Contents
- Floppy infant syndrome
- Floppy infant syndrome causes
- Floppy infant syndrome symptoms
- Floppy infant syndrome complications
- Floppy infant syndrome diagnosis
- Floppy infant syndrome differential diagnosis
- Floppy infant syndrome treatment
- Floppy infant syndrome prognosis
Floppy infant syndrome
Floppy infant syndrome also called rag-doll syndrome, floppy baby syndrome or neonatal hypotonia, is a medical term to describe a newborn infant with low muscle tone or generalized hypotonia that is present at birth (congenital hypotonia) affecting the muscles of the arms, legs, trunk, head, neck and face 1, 2, 3, 4, 5, 6, 7, 8. Hypotonia is a poor muscle tone resulting in floppiness. Hypotonia may or may not be associated with muscle weakness. An infant with hypotonia exhibits a floppy quality or “rag doll” feeling when he or she is held 9.
Each baby with floppy infant syndrome may experiences symptoms differently. Symptoms vary depending on the underlying cause of the floppy infant syndrome. The following are common symptoms associated with floppy infant syndrome:
- decreased muscle tone; muscles feel soft and doughy or “rag doll” feeling when he or she is held
- ability to extend limb beyond its normal limit
- failure to acquire motor-related developmental milestones (such as holding head up without support from parent, rolling over, sitting up without support, walking). Infants may lag behind in acquiring certain fine and gross motor developmental milestones that enable a baby to hold his or her head up when placed on the stomach, balance themselves, or get into a sitting position and remain seated without falling over.
- problems with feeding (inability to suck or chew for prolonged periods)
- shallow breathing
- mouth hangs open with tongue protruding (under-active gag reflex)
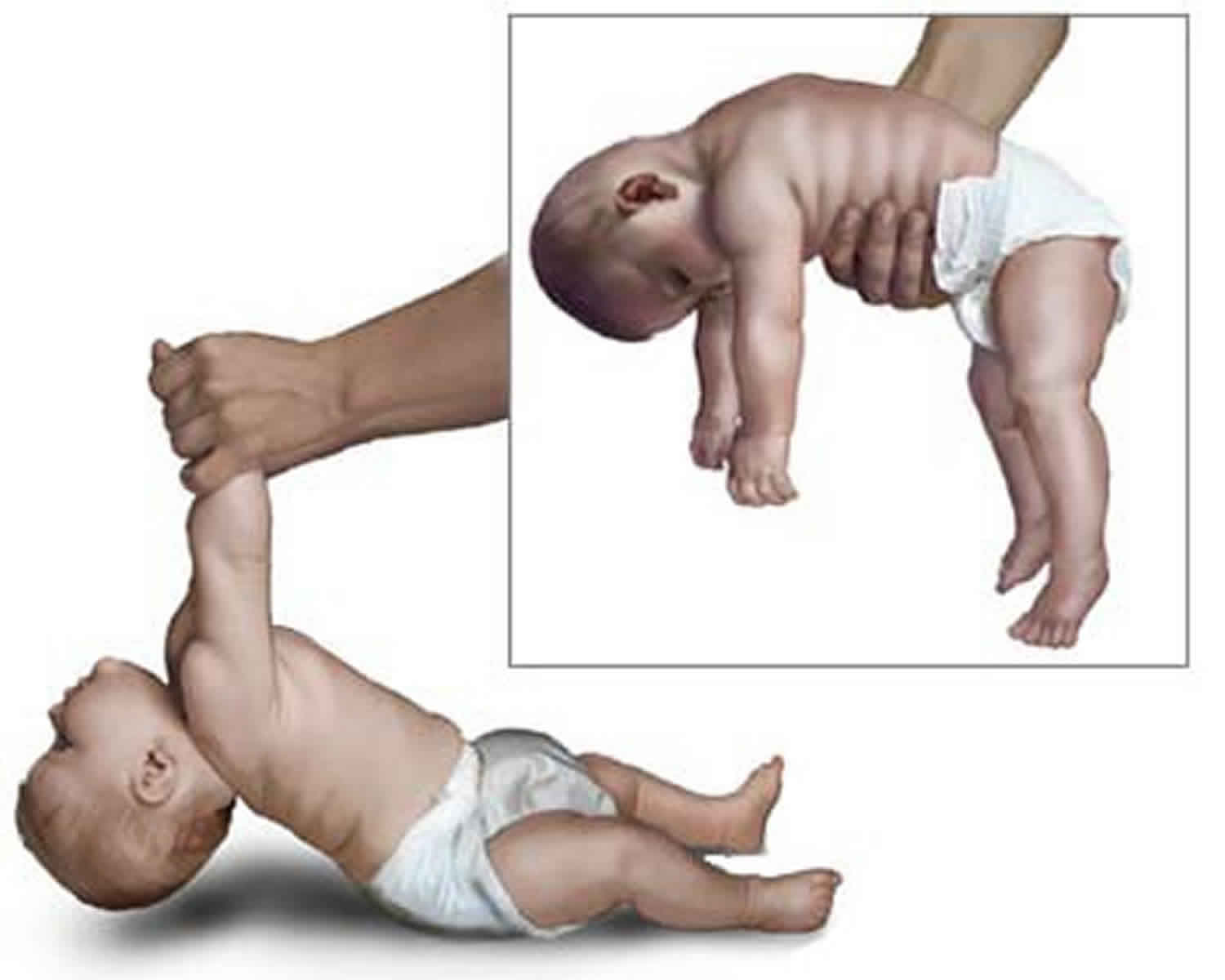
The examination for neonatal hypotonia is first carried out with the infant at rest, the head in midline position and the limbs moved passively. The truncal tone is assessed by suspending the infant with the abdomen held on the examiner’s hand (see Figures 1 and 2). While a normal full-term infant assumes a posture with straightened back, flexed limbs, and straight head, the hypotonic infant will drape over the examiner’s hand. Tone in the neck can be further assessed with the baby in the supine position and gently lifting up the baby by pulling both arms at the same time (see figure above). The hypotonic infant will generate little or no resistance to gravity and the head will assume a fully extended position. The shoulder girdle tone may be assessed by suspending the infant vertically while supporting the axillae. Full-term infants will adduct the arms and fix the shoulders, whereas hypotonic infants tend to slip through the examiners’ hands.
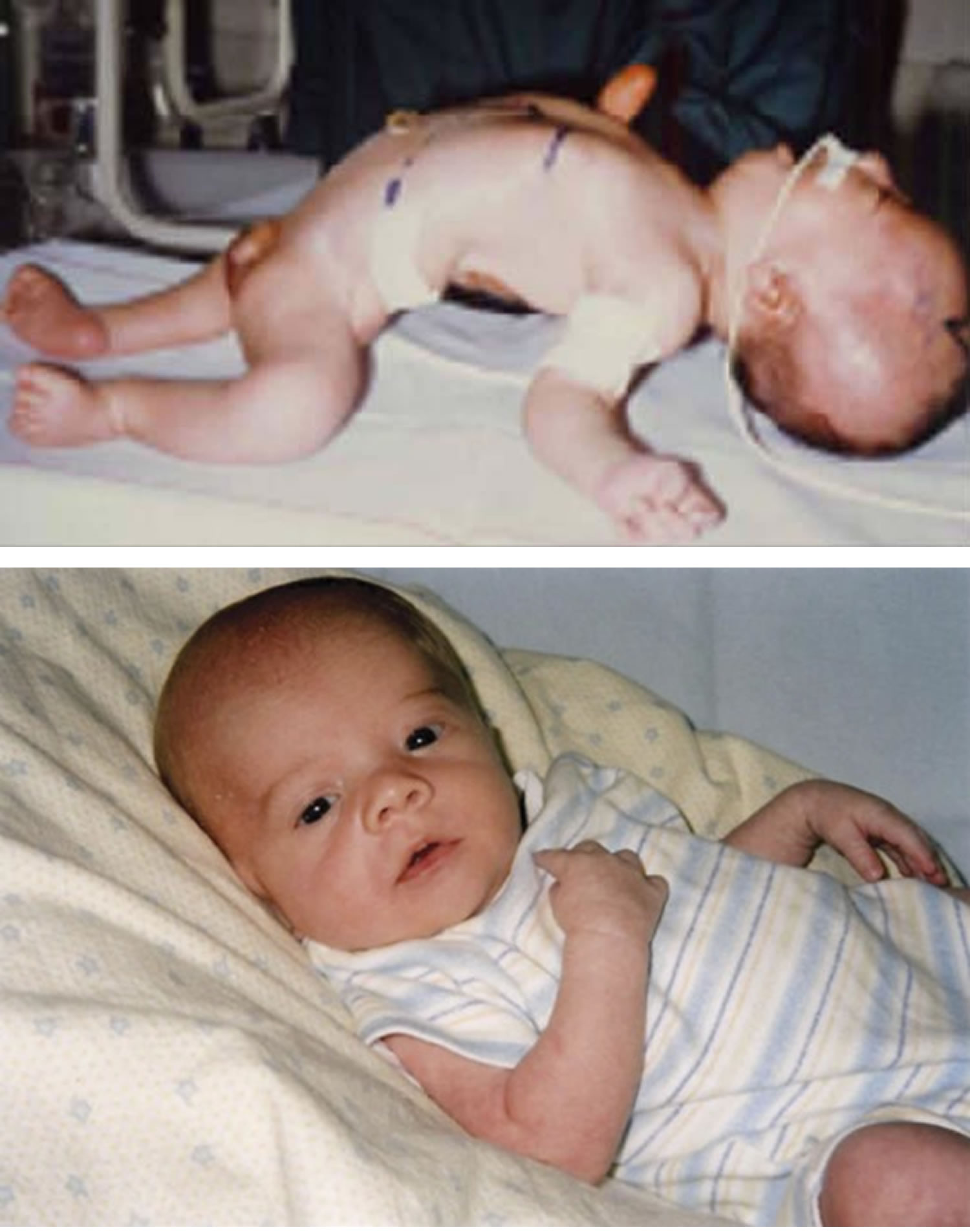
Figure 1. Floppy infant syndrome (rag-doll syndrome)
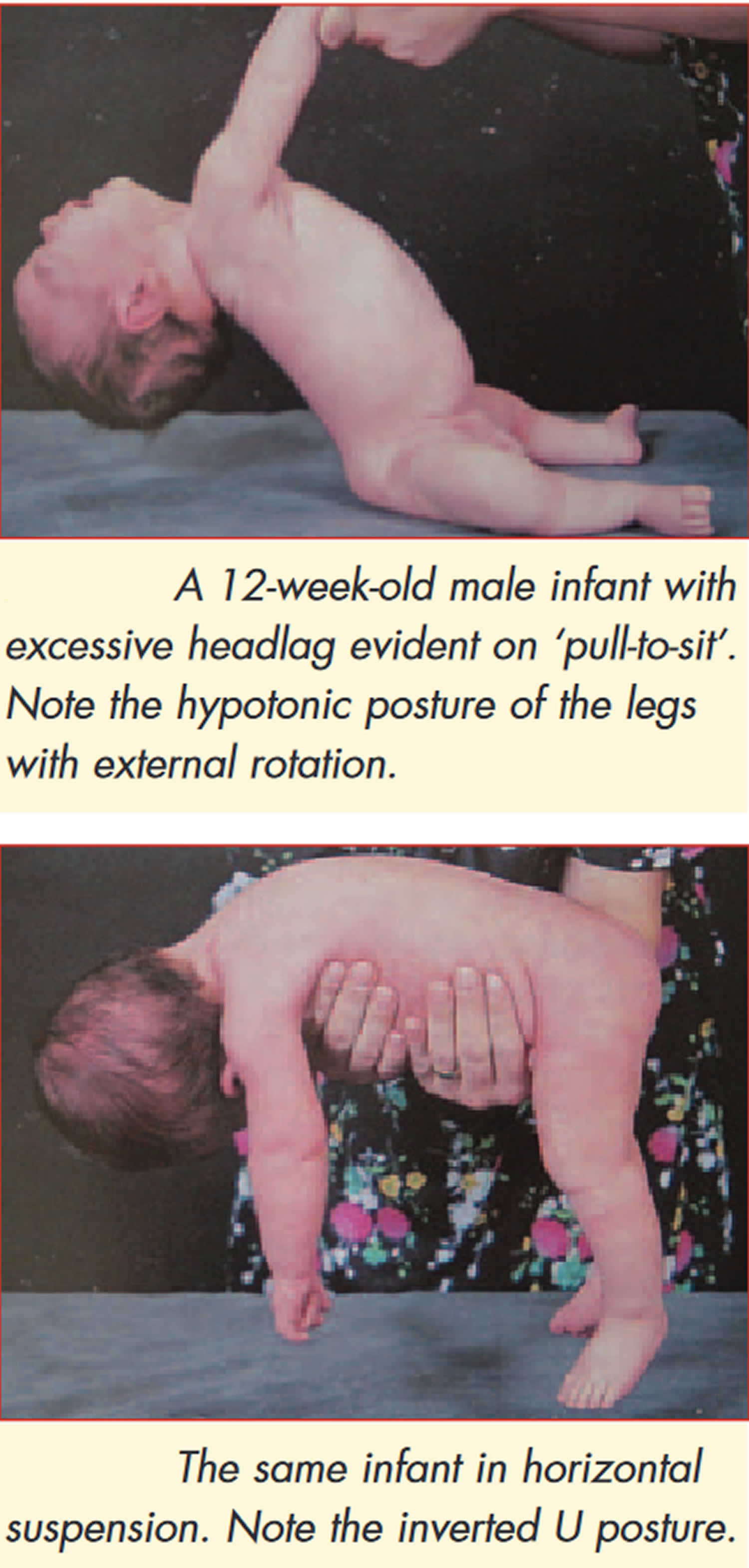
Figure 2. Physical examination findings in floppy infant syndrome (rag-doll syndrome)
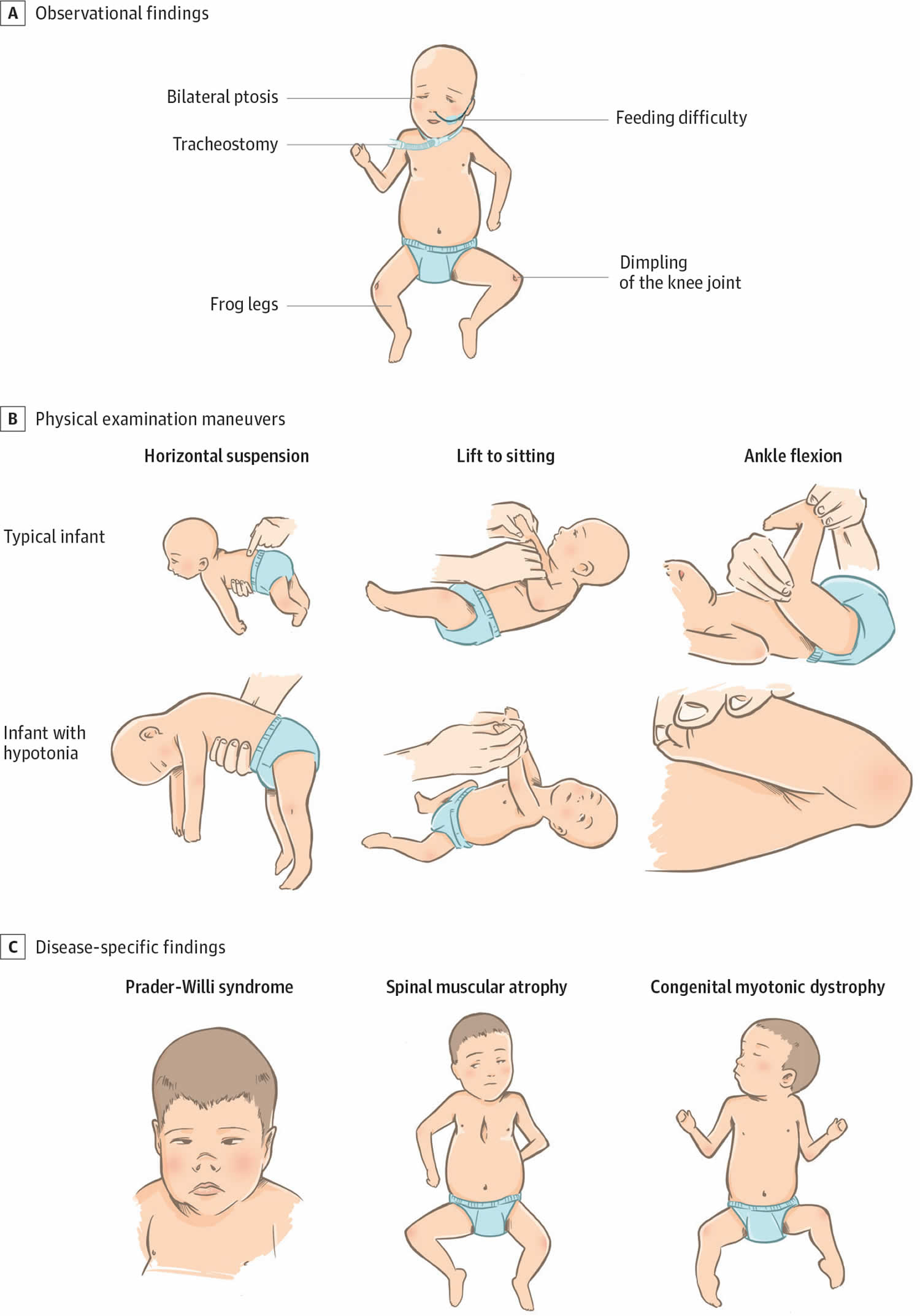
Footnotes:(A) Observational findings during examination of infants with primary hypotonia can include frog-leg positioning owing to low tone and paucity of movement, the presence of a nasogastric feeding tube owing to poor feeding, tracheostomy for respiratory failure, knee dimpling from restricted movements in utero, and ptosis with myopathic facies in neuromuscular junction disorders. (B) Physical examination maneuvers including horizontal suspension, lift to sitting position, and ankle flexion demonstrate differences between typical infants and infants with hypotonia. (C) Prader-Willi syndrome can be accompanied by narrow nasal bridge, narrowing of the forehead at the temples, almond-shaped eyes, thin upper lip, and down-turned corners of the mouth. Infants with spinal muscular atrophy can demonstrate bell-shaped chest, retractions with respirations, paucity of movement, and passive positioning with flexed hips and knees. Congenital muscular dystrophy is often associated with joint contractures.
[Source 4 ]The causes of floppy infant syndrome (rag-doll syndrome) are numerous and the diagnostic work-up in many instances can be complex. Although generalized neonatal hypotonia is easily recognizable clinically, it may be challenging for a doctor to determine the underlying cause of neonatal hypotonia. Therefore, always consult a pediatrician (a medical practitioner specializing in children and their diseases) for a diagnosis.
A variety of muscles, neuromuscular and central nervous system (brain and spinal cord) disorders can cause floppy infant syndrome (rag-doll syndrome) 10, 11. Several studies have shown that central nervous system (brain and spinal cord) disorders account for 60% to 80% of floppy infant syndrome (rag-doll syndrome) cases and that peripheral muscle and neuromuscular disorders occur in 15% to 30% floppy infant syndrome (rag-doll syndrome) cases 12, 13. Floppy infant syndrome (floppy baby syndrome) may also be a presenting feature of certain genetic disorders, metabolic diseases, endocrine problems, and acute or chronic illnesses 14. On long-term follow up, cerebral palsy and mental retardation turn out to be the 2 most common causes of floppy infant syndrome (rag-doll syndrome). In about 50% of the cases of hypotonia, the cause can be determined with a detailed history and physical examination 15.
Central nervous system (brain and spinal cord) causes include hypoxic encephalopathy, brain anomalies/insults, genetic/chromosomal syndromes, congenital or acquired infections, and disorders of metabolism. Peripheral muscle and neuromuscular causes include spinal muscular atrophy, myasthenia gravis, drug/toxin exposure, hereditary neuropathies, muscular dystrophies, congenital/metabolic myopathies, and congenital myotonic dystrophies 16, 15. Conditions where central and peripheral hypotonia may coexist are: familial dysautonomia, hypoxic–ischemic encephalopathy, infantile neuroaxonal degeneration, lipid storage diseases, lysosomal disorders and mitochondrial disorders 17.
Common causes of the floppy infant syndrome (rag-doll syndrome) include the following 18:
- Central nervous system (brain and spinal cord) disorders
- Cerebral palsy
- Neuromuscular disorders e.g., muscular dystrophy
- Connective tissue disorders
- Systemic illnesses
- Maternal medications
- Benign congenital hypotonia
- Down syndrome
- Prader-Willi syndrome
- Marfan syndrome
- Tay-Sachs disease
Determining the underlying cause is crucial for floppy infant syndrome (rag-doll syndrome) management and prognosis. Specific treatments are available for some diseases, but in general, treatment comprises providing supportive care with rehabilitation services, nutritional and respiratory support 19, 20, 14, 16, 15.
Figure 3. Lower motor neuron causes of floppy infant syndrome
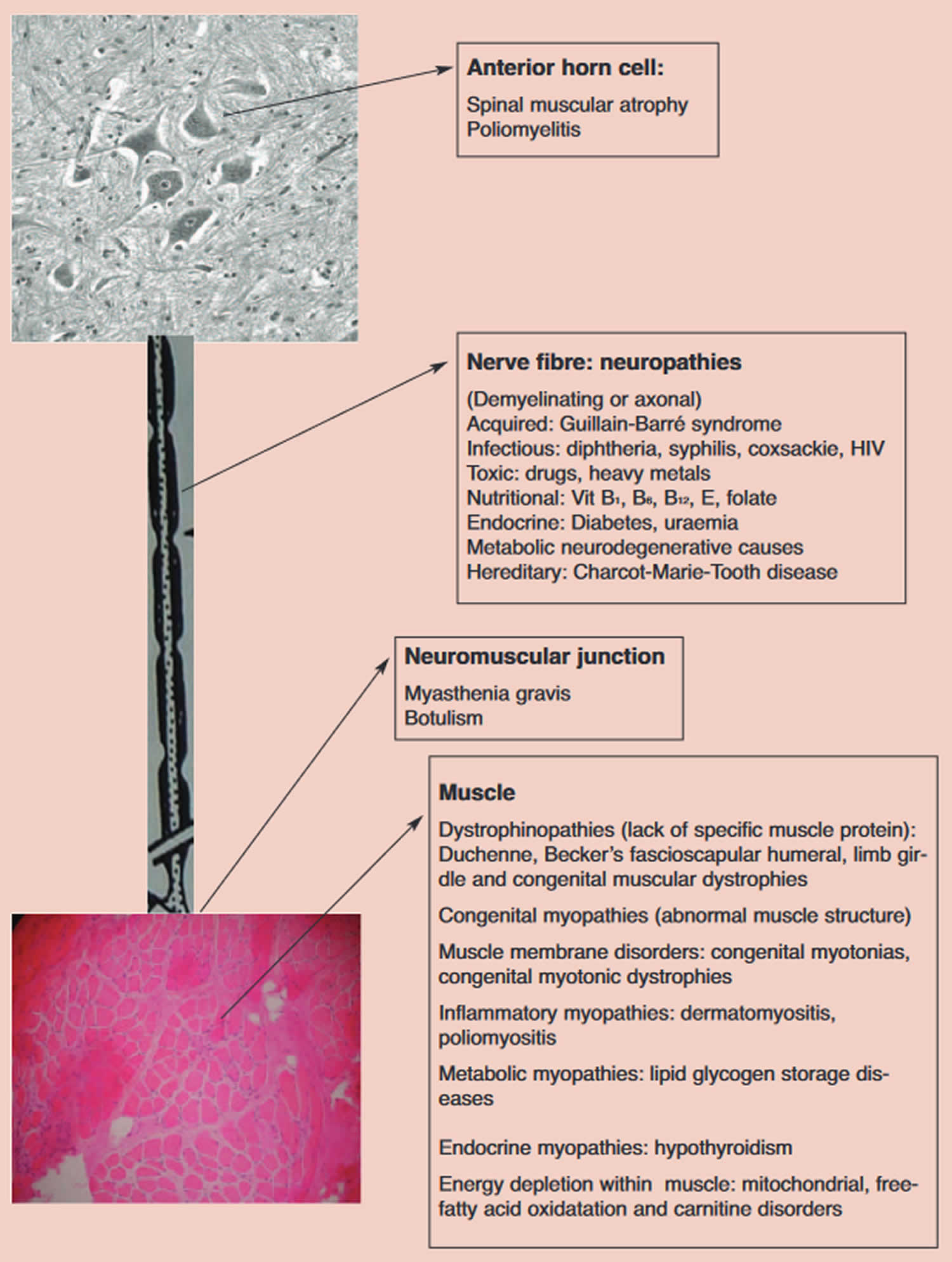
Figure 4. Floppy infant syndrome diagnosis
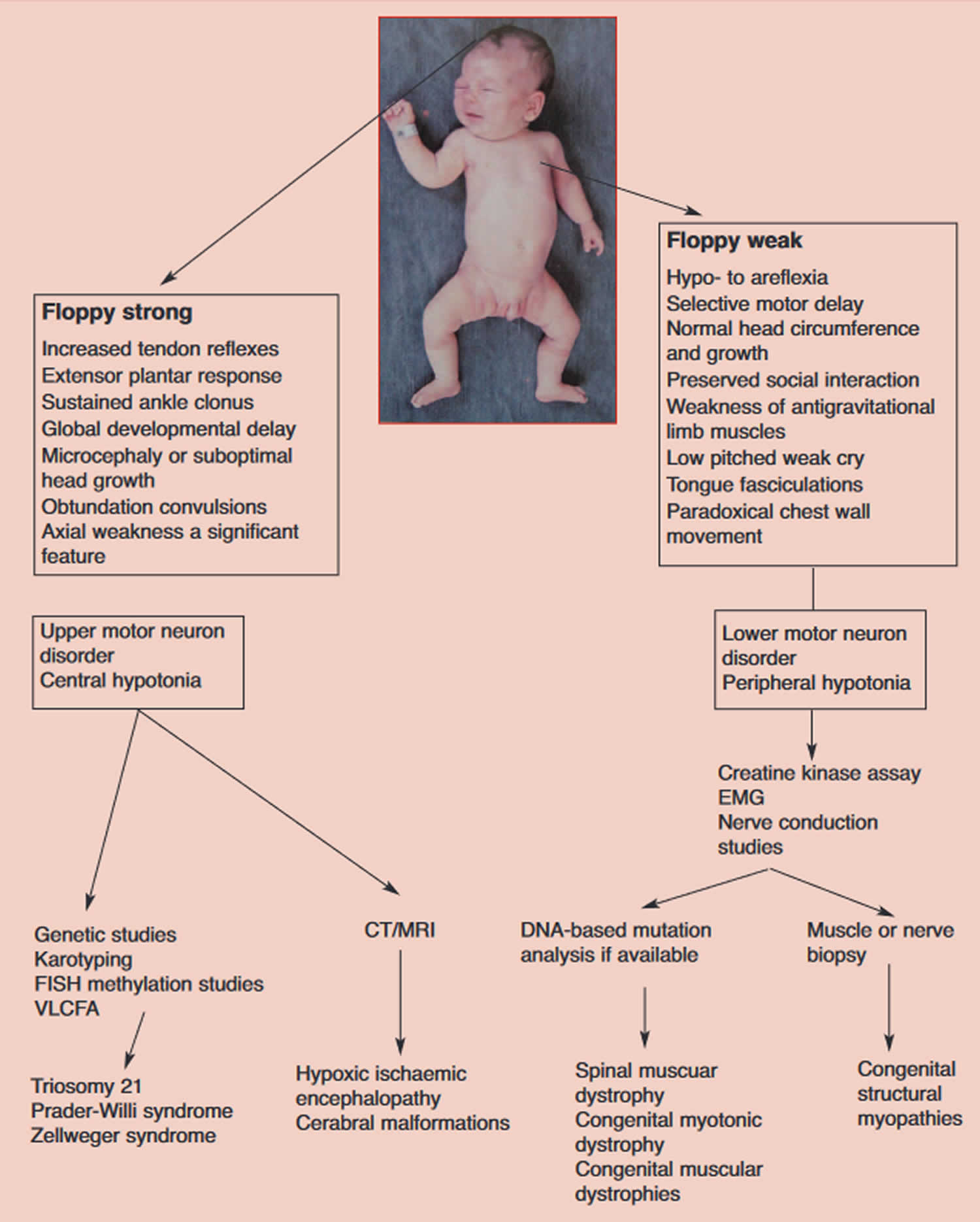
Abbreviations: EMG = electromyogram: CT= computed tomograph; MRI = magnetic resonance imaging; VLCFA = very long-chain fatty acids; FISH = fluorescent in situ hybridisation
[Source 1 ]Figure 5. Prader-Willi Syndrome and Cerebral Palsy
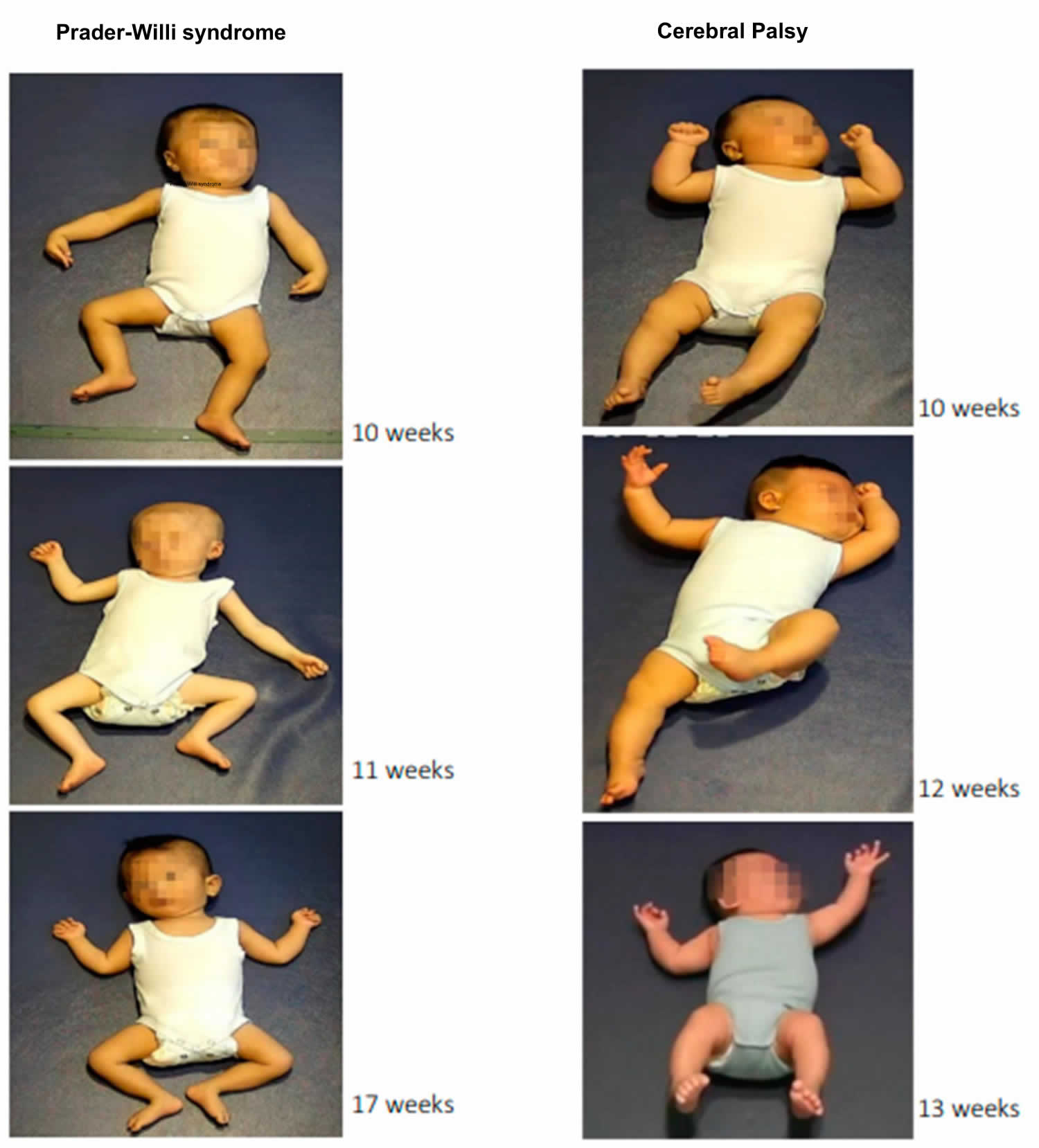
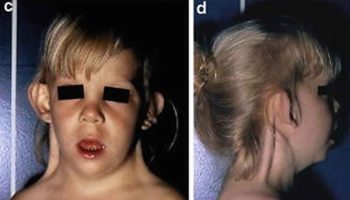
Figure 6. Zellweger syndrome (cerebrohepatorenal syndrome)
Footnote: Zellweger syndrome is a multisystem disorder resulting from peroxisome malfunction, abnormal development of the skull, face, ear, hands, and feet, as well as some mental retardation, hepatic interstitial fibrosis, renal and cortical cysts, and ocular anomalies. An example of a patient displaying facial features found in Zellweger syndrome. Note the high forehead, widely spaced eyes, broad nasal bridge, and mildly upturned nose.
[Source 22 ]Infants affected byfloppy infant syndrome can require prolonged life-supporting care, such as assisted ventilation and nutritional support. Furthermore, many infants require invasive testing, such as skin or muscle biopsy or electromyography, lumbar puncture, or invasive medical procedures (eg, tracheostomy and gastrostomy placement). Gene therapy is being developed to treat several causes of neonatal hypotonia, including spinal muscular atrophy (SMA), MTM1-related congenital myopathy, aromatic L-amino acid decarboxylase deficiency, giant axonal neuropathy, and metachromatic leukodystrophy 23, 24, 25, 26. Additional disease-modifying therapies, such as enzyme replacement therapy, are also used either in clinical practice (eg, for Pompe disease) or are under development (eg, for forms of neuronal ceroid lipofuscinosis). Furthermore, early diagnosis of a few genetic metabolic disorders, with prompt implementation of specific therapy, can greatly reduce the infant’s clinical condition.
Floppy infant syndrome key points 1:
- When evaluating a floppy infant, the first goal should be to distinguish whether the disorder is central (upper motor neuron lesion) or peripheral (lower motor neuron lesion) in origin.
- Hypotonia with weakness suggests a lower motor neuron lesion, whereas weakness is uncommon in disorders affecting the upper motor neuron except during the acute stage.
- Evaluation of tone and power should be delayed in the unwell, nutritionally compromised child.
- Muscle enzymes are rarely helpful in the floppy child, with the exception of the congenital muscular dystrophies and some of the structural congenital myopathies.
- EMG does not allow a definitive diagnosis but is indispensable in deciding whether there is weakness due to neuromuscular disease, or merely hypotonia from causes in other systems or parts of the nervous system.
- The most common of the neuromuscular disorders, spinal muscular atrophy (SMA), is now diagnosable by molecular genetic analysis (polymerase chain reaction [PCR]).
- Where clinical evaluation suggests complex multisystem involvement (i.e. hypotonia plus) inborn errors of metabolism should be excluded.
- Children with neuromuscular disorders deserve special attention when it comes to anaesthesia.
- The term ‘benign essential hypotonia’ or ‘hypotonia with a favorable outcome’ should be used with caution and only after compliance with strict diagnostic criteria.
Sometimes, low muscle tone is diagnosed shortly after birth during newborn health checks or at maternal and child health checks. In other cases, low muscle tone becomes evident later, usually when gross motor development delays become more obvious. If you are concerned about your child’s muscle tone, see your child’s doctor.
You may be referred to a pediatrician, who may examine and investigate for possible underlying causes of the low muscle tone. An occupational therapist or physiotherapist may become involved, to advise on treatment and activities to help improve your child’s muscle tone.
Floppy infant syndrome causes
A variety of muscles, neuromuscular and central nervous system (brain and spinal cord) disorders can cause floppy infant syndrome (rag-doll syndrome). Several studies have shown that central nervous system (brain and spinal cord) disorders account for 60% to 80% of floppy infant syndrome (rag-doll syndrome) cases and that peripheral muscle and neuromuscular disorders occur in 15% to 30% floppy infant syndrome (rag-doll syndrome) cases 12, 13. Floppy infant syndrome (floppy baby syndrome) may also be a presenting feature of certain genetic disorders, metabolic diseases, endocrine problems, and acute or chronic illnesses 14. On long-term follow up, cerebral palsy and mental retardation turn out to be the 2 most common causes of floppy infant syndrome (rag-doll syndrome). In about 50% of the cases of hypotonia, the cause can be determined with a detailed history and physical examination 15.
Central nervous system (brain and spinal cord) causes include hypoxic encephalopathy, brain anomalies/insults, genetic/chromosomal syndromes, congenital or acquired infections, and disorders of metabolism. Peripheral muscle and neuromuscular causes include spinal muscular atrophy, myasthenia gravis, drug/toxin exposure, hereditary neuropathies, muscular dystrophies, congenital/metabolic myopathies, and congenital myotonic dystrophies 16, 15. Conditions where central and peripheral hypotonia may coexist are: familial dysautonomia, hypoxic–ischemic encephalopathy, infantile neuroaxonal degeneration, lipid storage diseases, lysosomal disorders and mitochondrial disorders 17.
Central nervous system (brain and spinal cord) causes 27, 28, 29, 30, 5, 18:
- Cerebral insult
- Hypoxic ischemic encephalopathy. Hypoxic-ischemic encephalopathy is by far the most common cause of all hypotonia in newborns.
- Intracranial hemorrhage
- Brain malformations
- Chromosomal disorders
- Prader-Willi syndrome. Prader-Willi syndrome is among the differential diagnosis in the case of unexplained hypotonia in neonates 31, 32. The majority of cases (65%-75%) are due to a lack of expression of genes inherited paternally on chromosome 15. About one-fourth of cases are due to maternal disomy 15 (when both chromosomes are inherited from the mother, and there is an absence of paternal derived chromosome 15). Very few cases are because of a defect in the genetic imprinting. Patients with Prader-Willi syndrome have feeding difficulty, hypotonia, and failure to thrive in early infancy. This is replaced by excessive appetite and obesity in late infancy and childhood 33. Methylation analysis or a fluorescence in situ hybridization (FISH) can be used to make the diagnosis 14.
- Down syndrome (trisomy 21 ). Down syndrome (trisomy 21) is the most common chromosomal cause. Characteristic features include an up-slanting palpebral fissure, flat facial profile, single transverse palmar crease, and poor Moro reflex. Diagnosis can be confirmed with a high-resolution chromosome analysis or fluorescence in situ hybridization (FISH) 20, 14.
- Peroxisomal disorders
- Peroxisomal disorders (Zellweger’s syndrome). Peroxisomal disorders are a group of disorders that are a result of mutations in any of the 12 PEX genes, most common being the PEX 1 gene. These disorders can fall anywhere along the spectrum of severity. Zellweger syndrome is the most severe form, and infantile Refsum is the least severe form 20. They are inherited in an autosomal recessive manner. Patients usually present with significant hypotonia and poor feeding in the newborn period. There is involvement of the liver, kidneys, eyes, brain, bones, and adrenals. Diagnosis is based on the levels of very-long-chain fatty acids, with elevated levels of C26 and C26:1. Additionally, there is an elevated ratio of C26/C22 and C24/C22, along with elevation in the plasma levels of pristanic acid and phytanic acid 34, 22.
- Neonatal adrenoleukodystrophy
- Other genetic defects
- Familial dysautonomia
- Oculocerebrorenal syndrome (Lowe syndrome)
- Neurometabolic disorders
- Acid maltase deficiency
- Infantile GM1 gangliosidosis
- Drug effects (maternal benzodiazepines)
- Benign congenital hypotonia. Benign congenital hypotonia is a nonprogressive disorder of the neuromuscular system. It is commonly seen and is a diagnosis of exclusion. Muscle tone improves with increasing age, but the patient often has delayed milestone achievement. History and physical examination are not suggestive of a neurologic or a metabolic cause. Routine blood tests, including muscle enzyme results, are normal. It is associated with the risk of joint dislocations later in life, and parents must be counseled about it 14.
Peripheral causes 27, 28, 29, 30, 5, 18:
- Anterior horn cell disorders
- Spinal muscular atrophy (SMA). Spinal muscular atrophy (SMA) is an autosomal recessive disease that involves the anterior horn cells in the spinal cord and motor nuclei of the brainstem 35, 36. It is a progressive neuromuscular degenerative disorder that results in proximal muscle weakness and paralysis. There are 4 main types of SMA: SMA 1, SMA 2, SMA 3, and SMA 4, which are based on the age at which the disease presents (from pre/perinatal to adulthood) and the maximum motor function achieved 35, 36. Clinical features include symmetric proximal muscle weakness, atrophy, tongue fasciculations, and decreased to absent reflexes. These patients are also at increased risk of aspirations and failure to thrive 36. The weakness usually progresses to respiratory insufficiency and eventually, death 14. Patients have normal cognition regardless of the type 20.
- Traumatic myelopathy (especially following breech delivery)
- Hypoxic ischemic myelopathy
- Infantile neuronal degeneration
- Glycogen storage disease (Pompe’s disease). Pompe’s disease is a rare autosomal recessive disease, is caused by the deficiency of acid alpha-glucosidase deficiency. There is an accumulation of glycogen in the lysosomes in the heart and the skeletal muscle tissue. It can present as the classic infantile Pompe’s disease, which has less than 1% enzyme activity. It is seen in the neonatal period with marked hypotonia, failure to thrive, hepatomegaly, and often leads to death within the first year of life due to cardiac or respiratory causes if untreated. The electrocardiogram shows a short P-R interval and a wide QRS complex, which is a characteristic finding. A definitive diagnosis is made by a muscle biopsy, which shows a lower acid maltase enzyme activity 14. Enzyme replacement therapy is an approved treatment for infantile Pompe disease. Earlier treatment initiation has been associated with better outcomes 20, 37.
- Enterovirus infection (e.g., coxsackie, polio virus infection)
- Peripheral neuropathies
- Congenital hypomyelinating neuropathy
- Giant axonal neuropathy
- Charcot marie tooth disease
- Guillain-Barré syndrome
- Dejerine-Sottas disease
- Neuromuscular junction disorders
- Infant botulism is a potentially life-threatening disease caused by a bacterium called Clostridium botulinum (C. botulinum) caused by consumption of contaminated honey or corn syrup in which Clostridium botulinum is ingested, colonizes the gastrointestinal tract, and produces the toxin in situ 14. Infantile botulism differs from food-borne botulism in the sense that with food-borne botulism, there is ingestion of a preformed toxin in contrast to infantile botulism in which there is continued intra-intestinal production of toxin due to clostridial colonization of the large intestine. Infantile botulism usually occurs within six weeks to one year after birth, and the first symptom these infants present with is often constipation 38. According to Cagan et al. 39, the United States Centers for Disease Control and Prevention (CDC) reviewed the reported cases of botulism in all of its forms in the United States between 1899 and 1996 and found that 1442 cases of infant botulism were reported in 46 states between 1976 to 1996.
- Transient acquired neonatal myasthenia. Transient acquired neonatal myasthenia occurs in infants born to mothers with myasthenia gravis in which the acetylcholine receptor antibody (anti AChR) that causes myasthenia gravis crosses the placenta and exerts a blocking effect that is responsible for the interference with neuromuscular transmission 38. Neonatal transient myasthenia gravis is a self-limited disorder that may be potentially life-threatening if prompt and accurate diagnosis and supportive respiratory management are not initiated 40. There is a natural passive transfer of maternal antibodies that cross the placenta and bind to fetal motor-end plates, specifically against the nicotinic acetylcholine receptor (AChR) 41, 42. Transient neonatal myasthenia was reported in 12.26% of infants born to mothers with generalized myasthenia gravis before the discovery and use of acetylcholine receptor antibody (anti AChR) titers for diagnosis of acquired autoimmune myasthenia gravis 40. The anti-AChR antibodies that are passed onto the fetus through the placenta will passively induce the loss of AChRs, leading to impaired neuromuscular transmission causing muscle weakness as the primary symptom 43. The reduced number of acetylcholine receptors (AChRs) causes a decreased sensitivity to acetylcholine at the end-plate. Due to this mechanism, the end-plate potentials can be so low that threshold for activating the voltage-gated sodium channels is not reached, and consequently, no action potential is generated 41, 43. Other symptoms besides muscle weakness, include weak sucking, dysphagia, feeble cry, hypotonia, and, more rarely, respiratory difficulty, and are evident within the first two days of life and generally lasts 2-4 weeks 41. There also may be a slight delay after birth before the symptoms may appear 10. Constant clinical findings in the common form are poor sucking and generalized hypotonia, and other manifestations are weak cries, facial diparesis with an expressionless face, swallowing and sucking difficulties, and mild respiratory distress 40. Infants are typically severely hypotonic, causing neonatal transient myasthenia gravis to become a differential diagnosis for floppy infant syndrome; however, this hypotonia is typically not long-term. The generalized weakness is usually associated with facial diplegia and the pooling of oral secretions 10. Treatment is symptomatic; assisted ventilation, exchange transfusion, and intravenous immunoglobulins (IVIg) are rarely needed 41.
- Congenital myasthenic syndromes. Congenital myasthenic syndromes are due to the mutations in genes that are responsible for the structure and function of the neuromuscular junction 44, 20, 15. Congenital myasthenic syndromes can present at any time from birth to adulthood, though usually within the first two years of life, and result in a spectrum of diseases ranging from mild weakness to severe disability with life-threatening episodes 45. Infants presenting with the myasthenia syndrome share several features, including hypotonia, facial diplegia, ptosis, feeding difficulties, apnea, respiratory difficulties, generalized weakness, and a progressively weakening cry, making congenital myasthenia syndrome a differential diagnosis of floppy infant syndrome 38.
- Magnesium toxicity. A magnesium level of more than 1.15 mmol/L (2.8mg/dL) is defined as hypermagnesemia in infants. Elevated magnesium levels can be encountered in the newborn following the treatment of maternal eclampsia with magnesium sulfate or following the use of magnesium antacids in the newborn, resulting in an encephalopathic infant with hypotonia, depressed deep tendon reflexes, abdominal distension due to ileus and irregularities of cardiac rhythm 38. Neonatal hypermagnesemia can be caused by increased magnesium load such as maternal magnesium sulfate administration, newborn magnesium therapy, or decreased renal magnesium excretion due to prematurity or asphyxia 46. After administration, about 40% of plasma magnesium is protein-bound, and the unbound magnesium ion diffuses into the extravascular-extracellular space, into bone, and across the placenta and fetal membranes and into the fetus and amniotic fluid 47. Due to magnesium ions being present within the amniotic fluid, not only is the infant taking in the magnesium through fetal membranes but is also actively ingesting the magnesium through the ingestion of the amniotic fluid. Infants born to mothers with pre-eclampsia or eclampsia who received magnesium sulfate can have hypermagnesemia presenting with generalized hypotonia, apnea, bradycardia, feeding difficulty, and, in severe cases, respiratory distress and may even mimic septic shock 48. The combination of symptoms that the infants present with could be described as floppy infant syndrome. The feeding difficulty and respiratory distress could be allocated to the generalized hypotonia causing decreased muscle tone and, thus, decreased muscular movements. Magnesium is known to inactivate acetylcholine at the neuromuscular junction, especially in the respiratory muscles, and does not affect the brain directly 48, 10. At the neuromuscular junction, magnesium sulfate decreases the amount of acetylcholine liberated, diminishes the sensitivity of the end-plate to acetylcholine, and depresses the excitability of the muscle membrane thus, resulting in skeletal muscle weakness and respiratory distress 49. Calcium entry into the presynaptic terminal is necessary for acetylcholine release, and magnesium competitively blocks calcium entry 50.
- Aminoglycoside toxicity. Aminoglycosides are a mainstay of antimicrobial therapy for infants in cases in which infections are due to Gram-negative bacteria, accounting for up to 25% of all sepsis episodes in neonatal units 51. Aminoglycosides have a narrow therapeutic window, and close monitoring is required to minimize potential nephrotoxicity, ototoxicity, neuromuscular blockade 52. Aminoglycosides cause an antibiotic-induced neuromuscular blockage that can result in a decrease in the postjunctional acetylcholine sensitivity or a decrease in the release of acetylcholine in the presence of neomycin or gentamicin, specifically 53. Even within the category of aminoglycosides, gentamicin presents with the highest potency in terms of neuromuscular blocking activity 54. Out of the aminoglycosides, gentamicin, neomycin, streptomycin, tobramycin, and kanamycin have been reported to produce clinically significant muscle weakness on occasion in non-myasthenia gravis patients 53. Most evidence indicates that aminoglycosides possess both pre- and postjunctional blocking actions, but the reversal of the blockade by either calcium or anticholinesterase agents is unpredictable 55. Aminoglycoside toxicity is a concern of greater magnitude in premature infants and neonates, and this is mainly due to the renal system being immature and therefore, resulting in a prolonged serum half-life of the aminoglycosides. The prolonged serum half-life will manifest as nephrotoxicity, ototoxicity, and also cause neuromuscular blockade resulting in muscle weakness, generalized hypotonia – ultimately making aminoglycoside toxicity a differential diagnosis of floppy infant syndrome.
- Neonatal high levels of potassium (neonatal hyperkalemia). Hyperkalemia is a problem in which the amount of potassium in the blood is higher than normal. Hyperkalemia is present in up to 52% of premature infants with a birth weight of less than 1000g, and hyperkalemic infants are at a high risk of developing life-threatening cardiac arrhythmias 56. Non-oliguric hyperkalemia is characterized ours after by an excessive increase in serum potassium concentration at 24 hours after birth and is mainly due to the immature functioning of the sodium (Na+)/potassium (K+) pump 57. The early-onset hyperkalemia may have been caused by the accumulation of potassium ions transported through the placenta, the shift of potassium ions from the intracellular to the extracellular space in the infant due to the malfunctioning of the Na+/K+ pump and the inhibition of renal distal tube potassium ion secretion 58. Non-oliguric hyperkalemia of the premature infant is a result of a loss of potassium into the extracellular space to the extent that only occurs during the first days after birth in very immature infants, and it has been suggested that this potassium loss is secondary to an immature function of the Na+/K+ pump 56. Another explanation to the cause of the early onset of hyperkalemia is maternal hyperkalemia caused by hypermagnesemia since, usually, the fetal plasma K+ ion concentration is higher than the maternal plasma concentrations 58. The causes of hyperkalemia in infants are numerous, and therefore, to pinpoint one specific cause of hyperkalemia deems to be challenging. The causes of hyperkalemia in infancy include acute hemolysis, kidney disorders, and hormonal disorders 59.
- Skeletal muscle disorders (myopathies)
- Congenital muscular dystrophies are inherited disorders. The classification is based on the clinical features in the patients and certain biochemical defects. The presentation can vary from a mild disorder with survival into adulthood to a fatal disease with a poor prognosis. The 2 most common types are:
- Congenital muscular dystrophy type 1A where laminin-A2 is deficient in 30%-40% of patients. Clinical features include peripheral hypotonia, joint contractures, delayed motor milestone achievement, kyphoscoliosis, and respiratory problems. The disease begins at an early age during infancy or early childhood. Intelligence is normal in the majority of patients. Skeletal muscle biopsy would be significant for dystrophic changes. End-stage muscle disease has findings of increased muscle fibrosis 20.
- Ullrich congenital muscular dystrophy is another common cause of congenital muscular dystrophy. It presents with torticollis, kyphoscoliosis, distal joint hyperelasticity, and proximal joint contractures. Physiotherapy may improve scoliosis and contractures. Intelligence is often normal. Follicular hyperkeratosis may be an additional finding in some cases. Diagnosis is with genetic testing and biopsy of the skeletal muscle with immunohistochemical analysis 20.
- Metabolic myopathies
- Mitochrondrial myopathies
- Congenital myopathies. Congenital myopathies are a group of muscle diseases that are classified based on certain characteristic features. Congenital myopathies are present at birth and usually present with hypotonia, feeding problems, delay in milestone achievement, and muscle weakness 20.
- Central core/multi-mini core disease – usually does not progress or progresses very slowly. It is associated with malignant hyperthermia because of the defective ryanodine receptor. Biopsy of the muscle shows type 1 muscle fibers lacking oxidative enzyme activity 20.
- Nemaline myopathy presents with peripheral hypotonia at birth, scoliosis, and muscle weakness. A high arched palate and facial weakness are also noticed. 3 forms are inherited in an autosomal recessive manner, which includes the severe form seen in the neonatal period, the milder classic form, and the late-onset form 20.
- Myotubular myopathy mainly affects males. This should be suspected when there is neonatal hypotonia in a male, a positive family history of X-linked inheritance, macrocephaly, arachnodactyly, and cryptorchidism. The severe form presents with severe hypotonia, and patients often require respiratory support due to insufficiency. Prenatal history is significant for decreased fetal movements and polyhydramnios. Patients with the moderate form have a better prognosis. They achieve milestones earlier as compared to the severe form and mostly do not require respiratory support 20.
- Congenital muscular dystrophies are inherited disorders. The classification is based on the clinical features in the patients and certain biochemical defects. The presentation can vary from a mild disorder with survival into adulthood to a fatal disease with a poor prognosis. The 2 most common types are:
Floppy infant syndrome symptoms
Floppy infant syndrome or rag-doll syndrome is usually present at birth and is frequently diagnosed in early infancy. Each baby with floppy infant syndrome (rag-doll syndrome) may experiences symptoms differently. Symptoms vary depending on the underlying cause of the floppy infant syndrome. The following are common symptoms associated with floppy infant syndrome:
- decreased muscle tone; muscles feel soft and doughy or “rag doll” feeling when he or she is held
- ability to extend limb beyond its normal limit
- failure to acquire motor-related developmental milestones (such as holding head up without support from parent, rolling over, sitting up without support, walking). Infants may lag behind in acquiring certain fine and gross motor developmental milestones that enable a baby to hold his or her head up when placed on the stomach, balance themselves, or get into a sitting position and remain seated without falling over.
- problems with feeding (inability to suck or chew for prolonged periods)
- shallow breathing
- mouth hangs open with tongue protruding (under-active gag reflex).
- tendency for hip, jaw, and neck dislocations to occur.
A child with hypotonia may also have problems with speech or exhibit shallow breathing.
Indicators of Central Hypotonia
- Delay in motor milestones with relative normality of social and cognitive impairment;
- Family history of neuromuscular disorders/maternal myotonia;
- Reduced or absent spontaneous antigravity movements reduced or absent deep tendon jerks and increased range of joint mobility;
- Frog-leg posture or ‘jug-handle’ posture of arms in association with marked paucity of spontaneous movement;
- Myopathic facies (open mouth with tented upper lip, poor lip seal when sucking, lack of facial expression, ptosis and restricted ocular movements)
Indicators of Peripheral Hypotonia
- Social and cognitive impairment, in addition to motor delay;
- Dysmorphic features implying a syndrome or other organ malformations sometimes implying a syndrome;
- Fisting of hands;
- Normal or brisk tendon reflexes;
- Crossed adductor response or scissoring present upon vertical suspension;
- Features suggestive of an underlying spinal dysraphism;
- Seizures;
- History that is suggestive of hypoxic-ischemic encephalopathy, birth trauma or symptomatic hypoglycemia.
Floppy infant syndrome complications
Floppy infant syndrome complications are usually secondary to the problems associated with it. Mortality is highest under the age of 1 year, and those who survive beyond 1 year age often have developmental delays 60. The majority of the patients require nutritional support, and they may develop various micro/macro-nutrient deficiencies. Those requiring pulmonary support are more prone to respiratory infections in general and may develop pneumonia frequently. In some diseases, patients who are wheelchair-bound are more prone to fractures, osteopenia, development of contractures, and pressure ulcers even if adequate physical therapy is provided to them 61.
Floppy infant syndrome diagnosis
The task of evaluating infants with hypotonia is difficult and complex. In a hypotonic newborn, it is essential to proceed systematically to reach a correct diagnosis 16, 14, 62. Your child’s doctor will obtain a medical history for your family and your child and will perform a physical examination that will likely include a detailed muscle function and neurological examination. The latter, also called a neuro exam, may be performed with instruments, such as lights and reflex hammers, and usually does not cause any pain to the child.
- Assessment of motor and sensory skills
- Balance and coordination
- Mental status (the child’s level of awareness and interaction with the environment)
- Reflexes
- Functioning of the nerves
About 50% of the cases of hypotonia are successfully diagnosed with just a medical history and physical examination, including obtaining a family history, maternal obstetric history, clinical and neurological examinations 14. When looking at the causes of peripheral hypotonia, a diagnostic workup is based on an understanding of the anatomy of the motor unit as many causes of peripheral hypotonia can be localized to various compartments of the motor unit 10.
The initial assessment is aimed at ruling out systemic disorders. Examination for the abnormalities of deep tendon reflexes and laxity of ligaments helps in considering peripheral causes.
If suspected, sepsis must be ruled out with cultures of the blood, urine, and cerebrospinal fluid. Additionally, obtaining a complete blood count and comprehensive metabolic profile, including magnesium and a drug screen. If the neonate is found to have hepatosplenomegaly and/or calcification on a head ultrasound, then a workup for congenital infection should be initiated. It includes toxoplasmosis, rubella, cytomegalovirus, herpes simplex, and human immunodeficiency virus (TORCH) titers, urine CMV studies, and Zika virus testing. For assessment of the central causes of hypotonia, a workup should be done to look for genetic and metabolic disease as a cause. In the presence of dysmorphic features or congenital malformations, a chromosome analysis with routine karyotyping or specific testing for fluorescence in situ hybridization (FISH) can aid in the diagnosis of certain diseases such as Down syndrome and Prader-Willi syndrome.
Metabolic disorder screening is done when there is multisystemic involvement. Screening for carbohydrate and mitochondrial disorders with lactate and pyruvate levels may be done. Altered levels of plasma amino acids and urine organic acids may indicate an underlying amino acid disorder or organic acidemia. Acylcarnitine and total carnitine levels can be obtained for defects in the metabolism of organic acids and fatty acids. More specific testing may be done for peroxisomal disorders with very-long-chain fatty acids and plasmalogens. When suspecting peripheral hypotonia, a creatine kinase (CK) level should be done. Creatine kinase (CK) is typically very elevated (more than 10X normal) in muscular dystrophies.
Electrophysiologic studies can be used as a screening tool for peripheral causes of hypotonia. However, it may be difficult to perform and interpret in a neonate or a young infant. They can differentiate among disorders involving nerves, muscle, or neuromuscular junctions. If creatine kinase (CK) is mildly elevated (less than 10X normal) and electromyography (EMG) is myopathic, then a muscle biopsy will be needed. Muscle biopsy with immunohistochemical staining and electron microscopy can help differentiate between various muscular dystrophies and myopathies. Although an invasive procedure, it plays an important role in diagnosis and guiding the further workup. If the electromyography (EMG) is neuropathic, then testing for spinal muscular atrophy (SMA), hereditary sensorimotor neuropathy, and Dejerin-Sottas syndrome should be done.
Decrement or facilitation on electromyography (EMG) would suggest the disease of the neuromuscular junction. Radiologic evaluation with magnetic resonance imaging (MRI) and computed tomography (CT) of the brain can aid in the diagnosis of various causes of central hypotonia. Structural changes, migration defects, brain stem defects (Joubert syndrome), and abnormal signaling defects in the white matter and brain stem can be seen on neuroimaging. Magnetic resonance spectrometry can be useful in diagnosing certain metabolic disorders.
Medical history
The pre-, peri- and postnatal history is important. Enquire about the quality and quantity of fetal movements, breech presentation and the presence of either polyhydramnios or oligohydramnios. The incidence of breech presentation is higher in fetuses with neuromuscular disorders as turning requires adequate fetal mobility. Documentation of birth trauma, birth anoxia, delivery complications, low cord pH and Apgar scores are crucial as hypoxic-ischaemic encephalopathy remains an important cause of neonatal hypotonia. Neonatal seizures and an encephalopathic state offer further proof that the hypotonia is of central origin. The onset of the hypotonia is also important as it may distinguish between congenital and aquired aetiologies. Enquire about consanguinity and identify other affected family members in order to reach a definitive diagnosis, using a detailed family pedigree to assist future genetic counselling.
Neurological Examination
A detailed physical examination, along with a comprehensive neurological examination, must be done. There are two approaches to the diagnosis of infants with hypotonia (see Figure 2). The first is based on identifying the neuro-anatomical site of the lesion or insult. The second is to determine whether or not the hypotonia is accompanied by weakness 1. Careful neurological examination should, in most cases, localize the site of the lesion to the upper motor neuron (UMN) or lower motor neuron (LMN) unit 1. Useful clues are listed in Table 1.
Next assess whether the neonatal hypotonia is accompanied by weakness. Weakness is uncommon in upper motor neuron (UMN) hypotonia except in the acute stages. Hypotonia with profound weakness therefore suggests involvement of the lower motor neuron (LMN). Assessment of muscle power of infants is generally limited to inspection.
Useful indicators of weakness are 1:
- Ability to cough and clear airway secretions (‘cough test’). Apply pressure to the trachea and wait for a single cough that clears secretions. If more than one cough is needed to clear secretions, this is indicative of weakness.
- Poor swallowing ability as indicated by drooling and oropharyngeal pooling of secretions.
- The character of the cry — infants with consistent respiratory weakness have a weak cry.
- Paradoxical breathing pattern — intercostal muscles paralysed with intact diaphragm
- Frog-like posture and quality of spontaneous movements — poor spontaneous movements and the frog-like posture are characteristic of lower motor neuron (LMN) conditions.
Further confirmation of the last indicator can be obtained by means of informal neurological examination in the test positions. Excessive head lag will be evident on ‘pull to sit’. Minimal support with a sensation of ‘slipping through he hands’ during vertical suspension testing and inverted U position on ventral suspension are further indicators (see Figure 1).
A distinct pattern of weakness may favor certain causes 1:
- Axial weakness is a significant feature in central hypotonia.
- Generalised weakness with sparing of the diaphragm, facial muscles, pelvis and sphincters suggests anterior horn cell involvement.
- With myasthenic syndromes, the bulbar and oculomotor muscles exhibit a greater degree of involvement.
- Progressive proximal symmetrical weakness suggests a dystrophinopathy. Signs of proximal weakness in the older infant include a lordotic posture, Trendelenburg gait and Gower
sign. - A striking distribution of weakness of the face, upper arms and shoulders suggests fascioscapulohumeral muscular dystrophy.
- Distal muscle groups are predominantly affected with peripheral neuropathies. Signs suggesting distal weakness in an older infant would include weakness of hand grip, foot drop and a high stepping, slapping gait.
Tables 1 and 2 give a comprehensive approach to the clinical clues and investigations for the more common types of the floppy infant syndrome.
Once the lesion has been localized to the lower motor neuron (LMN) proceed with further neuroanatomical localization according to the compartments of the lower motor neuron (LMN). The
compartments and disease associations are listed in Figure 3 above.
Examine the tongue for size and fasciculations 1. Fasciculations, irregular twitching movements, generally indicate an abnormality of the anterior horn cells (Figure 3). Do not examine the tongue while the infant is crying. The co-existence of atrophy would strongly favor a denervative etiology. An ECG may be helpful in demonstrating baseline fasciculations of the intercostal muscles 1. Enlargement of the tongue may suggest a storage disorder such as Pompe’s disease 1.
The presence of a facial diplegia (myopathic facies) suggests either a congenital structural myopathy or myasthenia gravis 1. Respiratory and bulbar weakness can accompany both conditions. Fluctuation in strength would favor myasthenic syndromes 1.
The presence of facial dysmorphism should alert the doctor to the possibility of an underlying syndrome or genetic disorder. Conditions that need to be excluded include trisomy 21 (Down syndrome), Prader-Willi syndrome and Zellweger syndrome 1. At
birth the facial anomalies in Prader-Willi syndrome are often insignificant and the diagnosis is usually only established after onset of obesity (see Figure 5). Useful early markers include the presence of feeding difficulties, small hands and feet, almond shaped eyes, narrow bifrontal diameter and hypogonadism 1. Failure to identify electrophysiological abnormalities in an infant with profound hypotonia should prompt testing for Prader-Willi syndrome 1. This can be done by a DNA methylation study which will detect 99% of cases.
Zellweger syndrome presents in the newborn with typical facial dysmorphism including high forehead, wide patent fontanelles and sutures, shallow orbital ridges, epicanthus and micrognathia (see Figure 6). Neurological manifestations are dominated by severe hypotonia with depressed or absent tendon reflexes, and poor sucking and swallowing 1. Enlarged liver and liver dysfunction are consistent findings 1.
Where clinical evaluation suggests complex multisystem involvement (i.e. hypotonia plus) inborn errors of metabolism should be excluded 1. Referral to a tertiary center for metabolic investigations would then be indicated 1.
Table 1. Clinical features of Upper and Lower Motor Neuron causes of the floppy infant syndrome
| Upper Motor Neuron | Lower Motor Neuron |
|---|---|
| Hypotonia in infancy and spasticity in childhood | Hypotonia |
| Mild weakness or normal strength | Significant weakness |
| Increased tendon reflexes | Decreased or absent tendon reflexes |
| Abnormal reflexes (Babinski signs, ankle clonus) | No abnormal reflexes |
| No muscle atrophy | Muscle atrophy (may not be present in infancy) |
| Delayed developmental milestones | Only motor skills delayed |
| Signs and symptoms of CNS involvement | No CNS involvement |
Table 2. Clincical clues and investigations of the more common types of floppy infant syndrome

Floppy infant syndrome tests
Depending on what your child’s doctor suspects, the following tests may also be used to find out what’s causing your child’s muscle weakness:
- Magnetic resonance imaging (MRI): A diagnostic procedure that uses a combination of large magnets, radio frequencies, and a computer to produce detailed images of organs and structures within the body. This test is done to rule out any associated abnormalities of the spinal cord and nerves.
- Computerized tomography scan (CT or CAT scan): A diagnostic imaging procedure that uses a combination of x-rays and computer technology to produce cross-sectional images (often called “slices”), both horizontally and vertically, of the body. A CT scan shows detailed images of any part of the body, including the bones, muscles, fat, and organs. CT scans are more detailed than general X-rays.
- Blood tests
- EMG (electromyogram): A test used to evaluate nerve and muscle function
- EEG (electroencephalogram): A test that measures the electrical activity in the brain, called brain waves. An EEG measures brain waves through small button electrodes that are placed on your child’s scalp.
- Spinal tap (lumbar puncture). A spinal tap is done to measure the amount of pressure in the spinal canal and/or to remove a small amount of cerebral spinal fluid (CSF) for testing. Cerebral spinal fluid is the fluid that bathes your child’s brain and spinal cord.
- Karyotype (chromosomal analysis): Karyotyping is a test that allows doctors to examine chromosomes from a blood test that is used to determine whether the problem is the result of a genetic disorder.
- Muscle biopsy: a sample of muscle tissue is removed and examined under a microscope.
Evaluation of tone and power should be delayed in the systemically unwell nutritionally compromised child 1. Infective and biochemical parameters should first be normalized before attempts are made to examine tone and power.
Hypokalemia and hypophosphatemia in particular will cause reversible weakness 1. Serum albumin, calcium, phosphorus, alkaline phosphatase and thyroxine levels will provide confirmation of clinical suspicion in malnutrition, ricketts and hypothyroidism 1.
Suspected central cause investigations
For infants with central hypotonia, initial investigations include genetic testing and neuroimaging (CT or MRI scan). A karyotype is a must in the dysmorphic hypotonic child. This can be combined with FISH testing if Prader-Willi syndrome is suspected. Cranial MRI will identify patients with structural CNS malformations, neuronal migration defects and those with white matter
changes (congenital muscular dystrophies in particular) 1. Hypotonia may also be prominent in connective tissue diseases such as Ehlers Danlos syndrome or Marfan’s syndrome. An excessive range of joint mobility, unusual joint postures, the ability to do various contortions of the limbs and/or hyperelasticity of the skin are important diagnostic clues 1. Although metabolic disorders are well recognised as the cause for central hypotonia, because of the rarity of the conditions, the diagnostic yield is low. Very long-chain fatty acids (VLCFA) testing should be performed if a peroxisomal disorder such as Zellweger syndrome is suspected 1.
Table 3. Genetic Testing for Neonatal Hypotonia
| Test | Aneuploidy | Large intergenic deletions/duplications | Intragenic deletions/duplications | Monogenic SNVs and small I/Ds | Repeated element expansionsa | Methylation changes |
|---|---|---|---|---|---|---|
| Karyotype or fluorescence in situ hybridization (chromosome number and identity) | Optimaltest | Variable detection | Unable to detect | Unable to detect | Unable to detect | Unable to detect |
| Chromosomal microarray | Able to detect | Optimal test | Limited detection | Unable to detect | Unable to detect | May detect uniparental disomy or deletion leading to imprinting disorder |
| Next-generation sequencing-based gene panel | Unable to detect | Variable detection | Able to detect | Optimaltest | Unable to detect | Unable to detect |
| Methylation array or bisulfite sequencing (methylation state of DNA) | Unable to detect | Unable to detect | Unable to detect | Unable to detect | Unable to detect | Optimal test |
| Mitochondrial sequencing (mitochondrial genome sequencing) | Unable to detect | Unable to detect | Unable to detect | Optimaltest for mitochondrial genome SNV or I/Ds | Unable to detect | Unable to detect |
| Exome sequencing (autosomal coding sequences) | Able to detect | Able to detect | Able to detect | Optimal test | Limited detection | May detect uniparental disomy or deletion leading to imprinting disorder |
| Genome sequencing (coding and noncoding sequences) | Able to detect | Optimal test | Optimal test | Optimal test | Able to detect | May detect uniparental disomy or deletion leading to imprinting disorder |
Footnote: a Does not include targeted testing.
Abbreviations: I/D = insertion/deletion; SNV = single-nucleotide variant.
[Source 4 ]Suspected peripheral cause investigations
Major discoveries have been made in the genetic analysis of the muscular dystrophies, spinal muscular atrophies and hereditary neuropathies. This means that most neuromuscular conditions can now be diagnosed by molecular testing. Examples include spinal muscular atrophy (SMA) which can now be confirmed by polymerase chain reaction (PCR) testing for a deletion of the survivor motor neuron (SMN) gene and congenital myotonic dystrophy which can be diagnosed by demonstrating an expansion of cytosine thymine guanine (CTG) triplet repeats.
DNA Xp21 deletion testing (PCR assays) can confirm the diagnosis in 65% of males with Duchenne muscular dystrophy and 80% of males with Becker’s muscular dystrophy 1. Only in the remaining cases is muscle biopsy still advocated. Muscle enzymes levels (creatine kinase assay) are rarely helpful in the floppy infant, with the exception of muscle disorders where creatine kinase values are elevated, such as congenital muscular dystrophies and in some of the congenital structural myopathies 1.
Electromyography (EMG) should not be performed in isolation 1. It does not allow for a definitive diagnosis as there are virtually no waveforms that are pathognomonic for specific disease entities. In the floppy infant, EMG is indispensable in deciding whether
there is true weakness due to neuromuscular disease or merely hypotonicity from causes in other systems or other parts of the nervous system. The abnormalities detected may then assist in localizing the disease process to the neuron, neuromuscular junction or muscle. EMG is also useful to confirm a clinical suspicion of myotonia in the older child.
Nerve conduction studies are useful in the investigation of hereditary motor sensory neuropathies and distinguishing axonal from demyelinating disorders 1. Molecular DNA testing can then be used for specific demyelinating disorders. Commercial testing is not yet available for most axonal forms of hereditary neuropathies.
Muscle biopsy remains the investigation of choice in an infant with a suspected congenital structural myopathy, as none of the clinical features are truly pathognomonic. Even a creatine kinase within the reference range and normal EMG does not exclude the presence of a myopathy. Muscle biopsy is also indicated in infants with suspected limb-girdle muscular dystrophies (LGMD).
Floppy infant syndrome differential diagnosis
Floppy infant syndrome differential diagnosis includes a wide variety of causes, which may be related to central nervous system (CNS), anterior horn cell, peripheral nerve or muscle disorders, as well as connective tissue disorders or systemic illness.
Floppy infant syndrome treatment
Floppy infant syndrome treatment depends on the cause. In general, therapy is supportive and most of the time takes precedence over finding the underlying cause 5. It is tailored to the symptoms of the infant and may depend on the underlying cause 8. Rehabilitation is an important therapeutic consideration, with the aid of physical and occupational therapists. Regular physiotherapy will prevent contractures. Occupational therapy is important in facilitating activities of daily living. Vigorous treatment of respiratory infections is indicated. Annual flu vaccination is necessary 1. Annual orthopedic review is required to monitor for scoliosis and to exclude hip dislocation/subluxation.
Nutrition is of primary importance to maintain ideal body weight for the age and sex which is often achieved through the nasogastric tube or percutaneous gastrostomy 12. Maintenance of ideal weight is important, as excessive weight gain will exacerbate existing weakness.
Children with neuromuscular disorders deserve special attention when it comes to anaesthesia. The anaesthetist should be forewarned about the possibility of an underlying muscle disease even if the child has very mild or non-existing symptoms. A family history of muscle disease or mild hyper-creatine kinase-emia may be of importance. Muscle relaxants should only be used if essential because of their more profound and prolonged effect in myopathic children. All children with neuromuscular disease should also be considered potentially susceptible to malignant hypothermia (the strongest correlation is with central core disease) and implicating agents should therefore be avoided. Ethical considerations such as the appropriateness of cardiopulmonary resuscitation in the event of cardiac arrest or acute respiratory failure need to be addressed sensitively.
Respiratory care
Respiratory problems are a primary cause of sickness and/or hospital admission particularly in young children. There is an increased prevalence of sleep-related upper airway obstruction and lower airway disease, but the significance of symptoms is often underrecognized. Therefore, specialist investigation and treatment are often necessary but not often sought. In a teaching hospital in Australia, a retrospective chart review of 232 admissions of children with floppy syndrome over a 6.5-year period gave over half the causes for admission as respiratory problems. Of the total
admissions, 10% were ultimately admitted to the Pediatric Intensive Care Unit 5.
Regular physiotherapy is not popular with children, especially those with lower airway problems. However, it may be useful to teach parents how to perform physiotherapy so that it can be used when their child is unwell and so may tolerate it better. A large number of children may simply require oxygen, even if they have large airway problems. Non-invasive ventilation is relatively uncommon 63.
Floppy infant syndrome prognosis
Floppy infant syndrome prognosis depends on the underlying cause. In some cases, such as transient myasthenia gravis and infantile botulism, there is complete recovery with minimal to no complications. Hypotonia in sepsis or due to other illnesses like congestive heart failure also improves once the disease is treated. Certain conditions, such as spinal muscular atrophy (SMA) and Pompe disease (classic infantile type), can be fatal early in life. Having an interprofessional team approach towards the patient by providing rehabilitation services, nutritional support, home care services, and involving various specialists can alter the prognosis leading to better outcomes.
Patients labelled as ‘hypotonia with favorable outcome’ should fulfil all of the following criteria 1:
- early hypotonia, usually since birth
- active movements of limbs and normal tendon reflexes
- normal or mild motor retardation that improves later on
- normal muscle enzymes, EMG, motor nerve conduction studies and histology.
Some hypotonias are not progressive and are of an unknown origin (idiopathic), a condition known as benign congenital hypotonia.
- Central nervous system function and intelligence in children is normal.
- Children with benign congenital hypotonia may not experience developmental delay.
- Some children acquire gross motor skills (sitting, walking, running, jumping) more slowly than most.
Most children with benign congenital hypotonia will naturally improve over time, without any long-term impact on their physical strength and abilities. However, some people may experience muscle weakness into adulthood. A physiotherapist or occupational therapist can offer strategies and suggestions to help your child maximize their muscle tone.
Warm-up activities can improve your child’s endurance by helping to activate the muscles. It is important to encourage your child to do a warm-up activity every day. While these activities will not lead to a permanent change in your child’s muscle tone, they will help your child know what it is like to do everyday things using a more stable posture. Make the activities fun and exciting as this can help increase your child’s level of alertness and muscle tone.
For example, bouncing on a mini trampoline before sitting at a table to do a drawing may help your child to sit up straight rather than slump. If your child is slouching after they have started an activity, get them to stop what they are doing and have them do 10 star jumps to activate their muscles.
Squeezing and rolling playdough before writing may help your child to maintain their hold on the pencil and write for a longer period of time.
Your physical therapist or occupational therapist will recommend suitable warm-up activities, but some other suggestions include:
- Space hopper – encourage your child to bounce on a space hopper in the backyard or up and down the hallway.
- Running on the spot, stomping, star jumps, skipping with a skipping rope.
- Crawling activities, crab walks, bear walks, bunny hops – make it fun by setting up an obstacle course, relay or race.
- Tug of war – use a dressing gown cord or twist a bathroom towel to make a rope. Play tug-of-war with your child either sitting, standing or kneeling.
- Play ball games – catching, throwing, bouncing, aiming at targets.
- Newspaper scrunch – scrunch up sheets of newspaper into balls. Once a few balls have been made get your child to throw them into a bin or at a target.
- Tong relay – pick up small toys or objects with a pair of tongs and run and place them in a container.
- Spray bottles – water plants or make pictures by squirting water on the concrete.
- Arm wrestles – sit opposite your child with elbows on the table. Hold each other’s hands and encourage your child to push against your resistance.
- R van Toorn. Clinical approach to the floppy child. 2nd; 2001 http://www.cmej.org.za/index.php/cmej/article/view/1008/792[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Howell, R., Byrne, B., Darras, B. et al. Diagnostic challenges for Pompe disease: An under-recognized cause of floppy baby syndrome. Genet Med 8, 289–296 (2006). https://doi.org/10.1097/01.gim.0000204462.42910.b8[↩]
- Igarashi, Masanori MD. Floppy Infant Syndrome. Journal of Clinical Neuromuscular Disease 6(2):p 69-90, December 2004. https://journals.lww.com/jcnmd/abstract/2004/12000/floppy_infant_syndrome.3.aspx[↩]
- Morton SU, Christodoulou J, Costain G, Muntoni F, Wakeling E, Wojcik MH, French CE, Szuto A, Dowling JJ, Cohn RD, Raymond FL, Darras BT, Williams DA, Lunke S, Stark Z, Rowitch DH, Agrawal PB. Multicenter Consensus Approach to Evaluation of Neonatal Hypotonia in the Genomic Era: A Review. JAMA Neurol. 2022 Apr 1;79(4):405-413. doi: 10.1001/jamaneurol.2022.0067[↩][↩][↩]
- Kaur, Jaspreet & Punia, Sonu. (2016). FLOPPY INFANT SYNDROME: OVERVIEW. International Journal of Physiotherapy and Research. 4. 1554-1563. http://dx.doi.org/10.16965/ijpr.2016.134[↩][↩][↩][↩][↩]
- Low muscle tone. https://www.rch.org.au/kidsinfo/fact_sheets/Low_muscle_tone[↩]
- Chen, H. (2016). Floppy Infant. In: Atlas of Genetic Diagnosis and Counseling. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6430-3_95-2[↩]
- Madhok SS, Shabbir N. Hypotonia. [Updated 2022 Oct 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK562209[↩][↩]
- Muscle Weakness (Hypotonia). https://www.childrenshospital.org/conditions/muscle-weakness-hypotonia[↩]
- Miller VS, Delgado M, Iannaccone ST. Neonatal hypotonia. Semin Neurol. 1993 Mar;13(1):73-83. doi: 10.1055/s-2008-1041110[↩][↩][↩][↩][↩]
- Igarashi M. Floppy infant syndrome. J Clin Neuromuscul Dis. 2004 Dec;6(2):69-90. doi: 10.1097/00131402-200412000-00003[↩]
- Wijesekara DS. Clinical approach to a floppy infant. Sri Lanka Journal of Child Health 2013;41:211-216.[↩][↩][↩]
- Gowda V, Parr J, Jayawant S. Evaluation of the floppy infant, 2007.[↩][↩]
- Peredo DE, Hannibal MC. The floppy infant: evaluation of hypotonia. Pediatr Rev. 2009 Sep;30(9):e66-76. doi: 10.1542/pir.30-9-e66[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Kaler J, Hussain A, Patel S, Majhi S. Neuromuscular Junction Disorders and Floppy Infant Syndrome: A Comprehensive Review. Cureus. 2020 Feb 8;12(2):e6922. doi: 10.7759/cureus.6922[↩][↩][↩][↩][↩][↩]
- Sparks SE. Neonatal hypotonia. Clin Perinatol. 2015 Jun;42(2):363-71, ix. doi: 10.1016/j.clp.2015.02.008[↩][↩][↩][↩]
- Richer LP, Shevell MI, Miller SP. Diagnostic profile of neonatal hypotonia: an 11-year study. Pediatr Neurol. 2001 Jul;25(1):32-7. doi: 10.1016/s0887-8994(01)00277-6[↩][↩]
- Floppy infant syndrome. http://syllabus.cwru.edu/YearThree/neuroscience/NeurLrngObjectives/Floppy%20Baby.htm[↩][↩][↩]
- Harris SR. Congenital hypotonia: clinical and developmental assessment. Dev Med Child Neurol. 2008 Dec;50(12):889-92. doi: 10.1111/j.1469-8749.2008.03097.x[↩]
- Lisi EC, Cohn RD. Genetic evaluation of the pediatric patient with hypotonia: perspective from a hypotonia specialty clinic and review of the literature. Dev Med Child Neurol. 2011 Jul;53(7):586-99. doi: 10.1111/j.1469-8749.2011.03918.x[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Marschik-Zhang D, Wang J, Shen X, Zhu X, Gao H, Yang H, Marschik PB. Building Blocks for Deep Phenotyping in Infancy: A Use Case Comparing Spontaneous Neuromotor Functions in Prader-Willi Syndrome and Cerebral Palsy. J Clin Med. 2023 Jan 18;12(3):784. doi: 10.3390/jcm12030784[↩]
- Lee PR, Raymond GV. Child neurology: Zellweger syndrome. Neurology. 2013 May 14;80(20):e207-10. doi: 10.1212/WNL.0b013e3182929f8e[↩][↩]
- Waldrop MA, Karingada C, Storey MA, et al. Gene therapy for spinal muscular atrophy: safety and early outcomes. Pediatrics. 2020;146(3):e20200729. doi: 10.1542/peds.2020-0729[↩]
- Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. doi: 10.1126/science.1233158[↩]
- Jungbluth H, Muntoni F. Therapeutic aspects in congenital myopathies. Semin Pediatr Neurol. 2019;29:71–82. doi: 10.1016/j.spen.2019.01.004[↩]
- Gene Transfer Clinical Study in X-Linked Myotubular Myopathy (ASPIRO). https://clinicaltrials.gov/study/NCT03199469[↩]
- Lisi EC, Cohn RD. Genetic evaluation of the pediatric patient with hypotonia: perspective from a hypotonia specialty clinic and review of the literature. Dev Med Child Neurol. 2011;53(7):586–599. doi: 10.1111/j.1469-8749.2011.03918.x[↩][↩]
- Bodensteiner JB. The evaluation of the hypotonic infant. Semin Pediatr Neurol. 2008;15(1):10–20. doi: 10.1016/j.spen.2008.01.003[↩][↩]
- Fay AJ. Neuromuscular diseases of the newborn. Semin Pediatr Neurol. 2019;32:100771. doi: 10.1016/j.spen.2019.08.007[↩][↩]
- Peredo DE, Hannibal MC. The floppy infant: evaluation of hypotonia. Pediatr Rev. 2009;30(9):e66–e76. doi: 10.1542/pir.30.9.e66[↩][↩]
- Butler MG, Miller JL, Forster JL. Prader-Willi syndrome—clinical genetics, diagnosis and treatment approaches: an update. Curr Pediatr Rev. 2019;15(4):207–244. doi: 10.2174/1573396315666190716120925[↩]
- Tuysuz B, Kartal N, Erener-Ercan T, et al. Prevalence of Prader-Willi syndrome among infants with hypotonia. J Pediatr. 2014;164(5):1064–1067. doi: 10.1016/j.jpeds.2014.01.039[↩]
- Tuysuz B, Kartal N, Erener-Ercan T, Guclu-Geyik F, Vural M, Perk Y, Erçal D, Erginel-Unaltuna N. Prevalence of Prader-Willi syndrome among infants with hypotonia. J Pediatr. 2014 May;164(5):1064-7. doi: 10.1016/j.jpeds.2014.01.039[↩]
- Steinberg SJ, Raymond GV, Braverman NE, et al. Zellweger Spectrum Disorder. 2003 Dec 12 [Updated 2020 Oct 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1448[↩]
- D’Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011 Nov 2;6:71. doi: 10.1186/1750-1172-6-71[↩][↩]
- Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin. 2015 Nov;33(4):831-46. doi: 10.1016/j.ncl.2015.07.004[↩][↩][↩]
- Bay LB, Denzler I, Durand C, Eiroa H, Frabasil J, Fainboim A, Maxit C, Schenone A, Spécola N. Infantile-onset Pompe disease: Diagnosis and management. Arch Argent Pediatr. 2019 Aug 1;117(4):271-278. English, Spanish. doi: 10.5546/aap.2019.eng.271[↩]
- Prasad AN, Prasad C. The floppy infant: contribution of genetic and metabolic disorders. Brain Dev. 2003 Oct;25(7):457-76. doi: 10.1016/s0387-7604(03)00066-4[↩][↩][↩][↩]
- Cagan E, Peker E, Dogan M, Caksen H. Infant botulism. Eurasian J Med. 2010 Aug;42(2):92-4. doi: 10.5152/eajm.2010.25[↩]
- Papazian O. Transient neonatal myasthenia gravis. J Child Neurol. 1992 Apr;7(2):135-41. doi: 10.1177/088307389200700202. Erratum in: J Child Neurol 1992 Jul;7(3):325.[↩][↩][↩]
- Evoli A. Acquired myasthenia gravis in childhood. Curr Opin Neurol. 2010 Oct;23(5):536-40. doi: 10.1097/WCO.0b013e32833c32af[↩][↩][↩][↩]
- Vernet-der Garabedian B, Lacokova M, Eymard B, Morel E, Faltin M, Zajac J, Sadovsky O, Dommergues M, Tripon P, Bach JF. Association of neonatal myasthenia gravis with antibodies against the fetal acetylcholine receptor. J Clin Invest. 1994 Aug;94(2):555-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC296130/pdf/jcinvest00020-0095.pdf[↩]
- Gomez AM, Van Den Broeck J, Vrolix K, Janssen SP, Lemmens MA, Van Der Esch E, Duimel H, Frederik P, Molenaar PC, Martínez-Martínez P, De Baets MH, Losen M. Antibody effector mechanisms in myasthenia gravis-pathogenesis at the neuromuscular junction. Autoimmunity. 2010 Aug;43(5-6):353-70. doi: 10.3109/08916930903555943[↩][↩]
- Finlayson S, Beeson D, Palace J. Congenital myasthenic syndromes: an update. Pract Neurol. 2013 Apr;13(2):80-91. doi: 10.1136/practneurol-2012-000404[↩]
- Shillito P, Vincent A, Newsom-Davis J. Congenital myasthenic syndromes. Neuromuscul Disord. 1993 May;3(3):183-90. doi: 10.1016/0960-8966(93)90057-q[↩]
- Hyun HS, Choi HS, Kim JK, Ahn SY, Yoo HS, Kim ES, Chang YS, Park WS. Idiopathic severe hypermagnesemia in an extremely low birth weight infant on the first day of life. Korean J Pediatr. 2011 Jul;54(7):310-2. doi: 10.3345/kjp.2011.54.7.310[↩]
- Lu JF, Nightingale CH. Magnesium sulfate in eclampsia and pre-eclampsia: pharmacokinetic principles. Clin Pharmacokinet. 2000 Apr;38(4):305-14. doi: 10.2165/00003088-200038040-00002[↩]
- Kamity R. Common Problems in the Newborn Nursery. Cham: Springer International Publishing; 2019. Hypotonia in the newborn; pp. 171–182.[↩][↩]
- Role of neuromuscular junction monitoring in management of a postpartum eclamptic patient with iatrogenic hypermagnesemia. Parikh HG, Upasani CB. http://180.149.244.185:9898/iaforum/article/Role%20of%20Neuromuscular%20Junction%20Monitoring%20in%20Management%20of%20a%20Postpartum%20Eclamptic%20Patient%20with%20Iatrogenic%20Hypermagnesemia[↩]
- Krendel DA. Hypermagnesemia and neuromuscular transmission. Semin Neurol. 1990 Mar;10(1):42-5. doi: 10.1055/s-2008-1041252[↩]
- Kent A, Turner MA, Sharland M, Heath PT. Aminoglycoside toxicity in neonates: something to worry about? Expert Rev Anti Infect Ther. 2014 Mar;12(3):319-31. doi: 10.1586/14787210.2014.878648[↩]
- Aminoglycoside therapy in neonates. With particular reference to gentamicin. Young TE. Neoreviews. 2002;3:0.[↩]
- Torda T. The nature of gentamicin-induced neuromuscular block. Br J Anaesth. 1980 Mar;52(3):325-9. doi: 10.1093/bja/52.3.325[↩][↩]
- Paradelis AG, Triantaphyllidis C, Giala MM. Neuromuscular blocking activity of aminoglycoside antibiotics. Methods Find Exp Clin Pharmacol. 1980 Feb;2(1):45-51.[↩]
- Singh YN, Marshall IG, Harvey AL. Some effects of the aminoglycoside antibiotic amikacin on neuromuscular and autonomic transmission. Br J Anaesth. 1978 Feb;50(2):109-17. doi: 10.1093/bja/50.2.109[↩]
- Mildenberger E, Versmold HT. Pathogenesis and therapy of non-oliguric hyperkalaemia of the premature infant. Eur J Pediatr. 2002 Aug;161(8):415-22. doi: 10.1007/s00431-002-0986-9[↩][↩]
- Kwak JR, Gwon M, Lee JH, Park MS, Kim SH. Non-oliguric hyperkalemia in extremely low birth weight infants. Yonsei Med J. 2013 May 1;54(3):696-701. doi: 10.3349/ymj.2013.54.3.696[↩]
- Tanaka K, Mori H, Sakamoto R, Matsumoto S, Mitsubuchi H, Nakamura K, Iwai M. Early-onset neonatal hyperkalemia associated with maternal hypermagnesemia: a case report. BMC Pediatr. 2018 Feb 13;18(1):55. doi: 10.1186/s12887-018-1048-4[↩][↩]
- Rajpoot SK, Maggi C, Bhangoo A. Pseudohypoaldosteronism in a neonate presenting as life-threatening arrhythmia. Endocrinol Diabetes Metab Case Rep. 2014;2014:130077. doi: 10.1530/EDM-13-0077[↩]
- Birdi K, Prasad AN, Prasad C, Chodirker B, Chudley AE. The floppy infant: retrospective analysis of clinical experience (1990-2000) in a tertiary care facility. J Child Neurol. 2005 Oct;20(10):803-8. doi: 10.1177/08830738050200100401[↩]
- Pasrija D, Tadi P. Congenital Muscular Dystrophy. [Updated 2023 Jul 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK558956[↩]
- Bodensteiner JB. The evaluation of the hypotonic infant. Semin Pediatr Neurol. 2008 Mar;15(1):10-20. doi: 10.1016/j.spen.2008.01.003[↩]
- Dalen Y. Standing with and without vibration in children with severe cerebral palsy: Stockholm, 2011.[↩]




{kind=link}