Contents
What is liposarcoma
Liposarcoma is a group of very rare cancers that begin in your fat cells 1, 2, 3, 4, 5, 6. You can develop a liposarcoma in any part of your body, but these tumors typically form in the layer of fat just under your skin or in the deep soft tissues of your legs especially in your thigh or behind your knee or inside the back of your abdomen in an area called the “retroperitoneum” where, because of the vast amount of space, can effectively hide a tumor of substantial size and weight. Liposarcoma may also form in other parts of your body. Most liposarcomas are painless and tend to grow slowly, but some may grow quickly and spread to nearby tissue or to other parts of your body. Some individuals with liposarcoma may not have symptoms in the early stages, but as the tumor grows and advances to later stages, it can potentially compress other tissues and cause pain. The exact cause of liposarcoma is unknown, but genetic mutations that cause fat cells to multiply uncontrollably are involved. Liposarcoma is classified as a cancer (malignant) because of its potential to recur locally and spread to other areas of the body. The severity of disease depends on the subtype of the liposarcoma and the presenting stage of the primary tumor. Liposarcomas usually occur in adults between 50 and 65 years old, and are rare in children and adolescents. Liposarcomas are a type of soft tissue sarcoma or soft tissue cancer. Each year in the United States, about 2,000 people are diagnosed with liposarcoma. It affects men more than women, and more specifically middle-aged men ranging from 50 – 65 years of age. There is no specific ethnicity in which liposarcoma is more common.
Liposarcoma occurs when fat cells undergo genetic mutations, causing them to multiply uncontrollably and form tumors. Certain risk factors have been shown to predispose individuals to developing soft tissue sarcomas such as liposarcoma, including previous radiation therapy, familial cancer syndromes, inherited genetic conditions, damage to the lymph system, and long-term exposure to certain toxic chemicals such as vinyl chloride, a chemical used to make plastic. A benign lipoma may rarely develop into a liposarcoma.
Liposarcomas are mainly subclassified into five subtypes with different morphologic and behavioral spectrum 3, 7, 8. The types have similar symptoms but can require different treatments and have different prognoses or expected outcomes 9:
- Well-differentiated liposarcoma. Well-differentiated liposarcoma is the most common type of liposarcoma, representing about half of all liposarcoma cases. Well-differentiated liposarcoma is a slow-growing, low-grade malignant tumor, most commonly found in your arms or legs or retroperitoneum (back of the abdomen) 10, 11. Superficial well-differentiated liposarcoma lesions are usually located in the subcutaneous fat and are diagnosed as atypical lipomatous tumors (ALT) 12. Under microscopic examination, well-differentiated liposarcoma typically consists of mature adipocytes of variable size, encompassed by fibrous stroma exhibiting atypical spindle cells with hyperchromatic nuclei 13. Well-differentiated liposarcoma usually shows MDM2 (murine double minute 2) and CDK4 (cyclin-dependent kinase 4) expression 14, 15, 16. However, some cases with atypical morphology and immunophenotype require FISH for identification of MDM2 amplifications, in order to avoid misdiagnosis 17, 18, 19, 20, 21. While well-differentiated liposarcoma rarely metastasizes (spreads to other parts of the body), it has a tendency to recur locally after surgical removal, particularly in the retroperitoneum 22, 23. In rare cases, 17% of well-differentiated liposarcoma of the retroperitoneum and 6% of well-differentiated liposarcoma of the limbs can progress into dedifferentiated liposarcoma 24. Surgery is the most common and most reliable option for treatment of well-differentiated liposarcoma. In some cases, radiotherapy is used in addition to surgery. The current general consensus from researchers is that chemotherapy is not effective in well-differentiated liposarcoma 24.
- Dedifferentiated liposarcoma. Dedifferentiated liposarcoma is a type of cancer that develops when a well-differentiated liposarcoma or atypical lipomatous tumor (ALT) transforms into a more aggressive, non-lipogenic sarcoma 25, 26, 27, 28, 29, 30. Dedifferentiated liposarcoma is characterized by the presence of both well-differentiated and poorly differentiated tumor components. Dedifferentiated liposarcoma is more aggressive than well differentiated liposarcoma, with a higher risk of recurrence and metastasis, but its metastatic potential is related to the anatomic site of the tumor with retroperitoneal masses having a worse prognosis 27. Recurrence occurs in 40% to 75% of dedifferentiated liposarcoma cases and metastasis occurs in about 10–15% of cases 31. The genetic background of dedifferentiated liposarcoma is characterized by MDM2 (murine double minute 2) and CDK4 (cyclin-dependent kinase 4) amplification; therefore, immunohistochemical expression of the corresponding markers is used for diagnosis 26. A mutant TP53 immunophenotype, with hyperexpression of p53, is also reported in some studies 26, 32. Molecular analysis of this tumor is important, as new therapeutic targets have been identified 33. Surgery is the most common and most reliable option for treatment of differentiated liposarcoma 25, 26, 34, 31. Dedifferentiated liposarcomas in the are associated with a higher risk of recurrence, due to the difficulty of achieving complete surgical resection 34, 35, 36. Radiation therapy in sometimes used in patients with margins of less than 10mm after surgical resection 31. It’s not clear yet how suitable chemotherapy is as a treatment for differentiated liposarcoma, but an ongoing clinical trial is investigating this 31.
- Myxoid liposarcoma or myxoid/round cell liposarcoma. The term “round cell” refers to how tumor cells appear when viewed under a microscope. Myxoid liposarcoma or round cell liposarcomas is one of the most common subtype of liposarcoma making up about 30% of all liposarcoma cases 37. Myxoid liposarcoma is a type of liposarcoma mostly located in your arms and legs typically occurring in younger patients, with a variable behavior depending on the histological subtype. These tumors grow slowly, and they can spread to other parts of the body. Myxoid liposarcoma is more common in people aged 20 to 40 years old and is rarely seen in children under 10 years old. Myxoid liposarcoma tends to affect more males than females. Myxoid liposarcoma is a distinct entity with unique genetic and molecular features consisting of FUS-DDIT3 or EWSR1-DDIT3 fusion 38. Further studies on the FUS-DDIT3 fusion oncogene in myxoid liposarcoma suggest a link between this mutation and JAK-stat signaling in the stem cancerous cell 39, 40, 41. Currently, the most efficient treatment for myxoid liposarcoma comprises surgical resection with preoperative radiotherapy frequently administered, whereas high-risk lesions of the limbs or trunk may receive chemotherapy 42, 43, 44.
- Pleomorphic liposarcoma. Pleomorphic liposarcoma is a rare, aggressive, high-grade subtype of liposarcoma that typically arises in the deep soft tissues of your arms and legs and is characterized by the presence of abnormally shaped “pleomorphic” fat cells called lipoblasts mixed with undifferentiated pleomorphic sarcoma (UPS)-like areas 45, 46. Pleomorphic liposarcoma is the least common subtype of liposarcoma making up around 10% of all liposarcoma cases with a high rate of recurrence and poor outcomes. Pleomorphic liposarcoma is known for its rapid growth, a high rate of recurrence, and a tendency to spread (metastasize) to other parts of the body, most often the lungs, resulting in a poor prognosis. Diagnosis relies on the identification of these distinctive multivacuolated “pleomorphic lipoblasts” 45, 47. Surgery is the most common and reliable option for treatment of pleomorphic liposarcoma 48. Some larger tumors may be treated with chemotherapy before surgery. Radiation therapy (before or after surgery) may also be given if the surgical margin is narrow. Pleomorphic liposarcoma is usually a high-grade cancer that is less likely to respond well to treatment. Pleomorphic liposarcoma often forms in older adults with the median age at diagnosis being 70 years of age.
- Myxoid pleomorphic liposarcoma. Myxoid pleomorphic liposarcoma is an extremely rare and aggressive type of soft tissue cancer that most often forms in the middle of the chest (mediastinum) in children, adolescents, and young adults, though it can occur in adults and elsewhere in the body 49, 50, 51, 52. Myxoid pleomorphic liposarcoma is high grade cancer and can grow quickly and spread to other areas in the body. Studies suggest a link between it and conventional pleomorphic liposarcoma 49, 53. Myxoid pleomorphic liposarcoma is characterized by a combination of the distinct features of myxoid liposarcoma (myxoid stroma, chicken wire-like vasculature, lipoblasts) and high-grade pleomorphic liposarcoma (pleomorphic cells, high mitotic activity, and necrosis). Myxoid pleomorphic liposarcoma has a poor prognosis, with a high tendency for local recurrence, metastasis, and a significantly shorter survival rate compared to other liposarcoma subtypes.
Liposarcoma symptoms can vary depending on the location and type of liposarcoma you have. In general, liposarcoma develops in your arms or legs, particularly in your thighs or the back of your knees, but you can also develop liposarcoma in the back of your belly. Most liposarcomas grow very slowly and rarely cause pain. You may not notice any changes in your body unless you notice a large bump on your arm or leg that doesn’t go away or gets larger. A liposarcoma may cause pain if it presses on a nerve. Likewise, a liposarcoma may affect some of your organs. For example, a liposarcoma in your lungs may make it hard for you to breathe. For abdominal liposarcomas, symptoms can include abdominal pain, cramping, a feeling of fullness, loss of appetite, constipation, or unexplained weight loss.
If you have any of the these problems, see a doctor right away:
- A new lump or a lump that’s growing (anywhere on your body)
- Abdominal pain that’s getting worse
- Blood in your stool or vomit
- Black, tarry stools (when bleeding happens in the stomach or bowels, the blood can turn black as it’s digested, and it might make the stool very black and sticky)
Liposarcomas are usually found by a patient when a lump appears in the arms, legs, or torso. They can also be found during an investigation of other symptoms or during a routine operation. A diagnosis of liposarcoma may start with a visit to your doctor, who will examine you and then refer you to a specialist doctor. A specialist doctor will diagnose liposarcoma through a series of tests. These may include:
- Physical examination – looking at and feeling any lump
- Imaging tests. Imaging tests create pictures of the inside of your body. They might help show the size of the liposarcoma. Tests may include X-ray, CT scan and MRI. Sometimes a positron emission tomography scan (PET scan), is needed.
- Biopsy. A biopsy is a procedure to remove some cells for testing. The sample might be removed with a needle put through the skin. Or the sample might be taken during surgery to remove the cancer. The type of biopsy depends on the cancer’s location.
- Testing the cancers cells in a lab. The biopsy sample goes to a lab for testing. Doctors who specialize in analyzing blood and body tissue, called pathologists, test the cells to see if they’re cancerous. Other special tests give more details. Your health care team uses the results to understand your prognosis and create a treatment plan.
Treatment depends on the type and stage of the liposarcoma and may involve:
- Surgery. The goal of surgery is to remove all of the cancer cells. Whenever possible, surgeons work to remove the entire liposarcoma without damaging any surrounding organs. However, if a liposarcoma grows to involve nearby organs, removal of the entire liposarcoma may not be possible. In these situations, your doctor may recommend other treatments to shrink the liposarcoma first. That will make it easier to remove during an operation.
- Radiation therapy. Radiation therapy uses powerful energy beams to kill cancer cells. The energy can come from X-rays, protons or other sources. Radiation may be used after surgery to kill any cancer cells that remain. Radiation also may be used before surgery to shrink a tumor to make it more likely that surgeons can remove the entire tumor.
- Chemotherapy. Chemotherapy uses strong medicines to kill cancer cells. Some chemotherapy medicines are given through a vein and some are taken in pill form. Not all types of liposarcoma are sensitive to chemotherapy. Careful testing of your cancer cells can show whether chemotherapy is likely to help you. Chemotherapy may be used after surgery to kill any cancer cells that remain. It also may be used before surgery to shrink a tumor. Chemotherapy is sometimes combined with radiation therapy.
- Clinical trials. Clinical trials are studies of new treatments. These studies give you a chance to try the latest treatment options. The risk of side effects may not be known. Ask a member of your health care team whether you can participate in a clinical trial.
Figure 1. Retroperitoneal liposarcoma
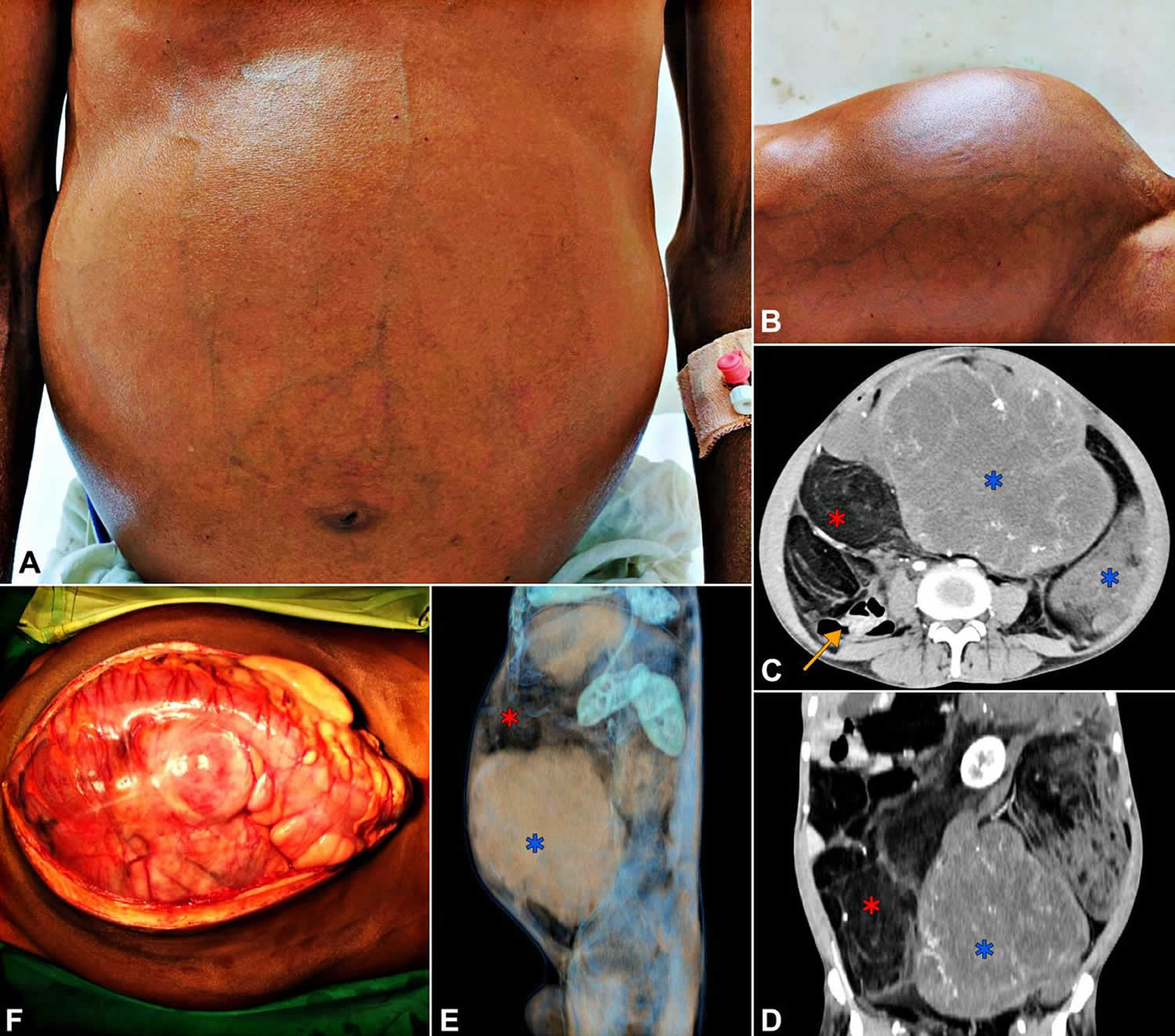
Footnotes: (A, B) Clinical photographs of the patient showing distended abdomen with prominent veins. (C, D, E) The contrast enhanced CT scan. (C) Axial section. (D) Coronal section. (E) Three-dimensional reconstruction with a sagittal view from the left side. Blue stars indicate the solid areas of the tumor (HU: 25–35). Red stars indicate the lipomatous portions (HU: −60 to −85). Yellow arrow in (D) shows the displaced bowel loops to the right. (F) Intraoperative photograph of the tumour after laparotomy.
[Source 54 ]Liposarcoma types
Liposarcomas are mainly subclassified into five subtypes with different morphologic and behavioral spectrum 3, 7, 8. The types have similar symptoms but can require different treatments and have different prognoses or expected outcomes 9.
Table 1. Liposarcoma types
| Liposarcoma subtype | Histopathology | Immunophenotype | Treatment |
|---|---|---|---|
| Well-Differentiated Liposarcoma | Lipogenic component with mature adipocytes of variable size Atypical spindle stromal cells | MDM2+, CDK+ DDIT3− S100+ CD34− p16+ p53 wildtype | Surgical resection with negative margins |
| Dedifferentiated Liposarcoma | Lipogenic component with an abrupt transition toward a non-lipogenic tumor area Necrosis [+/−] | MDM2+, CDK4+ DDIT3− S100+ CD34− p16+ p53 wildtype or mutant | Surgical resection Neoadjuvant radiotherapy Chemotherapy/targeted therapy |
| Myxoid Liposarcoma | Lipogenic areas Loose basophilic/myxoid stroma Necrosis [+/−] Atypical lipoblasts [+/−] Round cell component [+/−] | MDM2−, CDK4− DDIT3+ S100+ CD34− p16− p53 wildtype | Surgical resection Neoadjuvant or adjuvant radiotherapy Chemotherapy/targeted therapies |
| Pleomorphic Liposarcoma | High-grade undifferentiated sarcoma Pleomorphic lipoblasts Necrosis [+/−] | MDM2−, CDK4− DDIT3− S100+ CD34+ p16+ p53 mutant [hyperexpression] | Surgical resection or amputation Post-operative radiotherapy Chemotherapy |
| Myxoid Pleomorphic Liposarcoma | Myxoid liposarcoma-like areas Pleomorphic liposarcoma-like areas | MDM2−, CDK4− CD34+ p16+ p53 mutant [hyperexpression] Rb loss | Surgical resection with negative margins Benefits of radiotherapy and chemotherapy not well-established |
Well-differentiated liposarcoma
Well-differentiated liposarcoma is the most common type of liposarcoma, representing about half of all liposarcoma cases. Well-differentiated liposarcoma is a slow-growing, low-grade malignant tumor, most commonly found in your arms or legs or retroperitoneum (back of the abdomen) 10, 11. Superficial well-differentiated liposarcoma lesions are usually located in the subcutaneous fat and are diagnosed as atypical lipomatous tumors (ALT) 12. Under microscopic examination, well-differentiated liposarcoma typically consists of mature adipocytes of variable size, encompassed by fibrous stroma exhibiting atypical spindle cells with hyperchromatic nuclei 13. Well-differentiated liposarcoma usually shows MDM2 (murine double minute 2) and CDK4 (cyclin-dependent kinase 4) expression 14, 15, 16. However, some cases with atypical morphology and immunophenotype require FISH for identification of MDM2 amplifications, in order to avoid misdiagnosis 17, 18, 19, 20, 21. While well-differentiated liposarcoma rarely metastasizes (spreads to other parts of the body), it has a tendency to recur locally after surgical removal, particularly in the retroperitoneum 22, 23. In rare cases, 17% of well-differentiated liposarcoma of the retroperitoneum and 6% of well-differentiated liposarcoma of the limbs can progress into dedifferentiated liposarcoma 24. Surgery is the most common and most reliable option for treatment of well-differentiated liposarcoma. In some cases, radiotherapy is used in addition to surgery. The current general consensus from researchers is that chemotherapy is not effective in well-differentiated liposarcoma 24.
Dedifferentiated liposarcoma
Dedifferentiated liposarcoma is a type of cancer that develops when a well-differentiated liposarcoma or atypical lipomatous tumor (ALT) transforms into a more aggressive, non-lipogenic sarcoma 25, 26, 27, 28, 29, 30. Dedifferentiated liposarcoma is characterized by the presence of both well-differentiated and poorly differentiated tumor components. Dedifferentiated liposarcoma is more aggressive than well differentiated liposarcoma, with a higher risk of recurrence and metastasis, but its metastatic potential is related to the anatomic site of the tumor with retroperitoneal masses having a worse prognosis 27. Recurrence occurs in 40% to 75% of dedifferentiated liposarcoma cases and metastasis occurs in about 10–15% of cases 31. The genetic background of dedifferentiated liposarcoma is characterized by MDM2 (murine double minute 2) and CDK4 (cyclin-dependent kinase 4) amplification; therefore, immunohistochemical expression of the corresponding markers is used for diagnosis 26. A mutant TP53 immunophenotype, with hyperexpression of p53, is also reported in some studies 26, 32. Molecular analysis of this tumor is important, as new therapeutic targets have been identified 33. Surgery is the most common and most reliable option for treatment of differentiated liposarcoma 25, 26, 34, 31. Dedifferentiated liposarcomas in the are associated with a higher risk of recurrence, due to the difficulty of achieving complete surgical resection 34, 35, 36. Radiation therapy in sometimes used in patients with margins of less than 10mm after surgical resection 31. It’s not clear yet how suitable chemotherapy is as a treatment for differentiated liposarcoma, but an ongoing clinical trial is investigating this 31.
Myxoid liposarcoma or myxoid/round cell liposarcoma
The term “round cell” refers to how tumor cells appear when viewed under a microscope. Myxoid liposarcoma or round cell liposarcomas is one of the most common subtype of liposarcoma making up about 30% of all liposarcoma cases 37. Myxoid liposarcoma is a type of liposarcoma mostly located in your arms and legs typically occurring in younger patients, with a variable behavior depending on the histological subtype. These tumors grow slowly, and they can spread to other parts of the body. Myxoid liposarcoma is more common in people aged 20 to 40 years old and is rarely seen in children under 10 years old. Myxoid liposarcoma tends to affect more males than females. Myxoid liposarcoma is a distinct entity with unique genetic and molecular features consisting of FUS-DDIT3 or EWSR1-DDIT3 fusion 38. Further studies on the FUS-DDIT3 fusion oncogene in myxoid liposarcoma suggest a link between this mutation and JAK-stat signaling in the stem cancerous cell 39, 40, 41. Currently, the most efficient treatment for myxoid liposarcoma comprises surgical resection with preoperative radiotherapy frequently administered, whereas high-risk lesions of the limbs or trunk may receive chemotherapy 42, 43, 44.
Pleomorphic liposarcoma
Pleomorphic liposarcoma is a rare, aggressive, high-grade subtype of liposarcoma that typically arises in the deep soft tissues of your arms and legs and is characterized by the presence of abnormally shaped “pleomorphic” fat cells called lipoblasts mixed with undifferentiated pleomorphic sarcoma (UPS)-like areas 45, 46. Pleomorphic liposarcoma is the least common subtype of liposarcoma making up around 10% of all liposarcoma cases with a high rate of recurrence and poor outcomes. Pleomorphic liposarcoma is known for its rapid growth, a high rate of recurrence, and a tendency to spread (metastasize) to other parts of the body, most often the lungs, resulting in a poor prognosis. Diagnosis relies on the identification of these distinctive multivacuolated “pleomorphic lipoblasts” 45, 47. Surgery is the most common and reliable option for treatment of pleomorphic liposarcoma 48. Some larger tumors may be treated with chemotherapy before surgery. Radiation therapy (before or after surgery) may also be given if the surgical margin is narrow. Pleomorphic liposarcoma is usually a high-grade cancer that is less likely to respond well to treatment. Pleomorphic liposarcoma often forms in older adults with the median age at diagnosis being 70 years of age.
Myxoid pleomorphic liposarcoma
Myxoid pleomorphic liposarcoma is an extremely rare and aggressive type of soft tissue cancer that most often forms in the middle of the chest (mediastinum) in children, adolescents, and young adults, though it can occur in adults and elsewhere in the body 49, 50, 51, 52. Myxoid pleomorphic liposarcoma is high grade cancer and can grow quickly and spread to other areas in the body. Studies suggest a link between it and conventional pleomorphic liposarcoma 49, 53. Myxoid pleomorphic liposarcoma is characterized by a combination of the distinct features of myxoid liposarcoma (myxoid stroma, chicken wire-like vasculature, lipoblasts) and high-grade pleomorphic liposarcoma (pleomorphic cells, high mitotic activity, and necrosis). Myxoid pleomorphic liposarcoma has a poor prognosis, with a high tendency for local recurrence, metastasis, and a significantly shorter survival rate compared to other liposarcoma subtypes.
Liposarcoma causes
It’s not clear what causes liposarcoma 1, 3, 5, 6. Liposarcoma starts when fat cells get changes in their DNA. A cell’s DNA holds the instructions that tell the cell what to do. The changes turn the fat cells into cancer cells. The changes tell the cancer cells to grow quickly and make a lot of extra cells. The cancer cells keep living when healthy cells would die as part of their natural life cycle.
The cancer cells form a growth, called a tumor. In some types of liposarcoma, the cancer cells stay put. They continue making more cells, causing the tumor to get bigger. In other types of liposarcoma, the cancer cells might break away and spread to other parts of the body. When cancer spreads to other parts of the body, it’s called metastatic cancer.
Medical researchers have identified at least 20 different genetic mutations that cause liposarcoma. An abnormality of band 12q13 has been associated with the development of liposarcomas 56. The most common chromosomal translocation is the FUS-CHOP fusion gene, which encodes a transcription factor necessary for adipocyte differentiation 57. These and other distinct genetic aberrations may aid in the diagnosis of particular liposarcoma subtypes, and they can potentially be targets that can be exploited therapeutically 57. They don’t know all the reasons why these genes mutate, but researchers have identified some risk factors that increase your risk of developing liposarcoma:
- Radiation therapy for cancer.
- Exposure to workplace chemicals such as vinyl chloride.
- Certain inherited conditions.
Liposarcomas usually occur in adults between 50 and 65 years old, and are rare in children and adolescents. The mean patient age at onset is 50 years. Although liposarcomas account for about 17% of all soft tissue sarcomas, they are involved in only 4% of childhood soft tissue sarcomas. Cases of liposarcoma are reported in young adults and teenagers, but cases in children are rare 58.
Liposarcoma signs and symptoms
Liposarcoma symptoms depend on the part of the body where the cancer forms.
Liposarcoma in your arms and legs can cause:
- A growing lump of tissue under the skin.
- Pain.
- Swelling.
- Weakness of the affected limb.
Liposarcoma in your belly or abdomen, can cause:
- Abdominal pain.
- Abdominal swelling.
- Feeling full sooner when eating.
- Constipation.
- Blood in stool.
Most patients who are diagnosed with liposarcoma do not have any early symptoms and it can go unnoticed during the initial stages of the disease until the tumor has grown to a large enough size to compress neighboring tissues and cause pain or decreased function. Liposarcoma can sometimes be noticed as a deep-seated mass to touch. Liposarcoma usually appears as a well-circumscribed palpable mass as large as 10 cm in diameter. The mass tends to grow slowly over time. The lesion is commonly not tender on palpation. Liposarcoma, as with all other cancers, can also present with non-specific symptoms such as fevers, chills, fatigue, night sweats and weight loss 59. If the tumor is inside your abdomen in the retroperitoneal region, it can present with specific symptoms in the abdomen, including abdominal or flank pain, abdominal swelling, and constipation or the sensation of feeling full sooner than expected after eating 59.
Liposarcoma that resembles a skin tag has been reported but is an exceptionally rare event 60.
Liposarcoma complications
A liposarcoma is a malignant tumor. This means it’s cancerous and can spread to other parts of your body, including vital organs and tissues surrounding the original tumor. If left untreated, a liposarcoma can ultimately be life-threatening. Early detection and treatment of a liposarcoma are critically important.
Treatment complications
typically treat liposarcoma with surgery to remove the tumor and nearby healthy tissue. Any type of surgery may have complications. Your surgeon will discuss your specific situation, but some common surgery complications may include:
- Reaction to general anesthesia.
- Blood loss.
- Surgical wounds that don’t heal.
- Infection.
- Damage to organs or tissues affected by the tumor.
- Pain that isn’t managed by pain medication.
Liposarcoma diagnosis
Tests and procedures used to diagnose liposarcoma include:
- Imaging tests. Imaging tests create pictures of the inside of your body. They might help show the size of the liposarcoma. Tests may include X-ray, computed tomography (CT) scan and magnetic resonance imaging (MRI). Sometimes a positron emission tomography scan (PET scan), is needed. A PET scan is useful when your doctor thinks the cancer has spread but doesn’t know where. It can be used instead of many different x-rays because it scans your whole body. Often the PET scan is used with a CT scan (called a PET/CT scan). This helps decide if changes seen on the CT scan are cancer or something else. PET isn’t often used for sarcoma, but it can be helpful in some cases.
- Ultrasound. Ultrasound uses sound waves and their echoes to produce pictures of parts of the body. A small instrument called a transducer sends out sound waves and picks up the echoes as they bounce off the organs. A computer then converts the echoes into an image on a screen. Ultrasound may be done before a biopsy to see if a lump is a cyst, meaning if it has fluid in it and is likely not cancer, or if it’s solid and more likely a tumor. This test is often not needed if a CT or MRI was done.
- CT (computed tomography) scans. A CT scan uses x-rays to make detailed cross-sectional images of your body. This test is often done if the doctor suspects a soft tissue sarcoma in the chest, abdomen (belly), or the retroperitoneum (the back of the abdomen). This test is also used to see if the sarcoma has spread to the lungs, liver, or other organs. CT scans might be used to guide a biopsy needle into a tumor inside the body — the chest or abdomen, for example. This is called a CT-guided needle biopsy. You lie on the CT scanning table while a radiologist moves a biopsy needle toward the tumor. CT scans are repeated until the doctors are sure the needle is within the tumor.
- MRI (magnetic resonance imaging). MRI uses radio waves and strong magnets instead of x-rays to take pictures of the body. MRI scans are often part of the work-up of any tumor that could be a sarcoma. They’re often better than CT scans in evaluating sarcomas in the arms or legs. MRI provides a good picture of the extent of the tumor. It can show your health care team many things about the tumor, like where it is, how big it is, and sometimes even the type of tissue it comes from (like bone, fat, or muscle). MRIs are also very helpful in examining the brain and spinal cord.
- PET (positron emission tomography) scan. PET scans use a form of radioactive sugar called fluorodeoxyglucose (FDG) that’s put into your blood. Because cancers use glucose (sugar) at a higher rate than normal tissues, the radioactivity collects in the cancer. A scanner can then spot the radioactive deposits. A PET scan is useful when your doctor thinks the cancer has spread but doesn’t know where. It can be used instead of many different x-rays because it scans your whole body. Often the PET scan is used with a CT scan (called a PET/CT scan). This helps decide if changes seen on the CT scan are cancer or something else. PET isn’t often used for sarcoma, but it can be helpful in some cases.
- Biopsy. A biopsy is a procedure to remove some cells for testing. The sample might be removed with a needle put through the skin. Or the sample might be taken during surgery to remove the cancer. The type of biopsy depends on the cancer’s location.
- Testing the cancers cells in a lab. The biopsy sample goes to a lab for testing. Doctors who specialize in analyzing blood and body tissue, called pathologists, test the cells to see if they’re cancerous. Other special tests give more details. Your health care team uses the results to understand your prognosis and create a treatment plan.
Liposarcoma Stages
After someone is diagnosed with a liposarcoma, doctors will try to figure out if it has spread, and if so, how far. This process is called staging. The stage of a cancer describes how much cancer is in the body. It helps determine how serious the cancer is and how best to treat it. Doctors also use a cancer’s stage when talking about survival statistics.
The stages of liposarcomas range from stages I (1) through IV (4). As a rule, the lower the number, the less the cancer has spread. A higher number, such as stage IV, means cancer has spread more. And within a stage, an earlier letter means a lower stage. Although each person’s cancer experience is unique, cancers with similar stages tend to have a similar outlook and are often treated in much the same way.
The staging system most often used for soft tissue sarcomas is the American Joint Committee on Cancer (AJCC) TNM system, which is based on 4 key pieces of information:
- The extent of the tumor (T): How large is the cancer?
- The spread to nearby lymph nodes (N): Has the cancer spread to nearby lymph nodes?
- The spread (metastasis) to distant sites (M): Has the cancer spread to distant organs such as the lungs?
- The grade (G) of the cancer: How much do the sarcoma cells look like normal cells?
The grade is partly used to determine the stage of a sarcoma. The staging system divides sarcomas into 3 grades (1 to 3). The grade of a sarcoma helps predict how rapidly it will grow and spread. It’s useful in predicting a patient’s outlook and helps determine treatment options.
The grade of a sarcoma is determined using a system known as the French or FNCLCC system, and is based on 3 factors:
- Differentiation: Cancer cells are given a score of 1 to 3, with 1 being assigned when they look a lot like normal cells and 3 being used when the cancer cells look very abnormal. Certain types of sarcoma are given a higher score automatically.
- Mitotic count: How many cancer cells are seen dividing under the microscope; given a score from 1 to 3 (a lower score means fewer cells were seen dividing)
- Tumor necrosis: How much of the tumor is made up of dying tissue; given a score from 0 to 2 (a lower score means there was less dying tissue present).
Each factor is given a score, and the scores are added to determine the grade of the tumor. Sarcomas that have cells that look more normal and have fewer cells dividing are generally placed in a low-grade category. Low-grade tumors tend to be slow growing, slower to spread, and often have a better outlook (prognosis) than higher-grade tumors. Certain types of sarcoma are automatically given higher differentiation scores. This affects the overall score so much that they are never considered low grade. Examples of these include synovial sarcomas and embryonal sarcomas. Here’s what the grade numbers mean:
- GX: The grade cannot be assessed (because of incomplete information).
- Grade 1 (G1): Total score of 2 or 3
- Grade 2 (G2): Total score of 4 or 5
- Grade 3 (G3): Total score of 6, 7 or 8
Table 2. Trunk and Extremities Sarcoma Stages
| AJCC stage | Stage grouping | Trunk and Extremities Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm (T2) OR Larger than 10cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IV | Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. |
| OR | ||
| Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. | |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Table 3. Retroperitoneum Sarcoma Stages
| AJCC stage | Stage grouping | Retroperitoneum Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm OR Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| OR | ||
| Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. | |
| IV | Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Liposarcoma differential diagnosis
There are other diseases that can present very similarly to liposarcoma. Lipoma is a non-cancerous (benign) tumor that can feel like liposarcoma, but it is usually softer and feels like a mass directly below the skin rather than in deeper parts of the body. A benign lipoma may very rarely develop into a liposarcoma. Other soft tissue tumors, such as undifferentiated pleomorphic sarcoma, lipomatous hemangiopericytoma, non-lipogenic sarcoma and gastrointestinal stromal tumors, can also look like liposarcoma when initially evaluated under a microscope.
Late granulomatous reactions from silicone may appear in a site different from that of the injection and may cause an incorrect diagnosis of liposarcoma 62. Silicone implants for chin augmentation may create a tissue reaction that mimics a low-grade liposarcoma 63.
Myxofibrosarcoma, one of the most common soft tissue sarcomas of elderly patients, may histologically resemble pleomorphic liposarcoma 64.
Liposarcoma treatment
Liposarcoma treatment depends on the liposarcoma type, size, location and whether the cancer has spread and, if so, where it has spread. You may have a combination of more than one type of treatment for the disease. Treatments may include:
- Surgery: A surgeon removes the tumor and surrounding healthy tissue, including microscopic tumor cells.
- Radiation therapy: You may have radiation therapy to shrink the tumor before surgery, particularly if you have myxoid liposarcomas. You may also have this treatment after surgery to reduce the risk of the cancer coming back.
- Chemotherapy: Your doctor may use chemotherapy to treat tumors that surgery can’t remove.
Surgery is the main treatment used for liposarcoma, sometimes with additional radiation therapy or chemotherapy. Your surgeon will remove the tumor and will aim to take out an area of normal tissue around it too; this is known as taking a margin. It allows any cancer cells that are not visible to the naked eye to be removed along with the tumor, which can reduce the risk of the cancer coming back. In rare cases, surgical amputation may be performed. There are certain cases where it would be riskier to attempt to remove the tumor surgically. For example, this can occur if it is in the retroperitoneum where it is deeply embedded and involves multiple organs. Another example would be if the tumor were adjacent to vital structures, such as major blood vessels, where removing the tumor itself could pose a significant risk. If a tumor spreads in such a way that the mass cannot be completely removed with surgery, chemotherapy or radiation therapy can be considered to kill the rest of the tumor and reduce the risk of the cancer reoccurrence. In certain circumstances, chemotherapy and/or radiation therapy prior to surgery can be considered to shrink the tumor to a size where it can be successfully surgically removed.
After treatment, you will have regular follow-up appointments for several years. You should receive a follow-up schedule from your sarcoma clinical nurse specialist. The usual practice will include:
- A chance to discuss symptoms
- An examination to look for any signs of the sarcoma returning. This may include an MRI or ultrasound if required after examination
- A chest x-ray to rule out any secondary cancers occurring in the lungs
Liposarcoma prognosis
The prognosis of liposarcoma depends on several factors. The most important correlation with survival is associated with histologic subtypes, the grade of the tumor, the tumor’s location, and the status of surgical margins 65, 66, 67. A well-differentiated liposarcoma has been reported to have a 50% recurrence rate (these tumors are poorly circumscribed and locally recur after incomplete excision) with no risk of distant metastasis and an excellent five-year survival rate (75% to 100%) 68, 1. Although well-differentiated liposarcomas rarely metastasize, repeated local recurrences may cause the tumor to evolve into a higher grade of sarcoma or to dedifferentiate, in which case metastasis is possible 69. In comparison, myxoid liposarcoma and pleomorphic liposarcoma have a higher percentage of recurrence (up to 80%) and a poor to intermediate survival rate (ranging from 4 to 107 months) 1. Between 1973 and 2015, 1756 patients with extremity myxoid liposarcomas were evaluated. The 5- and 10-year overall survival rates of the entire cohort were 86.4% and 75.9%, respectively 70. Round-cell and poorly differentiated types have a poor prognosis. Each has a 5-year survival rate of about 50% because they recur locally and tend to metastasize quickly and widely, especially in poorly differentiated liposarcomas. The lungs and the liver are the most common sites of metastasis. Undifferentiated liposarcoma has a higher risk of distal metastasis 1. Positive surgical resection margin is associated with high local recurrence rates and poor survival 1. Although there are some case reports of using adjuvant (add-on) radiation therapy to decrease the rate of recurrence, the role of adjuvant chemotherapy and radiation therapy is under debate without a clear consensus 1.
Table 4. Liposarcoma subtypes and their prognosis
| Myxoid liposarcoma | Well-dedifferentiated liposarcoma (WDLPS) | Dedifferentiated liposarcoma (DDLPS) | Pleomorphic liposarcoma | Myxoid pleomorphic liposarcoma | |
|---|---|---|---|---|---|
| Characteristics | Continuum between pure myxoid/round cell morphology | Locally aggressive, well-differentiated adipocytic sarcoma | Malignant, progressing from well-differentiated to non-lipogenic tumour | Aggressive, undifferentiated sarcoma | Combination of myxoid & pleomorphic features |
| Constitutes how many % of liposarcoma | 20–30 % | 40–50 % | 15–20 % | 5–10 % | 15 % |
| Treatment options | Surgery, neoadjuvant/adjuvant radiotherapy, chemotherapy, targeted therapy | Surgery with negative margins | Neoadjuvant radiotherapy, surgery, targeted therapy | Surgery, post-operative radiotherapy, chemotherapy | Surgery with negative margins, benefits of chemotherapy & radiotherapy not well-confirmed |
| Median survival/mortality rate | 5-year survival rate: 80–90 % for pure myxoid subtype & 20–25 % for tumor containing high-grade round cells | Median time to death: 6–11 years | Mortality rate: ∼30 % | 5-year survival rate: 60 % | Median time to death: 2 years |
Is there a cure for liposarcoma?
That depends on the type of liposarcoma you have and if it’s spread. For example, if you have well-differentiated liposarcoma that hasn’t spread, your surgeon may be able to remove the entire tumor. If the tumor doesn’t come back, your doctor may consider you cured.
There are several types of liposarcoma with very different prognoses. If you have liposarcoma, your doctor is your best resource for information.
- Zafar R, Wheeler Y. Liposarcoma. [Updated 2023 Mar 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538265[↩][↩][↩][↩][↩][↩][↩]
- Creytens D. What’s new in adipocytic neoplasia? Virchows Arch. 2020;476(1):29–39.PMID: 31501988. doi: 10.1007/s00428-019-02652-3[↩]
- Suarez-Kelly, L. P., Baldi, G. G., & Gronchi, A. (2019). Pharmacotherapy for liposarcoma: current state of the art and emerging systemic treatments. Expert Opinion on Pharmacotherapy, 20(12), 1503–1515. https://doi.org/10.1080/14656566.2019.1618271[↩][↩][↩][↩]
- Ciongariu AM, Țăpoi DA, Dumitru AV, Enache V, Marin A, Creangă CA, Costache M. “Enhancing Liposarcoma Prognosis – A New Predictive Scoring System Integrating Histopathological Insights”. Cancer Manag Res. 2025 Feb 17;17:331-348. doi: 10.2147/CMAR.S504889[↩]
- Liposarcoma. https://emedicine.medscape.com/article/1102007-overview#a7[↩][↩]
- Lin ZC, Chang XZ, Huang XF, Zhang CL, Yu GS, Wu SY, Ye M, He JX. Giant liposarcoma of the esophagus: A case report. World J Gastroenterol. 2015 Sep 7;21(33):9827-32. doi: 10.3748/wjg.v21.i33.9827[↩][↩]
- Machhada A., Emam A., Colavitti G., Maggiani F., Coelho J.A., Ayre G., Mahrous A.M., Khundkar R., Wright T.C., Wilson P. Liposarcoma subtype recurrence and survival: A UK regional cohort study. J. Plast. Reconstr. Aesthetic Surg. 2022;75:2098–2107. doi: 10.1016/j.bjps.2022.02.023[↩][↩]
- Chamberlain F.E., Wilding C., Jones R.L., Huang P. Pazopanib in patients with advanced intermediate-grade or high-grade liposarcoma. Expert Opin. Investig. Drugs. 2019;28:505–511. doi: 10.1080/13543784.2019.1607291[↩][↩]
- The WHO Classification of Tumors Editorial Board . WHO Classification of Tumours of Soft Tissue and Bone. 5th ed. IARC Press; Lyon, France: 2020. pp. 36–48. https://publications.iarc.who.int/588[↩][↩]
- Warnick J., Lahiri R., Karanjia N., Fisher C., Bagwan I. Well Differentiated Liposarcoma Presenting as a Duodenal Polyp: A Case Report and Review of the Literature. Int. J. Surg. Pathol. 2023;31:307–311. doi: 10.1177/10668969221099628[↩][↩]
- Wang G.Y., Lucas D.R. Dedifferentiated Liposarcoma with Myofibroblastic Differentiation. Arch. Pathol. Lab. Med. 2018;142:1159–1163. doi: 10.5858/arpa.2018-0205-RA[↩][↩]
- Mashima E., Sawada Y., Saito-Sasaki N., Yamamoto K., Ohmori S., Omoto D., Yoshioka H., Yoshioka M., Okada E., Aoki T., et al. A Retrospective Study of Superficial Type Atypical Lipomatous Tumor. Front. Med. 2020;7:609515. doi: 10.3389/fmed.2020.609515[↩][↩]
- Lee A.T.J., Thway K., Huang P.H., Jones R.L. Clinical and Molecular Spectrum of Liposarcoma. J. Clin. Oncol. 2018;36:151–159. doi: 10.1200/JCO.2017.74.9598[↩][↩]
- Lu J., Wood D., Ingley E., Koks S., Wong D. Update on genomic and molecular landscapes of well-differentiated liposarcoma and dedifferentiated liposarcoma. Mol. Biol. Rep. 2021;48:3637–3647. doi: 10.1007/s11033-021-06362-5[↩][↩]
- Abdul Razak A.R., Bauer S., Suarez C., Lin C.C., Quek R., Hütter-Krönke M.L., Cubedo R., Ferretti S., Guerreiro N., Jullion A., et al. Co-Targeting of MDM2 and CDK4/6 with Siremadlin and Ribociclib for the Treatment of Patients with Well-Differentiated or Dedifferentiated Liposarcoma: Results from a Proof-of-Concept, Phase Ib Study. Clin. Cancer Res. 2022;28:1087–1097. doi: 10.1158/1078-0432.CCR-21-1291[↩][↩]
- Crago A.M., Dickson M.A. Liposarcoma: Multimodality Management and Future Targeted Therapies. Surg. Oncol. Clin. N. Am. 2016;25:761–773. doi: 10.1016/j.soc.2016.05.007[↩][↩]
- Narla S.L., Stephen P., Kurian A., Annapurneswari S. Well-differentiated liposarcoma of the breast arising in a background of malignant phyllodes tumor in a pregnant woman: A rare case report and review of literature. Indian J. Pathol. Microbiol. 2018;61:577–579. doi: 10.4103/IJPM.IJPM_238_17[↩][↩]
- Zhu H., Sun J., Wei S., Wang D., Brandwein M. Well-Differentiated Laryngeal/Hypopharyngeal Liposarcoma in the MDM2 Era Report of Three Cases and Literature Review. Head Neck Pathol. 2017;11:146–151. doi: 10.1007/s12105-016-0747-0[↩][↩]
- Eyermann C., Raguin T., Hemar P., Debry C. Well-differentiated, pedunculated liposarcoma of the hypopharynx. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2018;135:63–65. doi: 10.1016/j.anorl.2017.07.001[↩][↩]
- Lam T.C., Yuen H.K.L., Cheuk W. Primary Well-Differentiated Liposarcoma of the Orbit. Int. J. Surg. Pathol. 2021;29:406–407. doi: 10.1177/1066896920925168[↩][↩]
- Xue S., Liu Q.Y., Gou X.N., Zhao Y.W., Cheng Q., Kong L.F. [Well-differentiated/dedifferentiated liposarcoma associated with myxoid-like morphology: A clinicopathological and molecular genetic characteristics analysis of 34 cases] Zhonghua Bing Li Xue Za Zhi. 2024;53:168–173. doi: 10.3760/cma.j.cn112151-20231025-00302. In Chinese[↩][↩]
- Thway K. Well-differentiated liposarcoma and dedifferentiated liposarcoma: An updated review. Semin Diagn Pathol. 2019 Mar;36(2):112-121. doi: 10.1053/j.semdp.2019.02.006[↩][↩]
- Briski L.M., Jorns J.M. Primary Breast Atypical Lipomatous Tumor/Well-Differentiated Liposarcoma and Dedifferentiated Liposarcoma. Arch. Pathol. Lab. Med. 2018;142:268–274. doi: 10.5858/arpa.2016-0380-RSR2[↩][↩]
- Well-differentiated liposarcoma. https://sarcoma.org.uk/about-sarcoma/what-is-sarcoma/types-of-sarcoma/liposarcoma/well-differentiated-liposarcoma[↩][↩][↩][↩]
- Nishio J, Nakayama S, Nabeshima K, Yamamoto T. Biology and Management of Dedifferentiated Liposarcoma: State of the Art and Perspectives. J Clin Med. 2021 Jul 22;10(15):3230. doi: 10.3390/jcm10153230[↩][↩][↩][↩]
- Ciongariu A.M., Dumitru A.V., Cîrstoiu C., Crețu B., Sajin M., Țăpoi D.A., Ciobănoiu A.D., Bejenariu A., Marin A., Costache M. The Conundrum of Dedifferentiation in a Liposarcoma at a Peculiar Location: A Case Report and Literature Review. Medicina. 2023;59:967. doi: 10.3390/medicina59050967[↩][↩][↩][↩][↩][↩][↩][↩]
- Thway K., Jones R.L., Noujaim J., Zaidi S., Miah A.B., Fisher C. Dedifferentiated Liposarcoma: Updates on Morphology, Genetics, and Therapeutic Strategies. Adv. Anat. Pathol. 2016;23:30–40. doi: 10.1097/PAP.0000000000000101[↩][↩][↩][↩]
- Stock N. Tumeurs adipeuses [Adipocytic tumors] Ann. Pathol. 2015;35:41–53. doi: 10.1016/j.annpat.2014.12.001. In French[↩][↩]
- Kammerer-Jacquet S.F., Thierry S., Cabillic F., Lannes M., Burtin F., Henno S., Dugay F., Bouzillé G., Rioux-Leclercq N., Belaud-Rotureau M.A., et al. Differential diagnosis of atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma: Utility of p16 in combination with MDM2 and CDK4 immunohistochemistry. Hum. Pathol. 2017;59:34–40. doi: 10.1016/j.humpath.2016.08.009[↩][↩]
- Nishio J. Contributions of Cytogenetics and Molecular Cytogenetics to the Diagnosis of Adipocytic Tumors. J. Biomed. Biotechnol. 2011;2011:1–9. doi: 10.1155/2011/524067[↩][↩]
- Dedifferentiated liposarcoma. https://sarcoma.org.uk/about-sarcoma/what-is-sarcoma/types-of-sarcoma/liposarcoma/dedifferentiated-liposarcoma[↩][↩][↩][↩][↩][↩][↩][↩]
- Traweek R.S., Cope B.M., Roland C.L., Keung E.Z., Nassif E.F., Erstad D.J. Targeting the MDM2-p53 pathway in dedifferentiated liposarcoma. Front. Oncol. 2022;12:1006959. doi: 10.3389/fonc.2022.1006959[↩][↩]
- Bill K.L., Casadei L., Prudner B.C., Iwenofu H., Strohecker A.M., Pollock R.E. Liposarcoma: Molecular targets and therapeutic implications. Cell. Mol. Life Sci. 2016;73:3711–3718. doi: 10.1007/s00018-016-2266-2[↩][↩]
- Dubois-Silva A., Barbagelata-Lopez C. Retroperitoneal dedifferentiated liposarcoma. Intern. Emerg. Med. 2019;14:619–620. doi: 10.1007/s11739-018-2004-x[↩][↩][↩][↩]
- Bagaria S.P., Gabriel E., Mann G.N. Multiply recurrent retroperitoneal liposarcoma. J. Surg. Oncol. 2018;117:62–68. doi: 10.1002/jso.24929[↩][↩]
- Kilpatrick S.E. Dedifferentiated Liposarcoma: A Comprehensive Historical Review with Proposed Evidence-based Guidelines Regarding a Diagnosis in Need of Further Clarification. Adv. Anat. Pathol. 2021;28:426–438. doi: 10.1097/PAP.0000000000000314[↩][↩]
- Myxoid/Round Cell Liposarcoma. https://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/rare-soft-tissue-tumors/myxoid-round-cell-liposarcoma[↩][↩]
- Scapa J.V., Cloutier J.M., Raghavan S.S., Peters-Schulze G., Varma S., Charville G.W. DDIT3 Immunohistochemistry Is a Useful Tool for the Diagnosis of Myxoid Liposarcoma. Am. J. Surg. Pathol. 2021;45:230–239. doi: 10.1097/PAS.0000000000001564[↩][↩]
- Zullow H.J., Sankar A., Ingram D.R., Samé Guerra D.D., D’Avino A.R., Collings C.K., Lazcano R., Wang W.L., Liang Y., Qi J., et al. The FUS::DDIT3 fusion oncoprotein inhibits BAF complex targeting and activity in myxoid liposarcoma. Mol. Cell. 2022;82:1737–1750.e8. doi: 10.1016/j.molcel.2022.03.019[↩][↩]
- Dolatabadi S., Jonasson E., Andersson L., Luna Santamaría M., Lindén M., Österlund T., Åman P., Ståhlberg A. FUS-DDIT3 Fusion Oncoprotein Expression Affects JAK-STAT Signaling in Myxoid Liposarcoma. Front. Oncol. 2022;12:816894. doi: 10.3389/fonc.2022.816894[↩][↩]
- Mantilla J.G., Ricciotti R.W., Chen E.Y., Liu Y.J., Hoch B.L. Amplification of DNA damage-inducible transcript 3 (DDIT3) is associated with myxoid liposarcoma-like morphology and homologous lipoblastic differentiation in dedifferentiated liposarcoma. Mod. Pathol. 2019;32:585–592. doi: 10.1038/s41379-018-0171-y[↩][↩]
- Sugita S., Hasegawa T. Practical use and utility of fluorescence in situ hybridization in the pathological diagnosis of soft tissue and bone tumors. J. Orthop. Sci. 2017;22:601–612. doi: 10.1016/j.jos.2017.02.004[↩][↩]
- Lansu J., Bovée J.V.M.G., Braam P., van Boven H., Flucke U., Bonenkamp J.J., Miah A.B., Zaidi S.H., Thway K., Bruland Ø.S., et al. Dose Reduction of Preoperative Radiotherapy in Myxoid Liposarcoma: A Nonrandomized Controlled Trial. JAMA Oncol. 2021;7:e205865. doi: 10.1001/jamaoncol.2020.5865[↩][↩]
- Myxoid liposarcoma. https://sarcoma.org.uk/about-sarcoma/what-is-sarcoma/types-of-sarcoma/liposarcoma/myxoid-liposarcoma[↩][↩]
- Hadjimichael A.C., Bekos A., Tsukamoto S., Nitta Y., Righi A., Errani C., Mavrogenis A.F. Pleomorphic Liposarcoma Revisited. Orthopedics. 2023;46:e72–e80. doi: 10.3928/01477447-20220719-05[↩][↩][↩][↩]
- Gjorgova Gjeorgjievski S, Thway K, Dermawan JK, John I, Fisher C, Rubin BP, Jenkins S, Thangaiah JJ, Folpe AL, Fritchie KJ. Pleomorphic Liposarcoma: A Series of 120 Cases With Emphasis on Morphologic Variants. Am J Surg Pathol. 2022 Dec 1;46(12):1700-1705. doi: 10.1097/PAS.0000000000001962[↩][↩]
- Anderson W.J., Jo V.Y. Pleomorphic liposarcoma: Updates and current differential diagnosis. Semin. Diagn. Pathol. 2019;36:122–128. doi: 10.1053/j.semdp.2019.02.007[↩][↩]
- Pleomorphic liposarcoma. https://sarcoma.org.uk/about-sarcoma/what-is-sarcoma/types-of-sarcoma/liposarcoma/pleomorphic-liposarcoma[↩][↩]
- Creytens D., Folpe A.L., Koelsche C., Mentzel T., Ferdinande L., van Gorp J.M., Van der Linden M., Raman L., Menten B., Fritchie K., et al. Myxoid pleomorphic liposarcoma-a clinicopathologic, immunohistochemical, molecular genetic and epigenetic study of 12 cases, suggesting a possible relationship with conventional pleomorphic liposarcoma. Mod. Pathol. 2021;34:2043–2049. doi: 10.1038/s41379-021-00862-2[↩][↩][↩][↩]
- Dermawan JK, Hwang S, Wexler L, Tap WD, Singer S, Vanderbilt CM, Antonescu CR. Myxoid pleomorphic liposarcoma is distinguished from other liposarcomas by widespread loss of heterozygosity and significantly worse overall survival: a genomic and clinicopathologic study. Mod Pathol. 2022 Nov;35(11):1644-1655. doi: 10.1038/s41379-022-01107-6[↩][↩]
- Dermawan JK. Myxoid Pleomorphic Liposarcoma. Surg Pathol Clin. 2024 Mar;17(1):25-29. doi: 10.1016/j.path.2023.06.005[↩][↩]
- Fadaei S, Cordier F, Ferdinande L, Van Dorpe J, Creytens D. Myxoid pleomorphic liposarcoma. Histol Histopathol. 2024 Sep;39(9):1101-1108. doi: 10.14670/HH-18-724[↩][↩]
- Al Kindi A.H., Al Kindi F.A., Al Riyami M., Khalil E. Giant Mediastinal Myxoid Pleomorphic Liposarcoma. Sultan Qaboos Univ. Med. J. 2023;23:271–273. doi: 10.18295/squmj.12.2022.064[↩][↩]
- Tajudeen M, Dutta S, Bheemanathi S, Anandhi A. Pictorial essay on a case of giant retroperitoneal liposarcoma. BMJ Case Rep. 2020 Dec 12;13(12):e237607. doi: 10.1136/bcr-2020-237607[↩]
- Ciongariu AM, Țăpoi DA, Dumitru AV, Bejenariu A, Marin A, Costache M. Pleomorphic Liposarcoma Unraveled: Investigating Histopathological and Immunohistochemical Markers for Tailored Diagnosis and Therapeutic Innovations. Medicina (Kaunas). 2024 Jun 7;60(6):950. doi: 10.3390/medicina60060950[↩]
- Liposarcoma. https://emedicine.medscape.com/article/1102007-overview#a5[↩]
- Abbas Manji G, Singer S, Koff A, Schwartz GK. Application of molecular biology to individualize therapy for patients with liposarcoma. Am Soc Clin Oncol Educ Book. 2015:213-8. doi: 10.14694/EdBook_AM.2015.35.213[↩][↩]
- Vocks E, Worret WI, Burgdorf WH. Myxoid liposarcoma in a 12-year-old girl. Pediatr Dermatol. 2000 Mar-Apr;17(2):129-32. doi: 10.1046/j.1525-1470.2000.01731.x[↩]
- Liposarcoma. https://rarediseases.org/rare-diseases/liposarcoma[↩][↩]
- Paredes BE, Mentzel T. Atypical lipomatous tumor/”well-differentiated liposarcoma” of the skin clinically presenting as a skin tag: clinicopathologic, immunohistochemical, and molecular analysis of 2 cases. Am J Dermatopathol. 2011 Aug;33(6):603-7. doi: 10.1097/DAD.0b013e3181f1b226[↩]
- Soft Tissue Sarcoma Stages. https://www.cancer.org/cancer/types/soft-tissue-sarcoma/detection-diagnosis-staging/staging.html[↩][↩]
- Mustacchio V, Cabibi D, Minervini MI, Barresi E, Amato S. A diagnostic trap for the dermatopathologist: granulomatous reactions from cutaneous microimplants for cosmetic purposes. J Cutan Pathol. 2007 Mar;34(3):281-3. doi: 10.1111/j.1600-0560.2006.00607.x[↩]
- Gonçales ES, Almeida AS, Soares S, Oliveira DT. Silicone implant for chin augmentation mimicking a low-grade liposarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009 Apr;107(4):e21-3. doi: 10.1016/j.tripleo.2008.12.044[↩]
- Nascimento AF, Bertoni F, Fletcher CD. Epithelioid variant of myxofibrosarcoma: expanding the clinicomorphologic spectrum of myxofibrosarcoma in a series of 17 cases. Am J Surg Pathol. 2007 Jan;31(1):99-105. doi: 10.1097/01.pas.0000213379.94547.e7[↩]
- Hogg ME, Wayne JD. Atypical lipomatous tumor/well-differentiated liposarcoma: what is it? Surg Oncol Clin N Am. 2012 Apr;21(2):333-40. doi: 10.1016/j.soc.2011.12.007[↩]
- Rutkowski P, Trepka S, Ptaszynski K, Kołodziejczyk M. Surgery quality and tumor status impact on survival and local control of resectable liposarcomas of extremities or the trunk wall. Clin Orthop Relat Res. 2013 Mar;471(3):860-70. doi: 10.1007/s11999-012-2592-0[↩]
- Riva G, Sensini M, Corvino A, Garzaro M, Pecorari G. Liposarcoma of Hypopharynx and Esophagus: a Unique Entity? J Gastrointest Cancer. 2016 Jun;47(2):135-42. doi: 10.1007/s12029-016-9808-6[↩]
- Nishida Y, Tsukushi S, Nakashima H, Ishiguro N. Clinicopathologic prognostic factors of pure myxoid liposarcoma of the extremities and trunk wall. Clin Orthop Relat Res. 2010 Nov;468(11):3041-6. doi: 10.1007/s11999-010-1396-3[↩]
- Liposarcoma. https://emedicine.medscape.com/article/1102007-overview#a2[↩]
- Wu J, Qian S, Jin L. Prognostic factors of patients with extremity myxoid liposarcomas after surgery. J Orthop Surg Res. 2019 Mar 28;14(1):90. doi: 10.1186/s13018-019-1120-2[↩]
- Chong ZX, Ho WY, Yeap SK. Deciphering the roles of non-coding RNAs in liposarcoma development: Challenges and opportunities for translational therapeutic advances. Noncoding RNA Res. 2024 Nov 15;11:73-90. doi: 10.1016/j.ncrna.2024.11.005[↩]





{kind=link}