu J, Wang L, Lupo PJ, Hicks MJ, Bonilla DL, Dwyer KC, Berres ML, Poulikakos PI, Merad M, McClain KL, Wheeler DA, Allen CE, Parsons DW. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014 Nov 6;124(19):3007-15. doi: 10.1182/blood-2014-05-577825[↩]
- Milne P, Bigley V, Bacon CM, Néel A, McGovern N, Bomken S, Haniffa M, Diamond EL, Durham BH, Visser J, Hunt D, Gunawardena H, Macheta M, McClain KL, Allen C, Abdel-Wahab O, Collin M. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017 Jul 13;130(2):167-175. doi: 10.1182/blood-2016-12-757823[↩]
- Cooper SL, Jenrette JM. Rosai-Dorfman disease: management of CNS and systemic involvement. Clin Adv Hematol Oncol. 2012 Mar;10(3):199-202.[↩]
- Morgan NV, Morris MR, Cangul H, Gleeson D, Straatman-Iwanowska A, Davies N, Keenan S, Pasha S, Rahman F, Gentle D, Vreeswijk MP, Devilee P, Knowles MA, Ceylaner S, Trembath RC, Dalence C, Kismet E, Köseoğlu V, Rossbach HC, Gissen P, Tannahill D, Maher ER. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010 Feb 5;6(2):e1000833. doi: 10.1371/journal.pgen.1000833[↩][↩]
- Picarsic J, Jaffe R. Pathology of histiocytic neoplasms and related disorders. In: Abla O, Janka G, eds. Histiocytic Disorders. Zurich, Switzerland: Springer International Publishing; 2018:3-50.[↩][↩]
- Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, Ferrarini M, Abdel-Wahab O, Heaney ML, Scheel PJ, Feeley NK, Ferrero E, McClain KL, Vaglio A, Colby T, Arnaud L, Haroche J. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014 Jul 24;124(4):483-92. doi: 10.1182/blood-2014-03-561381[↩][↩]
- Al-Khateeb TH. Cutaneous Rosai-Dorfman Disease of the Face: A Comprehensive Literature Review and Case Report. J Oral Maxillofac Surg. 2016 Mar;74(3):528-40. doi: 10.1016/j.joms.2015.09.017[↩][↩][↩][↩][↩][↩]
- Sodhi KS, Suri S, Nijhawan R, Kang M, Gautam V. Rosai-Dorfman disease: unusual cause of diffuse and massive retroperitoneal lymphadenopathy. Br J Radiol. 2005 Sep;78(933):845-7. doi: 10.1259/bjr/23127241[↩]
- Rosai-Dorfman Disease. https://rarediseases.org/rare-diseases/rosai-dorfman-disease[↩][↩]
- Brenn T, Calonje E, Granter SR, Leonard N, Grayson W, Fletcher CD, McKee PH. Cutaneous rosai-dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002 Oct;24(5):385-91. doi: 10.1097/00000372-200210000-00001[↩]
- Sandoval-Sus JD, Sandoval-Leon AC, Chapman JR, Velazquez-Vega J, Borja MJ, Rosenberg S, Lossos A, Lossos IS. Rosai-Dorfman disease of the central nervous system: report of 6 cases and review of the literature. Medicine (Baltimore). 2014 May;93(3):165-175. doi: 10.1097/MD.0000000000000030[↩][↩][↩][↩]
- Taufiq M, Khair A, Begum F, Akhter S, Shamim Farooq M, Kamal M. Isolated Intracranial Rosai-Dorfman Disease. Case Rep Neurol Med. 2016;2016:1972594. doi: 10.1155/2016/1972594[↩][↩]
- Yetiser S, Cekin E, Tosun F, Yildirim A. Rosai-Dorfman disease associated with neurosensorial hearing loss in two siblings. Int J Pediatr Otorhinolaryngol. 2004 Aug;68(8):1095-100. doi: 10.1016/j.ijporl.2004.03.014[↩]
- Gupta K, Bagdi N, Sunitha P, Ghosal N. Isolated intracranial Rosai-Dorfman disease mimicking meningioma in a child: a case report and review of the literature. Br J Radiol. 2011 Jul;84(1003):e138-41. doi: 10.1259/bjr/15772106[↩]
- Nalini A, Jitender S, Anantaram G, Santosh V. Rosai Dorfman disease: case with extensive dural involvement and cerebrospinal fluid pleocytosis. J Neurol Sci. 2012 Mar 15;314(1-2):152-4. doi: 10.1016/j.jns.2011.10.002[↩][↩]
- Varan A, Şen H, Akalan N, Oğuz KK, Sağlam A, Akyüz C. Pontine Rosai-Dorfman disease in a child. Childs Nerv Syst. 2015 Jun;31(6):971-5. doi: 10.1007/s00381-015-2632-7[↩]
- Morandi X, Godey B, Riffaud L, Heresbach N, Brassier G. Isolated Rosai-Dorfman disease of the fourth ventricle. Case illustration. J Neurosurg. 2000 May;92(5):890. doi: 10.3171/jns.2000.92.5.0890[↩]
- Antuña Ramos A, Alvarez Vega MA, Alles JV, Antuña Garcia MJ, Meilán Martínez A. Multiple involvement of the central nervous system in Rosai-Dorfman disease. Pediatr Neurol. 2012 Jan;46(1):54-6. doi: 10.1016/j.pediatrneurol.2011.10.004[↩]
- Kozak B, Talbott J, Uzelac A, Rehani B. Rosai-Dorfman Disease Isolated to the Thoracic Epidural Spine. J Radiol Case Rep. 2015 Nov 30;9(11):6-16. doi: 10.3941/jrcr.v9i11.2629[↩]
- Tran HM, Chinichian S, Storkersen K, Tokuhara K. An Unusual Case of Extranodal Rosai-Dorfman Disease Manifesting as an Epibulbar Mass. Case Rep Ophthalmol. 2015 Oct 14;6(3):351-5. doi: 10.1159/000440994[↩]
- Chen HH, Zhou SH, Wang SQ, Teng XD, Fan J. Factors associated with recurrence and therapeutic strategies for sinonasal Rosai-Dorfman disease. Head Neck. 2012 Oct;34(10):1504-13. doi: 10.1002/hed.21832[↩][↩][↩]
- Vujhini SK, Kolte SS, Satarkar RN, Srikanth S. Fine needle aspiration diagnosis of Rosai-Dorfman Disease involving thyroid. J Cytol. 2012 Jan;29(1):83-5. doi: 10.4103/0970-9371.93239[↩]
- Ma YL, Liang ZP, Xu SE, Yang ZH, Peng Y, Sun XQ, Qin G. Rosai-Dorfman disease (RDD) in the paraglottic space: report of a case and review of literature. Int J Clin Exp Pathol. 2015 Oct 1;8(10):13532-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4680514[↩]
- Goupil de Bouillé J, de Muret A, Diot E, Dumont P, Plantier L, Diot P, Marchand-Adam S. Pulmonary manifestations revealing Rosai-Dorfman disease. Sarcoidosis Vasc Diffuse Lung Dis. 2015 Sep 14;32(3):275-7.[↩][↩]
- Boissière L, Patey M, Toubas O, Vella-Boucaud J, Perotin-Collard JM, Deslée G, Lebargy F, Dury S. Tracheobronchial Involvement of Rosai-Dorfman Disease: Case Report and Review of the Literature. Medicine (Baltimore). 2016 Feb;95(7):e2821. doi: 10.1097/MD.0000000000002821[↩]
- O’Gallagher K, Dancy L, Sinha A, Sado D. Rosai-Dorfman disease and the heart. Intractable Rare Dis Res. 2016 Feb;5(1):1-5. doi: 10.5582/irdr.2015.01047[↩]
- Krishnan A, Nassar A, Nieh PT. Rosai-Dorfman disease presenting as extranodal renal mass. Urology. 2005 Dec;66(6):1319. doi: 10.1016/j.urology.2005.06.103[↩]
- El Majdoub A, El Houari A, Chbani L, El Fatemi H, Khallouk A, Farih MH. Isolated localization of Rosai Dorfman disease as renal mass: a case report and review of literature. Pan Afr Med J. 2016 May 13;24:64. doi: 10.11604/pamj.2016.24.64.6291[↩]
- Sasaki K, Pemmaraju N, Westin JR, Wang WL, Khoury JD, Podoloff DA, Moon B, Daver N, Borthakur G. A single case of rosai-dorfman disease marked by pathologic fractures, kidney failure, and liver cirrhosis treated with single-agent cladribine. Front Oncol. 2014 Oct 29;4:297. doi: 10.3389/fonc.2014.00297[↩][↩][↩][↩][↩]
- Röcken C, Wieker K, Grote HJ, Müller G, Franke A, Roessner A. Rosai-Dorfman disease and generalized AA amyloidosis: a case report. Hum Pathol. 2000 May;31(5):621-4. doi: 10.1053/hp.2000.6705[↩]
- Harik L, Nassar A. Extranodal Rosai-Dorfman disease of the kidney and coexistent poorly differentiated prostatic adenocarcinoma. Arch Pathol Lab Med. 2006 Aug;130(8):1223-6. doi: 10.5858/2006-130-1223-ERDOTK[↩]
- Yamada S, Uemura M, Tokumoto M, Nagara T, Noguchi H, Hirahashi M, Nakano T, Tsuruya K, Kitazono T. Hypercalcemia induced by Rosai-Dorfman disease in a hemodialysis patient: histological evidence of extrarenal calcitriol overproduction. Intern Med. 2014;53(24):2783-7. doi: 10.2169/internalmedicine.53.3001[↩]
- Karaosmanoğlu AD, Onur MR, Shirkhoda A, Ozmen M, Hahn PF. Unusual benign solid neoplasms of the kidney: cross-sectional imaging findings. Diagn Interv Radiol. 2015 Sep-Oct;21(5):376-81. doi: 10.5152/dir.2015.14545[↩]
- Rodríguez Torres C, Riazuelo Fantova G, Escartín Martínez I, Castillo Escudero JJ, del Agua Arias C. Rosai-Dorfman disease with atypical intrascrotal involvement. Int J Urol. 2015 Aug;22(8):794-6. doi: 10.1111/iju.12805[↩]
- Flores-Carrillo VM, Santaella-Torres F, Sánchez-Martínez LC, Gómez-Lara MH, Arellano-Poblete M, López Segura-Rueda E, Villarroel-Noboa J. Enfermedad de Rosai-Dorfman en glándula suprarrenal. Informe de un caso clínico [Rosai-Dorfman disease presented with involvement of the adrenal gland. A clinical case reported]. Rev Med Inst Mex Seguro Soc. 2014 Mar-Apr;52(2):224-7.[↩]
- Zhao M, Li C, Zheng J, Yu J, Sha H, Yan M, Jin J, Sun K, Wang Z. Extranodal Rosai-Dorfman disease involving appendix and mesenteric nodes with a protracted course: report of a rare case lacking relationship to IgG4-related disease and review of the literature. Int J Clin Exp Pathol. 2013 Oct 15;6(11):2569-77. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3816829[↩][↩]
- Ide M, Asao T, Yoshida T, Hirato J, Shimura T, Morinaga N, Shitara Y, Ishizaki M, Kuwano H. Rosai-Dorfman disease of the colon presented as small solitary polypoid lesion. Rare Tumors. 2010 Mar 31;2(1):e2. doi: 10.4081/rt.2010.e2[↩]
- Shaikh F, Awan O, Mohiuddin S, Farooqui S, Khan SA, McCartney W. 18F-FDG PET/CT Imaging of Extranodal Rosai-Dorfman Disease with Hepatopancreatic Involvement – A Pictorial and Literature Review. Cureus. 2015 Dec 3;7(12):e392. doi: 10.7759/cureus.392[↩]
- Mantilla JG, Goldberg-Stein S, Wang Y. Extranodal Rosai-Dorfman Disease: Clinicopathologic Series of 10 Patients With Radiologic Correlation and Review of the Literature. Am J Clin Pathol. 2016 Feb;145(2):211-21. doi: 10.1093/ajcp/aqv029[↩]
- Patel MH, Jambhekar KR, Pandey T, Ram R. A rare case of extra nodal Rosai-Dorfman disease with isolated multifocal osseous manifestation. Indian J Radiol Imaging. 2015 Jul-Sep;25(3):284-7. doi: 10.4103/0971-3026.161459[↩]
- Paryani NN, Daugherty LC, O’Connor MI, Jiang L. Extranodal rosai-dorfman disease of the bone treated with surgery and radiotherapy. Rare Tumors. 2014 Dec 11;6(4):5531. doi: 10.4081/rt.2014.5531[↩][↩]
- Huang Q, Chang KL, Weiss LM. Extranodal Rosai-Dorfman disease involving the bone marrow: a case report. Am J Surg Pathol. 2006 Sep;30(9):1189-92. doi: 10.1097/01.pas.0000209846.52046.62[↩]
- Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system: a report of 11 cases. Mod Pathol. 2001 Mar;14(3):172-8. doi: 10.1038/modpathol.3880278[↩][↩]
- Wu M, Anderson AE, Kahn LB. A report of intracranial Rosai-Dorfman disease with literature review. Ann Diagn Pathol. 2001 Apr;5(2):96-102. doi: 10.1053/adpa.2001.23027[↩]
- Hollowell JP, Wolfla CE, Shah NC, Mark LP, Whittaker MH. Rosai-Dorman disease causing cervical myelopathy. Spine (Phila Pa 1976). 2000 Jun 1;25(11):1453-6. doi: 10.1097/00007632-200006010-00020[↩][↩]
- Kattner KA, Stroink AR, Roth TC, Lee JM. Rosai-Dorfman disease mimicking parasagittal meningioma: case presentation and review of literature. Surg Neurol. 2000 May;53(5):452-7; discussion 457. doi: 10.1016/s0090-3019(00)00197-x[↩]
- Deshpande AH, Nayak S, Munshi MM. Cytology of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Diagn Cytopathol. 2000 Mar;22(3):181-5. doi: 10.1002/(sici)1097-0339(20000301)22:3<181::aid-dc10>3.0.co;2-6[↩]
- Lee-Wing M, Oryschak A, Attariwala G, Ashenhurst M. Rosai-Dorfman disease presenting as bilateral lacrimal gland enlargement. Am J Ophthalmol. 2001 May;131(5):677-8. doi: 10.1016/s0002-9394(00)00864-3[↩]
- Di Rocco F, Garnett MR, Puget S, Pueyerredon F, Roujeau T, Jaubert F, Sainte-Rose C. Cerebral localization of Rosai-Dorfman disease in a child. Case report. J Neurosurg. 2007 Aug;107(2 Suppl):147-51. doi: 10.3171/PED-07/08/147[↩]
- Jurić G, Jakić-Razumović J, Rotim K, Zarković K. Extranodal sinus histiocytosis (Rosai-Dorfman disease) of the brain parenchyma. Acta Neurochir (Wien). 2003 Feb;145(2):145-9; discussion 149. doi: 10.1007/s00701-002-1031-5[↩]
- Hinduja A, Aguilar LG, Steineke T, Nochlin D, Landolfi JC. Rosai-Dorfman disease manifesting as intracranial and intraorbital lesion. J Neurooncol. 2009 Mar;92(1):117-20. doi: 10.1007/s11060-008-9733-z[↩]
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman Disease between Proliferation and Neoplasia. Cancers (Basel). 2022 Oct 27;14(21):5271. doi: 10.3390/cancers1421527[↩]
- Go R.S., Jacobsen E., Baiocchi R., Buhtoiarov I., Butler E.B., Campbell P.K., Coulter D.W., Diamond E., Flagg A., Goodman A.M., et al. Histiocytic Neoplasms, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021;19:1277–1303. doi: 10.6004/jnccn.2021.0053[↩][↩]
- Albano D, Bosio G, Bertagna F. 18F-FDG PET/CT Follow-up of Rosai-Dorfman Disease. Clin Nucl Med. 2015 Aug;40(8):e420-2. doi: 10.1097/RLU.0000000000000853[↩]
- Bonetti F., Chilosi M., Menestrina F., Scarpa A., Pelicci P.G., Amorosi E., Fiore-Donati L., Knowles D.M., 2nd Immunohistological analysis of Rosai-Dorfman histiocytosis. A disease of S-100 + CD1-histiocytes. Virchows Arch. A. 1987;411:129–135. doi: 10.1007/BF00712736[↩]
- Paulli M., Rosso R., Kindl S., Boveri E., Marocolo D., Chioda C., Agostini C., Magrini U., Facchetti F. Immunophenotypic characterization of the cell infiltrate in five cases of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) Hum. Pathol. 1992;23:647–654. doi: 10.1016/0046-8177(92)90320-3[↩]
- Aoyama K., Terashima K., Imai Y., Katsushima N., Okuyama Y., Niikawa K., Mukada T., Takahashi K. Sinus histiocytosis with massive lymphadenopathy. A histogenic analysis of histiocytes found in the fourth Japanese case. Acta Pathol. Jpn. 1984;34:375–388. doi: 10.1111/j.1440-1827.1984.tb07565.x[↩]
- Miettinen M., Paljakka P., Haveri P., Saxen E. Sinus histiocytosis with massive lymphadenopathy. A nodal and extranodal proliferation of S-100 protein positive histiocytes? Am. J. Clin. Pathol. 1987;88:270–277. doi: 10.1093/ajcp/88.3.270[↩]
- Garces S., Medeiros L.J., Patel K.P., Li S., Pina-Oviedo S., Li J., Garces J.C., Khoury J.D., Yin C.C. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod. Pathol. 2017;30:1367–1377. doi: 10.1038/modpathol.2017.55[↩][↩]
- Baraban E., Sadigh S., Rosenbaum J., Van Arnam J., Bogusz A.M., Mehr C., Bagg A. Cyclin D1 expression and novel mutational findings in Rosai-Dorfman disease. Br. J. Haematol. 2019;186:837–844. doi: 10.1111/bjh.16006[↩][↩][↩][↩]
- Garces S., Medeiros L.J., Marques-Piubelli M.L., Coelho Siqueira S.A., Miranda R.N., Cuglievan B., Sriganeshan V., Medina A.M., Garces J.C., Saluja K., et al. Cyclin D1 expression in Rosai-Dorfman disease: A near-constant finding that is not invariably associated with mitogen-activated protein kinase/extracellular signal-regulated kinase pathway activation. Hum. Pathol. 2022;121:36–45. doi: 10.1016/j.humpath.2021.12.013[↩][↩][↩]
- Abdulla Z., Turley H., Gatter K., Pezzella F. Immunohistological recognition of cyclin D1 expression by non-lymphoid cells among lymphoid neoplastic cells. APMIS. 2014;122:183–191. doi: 10.1111/apm.12123[↩]
- Wu J., Zhang Y., Sun L., Zhai Q. Cyclin D1 expression by histiocytes may mimic cyclin D1-positive proliferation centres of chronic lymphocytic leukaemia/small lymphocytic lymphoma. Pathol.-Res. Pract. 2018;214:72–75. doi: 10.1016/j.prp.2017.11.010[↩]
- Ravindran A., Goyal G., Go R.S., Rech K.L., Mayo Clinic Histiocytosis Working G. Rosai-Dorfman Disease Displays a Unique Monocyte-Macrophage Phenotype Characterized by Expression of OCT2. Am. J. Surg. Pathol. 2021;45:35–44. doi: 10.1097/PAS.0000000000001617[↩][↩][↩][↩]
- Mastropolo R., Close A., Allen S.W., McClain K.L., Maurer S., Picarsic J. BRAF-V600E-mutated Rosai-Dorfman-Destombes disease and Langerhans cell histiocytosis with response to BRAF inhibitor. Blood Adv. 2019;3:1848–1853. doi: 10.1182/bloodadvances.2019000093[↩][↩]
- Diamond E.L., Durham B.H., Haroche J., Yao Z., Ma J., Parikh S.A., Wang Z., Choi J., Kim E., Cohen-Aubart F., et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016;6:154–165. doi: 10.1158/2159-8290.CD-15-0913[↩]
- Matter M.S., Bihl M., Juskevicius D., Tzankov A. Is Rosai-Dorfman disease a reactve process? Detection of a MAP2K1 L115V mutation in a case of Rosai-Dorfman disease. Virchows Arch. 2017;471:545–547. doi: 10.1007/s00428-017-2173-4[↩]
- Jacobsen E., Shanmugam V., Jagannathan J. Rosai-Dorfman Disease with Activating KRAS Mutation—Response to Cobimetinib. N. Engl. J. Med. 2017;377:2398–2399. doi: 10.1056/NEJMc1713676[↩]
- Durham B.H., Lopez Rodrigo E., Picarsic J., Abramson D., Rotemberg V., De Munck S., Pannecoucke E., Lu S.X., Pastore A., Yoshimi A., et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat. Med. 2019;25:1839–1842. doi: 10.1038/s41591-019-0653-6[↩][↩]
- Lee L.H., Gasilina A., Roychoudhury J., Clark J., McCormack F.X., Pressey J., Grimley M.S., Lorsbach R., Ali S., Bailey M., et al. Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI Insight. 2017;2:e89473. doi: 10.1172/jci.insight.89473[↩]
- Wu K.J., Li S.H., Liao J.B., Chiou C.C., Wu C.S., Chen C.C. NRAS Mutations May Be Involved in the Pathogenesis of Cutaneous Rosai Dorfman Disease: A Pilot Study. Biology. 2021;10:396. doi: 10.3390/biology10050396[↩]
- Fatobene G., Haroche J., Helias-Rodzwicz Z., Charlotte F., Taly V., Ferreira A.M., Abdo A.N.R., Rocha V., Emile J.F. BRAF V600E mutation detected in a case of Rosai-Dorfman disease. Haematologica. 2018;103:e377–e379. doi: 10.3324/haematol.2018.190934[↩]
- Richardson T.E., Wachsmann M., Oliver D., Abedin Z., Ye D., Burns D.K., Raisanen J.M., Greenberg B.M., Hatanpaa K.J. BRAF mutation leading to central nervous system rosai-dorfman disease. Ann. Neurol. 2018;84:147–152. doi: 10.1002/ana.25281[↩]
- Chen J., Zhao A.L., Duan M.H., Cai H., Gao X.M., Liu T., Sun J., Liang Z.Y., Zhou D.B., Cao X.X., et al. Diverse kinase alterations and myeloid-associated mutations in adult histiocytosis. Leukemia. 2022;36:573–576. doi: 10.1038/s41375-021-01439-3[↩][↩][↩]
- Bruce-Brand C., Schneider J.W., Schubert P. Rosai-Dorfman disease: An overview. J. Clin. Pathol. 2020;73:697–705. doi: 10.1136/jclinpath-2020-206733[↩]
- Sacchi S., Artusi T., Selleri L., Temperani P., Zucchini P., Vecchi A., Emilia G., Torelli U. Sinus histiocytosis with massive lymphadenopathy: Immunological, cytogenetic and molecular studies. Blut. 1990;60:339–344. doi: 10.1007/BF01737849[↩]
- Pulsoni A., Anghel G., Falcucci P., Matera R., Pescarmona E., Ribersani M., Villiva N., Mandelli F. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Report of a case and literature review. Am. J. Hematol. 2002;69:67–71. doi: 10.1002/ajh.10008[↩]
- Löhr HF, Gödderz W, Wölfe T, Heike M, Knuth A, Meyer zum Büschenfelde KH, Dippold W. Long-term survival in a patient with Rosai-Dorfman disease treated with interferon-alpha. Eur J Cancer. 1995 Dec;31A(13-14):2427-8. doi: 10.1016/0959-8049(95)00375-4[↩]
- Alqanatish JT, Houghton K, Bond M, Senger C, Tucker LB. Rituximab treatment in a child with rosai-dorfman disease and systemic lupus erythematosus. J Rheumatol. 2010 Aug 1;37(8):1783-4. doi: 10.3899/jrheum.091275[↩]
- Pagel JM, Lionberger J, Gopal AK, Sabath DE, Loeb K. Therapeutic use of Rituximab for sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Am J Hematol. 2007 Dec;82(12):1121-2. doi: 10.1002/ajh.21024[↩]
- Gebhardt C, Averbeck M, Paasch U, Ugurel S, Kurzen H, Stumpp P, Simon JC, Treudler R. A case of cutaneous Rosai-Dorfman disease refractory to imatinib therapy. Arch Dermatol. 2009 May;145(5):571-4. doi: 10.1001/archdermatol.2008.597[↩][↩][↩]
- Mebazaa A, Trabelsi S, Denguezli M, Sriha B, Belajouza C, Nouira R. Extensive purely cutaneous Rosai-Dorfman disease responsive to acitretin. Int J Dermatol. 2007 Nov;46(11):1208-10. doi: 10.1111/j.1365-4632.2007.03234.x[↩]
- Aouba A, Terrier B, Vasiliu V, Candon S, Brousse N, Varet B, Hermine O. Dramatic clinical efficacy of cladribine in Rosai-Dorfman disease and evolution of the cytokine profile: towards a new therapeutic approach. Haematologica. 2006 Dec;91(12 Suppl):ECR52.[↩][↩][↩][↩]
- Simko SJ, Tran HD, Jones J, Bilgi M, Beaupin LK, Coulter D, Garrington T, McCavit TL, Moore C, Rivera-Ortegón F, Shaffer L, Stork L, Turcotte L, Welsh EC, Hicks MJ, McClain KL, Allen CE. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr Blood Cancer. 2014 Mar;61(3):479-87. doi: 10.1002/pbc.24772[↩][↩][↩][↩]
- Horneff G, Jürgens H, Hort W, Karitzky D, Göbel U. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): response to methotrexate and mercaptopurine. Med Pediatr Oncol. 1996 Sep;27(3):187-92. doi: 10.1002/(SICI)1096-911X(199609)27:3<187::AID-MPO10>3.0.CO;2-D[↩][↩][↩]
- Horneff G, Jürgens H, Hort W, Karitzky D, Göbel U. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): response to methotrexate and 6-mercaptopurine. Med Pediatr Oncol. 1996 Sep;27(3):187-92. doi: 10.1002/(SICI)1096-911X(199609)27:3<187::AID-MPO10>3.0.CO;2-D[↩]
- Le Guenno G, Galicier L, Uro-Coste E, Petitcolin V, Rieu V, Ruivard M. Successful treatment with azathioprine of relapsing Rosai-Dorfman disease of the central nervous system. J Neurosurg. 2012 Sep;117(3):486-9. doi: 10.3171/2012.5.JNS12148[↩][↩]
- Konca C, Özkurt ZN, Deger M, Akı Z, Yağcı M. Extranodal multifocal Rosai-Dorfman disease: response to 2-chlorodeoxyadenosine treatment. Int J Hematol. 2009 Jan;89(1):58-62. doi: 10.1007/s12185-008-0192-2[↩][↩][↩][↩]
- Maklad AM, Bayoumi Y, Tunio M, Alshakweer W, Dahar MA, Akbar SA. Steroid-resistant extranodal rosai-dorfman disease of cheek mass and ptosis treated with radiation therapy. Case Rep Hematol. 2013;2013:428297. doi: 10.1155/2013/428297[↩][↩][↩][↩]
- Toguri D, Louie AV, Rizkalla K, Franklin J, Rodrigues G, Venkatesan V. Radiotherapy for steroid-resistant laryngeal Rosai-Dorfman disease. Curr Oncol. 2011 Jun;18(3):e158-62. doi: 10.3747/co.v18i3.761[↩][↩][↩][↩]
- Baildam EM, Ewing CI, D’Souza SW, Stevens RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): response to acyclovir. J R Soc Med. 1992 Mar;85(3):179-80. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1294827/pdf/jrsocmed00113-0067.pdf[↩]
- Pulsoni A, Anghel G, Falcucci P, Matera R, Pescarmona E, Ribersani M, Villivà N, Mandelli F. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review. Am J Hematol. 2002 Jan;69(1):67-71. doi: 10.1002/ajh.10008[↩][↩][↩][↩][↩][↩][↩][↩]
- Forest F, N’guyen AT, Fesselet J, Metellus P, Bouvier C, de Paula AM, Roche PH, Figarella-Branger D. Meningeal Rosai-Dorfman disease mimicking meningioma. Ann Hematol. 2014 Jun;93(6):937-40. doi: 10.1007/s00277-013-1994-8[↩][↩][↩]
- Z’Graggen WJ, Sturzenegger M, Mariani L, Keserue B, Kappeler A, Vajtai I. Isolated Rosai-Dorfman disease of intracranial meninges. Pathol Res Pract. 2006;202(3):165-70. doi: 10.1016/j.prp.2005.11.004[↩][↩]
- Shulman S, Katzenstein H, Abramowsky C, Broecker J, Wulkan M, Shehata B. Unusual presentation of Rosai-Dorfman disease (RDD) in the bone in adolescents. Fetal Pediatr Pathol. 2011;30(6):442-7. doi: 10.3109/15513815.2011.618873[↩][↩]
- McPherson CM, Brown J, Kim AW, DeMonte F. Regression of intracranial rosai-dorfman disease following corticosteroid therapy. Case report. J Neurosurg. 2006 May;104(5):840-4. doi: 10.3171/jns.2006.104.5.840[↩][↩]
- Petrushkin H, Salisbury J, O’Sullivan E. Intralesional steroid for orbital manifestations of Rosai-Dorfman disease. Clin Exp Ophthalmol. 2015 Jul;43(5):483-5. doi: 10.1111/ceo.12476[↩][↩]
- Ottaviano G, Doro D, Marioni G, Mirabelli P, Marchese-Ragona R, Tognon S, Marino F, Staffieri A. Extranodal Rosai-Dorfman disease: involvement of eye, nose and trachea. Acta Otolaryngol. 2006 Jun;126(6):657-60. doi: 10.1080/00016480500452582[↩][↩]
- Sakallioglu O, Gok F, Kalman S, Atay AA, Kaya A, Duzova A, Gokcay E. Minimal change nephropathy in a 7-year-old boy with Rosai-Dorfman disease. J Nephrol. 2006 Mar-Apr;19(2):211-4.[↩][↩]
- Cooper SL, Arceci RJ, Gamper CJ, Teachey DT, Schafer ES. Successful Treatment of Recurrent Autoimmune Cytopenias in the Context of Sinus Histiocytosis With Massive Lymphadenopathy Using Sirolimus. Pediatr Blood Cancer. 2016 Feb;63(2):358-60. doi: 10.1002/pbc.25770[↩][↩][↩]
- Diamond EL, Durham BH, Dogan A, et al.. Phase 2 trial of single-agent cobimetinib for adults with BRAF-mutant and wildtype histiocytic disorders [abstract]. Blood 2017;130(suppl 1). Abstract 257.[↩][↩]
- Garces S, Medeiros LJ, Patel KP, Li S, Pina-Oviedo S, Li J, Garces JC, Khoury JD, Yin CC. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod Pathol. 2017 Oct;30(10):1367-1377. doi: 10.1038/modpathol.2017.55[↩]
- Tasso M, Esquembre C, Blanco E, Moscardó C, Niveiro M, Payá A. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) treated with 2-chlorodeoxyadenosine. Pediatr Blood Cancer. 2006 Oct 15;47(5):612-5. doi: 10.1002/pbc.20668[↩][↩][↩][↩]
- Visser J, Dyer MJS. Refractory, atypical Rosai-Dorfman disease successfully treated with a cytarabine-containing Langerhans cell histiocytosis treatment regimen – a case report. Pediatr Blood Cancer. 2014;61(11):2142.[↩][↩][↩]
- Scheel MM, Rady PL, Tyring SK, Pandya AG. Sinus histiocytosis with massive lymphadenopathy: presentation as giant granuloma annulare and detection of human herpesvirus 6. J Am Acad Dermatol. 1997 Oct;37(4):643-6. doi: 10.1016/s0190-9622(97)70186-5[↩][↩]
- Jabali Y, Smrcka V, Pradna J. Rosai-Dorfman disease: successful long-term results by combination chemotherapy with prednisone, 6-mercaptopurine, methotrexate, and vinblastine: a case report. Int J Surg Pathol. 2005 Jul;13(3):285-9. doi: 10.1177/106689690501300311[↩][↩][↩]
- Inoue S, Onwuzurike N. Venorelbine and methotrexate for the treatment of Rosai-Dorfman disease. Pediatr Blood Cancer. 2005 Jul;45(1):84-5; author reply 86. doi: 10.1002/pbc.20361[↩][↩][↩]
- Arnao V, Riolo M, Savettieri G, Aridon P. Mercaptopurine Treatment in an Adult Man with Orbital and Intracranial Rosai-Dorfman Disease. Case Rep Neurol Med. 2016;2016:1030478. doi: 10.1155/2016/1030478[↩][↩][↩]
- Rivera D, Pérez-Castillo M, Fernández B, Stoeter P. Long-term follow-up in two cases of intracranial Rosai-Dorfman Disease complicated by incomplete resection and recurrence. Surg Neurol Int. 2014 Feb 28;5:30. doi: 10.4103/2152-7806.128003[↩][↩]
- Liu P, Wang P, Du J, Zhang J. Successful treatment of refractory cutaneous Rosai-Dorfman disease with vincristine. J Dermatol. 2015 Jan;42(1):97-8. doi: 10.1111/1346-8138.12694[↩][↩]
- Chen E, Pavlidakey P, Sami N. Rosai-Dorfman disease successfully treated with thalidomide. JAAD Case Rep. 2016 Sep 28;2(5):369-372. doi: 10.1016/j.jdcr.2016.08.006[↩][↩]
- Rubinstein M, Assal A, Scherba M, Elman J, White R, Verma A, Strakhan M, Mohammadi F, Janakiram M. Lenalidomide in the treatment of Rosai Dorfman disease–a first in use report. Am J Hematol. 2016 Feb;91(2):E1. doi: 10.1002/ajh.24225[↩][↩]
- Petschner F, Walker UA, Schmitt-Gräff A, Uhl M, Peter HH. “Katastrophaler SLE” mit Rosai-Dorfman-Sinushistiozytose. Erfolgreiche Behandlung mit Anti-CD20/Rituximab [“Catastrophic systemic lupus erythematosus” with Rosai-Dorfman sinus histiocytosis. Successful treatment with anti-CD20/rutuximab]. Dtsch Med Wochenschr. 2001 Sep 14;126(37):998-1001. German. doi: 10.1055/s-2001-17109[↩][↩]
- Utikal J, Ugurel S, Kurzen H, Erben P, Reiter A, Hochhaus A, Nebe T, Hildenbrand R, Haberkorn U, Goerdt S, Schadendorf D. Imatinib as a treatment option for systemic non-Langerhans cell histiocytoses. Arch Dermatol. 2007 Jun;143(6):736-40. doi: 10.1001/archderm.143.6.736[↩][↩]
- Jacobsen E, Shanmugam V, Jagannathan J. Rosai-Dorfman Disease with Activating KRAS Mutation – Response to Cobimetinib. N Engl J Med. 2017 Dec 14;377(24):2398-2399. doi: 10.1056/NEJMc1713676[↩][↩]
- Cohen Aubart F, Emile JF, Carrat F, Charlotte F, Benameur N, Donadieu J, Maksud P, Idbaih A, Barete S, Hoang-Xuan K, Amoura Z, Haroche J. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood. 2017 Sep 14;130(11):1377-1380. doi: 10.1182/blood-2017-03-771873[↩][↩]
- Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, Wang Z, Choi J, Kim E, Cohen-Aubart F, Lee SC, Gao Y, Micol JB, Campbell P, Walsh MP, Sylvester B, Dolgalev I, Aminova O, Heguy A, Zappile P, Nakitandwe J, Ganzel C, Dalton JD, Ellison DW, Estrada-Veras J, Lacouture M, Gahl WA, Stephens PJ, Miller VA, Ross JS, Ali SM, Briggs SR, Fasan O, Block J, Héritier S, Donadieu J, Solit DB, Hyman DM, Baselga J, Janku F, Taylor BS, Park CY, Amoura Z, Dogan A, Emile JF, Rosen N, Gruber TA, Abdel-Wahab O. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 2016 Feb;6(2):154-65. doi: 10.1158/2159-8290.CD-15-0913[↩]
- Lima FB, Barcelos PS, Constâncio AP, Nogueira CD, Melo-Filho AA. Rosai-Dorfman disease with spontaneous resolution: case report of a child. Rev Bras Hematol Hemoter. 2011;33(4):312-4. doi: 10.5581/1516-8484.20110083[↩]
- Adeleye AO, Amir G, Fraifeld S, Shoshan Y, Umansky F, Spektor S. Diagnosis and management of Rosai-Dorfman disease involving the central nervous system. Neurol Res. 2010 Jul;32(6):572-8. doi: 10.1179/016164109X12608733393836[↩]
- Goyal G., Ravindran A., Young J.R., Shah M.V., Bennani N.N., Patnaik M.M., Nowakowski G.S., Thanarajasingam G., Habermann T.M., Vassallo R., et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348–357. doi: 10.3324/haematol.2019.219626[↩][↩]
- Gianfreda D, Nicastro M, Galetti M, Alberici F, Corradi D, Becchi G, Baldari G, De Filippo M, Ferretti S, Moroni G, Foti R, Di Gangi M, Jeannin G, Saffroy R, Emile JF, Buzio C, Vaglio A. Sirolimus plus prednisone for Erdheim-Chester disease: an open-label trial. Blood. 2015 Sep 3;126(10):1163-71. doi: 10.1182/blood-2015-01-620377[↩]
- Nadal M, Kervarrec T, Machet MC, Petrella T, Machet L. Cutaneous Rosai-Dorfman Disease Located on the Breast: Rapid Effectiveness of Methotrexate After Failure of Topical Corticosteroids, Acitretin and Thalidomide. Acta Derm Venereol. 2015 Jul;95(6):758-9. doi: 10.2340/00015555-2057[↩][↩]
- Le Guenno G, Galicier L, Fieschi C, Meignin V, Chabrol A, Oksenhendler E. Dramatic efficiency of pegylated interferon in sinus histiocytosis with massive lymphadenopathy. Br J Dermatol. 2011 Jan;164(1):213-5. doi: 10.1111/j.1365-2133.2010.10051.x[↩]
- Matter MS, Bihl M, Juskevicius D, Tzankov A. Is Rosai-Dorfman disease a reactve process? Detection of a MAP2K1 L115V mutation in a case of Rosai-Dorfman disease. Virchows Arch. 2017 Oct;471(4):545-547. doi: 10.1007/s00428-017-2173-4[↩]
- Vaiselbuh S.R., Bryceson Y.T., Allen C.E., Whitlock J.A., Abla O. Updates on histiocytic disorders. Pediatr. Blood Cancer. 2014;61:1329–1335. doi: 10.1002/pbc.25017[↩]
- Papo M., Cohen-Aubart F., Trefond L., Bauvois A., Amoura Z., Emile J.F., Haroche J. Systemic Histiocytosis (Langerhans Cell Histiocytosis, Erdheim-Chester Disease, Destombes-Rosai-Dorfman Disease): From Oncogenic Mutations to Inflammatory Disorders. Curr. Oncol. Rep. 2019;21:62. doi: 10.1007/s11912-019-0810-6[↩]
- Paryani N.N., Daugherty L.C., O’Connor M.I., Jiang L. Extranodal rosai-dorfman disease of the bone treated with surgery and radiotherapy. Rare Tumors. 2014;6:5531. doi: 10.4081/rt.2014.5531[↩]
- Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. An analysis of 14 deaths occurring in a patient registry. Cancer. 1984 Nov 1;54(9):1834-40. doi: 10.1002/1097-0142(19841101)54:9<1834::aid-cncr2820540911>3.0.co;2-f[↩][↩]
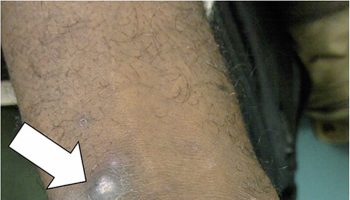
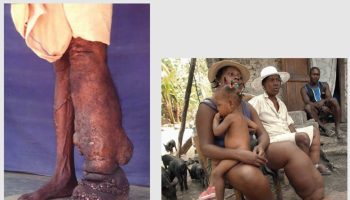
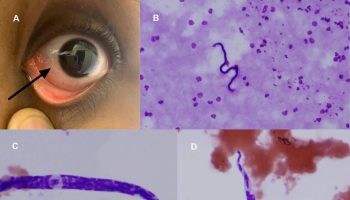
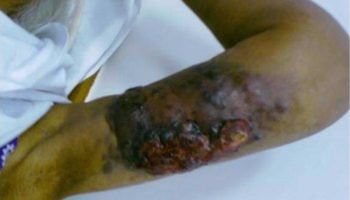
{kind=link}