Contents
Synovial sarcoma
Synovial sarcoma also known as synovial cell sarcoma is a rare cancer of the soft-tissues around joints in the arms or legs such as the knee, ankle or elbow in children and young adults younger than 30, but it can also occur almost anywhere in the body 1, 2, 3, 4, 5, 6. Synovial sarcoma most common locations are your hip, knee, ankle, and shoulder. Although many synovial sarcomas originate near joints, the name synovial sarcoma is a misnomer in that these cancers do not originate from intra-articular synovium per se, but from primitive mesenchymal cells 7. Synovial sarcomas most commonly present as soft tissue masses but cases of primary synovial sarcoma of bone have been reported 8. Synovial sarcomas are considered the most common soft tissue cancer of the foot 9, 10, 11.
Synovial sarcoma happens most often in older children and young adults with approximately more than half of synovial sarcoma occurring in the first two decades of life, but it can occur in older people too 12. Mean age of patients at diagnosis is approximately 30 years. The incidence of synovial sarcoma in children is 0.81 per 1,000,000 and 1.42 per 1,000,000 in adults with approximately 1000 patients diagnosed with synovial sarcoma in the United States each year 4. Synovial sarcoma affects both both males and females equally 13.
The majority (70%) of synovial sarcomas arise in the deep soft tissue of the lower and upper limbs, often around the joints 14. About 15% of synovial sarcomas arise in the trunk and 7% in the head and neck region 14. Unusual sites of synovial sarcomas include male and female external and internal reproductive organs, kidney, adrenal gland, retroperitoneum, stomach, small bowel, lung, heart, mediastinum, bone, brain, spinal cord and peripheral nerve.
Synovial sarcoma typically causes symptoms like a swelling (sometimes longstanding), which may be painful. The initial growth of synovial sarcoma is often slow, and a small circumscribed tumor may give the wrong impression of a benign lesion by clinical examination and imaging. Synovial sarcoma may have radiologically detectable stippled or spiculated forms of calcification. Synovial sarcoma with aggressive growth may erode or invade adjacent bone.
Synovial sarcoma symptoms can be slow to develop and may include:
- A lump or swelling, often painless at first. The first symptom of synovial sarcoma is usually a swelling or a lump under the skin. The lump may or may not hurt.
- Joint stiffness.
- Pain in the area of the tumor.
- Numbness or tingling when synovial sarcoma involves the nerve.
- Difficulty moving or a limited range of motion.
Synovial sarcoma is usually slow growing, so it can be years before a diagnosis is made. Given the insidious onset, younger age at presentation and atypical presenting symptoms, patients with synovial sarcoma may be initially clinically misdiagnosed with benign processes including myositis (muscle inflammation), synovitis (inflammation of the synovial membrane, a tissue lining the joint capsule that produces lubricating fluid), bursitis (inflammation of a bursa, a small, fluid-filled sac that cushions joints and reduces friction between bones, muscles, and tendons) or tendonitis (inflammation of a tendon) 1.
The exact cause of synovial sarcoma is unknown, but it is characterized by a specific genetic abnormality known as the translocation between chromosome X and 18 written as t(X;18), which fuses the SYT gene from chromosome 18 with the SSX1 (about 2 of 3 of cases), SSX2 (about 1 of 3 of cases), or SSX4 (rare cases) genes at Xp11.2 from the X chromosome and the production of SS18-SSX oncogenic fusion proteins which drive sarcomagenesis or sarcoma development 15, 16, 17, 18. This change in DNA can cause cells to grow uncontrollably, forming a tumor. Younger age is a risk factor for synovial sarcoma. And there is no way to prevent synovial sarcoma.
Synovial sarcoma diagnosis involves imaging scans such as CT or MRI scans to get a detailed view of the tumor and its extent and a biopsy to confirm the diagnosis.
The primary treatment for synovial sarcoma is surgery to remove the tumor, often with a margin of healthy tissue. The goal is to remove the cancer and some of the healthy tissue around it. This can sometimes mean the removal of an entire muscle or muscle group. In the past, surgery might have included removing an arm or leg, known as amputation. But medical advances have made amputation less likely. To lower the chances of the cancer returning, radiation therapy or chemotherapy might be used as well.
Other treatments, depending on the stage of the cancer, may include:
- Radiation therapy. Radiation therapy treats cancer with powerful energy beams such as x-rays, gamma rays, electron beams, or protons to destroy or damage cancer cells. During radiation therapy, you lie on a table while a machine moves around you. The machine directs radiation to precise points on the body. Radiation before surgery can shrink the cancer and make a successful surgery more likely. Radiation therapy after surgery can kill cancer cells that might still be there.
- Chemotherapy. Chemotherapy treats cancer with strong medicines to destroy cancer cells. For synovial sarcoma, chemotherapy might be used before or after surgery. It also may be used when cancer has spread to other parts of the body.
- Targeted therapy. Targeted therapy uses medicines that attack specific molecular targets within or on cancer cells, such as genes or proteins. This can cause cancer cells to die or stop growing. Targeted therapy medicines are being studied for advanced synovial sarcoma.
- Cell therapy. Cell therapy helps the immune system find and stop the cancer cells. This treatment involves taking some of your immune system cells and making them better at recognizing the cancer cells. Then the cells are put back in your body. This treatment can take months to set up. One cell therapy used for synovial sarcoma is afamitresgene autoleucel (Tecelra). It might be an option for treating advanced synovial sarcoma that hasn’t been helped by chemotherapy. Afamitresgene autoleucel (Tecelra), is an autologous T-cell receptor gene therapy that uses the patient’s own T-cells to treat advanced synovial sarcoma that cannot be surgically removed or has spread to other parts of the body 19. The T-cells are modified in the lab to recognize and target the MAGE-A4 antigen, which is present on some cancer cells. These modified T-cells are then grown in large numbers and infused back into the patient to help their immune system attack and destroy the cancer. Afamitresgene autoleucel received accelerated approval from the U.S. Food and Drug Administration (FDA) in August 2024, making it the first FDA-approved engineered cell therapy for solid tumors and the first new therapy for synovial sarcoma in over a decade.
- Clinical trials. Clinical trials are studies of new treatments. These studies provide a chance to try the latest treatment options. The side effects may not be known. Ask your healthcare team if there is a clinical trial available to take part in.
After treatment, you or your child will be monitored closely and will have regular follow-up appointments to check for symptoms and side-effects.
The follow-ups will usually include:
- A chance to discuss your symptoms
- An examination to look for any signs of synovial sarcoma returning. This may include scans such as CT, ultrasound or MRI scans
- A chest x-ray to rule out any secondary cancers occurring in the lungs
If you have any specific questions or concerns about you or your child’s treatment and follow up, you should contact your doctor.
Although synovial sarcoma has a similar clinical presentation in children and adults, there is a growing evidence that they have different prognosis (outcome), with children having significantly better survival rates 4, 5, 20. Using the registry data, Sultan et al 4 demonstrated the 5-year survival rate for children and adolescents to be 83% compared to 62% in adults. Similarly, Smolle et al 5 demonstrated an 89% five-year cancer specific survival rate in children compared to 75% in adults. Vlenterie et al 21 demonstrated a clear stepwise reduction in survival with age, regardless of tumor site, size and treatment.
Figure 1. Synovial sarcoma wrist
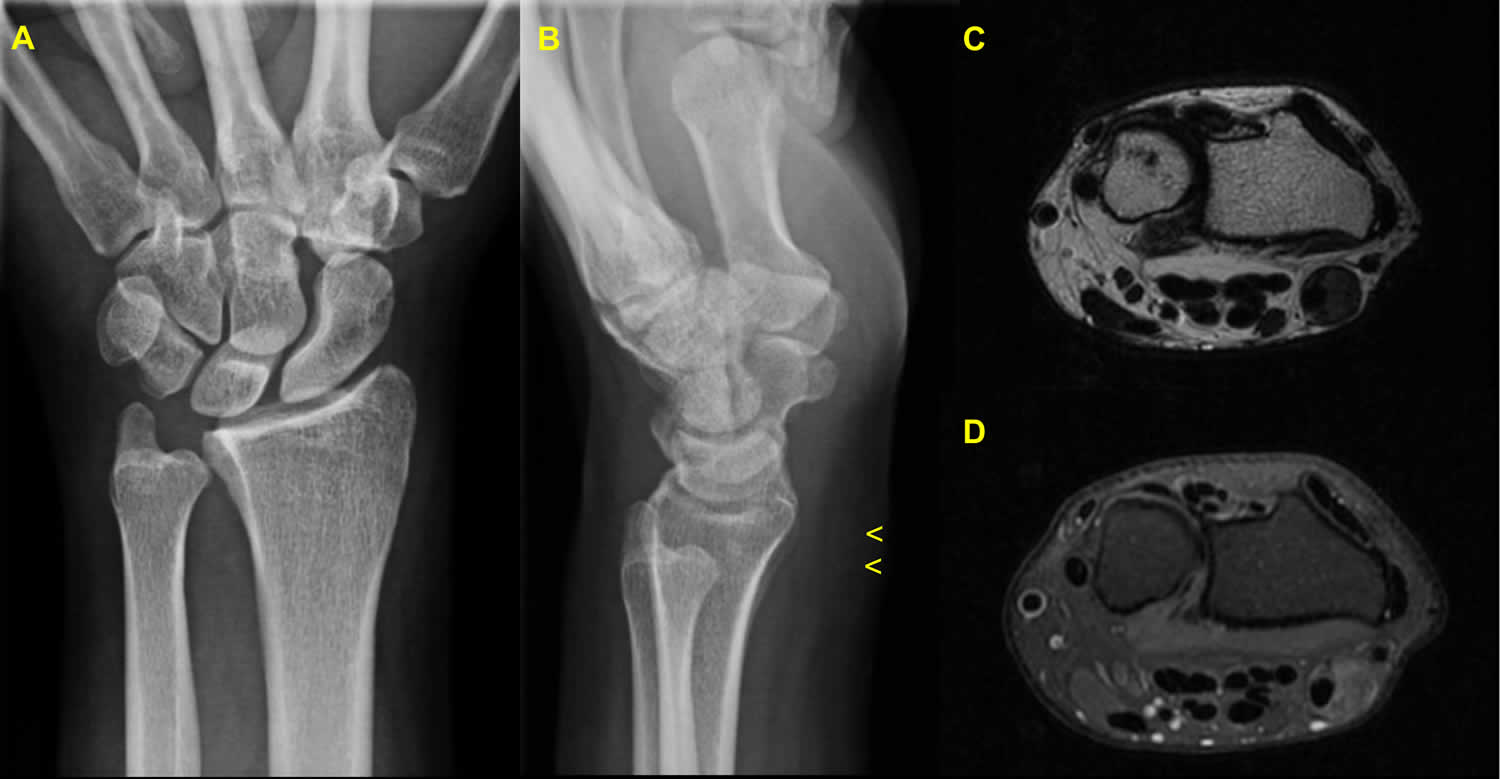
Footnotes: A 45-year-old male with synovial sarcoma in the left wrist. AP left wrist computed radiography (A) shows no significant abnormality. Lateral left wrist computed radiography (B): yellow arrowheads demonstrate a slight soft-tissue enlargement with no mineralization. A well-defined solid lesion on axial (C) T1 weighted image and axial T2 with fat-saturation (T2FS) weighted image of MRI.
[Source 11 ]Figure 2. Synovial sarcoma knee
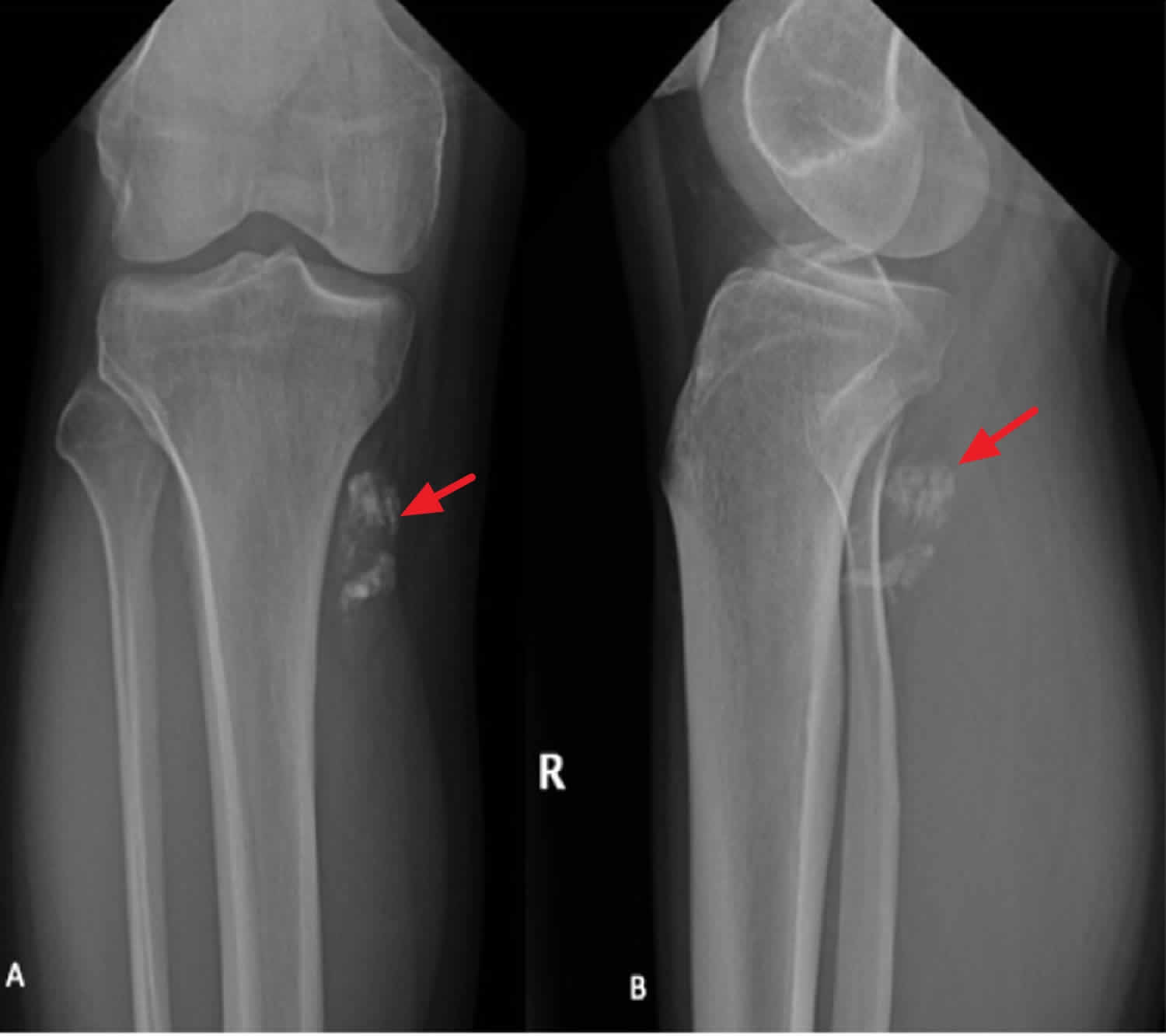
Footnotes: Plain X-ray anterior-posterior (AP) (A) and lateral (B) radiographs of the right (R) knee and lower leg in a 31-year-old male demonstrate coarse calcifications within the soft tissues adjacent to the posteromedial tibial plateau, corresponding to a biopsy proven synovial sarcoma. No significant articular abnormality. Adjacent bony structures appear unremarkable.
[Source 1 ]Figure 3. Synovial sarcoma knee MRI scan
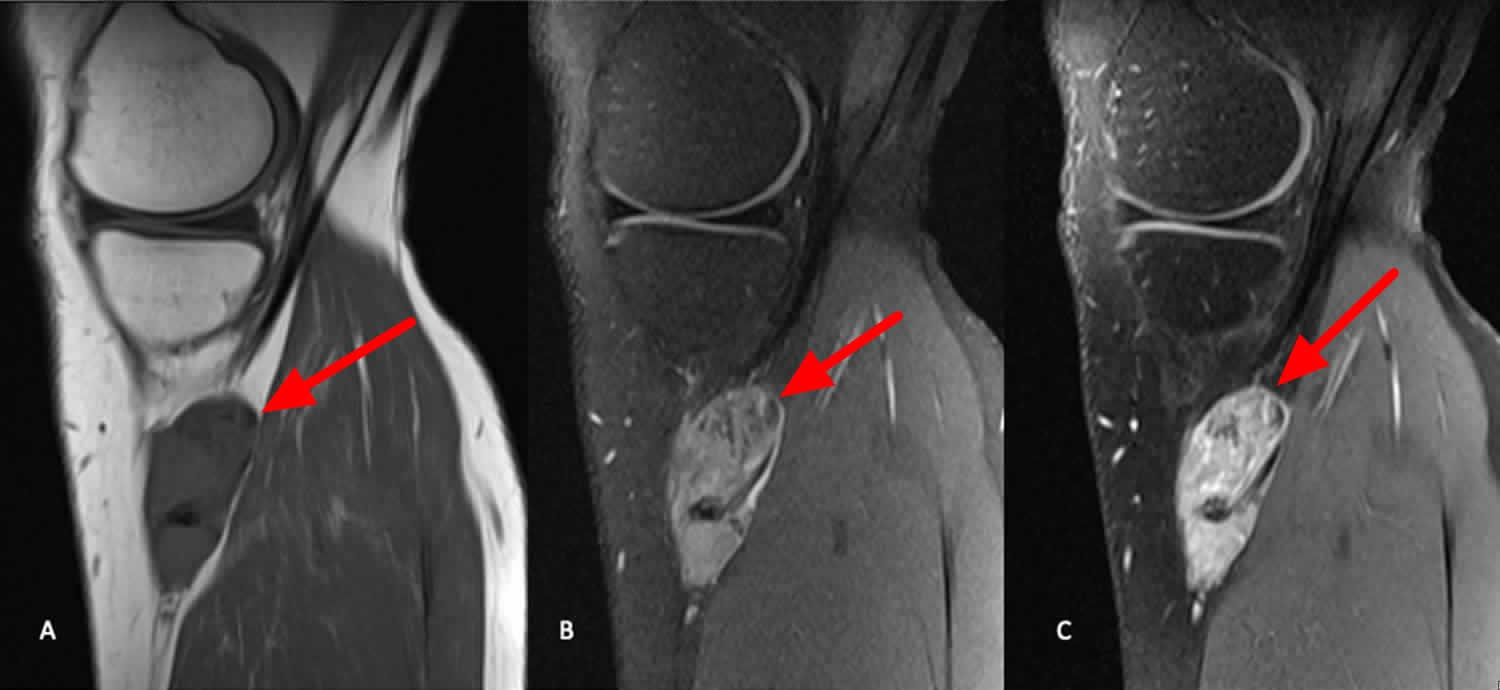
Footnotes: Sagittal MR images in the patient from Figure 2 above demonstrating a periarticular soft tissue mass situated posteromedial to the proximal tibia, in close relation to the pes anserine tendons. The mass demonstrates intermediate signal on T1 weighted images (A), and heterogeneously high signal on T2 weighted fat-saturated images (B). T1 weighted fat-saturated after gadolinium administration (C), demonstrates heterogeneously avid enhancement. Known areas of calcification are low signal on all sequences (arrow).
[Source 1 ]Figure 4. Synovial sarcoma leg
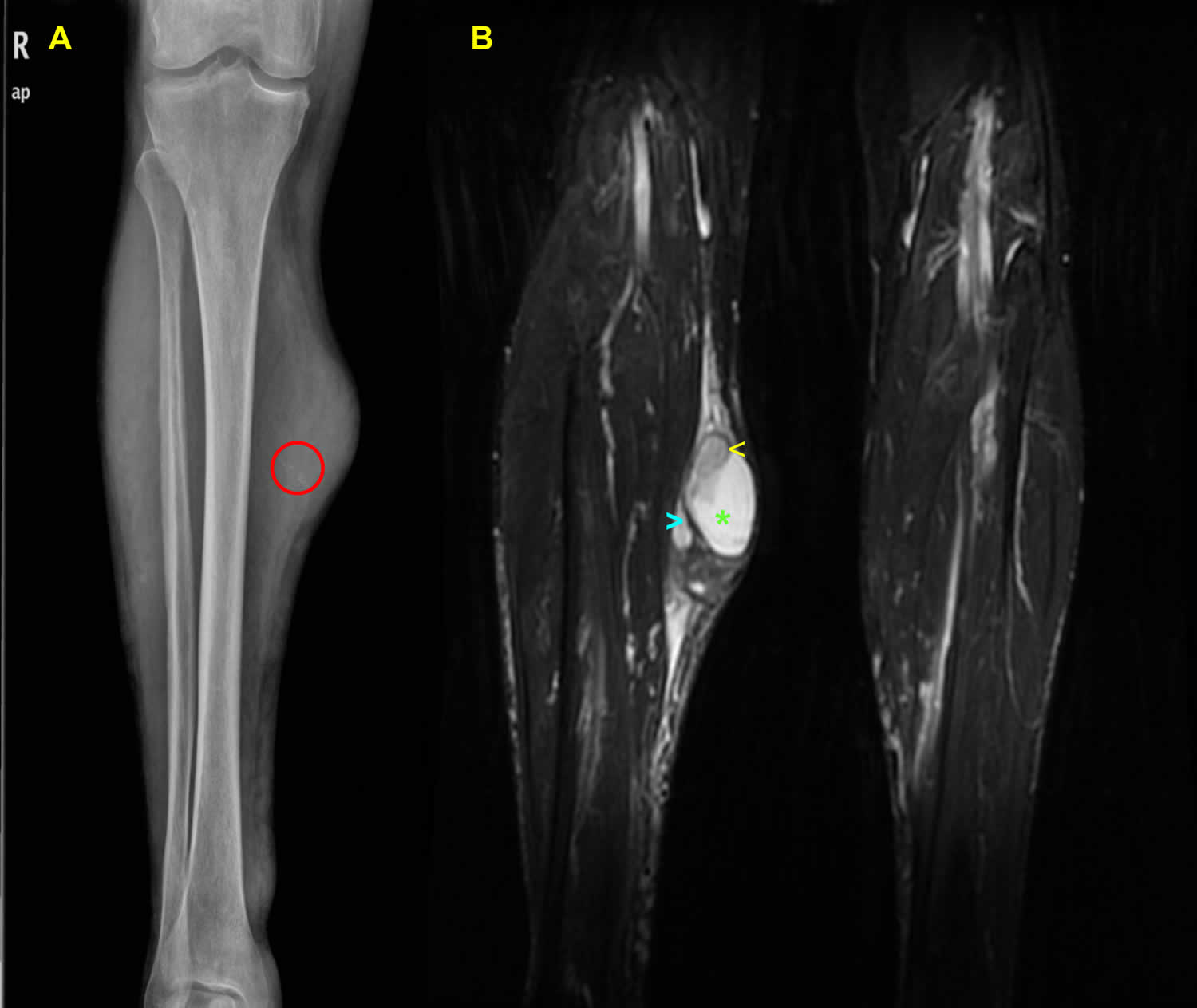
Footnotes: A 67-year-old male with synovial sarcoma in the right calf. Anterior-posterior (AP) right calf computed radiography (A) shows dystrophic calcification (red circled) in a soft-tissue mass. MRI T2 fat-saturation weighted image (B) demonstrate the so-called triple signal pattern alternating high (green asterisk), intermediate (yellow arrowhead) and low signal intensity septations (blue arrowhead).
[Source 11 ]Figure 5. Synovial sarcoma knee
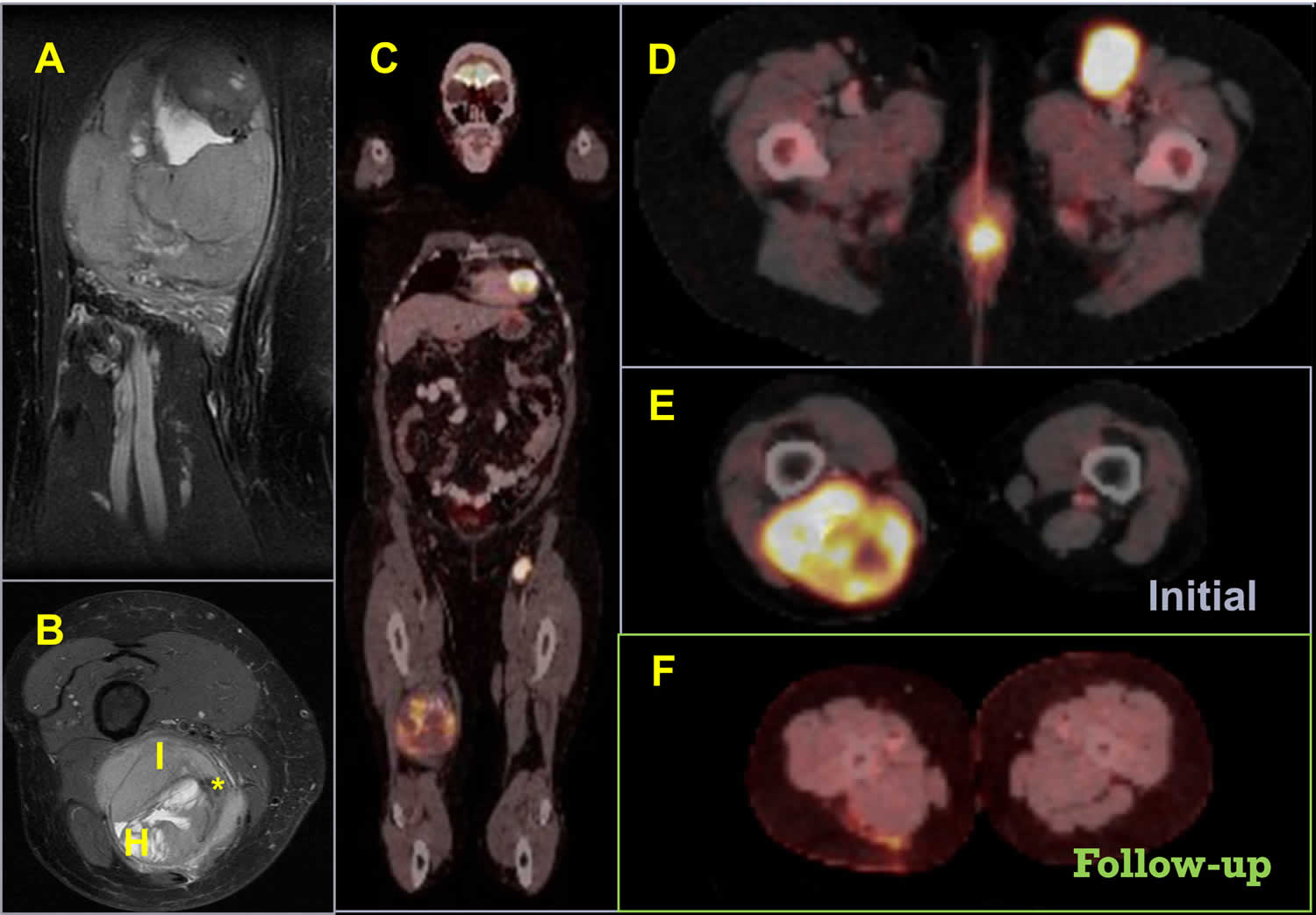
Footnotes: Synovial sarcoma in right popliteal fossa, a diamond-shaped, shallow depression located at the back of the knee joint that serves as a crucial passage for nerves and blood vessels between the thigh and leg. Coronal (A) and axial (B) T2-weighted imaging (T2WI) on MRI, heterogeneity of the lesion is shown with areas of hipo (*), iso (I) and hiperintensity (H) (triple sign). On initial PET-CT scan, high standardized uptake value (SUV) in popliteal fossa (C, E) with lymph node invasion biopsy proven. 3 months after chemotherapy (F), PET-CT scan confirmed good response.
[Source 11 ]Synovial sarcoma causes
The exact cause of synovial sarcoma is unknown, but it is characterized by a specific genetic abnormality known as the translocation between chromosome X and 18 written as t(X;18), which fuses the SYT gene from chromosome 18 with the SSX1 (about 2 of 3 of cases), SSX2 (about 1 of 3 of cases), or SSX4 (rare cases) genes at Xp11.2 from the X chromosome and the production of SS18-SSX oncogenic fusion proteins which drive sarcomagenesis or sarcoma development 15, 16, 17, 18. This change in DNA can cause cells to grow uncontrollably, forming a tumor. Younger age is a risk factor for synovial sarcoma. And there is no way to prevent synovial sarcoma.
Although many synovial sarcomas originate near joints, the name synovial sarcoma is a misnomer in that these cancers do not originate from intra-articular synovium per se, but from primitive mesenchymal cells 7. Synovial sarcomas most commonly present as soft tissue masses but cases of primary synovial sarcoma of bone have been reported 8. Synovial sarcomas are considered the most common soft tissue cancer of the foot 9, 10, 11.
The risk factors of synovial sarcoma remain unclear. Researchers have found that in radiotherapy of other cancers, specific inherited gene defects can increase the possibility of getting synovial sarcoma 22. Furthermore, other studies suggested that synovial sarcoma can be derived from undifferentiated cells, neural crest stem cells, pluripotent mesenchymal cells, and other parts of the body 23, 24, 25, 26.
Synovial sarcoma signs and symptoms
Signs and symptoms of synovial sarcoma depend on where the cancer starts. Most people notice a painless lump or bump that slowly gets bigger with the mean duration of symptoms before diagnosis is approximately 2 years 27, 5. The lump usually starts near the knee or ankle, but it can appear on any part of the body.
Synovial sarcoma symptoms may include:
- A lump or bump under the skin that slowly gets bigger.
- Joint stiffness.
- Pain.
- Swelling.
Synovial sarcoma that happens in the head or neck can cause other symptoms. These may include:
- Problems breathing.
- Difficulty swallowing.
- Changes in the way the voice sounds.
In comparison to other soft tissue sarcoma (cancer that forms in the soft tissues of the body), the duration of symptoms are long and patients may have pain or joint contractures that precede swelling 28. Synovial sarcoma is usually slow growing, so it can be years before a diagnosis is made. Given the insidious onset, younger age at presentation and atypical presenting symptoms, patients with synovial sarcoma may be initially clinically misdiagnosed with benign processes including myositis (muscle inflammation), synovitis (inflammation of the synovial membrane, a tissue lining the joint capsule that produces lubricating fluid), bursitis (inflammation of a bursa, a small, fluid-filled sac that cushions joints and reduces friction between bones, muscles, and tendons) or tendonitis (inflammation of a tendon) 1.
Synovial sarcoma complications
Compression of the nearby structures due to the “mass effect” of the growing tumor can lead to various compressive issues. Synovial sarcoma rarely metastasizes to the brain and can lead to seizures and other neurologic complications 2. Blood clot-formation can occur due to compressive effects on blood vessels or secondary to the presence of a tumor in the body. The complications from the use of chemotherapy include bone marrow suppression (a condition where the bone marrow produces fewer blood cells such as red blood cells, white blood cells, and platelets than normal can lead to anemia (fatigue), increased risk of infection (neutropenia), and bleeding problems due to low platelets), hair loss, nausea, vomiting, and dysgeusia (impaired sense of taste) 2.
Many adverse events are specific to the class of the drug used to treat synovial sarcoma. For example, anthracycline can cause heart complications, ifosfamide can cause nerve damage and kidney damage, pazopanib can cause heart and liver damage, and trabectedin can lead to heart, liver, and bone marrow toxicity.
Synovial sarcoma diagnosis
Synovial sarcoma is usually slow growing, so it can be years before a diagnosis is made. Sometimes, synovial sarcoma is diagnosed in error as a joint problem, such as arthritis or bursitis.
Tests and procedures used to diagnose synovial sarcoma include:
- Imaging tests. Imaging tests take pictures of the body. They can show where a synovial sarcoma is, how large it is and if it has spread to other areas of the body. Tests for synovial sarcoma might include MRI scans, X-rays and CT scans.
- Plain X-rays. Plain radiographs are not required for diagnosis but are typically performed as part of the initial workup and can identify adjacent bony remodeling, bone invasion or calcification of the soft tissue mass. Typically, synovial sarcoma presents as a well-defined or lobulated soft tissue mass on plain radiographs. Punctate calcifications, particularly around the periphery of the lesion, are visualized in one third of patients 29. Occasionally, more extensive calcification can be visualized and can mimic bone forming tumors including osteosarcoma and myositis ossificans 29.
- Magnetic resonance imaging (MRI) with and without contrast is the gold standard for diagnostic imaging for synovial sarcoma 29. MRI defines the local extent of the soft tissue mass and surrounding edema and provides excellent visualization of the mass with respect to the surrounding anatomy, which is critical for preoperative planning. The utilization of gadolinium contrast can differentiate between hemorrhagic or necrotic areas and areas of solid viable tumor. As with most soft tissue sarcoma, synovial sarcomas are typically heterogenous with low intensity on T1 and high intensity on T2-weighted images with post-gadolinium enhancement 29. Although synovial sarcomas generally present as a non-specific heterogenous mass, there are some unique features, which can aid in differentiation from other soft tissue sarcoma. Synovial sarcomas predominately present as well-defined, heterogeneously enhancing solid tumors that are multilobulated in nature 29. A triple signal intensity demonstrating areas of hyperintensity, isointensity and hypointensity indicating the mix of cystic and hemorrhagic areas, cellular elements and fibrotic areas can be characteristic 29. Smaller tumors, particularly those smaller than 5 cm in diameter, often show homogeneous enhancement which can be mistaken for a benign process 30. Several findings on MRI have been found to be predictive of high-grade lesions including the absence of calcifications and presence of hemorrhage and the triple signal intensity 31.
- Computed tomography (CT) with contrast can be utilized when an MRI is contraindicated or unavailable. Synovial sarcoma appears hypointense compared to muscle with heterogeneity in larger lesions 32. CT allows for better visualization of soft-tissue calcifications and local bone reaction 29.
- Biopsy. A biopsy is a procedure to remove a sample of tissue for testing in a lab. The tissue might be removed using a needle that is put through the skin and into the cancer 33. Options for biopsy include incisional biopsies, core needle biopsies and fine needle aspirations (FNA). Sometimes surgery is needed to get the tissue sample. The sample is tested in a lab to see if it is cancer. Other special tests give more details about the cancer cells. Your healthcare team uses this information to make a treatment plan. Historically, open incisional biopsies have been considered the gold standard for soft tissue lesions as they provide larger volumes of tissue. When compared to core needle biopsy and fine needle aspirations (FNA), incisional biopsy tend to have higher diagnostic accuracy but this comes with a higher rates of complications when compared to using a needle that is put through the skin and into the cancer techniques 34. Core needle biopsies retrieve more tissue than fine needle aspirations (FNA) and have higher diagnostic accuracy 35. Fine needle aspirations (FNA) are rarely used in soft tissue sarcoma due to the small quantity of sample material obtained and a limited ability to assess lesional architecture 34. Advances in diagnostic imaging has allowed for image guided percutaneous biopsies which has improved the diagnostic accuracy of these techniques 36. Given the lower morbidity and relatively high diagnostic accuracy of core needle biopsies, image guided core needle biopsies are the preferred method of biopsy, particularly for deeper tumors. When open biopsies are performed, the biopsy principles must be observed to reduce biopsy-related complications 37.
- Histologic subtypes. According to the International Classification of Diseases for Oncology, synovial sarcoma is divided into 3 distinct histologic subtypes: monophasic synovial sarcoma, biphasic synovial sarcoma, and poorly differentiated synovial sarcoma 38. Figure 6 below shows hematoxylin and eosin (H & E) staining of the three histologic variants of synovial sarcoma. Other synovial sarcoma variants include myxoid synovial sarcoma and ossifying/calcifying synovial sarcoma 39, 40.
- Monophasic synovial sarcoma contains uniform spindle cells.
- Biphasic synovial sarcoma consists of epithelial cells arranged into glandular structures with spindle cells arranged into fascicles, and spindles
- Poorly differentiated synovial sarcoma consists of round blue cells 41.
- The monophasic subtype and the biphasic subtype are the two most common subtypes, while the epithelioid cell subtype is rare. Histologically, synovial sarcoma comprised varying proportions of spindle and epithelial cell components. Due to the variable cellular and architectural morphology and resemblance to other neoplastic processes common to the region, the histopathological diagnosis of synovial sarcoma is very challenging 42. Immunohistochemistry plays a crucial role in histological diagnosis. synovial sarcoma is positive for epithelial markers, including cytokeratin, epithelial membrane antigen (EMA), and vimentin. synovial sarcoma is usually unfavorable for CD34 and FLI-1 43.
- Molecular Pathology. Synovial sarcoma is characterized by a pathognomonic translocation t(X:18) which is present in >95% of cases 44. The t(X:18) translocation leads to the expression of different SS18:SSX oncogenic fusion proteins, which drive sarcomagenesis or sarcoma development. Subtypes include SS18:SSX1 and SS18:SSX2 and less commonly SS18:SSX4 15. Both fluorescence in situ hybridization (FISH) and reverse transcription polymerase chain reaction (RT-PCR) testing have been validated in the diagnosis of this translocation 45. Almost all SS18-SSX2 synovial sarcomas show monophasic morphology and are significantly more common in women. Rare cases are associated with t(X;20) and SS18L1-SSX1 fusion transcript 46.
Figure 6. Synovial sarcoma histologic subtypes
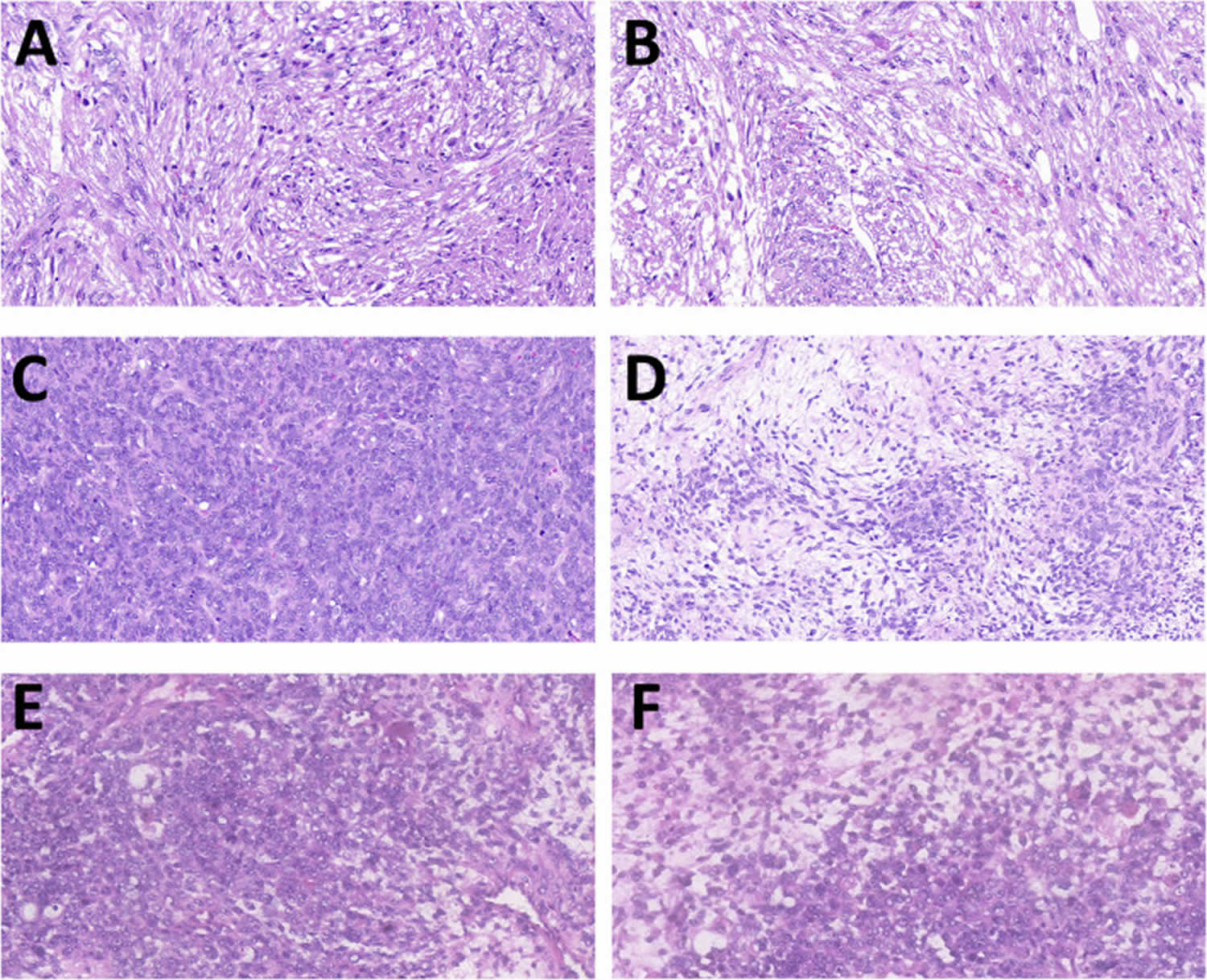
Footnotes: Hematoxylin and eosin staining of the three histologic variants of synovial sarcoma. (A,B) Slides of monophasic synovial sarcoma, made of spindle cells with moderate cytologic atypia and differentiated areas of variable cellularity. (C,D) Slides of biphasic synovial sarcoma composed by glandular-like structures with spindle cells without nuclear atypia. (E,F) Slides of poorly differentiated synovial sarcoma in its Ewing-like variant, with the presence of rosette-like structures (magnification: ×20).
[Source 6 ]Synovial sarcoma Stages
After someone is diagnosed with a synovial sarcoma, doctors will try to figure out if it has spread, and if so, how far. This process is called staging. The stage of a cancer describes how much cancer is in the body. It helps determine how serious the cancer is and how best to treat it. Doctors also use a cancer’s stage when talking about survival statistics.
The stages of synovial sarcoma range from stages I (1) through IV (4). As a rule, the lower the number, the less the cancer has spread. A higher number, such as stage IV, means cancer has spread more. And within a stage, an earlier letter means a lower stage. Although each person’s cancer experience is unique, cancers with similar stages tend to have a similar outlook and are often treated in much the same way.
The staging system most often used for soft tissue sarcomas is the American Joint Committee on Cancer (AJCC) TNM system, which is based on 4 key pieces of information:
- The extent of the tumor (T): How large is the cancer?
- The spread to nearby lymph nodes (N): Has the cancer spread to nearby lymph nodes?
- The spread (metastasis) to distant sites (M): Has the cancer spread to distant organs such as the lungs?
- The grade (G) of the cancer: How much do the sarcoma cells look like normal cells?
The grade is partly used to determine the stage of a sarcoma. The staging system divides sarcomas into 3 grades (1 to 3). The grade of a sarcoma helps predict how rapidly it will grow and spread. It’s useful in predicting a patient’s outlook and helps determine treatment options.
The grade of a sarcoma is determined using a system known as the French or FNCLCC (Fédération Nationale des Centres de Lutte Contre le Cancer) system, and is based on 3 factors:
- Differentiation: Cancer cells are given a score of 1 to 3, with 1 being assigned when they look a lot like normal cells and 3 being used when the cancer cells look very abnormal. Certain types of sarcoma are given a higher score automatically.
- Mitotic count: How many cancer cells are seen dividing under the microscope; given a score from 1 to 3 (a lower score means fewer cells were seen dividing). 0-9 mitosis/hpf = 1 pt, 10-19 mitosis/hpf = 2pts, ≥20 mitosis/hpf = 3 pts
- Tumor necrosis: How much of the tumor is made up of dying tissue; given a score from 0 to 2 (a lower score means there was less dying tissue present). No necrosis = 0 pts, less than 50% necrosis = 1pt, or ≥50% necrosis = 2 pts), differentiation (resembling normal adult mesenchymal tissue – 1pt, Sarcomas with definite histologic typing – 2pts, and undifferentiated – 3pts), and
Each factor is given a score, and the scores are added to determine the grade of the tumor. Sarcomas that have cells that look more normal and have fewer cells dividing are generally placed in a low-grade category. Low-grade tumors tend to be slow growing, slower to spread, and often have a better outlook (prognosis) than higher-grade tumors. Certain types of sarcoma are automatically given higher differentiation scores. This affects the overall score so much that they are never considered low grade. Examples of these include synovial sarcomas and embryonal sarcomas. Here’s what the grade numbers mean:
- GX: The grade cannot be assessed (because of incomplete information).
- Grade 1 (G1): Total score of 2 or 3
- Grade 2 (G2): Total score of 4 or 5
- Grade 3 (G3): Total score of 6, 7 or 8
Although sarcomas can be divided between stages I to IV as per the AJCC (American Joint Committee on Cancer) guidelines, they are either localized (stages 1 to 3) or metastatic (Stage 4). This oversimplified staging is essential in determining if the tumor is resectable (able to be completely removed by surgery) or not.
Table 1. Trunk and Extremities Sarcoma Stages
| AJCC stage | Stage grouping | Trunk and Extremities Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm (T2) OR Larger than 10cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IV | Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. |
| OR | ||
| Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. | |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Table 2. Retroperitoneum Sarcoma Stages
| AJCC stage | Stage grouping | Retroperitoneum Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm OR Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| OR | ||
| Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. | |
| IV | Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Synovial sarcoma treatment
Treatment of synovial sarcoma will depend on the type, location, and stage of the cancer, as well as your overall physical health 33. A patient with synovial sarcoma can either present with localized or metastatic disease. The only way to cure synovial sarcoma is to remove it with surgery, so surgery is part of the treatment for all synovial sarcomas whenever possible 48.
Treatment options for synovial sarcoma include:
- Surgery. Surgery is the main treatment for synovial sarcoma. The goal is to remove the cancer and some of the healthy tissue around it. This can sometimes mean the removal of an entire muscle or muscle group. In the past, surgery might have included removing an arm or leg, known as amputation. But medical advances have made amputation less likely. To lower the chances of the cancer returning, radiation therapy or chemotherapy might be used as well.
- Radiation therapy. Radiation therapy treats cancer with powerful energy beams such as x-rays, gamma rays, electron beams, or protons to destroy or damage cancer cells. During radiation therapy, you lie on a table while a machine moves around you. The machine directs radiation to precise points on the body. Radiation before surgery can shrink the cancer and make a successful surgery more likely. Radiation therapy after surgery can kill cancer cells that might still be there.
- Chemotherapy. Chemotherapy treats cancer with strong medicines to destroy cancer cells. For synovial sarcoma, chemotherapy might be used before or after surgery. It also may be used when cancer has spread to other parts of the body.
- Targeted therapy. Targeted therapy uses medicines that attack specific molecular targets within or on cancer cells, such as genes or proteins. This can cause cancer cells to die or stop growing. Targeted therapy medicines are being studied for advanced synovial sarcoma.
- Cell therapy. Cell therapy helps the immune system find and stop the cancer cells. This treatment involves taking some of your immune system cells and making them better at recognizing the cancer cells. Then the cells are put back in your body. This treatment can take months to set up. One cell therapy used for synovial sarcoma is afamitresgene autoleucel (Tecelra). It might be an option for treating advanced synovial sarcoma that hasn’t been helped by chemotherapy. Afamitresgene autoleucel (Tecelra), is an autologous T-cell receptor gene therapy that uses the patient’s own T-cells to treat advanced synovial sarcoma that cannot be surgically removed or has spread to other parts of the body 19. The T-cells are modified in the lab to recognize and target the MAGE-A4 antigen, which is present on some cancer cells. These modified T-cells are then grown in large numbers and infused back into the patient to help their immune system attack and destroy the cancer. Afamitresgene autoleucel received accelerated approval from the U.S. Food and Drug Administration (FDA) in August 2024, making it the first FDA-approved engineered cell therapy for solid tumors and the first new therapy for synovial sarcoma in over a decade.
- Clinical trials. Clinical trials are studies of new treatments. These studies provide a chance to try the latest treatment options. The side effects may not be known. Ask your healthcare team if there is a clinical trial available to take part in.
Generally, a localized synovial sarcoma is approached to achieve a microscopic negative margin (R0 resection) surgically 2. Where neoadjuvant chemotherapy (chemotherapy treatment given before surgery) is widely used in children, the use of neoadjuvant chemotherapy is controversial in adults 2.
Treating patients with locally advanced unresectable or metastatic synovial sarcoma aims at palliation and prolonging life 2. The therapy involves using chemotherapy, tyrosine kinase inhibitors (targeted therapy), or enrolling the patient in a clinical trial 2.
Surgery
Limb-sparing surgery also called limb-salvage surgery is the mainstay of treatment for patients with synovial sarcoma who present with localized disease. Amputation is rarely pursued and is reserved for patients where limb salvage surgery is impossible. The goal is to achieve a negative surgical margin or R0 margin of at least 1 to 2 cm in width 49. R0 margin (negative surgical margin) signifies a complete macroscopic and microscopic resection of the tumor, meaning no gross or microscopic tumor cells are found at the surgical margins after pathological examination. R0 margins (negative surgical margins) are of upmost importance as they predict both local recurrence and overall survival 50, 51, 52. While no specific guidelines exist regarding ideal negative margins in synovial sarcoma, surgical management is similar to that of other soft tissue sarcoma. For superficial tumors or small (<5 cm) deep tumors not intimately associated with critical structures, a wide excision with negative margins (1–2 cm) alone could be considered sufficient 20. In tumors that are closely associated with neurovascular structures or bone, the epineurium, adventitia or periosteum is utilized as the margin to allow for a functional limb postoperatively 53, 52. In these cases, very close or microscopically positive margins may occur and radiotherapy is essential to decrease local recurrence risk 54, 55. Carefully planned microscopically positive margins on a fixed structure (bone, nerve, blood vessel) have been shown to not increase the risk for local recurrence in setting of neoadjuvant radiation therapy (a cancer treatment that uses radiation therapy before surgery to shrink tumors, improve the chances of successful removal, and potentially make the surgery less invasive) 55. Given the heterogeneity and lack of standardization in the literature, recommendations regarding margin management must be taken into context of the individual patient. Both tumor and anatomic factors should be taken into consideration when determining acceptable surgical margins 56.
Alternatively, a fascial plane uninvolved by the tumor is an acceptable margin, regardless of the distance of the tumor from the fascia.
Due to its atypical presentation (slow-growing, painful mass), synovial sarcomas have a high rate of presentation following an unplanned excision, with up to 50% of patients presenting after unplanned excision 27, 57. Unplanned excisions of synovial sarcomas result in high rates of residual disease, particularly for larger and deeper tumors and increased risk for local recurrence even after re-excision 57, 58. In the case of referral following unplanned excision, patients should be re-staged, and the original histology reviewed at a sarcoma referral center. Tumor bed excision should be performed in patients with residual disease with the goal of complete tumor resection and negative margins 59. Typically, these resections are extensive in nature given that areas of potential contamination must be removed, with may necessitate reconstructive procedures 58. There is a lack of data to guide the utilization of radiotherapy in this population; however, radiotherapy is recommended as it would be in a primary presentation 59.
In select cases, limb-salvage techniques are not recommended and limb amputation may be required 48. Amputation is considered in patients with tumor location that necessitates excision of vital structures which would result in poor limb function 48. Patients who present following an unplanned excision may require limb amputation if there is extensive contamination of vital structures or major joints 48. Finally, older patients or those with extensive medical comorbidities may not be able to tolerate a major operation and limb amputation can be considered 48.
Radiation therapy
In patients with localized synovial sarcoma, radiation therapy is used either in the preoperative or postoperative setting. In patients with metastatic or those with locally advanced, unresectable tumors, radiation therapy can be given for palliative purposes (pain relief, cord compression) 60, 61.
Neoadjuvant (preoperative) or adjuvant (given after surgery) radiation therapy is recommended for larger tumors (>5 cm), or in any case where a close margin may be required to preserve a major neurovascular structure or bone 33. In large registry database studies, radiotherapy has been shown to improve local control and may have overall survival benefit in patients with synovial sarcoma 62, 63, 64. Radiation therapy can be administered pre or postoperatively with differing protocols. Preoperative (neoadjuvant) radiation therapy is associated with higher wound complication rates whereas postoperative (adjuvant) radiation therapy can case fibrosis and joint stiffness which may lead to worse long term functional outcomes 65. Regardless of timing, intensity-modulated radiation therapy (IMRT) is becoming the preferred method of radiation delivery in patients with soft tissue sarcoma 66, 33, 67. Intensity-modulated radiation therapy (IMRT) allows for a higher dose of radiation to more closely contour the tumor, which reduces the volume of radiation to the surrounding normal tissues. The utilization of preoperative intensity-modulated radiation therapy (IMRT) for soft tissue sarcoma has been shown to reduce wound complications and need for reconstructive soft-tissue flaps 68. Radiation therapy may be considered in isolation in patients with multiple medical comorbidities or patients with metastatic disease where the risks of surgery outweigh the potential benefits 69.
Localized Synovial Sarcoma
Intensity-modulated radiation therapy (IMRT) has become the preferred choice for delivering radiation 66.
Preoperative radiation therapy (Neoadjuvant radiation therapy): Delivered prior to surgery targeting a vascularized, well-oxygenated tumor which helps in the generation of free oxygen radicals.
- Advantages
- Smaller target and the need for a lower dose of radiation therapy (usually 50 Gy over 25 fractions)
- The option of delivering intra-operative radiation therapy or postoperative boost remains open (in case of R1 resection), and so does the prospect of postoperative radiation therapy in case of an R2 resection (gross residual disease).
- Long-term side effects are fewer.
- Disadvantages
- Wound healing can be compromised/ delayed.
- Potential delay in surgery (3 to 6 weeks cooling period after finishing radiation therapy)
Postoperative radiation therapy (Adjuvant radiation therapy)
- Advantages
- Lesser wound healing complications
- Radiation therapy dose can be tailored according to histology
- Disadvantages
- A higher dose is needed for long-term disease control (Up to 76 Gy may be required in patients with R2 resection)
- A wider area is irradiated.
- There is a higher risk of long-term complications like pathological fracture, lymphedema, fibrosis, and secondary sarcoma.
Systemic therapies
Systemic therapy is reserved for patients with metastatic or locally advanced unresectable synovial sarcoma and is thought to be more effective in younger patients 70, 71. Synovial sarcoma appears to be more chemosensitive, although there is still controversy surrounding which subgroups of patients benefit from systemic therapy 72. In general, anthracycline-based chemotherapy is first line for advanced soft tissue sarcoma and the addition of ifosfamide is dependent on the subtype of soft tissue sarcoma. Ifosfamide has well documented efficacy in synovial sarcoma in the palliative setting and should be considered in patients who undergo chemotherapy if the toxicities can be tolerated 73, 74. Spurrell et al 73 demonstrated median survival of 22 months in patients with advanced disease treated with a combination of doxorubicin and ifosfamide which was superior to either agent given in isolation.
Chemotherapy
In children and adolescents with intermediate or high-risk tumors (i.e., >5 cm, nodal involvement, positive margins), adjuvant (postoperative) or neoadjuvant (preoperative) chemotherapy is generally undertaken with the most common agents being ifosfamide and doxorubicin. In the absence of available randomized controlled trials, prospective multicentered cohort studies have demonstrated adequate response to chemotherapy 75. Recent data has demonstrated that pediatric or adolescent patients with low risk tumors (Grade 2 or Grade 3 < 5 cm) can be successfully treated with surgical intervention without systemic therapy 20.
The role of chemotherapy in adult patients with synovial sarcoma is less clear. Eilber et al 72 demonstrated that chemotherapy improved distant relapse-free survival in patients with high-risk synovial sarcoma. Their group also published a synovial sarcoma specific nomogram that supported survival benefit of ifosfamide-based chemotherapy for certain adult patient populations 70. Similarly, pooled data from 15 trials on advanced soft tissue sarcoma demonstrated significantly better response to chemotherapy and survival rates when compared to other soft tissue sarcoma 71. Contrary to this, the French Sarcoma Group recently demonstrated no overall survival benefit with adjuvant (postoperative) or neoadjuvant (preoperative) chemotherapy in adult patients with synovial sarcoma 76. However, this study included patients with low-risk tumor characteristics in which chemotherapy is unlikely to be of benefit.
Chemotherapy is also considered in metastatic or unresectable disease 71, 73.
- Anthracycline based regimens
- Either used alone or in combination with Ifosfamide
- The expected response rate is between 25% and 60%.
- Ifosfamide
- Has significant activity in patients with synovial sarcoma
- Primarily used in combination with anthracyclines.
- It can be used as a single agent for those not fit to receive anthracyclines.
- A recent phase III trial evaluated evofosfamide in combination with doxorubicin versus doxorubicin alone. In the subset of patients with synovial sarcoma, a numerical benefit in the OS was noted in patients receiving both doxorubicin and evofosfamide (22.1 months versus 9.4 months). Although evofosfamide is not approved in the US, this trial does reinforce the activity of Ifosfamide in synovial sarcoma 77.
- Trabectedin
Targeted Therapy
There has been interest in the development of targeted medical therapies in the treatment of synovial sarcoma 80. New agents including receptor tyrosine kinase inhibitors (TKIs), epigenetic modifiers and immunotherapies have been investigated in clinical trials. Thus far, only pazopanib, a receptor tyrosine kinase inhibitor is FDA approved for clinical use for patients with synovial sarcoma. Pazopanib has been investigated in patients with advanced synovial sarcoma and has demonstrated improved progression free survival in a Phase III trial 73, 81.
There has also been recent advances in cell-based therapies targeting the cancer testis antigen, NY-ESO-1c259. Early work examining the utility of adoptive T-cell therapy with autologous T cells that have been engineered to expressive NY-ESO-1, have been promising in patients with metastatic synovial sarcoma 82. However, although several larger agents have demonstrated preclinical success, clinical trials are needed to determine the role of these novel agents in the treatment of synovial sarcoma.
Multiple other molecular targets have been identified in patients with synovial sarcoma, including EZH2 (enhancer of zeste homolog 2), mTOR (mammalian target of rapamycin) pathways, and CDK 4/6 (cyclin D1-cyclin-dependent kinase 4/6) pathways. However, clinical trials investigating these pathways have not resulted in positive results.
Tyrosine Kinase Inhibitor (TKI)
A functional role of PDGFRA (platelet-derived growth factor alpha) has been described in synovial sarcoma. Hence inhibition of PDGFRA may help achieve a response in patients with synovial sarcoma. Pazopanib is a multi-targeted tyrosine kinase inhibitor (TKI) with several targets, including PDGFRA (platelet-derived growth factor alpha). Pazopanib is the only US FDA-approved TKI available in the market for patients with synovial sarcoma.
PALETTE trial 83 included 38 patients with synovial sarcoma. An improvement of 3 months was noted in the median Progression-Free Survival (the time a patient with a disease lives without their disease getting worse or death from any cause, and is used in cancer clinical trials to measure a treatment’s effectivenes) with pazopanib versus placebo (4.1 months versus one month). In patients with pleuro-pulmonary synovial sarcoma, a risk of pneumothorax exists. However, some patients can achieve a dramatic response using pazopanib.
Regorafenib
Regorafenib was evaluated in the phase II French trial REGOSARC 84. All patients in this trial were randomized between regorafenib and placebo. In the subgroup of synovial sarcoma, a Progression-Free Survival (the time a patient with a disease lives without their disease getting worse or death from any cause, and is used in cancer clinical trials to measure a treatment’s effectivenes) benefit of 4 months was noted (5.6 months versus one month).
Most recently, a novel small molecule tyrosine kinase inhibitor AL3818 (APROMIsynovial sarcoma trial) has shown a progression-free survival benefit compared to dacarbazine.
Promising Ongoing Trials
- NCT04044768 85. This phase 2 study evaluating ADP-A2M4 in HLA-A*02 eligible and MAGE-A4 positive subjects with metastatic or inoperable (advanced) synovial sarcoma. This trial uses the novel concept of genetically engineered T-cells to target MAGE-A4 positive cells in the context of HLA-A*02.
- NCT03016819 86. This is a phase 3 trial where a novel small molecule tyrosine kinase inhibitor AL3818 is being tested in patients with advanced synovial sarcoma (Advanced Alveolar Soft Part Sarcoma, Leiomyosarcoma, Synovial Sarcoma (APROMISS) trial). Preliminary results were presented at ASCO 2021, and authors reported a progression-free survival benefit compared to dacarbazine.
Synovial sarcoma prognosis
Synovial sarcoma is an aggressive cancer with a poor prognosis 2. By definition, synovial sarcoma is a high-grade cancer 87. With up to 13% of patients with synovial sarcoma having distant metastases at the time of diagnosis most commonly to the lungs 13. Historically, it was thought that synovial sarcoma had a tendency to spread or metastasize to your lymph nodes, necessitating the need for further advanced imaging and possible sentinel node biopsy 88. However, these findings were based on small case series and more recent Surveillance, Epidemiology and End Results (SEER) database findings suggest that the rates of lymph node metastases are in line with other soft tissue sarcoma and do not require additional workup beyond thorough physical examination 89.
Other factors that affect synovial cell sarcoma prognosis include 2:
- Younger age is a favorable prognostic factor 90, 91.
- Tumor size. A smaller size of the primary tumor is associated with better outcomes.
- Tumor site. Synovial sarcoma originating in the extremities has a much better prognosis 92.
- Perioperative radiation therapy is associated with a better prognosis 93.
- Bone invasion or nerve and blood vessel invasion is associated with poor outcomes 94.
- Unplanned resections are associated with poor outcomes.
There are several well documented key prognostic factors including tumor size, grade and anatomical location, patient age at diagnosis, negative surgical margins and adjuvant radiotherapy 52, 70, 95, 96, 21, 97. Osseous or neurovascular invasion, adult age, large tumour size and unplanned excision have been linked to worse prognosis 98, 95, 96, 21, 97. The role of the subtype of oncogene protein is conflicting and does not appear to have a definite impact on outcomes 44, 99, 100, 101. Recent data suggests that histologic subtype appears to be prognostic, with the biphasic subtype demonstrating the highest survival rates at both five and ten years 101. Synovial sarcomas with >20% poorly differentiated areas show more aggressive behavior. The best outcomes are seen with tumors with histologic features <6 mitoses/mm² and no necrosis 102.
Similar to other soft tissue sarcoma, tumor size and grade has repeatedly been shown to have prognostic value in patients with synovial sarcoma 63, 72, 96. In a cohort of 1189 patients, Naing et al 63 demonstrated that size predicted worse overall survival. Tumor location has been also been demonstrated to be of predictive value, with non-extremity based synovial sarcomas tending to have worse overall survival 13, 103. However, this may be in part due to the lack of early symptoms and later stage of presentation.
Synovial sarcoma survival rate
The expected 5-year survival is between 50% to 60% in adults, and 5-year metastasis-free survival is between 40% to 60% 2. The European Pediatric Soft Tissue Sarcoma Study Group reports a 90% 5-year survival 2. The current literature suggests the 5-year survival rates ranges from 59% to 75% 4, 13, 5, 81, 95.
There has been a trend for improved survival over time with early cohorts from the 1960s quoting a 25% to 51% 5-year survival rate. Local and metastatic relapse of soft-tissue sarcomas generally occur in the first two years following treatment and thus surveillance and follow-up is most intensive in this period 104. However, synovial sarcoma is unique in this regard in that it tends to recur much later. Krieg et al. 105 demonstrated that local recurrence occurred after a mean of 3.6 years (range 0.5 to 15 years) and metastases occurred at a mean of 5.7 years (range 0.5 to 16.3 years).
- Gazendam AM, Popovic S, Munir S, Parasu N, Wilson D, Ghert M. Synovial Sarcoma: A Clinical Review. Curr Oncol. 2021 May 19;28(3):1909-1920. doi: 10.3390/curroncol28030177[↩][↩][↩][↩][↩]
- Mangla A, Gasalberti DP. Synovial Cell Sarcoma. [Updated 2023 May 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK587366[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Stacchiotti S, Van Tine BA. Synovial Sarcoma: Current Concepts and Future Perspectives. J Clin Oncol. 2018 Jan 10;36(2):180-187. https://doi.org/10.1200/JCO.2017.75.194[↩]
- Sultan I., Rodriguez-Galindo C., Saab R., Yasir S., Casanova M., Ferrari A. Comparing children and adults with synovial sarcoma in the surveillance, epidemiology, and end results program, 1983 to 2005. Cancer. 2009;115:3537–3547. doi: 10.1002/cncr.24424[↩][↩][↩][↩][↩]
- Smolle M.A., Parry M., Jeys L., Abudu S., Grimer R. Synovial sarcoma: Do children do better? Eur. J. Surg. Oncol. 2019;45:254–260. doi: 10.1016/j.ejso.2018.07.006[↩][↩][↩][↩][↩]
- Quan H, Sreekissoon S, Wang Y. Synovial sarcoma of the head and neck: A review of reported cases on the clinical characteristics and treatment methods. Front Cell Dev Biol. 2023 Jan 6;10:1077756. doi: 10.3389/fcell.2022.1077756[↩][↩]
- Jo V.Y., Fletcher C.D.M. WHO Classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology. 2014;46:95–104. doi: 10.1097/PAT.0000000000000050[↩][↩]
- Caracciolo J.T., Henderson-Jackson E., Binitie O. Synovial sarcoma of bone: Sarcoma typically of soft Tissues presenting as a primary bone tumor. Radiol. Case Rep. 2018;14:204–207. doi: 10.1016/j.radcr.2018.10.026[↩][↩]
- McGrory J.E., Pritchard D.J., Arndt C.A., Nascimento A.G., Remstein E.D., Rowland C.M. Nonrhabdomyosarcoma soft tissue sarcomas in children: The mayo clinic experience. Clin. Orthop. Relat. Res. 2000;374:247–258. doi: 10.1097/00003086-200005000-00022[↩][↩]
- Deshmukh R., Mankin H.J., Singer S. Synovial sarcoma: The importance of size and location for survival. Clin. Orthop. Relat. Res. 2004;419:155–161. doi: 10.1097/00003086-200402000-00025[↩][↩]
- Synovial Sarcoma: Imaging findings and prognostic features. https://epos.myesr.org/poster/esr/ecr2018/C-0322[↩][↩][↩][↩][↩]
- Weiss S, Goldblum J. Malignant soft tissue tumors of uncertain type. In: S Weiss, J Goldblum, eds. Enzinger and Weiss’s Soft Tissue Tumors. St Louis, Missouri: CV Mosby; 2001: 1483-1571.[↩]
- Aytekin M.N., Öztürk R., Amer K., Yapar A. Epidemiology, incidence, and survival of synovial sarcoma subtypes: SEER database analysis. J. Orthop. Surg. 2020;28:2309499020936009. doi: 10.1177/2309499020936009[↩][↩][↩][↩]
- Synovial Sarcoma. https://curesarcoma.org/sarcoma-subtypes/synovial-sarcoma[↩][↩]
- Santos N.R.D., Bruijn D.R.H.D., Van Kessel A.G. Molecular mechanisms underlying human synovial sarcoma development. Genes Chromosomes Cancer. 2001;30:1–14. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1056>3.0.CO;2-G[↩][↩][↩]
- Mancuso T, Mezzelani A, Riva C, Fabbri A, Dal Bo L, Sampietro G, Perego P, Casali P, Zunino F, Sozzi G, Pierotti MA, Pilotti S. Analysis of SYT-SSX fusion transcripts and bcl-2 expression and phosphorylation status in synovial sarcoma. Lab Invest. 2000 Jun;80(6):805-13. doi: 10.1038/labinvest.3780085[↩][↩]
- Mezzelani A, Mariani L, Tamborini E, Agus V, Riva C, Lo Vullo S, Fabbri A, Stumbo M, Azzarelli A, Casali PG, Gronchi A, Sozzi G, Pierotti MA, Pilotti S. SYT-SSX fusion genes and prognosis in synovial sarcoma. Br J Cancer. 2001 Nov 16;85(10):1535-9. https://pmc.ncbi.nlm.nih.gov/articles/instance/2363950/pdf/85-6692088a.pdf[↩][↩]
- Crew AJ, Clark J, Fisher C, Gill S, Grimer R, Chand A, Shipley J, Gusterson BA, Cooper CS. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995 May 15;14(10):2333-40. https://pmc.ncbi.nlm.nih.gov/articles/instance/398342/pdf/emboj00034-0211.pdf[↩][↩]
- Afamitresgene Autoleucel. https://www.cancer.gov/about-cancer/treatment/drugs/afamitresgene-autoleucel[↩][↩]
- Ferrari A., Chi Y.-Y., Salvo G.L.D., Orbach D., Brennan B., Randall R.L., McCarville M.B., Black J.O., Alaggio R., Hawkins D.S., et al. Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma study group and the children’s oncology group. Eur. J. Cancer. 2017;78:1–6. doi: 10.1016/j.ejca.2017.03.003[↩][↩][↩]
- Vlenterie M., Ho V.K.Y., Kaal S.E.J., Vlenterie R., Haas R., Van der Graaf W.T.A. Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br. J. Cancer. 2015;113:1602–1606. doi: 10.1038/bjc.2015.375[↩][↩][↩]
- Ulusan S., Kizilkilic O., Yildirim T., Hurcan C., Bal N., Nursal T. Z. (2005). Radiological findings of primary retroperitoneal synovial sarcoma. Br. J. Radiol. 78 (926), 166–169. 10.1259/bjr/67990800[↩]
- De Logu F., Ugolini F., Caporalini C., Palomba A., Simi S., Portelli F., et al. (2020). TRPA1 expression in synovial sarcoma may support neural origin. Biomolecules 10 (10), 1446. 10.3390/biom10101446[↩]
- Carrillo R., Rodriguez-Peralto J. L., Batsakis J. G. (1992). Synovial sarcomas of the head and neck. Ann. Otol. Rhinol. Laryngol. 101 (4), 367–370. 10.1177/000348949210100415[↩]
- Machen S. K., Easley K. A., Goldblum J. R. (1999). Synovial sarcoma of the extremities: A clinicopathologic study of 34 cases, including semi-quantitative analysis of spindled, epithelial, and poorly differentiated areas. Am. J. Surg. Pathol. 23 (3), 268–275. 10.1097/00000478-199903000-00004[↩]
- Sturgis E. M., Potter B. O. (2003). Sarcomas of the head and neck region. Curr. Opin. Oncol. 15 (3), 239–252. 10.1097/00001622-200305000-00011[↩]
- Chotel F., Unnithan A., Chandrasekar C.R., Parot R., Jeys L., Grimer R.J. Variability in the presentation of synovial sarcoma in children. J. Bone Jt. Surg. Br. Vol. 2008;90:1090–1096. doi: 10.1302/0301-620X.90B8.19815[↩][↩]
- De Silva, M. V. Chandu, Barrett, Ann, Reid, Robin, Premonitory Pain Preceding Swelling: A Distinctive Clinical Presentation of Synovial Sarcoma which may Prompt Early Detection, Sarcoma, 7, 620502, 5 pages, 2003. https://doi.org/10.1080/13577140310001644788[↩]
- O’Sullivan P.J., Harris A.C., Munk P.L. Radiological features of synovial cell sarcoma. BJR. 2008;81:346–356. doi: 10.1259/bjr/28335824[↩][↩][↩][↩][↩][↩][↩]
- Liang C., Mao H., Tan J., Ji Y., Sun F., Dou W., Wang H., Wang H., Gao J. Synovial sarcoma: Magnetic resonance and computed tomography imaging features and differential diagnostic considerations. Oncol. Lett. 2015;9:661–666. doi: 10.3892/ol.2014.2774[↩]
- Tateishi U., Hasegawa T., Beppu Y., Satake M., Moriyama N. Synovial sarcoma of the soft tissues: Prognostic significance of imaging features. J. Comput. Assist. Tomogr. 2004;28:140–148. doi: 10.1097/00004728-200401000-00024[↩]
- Baheti A.D., Tirumani S.H., Sewatkar R., Shinagare A.B., Hornick J.L., Ramaiya N.H., Jagannathan J.P. Imaging features of primary and metastatic extremity synovial sarcoma: A single institute experience of 78 patients. BJR. 2015;88:20140608. doi: 10.1259/bjr.20140608[↩]
- Von Mehren M., Randall R.L., Benjamin R.S., Boles S., Bui M.M., Conrad E.U., Ganjoo K.N., George S., Gonzalez R.J., Heslin M.J., et al. Soft tissue sarcoma, version 2.2016, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2016;14:758–786. doi: 10.6004/jnccn.2016.0078[↩][↩][↩][↩]
- Kasraeian S., Allison D.C., Ahlmann E.R., Fedenko A.N., Menendez L.R. A comparison of fine-needle aspiration, core biopsy, and surgical biopsy in the diagnosis of extremity soft tissue masses. Clin. Orthop. Relat. Res. 2010;468:2992–3002. doi: 10.1007/s11999-010-1401-x[↩][↩]
- Yang Y.J., Damron T.A. Comparison of needle core biopsy and fine-needle aspiration for diagnostic accuracy in musculoskeletal lesions. Arch. Pathol. Lab. Med. 2004;128:759–764. doi: 10.5858/2004-128-759-CONCBA[↩]
- Narvani AA, Tsiridis E, Saifuddin A, Briggs T, Cannon S. Does image guidance improve accuracy of core needle biopsy in diagnosis of soft tissue tumours? Acta Orthop Belg. 2009 Apr;75(2):239-44.[↩]
- Mankin H.J., Mankin C.J., Simon M.A. The hazards of the biopsy, revisited. For the members of the musculoskeletal tumor society. JBJS. 1996;78:656–663. doi: 10.2106/00004623-199605000-00004[↩]
- Fiore M., Sambri A., Spinnato P., Zucchini R., Giannini C., Caldari E., et al. (2021). The Biology of synovial sarcoma: State-of-the-Art and future perspectives. Curr. Treat. Options Oncol. 22 (12), 109. 10.1007/s11864-021-00914-4[↩]
- Shukla P. N., Pathy S., Sen S., Purohit A., Julka P. K., Rath G. K. (2003). Primary orbital calcified synovial sarcoma: A case report. Orbit 22 (4), 299–303. 10.1076/orbi.22.4.299.17246[↩]
- Alabdulaaly L., AlDawood Z., Afshar S., Rahbar R., Al-Ibraheemi A., Woo S. B. (2021). Calcifying synovial sarcoma of the tongue with SS18 rearrangement: A rare variant in a rare location. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 132 (5), e186–e189. 10.1016/j.oooo.2020.08.016[↩]
- Jayasooriya P. R., Madawalagamage L. N., Mendis B. R., Lombardi T. (2016). Diagnostic approach to synovial sarcoma of the head and neck illustrated by two cases arising in the face and oral cavity. Dermatopathol. (Basel) 3 (1), 13–22. 10.1159/000444876[↩]
- Crowson M. G., Lalich I., Keeney M. G., Garcia J. J., Price D. L. (2015). Clinicopathologic factors and adjuvant treatment effects on survival in adult head and neck synovial cell sarcoma. Head. Neck 37 (3), 375–380. 10.1002/hed.23605[↩]
- Madabhavi I., Bhardawa V., Modi M., Patel A., Sarkar M. (2018). Primary synovial sarcoma (SS) of larynx: An unusual site. Oral Oncol. 79, 80–82. 10.1016/j.oraloncology.2018.02.016[↩]
- Stegmaier S., Leuschner I., Poremba C., Ladenstein R., Kazanowska B., Ljungman G., Scheer M., Blank B., Bielack S., Klingebiel T., et al. The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (cooperative weichteilsarkom studie) trials. Pediatric Blood Cancer. 2017;64:89–95. doi: 10.1002/pbc.26206[↩][↩]
- Amary M.F.C., Berisha F., Bernardi F.D.C., Herbert A., James M., Reis-Filho J.S., Fisher C., Nicholson A.G., Tirabosco R., Diss T.C., et al. Detection of SS18-SSX fusion transcripts in formalin-fixed paraffin-embedded neoplasms: Analysis of conventional RT-PCR, QRT-PCR and dual color FISH as diagnostic tools for synovial sarcoma. Mod. Pathol. 2007;20:482–496. doi: 10.1038/modpathol.3800761[↩]
- WHO . Soft Tissue and Bone Tumours. 5th ed. IARC Press; Lyon, France: 2020. Classification of Tumours Editorial.[↩]
- Soft Tissue Sarcoma Stages. https://www.cancer.org/cancer/types/soft-tissue-sarcoma/detection-diagnosis-staging/staging.html[↩][↩]
- Ghert M.A., Abudu A., Driver N., Davis A.M., Griffin A.M., Pearce D., White L., O’Sullivan B., Catton C.N., Bell R.S., et al. The indications for and the prognostic significance of amputation as the primary surgical procedure for localized soft tissue sarcoma of the extremity. Ann. Surg. Oncol. 2005;12:10–17. doi: 10.1007/s10434-004-1171-3[↩][↩][↩][↩][↩]
- Randall RL, Schabel KL, Hitchcock Y, Joyner DE, Albritton KH. Diagnosis and management of synovial sarcoma. Curr Treat Options Oncol. 2005 Nov;6(6):449-59. doi: 10.1007/s11864-005-0024-z[↩]
- Biau D.J., Ferguson P.C., Chung P., Griffin A.M., Catton C.N., O’Sullivan B., Wunder J.S. Local recurrence of localized soft tissue sarcoma: A new look at old predictors. Cancer. 2012;118:5867–5877. doi: 10.1002/cncr.27639[↩]
- Bilgeri A., Klein A., Lindner L.H., Nachbichler S., Knösel T., Birkenmaier C., Jansson V., Baur-Melnyk A., Dürr H.R. The effect of resection margin on local recurrence and survival in high grade soft tissue sarcoma of the extremities: How far is far enough? Cancers. 2020;12:2560. doi: 10.3390/cancers12092560[↩]
- Guadagnolo B.A., Zagars G.K., Ballo M.T., Patel S.R., Lewis V.O., Pisters P.W.T., Benjamin R.S., Pollock R.E. Long-term outcomes for synovial sarcoma treated with conservation surgery and radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2007;69:1173–1180. doi: 10.1016/j.ijrobp.2007.04.056[↩][↩][↩]
- Kawaguchi N., Ahmed A.R., Matsumoto S., Manabe J., Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin. Orthop. Relat. Res. 2004;419:165–172. doi: 10.1097/00003086-200402000-00027[↩]
- Gundle K.R., Gupta S., Kafchinski L., Griffin A.M., Kandel R.A., Dickson B.C., Chung P.W., Catton C.N., O’Sullivan B., Ferguson P.C., et al. An analysis of tumor- and surgery-related factors that contribute to inadvertent positive margins following soft tissue sarcoma resection. Ann. Surg. Oncol. 2017;24:2137–2144. doi: 10.1245/s10434-017-5848-9[↩]
- O’Donnell P.W., Griffin A.M., Eward W.C., Sternheim A., Catton C.N., Chung P.W., O’Sullivan B., Ferguson P.C., Wunder J.S. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120:2866–2875. doi: 10.1002/cncr.28793[↩][↩]
- Sambri A., Caldari E., Fiore M., Zucchini R., Giannini C., Pirini M.G., Spinnato P., Cappelli A., Donati D.M., Paolis M.D. Margin assessment in soft tissue sarcomas: Review of the literature. Cancers. 2021;13:1687. doi: 10.3390/cancers13071687[↩]
- Chandrasekar C.R., Wafa H., Grimer R.J., Carter S.R., Tillman R.M., Abudu A. The effect of an unplanned excision of a soft-tissue sarcoma on prognosis. J. Bone Jt. Surg. Br. Vol. 2008;90:203–208. doi: 10.1302/0301-620X.90B2.19760[↩][↩]
- Potter B.K., Adams S.C., Pitcher J.D., Temple H.T. Local recurrence of disease after unplanned excisions of high-grade soft tissue sarcomas. Clin. Orthop. Relat. Res. 2008;466:3093–3100. doi: 10.1007/s11999-008-0529-4[↩][↩]
- Pretell-Mazzini J., Barton M.D.J., Conway S.A., Temple H.T. Unplanned excision of soft-tissue sarcomas: Current concepts for management and prognosis. JBJS. 2015;97:597–603. doi: 10.2106/JBJS.N.00649[↩][↩]
- O’Sullivan B, Davis AM, Turcotte R, Bell R, Catton C, Chabot P, Wunder J, Kandel R, Goddard K, Sadura A, Pater J, Zee B. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet. 2002 Jun 29;359(9325):2235-41. doi: 10.1016/S0140-6736(02)09292-9[↩]
- Menon G, Mangla A, Yadav U. Leiomyosarcoma. [Updated 2024 Feb 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK551667[↩]
- Gingrich A.A., Marrufo A.S., Liu Y., Li C.-S., Darrow M.A., Monjazeb A.M., Thorpe S.W., Canter R.J. Radiotherapy is associated with improved survival in patients with synovial sarcoma undergoing surgery: A national cancer database analysis. J. Surg. Res. 2020;255:378–387. doi: 10.1016/j.jss.2020.05.075[↩]
- Naing K.W., Monjazeb A.M., Li C.-S., Lee L.-Y., Yang A., Borys D., Canter R.J. Perioperative radiotherapy is associated with improved survival among patients with synovial sarcoma: A SEER analysis. J. Surg. Oncol. 2015;111:158–164. doi: 10.1002/jso.23780[↩][↩][↩]
- Song S., Park J., Kim H.J., Kim I.H., Han I., Kim H.-S., Kim S. Effects of adjuvant radiotherapy in patients with synovial sarcoma. Am. J. Clin. Oncol. 2017;40:306–311. doi: 10.1097/COC.0000000000000148[↩]
- Davis A.M., O’Sullivan B., Turcotte R., Bell R., Catton C., Chabot P., Wunder J., Hammond A., Benk V., Kandel R., et al. Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother. Oncol. 2005;75:48–53. doi: 10.1016/j.radonc.2004.12.020[↩]
- Salerno KE, Alektiar KM, Baldini EH, Bedi M, Bishop AJ, Bradfield L, Chung P, DeLaney TF, Folpe A, Kane JM, Li XA, Petersen I, Powell J, Stolten M, Thorpe S, Trent JC, Voermans M, Guadagnolo BA. Radiation Therapy for Treatment of Soft Tissue Sarcoma in Adults: Executive Summary of an ASTRO Clinical Practice Guideline. Pract Radiat Oncol. 2021 Sep-Oct;11(5):339-351. doi: 10.1016/j.prro.2021.04.005[↩][↩]
- Wang J., Song Y., Liu X., Jin J., Wang W., Yu Z., Liu Y., Li N., Fang H., Ren H., et al. Comparison of outcome and toxicity of postoperative intensity-modulated radiation therapy with two-dimensional radiotherapy in patients with soft tissue sarcoma of extremities and trunk. Cancer Med. 2019;8:902–909. doi: 10.1002/cam4.1919[↩]
- O’Sullivan B., Griffin A.M., Dickie C.I., Sharpe M.B., Chung P.W.M., Catton C.N., Ferguson P.C., Wunder J.S., Deheshi B.M., White L.M., et al. Phase 2 study of preoperative image-guided intensity-modulated radiation therapy to reduce wound and combined modality morbidities in lower extremity soft tissue sarcoma. Cancer. 2013;119:1878–1884. doi: 10.1002/cncr.27951[↩]
- Ramu E.M., Houdek M.T., Isaac C.E., Dickie C.I., Ferguson P.C., Wunder J.S. Management of soft-tissue sarcomas; Treatment strategies, staging, and outcomes. Sicot-j. 2017;3:20. doi: 10.1051/sicotj/2017010[↩]
- Canter R.J., Qin L.-X., Maki R.G., Brennan M.F., Ladanyi M., Singer S. A synovial sarcoma-specific preoperative nomogram supports a survival benefit to ifosfamide-based chemotherapy and improves risk stratification for patients. Clin. Cancer Res. 2008;14:8191–8197. doi: 10.1158/1078-0432.CCR-08-0843[↩][↩][↩]
- Vlenterie M., Litière S., Rizzo E., Marréaud S., Judson I., Gelderblom H., Cesne A.L., Wardelmann E., Messiou C., Gronchi A., et al. Outcome of chemotherapy in advanced synovial sarcoma patients: Review of 15 clinical trials from the european organisation for research and treatment of cancer soft tissue and bone sarcoma group; Setting a new landmark for studies in this entity. Eur. J. Cancer. 2016;58:62–72. doi: 10.1016/j.ejca.2016.02.002[↩][↩][↩]
- Eilber F.C., Brennan M.F., Eilber F.R., Eckardt J.J., Grobmyer S.R., Riedel E., Forscher C., Maki R.G., Singer S. Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann. Surg. 2007;246:105–113. doi: 10.1097/01.sla.0000262787.88639.2b[↩][↩][↩]
- Spurrell E.L., Fisher C., Thomas J.M., Judson I.R. Prognostic Factors in Advanced Synovial Sarcoma: An Analysis of 104 Patients Treated at the Royal Marsden Hospital. Ann. Oncol. 2005;16:437–444. doi: 10.1093/annonc/mdi082[↩][↩][↩][↩]
- Sleijfer S., Ouali M., Van Glabbeke M., Krarup-Hansen A., Rodenhuis S., Cesne A.L., Hogendoorn P.C.W., Verweij J., Blay J.-Y. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: An exploratory, retrospective analysis on large series from the European organization for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC-STBSG) Eur. J. Cancer. 2010;46:72–83. doi: 10.1016/j.ejca.2009.09.022[↩]
- Ferrari A., Salvo G.L.D., Brennan B., Van Noesel M.M., Paoli A.D., Casanova M., Francotte N., Kelsey A., Alaggio R., Oberlin O., et al. Synovial sarcoma in children and adolescents: The european pediatric soft tissue sarcoma study group prospective trial (EpSSG NRSTS 2005) Ann. Oncol. 2015;26:567–572. doi: 10.1093/annonc/mdu562[↩]
- Italiano A., Penel N., Robin Y.-M., Bui B., Cesne A.L., Piperno-Neumann S., Tubiana-Hulin M., Bompas E., Chevreau C., Isambert N., et al. Neo/adjuvant chemotherapy does not improve outcome in resected primary synovial sarcoma: A study of the french sarcoma group. Ann. Oncol. 2009;20:425–430. doi: 10.1093/annonc/mdn678[↩]
- Tap WD, Papai Z, Van Tine BA, et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): an international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2017 Aug;18(8):1089-1103. doi: 10.1016/S1470-2045(17)30381-9. Epub 2017 Jun 23. Erratum in: Lancet Oncol. 2018 Feb;19(2):e78. doi: 10.1016/S1470-2045(18)30037-8[↩]
- Le Cesne A, Cresta S, Maki RG, Blay JY, Verweij J, Poveda A, Casali PG, Balaña C, Schöffski P, Grosso F, Lardelli P, Nieto A, Alfaro V, Demetri GD. A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur J Cancer. 2012 Nov;48(16):3036-44. doi: 10.1016/j.ejca.2012.05.012[↩]
- Sanfilippo R, Dileo P, Blay JY, Constantinidou A, Le Cesne A, Benson C, Vizzini L, Contu M, Baldi GG, Dei Tos AP, Casali PG. Trabectedin in advanced synovial sarcomas: a multicenter retrospective study from four European institutions and the Italian Rare Cancer Network. Anticancer Drugs. 2015 Jul;26(6):678-81. doi: 10.1097/CAD.0000000000000228[↩]
- Desar I.M., Fleuren E.D., Van der Graaf W.T. Systemic treatment for adults with synovial sarcoma. Curr. Treat. Options Oncol. 2018;19:1–17. doi: 10.1007/s11864-018-0525-1[↩]
- Trassard M., Le Doussal V., Hacène K., Terrier P., Ranchère D., Guillou L., Fiche M., Collin F., Vilain M.-O., Bertrand G. Prognostic Factors in Localized Primary Synovial Sarcoma: A Multicenter Study of 128 Adult Patients. J. Clin. Oncol. 2001;19:525–534. doi: 10.1200/JCO.2001.19.2.525[↩][↩]
- D’Angelo S.P., Melchiori L., Merchant M.S., Bernstein D., Glod J., Kaplan R., Grupp S., Tap W.D., Chagin K., Binder G.K. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 C259T cells in synovial sarcoma. Cancer Discov. 2018;8:944–957. doi: 10.1158/2159-8290.CD-17-1417[↩]
- van der Graaf WT, Blay JY, Chawla SP, et al. EORTC Soft Tissue and Bone Sarcoma Group; PALETTE study group. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012 May 19;379(9829):1879-86. doi: 10.1016/S0140-6736(12)60651-5[↩]
- Mir O, Brodowicz T, Italiano A, Wallet J, et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016 Dec;17(12):1732-1742. doi: 10.1016/S1470-2045(16)30507-1[↩]
- Spearhead 1 Study in Subjects With Advanced Synovial Sarcoma or Myxoid/Round Cell Liposarcoma. https://clinicaltrials.gov/study/NCT04044768[↩]
- Phase III Trial of Anlotinib, Catequentinib in Advanced Alveolar Soft Part Sarcoma, Leiomyosarcoma, Synovial Sarcoma (APROMISS). https://clinicaltrials.gov/study/NCT03016819[↩]
- Guillou L, Benhattar J, Bonichon F, Gallagher G, Terrier P, Stauffer E, Somerhausen Nde S, Michels JJ, Jundt G, Vince DR, Taylor S, Genevay M, Collin F, Trassard M, Coindre JM. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol. 2004 Oct 15;22(20):4040-50. https://doi.org/10.1200/JCO.2004.11.093[↩]
- Maduekwe U.N., Hornicek F.J., Springfield D.S., Raskin K.A., Harmon D.C., Choy E., Rosenberg A.E., Nielsen G.P., DeLaney T.F., Chen Y.-L. Role of sentinel lymph node biopsy in the staging of synovial, epithelioid, and clear cell sarcomas. Ann. Surg. Oncol. 2009;16:1356. doi: 10.1245/s10434-009-0393-9[↩]
- Jacobs A.J., Morris C.D., Levin A.S. Synovial sarcoma is not associated with a higher risk of lymph node metastasis compared with other soft tissue sarcomas. Clin. Orthop. Relat. Res. 2018;476:589–598. doi: 10.1007/s11999.0000000000000057[↩]
- Vlenterie M, Ho VK, Kaal SE, Vlenterie R, Haas R, van der Graaf WT. Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer. 2015 Dec 1;113(11):1602-6. doi: 10.1038/bjc.2015.375[↩]
- Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer. 2009 Aug 1;115(15):3537-47. doi: 10.1002/cncr.24424[↩]
- Ferrari A, Bisogno G, Alaggio R, Cecchetto G, Collini P, Rosolen A, Meazza C, Indolfi P, Garaventa A, De Sio L, D’Angelo P, Tamaro P, Casanova M, Carli M. Synovial sarcoma of children and adolescents: the prognostic role of axial sites. Eur J Cancer. 2008 Jun;44(9):1202-9. doi: 10.1016/j.ejca.2008.03.016[↩]
- Naing KW, Monjazeb AM, Li CS, Lee LY, Yang A, Borys D, Canter RJ. Perioperative radiotherapy is associated with improved survival among patients with synovial sarcoma: A SEER analysis. J Surg Oncol. 2015 Feb;111(2):158-64. doi: 10.1002/jso.23780[↩]
- Ferguson PC, Griffin AM, O’Sullivan B, Catton CN, Davis AM, Murji A, Bell RS, Wunder JS. Bone invasion in extremity soft-tissue sarcoma: impact on disease outcomes. Cancer. 2006 Jun 15;106(12):2692-700. doi: 10.1002/cncr.21949[↩]
- Lewis J.J., Antonescu C.R., Leung D.H., Blumberg D., Healey J.H., Woodruff J.M., Brennan M.F. Synovial sarcoma: A multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J. Clin. Oncol. 2000;18:2087–2094. doi: 10.1200/JCO.2000.18.10.2087[↩][↩][↩]
- Ferrari A., Gronchi A., Casanova M., Meazza C., Gandola L., Collini P., Lozza L., Bertulli R., Olmi P., Casali P.G. Synovial sarcoma: A retrospective analysis of 271 patients of all ages treated at a single institution. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2004;101:627–634. doi: 10.1002/cncr.20386[↩][↩][↩]
- Ferguson P.C., Griffin A.M., O’Sullivan B., Catton C.N., Davis A.M., Murji A., Bell R.S., Wunder J.S. Bone invasion in extremity soft-tissue sarcoma: Impact on disease outcomes. Cancer. 2006;106:2692–2700. doi: 10.1002/cncr.21949[↩][↩]
- Singer S., Baldini E.H., Demetri G.D., Fletcher J.A., Corson J.M. Synovial sarcoma: Prognostic significance of tumor size, margin of resection, and mitotic activity for survival. JCO. 1996;14:1201–1208. doi: 10.1200/JCO.1996.14.4.1201[↩]
- Takenaka S., Ueda T., Naka N., Araki N., Hashimoto N., Myoui A., Ozaki T., Nakayama T., Toguchida J., Tanaka K. Prognostic implication of SYT-SSX fusion type in synovial sarcoma: A multi-institutional retrospective analysis in Japan. Oncol. Rep. 2008;19:467–476. doi: 10.3892/or.19.2.467[↩]
- Guillou L., Benhattar J., Bonichon F., Gallagher G., Terrier P., Stauffer E., De Saint A.S.N., Michels J.-J., Jundt G., Vince D.R. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: A multicenter, retrospective analysis. J. Clin. Oncol. 2004;22:4040–4050. doi: 10.1200/JCO.2004.11.093[↩]
- Xiong L., Chen Z., Zhou Y., Li H., Xiao T. The survival and prognosis analysis of synovial sarcoma subtypes: A surveillance, epidemiology, and end results population-based analysis. Int. Orthop. 2020;44:2779–2786. doi: 10.1007/s00264-020-04708-5[↩][↩]
- Van de Rijn M., Barr F.G., Xiong Q.-B., Hedges M., Shipley J., Fisher C. Poorly differentiated synovial sarcoma: An analysis of clinical, pathologic, and molecular genetic features. Am. J. Surg. Pathol. 1999;23:106–112. doi: 10.1097/00000478-199901000-00012[↩]
- Ferrari A., Bisogno G., Alaggio R., Cecchetto G., Collini P., Rosolen A., Meazza C., Indolfi P., Garaventa A., De Sio L. Synovial sarcoma of children and adolescents: The prognostic role of axial sites. Eur. J. Cancer. 2008;44:1202–1209. doi: 10.1016/j.ejca.2008.03.016[↩]
- Wilson D.A., Gazendam A., Visgauss J., Perrin D., Griffin A.M., Chung P.W., Catton C.N., Shultz D., Ferguson P.C., Wunder J.S. Designing a rational follow-up schedule for patients with extremity soft tissue sarcoma. Ann. Surg. Oncol. 2020;27:1–9. doi: 10.1245/s10434-020-08240-z[↩]
- Krieg A.H., Hefti F., Speth B.M., Jundt G., Guillou L., Exner U.G., Von Hochstetter A.R., Cserhati M.D., Fuchs B., Mouhsine E., et al. Synovial sarcomas usually metastasize after >5 years: A multicenter retrospective analysis with minimum follow-up of 10 years for survivors. Ann. Oncol. 2011;22:458–467. doi: 10.1093/annonc/mdq394[↩]





{kind=link}