Contents
Propionic acidemia
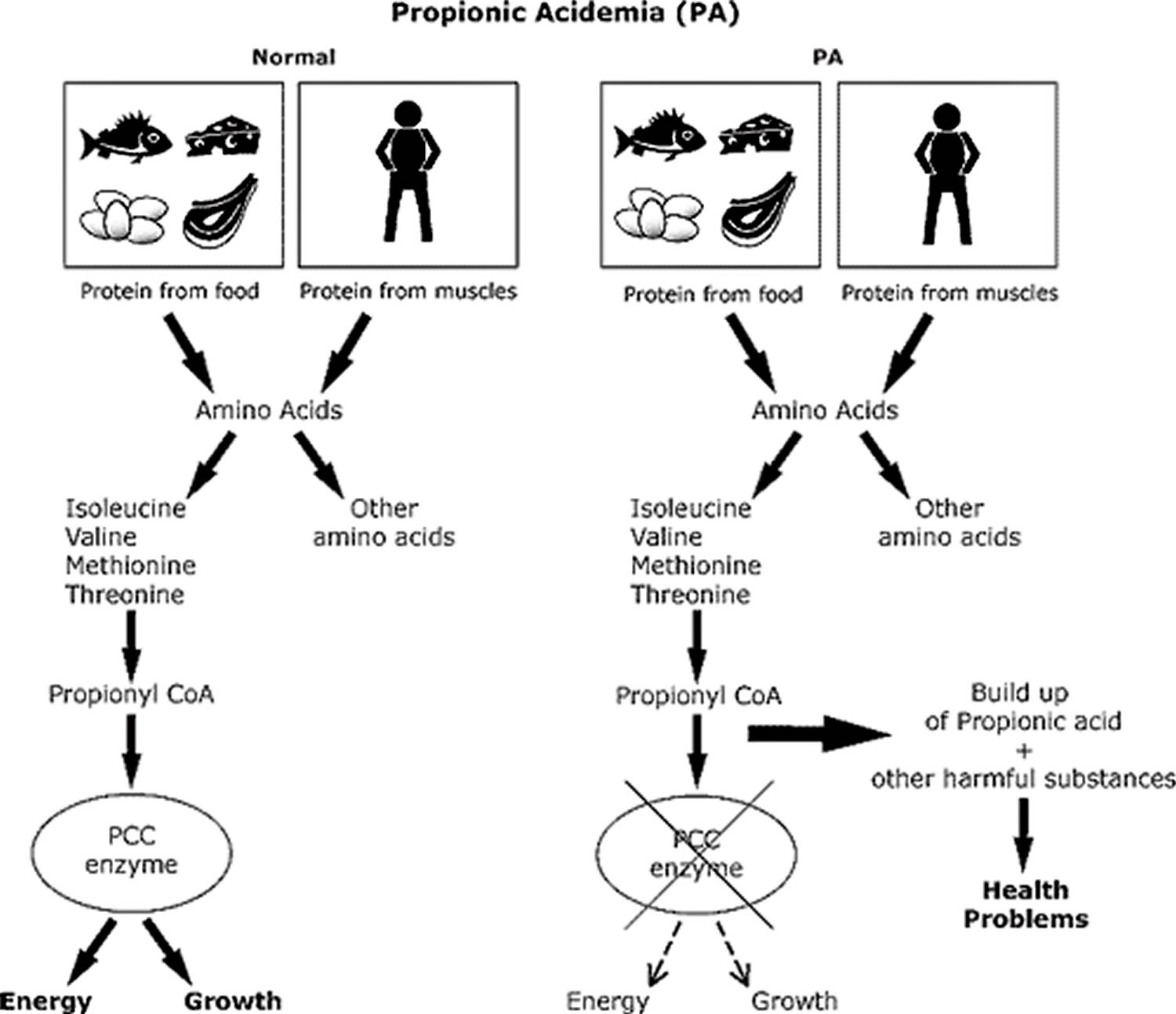
Propionic acidemia also known as propionyl-CoA carboxylase deficiency, is an inherited disorder in which the body is unable to process certain parts of proteins and lipids (fats) properly that is caused by deficiency of propionyl-CoA carboxylase, the enzyme that catalyzes the conversion of propionyl-CoA to methylmalonyl-CoA. Propionic acidemia is classified as an organic acid disorder, which is a condition that leads to an abnormal buildup of particular acids known as organic acids. Abnormal levels of organic acids in the blood (organic acidemia), urine (organic aciduria), and tissues can be toxic and can cause serious health problems. Signs and symptoms of propionic acidemia generally develop within the firsts few days after birth and may include poor feeding, vomiting, loss of appetite, weak muscle tone (hypotonia), and lack of energy (lethargy). Without early diagnosis and treatment, these symptoms may progress to more serious medical problems, such as heart abnormalities, seizures, coma, and possibly death. Propionic acidemia is caused by changes (mutations) in the PCCA and PCCB genes and is inherited in an autosomal recessive manner 1.
Propionic acidemia affects about 1 in 100,000 people in the United States 1. Propionic acidemia appears to be more common in several populations worldwide, including the Inuit population of Greenland, some Amish communities, and Saudi Arabians.
The spectrum of propionic acidemia ranges from neonatal-onset to late-onset disease 2.
- Neonatal-onset propionic acidemia, the most common form, is characterized by a healthy newborn with poor feeding and decreased arousal in the first few days of life, followed by progressive encephalopathy of unexplained origin. Without prompt diagnosis and management, this is followed by progressive encephalopathy manifesting as lethargy, seizures, or coma that can result in death. It is frequently accompanied by metabolic acidosis with anion gap, lactic acidosis, ketonuria, hypoglycemia, hyperammonemia, and cytopenias.
- Individuals with late-onset propionic acidemia may remain asymptomatic and suffer a metabolic crisis under catabolic stress (e.g., illness, surgery, fasting) or may experience a more insidious onset with the development of multiorgan complications including vomiting, protein intolerance, failure to thrive, hypotonia, developmental delays or regression, movement disorders, or cardiomyopathy.
- Isolated cardiomyopathy can be observed on rare occasion in the absence of clinical metabolic decompensation or neurocognitive deficits.
Manifestations of neonatal and late-onset propionic acidemia over time can include growth impairment, intellectual disability, seizures, basal ganglia lesions, pancreatitis, and cardiomyopathy. Other rarely reported complications include optic atrophy, hearing loss, premature ovarian insufficiency, and chronic renal failure.
In most cases, the features of propionic acidemia become apparent within a few days after birth. The initial symptoms include poor feeding, vomiting, loss of appetite, weak muscle tone (hypotonia), and lack of energy (lethargy). These symptoms sometimes progress to more serious medical problems, including heart abnormalities, seizures, coma, and possibly death.
Less commonly, the signs and symptoms of propionic acidemia appear during childhood and may come and go over time. Some affected children experience intellectual disability or delayed development. In children with this later-onset form of the condition, episodes of more serious health problems can be triggered by prolonged periods without food (fasting), fever, or infections.
Treatment generally includes a special diet and addressing symptoms during a metabolic crisis 3.
Does propionic acidemia carry risk factors for hearing loss?
Hearing loss is considered to be a rare complication associated with propionic acidemia. Very limited information is available on this subject and conclusions by different research studies are conflicting. Two different research studies noted an elevated incidence of deafness in patients with propionic acidemia. Although less common than other symptoms, one study stated that 13% of their participants reported impaired hearing ability. This study concluded that there is likely a connection between propionic acidemia and hearing loss 4. Another study from 2008 5 stated that according to present knowledge, no connection can be assumed between either of the two mutations that cause propionic acidemia and the severe sensorineural hearing loss that has been reported. No additional information was found about potentially elevated incidence of hearing loss with this condition; therefore, it is not known if hearing loss may possibly be a symptom of the underlying disorder, if it may be caused by metabolic crisis and subsequent neurologic deficits, or if it may be related in some other way (if at all).
Propionic acidemia causes
Mutations in the PCCA and PCCB genes cause propionic acidemia. The PCCA gene is located on the long arm (q) of chromosome 13 (13q22), and the PCCB gene has been mapped to the long arm of chromosome 3 (3q21-q22). These genes provide instructions for making two parts (subunits) of an enzyme called propionyl-CoA carboxylase, which plays a role in the normal breakdown of proteins. The propionyl CoA carboxylase enzyme is composed of two subunits, known as alpha and beta. The alpha subunit is regulated (encoded) by a gene known as the PCCA gene, and the beta subunit is encoded by the PCCB gene. Specifically, propionyl CoA carboxylase enzyme is required for the proper breakdown of the amino acids isoleucine, valine, threonine, and methionine, which are the building blocks of proteins. Propionyl-CoA carboxylase also helps break down certain types of fat and cholesterol in the body. Mutations in the PCCA or PCCB gene disrupt the function of the propionyl CoA carboxylase enzyme and prevent the normal breakdown of these molecules. As a result, a substance called propionyl-CoA and other potentially harmful compounds can build up to toxic levels in the body. This buildup damages the brain and nervous system, causing the serious health problems associated with propionic acidemia.
Propionic acidemia inheritance pattern
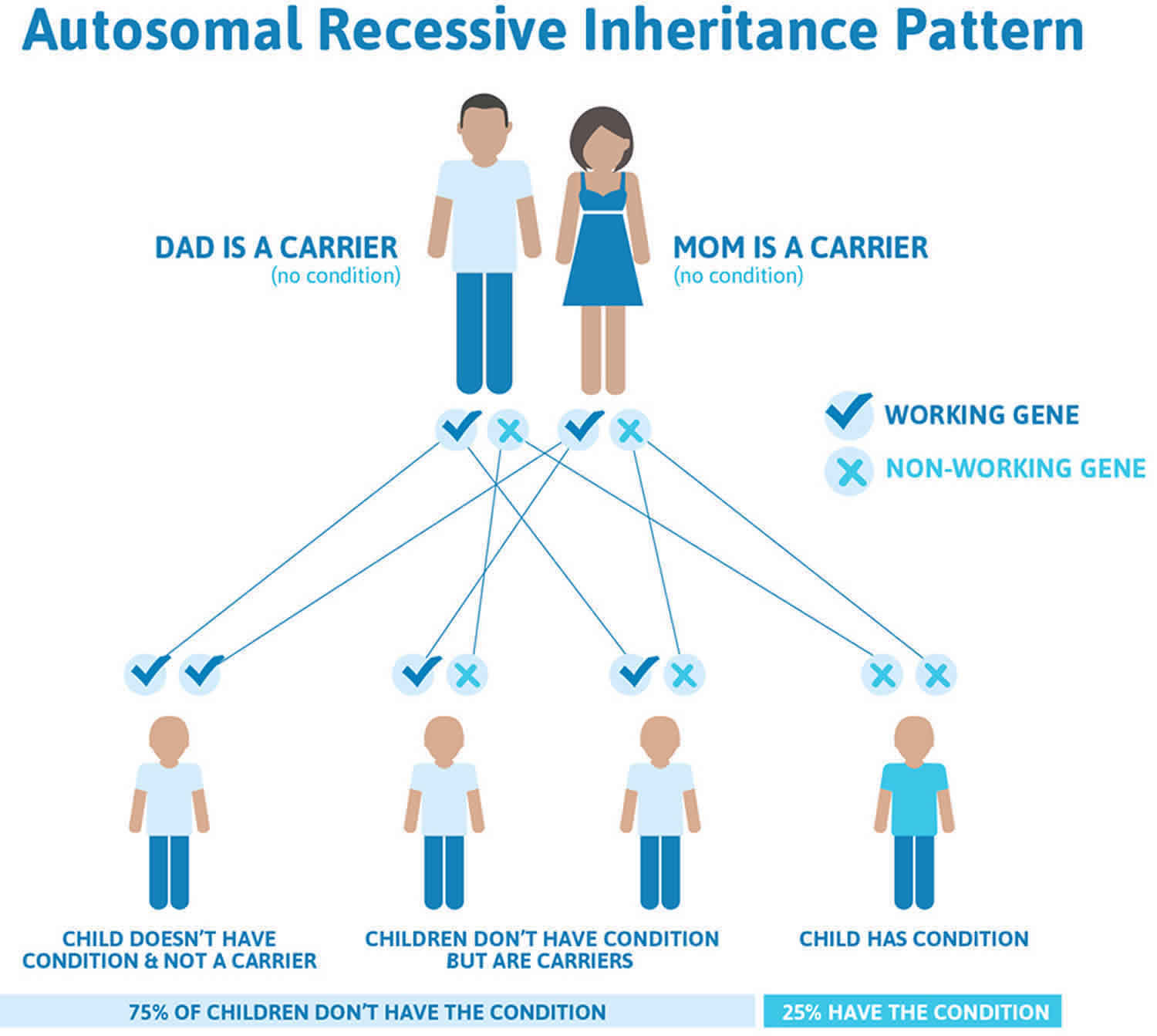
Propionic acidemia is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Propionic acidemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Propionic acidemia symptoms
In infants with propionic acidemia, symptoms most commonly develop within a few days after they are born (neonatal-onset). Such abnormalities may include poor feeding, loss of appetite, vomiting, lack of energy (lethargy), diminished muscle tone (hypotonia), failure to grow and gain weight at the expected rate (failure to thrive), and excessively low levels of bodily fluids (dehydration). Approximately 30 percent of affected infants symptoms can become more serious and may lead to heart abnormalities, seizures, coma, and possibly death. The recurrence or worsening (exacerbation) of symptoms may be associated with infection, constipation, or the consumption of high amounts of protein. In some affected infants, symptom episodes may be separated by periods of apparently normal health and development.
There is another form of propionic acidemia that is less common and the symptoms appear during childhood (late-onset). In such cases, affected infants may not experience sudden, acute episodes of ketoacidosis and may tend to come to medical attention due to mental retardation. Because breast milk has a lower, protein content than cow’s milk or formulas, some researchers suggest that breastfeeding may contribute to a later onset of symptoms and milder disease in some affected infants. The symptoms are similar to the neonatal-onset form of this disorder, but the symptoms may also come and go over time. More serious health problems can be triggered by prolonged periods without food (fasting), fever, or infections. Some children with this form of propionic acidemia experience intellectual disability, delayed development, or developmental regression (where the child looses skills over time).
In infants with propionic acidemia, episodes are associated with certain characteristic findings that are apparent upon laboratory testing. These findings may include excessive levels of acids in the blood and bodily tissues (acidosis); increased levels of acids in the blood and bodily tissues (acidosis); increased levels of the amino acid glycine in bodily fluids (hyperglycinemia); abnormal accumulations of certain chemical substances (ketone bodies) in bodily tissues and fluids due to excessive breakdown of fats (ketosis); and high levels of ammonia in the blood (hyperammonemia). In addition, there may be low levels of circulating platelets (thrombocytopenia) and certain white blood cells (neutropenia). Without appropriate treatment, episodes of vomiting, lethargy, dehydration, and acidosis and ketosis (ketoacidosis) may lead to a state of unconsciousness (coma) and potentially life-threatening complications.
Propionic acidemia diagnosis
In some cases, propionic acidemia may be diagnosed before birth (prenatally) by measuring the concentration of the characteristic metabolites in amniotic fluid or the activity of the propionyl CoA carboxylase enzyme in fluid or tissue samples obtained from the fetus or uterus during pregnancy (amniocentesis or chorionic villus sampling [CVS]). During amniocentesis, a sample of fluid surrounding the developing fetus is removed and analyzed. CVS (chorionic villus sampling) involves the removal and examination of tissue from a portion of the placenta. This disorder can be identified at birth through expanded newborn screening with tandem mass spectrometry.
In most affected infants, chorionic villus sampling is diagnosed or confirmed in the first weeks of life, based upon a thorough clinical evaluation, a detailed patient and family history, and a variety of specialized tests including expanded newborn screening showing elevated C3 (propionylcarnitine). Laboratory studies (assays) are typically conducted on certain white blood cells (leukocytes) or cultured skin cells (fibroblasts) to confirm deficient activity of the propionyl CoA carboxylase enzyme. Additional laboratory studies may reveal excessive levels of acids and increased accumulations of ketone bodies in bodily tissues and fluids (ketoacidosis); increased levels of glycine in the blood and urine (hyperglycinemia and hyperglycinuria); high levels of ammonia in the blood (hyperammonemia); and/or decreased levels of circulating platelets and white blood cells (thrombocytopenia and neutropenia).
Testing of urine organic acids in persons who are symptomatic or those detected by newborn screening reveals elevated 3-hydroxypropionate and the presence of methylcitrate, tiglylglycine, propionylglycine, and lactic acid. Testing of plasma amino acids reveals elevated glycine. Confirmation of the diagnosis relies on detection of biallelic pathogenic variants in PCCA or PCCB gene or of deficient propionyl CoA carboxylase enzymatic activity. In individuals with equivocal molecular genetic test results, a combination of enzymatic and molecular diagnostics may be necessary.
Propionic acidemia treatment
Most patients with propionyl coenzyme A (CoA) carboxylase deficiency (propionic acidemia) are so ill at presentation that they have already been admitted to a hospital, which should facilitate appropriate diagnosis and early treatment. During acute episodes, the treatment of infants with propionic acidemia may require fluid and electrolyte therapy; measures to ensure appropriate nutritional and caloric intake (e.g., parenteral hyperalimentation); administration of certain medications to prevent or treat bacterial infection; and other supportive measures as required. In infants with severe disease (e.g., severe acidosis, hyperammonemia), treatment may require procedures that remove excess waste products from the blood (hemodialysis or peritoneal dialysis). During hemodialysis, waste products are removed by filtering the blood through an artificial kidney machine. Peritoneal dialysis is a technique during which the peritoneum is used as a natural filtering membrane. The peritoneum is the two-layered membrane that lines the abdominal wall and covers abdominal organs. In addition, until the diagnosis is confirmed, physicians may administer biotin, a B complex vitamin that plays a role in the metabolism of certain fatty and amino acids.
Ketoacidosis is best treated with increased carbohydrate calories, bicarbonate replacement, and increased fluids to enhance excretion. In severely ill patients, metabolic reversal can be expedited by an insulin drip, but this should only be administered in an intensive care setting.
Reinitiate protein feeding to a level of protein no greater than 1.5 g/kg/d after the patient’s condition has normalized. From this point, the patient should be under the care of a biochemical geneticist who may prescribe a special diet prior to discharge.
Long-term treatment includes administration of a low-protein diet, possibly in combination with artificial (synthetic) proteins that are low in certain amino acids (i.e., propionate precursors, e.g., isoleucine, valine, threonine, and methionine). Infants and children with the disorder may develop secondary deficiency of carnitine, a substance that plays a role in metabolism and the proper use of fatty acids. In such cases, therapy includes administration of the amino acid derivative L-carnitine.
Early intervention may be important in ensuring that children with propionic acidemia reach their potential. Special services that may be beneficial to affected children may include special remedial education and other medical, social, and/or vocational services.
Children with propionic acidemia need to eat more starchy foods and drink more fluids when they are ill – even if they aren’t hungry – or they could have a metabolic crisis. In addition, they should avoid eating protein during any illness.
Many children with propionic acidemia need to be treated in the hospital during illness to avoid serious health problems. Ask your metabolic doctor if you should carry a special travel letter with medical instructions for your child’s care.
Genetic counseling will also be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Regular blood and urine tests
- Tracking of ketones: Your child will have periodic urine tests to check the level of ketones. These can be done at home or at the doctor’s office. Ketones are substances formed when body fat is broken down for energy. This can happen after going without food for long periods of time, as the result of an illness, or during periods of heavy exercise. Ketones in the urine may signal the start of a metabolic crisis.
- Blood tests: Your child will have regular blood tests to measure the levels of amino acids. Urine tests may also be done. Your child’s diet and medication may need to be adjusted based on the results of these tests.
Propionic acidemia liver transplant
Liver transplant surgery is an optional treatment for people with propionic acidemia. The propionyl coenzyme A carboxylase enzyme that causes propionic acidemia is located in the liver. Because of this, some children with propionic acidemia have had liver transplantation surgery (removal of their liver and replacement with a donor liver) to treat their propionic acidemia symptoms.
This major surgical procedure is associated with risks, and individuals who have had a liver transplant must take medication for the rest of their lives to prevent their body from rejecting the donor liver. However, even with a successful liver transplantation people with propionic acidemia may still need to have a restricted diet.
Many factors must be considered before surgery and this option should be discussed thoroughly with your child’s physicians.
Propionic acidemia diet
Because the usual major metabolic precursors of propionic acidemia are the essential amino acids (isoleucine, valine, threonine, methionine), halt all protein ingestion and emphasize alternative sources of calories on a temporary basis. Several commercially produced formulas are available that provide a protein supplement without any of the 4 amino acids that result in propionate production. However, because they are all essential in humans, closely monitored quantities of isoleucine, valine, threonine, and methionine must be added. For this reason, collaboration between the biochemical geneticist and the nutritionist is imperative.
1. Low-protein diet, medical foods and medical formula
Low-protein diet
A food plan low in the amino acids leucine, valine, methionine, and threonine, with limited amounts protein is often recommended. Most food in the diet will be carbohydrates (bread, cereal, pasta, fruit, vegetables, etc.). Carbohydrates give the body many types of sugar that can be used as energy. Eating a diet high in carbohydrates and low in protein can help prevent metabolic crises.
Foods high in protein that may need to be avoided or limited include:
- milk and dairy products
- meat and poultry
- fish
- eggs
- dried beans and legumes
- nuts and peanut butter
Many vegetables and fruits have only small amounts of protein and can be eaten in carefully measured amounts. Do not remove all protein from the diet. Children with propionic acidemia need a certain amount of protein to grow properly.
Your dietician will create a food plan that contains the right amount of protein, nutrients, and energy to keep your child healthy. Your child will need to be on a special food plan throughout his or her life.
Medical formula and foods
In addition to a low-protein diet, your child may be given a special medical formula. This formula contains the correct amount of protein and nutrients needed for normal growth and development. Your metabolic doctor and dietician will tell you what type of formula is best and how much to use.
There are also medical foods such as special low protein flours, pastas, and rice that are made especially for people with organic acid disorders. Your dietician will tell you how to use these foods as part of your child’s diet.
Some states offer help with payment, or require private insurance to pay for the formula and other special medical foods.
2. Avoid going a long time without food
Infants and young children with propionic acidemia need to eat frequently to prevent a metabolic crisis. Your metabolic doctor will tell you how often your child needs to be fed. In general, it is often suggested that infants be fed every four to six hours. Some babies need to eat even more frequently than this. It is important that infants be fed during the night. They may need to be woken up to eat if they do not wake up on their own. Your metabolic doctor and dietician will give you an appropriate feeding plan for your infant. Your doctor will also give you a ‘sick day’ plan, tailored to your child’s needs, for you to follow during illnesses or other times when your child will not eat.
Your metabolic doctor will continue to advise you on how often your child should eat as he or she gets older.
3. Medication
Children with propionic acidemia may benefit by taking L-carnitine. This is a safe and natural substance that helps the body make energy. It also helps get rid of harmful wastes. L-carnitine is part of the usual treatment for propionic acidemia. Your doctor will tell you how much your child needs. Unless you are advised otherwise, use only L-carnitine prescribed by your doctor.
Certain antibiotics, taken by mouth, can help reduce the amount of propionic acid in the intestines. Your doctor will decide if your child needs antibiotics and, if so, what type.
Some children may be given biotin supplements by mouth. Biotin is a type of B vitamin that helps the body make energy from food. Biotin has not been proven to help in propionic acidemia. But, your doctor may talk with you about trying this supplement to see if it is of benefit to your child.
Children who are having symptoms of a metabolic crisis should be treated in the hospital. During a metabolic crisis, your child may be given medications such as bicarbonate by IV to help reduce the acid levels in the blood. Glucose is often given by IV to prevent the breakdown of protein and fat stored in the body.
Do not use any medication or supplement without first checking with your doctor or metabolic doctor.
Propionic acidemia prognosis
Babies who receive prompt and ongoing treatment before they experience a metabolic crisis may have normal growth and development. In general, the earlier treatment is started, the better the outcome. However, even with treatment, some children children have life-long learning problems, cognitive deficiency, seizures, or problems with involuntary movements, even with treatment 6. Children with propionic acidemia often have more infections than usual. These need to be treated promptly to avoid a metabolic crisis. Children with a neonatal-onset and course generally have a guarded prognosis, meaning they are typically stable but there is a potential for serious issues, especially if they have a metabolic crisis. In severe cases, survival may be threatened, particularly in the presence of severe brain damage 7.
Propionic acidemia life expectancy
Although less-severely affected patients have been reported, most individuals with propionic acidemia have a classic presentation and course and a guarded prognosis. Survival is in question, and significant brain damage is likely.
- Propionic acidemia. https://ghr.nlm.nih.gov/condition/propionic-acidemia[↩][↩]
- Shchelochkov OA, Carrillo N, Venditti C. Propionic Acidemia. 2012 May 17 [Updated 2016 Oct 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK92946[↩]
- Genetics of Propionic Acidemia (Propionyl CoA Carboxylase Deficiency). https://emedicine.medscape.com/article/948084-overview[↩]
- Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet Journal of Rare Diseasesvolume 8, Article number: 6 (2013). https://ojrd.biomedcentral.com/track/pdf/10.1186/1750-1172-8-6[↩]
- Brosch S, Rauffeisen A, Baur M, Michels L, Trefz FK, Pfister M. Propionic acidemia and sensorineural hearing loss: is there a connection at the molecular genetics level?. HNO. January 2008; 56(1):37-42.[↩]
- Propionic Acidemia. http://www.newbornscreening.info/Parents/organicaciddisorders/PA.html[↩]
- Genetics of Propionic Acidemia (Propionyl CoA Carboxylase Deficiency) Treatment & Management. https://emedicine.medscape.com/article/948084-treatment[↩]





{kind=link}