Contents
- Combined malonic and methylmalonic aciduria
- Combined malonic and methylmalonic aciduria causes
- Combined malonic and methylmalonic aciduria signs and symptoms
- Combined malonic and methylmalonic aciduria complications
- Combined malonic and methylmalonic aciduria diagnosis
- Combined malonic and methylmalonic aciduria treatment
- Combined malonic and methylmalonic aciduria prognosis
Combined malonic and methylmalonic aciduria
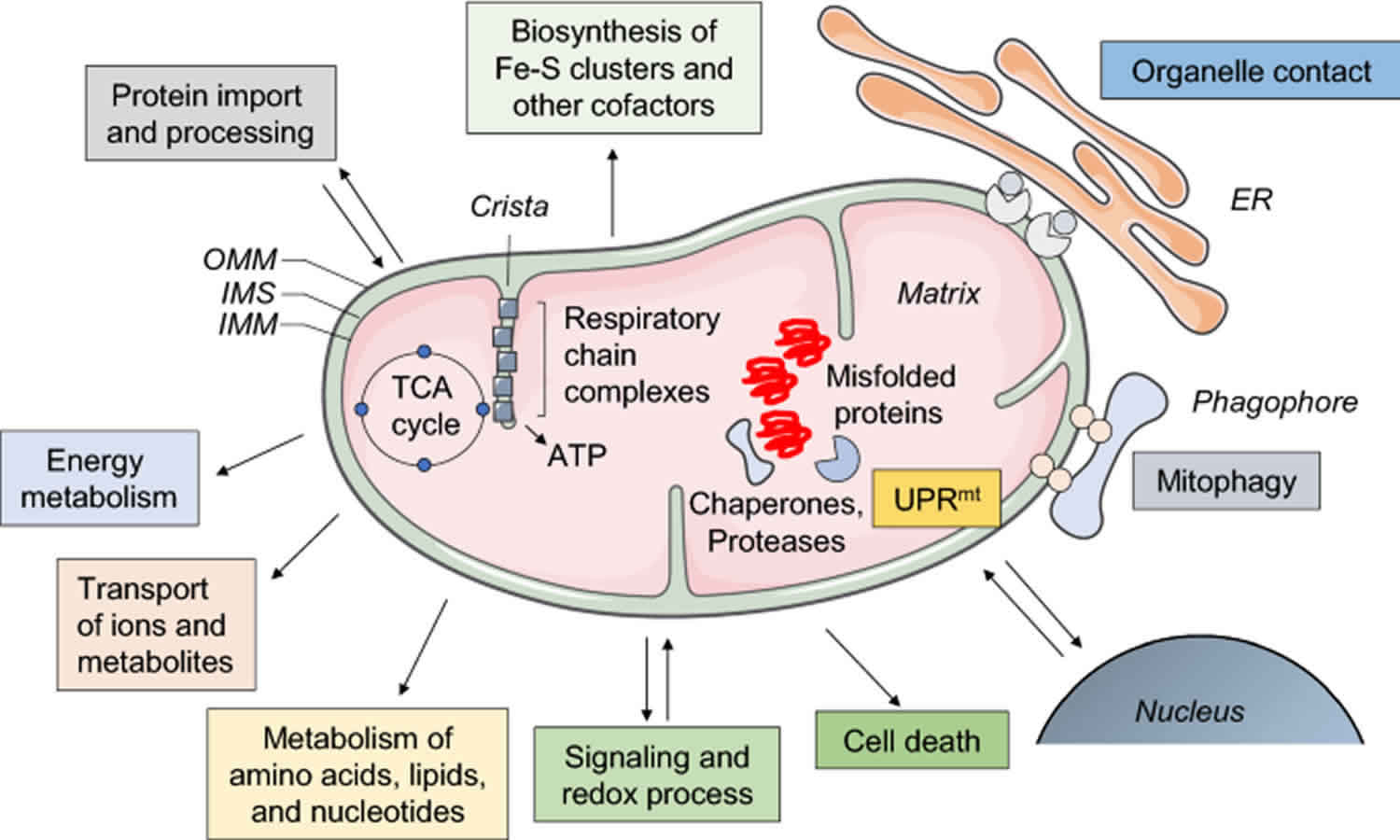
Combined malonic and methylmalonic aciduria also called “CMAMMA” is an inherited condition in which malonic acid (MA) and methylmalonic acid (MMA) accumulate in the blood and urine of affected individuals 1, 2, 3, 4, 5, 6, 7. Combined malonic and methylmalonic aciduria (CMAMMA) is biochemically characterized by elevated concentration of the diagnostic compounds malonic acid (MA) and methylmalonic acid (MMA) in both urine and plasma. A distinguishing feature of combined malonic and methylmalonic aciduria (CMAMMA) is higher levels of methylmalonic acid (MMA) than malonic acid (MA) in the urine, although both are elevated. Methylmalonic acid (MMA) excretion in patients with CMAMMA has been reported to be 8–194 times normal, malonic acid (MA) excretion is typically present at relatively lower levels 3–91 times normal excretion 8, 3. Plasma malonic acid (MA)/methylmalonic acid (MMA) ratio can serve as fast diagnostic tool for detection of CMAMMA 9.
Mutations in the ACSF3 gene cause combined malonic and methylmalonic aciduria (CMAMMA) 10, 5. ACSF3 gene provides instructions for making mitochondrial malonyl-CoA synthetase and methylmalonyl-CoA synthetase enzyme that play a role in the formation (synthesis) of fatty acids. Fatty acids are building blocks used to make fats (lipids). The ACSF3 enzyme performs a chemical reaction that converts malonic acid to malonyl-CoA, which is the first step of fatty acid synthesis in cellular structures called mitochondria. Based on this activity, the enzyme is classified as a malonyl-CoA synthetase. The ACSF3 enzyme (malonyl-CoA synthetase) adds a coenzyme A moiety to both malonic acid (MA) and methylmalonic acid (MMA), and patients with mutations in ACSF3 gene excrete more methylmalonic acid (MMA) than malonic acid (MA) 8. The ACSF3 enzyme also converts methylmalonic acid to methylmalonyl-CoA, making it a methylmalonyl-CoA synthetase as well. So far, the mechanism of malonic and methylmalonic aciduria (CMAMMA) symptoms development has not yet been worked out, although the involvement of the accumulating metabolites malonic acid (MA) and methylmalonic acid (MMA) has been proposed 11, 1.
People with combined malonic and methylmalonic aciduria (CMAMMA) can have a wide variety of symptoms, ranging from asymptomatic to severe 8, 3, 12, 13, 14. Children with combined malonic and methylmalonic aciduria (CMAMMA) can suffer from metabolic acidosis (ketoacidosis), low blood glucose (hypoglycemia), involuntary muscle tensing (dystonia), weak muscle tone (hypotonia), developmental delay and a failure to gain weight and grow (failure to thrive) and coma 3, 6, 5. Some affected children have an unusually small head size (microcephaly). Other people with CMAMMA do not develop signs and symptoms until adulthood. In adults, symptoms may include seizures, memory loss, a decline in thinking ability and neurological problems that can mimic Alzheimer’s disease and multiple sclerosis 3.
Combined malonic and methylmalonic aciduria (CMAMMA) appears to be a rare disease 5. The incidence of CMAMMA due to mutations in the ACSF3 gene has been estimated at approximately 1 in 30,000 3. However, at present only 11 patients with mutations (missense, in frame deletion, and nonsense mutations) in the ACSF3 gene have been reported in the medical literature 8, 3.
There is no cure for combined malonic and methylmalonic aciduria, since it is a genetic condition. Combined malonic and methylmalonic aciduria treatment is usually given to manage the signs and symptoms and any complication that develops. A consultation with a nutrition expert may be recommended for individual CMAMMA.
Figure 1. combined malonic and methylmalonic aciduria (CMAMMA) brain MRI
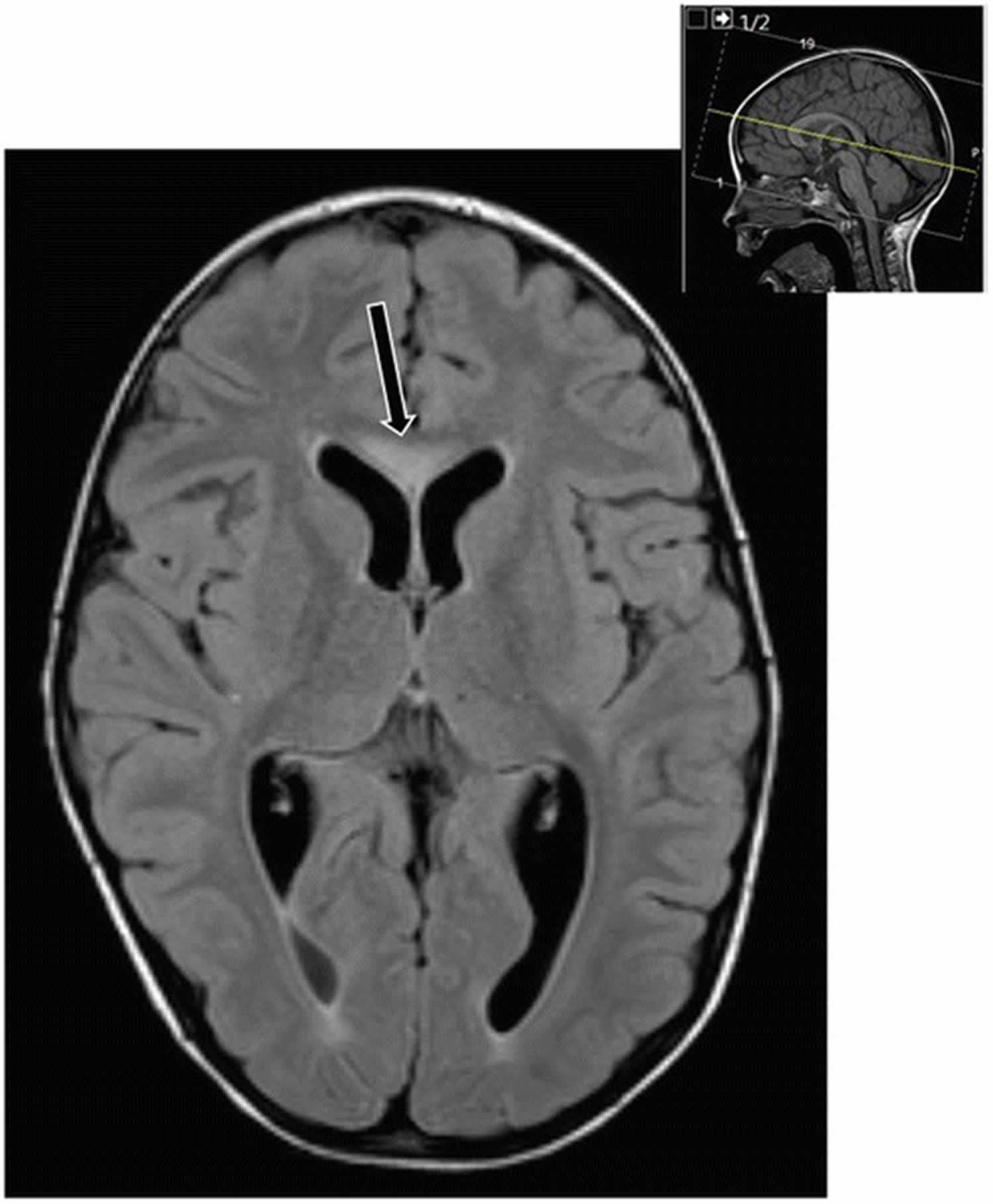
Footnote: Sagittal T2 flair showing hyperintense lesion in the genu of corpus callosum
[Source 9 ]Combined malonic and methylmalonic aciduria causes
Mutations in the ACSF3 gene cause combined malonic and methylmalonic aciduria (CMAMMA) 10, 5. ACSF3 gene provides instructions for making mitochondrial malonyl-CoA synthetase and methylmalonyl-CoA synthetase enzyme that play a role in the formation (synthesis) of fatty acids. Fatty acids are building blocks used to make fats (lipids). The ACSF3 enzyme performs a chemical reaction that converts malonic acid to malonyl-CoA, which is the first step of fatty acid synthesis in cellular structures called mitochondria. Based on this activity, the enzyme is classified as a malonyl-CoA synthetase. The ACSF3 enzyme (malonyl-CoA synthetase) adds a coenzyme A moiety to both malonic acid (MA) and methylmalonic acid (MMA), and patients with mutations in ACSF3 gene excrete more methylmalonic acid (MMA) than malonic acid (MA) 8. The ACSF3 enzyme also converts methylmalonic acid to methylmalonyl-CoA, making it a methylmalonyl-CoA synthetase as well.
Fatty acid synthesis occurs through two pathways, one of which takes place in cellular structures called mitochondria. Mitochondria convert the energy from food into a form that cells can use, and fatty acid synthesis in these structures is thought to be important for their proper functioning. The ACSF3 enzyme is found only in mitochondria and is involved in mitochondrial fatty acid synthesis.
Combined malonic and methylmalonic aciduria inheritance pattern
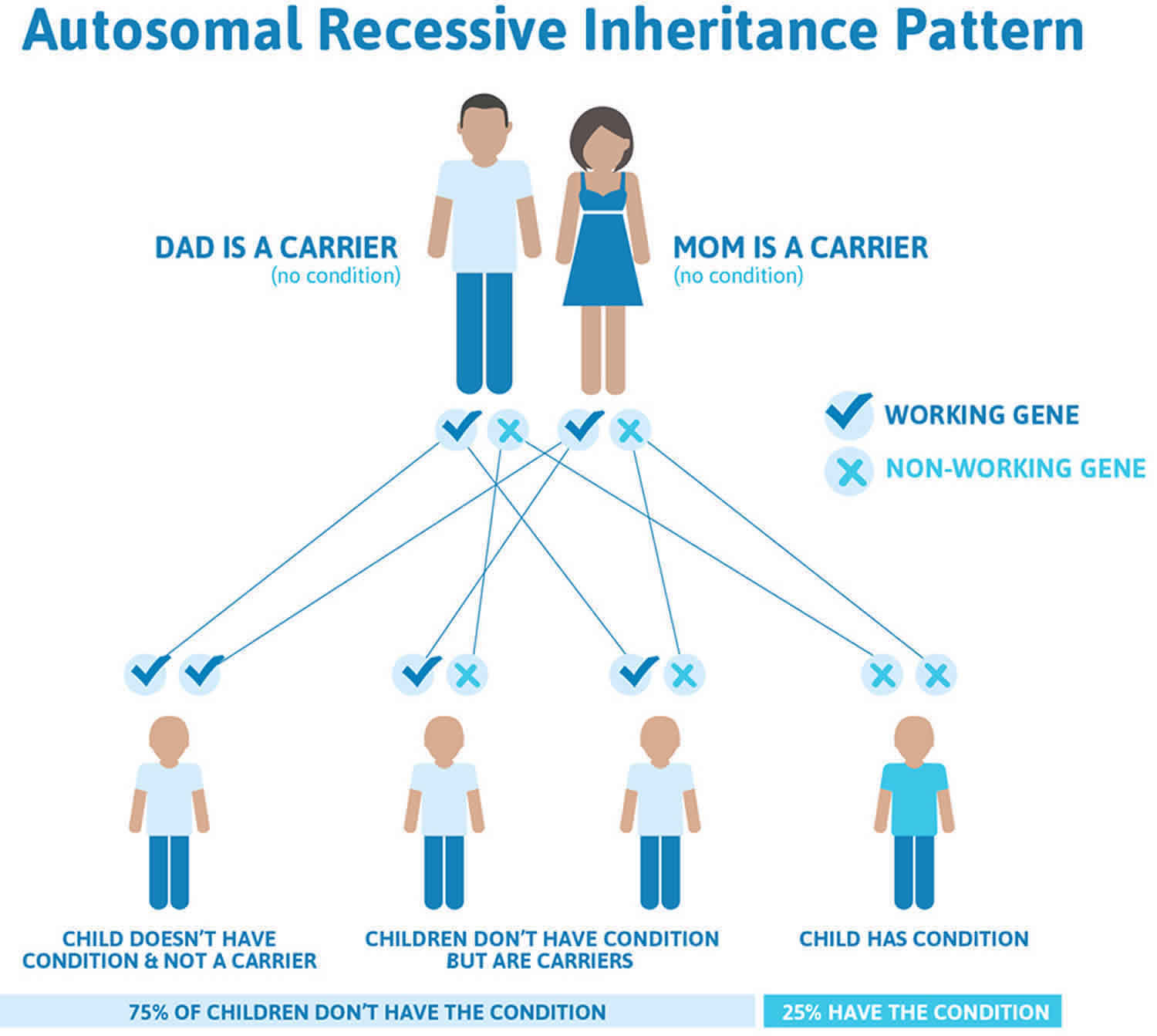
Combined malonic and methylmalonic aciduria (CMAMMA) is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Autosomal recessive conditions are traits or disorders that occur when two copies of an abnormal gene have been inherited on a non-sex chromosome. If both parents have an autosomal recessive condition, there is a 100% likelihood of passing on the mutated genes to their children. If, however, only one mutant copy of the gene is inherited, the individual will be a carrier of the condition, but will not be present with any symptoms. Children born to two carriers, have a 25% chance of being homozygous dominant (unaffected), a 50% chance of being heterozygous (carrier), and a 25% chance of being homozygous recessive (affected).
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Combined malonic and methylmalonic aciduria autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Combined malonic and methylmalonic aciduria signs and symptoms
People with combined malonic and methylmalonic aciduria (CMAMMA) can have a wide variety of symptoms, ranging from asymptomatic to severe 8, 3, 12, 13, 14.
The signs and symptoms of combined malonic and methylmalonic aciduria (CMAMMA) may vary among affected individuals in type, severity and age of onset, and may include the following:
- Dehydration
- Diarrhea
- Generalized clonic seizures
- Global developmental delay
- Ketoacidosis
- Methylmalonic aciduria
- Vomiting
Children with combined malonic and methylmalonic aciduria (CMAMMA) can suffer from metabolic acidosis (ketoacidosis), low blood glucose (hypoglycemia), involuntary muscle tensing (dystonia), weak muscle tone (hypotonia), developmental delay and a failure to gain weight and grow (failure to thrive) and coma 3, 6, 5. Some affected children have an unusually small head size (microcephaly). Other people with CMAMMA do not develop signs and symptoms until adulthood. In adults, symptoms may include seizures, memory loss, a decline in thinking ability and neurological problems that can mimic Alzheimer’s disease and multiple sclerosis 3.
Table 1. Symptoms and signs of patients with combined malonic and methylmalonic aciduria from mutation in the ACSF3 gene
| Symptoms and signs | Frequency |
|---|---|
| T2 hyperintensities | 4 |
| Increased signal on T2 of the genu of the corpus callosum | 1 |
| Diffusely increased T2 signal in the white matter | 1 |
| Extrapyramidal tract involvement | 3 |
| Oculogyric crises | 2 |
| Encephalopathy | 3 |
| Loss of speech/mutism | 3 |
| Mutism | 2 |
| Psychiatric symptoms | 3 |
| Catatonia | 2 |
| Memory problems | 3 |
| Neurodegeneration of the brain and spinal cord | 2 |
| Increased T2 signal in the cervical myelum (c2–c7) | 1 |
| Pyramidal tract involvement | 2 |
| Opisthotonus | 2 |
| Acidosis/ketoacidosis | 2 |
| Axial hypotonia | 2 |
| Psychomotor delay | 2 |
| Poor weight gain/failure to thrive | 2 |
| Seizure | 1 |
| Complex partial seizures | 1 |
| Speech delay | 1 |
| Elevated transaminases | 1 |
| Failure to thrive | 1 |
| Hypoglycemia | 1 |
| Incontinence | 1 |
| Microcephaly | 1 |
| Ocular migraine | 1 |
Combined malonic and methylmalonic aciduria complications
The complications of combined malonic and methylmalonic aciduria may include:
- Inadequate weight gain
- Rosk of falls and injuries due to seizures
- Low blood sugar
- Damage to tissues and organs due to highly acidic blood
- Memory lapses
- Confusion
- Decline in coherant thinking
- Behavioral abnormalities
- Swelling in the brain due to severe ketoacidosis
- Failure to thrive
- Coma
Complications may occur with or without treatment, and in some cases, due to treatment also.
Combined malonic and methylmalonic aciduria diagnosis
Combined malonic and methylmalonic aciduria is diagnosed on the basis of the following information:
- Complete physical examination
- Thorough medical history evaluation
- Assessment of signs and symptoms
- Laboratory tests
- Imaging studies
- Biopsy studies, if necessary
- Molecular genetic testing to check for or confirm mutation(s) in the ACSF3 gene
Many clinical conditions may have similar signs and symptoms. Your healthcare provider may perform additional tests to rule out other clinical conditions to arrive at a definitive diagnosis.
Combined malonic and methylmalonic aciduria treatment
There is no cure for combined malonic and methylmalonic aciduria, since it is a genetic condition. Combined malonic and methylmalonic aciduria treatment is usually given to manage the signs and symptoms and any complication that develops. A consultation with a nutrition expert may be recommended for individual CMAMMA.
Combined malonic and methylmalonic aciduria prognosis
Combined malonic and methylmalonic aciduria prognosis remains to be fully defined which is dependent on the severity of the signs and symptoms and associated complications 2. Typically, combined malonic and methylmalonic aciduria prognosis prognosis may be assessed on a case-by-case basis. Some reports describe combined malonic and methylmalonic aciduria (CMAMMA) individuals with severe symptoms and signs such as metabolic acidosis, seizures, developmental delay, gastrointestinal disease, failure to thrive, heart muscle problem (cardiomyopathy), dysmorphic facial features, memory problems, psychiatric problems and/or cognitive decline especially in older patients; others document a benign clinical course in affected adults and children 12, 13, 14, 8, 3. A cross-sectional multicenter retrospective study identified 25 patients (6 months to 30 years old) with combined malonic and methylmalonic aciduria (CMAMMA) due to ACSF3 mutations have favorable clinical course regardless of treatment strongly suggestive of the benign condition of CMAMMA 15.
- Tucci S. Brain metabolism and neurological symptoms in combined malonic and methylmalonic aciduria. Orphanet J Rare Dis. 2020 Jan 22;15(1):27. doi: 10.1186/s13023-020-1299-7[↩][↩]
- Gabriel, M.C., Rice, S.M., Sloan, J.L., Mossayebi, M.H., Venditti, C.P. and Al-Kouatly, H.B. (2021), Considerations of expanded carrier screening: Lessons learned from combined malonic and methylmalonic aciduria. Mol. Genet. Genomic Med., 9: e1621. https://doi.org/10.1002/mgg3.1621[↩][↩]
- Sloan JL, Johnston JJ, Manoli I, Chandler RJ, Krause C, Carrillo-Carrasco N, Chandrasekaran SD, Sysol JR, O’Brien K, Hauser NS, Sapp JC, Dorward HM, Huizing M; NIH Intramural Sequencing Center Group; Barshop BA, Berry SA, James PM, Champaigne NL, de Lonlay P, Valayannopoulos V, Geschwind MD, Gavrilov DK, Nyhan WL, Biesecker LG, Venditti CP. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat Genet. 2011 Aug 14;43(9):883-6. doi: 10.1038/ng.908[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Levtova A, Waters PJ, Buhas D, Lévesque S, Auray-Blais C, Clarke JTR, Laframboise R, Maranda B, Mitchell GA, Brunel-Guitton C, Braverman NE. Combined malonic and methylmalonic aciduria due to ACSF3 mutations: Benign clinical course in an unselected cohort. J Inherit Metab Dis. 2019 Jan;42(1):107-116. doi: 10.1002/jimd.12032[↩]
- Combined malonic and methylmalonic aciduria. https://medlineplus.gov/genetics/condition/combined-malonic-and-methylmalonic-aciduria[↩][↩][↩][↩][↩][↩]
- Combined malonic and methylmalonic acidemia. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=289504[↩][↩][↩]
- Combined malonic and methylmalonic acidemia. https://rarediseases.info.nih.gov/diseases/10818/combined-malonic-and-methylmalonic-aciduria[↩]
- Alfares A, Nunez LD, Al-Thihli K, et al. Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. J Med Genet. 2011;48:602–605. doi: 10.1136/jmedgenet-2011-100230[↩][↩][↩][↩][↩][↩][↩]
- de Sain-van der Velden MG, van der Ham M, Jans JJ, Visser G, Prinsen HC, Verhoeven-Duif NM, van Gassen KL, van Hasselt PM. A New Approach for Fast Metabolic Diagnostics in CMAMMA. JIMD Rep. 2016;30:15-22. doi: 10.1007/8904_2016_531[↩][↩][↩]
- Witkowski A, Thweatt J, Smith S. Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J Biol Chem. 2011;286(39):33729–33736. doi: 10.1074/jbc.M111.291591[↩][↩]
- Wehbe Z, Behringer S, Alatibi K, Watkins D, Rosenblatt D, Spiekerkoetter U, Tucci S. The emerging role of the mitochondrial fatty-acid synthase (mtFASII) in the regulation of energy metabolism. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(11):1629–1643. doi: 10.1016/j.bbalip.2019.07.012[↩]
- de Sain-van der Velden MG, van der Ham M, Jans JJ, Visser G, Prinsen HC, Verhoeven-Duif NM, van Gassen KL, van Hasselt PM. A new approach for fast metabolic diagnostics in CMAMMA. JIMD Rep. 2016;30:15–22. doi: 10.1007/8904_2016_531[↩][↩][↩]
- Pupavac M, Tian X, Chu J, Wang G, Feng Y, Chen S, Fenter R, Zhang VW, Wang J, Watkins D, et al. Added value of next generation gene panel analysis for patients with elevated methylmalonic acid and no clinical diagnosis following functional studies of vitamin B12 metabolism. Mol Genet Metab. 2016;117(3):363–368. doi: 10.1016/j.ymgme.2016.01.008[↩][↩][↩]
- Reid ES, Papandreou A, Drury S, Boustred C, Yue WW, Wedatilake Y, Beesley C, Jacques TS, Anderson G, Abulhoul L, et al. Advantages and pitfalls of an extended gene panel for investigating complex neurometabolic phenotypes. Brain. 2016;139(11):2844–2854. doi: 10.1093/brain/aww221[↩][↩][↩]
- Levtova, A, Waters, PJ, Buhas, D, et al. Combined malonic and methylmalonic aciduria due to ACSF3 mutations: Benign clinical course in an unselected cohort. J Inherit Metab Dis. 2019; 42: 107–116. https://doi.org/10.1002/jimd.12032[↩]
{kind=link}