Contents
- Blood clotting disorders
- Blood coagulation physiology
- Blood clotting disorders list
- Bleeding disorders (Coagulopathy)
- Hemophilias
- Von Willebrand disease
- Inherited platelet disorders
- Factor 1 (fibrinogen) deficiency
- Factor 2 (prothrombin) deficiency
- Factor 5 deficiency
- Combined factor 5 and factor 8 deficiency
- Factor 7 deficiency
- Factor 10 deficiency
- Factor 11 deficiency
- Factor 12 deficiency
- Factor 13 deficiency
- Combined deficiency of vitamin K-dependent clotting factors
- Disseminated intravascular coagulation (DIC)
- Liver disease
- Clotting disorders (thrombophilia)
- Blood clotting disorders diagnosis
- Blood clotting disorders treatment
Blood clotting disorders
Blood clotting disorders are dysfunctions in the body’s ability to control the formation of blood clots where your blood may not clot enough, which can lead to too much bleeding (hemorrhage), or your blood may form blood clots even without an injury (thrombosis) 1, 2.
Blood clotting disorders are also known as bleeding disorders (coagulation disorders or coagulopathy) or clotting disorders (thrombophilia).
- Bleeding disorders (coagulation disorders or coagulopathy) means that people bruise and bleed too easily. In people with bleeding disorders, the blood clotting process doesn’t work properly, with the result that they can bleed for longer than normal, and some people may experience spontaneous bleeding into joints, muscles, or other parts of their bodies which can lead to developmental and permanent mobility issues. Bleeding disorders (coagulation disorders or coagulopathy) can result from disorders of:
- Blood clotting (coagulation) system. Sometimes there is an abnormality of blood coagulation that increases the risk of clotting (called thrombophilia).
- Blood vessels
- Platelets (cell-like particles that help in the clotting system)
- Clotting disorders, also called hypercoagulable state or thrombophilia, occur when the body is unable to make sufficient amounts of the proteins that are needed to help the blood clot, stopping bleeding. These proteins are called clotting factors (coagulation factors). All clotting factors are made in your liver. The liver requires vitamin K to make some of the clotting factors.
Blood clotting disorders can be inherited (meaning that you are born with the condition) or acquired (meaning you develop the condition as the result of another illness or injury). Acquired blood clotting disorders include antiphospholipid syndrome (APS) and disseminated intravascular coagulation (DIC).
The most common inherited bleeding disorder in which your blood doesn’t clot properly is von Willebrand disease. People with the von Willebrand disease have low levels of von Willebrand factor (vWF), a plasma protein that helps the initial adhesion of platelets at sites of vascular injury and also binds and stabilizes blood clotting factor 8 (FVIII) in the circulation. Therefore, defects in vWF can cause bleeding by impairing platelet adhesion or by reducing the concentration of blood clotting factor 8 (FVIII). If you have von Willebrand disease, you might have:
- Excessive bleeding from an injury or after surgery or dental work
- Frequent nosebleeds that don’t stop within 10 minutes
- Heavy or long menstrual bleeding
- Heavy bleeding during labor and delivery
- Blood in your urine or stool
- Easy bruising or lumpy bruises
Women with the von Willebrand disease menstrual signs and symptoms might include:
- Blood clots greater than 1 inch (2.5 centimeters) in diameter in your menstrual flow
- The need to change your menstrual pad or tampon more than once an hour
- The need to use double sanitary protection for menstrual flow
- Symptoms of anemia, including tiredness, fatigue or shortness of breath.
Blood clots (thrombi) can cause many health problems. Symptoms of blood clots depend on where in the body they form. Typically, a blood clot (thrombus) will form in the veins and appear in the legs or lungs. Blood clots in the legs can cause deep vein thrombosis (DVT). Blood clots in the lungs can cause a pulmonary embolism (PE). A blood clot that becomes lodged in a lung artery can cause lung damage, organ damage or death. It is rare for blood clots to form in the arteries where it is called an arterial thrombosis. When they do, they can lead to heart attack or stroke.
The most common inherited coagulation disorders in which the blood doesn’t clot are the hemophilias. Hemophilia occurs when a clotting factor is missing or levels of the clotting factor are low. Clotting factors are proteins in the blood that work with cells known as platelets to form clots. People with hemophilia experience prolonged bleeding or oozing following an injury, surgery, or having a tooth pulled. In severe cases of hemophilia, continuous bleeding occurs after minor trauma or even when there is no obvious injury sometimes called spontaneous bleeding. Serious complications can result from bleeding into the joints, muscles, brain, or other internal organs. Milder forms of hemophilia do not necessarily involve spontaneous bleeding, and the condition may not become apparent until abnormal bleeding occurs following surgery or a serious injury.
There are several different types of hemophilia. The most common are:
- Hemophilia A also called Classic hemophilia or factor 8 deficiency, is the most common form of hemophilia that is caused by a lack or decrease of blood clotting factor 8 (factor VIII). The Centers for Disease Control and Prevention (CDC) estimates about 10 in 100,000 people have hemophilia A.
- Hemophilia B also called Christmas disease or factor 9 deficiency, is caused by a lack or decrease of blood clotting factor 9 (factor IX). The CDC estimates about 3 in 100,000 people in the U.S. have hemophilia B.
- Hemophilia C is very rare affecting 1 in 100,000 people and is due to deficiency of factor 11 (factor XI).
Although hemophilia A and hemophilia B have very similar signs and symptoms, they are caused by variants also known as mutations in different genes. People with an unusual form of hemophilia B, known as hemophilia B Leyden, experience episodes of excessive bleeding in childhood but have few bleeding problems after puberty.
Some people develop hemophilia with no family history of the disorder. This is called acquired hemophilia. Acquired hemophilia is a variety of the condition that occurs when a person’s immune system attacks clotting factor 8 (factor VIII) or factor 9 (factor IX) in the blood. It can be associated with:
- Pregnancy
- Autoimmune conditions
- Cancer
- Multiple sclerosis
- Drug reactions
People with blood clotting disorders should be followed by a treatment centre that specializes in the diagnosis and treatment of bleeding disorders, as they are most likely to offer the best care and information.
If you think you may have a blood clotting disorder, your doctor will ask about your family and medical history. They may also run tests to be sure of the diagnosis. If you have a blood clotting disorder, you may need medicine to stop the blood from clotting. Your doctor may also talk to you about ways to prevent blood clots and to stay healthy.
What are clotting factors?
Clotting factors are proteins in the blood that control bleeding. Coagulation factors circulate in your blood in an inactive form. When a blood vessel is injured, the walls of the blood vessel contract to limit the flow of blood to the damaged area as process known as vasoconstriction. Then, small blood cells called platelets stick to the site of injury and spread along the surface of the blood vessel to stop the bleeding. At the same time, chemical signals are released from small sacs inside the platelets that attract other platelets to the area and make them clump together to form what is called a platelet plug.
On the surface of these activated platelets, many different clotting factors work together in a series of complex chemical reactions known as the coagulation cascade to form a fibrin clot (see Figure 3). The clot acts like a mesh to stop the bleeding. The coagulation cascade is a series of pathways that converge on factor X (factor 10), a clotting factor essential for fibrin release and clot formation.
When a blood vessel is injured, the coagulation cascade is initiated, and each coagulation factor is activated in a specific order to lead to the formation of the blood clot. Coagulation factors are identified with Roman numerals (i.e., factor VIII or FVIII). The addition of the letter a (i.e., factor VIIIa) signifies the activated form of the clotting factor.
If any of the clotting factors are missing or are not working properly, the coagulation cascade is blocked. When this happens, the blood clot does not form, and the bleeding continues longer than it should.
In people with a clotting factor deficiency, one (or very rarely, more than one) clotting factor is missing, present at a low level, or not working properly. This impacts the normal process of blood clotting, making it difficult for the blood to form a clot.
Is a blood clotting disorder dangerous?
Yes, blood clotting disorders can be dangerous, especially when you don’t get treatment. People with clotting disorders have an increased risk of getting a blood clot in their:
- Arteries (blood vessels that carry blood away from your heart).
- Veins (blood vessels that carry blood to your heart).
Another name for a clot inside a blood vessel is a thrombus or an embolus.
Blood clots in your veins can travel through your bloodstream and cause:
- Deep vein thrombosis (a blood clot in the veins of your pelvis, leg, arm, liver, intestines or kidneys).
- A pulmonary embolus (blood clot in your lungs).
Blood clots in your arteries can increase your risk for:
- Stroke.
- Heart attack.
- Severe leg pain.
- Difficulty walking.
- Loss of an arm or leg.
Can a blood clotting disorder cause miscarriage?
Yes, it’s possible to have a miscarriage if you have a blood clot disorder like antiphospholipid syndrome (APS). Antiphospholipid syndrome (APS) increases your blood clot risk, especially if you’ve had blood clots before. Higher blood volume and pressure during pregnancy play a role in making you five times more likely to develop a blood clot, even if you don’t have a blood clotting disorder.
What are platelets?
Platelets are small disc-shaped cells that circulate in the blood. They play an important role in the formation of blood clots to help stop bleeding and in the repair of damaged blood vessels.
When a blood vessel is injured, platelets begin the process to stop the bleeding by forming what is called a platelet plug. This happens in three stages (see Figures 1 and 2):
- Adhesion: platelets stick to the damaged area and spread along the surface of the blood vessel to stop the bleeding.
- Secretion: as they do this, the platelets become “activated” and chemical signals are released from small packets called granules inside the platelets.
- Aggregation: these chemicals attract other platelets to the site of injury and make them clump together to form the platelet plug that becomes strengthened by fibrin.
As the platelet plug is forming, proteins called clotting factors are also recruited to the site of injury. These clotting factors work together on the surface of the platelets to strengthen the platelet plug by forming a mesh called a fibrin clot.
Platelets have several components, such as receptors on their surface and internal granules, that all contribute to clot formation.
- Receptors. Receptors are proteins on the surface of the platelets that help the platelet interact with, and respond to, chemicals, proteins, or other blood cells.
- Granules. Granules are small packets inside the platelets where proteins and other chemicals are stored. The contents of the granules are released from the platelet during the secretion phase of activation. These molecules act as signals to recruit more platelets and other cells to the site of injury to stop the bleeding. There are two types of granules: alpha granules and dense granules. The contents differ between the alpha and the dense granules, and they work in different ways to recruit more platelets, activate the clotting factors, and stop the bleeding.
Blood coagulation physiology
Blood coagulation is based on hemostasis, is a process to prevent and stops bleeding that involves multiple interlinked steps culminating into the formation of a “plug” that closes up the damaged site of the blood vessel controlling the bleeding 3.
Hemostasis consists of four main stages 3:
- Constriction of the blood vessel (vasoconstriction). Vasoconstriction (constriction of the blood vessel) refers to contraction of smooth muscles (vascular spasm) in the tunica media layer of endothelium (blood vessel wall) (Figure 1). At the site of the disrupted endothelial lining, the extracellular matrix (ECM) or collagen becomes exposed to the blood components, promoting platelet adhesion and aggregation 4, 5, 6
- Formation of a temporary “platelet plug”. Platelet activators, such as platelet activating factor and thromboxane A2 (TXA2), activate platelets in the bloodstream, leading to attachment of platelets’ membrane receptors (e.g. glycoprotein IIb/IIIa) to extracellular matrix proteins (e.g. von Willebrand factor) on cell membranes of damaged endothelial cells and exposed collagen at the site of injury. The adhered platelets aggregate and form a temporary “platelet plug” to stop bleeding (Figure 2). This process is often called “primary hemostasis” 7.
- Activation of the coagulation cascade. Coagulation cascade is a series of enzymatic reactions that lead to the formation of a stable blood clot. The endothelial cells release substances like tissue factor, which triggers the extrinsic pathway of the coagulation cascade. This is called as “secondary hemostasis”. Approximately 50 significant substances affect the blood coagulation mechanisms. The blood coagulation cascade of secondary hemostasis mainly consists of two main pathways: (i) Intrinsic pathway (contact activation pathway) (ii) Extrinsic (tissue factor pathway). The blood clotting process can be classified into three important steady steps as follows: (i) involvement of a complex cascade, triggering the chemical reactions that are mediated by the coagulation factors that respond to form fibrin strands for consolidating the platelet plugs; (ii) the conversion of prothrombin (PT) into thrombin, which is catalyzed by the prothrombin activator; and (iii) conversion of fibrinogen into fibrin, which eventually enmeshes the plasma, platelets and blood cells to build a firmer clot (Figure 3) 8, 9, 10.
- Formation of “fibrin plug” or the final clot. Near the end of the extrinsic pathway, after thrombin completes conversion of fibrinogen into fibrin, factor XIIIa (plasma transglutaminase; activated form of fibrin-stabilizing factor) promotes fibrin cross-linking, and subsequent stabilization of fibrin, leading to the formation of a fibrin clot (final blood clot), which temporarily seals the wound to allow wound healing until its inner part is dissolved by fibrinolytic enzymes, while the clot’s outer part is shed off.
Clot Resolution (Tertiary Hemostasis)
After the fibrin clot is formed, clot retraction occurs and then clot resolution starts, and these two process are together called “tertiary hemostasis” 11, 12. Activated platelets contract their internal actin and myosin fibrils in their cytoskeleton, which leads to shrinkage of the clot volume. Plasminogen activators, such as tissue plasminogen activator (t-PA), activate plasminogen into plasmin, which promotes lysis of the fibrin clot; this restores the flow of blood in the damaged or obstructed blood vessels 13, 3.
Figure 1. Vasoconstriction phase of blood coagulation pathway
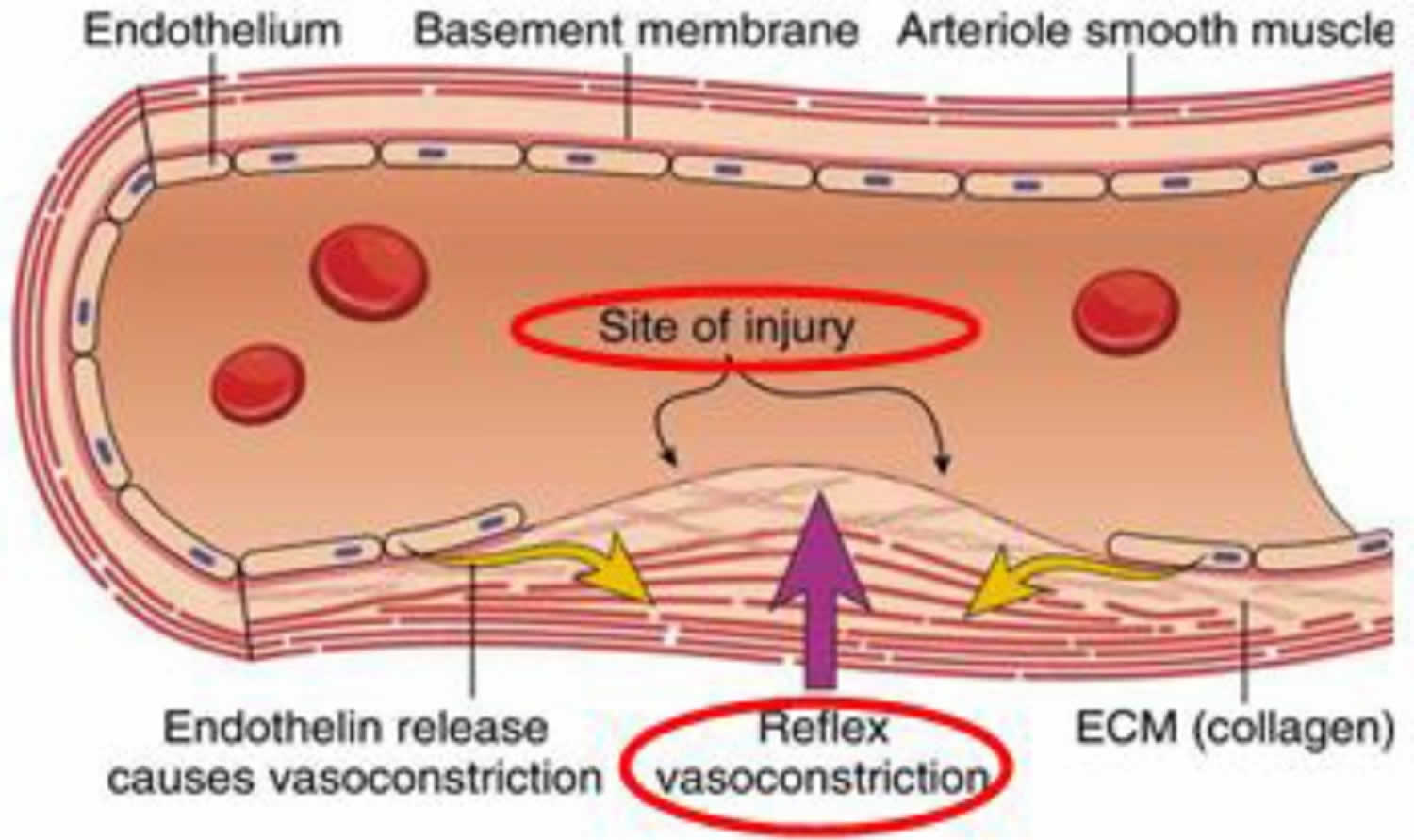
Footnotes: Vascular spasm occurs whenever there is an injury or damage to the blood vessels. This will trigger a vasoconstriction, which could eventually stop the blood flow. This reaction can be responded within 30 minutes, and is localized to the injured area. At this stage, exposed collagen fibers will release ATP and other inflammatory mediators to recruit macrophages. In addition, at the site of the disrupted endothelial lining, the extracellular matrix (ECM) or collagen becomes exposed to the blood components, promoting platelet to adhere, activate and aggregate to form a platelet plug, sealing off the injured area 4, 5.
[Source 14 ]Figure 2. Platelet plug formation phase of blood coagulation pathway
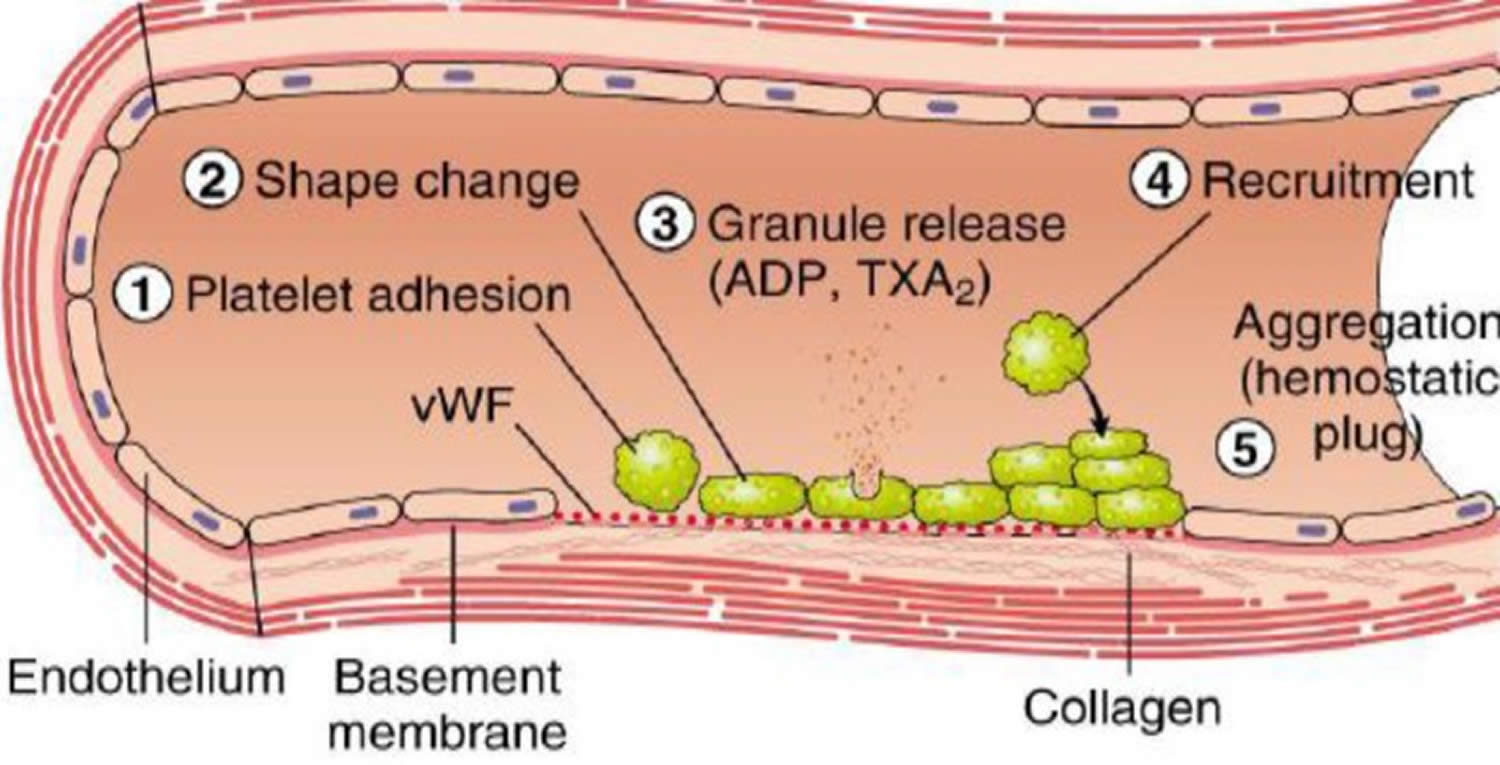
Footnotes: Injuries on the endothelial cells highly exposes to thrombogenic, subendothelial extracellular matrix (ECM) to ease platelet adherences and activation. Platelet activation triggers platelet shape changes by releasing secretory granules. Released secretary granules will recruit additional platelets to form the platelet plug, which is referred to as primary hemostasis 4.
[Source 14 ]Figure 3. Blood coagulation cascade
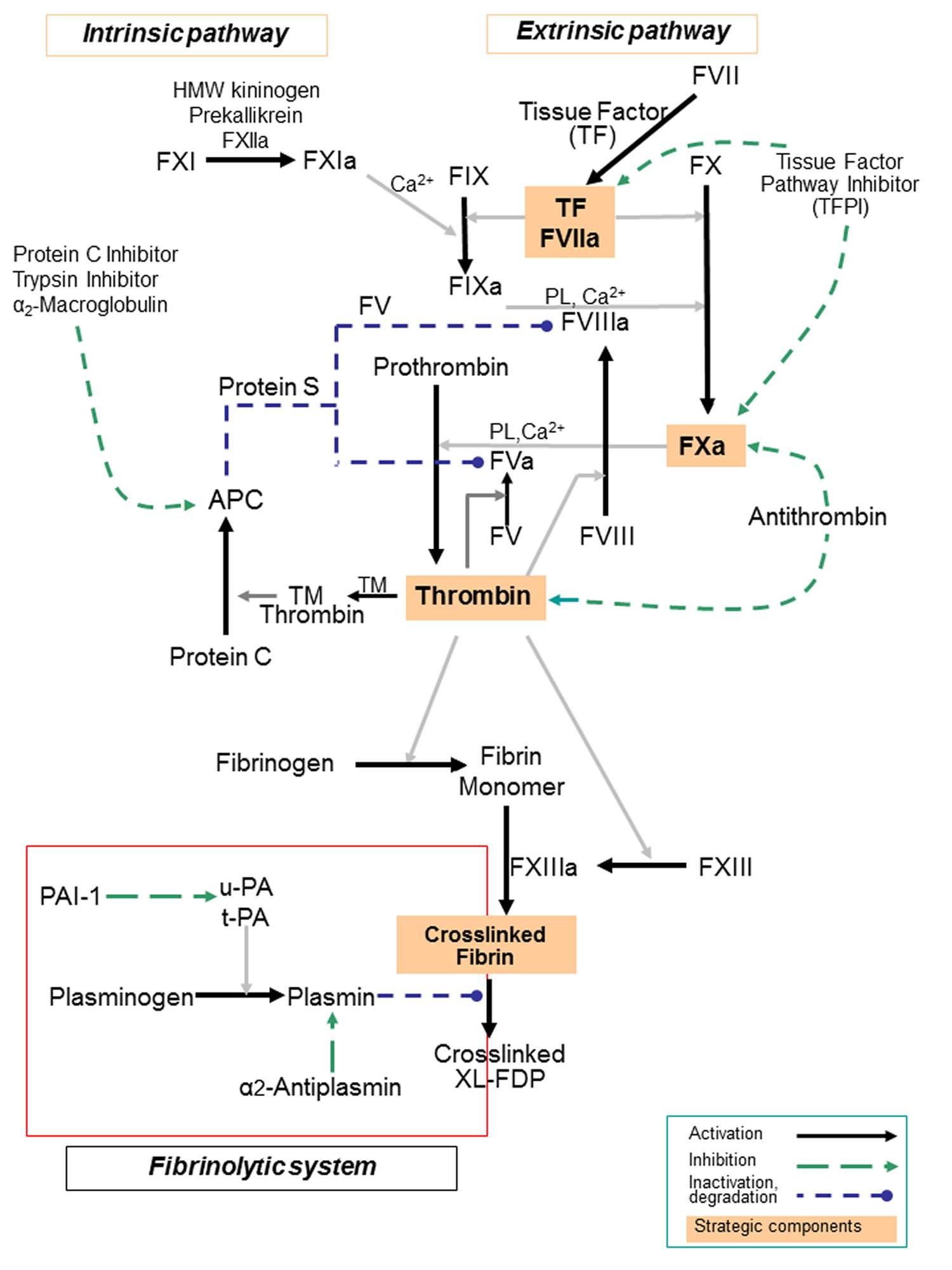
Footnotes: Blood coagulation pathway showing intrinsic pathway and extrinsic pathway model of coagulation, reflected in the laboratory measurements of the activated partial thromboplastin time (APTT is a reflection of the intrinsic pathway), prothrombin time (PT is a reflection of the extrinsic pathway) and thrombin clotting time (TCT is a measure of the final step in the coagulation pathway, the conversion of fibrinogen to fibrin via the action of thrombin). It is now understood that coagulation tests such as the prothrombin time (PT) and activated partial thromboplastin time (APTT) are based on what happens artificially in the test setting (in vitro) and therefore do not necessarily reflect what actually happens in your body (in vivo). Nevertheless, they can be used to evaluate certain components of the hemostasis system. The prothrombin time (PT) and activated partial thromboplastin time (APTT) tests each evaluate coagulation factors that are part of different groups of chemical reaction pathways in the cascade, called the intrinsic, extrinsic, and common pathways.
- Isolated prolongations of activated partial thromboplastin time (APTT) should prompt considerations of factor VIII, IX, XI or XII deficiency. Of these, factor VIII deficiency (hemophilia A) and factor IX deficiency (hemophilia B) can cause severe bleeding tendencies depending on the factor levels. Factor XI deficiency (hemophilia C) has a variable phenotype ranging from asymptomatic to severe bleeding. Factor XII deficiency is not associated with a bleeding disorder.
- Isolated prolongations of the prothrombin time (PT) are most often due to factor VII deficiency.
- Deficiencies in factors V, X, thrombin and fibrinogen prolong both the APTT and the PT, as they are in the common pathway.
- Isolated prolongations of thrombin clotting time (TCT) should prompt considerations of sensitive to deficiencies in fibrinogen and drugs such as direct and indirect thrombin inhibitors.
Blood clotting disorders list
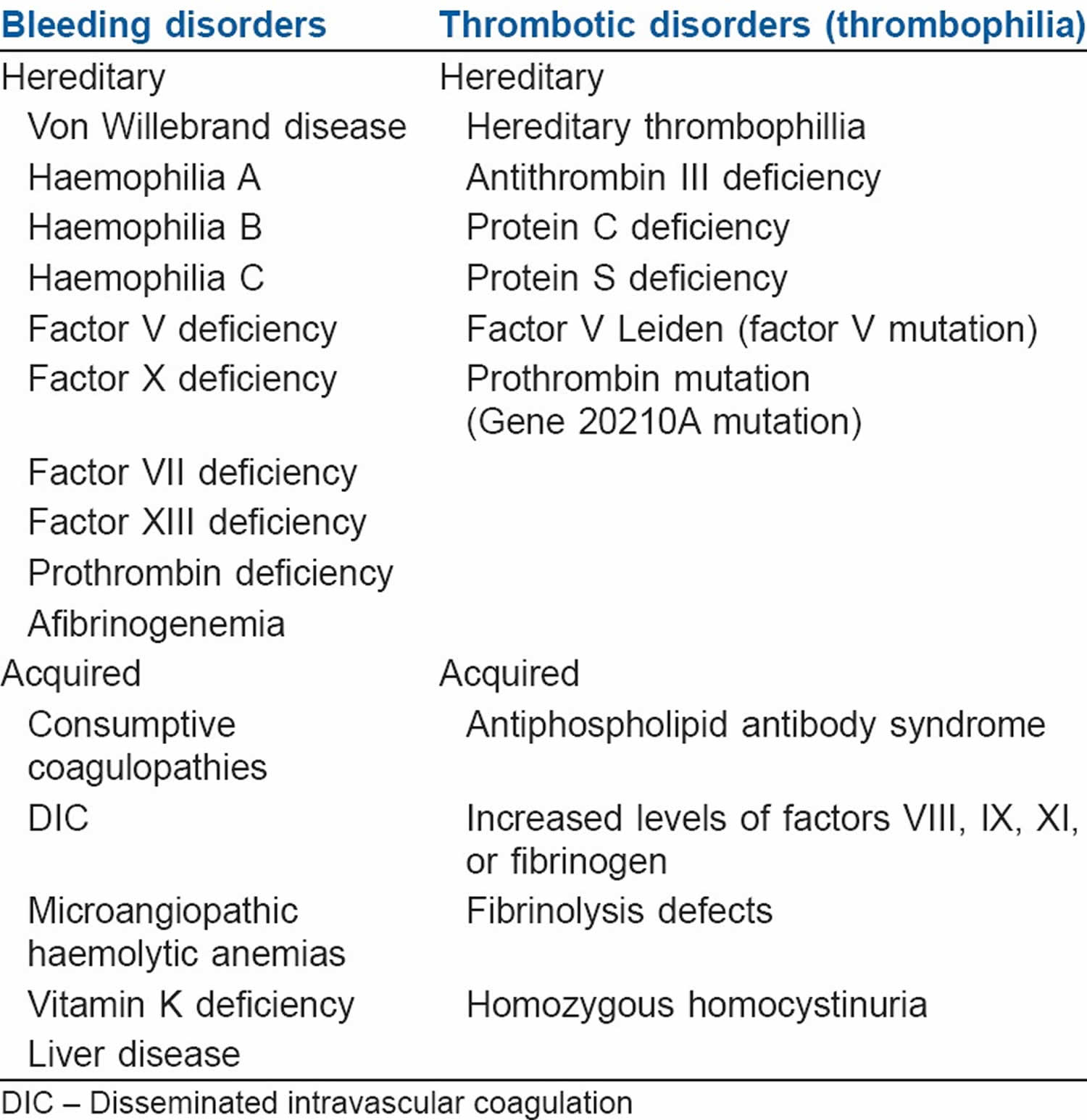
Bleeding disorders (Coagulopathy)
In people with bleeding disorders, the clotting process doesn’t work properly. As a result, people with bleeding disorders can bleed for longer than normal, and some may experience spontaneous bleeding into joints, muscles, or other parts of their bodies. Most bleeding disorders are present at birth (i.e., inherited). Rarely, some people develop (or acquire) a bleeding disorder later in life.
The most common bleeding disorder is von Willebrand disease, where 1 out of 1,000 individuals are affected and require medical attention for bleeding 15. However, it is generally less severe than other bleeding disorders. Many people with von Willebrand disease may not know that they have the disorder because their bleeding symptoms are very mild.
Hemophilia is a bleeding disorder that affects approximately 1,125,000 men worldwide 15. People with hemophilia do not have enough clotting factor VIII (hemophilia A) or factor IX (hemophilia B) in their blood. As a result, they can bleed for longer than normal.
Rare clotting factor deficiencies are bleeding disorders in which one or more of the other clotting factors (i.e., factors I, II, V, V+VIII, VII, X, XI, XII, or XIII) is missing or not working properly. Less is known about these disorders because they are diagnosed so rarely. All factor deficiencies are rare diseases, and rare clotting factor deficiencies are considered ultra-rare diseases, because they affect even fewer people.
Humans have twenty-three pairs of chromosomes: twenty-two pairs of autosomal chromosomes (also called autosomes) and one pair of sex chromosomes (X or Y). Unlike hemophilia, which is due to mutations on the X chromosome, rare clotting factor deficiencies are due to mutations on the autosomes.
Majority of clotting factors are synthesized in your liver therefore severe liver disease is associated with bleeding disorders (coagulopathy). Since your liver is also involved in the clearance of activated clotting factors and fibrinolytic products, it may predispose to disseminated intravascular coagulation (DIC). Management of bleeding secondary to liver disease is based on the laboratory values of various coagulation tests.
Hypothermia is also associated with anticoagulatory effects, which are more pronounced in the presence of acidosis. The effects may result from platelet dysfunction in mild hypothermia (below 95°F [35°C]) to decreased synthesis of clotting enzymes and plasminogen activator inhibitors when temperatures is less than 91.4°F (33°C) 16.
Finally, inherited platelet disorders are conditions in which platelets don’t work the way they should, resulting in a tendency to bleed or bruise.
Hemophilias
Hemophilia is a rare bleeding disorder in which the blood does not clot properly because it doesn’t have enough blood-clotting proteins (clotting factors). If you have hemophilia, you might bleed for a longer time after an injury than you would if your blood clotted properly. This can lead to problems with bleeding too much after an injury or surgery. You can also have sudden bleeding inside your body, such as in your joints, muscles, and organs. The most significant symptom of hemophilia is unusual or excessive bleeding or bruising.
There are several different types of hemophilia. The most common are:
- Hemophilia A also called Classic hemophilia or factor 8 deficiency, is the most common form of hemophilia that is caused by a lack or decrease of blood clotting factor 8 (factor VIII). The Centers for Disease Control and Prevention (CDC) estimates about 10 in 100,000 people have hemophilia A.
- Hemophilia B also called Christmas disease or factor 9 deficiency, is caused by a lack or decrease of blood clotting factor 9 (factor IX). The CDC estimates about 3 in 100,000 people in the U.S. have hemophilia B.
- Hemophilia C is very rare affecting 1 in 100,000 people and is due to deficiency of factor 11 (factor XI).
The severity of the bleeding tendency in people with hemophilia is directly related to the levels of the clotting factors. The levels of blood clotting factor 8 (factor VIII) need to be assessed in the preoperative period and human or recombinant factor VIII concentrates are transfused to keep the factor VIII levels 100% in the perioperative period 17.
Is hemophilia a serious illness?
It can be. People with severe hemophilia may develop life-threatening bleeding, but they’re more likely to develop bleeding in their muscles and joints.
Hemophilia causes
Most types of hemophilia are inherited. They are caused by change in one of the genes also called a mutation that provides instructions for making the clotting factor proteins. The change may mean that the clotting proteins don’t work properly or that they are missing altogether. These genes that provide instructions for making the clotting factor proteins are on the X chromosome (a sex chromosome).
- Every person receives one set of chromosomes from their biological mother and one set of chromosomes from their biological father. A mother will always pass an X chromosome to her offspring. The father will determine the assigned sex at birth by providing either an X or a Y chromosome.
- People who are born male have one X chromosome from the mother and one Y chromosome from the father, in other word males have XY sex chromosome. Males can get hemophilia if their X chromosome has the gene mutation.
- People who are born female have two X chromosomes, one from the father and one from the mother i.e., XX sex chromosome. They usually only get hemophilia if:
- Both their X chromosomes have the gene change
- OR
- One X chromosome has the gene change and the other X chromosome is missing or inactive.
This means that hemophilia almost always occurs in boys and is passed from mother to son through one of the mother’s genes on her X chromosome. Most women with the defective gene on one X chromosome are called “carriers” of hemophilia because they carry hemophilia but may not have any signs or symptoms of hemophilia. That’s because there’s a normal factor gene on their second X chromosome. However, some carriers of hemophilia can have bleeding symptoms if their clotting factors are moderately decreased. Women “carrier” of hemophilia can pass the gene change on to their children. There’s a 50% chance that any children of women “carrier” of hemophilia — boys or girls — will inherit hemophilia. Boys who do inherit hemophilia are more likely to have severe symptoms. That’s because they don’t get a healthy X chromosome from their father.
Some people develop hemophilia with no family history of the disorder. This is called acquired hemophilia. It is rare. Acquired hemophilia happens when your body makes specialized proteins called autoantibodies that attack and disable clotting factor 8 or 9 in the blood. This can happen because of pregnancy, immune system disorders, cancer, or allergic reactions to certain medicines. Sometimes the cause is unknown.
Acquired hemophilia can be associated with:
- Pregnancy
- Autoimmune conditions
- Cancer
- Multiple sclerosis
- Drug reactions.
Who is at risk for hemophilia?
Hemophilia is much more common in people who were born male since they can get it with a change to the gene on one X chromosome. People who have a family history of hemophilia are also at higher risk.
Hemophilia signs and symptoms
Signs and symptoms of hemophilia vary, depending on your level of clotting factors. If your clotting-factor level is mildly reduced, you might bleed only after surgery or trauma. If your clotting factor deficiency is severe, you can bleed easily for seemingly no reason.
The signs and symptoms of hemophilia are:
- Unexplained and excessive bleeding from cuts or injuries, or after surgery or dental work
- Bleeding into the joints. This can cause swelling and pain or tightness in the joints. It often affects the knees, elbows, and ankles.
- Bleeding into the skin or bruising.
- Bleeding into the muscle and soft tissue, which can cause a build-up of blood in the area (called a hematoma).
- Bleeding of the mouth and gums, including bleeding that is hard to stop after you lose a tooth.
- Bleeding after circumcision.
- Unusual bleeding after vaccinations.
- Bleeding in the head of an infant after a difficult delivery.
- Blood in the urine or stool.
- Frequent and hard-to-stop nosebleeds.
- In infants, unexplained irritability
In some cases, severe hemophilia may cause bleeding in the brain. This may cause brain damage and can be life-threatening.
A simple bump on the head can cause bleeding into the brain for some people who have severe hemophilia. This rarely happens, but it’s one of the most serious complications that can occur. Signs and symptoms of bleeding into the brain include:
- Painful, prolonged headache
- Repeated vomiting
- Sleepiness or lethargy
- Double vision
- Sudden weakness or clumsiness
- Convulsions or seizures
Hemophilia complications
Complications of hemophilia can include:
- Deep internal bleeding. Bleeding that occurs in deep muscle can cause the limbs to swell. The swelling can press on nerves and lead to numbness or pain. Depending on where the bleeding occurs, it could be life-threatening.
- Bleeding into the throat or neck. This can affect a person’s ability to breathe.
- Damage to joints. Internal bleeding can put pressure on the joints, causing severe pain. Left untreated, frequent internal bleeding can cause arthritis or destruction of the joint.
- Infection. If the clotting factors used to treat hemophilia come from human blood, there’s an increased risk of viral infections such as hepatitis C. Because of donor screening techniques, the risk is low.
- Adverse reaction to clotting factor treatment. In some people with severe hemophilia, the immune system has a negative reaction to the clotting factors used to treat bleeding. When this happens, the immune system develops proteins that keep the clotting factors from working, making treatment less effective.
Hemophilia diagnosis
Severe cases of hemophilia usually are diagnosed within the first year of life. Mild forms might not be apparent until adulthood. Some people learn they have hemophilia after they bleed excessively during a surgical procedure.
To find out if you have hemophilia, your doctor will:
- Ask about your medical history, including your symptoms and other health conditions you may have.
- Ask about your family history, to find out if you have relatives who have or had hemophilia.
- Do a physical exam to look for signs of hemophilia, such as bruising.
- Do certain blood tests to show if your blood is clotting properly. If it does not, then you will have clotting factor tests to diagnose the cause of the bleeding disorder. These blood tests would show the type of hemophilia, the clotting-factor deficiency and the severity of the hemophilia.
For people with a family history of hemophilia, genetic testing might be used to identify carriers to make informed decisions about becoming pregnant.
There is genetic testing for the factor VIII (factor 8) and factor IX (factor 9) genes. This testing may be used in people who have a family history of hemophilia to:
- Identify people who are carriers before they make decisions about pregnancy
- Test a fetus for hemophilia during pregnancy. However, the testing poses some risks to the fetus. Discuss the benefits and risks of testing with your doctor.
- Test a newborn for hemophilia
Hemophilia treatment
The best way to treat hemophilia is to replace the missing clotting factor so that your blood can clot properly. This is usually done by injecting replacement clotting factor into a vein. The replacement clotting factor may be made from donated human blood. Or it may be made in a lab; this man-made clotting factor is called a recombinant clotting factor. Replacement clotting factor can help treat a bleeding episode. In more severe cases of hemophilia, you might get the factor on a regular basis to prevent bleeding. You can learn how to inject the factor so that you can do it yourself at home.
The replacement clotting factor therapy can be given to treat a bleeding episode in progress. It can also be given on a regular schedule at home to help prevent bleeding episodes. Some people receive continuous replacement therapy.
There are other medicines to treat hemophilia. They may work by releasing factor VIII (factor 8) from where it is stored in the body tissues, replacing the function of factor VIII (factor 8), or preventing clots from breaking down.
- Desmopressin. In some forms of mild hemophilia, this hormone can stimulate the body to release more clotting factor. It can be injected slowly into a vein or used as a nasal spray.
- Emicizumab (Hemlibra) is a newer drug that doesn’t include clotting factors. This drug can help prevent bleeding episodes in people with hemophilia A.
- Clot-preserving medications, also known as anti-fibrinolytics, help prevent blood clots from breaking down.
- Fibrin sealants can be applied directly to wound sites to promote clotting and healing. Fibrin sealants are especially useful for dental work.
First aid for minor cuts. Using pressure and a bandage will generally take care of the bleeding. For small areas of bleeding beneath the skin, use an ice pack. Ice pops can be used to slow down minor bleeding in the mouth.
If bleeding has damaged your joints, physical therapy may help them function better. Physical therapy can ease signs and symptoms if internal bleeding has damaged your joints. Severe damage might require surgery.
To avoid excessive bleeding and protect your joints:
- Exercise regularly. Activities such as swimming, bicycle riding and walking can build muscles while protecting joints. Contact sports — such as football, hockey or wrestling — are not safe for people with hemophilia.
- Avoid certain pain medications. Drugs that can make bleeding worse include aspirin and ibuprofen (Advil, Motrin IB, others). Instead, use acetaminophen (Tylenol, others), which is a safer alternative for mild pain relief.
- Avoid blood-thinning medications. Medications that prevent blood from clotting include heparin, warfarin (Jantoven), clopidogrel (Plavix), prasugrel (Effient), ticagrelor (Brilinta), rivaroxaban (Xarelto), apixaban (Eliquis), edoxaban (Savaysa) and dabigatran (Pradaxa).
- Practice good dental hygiene. The goal is to prevent tooth and gum disease, which can lead to excessive bleeding.
- Get vaccinations. People with hemophilia should receive recommended vaccinations at the appropriate ages, as well as hepatitis A and B. Requesting use of the smallest gauge needle and having pressure or ice applied for 3 to 5 minutes after the injection can reduce the risk of bleeding.
- Protect your child from injuries that could cause bleeding. Kneepads, elbow pads, helmets and safety belts all help prevent injuries from falls and other accidents. Keep your home free of furniture with sharp corners.
Hemophilia prognosis
If you have hemophilia, you’ll need medical treatment for the rest of your life. How much treatment you’ll need depends on your condition type, severity and if you develop inhibitors.
Hemophilia life expectancy
According to 2012 data from the World Federation of Hemophilia, the lifespan for men and people with hemophilia is about 10 years fewer than the lifespan for men/people without hemophilia. The federation also states that children diagnosed with and treated for hemophilia have a normal life expectancy.
But everyone is different. What’s true for one person with hemophilia may not be true for others. If you or your child has hemophilia, ask your provider what you can expect. They know your/your child’s situation, including overall health, and they’re your best resource for information.
Von Willebrand disease
Von Willebrand disease is a bleeding disorder that slows the blood clotting process, causing prolonged bleeding after an injury. People with this condition often experience easy bruising, long-lasting nosebleeds, and excessive bleeding or oozing following an injury, surgery, or dental work. Mild forms of von Willebrand disease may become apparent only when abnormal bleeding occurs following surgery or a serious injury. People with this condition who have menstrual periods typically have heavy or prolonged bleeding during menstruation (menorrhagia), and some may also experience reproductive tract bleeding during pregnancy and childbirth. In severe cases of von Willebrand disease, heavy bleeding occurs after minor trauma or even in the absence of injury (spontaneous bleeding). Symptoms of von Willebrand disease may change over time. Increased age, pregnancy, exercise, and stress may cause bleeding symptoms to become less frequent.
Von Willebrand disease is divided into three types. Type 1 has one subtype (1C), and type 2 is divided into four subtypes (2A, 2B, 2M, and 2N). Type 1 is the most common of the three types, accounting for 75 percent of affected individuals. Type 1 is typically mild, but some people are severely affected. Type 2 accounts for about 15 percent of cases. This type is usually of intermediate severity. Type 3 is the rarest form of the condition, accounting for about 5 percent of affected individuals, and is usually the most severe.
Another form of the disorder, acquired von Willebrand syndrome, is not caused by inherited gene variants (also called mutations). Acquired von Willebrand syndrome is typically seen in people with other disorders, such as diseases that affect bone marrow or immune cell function. This rare form of the condition is characterized by abnormal bleeding into the skin and other soft tissues, usually beginning in adulthood.
Von Willebrand disease cause
Von Willebrand disease is caused by a deficiency or poor functioning of von Willebrand factor (vWF) a protein that plays a key role in blood clotting. Von Willebrand factor helps blood platelets clump together and stick to the blood vessel wall, which is necessary for normal blood clotting.
The usual cause of von Willebrand disease is an inherited abnormal VWF gene that controls von Willebrand factor (vWF). The VWF gene provides instructions for making a blood clotting protein called von Willebrand factor (vWF), which is essential for the formation of blood clots. When you have low levels of von Willebrand factor (vWF) protein or it doesn’t work as it should, small blood cells called platelets cannot stick together properly nor attach themselves normally to the blood vessel walls when an injury has occurred. This interferes with the clotting process and can sometimes cause uncontrolled bleeding.
Many people with von Willebrand disease also have low levels of factor VIII (factor 8), another protein that helps in clotting. Factor VIII (factor 8) is involved in another inherited clotting disorder called hemophilia. But unlike hemophilia, which mainly affects males, von Willebrand disease affects males and females and is usually milder.
Von Willebrand disease is divided into three types based on the amount of von Willebrand factor (vWF) that is produced or its ability to function:
- Type 1 von Willebrand disease is the most common of the three types, accounting for 75 percent of affected individuals. People with type 1 von Willebrand disease have varying amounts of von Willebrand factor (vWF) in their bloodstream. Type 1 is typically mild, but some people are severely affected. Some people with a mild case of type 1 never experience a prolonged bleeding episode. In type 1C, the amount of von Willebrand factor is low because the protein is removed from the body more quickly than usual.
- Type 2 von Willebrand disease accounts for about 15 percent of cases. This type is usually of intermediate severity. People with type 2 von Willebrand disease can have bleeding episodes of varying severity depending on the extent of the von Willebrand factor (vWF) abnormalities, but the bleeding episodes are typically similar to those seen in type 1. Type 2 von Willebrand disease is divided into four subtypes (2A, 2B, 2M, and 2N).
- In type 2A the von Willebrand factor (vWF) is not the right size and cannot help form clots.
- In type 2B the von Willebrand factor (vWF) forms clots too easily. The clots are removed from the body, so there is not enough von Willebrand factor available when it is needed for clotting.
- In types 2M and 2N the von Willebrand factor (vWF) is unable to interact with other structures or proteins needed to form blood clots.
- Type 3 von Willebrand disease is the rarest form of the condition, accounting for about 5 percent of affected individuals, and have severe bleeding episodes.
Von Willebrand disease can have different inheritance patterns. Most cases of type 1 and type 2 von Willebrand disease are inherited in an “autosomal dominant pattern”, which means one copy of the altered gene in each cell is sufficient to cause the disease. If you have the mutated gene for von Willebrand disease, you have a 50% chance of transmitting this gene to your children.
Type 3, some cases of type 2, and a small number of type 1 cases of von Willebrand disease are inherited in an “autosomal recessive pattern”, which means both of your parents have to pass a mutated gene to you cause the disease. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
If you plan to have children and have a family history of von Willebrand disease, consider genetic counseling. If you carry the gene for von Willebrand disease, you can pass it on to your offspring, even if you don’t have symptoms.
Rarely, von Willebrand disease can develop later in life in people who didn’t inherit an affected gene from a parent. This is known as acquired von Willebrand syndrome, and it’s likely caused by an underlying medical condition. Acquired von Willebrand syndrome is typically seen in people with other disorders, such as diseases that affect bone marrow or immune cell function. This rare form of the condition is characterized by abnormal bleeding into the skin and other soft tissues, usually beginning in adulthood.
Von Willebrand disease signs and symptoms
Many people with von Willebrand disease don’t know it because the signs are mild or absent. The most common sign of the condition is abnormal bleeding.
There are three main types of the disease. The amount of bleeding varies from one person to another, depending on the type and severity of the disease.
If you have von Willebrand disease, you might have:
- Excessive bleeding from an injury or after surgery or dental work
- Frequent nosebleeds that don’t stop within 10 minutes
- Heavy or long menstrual bleeding
- Heavy bleeding during labor and delivery
- Blood in your urine or stool
- Easy bruising or lumpy bruises
Women menstrual signs and symptoms might include:
- Blood clots greater than 1 inch (2.5 centimeters) in diameter in your menstrual flow
- The need to change your menstrual pad or tampon more than once an hour
- The need to use double sanitary protection for menstrual flow
- Symptoms of anemia, including tiredness, fatigue or shortness of breath.
Note: Most women with heavy or prolonged menstrual bleeding do not have von Willebrand disease.
Von Willebrand disease complications
Rarely, von Willebrand disease can cause uncontrollable bleeding, which can be life-threatening. Other complications of von Willebrand disease can include:
- Anemia. Heavy menstrual bleeding can cause iron deficiency anemia.
- Swelling and pain. This can be a result of abnormal bleeding in the joints or soft tissue.
Von Willebrand disease diagnosis
Mild forms of von Willebrand disease can be difficult to diagnose because bleeding is common, and, for most people, doesn’t indicate a disease. Low von Willebrand factor levels and bleeding do not always mean you have von Willebrand disease. However, if your doctor suspects you have a bleeding disorder, he or she might refer you to a blood disorders specialist (hematologist).
To evaluate you for von Willebrand disease, your doctor will likely ask you detailed questions about your medical history and check for bruises or other signs of recent bleeding.
Your doctor will also likely recommend the following blood tests:
- Von Willebrand factor antigen. This determines the level of von Willebrand factor in your blood by measuring a particular protein.
- Von Willebrand factor activity. There are a variety of tests to measure how well the von Willebrand factor works in your clotting process.
- Factor VIII (factor 8) clotting activity. This shows whether you have abnormally low levels and activity of factor VIII (factor 8).
- Von Willebrand factor multimers. This evaluates the structure of von Willebrand factor in your blood, its protein complexes and how its molecules break down. This information helps identify the type of von Willebrand disease you have.
- Bleeding time
- Blood typing
- Platelet function analysis
- Platelet count
- Ristocetin cofactor test.
The results of these tests can fluctuate in the same person over time due to factors such as stress, exercise, infection, pregnancy and medications. So you might need to repeat some tests.
If you have von Willebrand disease, your doctor might suggest that family members undergo tests to determine if this condition runs in your family.
Von Willebrand disease treatment
Even though von Willebrand disease has no cure, treatment can help prevent or stop bleeding episodes. Your doctor might suggest one or more of the following treatments to increase your von Willebrand factor, strengthen blood clots or control heavy menstrual bleeding:
- Desmopressin. Desmopressin is available as an DDAVP (desamino-8-arginine vasopressin) injection. DDAVP is a man-made hormone that controls bleeding by stimulating your body to release more of the von Willebrand factor (vWF) stored in the lining of your blood vessels. Many doctors consider desmopressin (DDAVP) the first treatment for managing von Willebrand disease. It can be used before minor surgical procedures to help control bleeding. You might be given a trial of desmopressin to make sure it’s effective for you. However, desmopressin (DDAVP) does not work for all types of von Willebrand disease. Tests should be done to determine what type of von Willebrand you have. If you are going to have surgery, your doctor may give you DDAVP before surgery to see if your von Willebrand factor levels increase.
- Replacement therapies. These include infusions of concentrated blood-clotting factors containing von Willebrand factor and factor VIII (factor 8). Your doctor might recommend them if DDAVP isn’t an option for you or has been ineffective. Another replacement therapy approved by the Food and Drug Administration for treating adults 18 and older is a genetically engineered (recombinant) von Willebrand factor product. Because recombinant factor is made without plasma, it can reduce the risk of a viral infection or allergic reaction.
- Alphanate (antihemophilic factor) is approved to treat or prevent bleeding episodes in people with von Willebrand disease who must have surgery or any other invasive procedure. Alphanate is also used to treat or prevent bleeding episodes in people with hemophilia A.
- Oral contraceptives. In addition to preventing pregnancy, these drugs can help control heavy bleeding during menstrual periods. The estrogen hormones in birth control pills can boost von Willebrand factor and factor VIII activity.
- Clot-stabilizing medications. These anti-fibrinolytic medications — such as aminocaproic acid (Amicar) and tranexamic acid (Cyklokapron, Lysteda) — can help stop bleeding by slowing the breakdown of blood clots. Doctors often prescribe these drugs before or after a surgical procedure or tooth extraction.
- Drugs applied to cuts. A fibrin sealant (Tisseel) placed on a cut helps curtail bleeding. This is applied like glue using a syringe. There are also over-the-counter products to stop nosebleeds.
If your condition is mild, your doctor might recommend treatment only when you’re having surgery or dental work or when you’ve had a trauma, such as a car accident.
Blood plasma or certain factor VIII (factor 8) preparations may also be used to decrease bleeding.
Von Willebrand disease prognosis
Bleeding may decrease during pregnancy. Women who have von Willebrand disease usually do not have excessive bleeding during childbirth.
von Willebrand disease is passed down through families. Genetic counseling may help prospective parents understand the risk for their children.
Inherited platelet disorders
Inherited (i.e., passed down from parent to child) platelet function disorders are conditions in which the platelets do not work the way they should. If the platelet plug does not form properly, bleeding can continue for longer than normal. People with platelet function disorders tend to bruise or bleed more easily than normal. Platelet function disorders can be caused by a problem with one of the receptors, with the granules, or with activation processes inside the platelets, and may not be associated with a low platelet count.
Bernard-Soulier syndrome
Bernard-Soulier syndrome is an inherited platelet function disorder caused by an abnormality in the receptor for von Willebrand factor (vWF). This receptor is also called Glycoprotein (GP) Ib/IX/V. Receptors are proteins on the surface of the platelets that help them interact with, and respond to, other blood cells or substances. Since the von Willebrand factor (vWF) receptor is absent or does not work properly, the platelets cannot bind to von Willebrand factor (vWF) and do not stick to the injured blood vessel wall the way they should. As a result, the platelet plug does not form normally.
Bernard-Soulier syndrome is an autosomal recessive disorder, meaning that both parents carry a genetic change (even though they themselves do not usually have the disorder, they may have mild bleeding symptoms), and pass this changed gene on to their child. Bernard-Soulier syndrome affects both males and females.
People with Bernard-Soulier syndrome should not take aspirin (acetylsalicylic acid), other nonsteroidal anti-inflammatory drugs (such as ibuprofen and naproxen), or blood thinners (anticoagulants), as these can worsen bleeding symptoms.
Bernard-Soulier syndrome symptoms
The symptoms of Bernard-Soulier syndrome vary from one individual to another. Signs of Bernard-Soulier syndrome are usually first noticed during childhood.
People with Bernard-Soulier syndrome may experience:
- Easy bruising
- Nose bleeds (epistaxis)
- Bleeding gums
- Heavy or prolonged menstrual bleeding (menorrhagia), bleeding during ovulation, or bleeding during or after childbirth
- Abnormal bleeding during or after surgery, circumcision, or dental work
- Rarely, vomiting blood or passing blood in bowel movements due to bleeding from the intestines (gastrointestinal hemorrhage)
Bernard-Soulier syndrome may cause more problems for women than men in early adulthood because of the risk of bleeding associated with menstruation and childbirth.
Bernard-Soulier syndrome diagnosis
Bernard-Soulier syndrome diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. The diagnosis of Bernard-Soulier syndrome requires a careful medical history and a series of laboratory tests.
In people with Bernard-Soulier syndrome:
- There are usually fewer platelets circulating in the blood, so the platelet count is low.
- The bleeding time (a standardized test that measures the time it takes for a small cut to stop bleeding) is longer than normal. This test may be difficult to perform in young children and is rarely used where more specific tests are available.
- The closure time (the time it takes for the platelet plug to form in a sample of blood) is longer than normal. This screening test is performed using a special instrument called a platelet function analyser (PFA-100/200®). It is more commonly used than the bleeding time test but may not be used where more specific tests are available.
- Examination of blood spread on a glass slide, using a microscope, identifies platelets that are larger than normal.
- Platelets do not clump together normally in a laboratory test called ristocetin-induced platelet aggregation.
- A test completed using a special technology called flow cytometry reveals fewer or absent VWF receptors (GPIb/IX/V) on the platelet surface. This is a conclusive diagnostic test.
- Genetic testing provides confirmation.
Note: Some tests are not available in all centers.
Bernard-Soulier syndrome is sometimes misdiagnosed as immune thrombocytopenia (ITP), an acquired platelet disorder in which the platelet count is low.
Bernard-Soulier syndrome treatment
Most people with Bernard-Soulier syndrome need treatment before surgical procedures (including dental work) or after injury or accidents. Some people will need treatment for severe nose bleeds or other bleeding symptoms, such as heavy menstrual bleeding.
When needed, Bernard-Soulier syndrome may be treated with:
- Antifibrinolytic drugs (drugs such as tranexamic acid that stabilize blood clots)
- Recombinant factor VIIa
- Desmopressin
- Fibrin sealants
- Hormonal suppressive therapy (birth control medications) and/or levonorgestrel-releasing intrauterine device/system to control heavy menstrual bleeding
- Iron replacement, as needed, to treat anemia caused by excessive or prolonged bleeding
- Platelet transfusions for severe bleeding episodes may sometimes be required
Glanzmann thrombasthenia
Glanzmann thrombasthenia is an inherited platelet function disorder caused by an abnormality in the receptor for fibrinogen also called GPIIb/IIIa. Receptors are proteins on the surface of the platelets that help the platelet interact with, and respond to, other blood cells or substances. Since the fibrinogen receptor is absent or does not work properly, the platelets do not stick to each other at the site of injury. As a result, the platelet plug does not form normally.
Glanzmann thrombasthenia is an autosomal recessive disorder, meaning that both parents carry a genetic change (even though they themselves do not usually have the disorder), and pass this changed gene on to their child. Glanzmann thrombasthenia affects both males and females.
People with Glanzmann thrombasthenia should not take aspirin (acetylsalicylic acid), other nonsteroidal anti-inflammatory drugs (such as ibuprofen and naproxen), or blood thinners (anticoagulants), as these can worsen bleeding symptoms.
Glanzmann thrombasthenia symptoms
Symptoms of Glanzmann thrombasthenia vary from one individual to another and range from mild to potentially life-threatening bleeding. Signs of Glanzmann thrombasthenia are usually first noticed during childhood.
People with Glanzmann thrombasthenia may experience:
- Easy bruising
- Nose bleeds (epistaxis)
- Bleeding gums
- Heavy or prolonged menstrual bleeding (menorrhagia), bleeding during ovulation, or bleeding during or after childbirth
- Abnormal bleeding during or after surgery, circumcision, or dental work
- Rarely, vomiting blood or passing blood in bowel movements due to bleeding from the intestines (gastrointestinal hemorrhage) or genitourinary tract (kidneys, ureters, bladder, and urethra)
Glanzmann thrombasthenia may cause more problems for women than men in early adulthood because of the risk of bleeding associated with menstruation and childbirth.
Glanzmann thrombasthenia diagnosis
Glanzmann thrombasthenia diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. The diagnosis of Glanzmann thrombasthenia requires a careful medical history and a series of laboratory tests.
In people with Glanzmann thrombasthenia:
- There is a normal number of platelets circulating in the blood. The platelet count is normal.
- The bleeding time (a standardized test that measures the time it takes for a small cut to stop bleeding) is longer than normal. This test may be difficult to perform in young children and is rarely used where more specific tests are available.
- The closure time (the time it takes for the platelet plug to form in a sample of blood) is longer than normal. This screening test is performed using a special instrument called a platelet function analyser (PFA-100/200®). It is more commonly completed than the bleeding time test but may not be used where more specific tests are available.
- Platelets do not clump together normally when exposed to chemicals that activate platelets. This test is called platelet aggregation.
- A test completed using a special technology called flow cytometry reveals fewer or absent fibrinogen receptors (GPIIb/IIIa) on the platelet surface. This is a conclusive diagnostic test.
- Genetic testing provides confirmation.
Note: Some tests are not available in all centers.
Glanzmann thrombasthenia treatment
Most people with Glanzmann thrombasthenia need treatment before surgical procedures (including dental work) or after injury or accidents. Some people will need treatment for severe nose bleeds or other bleeding.
When needed, Glanzmann thrombasthenia may be treated with:
- Antifibrinolytic drugs (drugs such as tranexamic acid that stabilize blood clots)
- Recombinant factor VIIa
- Fibrin sealants
- Hormonal suppressive therapy (birth control medications) and/or levonorgestrel-releasing intrauterine device/system to control heavy menstrual bleeding
- Iron replacement, as needed, to treat anemia caused by excessive or prolonged bleeding
- Platelet transfusions for severe bleeding episodes may be required
Platelet granule disorders
Platelet granule disorders are a diverse group of inherited disorders. Some are caused by an absence of platelet granules or their contents, but the most common are caused by a failure of the platelets to release their granule contents into the bloodstream.
Platelet granules are small packets inside the platelets in which proteins and other chemicals are stored. The contents of the granules are released during the secretion phase of platelet activation, acting as chemical signals to recruit more platelets and other cells to the site of injury to stop the bleeding. There are two types of granules: alpha granules and dense granules.
The way that platelet granule disorders are inherited (i.e., passed down from parent to child) is less consistent than for other types of platelet disorders and varies from one individual to the next.
- Secretion defects are a diverse group of disorders caused by an abnormality in the secretion mechanism. Even though the granules are present, their contents are not released into the bloodstream when the platelets are activated.
- Dense granule deficiency is a platelet function disorder caused by a lack of dense granules and the chemicals normally stored inside them. Without these chemicals, platelets are not activated properly, and the injured blood vessel does not constrict to help stop bleeding. This platelet disorder can occur as part of inherited syndromes such as Hermansky-Pudlak and Chediak-Higashi syndromes.
- Gray platelet syndrome is a very rare platelet function disorder caused by a lack of alpha granules and the chemicals and proteins normally stored inside them. Without these chemicals, platelets cannot stick to the blood vessel wall, clump together, or repair the injured blood vessel.
People with platelet granule disorders should not take aspirin (acetylsalicylic acid), other nonsteroidal anti-inflammatory drugs (such as ibuprofen and naproxen), or blood thinners (anticoagulants), as these can worsen bleeding symptoms, unless prescribed for a specific reason by a physician familiar with their disorder.
Women with inherited platelet function disorders should receive genetic counseling about the risks of having an affected child well in advance of a planned pregnancy and should be seen by an obstetrician as soon as they suspect they are pregnant. The obstetrician should work closely with the staff of the bleeding disorder treatment centre to provide the best care during pregnancy and childbirth, and to minimize potential complications for both the mother and newborn.
The main risk related to pregnancy and delivery is postpartum hemorrhage. Bleeding disorders are associated with an increased risk of bleeding, both immediately after and for several weeks following delivery. Women with platelet function disorders should therefore work with their doctors (both bleeding disorder specialist and the obstetrician) to develop an individual delivery plan. This plan should address all stages of labour, including delivery of the placenta, to reduce the risk and severity of bleeding. Treatment is different for each woman and depends on her personal and family history of bleeding symptoms, the diagnosis and severity of the inherited platelet function disorder, and the mode of delivery. Women with platelet function disorders should be advised to contact their health care provider immediately if postpartum bleeding is excessive.
In some circumstances, infants born to women with inherited platelet function disorders may also be at risk of inheriting the disorder and experience bleeding. Difficult and prolonged labour, and deliveries that require instrumentation, such as the use of forceps or vacuum extraction, should be avoided.
Platelet granule disorders symptoms
Symptoms of platelet granule disorders vary from one individual to the next.
People with granule disorders may experience:
- Easy bruising
- Nose bleeds (epistaxis)
- Bleeding gums
- Heavy or prolonged menstrual bleeding (menorrhagia), bleeding during ovulation, or bleeding during or after childbirth
- Abnormal bleeding during or after surgery, circumcision, or dental work.
Women with inherited platelet function disorders may experience more symptoms than men because of the risk of bleeding associated with menstruation and childbirth. Girls may experience heavy bleeding when they begin to menstruate.
Women with inherited platelet function disorders may have heavier and/or longer menstrual flow that could lead to iron deficiency (low levels of iron, which results in weakness and fatigue) and/or anemia (low levels of red blood cells).
Platelet granule disorders diagnosis
Platelet granule disorders diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. The diagnosis of platelet granule disorders requires a careful medical history and a series of laboratory tests.
In people with platelet granule disorders:
- Platelets may not clump together normally when exposed to chemicals that activate platelets. This test is called platelet aggregation. Platelet aggregation tests are the most useful way to diagnose these disorders.
- Granules can be evaluated using a high-power microscope called an electron microscope.
- The bleeding time (a standardized test that measures the time it takes for a small cut to stop bleeding) is longer than normal. This test may be difficult to perform in young children and is rarely used where more specific tests are available.
Platelet granule disorders treatment
Many people with platelet granule disorders need treatment before surgical procedures (including dental work) or after injury or accidents.
When needed, people with platelet granule disorders may be treated with:
- Antifibrinolytic drugs (drugs such as tranexamic acid that stabilize blood clots)
- Desmopressin (may not be useful in alpha granule deficiency)
- Hormonal suppressive therapy (birth control medications) and/or levonorgestrel-releasing intrauterine device/system to control excessive menstrual bleeding
- Iron replacement, as needed, to treat anemia caused by excessive or prolonged bleeding
- Fibrin sealants
- Platelet transfusions for severe bleeding episodes may be required.
Factor 1 (fibrinogen) deficiency
Factor 1 deficiency, also called fibrinogen deficiency, is an umbrella term for several related disorders, known as congenital fibrinogen defects. Factor 1 (fibrinogen) deficiency is an inherited bleeding disorder that is caused by a problem with factor 1. Because the body produces less fibrinogen (factor 1) than it should, or because the fibrinogen is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
- Afibrinogenemia (a complete lack of fibrinogen) and hypofibrinogenemia (low levels of fibrinogen) are an extremely rare inherited bleeding disorder where the amount of fibrinogen in the blood is abnormal (quantitative defect). Afibrinogenemia is rather well tolerated and may manifest as subcutaneous hematoma or umbilical hematoma at birth. The clinical findings are variable in childhood and adults 18.
- Dysfibrinogenemia is a qualitative defect in which fibrinogen does not work the way it should.
- Hypodysfibrinogenemia is a combined defect that involves both low levels of fibrinogen and impaired function.
Afibrinogenemia is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child.
Hypofibrinogenemia, dysfibrinogenemia, and hypodysfibrinogenemia can be either recessive (both parents carry the gene) or dominant (only one parent carries and transmits the gene).
All types of factor 1 deficiency affect both males and females.
Factor 1 (fibrinogen) deficiency symptoms
The symptoms of factor 1 deficiency differ depending on which form of the disorder a person has.
Afibrinogenemia (a complete lack of fibrinogen)
Given that fibrinogen is a central player of coagulation, the complete absence of fibrinogen leads to severe hemostatic defects, from life-threatening bleeding to thrombosis. Bleeding events are the most frequent symptoms – these are usually not predictable and can be severe and life-threatening hemorrhages.
People with afibrinogenemia (a complete lack of fibrinogen) may experience bleeding such as:
- nosebleeds (epistaxis)
- easy bruising
- bleeding from the umbilical cord stump at birth
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- abnormal bleeding during or after injury, surgery, or childbirth
- abnormal bleeding after circumcision
- bleeding into joints (hemarthrosis) or muscle, mostly post-trauma
- bleeding into bone
- heavy or prolonged menstrual bleeding (menorrhagia)
- problems during pregnancy (including miscarriage, placental abruption, and postpartum hemorrhage)
- bleeding in the gut (gastrointestinal hemorrhage)
- formation of blood clots (thrombosis). Although the mechanism is largely unknown, this may be due to unstable platelet clots due to lack of fibrin
- bleeding in the central nervous system (the brain and spinal cord)
- bleeding in the peritoneum (from ovary cyst rupture or spontaneous spleen rupture).
Incidence of thrombotic events is higher in young children.
Hypofibrinogenemia (low levels of fibrinogen)
Symptoms are like those seen in afibrinogenemia (low levels of fibrinogen). As a rule, the less factor 1 a person has in his/her blood, the more frequent and/or severe the symptoms.
Dysfibrinogenemia (a qualitative defect in which fibrinogen does not work the way it should)
Symptoms depend on how the fibrinogen (which is present in normal quantities) is functioning. Some people have no symptoms at all. Other people experience bleeding (like those seen in afibrinogenemia) and others show signs of thrombosis (abnormal blood clots in blood vessels) instead of bleeding.
Hypodysfibrinogenemia
Symptoms are variable and depend on the amount of fibrinogen that is produced and how it is functioning.
Factor 1 (fibrinogen) deficiency diagnosis
Factor 1 (fibrinogen) deficiency diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. Factor 1 deficiency is diagnosed by a variety of blood tests, including a specific test that measures the amount of fibrinogen in the blood. However, low fibrinogen levels or abnormal function may be a sign of another disease, such as liver or kidney disorders, which should be ruled out before a bleeding disorder is diagnosed.
Factor 1 (fibrinogen) deficiency treatment
There are three treatments available for factor 1 deficiency. All are made from human plasma.
- Factor 1 (fibrinogen) concentrate
- Cryoprecipitate / pathogen-reduced cryoprecipitate
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
Treatment may also be given to prevent the formation of blood clots, as this complication can occur after fibrinogen replacement therapy.
Many people who have hypofibrinogenemia or dysfibrinogenemia do not need treatment. Excessive menstrual bleeding in women with factor I deficiency may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Factor 2 (prothrombin) deficiency
Factor 2 (prothrombin) deficiency is very rare. Factor 2 (prothrombin) deficiency is an inherited bleeding disorder that is caused by a problem with factor II (factor 2). Because the body produces less prothrombin than it should, or because the prothrombin is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
Factor 2 (prothrombin) deficiency is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females.
Factor 2 (prothrombin) deficiency may also be inherited with other factors deficiencies.
Acquired factor 2 (prothrombin) deficiency is more common than the inherited form. Factor 2 (prothrombin) deficiency can also be acquired later in life as a result of liver disease, vitamin K deficiency, or certain medications such as the blood-thinning drug warfarin.
Factor 2 (prothrombin) deficiency symptoms
The symptoms of factor 2 (prothrombin) deficiency are different for everyone. Generally, the less factor II (factor 2) a person has in his/her blood, the more frequent and/or severe the symptoms.
People with factor 2 (prothrombin) deficiency may experience bleeding such as:
- nosebleeds (epistaxis)
- easy bruising
- heavy or prolonged menstrual bleeding (menorrhagia)
- bleeding into joints (hemarthrosis)
- muscle bleeds
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- bleeding in the gut (gastrointestinal hemorrhage)
- bleeding from the umbilical cord stump at birth
- abnormal bleeding during or after injury, surgery, or childbirth
- bleeding in the central nervous system (the brain and spinal cord)
- blood in urine (hematuria)
Factor 2 (prothrombin) deficiency diagnosis
Factor 2 (prothrombin) deficiency diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. Factor 2 (prothrombin) deficiency is diagnosed by a variety of blood tests. The doctor will need to test for prothrombin time (PT) and activated partial thromboplastin (aPTT). If both tests have prolonged time, then they will need to measure the blood level of factors I, II, V, VII, IX, and X.
Factor 2 (prothrombin) deficiency treatment
There are two treatments available for factor 2 (prothrombin) deficiency. Both are made from human plasma.
- Prothrombin complex concentrate (PCC) containing factor 2 (factor II)
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
Excessive menstrual bleeding in women with factor 2 (prothrombin) deficiency may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Factor 5 deficiency
Factor 5 deficiency (factor V deficiency) is very rare. Factor 5 deficiency (factor V deficiency) is an inherited bleeding disorder that is caused by a problem with factor V (factor 5). Because the body produces less factor 5 (factor V) than it should, or because the factor V is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
Factor 5 deficiency is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females.
Factor 5 deficiency symptoms
The symptoms of factor 5 deficiency (factor V deficiency) are generally mild, and some people may experience no symptoms at all. However, children with a severe deficiency of factor V (factor 5) may bleed at a very young age. Some people with factor 5 deficiency (factor V deficiency) have experienced bleeding in the central nervous system (the brain and spinal cord) very early in life.
People with factor 5 deficiency (factor V deficiency) may experience bleeding such as:
- nosebleeds (epistaxis)
- easy bruising
- heavy or prolonged menstrual bleeding (menorrhagia)
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- bleeding in the gut (gastrointestinal hemorrhage)
- muscle bleeds
- abnormal bleeding during or after injury, surgery, or childbirth
- bleeding into joints (hemarthrosis)
- bleeding in the central nervous system (the brain and spinal cord)
Factor 5 deficiency diagnosis
Factor 5 deficiency (factor V deficiency) diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. Factor 5 deficiency (factor V deficiency) is diagnosed by a variety of blood tests. The doctor will need to test for prothrombin time (PT) and activated partial thromboplastin (aPTT). If both tests have prolonged time, then they will need to measure the blood level of factors I, II, V, VII, IX, and X. People with abnormal levels of factor V (factor 5) should also have their factor VIII (factor 8) levels checked to rule out combined factor V and factor VIII deficiency, which is a completely separate disorder.
Factor 5 deficiency treatment
Treatment for factor 5 deficiency (factor V deficiency) is usually only needed for severe bleeds or before surgery. Fresh frozen plasma (FFP) or pathogen-reduced FFP is the usual treatment because there is no concentrate containing only factor V (factor 5). Platelet transfusions, which contain factor V (factor 5), are also sometimes an option.
Excessive menstrual bleeding in women with factor 5 deficiency (factor V deficiency) may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Combined factor 5 and factor 8 deficiency
Combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) is an inherited bleeding disorder that is caused by low levels of factors 5 and 8 (factors V and VIII). Because the amount of these factors in the body is lower than normal, the clotting reaction is blocked prematurely, and the blood clot does not form. The combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) is a completely separate deficiency from factor 5 deficiency (factor V deficiency) and factor 8 deficiency (factor VIII deficiency or hemophilia A).
Combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females. The deficiency is very rare, and very rarely, factor 8 deficiency (factor VIII deficiency) could be inherited separately from only one parent.
In most cases, the disorder is caused by a single gene defect that affects the body’s ability to transport factor V and factor VIII outside the cell and into the bloodstream, and not by a problem with the gene for either factor.
Combined factor 5 and factor 8 deficiency symptoms
The combination of factor 5 (factor V) and factor 8 (factor VIII) deficiency does not seem to cause more bleeding than if only one or the other of the factors were affected. The symptoms of combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) are generally mild.
People with combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) may experience bleeding such as:
- bleeding from the skin
- heavy or prolonged menstrual bleeding (menorrhagia)
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- bleeding after circumcision
- abnormal bleeding during or after injury, surgery, or childbirth
- nosebleeds (epistaxis)
- bleeding into joints (hemarthrosis)
- muscle bleeds
Combined factor 5 and factor 8 deficiency diagnosis
Combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. Combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) is diagnosed by a variety of blood tests to determine if the levels of both factors are lower than normal.
Combined factor 5 and factor 8 deficiency treatment
There are three treatments available for combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency).
- Factor VIII (factor 8) concentrate
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
- Desmopressin
Excessive menstrual bleeding in women with combined factor 5 and factor 8 deficiency (combined factor V and factor VIII deficiency) may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Factor 7 deficiency
Factor 7 deficiency (factor VII deficiency) is very rare. Factor 7 deficiency (factor VII deficiency) is an inherited bleeding disorder that is caused by a problem with factor VII (factor 7). Because the body produces less factor 7 (factor VII) than it should, or because the factor 7 (factor VII) is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
Factor 7 deficiency (factor VII deficiency) is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females. Factor 7 deficiency (factor VII deficiency) may be inherited with other factor deficiencies. It can also be acquired later in life as a result of liver disease, vitamin K deficiency, or certain medications such as the blood-thinning drug warfarin.
Factor 7 deficiency symptoms
The symptoms of factor 7 deficiency (factor VII deficiency) are different for everyone. As a rule, the less factor 7 (factor VII) a person has in his/her blood, the more frequent and/or severe the symptoms. People with very low levels of factor 7 (factor VII) can have very serious symptoms.
People with factor 7 deficiency (factor VII deficiency) may experience bleeding such as:
- nosebleeds (epistaxis)
- easy bruising
- heavy or prolonged menstrual bleeding (menorrhagia)
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- bleeding in the head (newborns)
- heavy bleeding at circumcision
- bleeding in the gut (gastrointestinal bleeding)
- bleeding into joints (hemarthrosis)
- muscle bleeds
- bleeding in the central nervous system (the brain and spinal cord)
- abnormal bleeding during or after injury, surgery, or childbirth
- blood in urine (hematuria)
- bleeding from the umbilical cord stump at birth
Factor 7 deficiency diagnosis
Factor 7 deficiency (factor VII deficiency) diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center.
Factor 7 deficiency treatment
There are several treatments available for factor 7 deficiency (factor VII deficiency).
- Recombinant factor VIIa concentrate (rFVIIa)
- Factor VII concentrate
- Prothrombin complex concentrate (PCC) containing factor VII
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
Excessive menstrual bleeding in women with factor 7 deficiency (factor VII deficiency) may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Factor 10 deficiency
Factor 10 deficiency (factor X deficiency) is one of the rarest inherited clotting disorders. Factor 10 deficiency (factor X deficiency) is an inherited bleeding disorder that is caused by a problem with factor X (factor 10). Because the body produces less factor 10 (factor X) than it should, or because the factor 10 (factor X) is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
Factor 10 deficiency (factor X deficiency) is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females.
Factor 10 deficiency (factor X deficiency) may also be inherited with other factor deficiencies.
Factor 10 deficiency symptoms
Generally, the less factor 10 (factor X) a person has in his/her blood, the more frequent and/or severe the symptoms. People with severe factor 10 deficiency (factor X deficiency) can have serious bleeding episodes.
People with factor 10 deficiency (factor X deficiency) may experience bleeding such as:
- nosebleeds (epistaxis)
- easy bruising
- bleeding in the gut (gastrointestinal hemorrhage)
- bleeding into joints (hemarthrosis)
- muscle bleeds
- bleeding from the umbilical cord stump at birth
- bleeding from the mouth, particularly after dental surgery or tooth extraction
- bleeding during or after surgery or injury
- heavy or prolonged menstrual bleeding (menorrhagia)
- bleeding after circumcision
- abnormal or prolonged bleeding after childbirth
- first-trimester miscarriage (spontaneous abortion)
- blood in urine (hematuria)
- bleeding in the central nervous system (the brain and spinal cord)
Factor 10 deficiency diagnosis
Factor 10 deficiency (factor X deficiency) diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center.
Factor 10 deficiency treatment
There are three treatments available for factor 10 deficiency (factor X deficiency) .
- Plasma-derived factor 10 (factor X) concentrate
- Prothrombin complex concentrate (PCC) containing factor 10 (factor X)
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
Excessive menstrual bleeding in women with factor 10 deficiency (factor X deficiency) may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Factor 11 deficiency
Factor 11 deficiency (factor XI deficiency) also called hemophilia C is an inherited bleeding disorder that is caused by a problem with factor XI (factor 11). Because the body produces less factor 11 (factor XI) than it should, or because the factor 11 (factor XI) is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
Factor 11 deficiency (factor XI deficiency) differs from hemophilia A and B in that there is no bleeding into joints and muscles. Factor 11 deficiency (factor XI deficiency) is the most common of the rare clotting factor deficiencies and the second most common bleeding disorder that impacts women after von Willebrand disease.
Factor 11 deficiency (factor XI deficiency) inheritance has an autosomal recessive pattern; however, some people have inherited factor 11 deficiency (factor XI deficiency) when only one parent carries the gene. The disorder is most common in Ashkenazi Jews (Jews of Eastern European ancestry).
Factor 11 deficiency symptoms
Most people with factor 11 deficiency (factor XI deficiency) will have little or no symptoms at all. The relationship between the amount of factor 11 (factor XI) in a person’s blood and the severity of his/her symptoms is unclear; people with only a mild deficiency in factor 11 (factor XI) can have serious bleeding episodes. Symptoms of factor 11 deficiency (factor XI deficiency) vary widely, even among family members, which can make it difficult to diagnose.
People with factor 11 deficiency (factor XI deficiency) may experience bleeding such as:
- nosebleeds (epistaxis)
- easy bruising
- heavy or prolonged menstrual bleeding (menorrhagia)
- abnormal bleeding during or after injury, surgery, or childbirth
- bleeding in the gut (gastrointestinal hemorrhage)
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- blood in the urine (hematuria)
Factor 11 deficiency diagnosis
Factor 11 deficiency (factor XI deficiency) diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center.
Factor 11 deficiency treatment
There are several treatments available to help control bleeding in people with factor 11 deficiency (factor XI deficiency).
- Factor 11 (factor XI) concentrate
- Antifibrinolytic drugs
- Fibrin glue
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
Excessive menstrual bleeding in women with factor 11 deficiency (factor XI deficiency) may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Factor 12 deficiency
Factor 12 deficiency (factor XII deficiency) is very rare. Factor 12 deficiency (factor XII deficiency) is an inherited bleeding disorder that is caused by a problem with factor XII. Because the body produces less factor XII than it should, or because the factor XII is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
Factor 12 deficiency (factor XII deficiency) is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females.
Factor 12 deficiency symptoms
Most people with factor 12 deficiency (factor XII deficiency) do not experience many symptoms, even after major surgery. However, some experience poor wound healing.
Factor 12 deficiency diagnosis
Factor 12 deficiency (factor XII deficiency) is often diagnosed accidentally during a routine coagulation blood test done prior to surgery or due to family history. Factor 12 deficiency (factor XII deficiency) diagnosis is made by prothrombin time (PT) and activated partial thromboplastin (aPTT) tests, followed by a factor 12 (factor XII) assay to confirm the diagnosis.
Factor 12 deficiency treatment
Treatment is usually not needed for people with factor 12 deficiency (factor XII deficiency) as symptoms are usually mild or non-existent. However, it is still important to notify health care providers or dental professionals before procedures.
Factor 13 deficiency
Factor 13 deficiency (factor XIII deficiency) is very rare. Factor 13 deficiency (factor XIII deficiency) is an inherited bleeding disorder that is caused by a problem with factor XIII (factor 13). Because the body produces less factor 13 (factor XIII) than it should, or because the factor 13 (factor XIII) is not working properly, the clotting reaction is blocked prematurely, and the blood clot does not form.
Factor 13 deficiency (factor XIII deficiency) is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females.
Factor 13 deficiency symptoms
Most people with factor 13 deficiency (factor XIII deficiency) experience symptoms from birth, often bleeding from the umbilical cord stump. Symptoms tend to continue throughout life. Generally, the less factor 13 (factor XIII) a person has in his/her blood, the more frequent and/or severe the symptoms.
People with factor 13 deficiency (factor XIII deficiency) may experience bleeding such as:
- bleeding from the umbilical cord stump at birth
- nosebleeds (epistaxis)
- easy bruising
- bleeding into joints (hemarthrosis)
- bleeding in the central nervous system (the brain and spinal cord)
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- poor wound healing and abnormal scar formation
- bleeding in soft tissue
- problems during pregnancy (including recurrent miscarriages)
- bleeding after circumcision
- abnormal bleeding during or after injury or surgery
- heavy or prolonged menstrual bleeding (menorrhagia)
- blood in urine (hematuria)
- bleeding in the gut (gastrointestinal hemorrhage)
- muscle bleeds
- bleeding in the spleen, lungs, ears, or eyes
Factor 13 deficiency diagnosis
Factor 13 deficiency (factor XIII deficiency) diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center because it’s difficult to diagnose. Standard blood clotting tests do not detect factor 13 deficiency (factor XIII deficiency), and many laboratories are not equipped with more specialized tests that measure the amount of factor 13 (factor XIII) in a blood sample or how well factor 13 (factor XIII) is working. The high rate of bleeding at birth usually leads to early diagnosis.
Factor 13 deficiency treatment
There are several treatments available to help control bleeding in people with factor 13 deficiency (factor XIII deficiency).
- Factor 13 (factor XIII) concentrate
- Cryoprecipitate / pathogen-reduced cryoprecipitate
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
Excessive menstrual bleeding in women with factor 13 deficiency (factor XIII deficiency) may be controlled with antifibrinolytic drugs, or with hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) in women who do not wish to conceive.
Combined deficiency of vitamin K-dependent clotting factors
Combined deficiency of vitamin K-dependent clotting factors (VKCFD) is very rare. Inherited combined deficiency of the vitamin K-dependent clotting factors (VKCFD) is a very rare inherited bleeding disorder that is caused by a problem with clotting factors II, VII, IX, and X. To continue the chain reaction of the coagulation cascade, these four factors need to be activated in a chemical reaction that involves vitamin K. When this reaction does not happen the way it should, the clotting reaction is blocked and the blood clot does not form.
Combined deficiency of vitamin K-dependent clotting factors (VKCFD) is an autosomal recessive disorder, which means that both parents must carry the defective gene to pass it on to their child. It also means that the disorder affects both males and females.
Combined deficiency of vitamin K-dependent clotting factors (VKCFD) is very rare. can also be acquired later in life as a result of disorders of the bowel, liver disease, dietary vitamin K deficiency, or certain medications such as the blood-thinning drug warfarin. Acquired combined deficiency of vitamin K-dependent clotting factors (VKCFD) is very rare. is more common than the inherited form. Some newborn babies have a temporary vitamin K deficiency, which can be treated with supplements at birth.
Combined deficiency of vitamin K-dependent clotting factors symptoms
The symptoms of combined deficiency of vitamin K-dependent clotting factors (VKCFD) vary a great deal from one individual to another but are generally mild. The first symptoms may appear at birth or not until later in life. Symptoms at birth must be differentiated from the acquired deficiency. People with severe deficiencies can have serious bleeding episodes, but the more serious symptoms are generally rare and only occur in those individuals with very low factor levels.
People with combined deficiency of vitamin K-dependent clotting factors (VKCFD) may experience bleeding such as:
- bleeding from the umbilical cord stump at birth
- bleeding into joints (hemarthrosis)
- bleeding in soft tissue and muscle
- bleeding in the gut (gastrointestinal hemorrhage)
- easy bruising
- excessive bleeding after surgery
- bleeding in the brain (intracranial hemorrhage)
- skeletal abnormalities and mild hearing loss (in severe cases)
Combined deficiency of vitamin K-dependent clotting factors diagnosis
Combined deficiency of vitamin K-dependent clotting factors (VKCFD) diagnosis should be performed by a specialist at a hemophilia/bleeding disorders treatment center. Combined deficiency of vitamin K-dependent clotting factors (VKCFD) is diagnosed by a variety of blood tests. Care should be taken, particularly in newborns, to exclude causes of acquired vitamin K deficiency or exposure to certain medications.
Combined deficiency of vitamin K-dependent clotting factors treatment
There are three treatments available for combined deficiency of vitamin K-dependent clotting factors (VKCFD).
- Vitamin K supplementation
- Prothrombin complex concentrates (PCC)
- Fresh frozen plasma (FFP) / pathogen-reduced FFP
Disseminated intravascular coagulation (DIC)
Disseminated intravascular coagulation (DIC) also called consumptive coagulopathy that causes abnormal blood clotting throughout your body’s blood vessels causing organ damage and uncontrollable bleeding. Disseminated intravascular coagulation (DIC) is a complication of different serious medical conditions that can be life-threatening if you don’t receive treatment.
Multiple medical conditions can lead to the development of disseminated intravascular coagulation (DIC) either through a systemic inflammatory response or the release of procoagulants into the bloodstream.
Disseminated intravascular coagulation (DIC) is usually caused by inflammation from an infection, injury, or illness. Some common causes include 19, 20, 21, 22, 23, 24, 25:
- Sepsis: Sepsis is a body-wide response to infection that causes inflammation. Sepsis is the most common risk factor for disseminated intravascular coagulation (DIC) and disseminated intravascular coagulation (DIC) has been estimated to occur in up to 30% to 50% of cases of severe sepsis. Classically, disseminated intravascular coagulation (DIC) has been associated with gram-negative bacteria sepsis, though the prevalence of this disorder in sepsis due to gram-positive organisms may, in fact, be similar 26. Other causes of sepsis, including parasites, can also lead to disseminated intravascular coagulation (DIC).
- Major damage to organs or tissues: This may be caused by cirrhosis of the liver, pancreatitis, severe injury, burns, or major surgery.
- Severe immune reactions: Your body may overreact because of a failed blood transfusion, rejection of an organ transplant, or a toxin such as snake venom.
- Serious pregnancy-related problems: These include the placenta separating from the uterus before delivery, placental abruption, amniotic fluid entering the bloodstream (amniotic fluid embolism) or serious bleeding during or after delivery.
- Cancer. Up to 20% of patients with metastasized adenocarcinoma or lymphoproliferative disease also suffer from disseminated intravascular coagulation (DIC), in addition to one to five percent of patients with chronic diseases like solid tumors and aortic aneurysms.
Risk factors for disseminated intravascular coagulation (DIC) include:
- Blood transfusion reaction
- Cancer, especially certain types of leukemia
- Inflammation of the pancreas (pancreatitis)
- Infection in the blood, especially by bacteria or fungus
- Liver disease
- Pregnancy complications such as placenta that is left behind after delivery; hemolysis, elevated liver enzymes, and low platelet count (HELLP syndrome)
- Recent surgery or anesthesia
- Severe immune system reactions to organ transplants
- Severe tissue injury (as in burns and head injury)
- Large hemangioma (a blood vessel that is not formed properly)
About 15.5% of cases of disseminated intravascular coagulation (DIC) have also been linked to complications occurring after surgery 26.
To understand what causes disseminated intravascular coagulation (DIC), it may help to understand how your body makes blood clots. Your liver makes clotting factors (proteins) that stick to platelets in your blood to form a blood clot. Normally, blood clots stop or slow bleeding and start your body’s healing process. But in disseminated intravascular coagulation (DIC), your body develops more blood clots than you need.
Disseminated intravascular coagulation (DIC) progresses through two stages: overactive clotting followed by bleeding 25.
- In stage one, overactive clotting leads to blood clots throughout the blood vessels. The clots can reduce or block blood flow, which can damage organs.
- In stage two, as DIC progresses, the overactive clotting uses up platelets and clotting factors that help the blood to clot. Without these platelets and clotting factors, DIC leads to bleeding just beneath the skin, in the nose or mouth, or deep inside the body.
Without treatment, DIC can cause:
- Stroke.
- Shock.
- Excessive bleeding.
- Acute respiratory distress syndrome (ARDS)
- Bleeding in the gastrointestinal tract
- Heart attack
- Venous thromboembolism
Disseminated intravascular coagulation (DIC) treatment starts with treating the underlying condition or injury that triggered blood clot development. Doctors also take steps to reduce bleeding with:
- Plasma transfusions to replace blood clotting factors affected by DIC.
- Clotting factor replacement therapy
- Plasma transfusion
- Platelet transfusion
- Transfusions of red blood cells and/or platelets.
- Anticoagulant medications (blood thinners).
Supportive treatments may include:
- Plasma transfusions to replace blood clotting factors if a large amount of bleeding is occurring.
- Blood thinner medicine (heparin) to prevent blood clotting if a large amount of clotting is occurring.
Disseminated intravascular coagulation (DIC) symptoms
Symptoms of disseminated intravascular coagulation (DIC) may include any of the following:
- Bleeding, from many sites in your body
- Bleeding at wound sites or from the nose, gums, or mouth
- Blood in the stool or urine
- Blood clots
- Bruising in small dots or larger patches on the body
- Drop in blood pressure
- Shortness of breath
- Chest pain
- Confusion, memory loss or change of behavior
- Fever
- Pain, redness, warmth, and swelling of the leg
Disseminated intravascular coagulation (DIC) diagnosis
Your doctor will diagnose disseminated intravascular coagulation (DIC) based on your medical history, a physical exam, and tests to find sepsis, cancer or other medical conditions that cause disseminated intravascular coagulation (DIC). Your doctor will also look for the cause of disseminated intravascular coagulation (DIC), because it does not occur on its own.
Your doctor may order the following blood tests:
- Complete blood count (CBC) with blood smear exam.
- Partial thromboplastin time (PTT).
- Prothrombin time (PT) test.
- Fibrinogen test.
- D-dimer test.
Your doctor may suggest other tests or procedures to find out whether a different condition is causing your symptoms. These tests may include:
- ADAMTS13 testing to check blood levels and activity of this protein, which can be low in a condition called thrombotic thrombocytopenic purpura
- Liver biopsy and liver function tests to check for cirrhosis or chronic liver disease, which may have symptoms like DIC
Disseminated intravascular coagulation (DIC) treatment
Disseminated intravascular coagulation (DIC) treatment starts with treating the underlying condition or injury that triggered blood clot development. Doctors also take steps to reduce bleeding with:
- Plasma transfusions to replace blood clotting factors affected by DIC.
- Clotting factor replacement therapy
- Plasma transfusion
- Platelet transfusion
- Transfusions of red blood cells and/or platelets.
- Anticoagulant medications (blood thinners).
Disseminated intravascular coagulation (DIC) prognosis
Disseminated intravascular coagulation (DIC) prognosis (outcome) depends on what is causing the disorder. Supportive treatments like anticoagulants and blood and platelet transfusions may stop blood clots from forming. DIC can be life threatening. Disseminated intravascular coagulation can quickly lead to multiorgan failure and death, particularly if early recognition and treatment fail to occur.
Liver disease
Majority of clotting factors are synthesized in your liver therefore severe liver disease or liver failure (acute liver failure & chronic liver failure) is associated with bleeding disorders (coagulopathy). Since your liver is also involved in the clearance of activated clotting factors and fibrinolytic products, it may predispose to disseminated intravascular coagulation (DIC). Management of bleeding secondary to liver disease is based on the laboratory values of various coagulation tests.
Your liver performs many important functions, including:
- Making clotting factors that aid in clotting, transporting oxygen and supporting your immune system.
- Manufacturing bile, a substance needed to help digest food.
- Helping your body store sugar (glucose) in the form of glycogen.
- Ridding your body of harmful substances in the bloodstream, including drugs and alcohol.
- Breaking down saturated fat and producing cholesterol.
Liver failure occurs when your liver isn’t working well enough to perform these tasks. Liver failure can be a life-threatening emergency that requires immediate medical attention.
Many different diseases and conditions cause liver failure, including Hepatitis B and C, non-alcohol related fatty liver disease (NAFLD), alcohol abuse and hemochromatosis.
In many cases, chronic liver failure results from cirrhosis. Cirrhosis is the scarring of your liver from repeated or long-lasting injury, such as from drinking alcohol excessively over a long period of time or chronic hepatitis infection. As scar tissue replaces healthy liver tissue, your liver loses its ability to function.
Liver failure is most often caused by:
- Viral infections, such as hepatitis A, hepatitis B, and hepatitis C.
- The overuse of certain drugs or toxins, like acetaminophen (Tylenol), and the use of other medications (including certain antibiotics, antidepressants, anti-seizure medications, man-made hormones and antifungal drugs) and herbs (green tea extract and kava).
- Metabolic (biologic) or vascular (vessels that carry fluids, such as arteries) disorders, such as Wilson disease and autoimmune hepatitis.
- Too much alcohol
- Liver cancer
- Inherited diseases, such as hemochromatosis and Wilson disease.
Factors that may increase your risk of liver disease include:
- Heavy alcohol use
- Obesity
- Type 2 diabetes
- Tattoos or body piercings
- Injecting drugs using shared needles
- Blood transfusion before 1992
- Exposure to other people’s blood and body fluids
- Unprotected sex
- Exposure to certain chemicals or toxins
- Family history of liver disease
The outcome of a liver disease vary, depending on the cause of your liver problems. Untreated liver disease may progress to liver failure, a life-threatening condition that may eventually result in death.
Liver disease causes
Liver disease has many causes.
- Infection. Parasites and viruses can infect the liver, causing inflammation that reduces liver function. The viruses that cause liver damage can be spread through blood or semen, contaminated food or water, or close contact with a person who is infected. The most common types of liver infection are hepatitis viruses, including:
- Hepatitis A
- Hepatitis B
- Hepatitis C
- Immune system abnormality. Diseases in which your immune system attacks certain parts of your body (autoimmune) can affect your liver. Examples of autoimmune liver diseases include:
- Autoimmune hepatitis
- Primary biliary cholangitis
- Primary sclerosing cholangitis
- Genetics. An abnormal gene inherited from one or both of your parents can cause various substances to build up in your liver, resulting in liver damage. Genetic liver diseases include:
- Hemochromatosis
- Wilson’s disease
- Alpha-1 antitrypsin deficiency
- Cancer and other growths. Examples include:
- Liver cancer
- Bile duct cancer
- Liver adenoma
- Additional, common causes of liver disease include:
- Chronic alcohol abuse
- Fat accumulation in the liver (nonalcoholic fatty liver disease)
- Certain prescription or over-the-counter medications
- Certain herbal compounds
Liver disease prevention
To prevent liver disease:
- Don’t drink alcohol or drink alcohol in moderation. For healthy adults, that means up to one drink a day for women and up to two drinks a day for men. Heavy or high-risk drinking is defined as more than eight drinks a week for women and more than 15 drinks a week for men.
- Avoid risky behavior. Use a condom during sex. If you choose to have tattoos or body piercings, be picky about cleanliness and safety when selecting a shop. Seek help if you use illicit intravenous drugs, and don’t share needles to inject drugs.
- Get vaccinated. If you’re at increased risk of contracting hepatitis or if you’ve already been infected with any form of the hepatitis virus, talk to your doctor about getting the hepatitis A and hepatitis B vaccines.
- Use medications wisely. Take prescription and nonprescription drugs only when needed and only in recommended doses. Don’t mix medications and alcohol. Talk to your doctor before mixing herbal supplements or prescription or nonprescription drugs.
- Avoid contact with other people’s blood and body fluids. Hepatitis viruses can be spread by accidental needle sticks or improper cleanup of blood or body fluids.
- Keep your food safe. Wash your hands thoroughly before eating or preparing foods. If traveling in a developing country, use bottled water to drink, wash your hands and brush your teeth.
- Take care with aerosol sprays. Make sure to use these products in a well-ventilated area, and wear a mask when spraying insecticides, fungicides, paint and other toxic chemicals. Always follow the manufacturer’s instructions.
- Protect your skin. When using insecticides and other toxic chemicals, wear gloves, long sleeves, a hat and a mask so that chemicals aren’t absorbed through your skin.
- Maintain a healthy weight. Obesity can cause nonalcoholic fatty liver disease.
Liver disease symptoms
Liver failure can take years to develop. The symptoms of liver failure often look like symptoms of other medical conditions, which can make it hard to diagnose in its early stages. Symptoms get worse as your failing liver continues to get weaker.
Sometimes, your liver fails suddenly, which is known as acute liver failure. People with acute liver failure may have the following symptoms:
- Bleeding.
- Changes in mental status.
- Musty or sweet breath odor.
- Movement problems.
- Loss of appetite.
- General feeling of being unwell.
- Jaundice.
Chronic liver failure, or liver failure that occurs over many years, may cause:
- Fatigue
- Nausea
- Loss of appetite
- Diarrhea
- Vomiting blood
- Blood in the stool
As liver failure advances, symptoms become more severe. In later stages, symptoms of liver failure may include:
- Jaundice (yellowing of your skin and eyes).
- Extreme tiredness.
- Disorientation (confusion and uncertainty).
- Fluid buildup in your abdomen and extremities (arms and legs).
Liver disease diagnosis
Doctors diagnose liver disease and liver failure based on your symptoms, your medical history and the results of tests (blood tests, urine tests, abdominal imaging and liver biopsy).
Tests and procedures used to diagnose liver disease and liver failure include:
- Blood tests. Blood tests are done to determine how well your liver works. A prothrombin time (PT) test measures how long it takes your blood to clot. With acute liver failure, blood doesn’t clot as quickly as it should.
- Imaging tests. Your doctor may recommend an ultrasound exam to look at your liver. Such testing may show liver damage and help determine the cause of your liver problems. Your doctor may also recommend abdominal computerized tomography (CT) scanning or magnetic resonance imaging (MRI) to look at your liver and blood vessels. These tests can look for certain causes of acute liver failure, such as Budd-Chiari syndrome or tumors. They may be used if your provider suspects a problem and ultrasound testing is negative.
- Examination of liver tissue. Your doctor may recommend removing a small piece of liver tissue (liver biopsy). Doing so may help your provider understand why your liver is failing. Since people with acute liver failure are at risk of bleeding during biopsy, a transjugular liver biopsy may be performed. This procedure involves making a tiny incision on the right side of your neck. A thin tube (catheter) is then inserted into a neck vein, through the heart and into a vein exiting the liver. A needle is then threaded through the catheter to retrieve a sample of liver tissue.
Liver disease treatment
Treatment for liver disease depends on your diagnosis. Some liver problems can be treated with lifestyle modifications, such as stopping alcohol use or losing weight, typically as part of a medical program that includes careful monitoring of liver function. Other liver problems may be treated with medications or may require surgery.
Treatment for liver disease that causes or has led to liver failure is usually treated by specialists called hepatologists and may ultimately require a liver transplant.
Treatment of liver failure depends on whether it is acute or chronic.
For acute (sudden) liver failure, treatment includes:
- Intravenous (IV) fluids to maintain blood pressure.
- Medications such as laxatives or enemas to help flush toxins (poisons) out.
- Blood glucose (sugar) monitoring. Your provider will give you glucose if your blood sugar drops.
You may also receive a blood transfusion if you are bleeding excessively, or a breathing tube to help you breathe.
For chronic liver failure, treatment includes changes to your diet and lifestyle, including:
- Avoiding alcohol or medications that can harm your liver.
- Eating less of certain foods, including red meat, cheese and eggs.
- Weight loss and management of metabolic risk factors, including high blood pressure and diabetes.
- Cutting down on salt in your diet (including not adding salt to food).
In both acute and chronic liver failure, your doctor may recommend a liver transplant. Before transplantation, doctors thoroughly screen transplant candidates to make sure a new organ might help them before placing them on organ waiting lists.
During the transplantation surgery, a healthy liver from a living or deceased donor replaces a damaged or diseased liver. Some transplant centers are able to replace a damaged liver with a portion of a healthy liver because the liver can regenerate, or grow back.
Clotting disorders (thrombophilia)
Thrombophilia is a blood disorder that makes the blood in your veins and arteries more likely to clot. Doctors call this a “hypercoagulable” condition because your blood coagulates or clots more easily. Thrombophilia can be an inherited (meaning that you are born with the condition) or acquired (meaning you develop the condition as the result of another illness or injury) tendency to form blood clots in arteries and veins.
Normally, your body makes a blood clot when you cut your finger with a knife, for example. The blood clot stops the bleeding. Later, your body breaks the clot apart when it doesn’t need it anymore. When you have thrombophilia, your body makes too many blood clots or doesn’t break down the old ones.
Blood clots can cause clogs or blockages in your veins or arteries. This can hurt your major organs or cause a stroke or heart attack because your blood vessels carry the oxygen your cells need. A clog in your blood vessel keeps blood from getting to your cells.
Blood thinners can prevent or treat blood clots, whether you have an inherited or acquired type of thrombophilia.
There are two types of thrombophilia: the kind you’re born with (genetic) and the kind you get (acquired) in other ways.
- Acquired thrombophilia, which is more common than the inherited kind, comes from a variety of things, like medicines, your lifestyle or diseases. The most common acquired thrombophilia is antiphospholipid syndrome. It’s also the most aggressive thrombophilia. Acquired blood clotting disorders include:
- Antiphospholipid syndrome (APS).
- Disseminated intravascular coagulation (DIC).
- Cancer (one of the most common causes).
- Some medications that treat cancer.
- Recent trauma or surgery.
- Central venous catheter placement.
- Obesity.
- Pregnancy.
- Supplemental estrogen use, including oral contraceptive pills (birth control pills).
- Hormone replacement therapy.
- Not moving your body for a long time because of bed rest or long plane rides.
- Heart attack, congestive heart failure, stroke and other illnesses that lead to decreased activity.
- Heparin-induced thrombocytopenia (decreased platelets in your blood from heparin or low molecular weight heparin preparations).
- Autoimmune disorders.
- Antiphospholipid antibody syndrome.
- Previous history of deep vein thrombosis or pulmonary embolism.
- Myeloproliferative disorders such as polycythemia vera or essential thrombocytosis.
- Paroxysmal nocturnal hemoglobinuria.
- Inflammatory bowel syndrome.
- Not having enough folate or other B vitamins.
- HIV, sepsis or other infections.
- Nephrotic syndrome (too much protein in your urine).
- Genetic (inherited) thrombophilia is the type you get from one or both of your parents. The affected gene causes your body to make certain clot-forming proteins that don’t work the way they should. Some genetic issues make you unable to produce enough of a protein you need to stop clotting. You may have inherited thrombophilia if you’ve had miscarriages or blood clots before age 40. You may have a relative who had blood clots, too. Types of genetic thrombophilia include:
- Factor V Leiden thrombophilia: The most common type of genetic thrombophilia (affecting 1% to 5% of the population). People with this type have a higher risk of getting a first-event deep vein thrombosis (DVT), but probably not a higher risk for more blood clots after the first one.
- Prothrombin thrombophilia: The second most common type of genetic thrombophilia, affecting 1% to 5% of the general population. People with this type have a higher risk of first-event pulmonary embolism, deep vein thrombosis (DVT) or miscarriage, but probably not a higher risk for more blood clots after the first one.
- Protein C deficiency: Less common type, affecting less than 1% of people. This type of thrombophilia puts you at a higher risk of repeated blood clots. If you got it from both parents, it can be life-threatening.
- Protein S deficiency: Less common type, affecting less than 1% of people. An even rarer form (from both parents) can cause a life-threatening clotting issue in infants.
- Protein Z deficiency: This type can increase your risk of thromboembolisms and pregnancy complications (miscarriage and preeclampsia).
- Antithrombin deficiency: Less common type, affecting 1 in 500 to 5,000 people. But those who have it have a higher risk of blood clots than people with other inherited blood clotting disorders. More than 80% of people with this type of thrombophilia get at least one blood clot by age 50.
Thrombophilia risk factors include:
- Having overweight.
- Being pregnant.
- Using tobacco products.
- Having atherosclerosis, cancer, diabetes, HIV or certain heart problems.
- Not moving your body for a long period of time.
- Having surgery or being in the hospital.
- Taking birth control pills containing estrogen.
- Taking hormone replacement therapy containing estrogen.
- Having a family history of blood clots.
- Being an older adult.
- Having unexplained miscarriages.
- Having more than one blood clot by age 40.
Clotting disorders (thrombophilia) treatment for acquired or inherited types of the condition may include compression stockings for your legs or medicine to prevent or break up a blood clot. Some people may need surgery to remove a blood clot. You usually only need treatment if you have a blood clot, such as deep vein thrombosis (DVT) or pulmonary embolism (PE).
Blood clots are usually treated in the same way, whether or not you have thrombophilia.
The main treatment for blood clots is anticoagulant medicine.
If you’re at high risk of blood clots, you may need to take this medicine regularly for several months or years.
There’s an increased risk of blood clots during pregnancy with some types of thrombophilia. Speak to your doctor if you’re pregnant or planning to get pregnant and you have thrombophilia.
Clotting disorders (thrombophilia) prevention
You can’t prevent thrombophilia that you got from your parents, but you may be able to prevent some acquired thrombophilias.
If you’re at a high risk of blood clots, your doctor may give you:
- Heparin after surgery if you’re at risk for a venous thromboembolism (VTE).
- Antithrombin injection before and after surgery if you don’t have enough antithrombin.
- Compression stockings or an intermittent pneumatic compression device if you’re at risk of a venous thromboembolism (VTE).
- Alternatives to standard birth control pills, like certain intrauterine devices or pills that only have progestogen.
- A dose of heparin before a long flight.
You can lower your risk of acquired thrombophilia on your own by:
- Avoiding tobacco products.
- Staying at a weight that’s healthy for you.
- Walking around every hour or two on a long flight or car ride.
- Avoiding medicines that contain estrogen.
- Getting up and walking as soon after surgery as you can.
- Making exercise part of your routine.
- Getting treatment for medical conditions that can cause thrombophilia.
- Following your provider’s instructions for taking a blood thinner medication.
- Making sure you’re up to date with all cancer screenings your provider recommends for you. Cancer is a strong risk factor for developing blood clots.
Clotting disorders (thrombophilia) symptoms
You may not feel any thrombophilia symptoms unless you get a blood clot. Blood clot symptoms differ in various parts of your body:
- Blood clot in Brain symptoms may include:
- Stroke (ischemic stroke). It happens when a major blood vessel in the brain is blocked. It may be blocked by a blood clot. Or it may be blocked by a buildup of fatty deposit and cholesterol. Symptoms of stroke include:
- Trouble speaking and understanding what others are saying. A person having a stroke may be confused, slur words or may not be able to understand speech.
- Numbness, weakness or paralysis in the face, arm or leg. This often affects just one side of the body. The person can try to raise both arms over the head. If one arm begins to fall, it may be a sign of a stroke. Also, one side of the mouth may droop when trying to smile.
- Problems seeing in one or both eyes. The person may suddenly have blurred or blackened vision in one or both eyes. Or the person may see double.
- Headache. A sudden, severe headache may be a symptom of a stroke. Vomiting, dizziness and a change in consciousness may occur with the headache.
- Trouble walking. Someone having a stroke may stumble or lose balance or coordination.
- Seizures.
- Sudden headache.
- Difficulty talking or seeing.
- Feeling weak on one side of your body.
- Stroke (ischemic stroke). It happens when a major blood vessel in the brain is blocked. It may be blocked by a blood clot. Or it may be blocked by a buildup of fatty deposit and cholesterol. Symptoms of stroke include:
- Blood clot in Heart symptoms may include:
- Shortness of breath.
- Chest pain.
- Painful left arm.
- Sweating.
- Lightheadedness.
- Nausea.
- Blood clot in Lungs symptoms may include:
- Fast breathing.
- Fast heart rate.
- Shortness of breath.
- Painful deep breathing.
- Chest pain.
- Blood clot in Belly symptoms may include:
- Nausea.
- Throwing up.
- Pain in your belly.
- Blood clot in Leg or arm symptoms may include:
- Swelling.
- Pain.
- Warm feeling.
Clotting disorders (thrombophilia) complications
Blood clots can travel all over your body, limiting or blocking blood flow to your organs. This can cause serious problems in your:
- Lungs (pulmonary embolism).
- Heart (heart attack).
- Brain (stroke).
- Kidneys (kidney failure).
- Leg or arm veins (deep vein thrombosis or DVT).
- Leg and pelvis arteries (peripheral artery disease or PAD).
- Developing fetus (miscarriage).
Clotting disorders (thrombophilia) diagnosis
Your doctor can make a thrombophilia diagnosis with:
- Your medical history.
- A physical exam.
- Blood tests to check for a genetic cause of thrombophilia.
- Tests that show what’s going on in your body.
Your doctor may order thrombophilia testing that includes:
- Angiograms or venograms (X-rays that read a dye injection in your blood vessels).
- Ultrasound (using sound waves).
- Computed tomography (CT) scan (using X-rays and a computer).
- Magnetic resonance imaging (MRI).
Clotting disorders (thrombophilia) treatment
Clotting disorders (thrombophilia) treatment for acquired or inherited types of the condition may include compression stockings for your legs or medicine to prevent or break up a blood clot. Some people may need surgery to remove a blood clot.
You usually only need treatment if you have a blood clot, such as deep vein thrombosis (DVT) or pulmonary embolism (PE).
Blood clots are usually treated in the same way, whether or not you have thrombophilia.
The main treatment for blood clots is anticoagulant medicine.
If you’re at high risk of blood clots, you may need to take this medicine regularly for several months or years.
There’s an increased risk of blood clots during pregnancy with some types of thrombophilia. Speak to your doctor if you’re pregnant or planning to get pregnant and you have thrombophilia.
Clotting disorders (thrombophilia) medications
Doctors usually use medications such as:
- Blood thinners (anticoagulants) like heparin, warfarin (Coumadin or Jantoven) or newer blood thinners like rivaroxaban or apixaban.
- Heparin is a fast-acting anticoagulant that may be used short-term in the hospital.
- Slower acting warfarin (Jantoven) may be used over a longer term. Warfarin is a powerful blood-thinning medicine, so you need to take it exactly as directed and watch for side effects. You also need regular blood tests to monitor warfarin’s effects.
- Several newer blood-thinning medicines include dabigatran (Pradaxa), rivaroxaban (Xarelto), apixaban (Eliquis) and edoxaban (Savaysa). They work faster than warfarin and usually don’t require regular blood tests or monitoring by your healthcare professional. These medicines also are associated with a lower risk of bleeding complications compared to warfarin.
- Thrombolytics (clot-dissolving drugs that doctors use only in emergencies). Thrombolytics (clot-dissolving drugs) you get through an IV can dissolve clots quickly. An IV thrombolytics (clot-dissolving drugs) that can break up a clot has to be given within 4.5 hours from when symptoms began. The sooner the medicine is given, the better. Quick treatment improves your chances of survival and may reduce complications.
It’s important to know that blood thinners (anticoagulants) don’t dissolve blood clots. They stabilize the blood clots so they don’t move or get bigger, and allow your body’s natural resources to absorb the clot over time.
Side effects of blood thinners (anticoagulants) may include:
- Bleeding too much when you get a cut.
- Having chills.
- Losing hair.
- Having nosebleeds.
- Having pain in your belly.
Clotting disorders (thrombophilia) prognosis
Although your doctor can’t cure genetic thrombophilia, they can order medicine like blood thinners for you to take for life. This medicine will help you manage your thrombophilia.
Nearly 90% of people who have thrombophilia never get a blood clot, but some people get one or more serious clots.
If you inherited thrombophilia, you’ll have it for life. Other kinds of thrombophilia can improve when you treat the condition that caused it.
Blood clotting disorders diagnosis
Many conditions can increase your risk of developing either blood clots (thrombosis) or excessive bleeding (hemorrhage). A doctor will start by doing a complete personal and family medical history and physical examination. Your doctor will also order blood tests to diagnose your condition.
You may be a candidate for screening for coagulation disorders if you have:
- A family history of abnormal blood clotting.
- Abnormal blood clotting at a young age (younger than age 50).
- Thrombosis in unusual locations, such as veins in your arms, liver, intestines, kidney or brain.
- Blood clots that occur without a clear cause.
- Blood clots that keep coming back.
- A history of frequent miscarriages.
- Stroke at a young age.
Blood tests
Blood tests can help your doctor evaluate your condition.
Blood clot disorder tests include:
- Prothrombin time (PT)-INR Test: A prothrombin time (PT) is a test used to help detect and diagnose a bleeding disorder or excessive clotting disorder. A prothrombin time (PT) test evaluates the coagulation factors VII, X, V, II, and I (fibrinogen). The international normalized ratio (INR) is calculated from a prothrombin time (PT) result and is used to monitor how well the blood-thinning medication (anticoagulant) such as warfarin (Coumadin) is working to prevent blood clots. Your results will help your doctor figure out how fast your blood is clotting and whether you need a different dose.
- Activated partial thromboplastin time (aPTT) also known as partial thromboplastin time (PTT): Activated partial thromboplastin time (aPTT) is a screening test that helps evaluate a person’s ability to appropriately form blood clots. It measures the number of seconds it takes for a clot to form in a sample of blood after substances (reagents) are added. The activated partial thromboplastin time (aPTT) assesses the amount and the function of certain proteins in the blood called coagulation or clotting factors that are an important part of blood clot formation. The activated partial thromboplastin time (aPTT) is used to evaluate the coagulation factors XII, XI, IX, VIII, X, V, II (prothrombin), and I (fibrinogen) as well as prekallikrein (PK) and high molecular weight kininogen (HK). Your doctor uses this test to monitor your condition if you’re taking heparin. With a activated partial thromboplastin time (aPTT), your result is compared to a normal reference interval for clotting time. When your partial thromboplastin time (PTT) takes longer than normal to clot, the PTT is considered “prolonged.”
- Fibrinogen test. Fibrinogen testing measures the amount of fibrinogen in your blood and assesses whether your fibrinogen can properly form blood clots. Fibrinogen is a protein made by your liver that circulates in your blood. When an injury occurs and bleeding needs to be stopped, fibrinogen works with other clotting factors and platelets to form a blood clot, which is a mass of blood cells, platelets, and proteins that cluster together to stop bleeding. Two methods for testing fibrinogen may be used simultaneously:
- Fibrinogen antigen test: This test measures the amount (quantity) of fibrinogen in a blood sample. It is generally reported in milligrams per deciliter (mg/dL). The fibrinogen antigen test is only needed when the fibrinogen activity test result is abnormal and is therefore typically ordered to help determine if the low activity is due to low quantity or abnormal function of fibrinogen.
- Fibrinogen activity test: This test evaluates how much time it takes for fibrinogen to form a clot. Thrombin is added to a prepared blood sample to stimulate the coagulation process in a test tube. The amount of fibrinogen incorporated into the blood clot is called active (or functional) fibrinogen. Most labs offer a clotting-based activity test (functional) and are also reported as milligrams per deciliter (mg/dL).
- Complete blood count (CBC). Doctors use this test to measure and study blood cells.
Tests used to help diagnose inherited coagulation disorders include:
- Genetic tests, including factor V Leiden, activated protein C resistance and prothrombin gene mutation (G20210A).
- Von Willebrand factor antigen: This test measures the amount of von Willebrand factor protein in your bloodstream.
- Ristocetin cofactor: This test evaluates von Willebrand factor activity.
- Von Willebrand factor multimers: This test measures the factor’s structure.
- Antithrombin activity.
- Protein C activity.
- Protein S activity.
- Homocysteine test.
Other tests that help diagnose acquired coagulation disorders include tests for:
- Things that are part of the antiphospholipid antibody syndrome.
- Heparin antibodies (in people who develop low platelet counts while exposed to heparin).
A health care practitioner will evaluate the results of these blood tests to help rule out or determine the cause of bleeding or clotting disorder.
Testing can help:
- Identify whether you’re at risk for further clotting.
- Determine the right course and length of treatment to prevent future clots.
- Identify relatives who don’t currently have symptoms but may be at risk.
A specialized coagulation laboratory should do the tests. A pathologist or clinician with expertise in coagulation, vascular medicine or hematology should interpret them.
Ideally, the tests should be done when you aren’t having an acute clotting event.
Blood clotting disorders treatment
Blood clotting disorders treatment depends on the underlying cause and severity of your condition.
Your treatment also depends on:
- How you’ve responded to previous therapy
- Your other medications and conditions
Clotting disorders (thrombophilia) treatment
When a blood clot develops in your vein or artery you’ll need medicines that help prevent blood clots called anticoagulants or blood thinners to decrease your blood’s ability to clot and prevent additional clots from forming. Although anticoagulants are called blood thinners, these medicines do not really thin your blood. Instead, they decrease your blood’s ability to clot by interfering with the normal clotting processes. Just like their name suggests, anticoagulants or blood thinners prevent or undo coagulation, the process where your blood solidifies to form a clot. Depending on the type of anticoagulant, the clotting process disruption happens in different ways. Furthermore, anticoagulants or blood thinners do not break up clots that you already have. But they can stop those clots from getting bigger.
Decreased clotting keeps fewer harmful blood clots from forming and from blocking your blood vessels. It’s important to treat blood clots, because blood clots in your blood vessels and heart can cause heart attacks, strokes, and blockages.
Anticoagulants come in many different forms, including injections, intravenous (IV) drugs and medications you take by mouth. The most commonly prescribed anticoagulant are warfarin (Coumadin) and heparin (intravenous anticoagulant). Warfarin (Coumadin or Jantoven), comes as a tablet you swallow. Heparin, a liquid medication you get through an IV in your vein or from an injection in the hospital.
Types of anticoagulants
IV and injectable anticoagulants
Heparin and its derivatives
Heparin is a medication that inhibits clotting by activating your body’s anti-clotting processes. One of the anticlotting processes uses a type of blood protein called antithrombin. Heparin works by activating antithrombin, and then antithrombin keeps other parts of the clotting process from working normally.
Heparin comes in two different types, and there is a third medication that is closely related:
- Unfractionated heparin (UFH). Unfractionated heparin (UFH) is stronger and fast-acting. This is because unfractionated heparin (UFH) has a longer molecule, which means it’s long enough to help wrap around both antithrombin and thrombin, a protein that promotes clotting, holding them together. This neutralizes both proteins, further preventing clotting. Unfractionated heparin (UFH) also needs constant monitoring with lab tests. That’s because its effectiveness depends on its dosage, and the needed dosage can be very different from person to person. Too little won’t do enough to prevent clotting, and too much will create a risk of bleeding.
- Low-molecular-weight heparin (LMWH). Low-molecular-weight heparin (LMWH) has shorter molecules, which means it can only attach to antithrombin. This also means the effects are longer-lasting, more predictable and low-molecular-weight heparin (LMWH) doesn’t need the close monitoring required with unfractionated heparin (UFH).
- Fondaparinux. Fondaparinux is a synthetic medication that works similarly to heparin. Like heparin, fondaparinux activates antithrombin but acts over a much longer period. However, it’s not as strong as unfractionated heparin (UFH) or low-molecular-weight heparin (LMWH), so it’s most often used to prevent clots rather than treat clotting problems that are already happening (unless given along with other medications).
Direct thrombin inhibitors
Thrombin inhibitors work by attaching to thrombin, keeping it from assisting clotting processes. They are often used as alternatives to heparin and its variants, especially to prevent the formation of clots after certain medical procedures. These include argatroban, desirudin and bivalirudin.
Oral anticoagulants
Warfarin (vitamin K antagonist)
Warfarin (Coumadin or Jantoven) is a vitamin K antagonist, meaning it blocks the use of vitamin K, a key ingredient in the clotting process. However, a major drawback of warfarin (Coumadin) is that it needs careful dosing and regular lab testing to prevent complications. When the dosage isn’t precise enough, it can lead to severe bleeding.
In some cases, certain conditions mean that warfarin is the only anticoagulant that you can take. These include:
- Diseases affecting the mitral valve of your heart.
- Having a mechanical heart valve.
- End-stage kidney disease.
Direct oral anticoagulants
Newer types of anticoagulants (blood thinners) are also available and are becoming increasingly common. These medications can all be taken regularly without regular lab testing and are often used when warfarin isn’t an option.
Newer types of anticoagulants (blood thinners) include:
- Rivaroxaban (Xarelto)
- Apixaban (Eliquis)
- Dabigatran (Pradaxa)
- Edoxaban (Lixiana)
- Betrixaban (Bevyxxa)
One medication, dabigatran (Pradaxa), is a thrombin inhibitor similar to the IV thrombin inhibitors listed earlier (e.g., argatroban, desirudin and bivalirudin). Other medications, apixaban (Eliquis), edoxaban (Lixiana) and betrixaban (Bevyxxa), are all inhibitors of factor Xa (factor 10A), a key clotting component.
Warfarin and the newer alternatives are taken as tablets or capsules. The most common side effect risk with any anticoagulant is bleeding. Direct oral anticoagulants can sometimes cause indigestion or bleeding in your gastrointestinal tract. And depending on the medication used, other potential risks exist.
Your doctor will talk to you about the benefits and risks of these medications. This information, along with your diagnosis, will help determine the type of anticoagulant medication you will take, how long you will need to take it, and the type of follow-up monitoring you need.
As with any medication, it’s important to know how and when to take your anticoagulant according to your doctor’s guidelines and to have frequent blood tests. You shouldn’t take warfarin if you’re pregnant or planning to become pregnant. Warfarin can damage an embryo or fetus, so it shouldn’t be taken during pregnancy. However, warfarin is safe when breastfeeding because it can’t be passed through breast milk. If you are pregnant or planning to become pregnant, ask your doctor about switching to a different type of anticoagulant medication, especially during the first trimester and before delivery.
Bleeding disorders (coagulopathy) treatment
When you have a bleeding disorder (coagulopathy) you’ll need to replace the missing blood clotting factor so that your blood can clot properly. For example, doctors treat hemophilia by boosting clotting factor levels or replacing missing clotting factors (replacement therapy). In replacement therapy, you receive human plasma concentrates or lab-made (recombinant) clotting factors. In general, only people with severe hemophilia need regular replacement therapy. People with mild or moderate hemophilia who need surgery may receive replacement therapy. They may also receive antifibrinolytics, a medication that keeps blood clots from breaking down. Blood factor concentrates are made from donated human blood that’s been treated and screened to reduce the risk of transmitting infectious diseases, such as hepatitis and HIV. People receive replacement factors via intravenous infusion (IV). If you have severe hemophilia and frequent bleeding episodes, your healthcare provider may prescribe prophylactic factor infusions to prevent bleeding.
For von Willebrand disease your doctor might suggest one or more of the following treatments to increase your von Willebrand factor, strengthen blood clots or control heavy menstrual bleeding:
- Desmopressin. Desmopressin is available as an DDAVP (desamino-8-arginine vasopressin) injection. DDAVP is a man-made hormone that controls bleeding by stimulating your body to release more of the von Willebrand factor (vWF) stored in the lining of your blood vessels. Many doctors consider desmopressin (DDAVP) the first treatment for managing von Willebrand disease. It can be used before minor surgical procedures to help control bleeding. You might be given a trial of desmopressin to make sure it’s effective for you. However, desmopressin (DDAVP) does not work for all types of von Willebrand disease. Tests should be done to determine what type of von Willebrand you have. If you are going to have surgery, your doctor may give you DDAVP before surgery to see if your von Willebrand factor levels increase.
- Replacement therapies. These include infusions of concentrated blood-clotting factors containing von Willebrand factor and factor VIII (factor 8). Your doctor might recommend them if DDAVP isn’t an option for you or has been ineffective. Another replacement therapy approved by the Food and Drug Administration for treating adults 18 and older is a genetically engineered (recombinant) von Willebrand factor product. Because recombinant factor is made without plasma, it can reduce the risk of a viral infection or allergic reaction.
- Factor concentrates. When they are available, factor concentrates are the ideal and safest treatment for rare bleeding disorders. Unfortunately, individual concentrates are available only for factors I, VII, X, XI, and XIII. Factor concentrates for rare bleeding disorders are usually made from human plasma and are treated to eliminate viruses like HIV, and hepatitis B and C. Recombinant factor VIII and recombinant factor VIIa are also available. They are made in the laboratory and not from human plasma, so they carry no risk of infectious disease. Factor concentrates are administered intravenously.
- Prothrombin complex concentrate (PCC). Prothrombin complex concentrate (PCC) is made from human plasma and contains a mixture of vitamin K-dependent coagulation factors, including factors II, VII, IX, and X (however, some products do not contain all four factors) and proteins C and S. Prothrombin complex concentrates (PCCs) are isolated from the cryoprecipitate supernatant of large plasma pools after removal of antithrombin and factor XI. It is treated to eliminate viruses like HIV, and hepatitis B and C. Prothrombin complex concentrate (PCC) is suitable for individual deficiencies of factor II and X as well as inherited combined deficiency of the vitamin K-dependent clotting factors (VKCFD). Some PCCs have been reported to cause potentially dangerous blood clots (thrombosis). Prothrombin complex concentrate (PCC) is administered intravenously.
- Fresh frozen plasma (FFP). Plasma is the portion of blood that contains all the clotting factors, as well as other blood proteins. Fresh frozen plasma (FFP) is used to treat rare bleeding disorders when concentrates of the specific factor that is missing, are not available. Fresh frozen plasma (FFP) is the usual treatment for factor 5 deficiency (factor V deficiency). However, fresh frozen plasma (FFP) usually does not undergo viral inactivation, so the risk of transmission of infectious diseases is higher. Viral-inactivated fresh frozen plasma (FFP) is available in some countries and is preferable. Circulatory overload is a potential problem with this treatment: since the concentration of each coagulation factor in FFP is low, a large volume of it must be given over several hours to achieve an adequate rise in factor level. This large amount of fresh frozen plasma (FFP) can overload the circulatory system and stress the heart. Other complications of treatment with FFP can occur, particularly allergic reactions or lung problems (transfusion-related lung injury [TRALI]). Risk of TRALI (transfusion-related lung injury) can be reduced by avoiding the transfusion of FFP collected from a previously pregnant female. These problems are much less common if viral-inactivated pooled FFP or pathogen-reduced FFP is used. Fresh frozen plasma (FFP) is administered intravenously.
- Cryoprecipitate. Made from human plasma, cryoprecipitate contains factor 8 (factor VIII), fibrinogen (factor 1), and a few other proteins important for blood clotting. Non-pathogen-reduced cryoprecipitate does not undergo viral inactivation and should only be used when clotting factor concentrate is not available. Pathogen-reduced or viral-inactivated cryoprecipitate can be prepared by blood transfusion centers and should be used in the absence of clotting factor concentrates. It contains higher concentrations of some (but not all) coagulation factors than FFP, so less volume is needed. It is only suitable for a few deficiencies. Cryoprecipitate is administered intravenously.
- Platelet transfusions. Platelets are small blood cells that are involved in the formation of blood clots and the repair of damaged blood vessels. Certain clotting factors, including factor 5 (factor V), are stored in small sacs inside platelets. Platelet transfusions are sometimes used to treat factor 5 deficiency (factor V deficiency).
- Alphanate (antihemophilic factor) is approved to treat or prevent bleeding episodes in people with von Willebrand disease who must have surgery or any other invasive procedure. Alphanate is also used to treat or prevent bleeding episodes in people with hemophilia A.
- Oral contraceptives. In addition to preventing pregnancy, these drugs can help control heavy bleeding during menstrual periods. The estrogen hormones in birth control pills can boost von Willebrand factor and factor 8 (factor VIII) activity.
- Hormonal contraceptives. Hormonal contraceptives (such as birth control pills, or levonorgestrel releasing intrauterine device/system [IUD or IUS]) can help control menstrual bleeding in women who do not wish to conceive.
- Clot-stabilizing medications. These anti-fibrinolytic medications — such as aminocaproic acid (Amicar) and tranexamic acid (Cyklokapron, Lysteda) — can help stop bleeding by slowing the breakdown of blood clots. Antifibrinolytic drugs are also very useful in many situations, such as during dental work, but are not effective for major internal bleeding or surgery. Doctors often prescribe these drugs before or after a minor surgical procedure or tooth extraction. Antifibrinolytic drugs are particularly useful for patients with factor 9 deficiency (factor XI deficiency). They are also used to help control excessive menstrual bleeding. Antifibrinolytic drugs can be administered orally or by injection. Antifibrinolytic drugs should be avoided for urinary bleeding.
- Vitamin K. Treatment with vitamin K (either in pill form or by injection) can help control symptoms of inherited combined deficiency of the vitamin K-dependent clotting factors (VKCFD). However, not everyone responds to this type of treatment. People who do not respond to vitamin K and have a bleed or need surgery will need factor replacement.
- Drugs applied to cuts. A fibrin sealant (Tisseel) placed on a cut helps curtail bleeding. This is applied like glue using a syringe. There are also over-the-counter products to stop nosebleeds. Fibrin glue can be used to treat external wounds and during dental work, such as a tooth extraction. It is not used for major bleeding or surgery. It is applied directly to the bleeding site.
If your condition is mild, your doctor might recommend treatment only when you’re having surgery or dental work or when you’ve had a trauma, such as a car accident.
- What Are Blood Clotting Disorders? https://www.nhlbi.nih.gov/health/clotting-disorders[↩]
- Overview of Coagulation Disorders. https://www.msdmanuals.com/en-au/professional/hematology-and-oncology/coagulation-disorders/overview-of-coagulation-disorders[↩]
- LaPelusa A, Dave HD. Physiology, Hemostasis. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK545263[↩][↩][↩]
- Kumar, V., Abbas A.K., Aster, J.C. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Saunders Elsevier; 2009.[↩][↩][↩]
- Gale AJ. Continuing education course #2: current understanding of hemostasis. Toxicol Pathol. 2011 Jan;39(1):273-80. doi: 10.1177/0192623310389474[↩][↩]
- Periayah MH, Halim AS, Mat Saad AZ. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int J Hematol Oncol Stem Cell Res. 2017 Oct 1;11(4):319-327[↩]
- Primary and secondary hemostasis, regulators of coagulation, and fibrinolysis: Understanding the basics. SickKids Handbook of Pediatric Thrombosis and Hemostasis. Karger Medical and Scientific Publishers. ISBN 978-3-318-03026-6.[↩]
- LefkowitzJB . Hemostasis Physiology. JB Lippincott co: Philadelphia, Pa; 2006. Chapter 1: Coagulation pathway and physiology; pp. 3–12.[↩]
- Pallister CJ, Watson MS. Haematology. 2ndedn. UK: Scion Publishing Ltd; 2010. pp. 336–347.[↩]
- Hall JE, Guyton AC. Guyton and Hall Textbook of Medical Physiology. 12th edn. Philadelphia, PA: Saunders Elsevier; 2011.[↩]
- Hoffman M. Remodeling the blood coagulation cascade. J Thromb Thrombolysis. 2003 Aug-Oct;16(1-2):17-20. doi: 10.1023/B:THRO.0000014588.95061.28[↩]
- Hoffman MM, Monroe DM. Rethinking the coagulation cascade. Curr Hematol Rep. 2005 Sep;4(5):391-6.[↩]
- Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth. 2014 Sep;58(5):515-23. doi: 10.4103/0019-5049.144643[↩][↩]
- Periayah MH, Halim AS, Mat Saad AZ. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int J Hematol Oncol Stem Cell Res. 2017 Oct 1;11(4):319-327. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5767294[↩][↩]
- Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020: 26(Suppl 6): 1-158. https://doi.org/10.1111/hae.14046[↩][↩]
- Polderman HK. Hypothermia and coagulation. Crit Care. 2012;16:A20.[↩]
- Martlew VJ. Peri-operative management of patients with coagulation disorders. Br J Anaesth. 2000 Sep;85(3):446-55. doi: 10.1093/bja/85.3.446[↩]
- Roberts HR, Stinchcombe TE, Gabriel DA. The dysfibrinogenaemias. Br J Haematol. 2001 Aug;114(2):249-57. doi: 10.1046/j.1365-2141.2001.02892.x[↩]
- Passmore MR, Obonyo NG, Byrne L, Boon AC, Diab SD, Dunster KR, Fung YL, Spanevello MM, Fauzi MH, Pedersen SE, Simonova G, Anstey CM, Shekar K, Tung JP, Maitland K, Fraser JF. Fluid resuscitation with 0.9% saline alters haemostasis in an ovine model of endotoxemic shock. Thromb Res. 2019 Apr;176:39-45. doi: 10.1016/j.thromres.2019.02.015[↩]
- Gonzalez MG, Wei RM, Hatch KD, Gries LM, Hill MG. A Novel Treatment for Massive Hemorrhage after Maternal Trauma in Pregnancy. AJP Rep. 2019 Jan;9(1):e27-e29. doi: 10.1055/s-0039-1678735[↩]
- Nolan S, Czuzoj-Shulman N, Abenhaim HA. Obstetrical and newborn outcomes among women with acute leukemias in pregnancy: a population-based study. J Matern Fetal Neonatal Med. 2020 Oct;33(20):3514-3520. doi: 10.1080/14767058.2019.1579188[↩]
- Karakike E, Giamarellos-Bourboulis EJ. Macrophage Activation-Like Syndrome: A Distinct Entity Leading to Early Death in Sepsis. Front Immunol. 2019 Jan 31;10:55. doi: 10.3389/fimmu.2019.00055[↩]
- Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999 Aug 19;341(8):586-92. doi: 10.1056/NEJM199908193410807[↩]
- Andrew Retter, Beverley J. Hunt; Consumptive coagulopathy in the ICU. Hematology Am Soc Hematol Educ Program 2023; 2023 (1): 754–760. doi: https://doi.org/10.1182[↩]
- Disseminated Intravascular Coagulation (DIC). https://www.nhlbi.nih.gov/health/disseminated-intravascular-coagulation[↩][↩]
- Costello RA, Nehring SM. Disseminated Intravascular Coagulation. [Updated 2023 Jan 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441834[↩][↩]





{kind=link}