Contents
- What is gonadal dysgenesis
- 46XX complete gonadal dysgenesis
- 46 XY complete gonadal dysgenesis (Swyer syndrome)
- 46 XY complete gonadal dysgenesis (Swyer syndrome) causes
- 46 XY complete gonadal dysgenesis (Swyer syndrome) inheritance pattern
- 46 XY complete gonadal dysgenesis (Swyer syndrome) signs and symptoms
- 46 XY complete gonadal dysgenesis (Swyer syndrome) diagnosis
- 46 XY complete gonadal dysgenesis (Swyer syndrome) treatment
- Mixed gonadal dysgenesis
- Partial gonadal dysgenesis
- Gonadal dysgenesis causes
- Gonadal dysgenesis signs and symptoms
- Gonadal dysgenesis diagnosis
- Gonadal dysgenesis treatment
- Gonadal dysgenesis prognosis
What is gonadal dysgenesis
Gonadal dysgenesis also called gonadal dysgenesis syndrome is a medical term for abnormal development of gonadal tissues, a genetic condition or a subset of disorders of sexual development (DSD) where the gonads, ovaries or testes, do not develop properly, resulting in streak gonads (fibrous tissue), preventing the development of secondary sex characteristics and infertility 1, 2, 3, 4, 5. Gonadal dysgenesis are characterized by variable degrees of immaturity or dysfunction of the gonads (ovary or testis), which can manifest in a wide range of genital ambiguity. Gonadal dysgenesis signs and symptoms include a lack of puberty, missed periods (amenorrhea), underdeveloped breasts, and sometimes ambiguous or female external genitalia. Gonadal dysgenesis is a genetic condition caused by errors in cell division or alterations in genetic material, leading to a loss of gonadal (ovaries or testes) development early in fetal life. The development of gonadal dysgenesis begins early either at fertilization or shortly after in the early stages of the embryo and fetus 1.
Gonadal dysgenesis can be classified into different types depending on the gonadal morphology 6, 7:
- Complete gonadal dysgenesis that occurs when gonads are completely absent and exist as streak gonads.
- 46 XX complete gonadal dysgenesis also called XX female gonadal dysgenesis, follicular stimulating hormone-resistant ovaries, hypergonadotropic ovarian dysgenesis or ovarian dysgenesis, is a primary ovarian defect leading to premature ovarian failure in otherwise normal 46XX females as a result of failure of the gonads to develop or due to resistance to gonadotrophin stimulation.
- 46 XY complete gonadal dysgenesis, also called Swyer syndrome or 46XY pure gonadal dysgenesis, is a condition that occurs in individuals with XY male sex chromosomes in each of their cells, but has female external genitalia and streak gonads (ovaries or testes), which have a high risk of becoming cancerous. People with Swyer syndrome or 46XY complete gonadal dysgenesis have an XY chromosomal makeup (as males normally do) but have female external genitalia and some female internal reproductive structures. Individuals with Swyer syndrome or 46XY complete gonadal dysgenesis usually have a vagina, uterus and fallopian tubes, but their gonads (ovaries or testes) are not functional. Instead, their gonads (ovaries or testes) are small and underdeveloped and contain little gonadal tissue. These structures are called “streak gonads”. The streak gonadal tissue is at risk of developing cancer that is often hard-to-detect, so it is usually removed surgically.
- Mixed gonadal dysgenesis, a form of gonadal dysgenesis that occurs in individuals with a Y chromosome mosaicism, where one gonad is a streak gonad and the other is a partially developed testicle. Chromosome mosaicism is when someone has two or more sets of cells that differ genetically from one another. It happens when an error in cell division occurs after a single fertilized egg (a zygote) has begun to divide, leading to some cells having a different chromosome number than others. When cells divide and multiply, they make an exact copy of their DNA, and then split the copies between each of the resulting two cells. If there is a mistake anywhere in this process—for example, a DNA copy that contains a mistake or DNA that is split unevenly, the resulting cells would be different. In some cases, the abnormal cell may simply die; but, if it survives, the result is mosaicism. If this occurs early in development, as many as 50% of a person’s cells could be abnormal, which would be a high level of mosaicism. If the mistake happens later, however, a smaller percentage of cells would be abnormal, resulting in a lower level of mosaicism. The impact of mosaicism is typically determined by the severity of the mistake in the DNA, as well as the degree of mosaicism.
- Partial gonadal dysgenesis that occurs when the gonads (ovaries or testes) partially form but do not produce enough sex hormones, leading to ambiguous or nonbinary external genitalia. The most common karyotype seen in partial gonadal dysgenesis is 45,X or 46,XY, but 46,XY and other forms of mosaicism involving the Y chromosome also can be seen.
Genes are sequences of DNA that are found on a specific location of a chromosome and are the basic unit of inheritance. Genes determine a particular characteristic or trait in a person. Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Chromosomes contain the genetic instructions for how your body develops and functions. People usually have 46 chromosomes in each cell. Pairs of human chromosomes are numbered from 1 through 22 and called autosomes. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male or female reproductive structures. Females typically have two X chromosomes (46,XX karyotype), while males typically have one X chromosome and one Y chromosome (46,XY karyotype).
The most notable of gonadal dysgenesis is Turner syndrome also known as monosomy X or 45X syndrome, a genetic disorder affecting 1 in every 2500 live female births, occurring when one X chromosome is partially or completely missing in females 8, 9, 10, 11. Turner syndrome clinical features are highly variable and can differ significantly from one person to another 8, 9, 10, 11. Turner syndrome common symptoms include short stature, premature ovarian failure, which can result in failure to attain puberty or delayed puberty, infertility, and various other health issues, including heart and kidney abnormalities and learning difficulties. Most people with Turner syndrome are infertile. A variety of additional symptoms can occur including abnormalities of the eyes and ears and skeletal malformations. Intelligence is usually normal, but affected individuals may experience certain learning disabilities. Turner syndrome may be diagnosed before birth or shortly after birth or during early childhood. However, in some cases, Turner syndrome may not be diagnosed until well into adulthood, often as an incidental finding. Most Turner syndrome cases do not run in families and appear to occur randomly for no apparent reason (sporadically) 12. While there is no cure, treatments like growth hormone and estrogen therapy can help manage symptoms and improve quality of life. 13.
Gonadal dysgenesis is diagnosed through a combination of physical exams, genetic testing, hormone levels, and imaging, often identified in adolescence due to absent puberty. Key diagnostic indicators include delayed or absent puberty, lack of menstruation, and a tall stature. A doctor may order a pelvic ultrasound, blood tests for hormones like follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and a genetic analysis (karyotype) to determine the underlying cause.
Many individuals with gonadal dysgenesis are diagnosed around puberty due to the lack of secondary sexual development. In some cases, gonadal dysgenesis can be suspected during pregnancy through non-invasive prenatal screening or identified via chorionic villus sampling or amniocentesis. Some forms of gonadal dysgenesis, particularly mixed gonadal dysgenesis, may be diagnosed earlier due to ambiguous genitalia.
Patients with gonadal dysgenesis who have a Y-chromosome or Y-chromosome material are at increased risk for developing germ cell tumors (a type of tumor that can be either benign or cancer, arising from the cells that normally develop into eggs or sperm) such as gonadoblastoma or carcinoma in situ (CIS), with the potential for malignant transformation to dysgerminoma or seminoma, respectively 14, 15, 16. The term gonadoblastoma was first introduced by Scully in 1953 and is the most common germ cell tumor seen in patients with XY gonadal dysgenesis 17. A benign germ cell ovarian tumor composed of germ cells and sex cord stromal cells, a gonadoblastoma almost always arises from a dysgenetic gonad with a Y chromosome 18. Gonadoblastoma usually presents in the second decade, but cases occurring in early infancy have been reported 19. In 50% to 60% of cases, gonadoblastomas are associated with malignant germ cell tumors, most commonly dysgerminomas. The prognosis is favorable when the gonadoblastoma is associated with dysgerminoma, but unfavorable when associated with other germ cell tumors including yolk sac tumors, seminomas, immature teratomas, embryonal carcinomas, or choriocarcinomas 20. Carcinoma in situ (CIS) also known as intratubular germ cell neoplasia unclassified, is the common precursor for testicular germ cell tumors including seminomas, embyronal carcinomas, teratomas, and yolk cell tumors 15. Gonadal dysgenesis is a known risk factor for carcinoma in situ (CIS) 21. The natural history of untreated carcinoma in situ (CIS) is a 40% estimated risk of progression to invasive cancer within three years and a 50% estimated risk of progression within five years 22.
To prevent the development of cancer in patients with XY gonadal dysgenesis, the surgical removal of the gonads (ovaries in females and the testes in males) typically is recommended, but debate ensues concerning which patients require surgery and the appropriate timing 23. Furthermore, no standard approach or guidelines have been established for the diagnostic workup and management of these patients. Given the irreversible nature of gonadectomy (surgical removal of the gonads), certain ethical considerations must be taken into account in addition to determining each individual patient’s risk for developing a cancer. Both risks and benefits are involved in either retaining or removing gonads, and a general, benefit-based principle of intervening only when the benefits are reliably judged to outweigh the risks should be maintained 24, 25, 26. Benefits of undergoing a surgical removal of the gonads would include decreasing the risk of developing a gonadal cancer. In the case of a patient with XY partial gonadal dysgenesis who is assigned a female sex, the function of gonads at puberty may cause unwanted development of male physical characteristics such as muscle bulk, body hair, and deep voice in a female (virilization), rendering a gonadectomy psychosocially beneficial. In contradistinction, certain benefits may be associated with retaining the gonad. Surgical procedures can lead to associated sickness, and for situations with lower risks of development of a cancer, it may be reasonable to wait until the patient has reached the capacity for developmentally appropriate acceptance or can legally consent before being subjected to such risk 25, 26. In addition, for patients with XY partial gonadal dysgenesis and a male sex assignment, the gonads may have partially functioning testicular tissue that could be a source of hormone production through puberty and potential fertility. Overall, the decision for performing a gonadectomy must be made on a case-by-case basis based on the best interest of the patient.
Figure 1. Human karyotype (an individual’s collection of chromosomes)
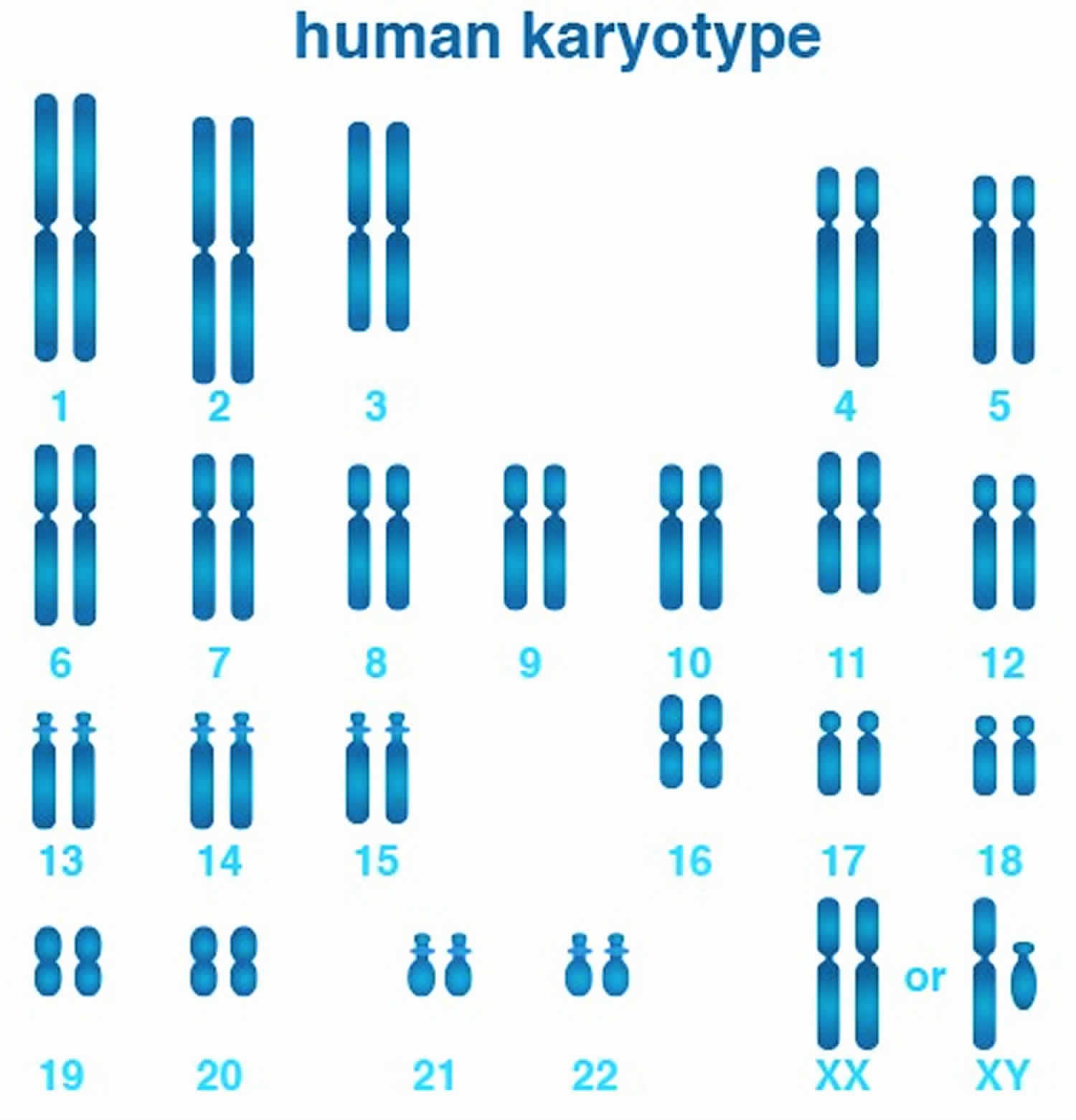
Figure 2. Female karyotype (normal female chromosomes)
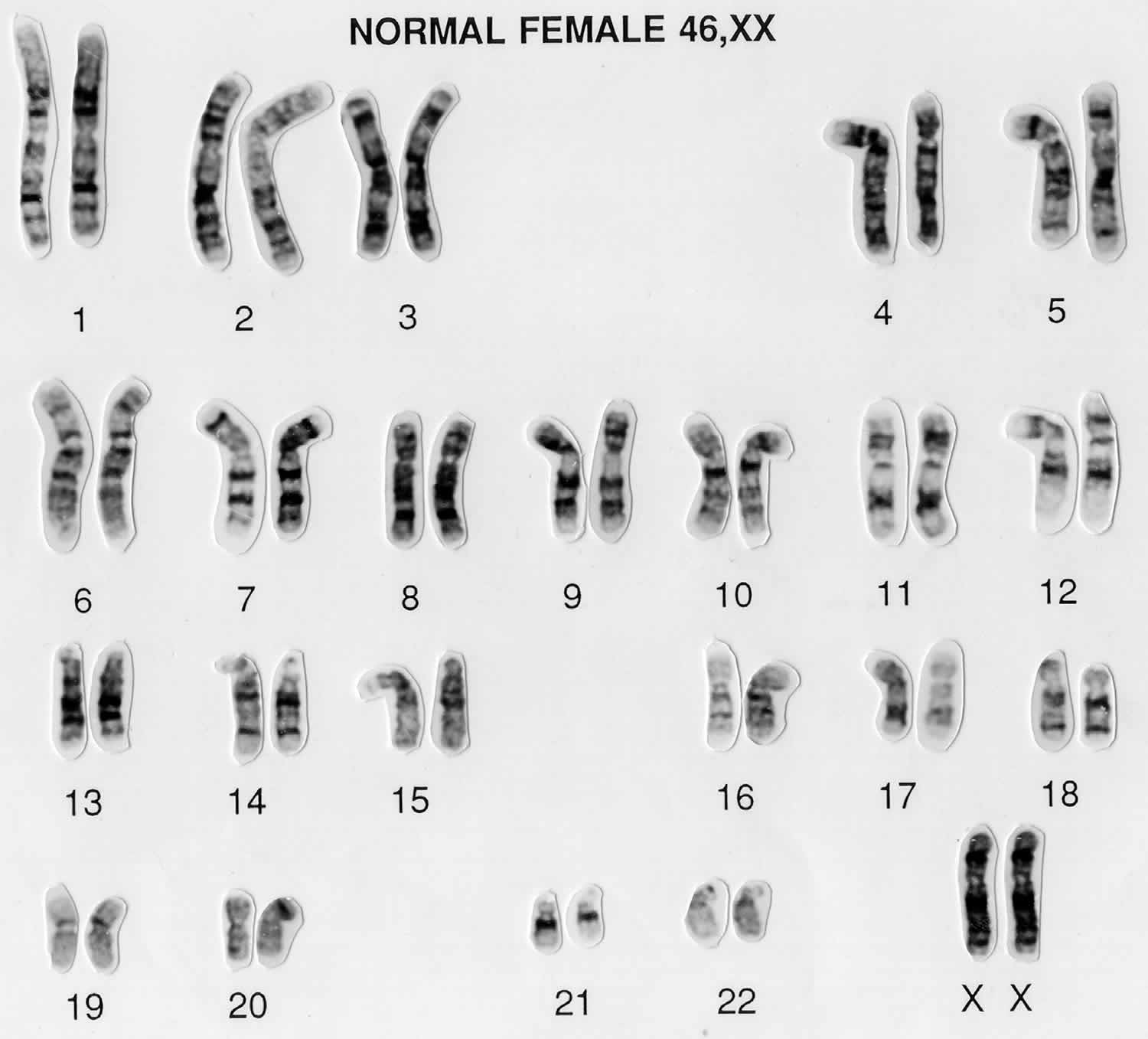
Figure 3. Male karyotype (normal male chromosomes)
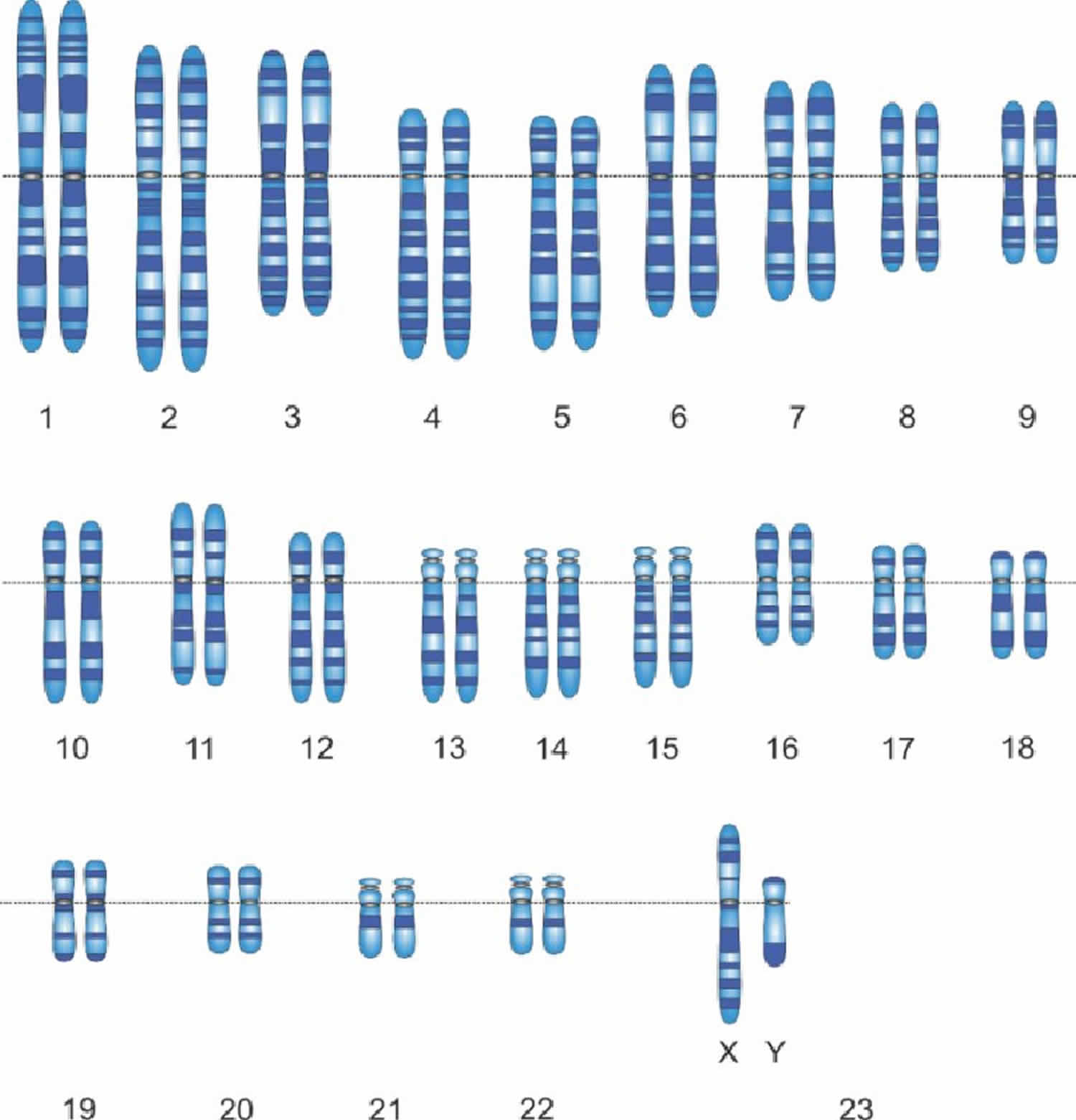
Figure 4. DNA structure
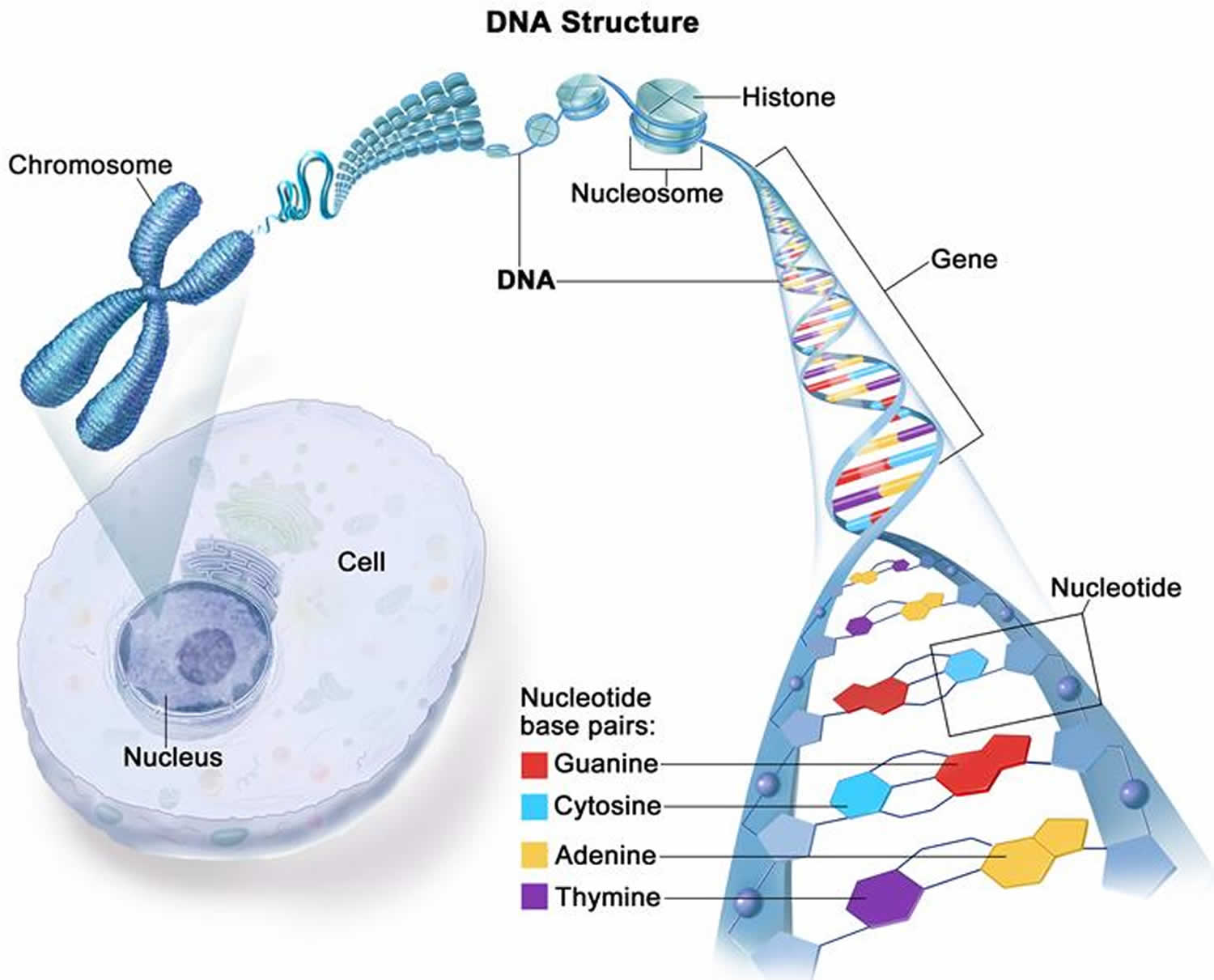
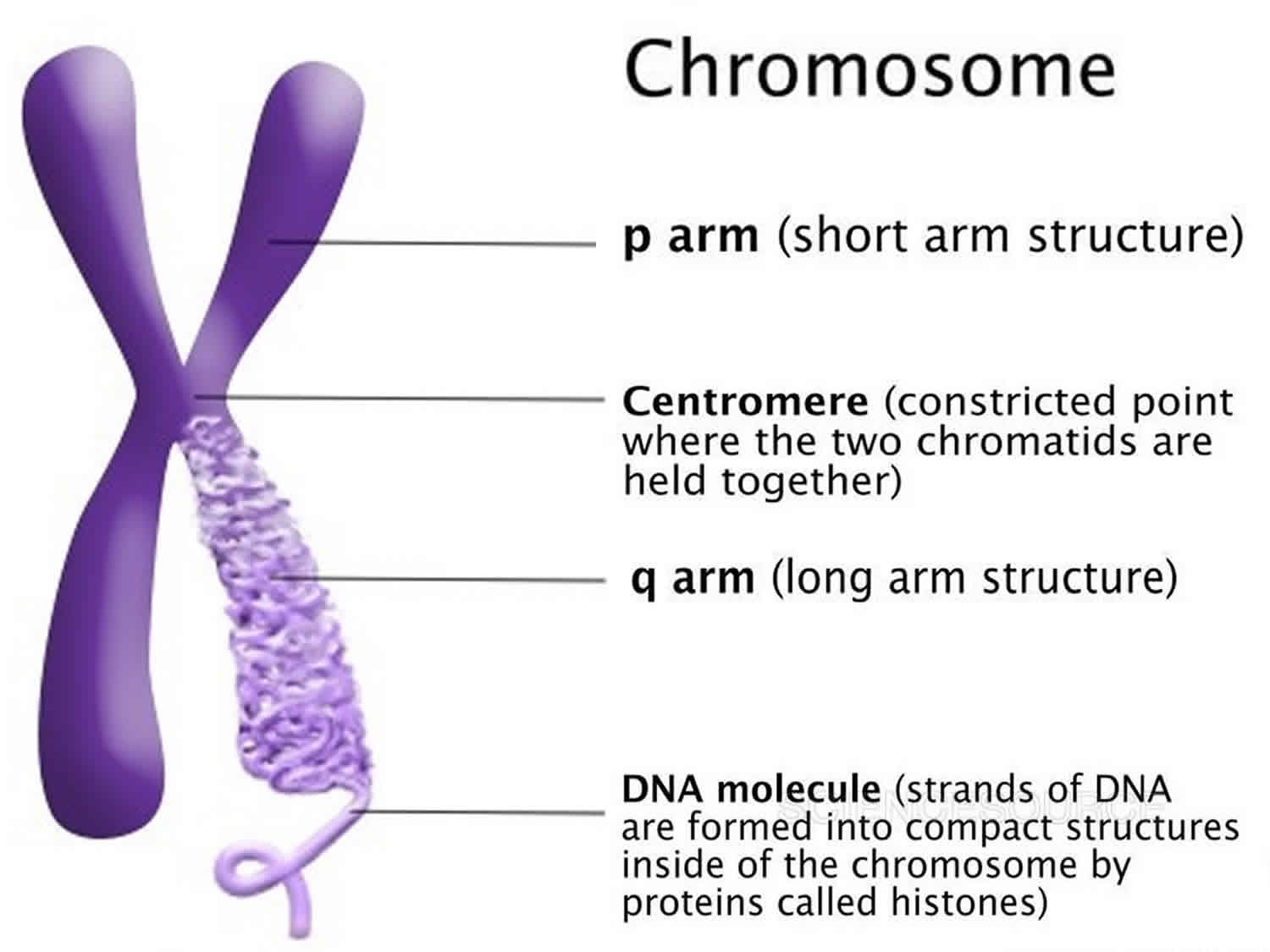
Figure 5. Chromosome structure
How many chromosomes do people have?
In humans, each cell normally contains 23 pairs of chromosomes, for a total of 46 chromosomes. One-half of each pair of chromosomes is inherited from an individual’s mother and the other half of the pair is inherited from an individual’s father. Twenty-two of the 23 pairs of chromosomes are called autosomes, look the same in both males and females. The 23rd pair, are the sex chromosomes, differ between males and females. Females have two copies of the X chromosome (XX), while males have one X and one Y chromosome (XY). The sex chromosomes determine a person’s sex; normal males have one X and one Y chromosome (XY) (see Figure 3 above) while normal females have two X chromosomes (XX) (see Figure 2 above).
The 22 autosomes are numbered by size. The other two chromosomes, X and Y, are the sex chromosomes. This picture of the human chromosomes lined up in pairs is called a karyotype (see Figure 1).
What is a gene?
A gene is the basic physical and functional unit of inheritance. Genes are passed from parents to offspring and contain the information needed to specify traits. Genes are pieces of DNA and most genes contain the information for making a specific protein. Some genes act as instructions to make molecules called proteins. However, many genes do not code for proteins. In humans, genes vary in size from a few hundred DNA bases to more than 2 million bases. Genes are arranged, one after another, on structures called chromosomes. Each chromosome contains many genes. A chromosome contains a single, long DNA molecule, only a portion of which corresponds to a single gene. The Human Genome Project estimated that humans have between 20,000 and 25,000 genes arranged on their chromosomes.
Every person has two copies of each gene, one inherited from each parent. Most genes are the same in all people, but a small number of genes (less than 1 percent of the total) are slightly different between people. Alleles are forms of the same gene with small differences in their sequence of DNA bases. These small differences contribute to each person’s unique physical features.
Scientists keep track of genes by giving them unique names. Because gene names can be long, genes are also assigned symbols, which are short combinations of letters (and sometimes numbers) that represent an abbreviated version of the gene name. For example, a gene on chromosome 7 that has been associated with cystic fibrosis is called the cystic fibrosis transmembrane conductance regulator; its symbol is CFTR.
46XX complete gonadal dysgenesis
46XX complete gonadal dysgenesis also called XX female gonadal dysgenesis, follicular stimulating hormone-resistant ovaries, hypergonadotropic ovarian dysgenesis or ovarian dysgenesis, is a primary ovarian defect leading to premature ovarian failure in otherwise normal 46,XX females as a result of failure of the gonads to develop or due to resistance to gonadotrophin stimulation 27.
Individuals with 46XX complete gonadal dysgenesis are born with typical female sex characteristics. However, affected individuals present during adolescence or young adulthood with either delayed or absent puberty resulting in primary or sometimes secondary amenorrhea which is absence of a menstrual period with primary amenorrhea is where a period never started by age 16 and secondary amenorrhea is where a period stops for three or more consecutive months. The internal and external genitalia are typically developed. 46XX complete gonadal dysgenesis or hypergonadotropic ovarian dysgenesis accounts for about half the cases of primary amenorrhea 28.
46,XX gonadal dysgenesis can occur as part of Perrault syndrome, which is a rare, autosomal recessive multisystem genetic disorder characterized by sensorineural hearing loss in both males and females, and ovarian dysfunction in females (primarily ovarian dysgenesis) 29, 30, 31, 32. Affected females experience delayed or absent puberty, leading to primary amenorrhea. Some individuals, regardless of sex, may also have neurological symptoms like developmental delays, intellectual disability, and cerebellar ataxia.
46XX complete gonadal dysgenesis can also occur as part of other rare syndromes such as lung fibrosis-immunodeficiency-46XX gonadal dysgenesis syndrome, a rare and fatal syndrome characterized by immune deficiency, gonadal dysgenesis, and fatal rapidly progressive lung fibrosis 33, 34, 35. Lung fibrosis-immunodeficiency-gonadal dysgenesis has been described in a small number of cases, such as two sisters from consanguineous parents, who had normal female karyotypes (46,XX) but severe immune abnormalities and fatal lung disease. The exact genetic cause is not yet identified.
46XX complete gonadal dysgenesis management should include hormone replacement therapy (HRT) to induce puberty, develop secondary sexual characteristics, and provide long-term health benefits. Calcium and vitamin D supplements are often recommended throughout life to support bone health. Psychological support should also be offered to patients and their families in a difference/disorder of sexual development-center setting. Infertility is an important management issue; however, pregnancy may be feasible through zygote egg donation.
46 XX complete gonadal dysgenesis causes
46 XX complete gonadal dysgenesis is caused by genetic mutations of ovarian development. Genetic mutations can be hereditary, when parents pass them down to their children, or they may occur randomly when cells are dividing. Genetic mutations may also result from contracted viruses, environmental factors, such as ultraviolet (UV) radiation from sunlight exposure, or a combination of any of these. Although the underlying cause remains unknown in most cases, several genes have been implicated including homozygous or compound heterozygous inactivating mutations of the follicle-stimulating hormone receptor gene (FSHR; 2p21-p16) of the ovary, mutations in the BMP15 gene (Xp11.2) and mutations in the NR5A1 gene (9q33), amongst others, whose functions are relevant for ovarian development 36, 37, 38. Inactivating mutations in the FSHR gene coding for the follicle-stimulating hormone receptor is mainly in the Finnish population 1. Mutation in follicle-stimulating hormone receptor (FSHR) of the ovary leads to decreased or no signaling leading to decreased follicle formation in the ovary causing the follicles to remain in the primary or antral stage. The depletion of the follicles is also seen with FSHR mutation 39.
Studies have tried to find other genes that might have specific links to 46, XX gonadal dysgenesis, and the gene BMP15 appears to be a possible candidate. The BMP15 gene encodes the bone morphogenetic protein (BMP) which is part of transforming growth factor-beta made by oocytes involved in folliculogenesis 40, 41. The products of BMP15 gene also stimulate granulosa cell proliferation. The transforming growth factor-beta proteins first become translated into pre-proteins with a single peptide, as well as a pro and mature region. The individual parts of the preprotein are required for proper posttranslational processing to produce active proteins. The lack of these active proteins is believed to have connections to decreased follicle production as well as depletion of ovarian follicles altogether 39. A rare amino acid substitution at p.A180T in BMP15 was present in a small number of patients with ovarian failure in several studies; additional studies have found that this amino acid substitution may be a rare polymorphism 39. However, a definite link to 46 XX complete gonadal dysgenesis remains unconfirmed 1.
Cases of 46 XX complete gonadal dysgenesis have also been found in congenital adrenal hyperplasia (CAH) most often are caused by mutations in the CYP21A2 gene 42. The CYP21A2 gene provides instructions for making an enzyme called 21-hydroxylase, which is part of the cytochrome P450 family of enzymes 43. Cytochrome P450 enzymes are involved in many processes in the body, such as assisting with reactions that break down drugs and helping to produce cholesterol, certain hormones, and fats (lipids) 43. The 21-hydroxylase enzyme is found in your adrenal glands, which are located on top of your kidneys and produce a variety of hormones that regulate many essential functions in the body. The 21-hydroxylase enzyme plays a role in producing hormones called cortisol and aldosterone. Cortisol helps maintain blood sugar (glucose) levels, protects the body from stress, and suppresses inflammation. Aldosterone is sometimes called the salt-retaining hormone because it regulates the amount of salt retained by the kidneys. The retention of salt affects fluid levels in the body and blood pressure.
The 21-hydroxylase enzyme typically catalyzes the hydroxylation of 17-hydroxyprogesterone to 11 deoxycortisol. When the 21-hydroxylase enzyme is deficient, it decreases the production of cortisol which increases adrenocorticotropic hormone (ACTH), a hormone produced by your pituitary gland that tells your adrenal glands to make cortisol which then increases cholesterol uptake by cells and increases the production of pregnenolone. The increased steroid precursors shunt toward testosterone production leading to development of male physical characteristics such as muscle bulk, body hair, and deep voice in a female or precociously in a boy, typically as a result of excess androgen production 42.
46 XX complete gonadal dysgenesis signs and symptoms
The types of symptoms experienced, and their intensity, may vary among people with 46XX complete gonadal dysgenesis, hypergonadotropic ovarian dysgenesis or ovarian dysgenesis. Your experience may be different from others. Symptoms of this disease may start to appear as a teenager and as an adult.
46XX complete gonadal dysgenesis or hypergonadotropic ovarian dysgenesis causes premature ovarian failure (POF) that is characterized by lack of spontaneous pubertal development, primary amenorrhea, uterine hypoplasia, and hypergonadotropic hypogonadism (primary hypogonadism) as a result of streak gonads.
Elliott et al 44 reported 46XX complete gonadal dysgenesis in 3 sisters who had normal stature and sex chromatin but had never menstruated and had severe osteoporosis. The parents were first cousins in the case of the 2 affected sisters (with normal stature and sex-chromatin positivity) reported by Klotz et al. 45. Christakos et al. 46 observed gonadal dysgenesis in 3 sisters whose parents were second cousins. Each had a normal female 46,XX karyotype. Somatic features of Turner syndrome were not found. All 3 had elevated gonadotropins, and laparotomy on the 2 older sisters showed streak gonads and unstimulated mullerian structures. Gonadal dysgenesis, often with somatic abnormalities, has been reported in siblings by several other authors and in some of these reports the parents were consanguineous. Simpson et al 47 pointed out that only affected siblings have been described and parental consanguinity is frequent. Vesely et al 48 reported 3 affected sisters and expressed the opinion that only the family reported by Elliott et al 44 was similar in having sisters above 152 cm in height, with no associated congenital anomalies. Aleem 49 described affected sisters, aged 16 and 17, who presented with secondary amenorrhea.
In a nationwide population-based study of women born between 1950 and 1976 in Finland, Aittomaki 50 identified 75 patients with XX gonadal dysgenesis. In 1 family, 4 daughters were affected; in 6 families, 2 daughters were affected; and 57 cases were isolated. In 1 additional family, there were 2 affected females in successive generations. Consanguinity was detected in 8 of 66 families (12%). When only females were considered, the segregation analysis yielded a proportion of 0.23 affected. The relatively high incidence of 1 in 8,300 liveborn girls implied a high gene frequency in the Finnish population. The geographic distribution was highly uneven, with most families originating in the sparsely populated north-central part of Finland. The findings supported the existence of an autosomal recessive XX gonadal dysgenesis gene, which Aittomaki 50 symbolized ODG1 (for ovarian dysgenesis-1), that is highly enriched in Finland. This is one of the examples of ‘Finnish diseases’ of which some 30 cases have been defined 51.
Aittomaki et al 52 studied 22 Finnish women with ovarian dysgenesis and the A189V mutation in the FSHR gene. All had primary amenorrhea, and pubertal development was reported as delayed in 6. Gonadotropin levels were elevated in all. Pelvic ultrasound, performed in 12 patients, showed streak or hypoplastic ovaries, with identifiable ovarian tissue always present. Ovarian biopsy was obtained in 9 of the patients and revealed consistently hypoplastic histology. Follicles were present in all ovaries, but the number of follicles was very low, and there was much more fibrosis compared to the normal prepubertal ovary. The authors noted that the birthplaces of mutation carriers (parents of patients) were almost entirely within the more lately settled parts of Finland, consistent with a recent founder effect, suggesting that enrichment of the A189V variant occurred mainly during the past 300 to 500 years 52.
46 XX complete gonadal dysgenesis diagnosis
During your appointment, your doctor will perform a pelvic exam to check for any problems with your reproductive organs. If you’ve never had a period, your doctor may examine your breasts and genitals to see if you’re experiencing the normal changes of puberty.
A variety of blood tests may be necessary:
- Pregnancy test. This will probably be the first test your doctor suggests, to rule out or confirm a possible pregnancy.
- Thyroid function test. Measuring the amount of thyroid-stimulating hormone (TSH) in your blood can determine if your thyroid is working properly.
- Ovary function test. Measuring the amount of follicle-stimulating hormone (FSH) in your blood can determine if your ovaries are working properly.
- Prolactin test. Low levels of the hormone prolactin may be a sign of a pituitary gland tumor.
- Male hormone test. If you’re experiencing increased facial hair and a lowered voice, your doctor may want to check the level of male hormones in your blood.
- Blood tests to look at chromosomes. This can show the genetic sex: typically, XX or XY. These blood tests also can show conditions caused by a gene change that affects sex organ development.
Imaging tests
Depending on your signs and symptoms — and the result of any blood tests you’ve had — your doctor might recommend one or more imaging tests, including:
- Ultrasound of the pelvis and belly to look for testicles that haven’t descended, a uterus or a vagina. If you have never had a period, your doctor may suggest an ultrasound test to check for any abnormalities in your reproductive organs.
- Magnetic resonance imaging (MRI). MRI uses radio waves with a strong magnetic field to produce exceptionally detailed images of soft tissues within the body. Your doctor may order an MRI to check for a pituitary tumor.
- X-ray studies using a contrast dye to help give a clear picture of pelvic and belly structures.
If other testing reveals no specific cause, your doctor may recommend a hysteroscopy — a test in which a thin, lighted camera is passed through your vagina and cervix to look at the inside of your uterus. And sometimes minimally invasive surgery (laparoscopy) is needed to collect a tissue sample of your ovarian tissue. This surgery is done through one or more small cuts using tiny cameras and surgical tools.
46 XX complete gonadal dysgenesis treatment
Treatment for 46XX gonadal dysgenesis involves hormone replacement therapy to induce puberty and managing your long-term health problems, psychological support, and a discussion about fertility options like egg donation 53. Estrogen and progesterone are key components of hormone therapy to develop secondary sexual characteristics and protect your bone and cardiovascular health.
Hormone replacement therapy (HRT)
Hormone replacement therapy is used to induce puberty, develop secondary sexual characteristics, and provide long-term health benefits.
- Estrogen: Administered to help the body go through puberty and menstrual cycles. It is essential for protecting against bone density loss (osteopenia/osteoporosis) and maintaining cardiovascular and urogenital health.
- Progesterone: Added after 12 months of estrogen or after menstruation begins, to protect against endometrial hyperplasia and potential cancer risk.
- Other options: Combined oral contraceptives can also be used.
Bone health
- Supplementation with calcium and vitamin D are often recommended throughout life to support bone health.
- Your doctor will also monitor for and manage any bone loss. Bisphosphonates may be considered for severe bone loss that doesn’t improve with hormone replacement therapy.
Fertility and pregnancy
Infertility is a consequence of 46XX gonadal dysgenesis. Pregnancy may be possible with the use of donated eggs. After proper endometrial stimulation, an embryo from a donated egg can be implanted.
Psychological and emotional support
Psychological support is a crucial part of management for patients and their families, especially regarding infertility concerns. With appropriate management, the physiological and clinical outcome for patients is good.
Other considerations
- Early diagnosis: Early diagnosis is vital for promptly starting treatment, providing support, and reducing long-term health risks associated with hormone deficiency.
- Regular monitoring: Lifelong follow-up care is recommended to monitor hormone levels and overall health.
46 XY complete gonadal dysgenesis (Swyer syndrome)
46XY complete gonadal dysgenesis also known as Swyer syndrome or 46XY pure gonadal dysgenesis is a condition that occurs in individuals with “XY” male sex chromosomes in each of their cells, but has female external genitalia and streak gonads (ovaries or testes), which have a high risk of becoming cancerous. People with Swyer syndrome or 46XY complete gonadal dysgenesis have an XY chromosomal makeup (as males normally do) but have female external genitalia and some female internal reproductive structures. Individuals with Swyer syndrome or 46XY complete gonadal dysgenesis usually have a vagina, uterus and fallopian tubes, but their gonads (ovaries or testes) are not functional. Instead, their gonads (ovaries or testes) are small and underdeveloped and contain little gonadal tissue. These structures are called “streak gonads”. The streak gonadal tissue is at risk of developing cancer that is often hard-to-detect, so it is usually removed surgically.
Individuals with Swyer syndrome lack sex glands ( testicles or ovaries). Instead of sex glands, individuals with Swyer syndrome have “gonadal streaks”, in which the ovaries do not develop properly (aplasia) and are replaced by functionless scar (fibrous) tissue. Because they lack ovaries, girls with Swyer syndrome do not produce sex hormones and will not undergo puberty (unless treated with hormone replacement therapy).
46XY complete gonadal dysgenesis or Swyer syndrome is classified as a disorder of sex development (DSD), which encompasses any disorder in which chromosomal, gonadal or anatomic sex development is abnormal. Swyer syndrome is a rare disorder characterized by the failure of the sex glands (i.e., testicles or ovaries) to develop. Mutations in several different genes are known to cause Swyer syndrome. Swyer syndrome can occur as the result of a new gene mutation or can be inherited in an autosomal dominant, autosomal recessive, X-linked or Y-linked manner.
Swyer syndrome may be identified before birth, at birth, or later when a child does not go through puberty as usual. Because they do not have functional ovaries that produce hormones, affected individuals often begin hormone replacement therapy during early adolescence to start puberty, causing the breasts and uterus to grow, and eventually leading to menstruation. Hormone replacement therapy is also important for bone health and helps reduce the risk of low bone density (osteopenia) and fragile bones (osteoporosis). Women with Swyer syndrome do not produce eggs (ova), but if they have a uterus, they may be able to become pregnant with a donated egg or embryo.
Swyer syndrome also called 46XY complete gonadal dysgenesis (lack of development of the gonads), is a condition in which people with one X chromosome and one Y chromosome (normally present in males) have a female appearance. People usually have 46 chromosomes in each cell. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male or female sex characteristics. Girls and women typically have two X chromosomes (46,XX karyotype), while boys and men usually have one X chromosome and one Y chromosome (46,XY karyotype). In Swyer syndrome, individuals with one X chromosome and one Y chromosome in each cell, the pattern typically found in boys and men, have female reproductive structures. People with Swyer syndrome have typical female external genitalia. The uterus and fallopian tubes are normally-formed, but the gonads (ovaries or testes) are not functional; affected individuals have undeveloped clumps of tissue called streak gonads. Streak gonads often become cancerous, so they are usually surgically removed as early as possible. In addition to removal of streak gonads, treatment may include hormone replacement therapy from puberty onward.
People with Swyer syndrome are typically raised as females, have a female gender identity, and have typical female external genitalia. The uterus and Fallopian tubes may be normal, or they may be small or underdeveloped 54. Because they do not have functional ovaries, affected individuals usually begin hormone replacement therapy during adolescence to induce menstruation and development of female secondary sex characteristics such as breast enlargement and uterine growth. Hormone replacement therapy also helps reduce the risk of reduced bone density (osteopenia and osteoporosis). Women with Swyer syndrome do not produce eggs (ova) and are infertile, but they may be able to become pregnant with a donated egg or embryo 55.
Swyer syndrome may be caused by mutations in any of several genes. The inheritance pattern depends on the responsible gene 56.
Swyer syndrome usually affects only sexual development; such cases are called isolated Swyer syndrome. However, depending on the genetic cause, Swyer syndrome may also occur along with health conditions such as nerve problems (neuropathy) or as part of a syndrome such as campomelic dysplasia, which causes severe skeletal abnormalities.
Swyer syndrome occurs in approximately 1 in 80,000 people 57. Another estimate placed the incidence of Swyer syndrome (complete gonadal dysgenesis) and partial gonadal dysgenesis combined at 1 in 20,000 births. Genital anomalies in general occur in approximately 1 in 4,500 births.
The treatment of a person with Swyer syndrome may depend on the specific characteristics that each person has. Some people need surgery to repair the external genitalia and to create and/or enlarge the vagina. Hormone replacement therapy (HRT) is typically needed from puberty onward and usually includes estrogen and progesterone 58. In addition to helping with normal development of secondary sexual characteristics, hormone replacement therapy can help prevent bone loss and thinning (osteoporosis) later in life. Abdominal dysgenetic gonads (testes or ovaries with abnormal development) or streak gonads, which are common in people with Swyer syndrome, are at increased risk for gonadal tumors such as gonadoblastoma and should be surgically removed. Although women with Swyer syndrome are infertile, they may become pregnant and carry to term through egg donation 58.
46 XY complete gonadal dysgenesis (Swyer syndrome) causes
In most cases of 46XY complete gonadal dysgenesis or Swyer syndrome, the exact cause of the disorder is unknown 59, 60, 61, 62. Researchers believe that disruptions or changes (mutations) of a gene or genes that are involved in normal sex differentiation of a fetus with an XY chromosomal makeup cause Swyer syndrome.
In approximately 15-20 percent of patients, 46XY complete gonadal dysgenesis or Swyer syndrome occurs due to mutations of the sex-determining region Y (SRY) gene on the Y chromosome or deletion of the segment of the Y chromosome containing the SRY gene 63, 64. The SRY gene, located on the Y chromosome, provides instructions for making the sex-determining region Y protein. This protein is a transcription factor, which means it attaches (binds) to specific regions of DNA and helps control the activity of particular genes. The sex-determining region Y (SRY) protein starts processes that are involved in male sexual development. These processes cause a fetus to develop male gonads (testes) and prevent the development of female reproductive structures (uterus and fallopian tubes). SRY gene mutations that cause Swyer syndrome prevent production of the sex-determining region Y (SRY) protein or result in the production of a nonfunctioning protein. A fetus whose cells do not produce functional sex-determining region Y (SRY) protein will not develop testes but will develop a uterus and fallopian tubes, despite having a typically male karyotype (male sex XY chromosomes).
Since only 15-20 percent of individuals with 46XY complete gonadal dysgenesis or Swyer syndrome have a mutation of the SRY gene, researchers believe that defects involving other genes can also cause the disorder. These other genes are all suspected to play a role in the promoting the development of the testes and, ultimately, the differentiation of an XY fetus into a male. Mutations in the Map3K1 are also a common cause of Swyer syndrome.
Some women with Swyer syndrome have mutations in the NROB1 gene on the X chromosome. Investigators have linked other cases of Swyer syndrome to mutations of the desert hedgehog (DHH) gene located on chromosome 12. Mutations in the DEAH37 gene have been identified as a common cause. A few rare cases have been associated with mutations in the steroidogenic factor 1 (SF1 or NR5A1) gene, the protein Wnt-4 (WNT4) gene, SOX9, GATA4, FOG2, NR5A1, WT1 and the CBX2, GATA4 and WWOX genes, many of which code for transcription factors 42, 61, 63. The DHH gene provides instructions for making a protein that is important for early development of tissues in many parts of the body. The NR5A1 gene provides instructions for producing another transcription factor called the steroidogenic factor 1 (SF1). This protein helps control the activity of several genes related to the production of sex hormones and sexual differentiation. Mutations in the DHH and NR5A1 genes affect the process of sexual differentiation, preventing affected individuals with a typically male karyotype from developing testes and causing them to develop a uterus and fallopian tubes.
Mutations in MAP3K1 gene have been found in 13% to 18% of patients with 46, XY gonadal dysgenesis. The MAP3K1 gene provides instructions for making a protein that helps regulate signaling pathways that control various processes in the body. These include the processes of determining sexual characteristics before birth. The mutations in MAP3K1 gene that cause Swyer syndrome were found to cause a gain of function effects including phosphorylation of targets leading to decreased expression of SOX9. SOX9 is vital for testes development and is linked to beta-catenin another factor in sexual development 65. Mutations in MAP3K1 gene decrease signaling that leads to male sexual differentiation and enhance signaling that leads to female sexual differentiation, preventing the development of testes and allowing the development of a uterus and fallopian tubes.
Researchers believe that additional, as yet unidentified, genes may also be associated with the development of Swyer syndrome.
Changes affecting other genes have also been identified in a small number of people with Swyer syndrome. Nongenetic factors, such as hormonal medications taken by the mother during pregnancy, have also been associated with this condition. However, in most individuals with Swyer syndrome, the cause is unknown.
Some cases of Swyer syndrome are not believed to be inherited, but rather the result of a new genetic mutation (de novo mutation) or abnormality that occurs for unknown reasons (spontaneously). However, some individuals with Swyer syndrome due to mutation of the SRY gene have had fathers and some even brothers who also have the SRY mutation on the Y chromosome. It is not known why, in these cases, the fathers and/or brothers did not develop Swyer syndrome. Researchers speculate that other genes and/or factors in combination with a mutation of the SRY gene may be necessary for the development of Swyer syndrome in these patients.
Cases of Swyer syndrome due to mutation of the NROB1 gene may be inherited in an X-linked pattern. X-linked genetic disorders are conditions caused by an abnormal gene on the X chromosome. Females usually have two X chromosomes (XX sex chromosomes) and one of the X chromosomes is “turned off” and all of the genes on that chromosome are inactivated. Females who have a disease gene present on one of their X chromosomes usually do not display symptoms of the disorder because it is usually the X chromosome with the abnormal gene that is “turned off”. However, because women with Swyer syndrome have an XY chromosomal makeup and lack a second X chromosome, they will express symptoms associated with a defect on their one X chromosome.
According to the medical literature, some cases of Swyer syndrome appear to follow autosomal dominant or recessive inheritance. Autosomal inheritance refers to the transmission of genes located on the 22 pairs of non-sex chromosomes (autosomes). There are two main types: autosomal dominant, where only one copy of a mutated gene from one parent is needed to cause a condition, and autosomal recessive, where both copies of a gene must be mutated for the condition to appear. Both males and females are equally affected in autosomal inheritance, as it does not involve the sex chromosomes. Mutations of the WNT4, MAP3K1 or the SF1 (NR5A1) genes may be inherited in as autosomal dominant pattern. Mutation of the DHH gene may be inherited in an autosomal recessive manner.
Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease. The abnormal gene can be inherited from either parent or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females. In some individuals, the disorder is due to a spontaneous (de novo) genetic mutation that occurs in the egg or sperm cell. In such situations, the disorder is not inherited from the parents.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Affected individuals are encouraged to seek genetic counseling for answers to any questions regarding the complex genetic factors involved in Swyer syndrome.
46 XY complete gonadal dysgenesis (Swyer syndrome) inheritance pattern
Most cases of Swyer syndrome are not inherited; they occur in people with no history of the condition in their family. These cases result either from nongenetic causes or from new (de novo) mutations in a gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development.
SRY-related Swyer syndrome is usually caused by a new mutation. However, some individuals with Swyer syndrome inherit an altered SRY gene from an unaffected father who is mosaic for the mutation. Mosaic means that an individual has the mutation in some cells (including some reproductive cells) but not in others. In rare cases, a father may carry the mutation in every cell of the body but also has other genetic variations that prevent him from being affected by the condition. Because the SRY gene is on the Y chromosome, Swyer syndrome caused by SRY gene mutations is described as having a Y-linked inheritance pattern.
When Swyer syndrome is associated with an MAP3K1 or NR5A1 gene mutation, the condition is also usually caused by a new mutation. In the rare inherited cases, the mutation may be inherited from either parent, since these genes are not on the Y chromosome. In these cases, the condition has an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the condition.
Cases of Swyer syndrome due to mutation of the NROB1 gene may be inherited in an X-linked pattern. X-linked genetic disorders are conditions caused by an abnormal gene on the X chromosome. Females usually have two X chromosomes and one of the X chromosomes is “turned off” and all of the genes on that chromosome are inactivated. Females who have a disease gene present on one of their X chromosomes usually do not display symptoms of the disorder because it is usually the X chromosome with the abnormal gene that is “turned off”. However, because women with Swyer syndrome have an XY chromosomal makeup and lack a second X chromosome, they will express symptoms associated with a defect on their one X chromosome.
Swyer syndrome caused by mutations in the DHH gene is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition are carriers of one copy of the altered gene. Female carriers of a DHH gene mutation generally have typical sex development. Male carriers of a DHH gene mutation may also be unaffected, or they may have genital differences such as the urethra opening on the underside of the penis (hypospadias).
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
46 XY complete gonadal dysgenesis (Swyer syndrome) signs and symptoms
Most individuals with Swyer syndrome do not experience any outward symptoms until their early teens when they fail to begin having a period (primary amenorrhea). At this point, it is usually discovered that these girls lack ovaries and, therefore, do not have sex hormones (estrogen or progesterone) that are required to undergo puberty. When hormone replacement therapy is started, these girls will develop enlarged breasts, underarm and pubic hair, regular menstrual cycles and other aspects of normal development during puberty.
Women with Swyer syndrome may be tall and often have a small uterus and a slightly enlarged clitoris in comparison to most women. Because women with Swyer syndrome lack ovaries, they are infertile. However, they can become pregnant through the implantation of donated eggs.
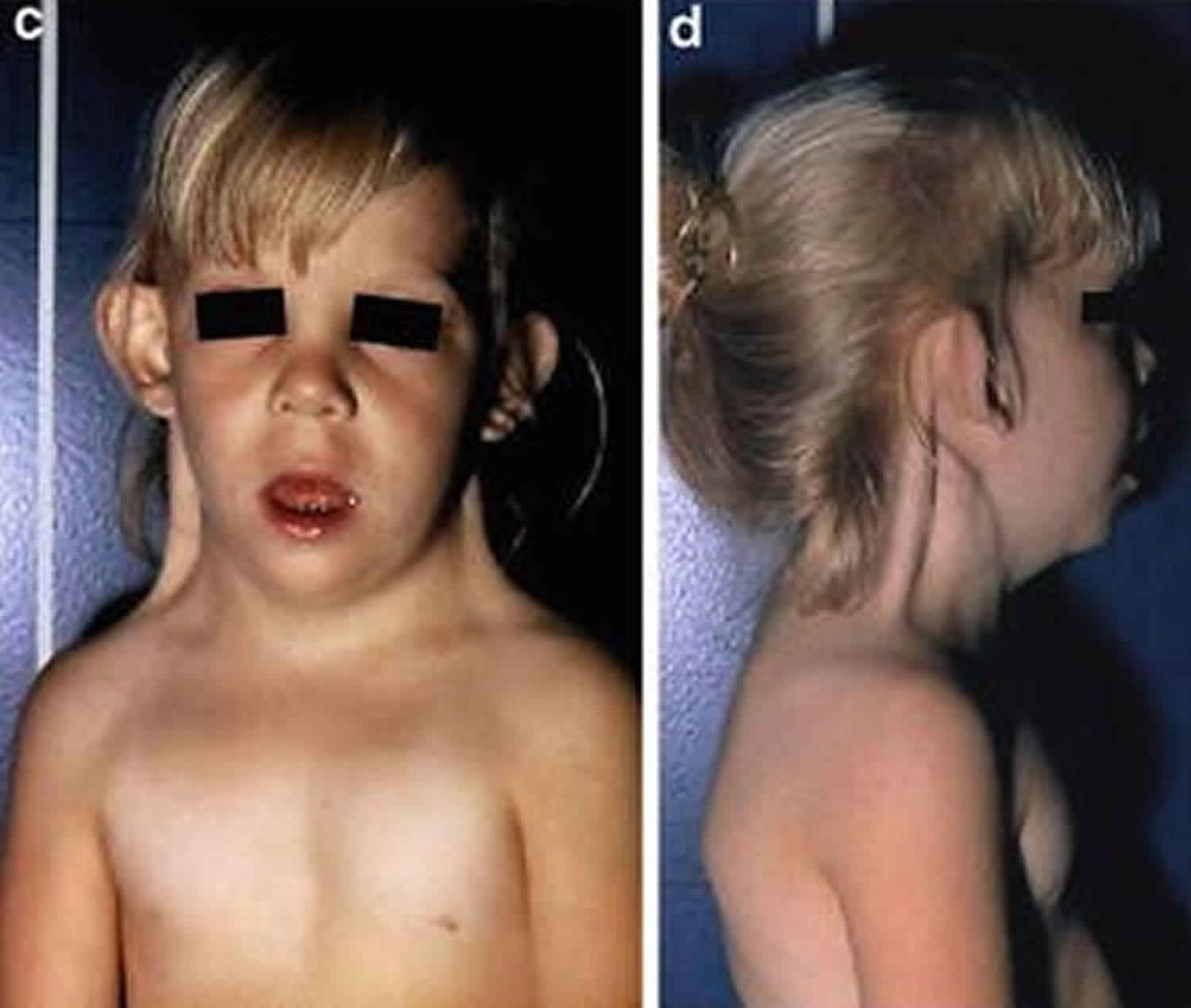
A chief medical concern of women with Swyer syndrome is an increased risk of developing cancer of the underdeveloped gonadal tissue. Approximately 30 percent of women with Swyer syndrome develop a tumor that arises from the cells that forms the testes or ovaries (gonadal tumor). The most common gonadal tumor in women with Swyer syndrome is a gonadoblastoma, a benign (non-cancerous) tumor that occurs exclusively in people with defective development of the gonads. A gonadoblastoma usually does not become malignant or spread. Gonadoblastomas, however, may be precursors to the development of a malignant (cancerous) tumor such as a dysgerminoma, which has also been reported to occur with greater frequency in women with Swyer syndrome than in the general population.
Gonadal tumors can develop at any age including during childhood before a diagnosis of Swyer syndrome is even suspected.
46 XY complete gonadal dysgenesis (Swyer syndrome) diagnosis
A diagnosis of Swyer syndrome is made based upon a thorough clinical evaluation, a detailed patient history, identification of characteristic findings (e.g., no periods, streak gonads) and a variety of tests including chromosomal analysis. For example, a specific technique called fluorescent in situ hybridization (FISH) can be used to determine a person’s karyotype. A karyotype is a visual representation of a person’s chromosomal makeup, (i.e., the 46 chromosomes in a cell). These 46 chromosomes are broken down into 22 matched pairs (each pair has one chromosome received from the father and one receive from the mother). The sex chromosomes are seen as a separate pair, either XX or XY. A diagnosis of Swyer syndrome is usually made when young adults are tested for delayed puberty.
Molecular genetic testing can determine whether one of the specific gene mutations that are associated with Swyer syndrome is present in an affected individual.
Evaluation of immediate family members of an affected person can be helpful in determining if the condition is sporadic or inherited in that family.
46 XY complete gonadal dysgenesis (Swyer syndrome) treatment
The treatment of Swyer syndrome may require the coordinated efforts of a team of specialists. Pediatricians, pediatric endocrinologists, geneticists, urologists or gynecologists, psychologists or psychiatrists, social workers and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment.
Swyer syndrome is treated with hormonal replacement therapy including replacing estrogen and progesterone that is usually begun from puberty onward. In addition to helping with normal development of secondary sexual characteristics, hormone replacement therapy can also help prevent bone loss and thinning (osteoporosis) later during life.
Streak gonads are usually removed surgically because they place affected individuals at an increased risk of developing a gonadal tumor.
Individuals with SF1 mutations may have adrenal insufficiency. This should be investigated and treated, if present.
Genetic counseling is recommended for affected individuals and their families. Other treatment is symptomatic and supportive.
Although women with Swyer syndrome are infertile, they may become pregnant and carry to term through the use of donated eggs.
Mixed gonadal dysgenesis
A form that occurs in individuals with a Y chromosome mosaicism, where one gonad is a streak gonad and the other is a partially developed testicle.
Partial gonadal dysgenesis
Occurs when gonads partially form but do not produce enough sex hormones, leading to ambiguous or nonbinary external genitalia.
Gonadal dysgenesis causes
Gonadal dysgenesis can be due to either structural or numerical anomalies of the sex chromosomes or mutations in the genes involved in the development of the gonad 1, 2, 3, 4, 5. Sex chromosomes carry the genes that initiate development pathways when a baby is growing and developing in utero. When the pathways to develop gonads are interrupted or altered the body is unable to develop them in a typical way. Without functioning gonads the body is unable to produce hormones or gametes, which can impact physical development. Some kinds of gonadal dysgenesis can be inherited, such as Swyer syndrome. This can be inherited in a number of ways depending on the specific genetic change at play. Inheritance may be autosomal dominant, autosomal recessive, X chromosome-linked or Y chromosome-linked.
Gonadal dysgenesis signs and symptoms
Signs and symptoms of gonadal dysgenesis vary but include ambiguous genitalia where an infant’s external genitals do not appear clearly male or female at birth, delayed or absent puberty, and a lack of menstruation (primary amenorrhea). Some individuals may also have an average female appearance but experience these pubertal delays, while others may have associated conditions like hearing loss or kidney problems.
Signs and symptoms at birth or in infancy
- Ambiguous or atypical appearance of the genitals
- Being born with two different gonads (in mixed gonadal dysgenesis)
Signs and symptoms in adolescence or adulthood
- Delayed or absent puberty
- Lack of menstruation (primary amenorrhea)
- Failure to develop breasts
- Taller than average stature (depending on the specific type)
- Infertility (inability to produce eggs or sperm)
Associated conditions
- Hearing loss: Some forms of XX gonadal dysgenesis are linked to Perrault syndrome, which includes sensorineural hearing loss.
- Kidney issues: Kidney disease or abnormalities in kidney development can be associated with gonadal dysgenesis.
- Other developmental abnormalities: Some cases can involve other malformations, such as a small head size, abnormal eye movements, or poor motor skills.
- Heart disease: In some XY gonadal dysgenesis cases, there may be an increased suspicion for a GATA4 mutation, which can also be linked to congenital heart disease.
- Autoimmune disorders: Individuals with some forms, such as Turner syndrome, may be more susceptible to autoimmune thyroiditis.
Gonadal dysgenesis diagnosis
Doctors will look for signs like a tall stature, absent breast development, and lack of pubic or body hair. Most patients with gonadal dysgenesis will begin a necessary workup for their symptoms, including an follicle-stimulating hormone (FSH), luteinizing hormone (LH) level and estradiol to assess gonadal function and serum electrolytes/chemistry levels. These tests mainly aim at excluding a salt-wasting form of congenital adrenal hyperplasia (CAH). Basic values to be determined include 17-hydroxyprogesterone, cortisol, estradiol, androstenedione, testosterone, and dihydrotestosterone. 17-hydroxyprogesterone levels should not be measured until day 3 or 4, because the stress of delivery may result in physiologic elevation of its levels in the first two days of life.
In the absence of palpable testes, markedly elevated luteinizing hormone (LH) levels or lack of testosterone elevation in response to human chorionic gonadotropin (hCG) stimulation can indicate the absence of normally functioning testicular tissue in patients with nonpalpable gonads. The human chorionic gonadotropin (hCG) stimulation test can also help to diagnose 5-alpha-reductase deficiency and distinguish impaired testosterone synthesis from androgen insensitivity. The human chorionic gonadotropin (hCG) stimulation test is not needed in the first 3 months of life, when there is a natural surge of gonadotropin with a subsequent increase in the testosterone level. Luteinizing hormone (LH), follicle-stimulating hormone (FSH), and testosterone levels are most informative at one week of life.
Other less frequently ordered tests include 5-alpha-reductase type II levels and antimüllerian hormone (AMH)/müllerian-inhibiting substance (MIS) levels. Androgen-receptor characterization is performed in specialized laboratories.
Imaging Studies
A pelvic ultrasound or MRI helps to visualize the internal reproductive anatomy and the size and location of the gonads as well as the perineal region and of the entire urinary tract, is recommended 66.
Pelvic ultrasound is helpful for precisely defining the müllerian anatomy, and it can be helpful for evaluating the location and anatomy of nonpalpable gonads. A cyst in the gonad could be suggestive of ovotestis.
Renal-bladder ultrasound is recommended in view of the high prevalence of congenital anomalies of the kidney and urinary tract in patients with gonadal dysgenesis. The newborn adrenal glands may be enlarged in patients with congenital adrenal hyperplasia (CAH) and usually depict a cribriform appearance. However, it is important to note that normal-appearing adrenal glands on ultrasound do not exclude congenital adrenal hyperplasia (CAH). Ambiguous genitalia, enlarged adrenal glands, and the presence of müllerian structures in a newborn are virtually pathognomonic for congenital adrenal hyperplasia (CAH).
Genitography can help determine the ductal anatomy. In a neonate with ambiguous genitalia, a catheter can be inserted into the distal urogenital sinus (urethra). Contrast is injected to outline the internal ductal anatomy. Findings may indicate normal urethral anatomy, an enlarged utricle in a male, or a common urogenital sinus in a female and can help define the level of vaginal and urethral confluence. Contrast-enhanced ultrasound genitography may be used as an alternative 67. For operative planning, some surgeons prefer endoscopic evaluation to genitography.
Magnetic resonance imaging (MRI) is usually not indicated, but it may help identify the internal anatomy, intra-abdominal gonads, or an associated suspected spinal cord pathology. Cost and the need for general anesthesia are obvious limitations on its use.
Cystourethroscopy before or during the definitive reconstructive surgical procedure may help better define the anatomy of the urogenital sinus and determine the exact location of the confluence where the vagina meets the urethra (ie, high, intermediate, or low).
Prenatal diagnosis may be pursued in cases of abnormal maternal triple or quadruple screens, or if the fetus presents with atypical features in the womb. All fetal testing, including sonographic examination of the fetal genitalia and fetal karyotyping by amniocentesis or free fetal DNA, should be documented 68. For example, Turner syndrome may present prenatally as coaction of the aorta, intrauterine growth restriction, oligohydramnios, polyhydramnios, increased nuchal translucency, cystic hygroma, or nonimmune hydrops 13. Although these may be signs of Turner syndrome, a definitive diagnosis is only by a karyotype. A karyotype involves sending whole blood in a sodium heparin tube to the lab for testing, which may take up to a week, but a rushed result utilizing fluorescence in situ hybridization can identify monosomy X in under 24 hours 69.
Genetic testing
Chromosomal analysis is a basic examination. A karyotype is performed to analyze the chromosomes, which is crucial for determining the specific type of gonadal dysgenesis. Chromosomal analysis allows for the differentiation of 46,XX gonadal dysgenesis and 46,XX gonadal dysgenesis, which can guide further diagnostic testing. For individuals with a Y chromosome, further genetic testing may check for mutations in specific genes like the SRY gene. Molecular cytogenetic examinations can detect specific structural rearrangements. For example, fluorescence in-situ hybridization (FISH) can help define the sex-determining region of the Y chromosome (SRY) in some cases of testicular gonadal dysgenesis. Array-comparative genomic hybridization can detect microdeletions or duplications and is important in cases of XY gonadal dysgenesis or the Mayer-Rokitansky-Kuster-Hauser syndrome. Single gene testing, whole exome sequencing, and whole genome sequencing are sometimes indicated in patients with gonadal dysgenesis. Advanced genetic testing is optional, based on initial evaluation, family preference, availability, and cost.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Laparoscopy
Sometimes a minimally invasive surgery (laparoscopy) is needed to biopsy of intra-abdominal gonads. This surgery is done through one or more small cuts using tiny cameras and surgical tools. Ideally, bilateral deep and longitudinal biopsies are required for optimal histologic evaluation. Furthermore, laparoscopy can help delineate the internal duct anatomy without the morbidity associated with open exploration. Currently, open exploration is rarely used to help identify the internal duct anatomy and obtain biopsy specimens of gonadal tissue for histologic characterization. Removal of the gonads and reproductive organs should be avoided until a diagnosis is made and a gender has been assigned. Histologic analysis of gonadal biopsy specimens may identify ovarian tissue, testicular tissue, ovotestes, or streak gonads.
Other tests
Depending on the clinical presentation, other tests may be performed, such as a biopsy and blood tests for tumor markers (like alpha-fetoprotein (AFP), lactate dehydrogenase (LDH), and beta human chorionic gonadotropin hormone (beta-hCG)) if a gonadal mass is suspected
In all patients with suspected gonadal dysgenesis, determination of whether a Y chromosome is present, including mosaic as well as partial and fragment Y chromosomal forms, is necessary due to risks of gonadoblastoma and germ cell tumor associated with the retained gonads 14, 15, 16, 17. After the initial analysis and karyotype, a specialist, most likely an endocrinologist, may perform further testing for specific gene mutations to try to determine the cause of the patient’s gonadal dysgenesis as well as gain an understanding of what other symptoms they may develop based on specific genetic alterations. The specific genetic testing to be performed is determined by the clinical presentation of the patient and their symptomatic presentation. The genes with known associations to their symptoms can be tested. In addition to the above testing, patients who have evidence of a Y chromosome, as well as evidence of a mass on the gonad, will need to have a biopsy performed as well as tumor markers including alpha-fetoprotein (AFP), lactate dehydrogenase (LDH), and beta human chorionic gonadotropin hormone (beta-hCG) to determine the presence of gonadoblastoma or germ cell tumor 3 Although genetic testing is an essential part of the diagnosis and management of gonadal dysgenesis, the patients will undergo a multitude of testing during their lifetime to aid in the management of their condition. This testing can include basic labs, i.e., complete blood count (CBC), complete metabolic panel (CMP), echocardiography, and renal ultrasound, to ensure all the patient’s associated symptoms are addressed and managed.
Early recognition can help prevent growth failure and hearing problems as well as better manage chronic kidney and heart conditions associated with Turner syndrome. A karyotype in an older child is necessary when there is a high suspicion of Turner syndrome. For example, if a child has a growth delay or lack of puberty and secondary sexual characteristics, or if a child exhibited other signs such as a webbed neck or widely spaced nipples, it would serve as an indication for a karyotype. If high suspicion exists of a Turner syndrome diagnosis and initial blood karyotype is normal, another tissue should be tested, such as skin, to determine the child’s genetic make-up. All patients should also have analysis to determine if there is any presence of a Y chromosome, due to the risk of gonadoblastoma and germ cell tumor. Analysis of a Y chromosome should be performed in the prenatal period as well 13. Similar to Turner syndrome, testing is also indicated for 46 XY complete gonadal dysgenesis when clinical findings suspect the diagnosis. A patient with delayed puberty or amenorrhea that has elevated basal luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels and other causes for amenorrhea and delayed puberty ruled out can have a karyotype performed. Pelvic ultrasound or MRI may also reveal streak gonads, which would indicate a karyotype 3. 46 XX gonadal dysgenesis will present similarly to 46 XY gonadal dysgenesis with delayed puberty, amenorrhea, increased luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels, and streak gonads may be present; again, clinical suspicion is the leading indication for testing in this disorder.
Gonadal dysgenesis treatment
The treatment of gonadal dysgenesis requires the coordinated efforts of a team of specialists in order to provide effective diagnosis, treatment, and support. Pediatricians, pediatric endocrinologists, geneticists, urologists or gynecologists, psychologists or psychiatrists, social workers and other healthcare professionals as needed to systematically and comprehensively plan the treatment.
Treatment for gonadal dysgenesis involves hormone replacement therapy (HRT), which is essential for pubertal development and long-term health. It may also include surgery to remove the gonads (gonadectomy) to reduce the risk of tumors, and psychological and fertility support. The specific hormonal treatment (e.g., estrogen and progesterone or testosterone) is chosen based on an individual’s identity and goals for puberty.
Gender Assignment
Assigning a gender on the basis of anatomic (phenotype) and biologic criteria without being certain of the individual’s ultimate gender identity is one of the major challenges of disorders of sex development (DSDs) management. Because each patient is unique, as is each situation, the aim of the multidisciplinary team should be to provide individualized treatment. The therapeutic approaches to the spectrum of disorders of sex development (DSDs) vary widely, and it would be an oversimplification to generalize the management of these conditions. After a diagnosis is made and the anatomy is defined, the multidisciplinary team should have a thorough and candid discussion with the patient and family regarding future potential sexual function, fertility, and malignancy risk.
In 46,XX overvirilized females, gender assignment is typically female. These patients have normal ovaries and female internal organs with normal reproductive potential. In patients with 46,XY gonadal dysgenesis, however, gender assignment is more complex and relies on several factors, including penile length, androgen insensitivity, and future fertility potential.
In children, decisions about disorders of sex development (DSDs) management are made by parents on behalf of their children. The American Medical Association (AMA) continues to support parents’ rights in making shared and informed decisions on behalf of their children while also supporting children’s involvement in their own care and right to self-determination when possible. Parent counseling should include all treatment options, including medical management, surgical intervention, gender assignment, and nonintervention. The goal should be the development of happy and healthy children and adults throughout their lives. Treatment decisions should be made jointly between parents and the multidisciplinary team.
Hormone replacement therapy (HRT)
- Purpose: To induce puberty, develop secondary sex characteristics, and maintain bone health.
- Feminizing puberty: Individuals raised as girls typically receive estrogen and progesterone. Progesterone should be given with estrogen if a uterus is present to lower the risk of endometrial carcinoma.
- Masculinizing puberty: Individuals raised as males may receive testosterone.
- Decision-making: The choice of hormones should be made in consultation with a doctor, considering personal preference, identity, and potential side effects.
Surgery
- Gonadectomy: Surgery to remove the gonads is often recommended to lower the risk of developing tumors, such as gonadoblastoma, which is a risk in individuals with gonadal dysgenesis or if sex hormone production is discordant with the chosen sex. Gonadectomy at an early age for conditions associated with an increased risk of malignancy at an early age remains an ethical dilemma. Regular screening must be guaranteed for patients with increased gonadal malignancy risk who retain their gonads into adulthood. If gonadectomy is considered, cryopreservation should be discussed if vital spermatozoa are found 70.
- Surgery for virilized females: The surgical procedure for a virilized female, termed feminizing genitoplasty, includes separation of the urethra and vagina, vaginoplasty, labioplasty, and clitoroplasty 71. In modern management of disorders of sex development (DSDs), clitoroplasty is a highly debatable practice and is rarely done except in highly virilized female patients with Prader stage 4 or 5. The optimal timing of feminizing genitoplasty (ie, early or delayed) remains to be determined and requires shared decision-making with the family 72, 73.
- Surgery for undervirilized males: Undervirilized males typically have hypospadias (a congenital condition in males in which the opening of the urethra is on the underside of the penis instead of the tip) requiring surgical reconstruction, undescended testes requiring orchiopexy, or both. The limited data available on the ideal timing of hypospadias repair in patients with disorders of sex development (DSDs) is derived mainly from the hypospadias literature and from expert opinions. It may be reasonable to advocate early surgery (ie, between the ages of 6 and 18 months) unless gender assignment is questionable 74. Gender reassignment may be considered in patients with 46,XY gonadal dysgenesis and genital inadequacy 71.
- Timing: The timing of surgery can depend on the specific diagnosis and gender assignment.
The gender assigned by the physician and family may not correlate with the gender preferred by the patient upon reaching adulthood. Therefore, some guidelines and advocacy groups recommend delaying surgical intervention until patients are able to make their own decisions about it. Various activists and some healthcare professionals have even called for a moratorium on gender reassignment and genital surgeries in childhood until studies have been completed on the long-term effects of such surgeries. Several long-term follow-up studies are being conducted, including a study by the North American Task Force on Intersexuality.
A critical issue is the timing of genital reconstruction. Some favor early surgery to restore a more normal visible anatomy and avoid ambiguity (which is often the parents’ wish), whereas others favor performing reconstruction later, when the individual is able to express his or her own preferences. Supporters of the first option claim that early genital surgery is technically easier and possibly has less psychological impact than later surgery. Genital surgery in adolescence carries a much higher physical and psychological morbidity, and fewer surgeons have substantial experienced with genital surgery in this age group. It is important to note that opponents of early surgery have no evidence that late surgery is better.
Although most of these controversies remain unresolved at present, one point that has been established is that treatment must be individualized. Attempts to develop broad management policies for such a disparate spectrum of anomalies would be ill-conceived and would disregard the multidisciplinary knowledge that has accrued over the past few decades.
Other supportive care
- Psychological support: A psychologist can help individuals and families cope with the lifelong aspects of the condition and develop a healthy sense of self.
- Fertility counseling: Individuals may need counseling about fertility options, such as using donor eggs, as gonadal dysgenesis often leads to infertility.
- Nutritional supplements: Calcium and vitamin D may be recommended to help maintain bone health.
Gonadal dysgenesis prognosis
Most people with gonadal dysgenesis live full, happy, and healthy lives. However, there are some health considerations to be aware of.
- Low hormone risks: These include loss of bone density (osteopenia) and osteoporosis (a condition where bones become weak, porous, and fragile, making them more susceptible to fractures from even minor falls) and have other health and wellbeing impacts to cognition, mood and sex drive.
- Infertility or impaired fertility.
- Gonadal cancers.
There are a range of treatments available to address and manage these conditions should they arise.
- Breehl L, Caban O. Genetics, Gonadal Dysgenesis. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539886[↩][↩][↩][↩][↩]
- Rocha VB, Guerra-Júnior G, Marques-de-Faria AP, de Mello MP, Maciel-Guerra AT. Complete gonadal dysgenesis in clinical practice: the 46,XY karyotype accounts for more than one third of cases. Fertil Steril. 2011 Dec;96(6):1431-4. doi: 10.1016/j.fertnstert.2011.09.009[↩][↩]
- McCann-Crosby B, Mansouri R, Dietrich JE, McCullough LB, Sutton VR, Austin EG, Schlomer B, Roth DR, Karaviti L, Gunn S, Hicks MJ, Macias CG. State of the art review in gonadal dysgenesis: challenges in diagnosis and management. Int J Pediatr Endocrinol. 2014;2014(1):4. doi: 10.1186/1687-9856-2014-4[↩][↩][↩][↩]
- Lee PA, Houk CP, Ahmed SF. Consensus statement on management of intersex disorders: international consensus conference on intersex. Pediatrics. 2006;118:e488–e500. doi: 10.1542/peds.2006-0738[↩][↩]
- MacLaughlin DT, Donahoe PK. Sex determination and differentiation. N Engl J Med. 2004;350:367–378. doi: 10.1056/NEJMra022784[↩][↩]
- Ostrer H. 46,XY disorder of Sex development and 46.XY complete gonadal dysgenesis. Seattle: GeneReviewsSeries; 1993.[↩]
- Fallat ME, Donahoe PK. Intersex genetic anomalies with malignant potential. Curr Opin Pediatr. 2006;18:305–311. doi: 10.1097/01.mop.0000193316.60580.d7[↩]
- Sharma L, Shankar Kikkeri N. Turner Syndrome. [Updated 2025 Jun 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK554621[↩][↩]
- Turner Syndrome. https://rarediseases.org/rare-diseases/turner-syndrome/[↩][↩]
- Leng XF, Lei K, Li Y, Tian F, Yao Q, Zheng QM, Chen ZH. Gonadal dysgenesis in Turner syndrome with Y-chromosome mosaicism: Two case reports. World J Clin Cases. 2020 Nov 26;8(22):5737-5743. doi: 10.12998/wjcc.v8.i22.5737[↩][↩]
- Sybert VP, McCauley E. Turner’s syndrome. N Engl J Med. 2004 Sep 16;351(12):1227-38. doi: 10.1056/NEJMra030360[↩][↩]
- Gravholt CH, Andersen NH, Conway GS, et al. International Turner Syndrome Consensus Group. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017 Sep;177(3):G1-G70. doi: 10.1530/EJE-17-0430[↩]
- Chacko E, Graber E, Regelmann MO, Wallach E, Costin G, Rapaport R. Update on Turner and Noonan syndromes. Endocrinol Metab Clin North Am. 2012 Dec;41(4):713-34. doi: 10.1016/j.ecl.2012.08.007[↩][↩][↩]
- Giwercman A, Berthelsen JG, Muller J. Screening for carcinoma-in-situ of the testis. Int J Androl. 1987;10:173–180. doi: 10.1111/j.1365-2605.1987.tb00180.x[↩][↩]
- Skakkebaek NE, Berthelsen JG, Giwercman A. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int J Androl. 1987;10:19–28. doi: 10.1111/j.1365-2605.1987.tb00161.x[↩][↩][↩]
- Verp MS, Simpson JL. Abnormal sexual differentiation and neoplasia. Cancer Genet Cytogenet. 1987;25:191–218. doi: 10.1016/0165-4608(87)90180-4[↩][↩]
- Scully RE. Gonadoblastoma; a gonadal tumor related to the dysgerminoma (seminoma) and capable of sex-hormone production. Cancer. 1953 May;6(3):455-63. doi: 10.1002/1097-0142(195305)6:3<455::aid-cncr2820060303>3.0.co;2-u[↩][↩]
- Scully RE. Gonadoblastoma. A review of 74 cases. Cancer. 1970 Jun;25(6):1340-56. doi: 10.1002/1097-0142(197006)25:6<1340::aid-cncr2820250612>3.0.co;2-n[↩]
- Hung W, Randolph JG, Chandra R. Gonadoblastoma in dysgenetic testis causing male pseudohermaphroditism in newborn. Urology. 1981;17:584–587. doi: 10.1016/0090-4295(81)90081-9[↩]
- Hart WR, Burkons DM. Germ cell neoplasms arising in gonadoblastomas. Cancer. 1979 Feb;43(2):669-78. doi: 10.1002/1097-0142(197902)43:2<669::aid-cncr2820430239>3.0.co;2-h[↩]
- Dieckmann KP, Pichlmeier U. Clinical epidemiology of testicular germ cell tumors. World J Urol. 2004;22:2–14. doi: 10.1007/s00345-004-0398-8[↩]
- von der Maase H, Rorth M, Walbom-Jorgensen S. Carcinoma in situ of contralateral testis in patients with testicular germ cell cancer: study of 27 cases in 500 patients. Br Med J. 1986;293:1398–1401. doi: 10.1136/bmj.293.6559.1398[↩]
- Cools M, Drop SL, Wolffenbuttel KP. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev. 2006;27:468–484. doi: 10.1210/er.2006-0005[↩]
- Wiesemann C, Ude-Koeller S, Sinnecker GH. Ethical principles and recommendations for the medical management of differences of sex development (DSD)/intersex in children and adolescents. Eur J Pediatr. 2010;169:671–679. doi: 10.1007/s00431-009-1086-x[↩]
- Gillam LH, Hewitt JK, Warne GL. Ethical principles for the management of infants with disorders of sex development. Horm Res Paediatr. 2010;74:412–418. doi: 10.1159/000316940[↩][↩]
- Maharaj NR, Dhai A, Wiersma R. Intersex conditions in children and adolescents: surgical, ethical, and legal considerations. J Pediatr Adolesc Gynecol. 2005;18:399–402. doi: 10.1016/j.jpag.2005.09.005[↩][↩]
- OVARIAN DYSGENESIS 1; ODG1. https://www.omim.org/entry/233300[↩]
- Timmreck LS, Reindollar RH. Contemporary issues in primary amenorrhea. Obstet Gynecol Clin North Am. 2003 Jun;30(2):287-302. doi: 10.1016/s0889-8545(03)00027-5[↩]
- Li T, Faridi R, Newman WG, et al. Perrault Syndrome Overview. 2014 Sep 25 [Updated 2025 May 8]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK242617[↩]
- Ameen KH, Pinninti R. A rare cause for primary amenorrhea: Sporadic perrault syndrome. Indian J Endocrinol Metab. 2012 Sep;16(5):843-5. doi: 10.4103/2230-8210.100677[↩]
- Ameen KH, Pinninti R. A rare cause for primary amenorrhoea. J Hum Reprod Sci. 2012 May;5(2):218-20. doi: 10.4103/0974-1208.101026. Retraction in: J Hum Reprod Sci. 2015 Oct-Dec;8(4):247. doi: 10.4103/0974-1208.170422[↩]
- Weinberg-Shukron A, Renbaum P, Kalifa R, Zeligson S, Ben-Neriah Z, Dreifuss A, Abu-Rayyan A, Maatuk N, Fardian N, Rekler D, Kanaan M, Samson AO, Levy-Lahad E, Gerlitz O, Zangen D. A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis. J Clin Invest. 2015 Nov 2;125(11):4295-304. doi: 10.1172/JCI83553[↩]
- Lung fibrosis-immunodeficiency-46,XX gonadal dysgenesis syndrome. https://www.ncbi.nlm.nih.gov/medgen/461506[↩]
- Somech R, Somers GR, Chitayat D, Grunebaum E, Atkinson A, Kolomietz E, Roifman CM. Fatal lung fibrosis associated with immunodeficiency and gonadal dysgenesis in 46XX sisters–a new syndrome. Am J Med Genet A. 2008 Jan 1;146A(1):8-14. https://doi.org/10.1002/ajmg.a.32014[↩]
- Röpke A, Pelz AF, Volleth M, Schlösser HW, Morlot S, Wieacker PF. Sex chromosomal mosaicism in the gonads of patients with gonadal dysgenesis, but normal female or male karyotypes in lymphocytes. Am J Obstet Gynecol. 2004 Apr;190(4):1059-62. doi: 10.1016/j.ajog.2003.09.053[↩]
- Zinn AR, Tonk VS, Chen Z, Flejter WL, Gardner HA, Guerra R, Kushner H, Schwartz S, Sybert VP, Van Dyke DL, Ross JL. Evidence for a Turner syndrome locus or loci at Xp11.2-p22.1. Am J Hum Genet. 1998 Dec;63(6):1757-66. https://pmc.ncbi.nlm.nih.gov/articles/instance/1377648/pdf/9837829.pdf[↩]
- Simpson JL, Rajkovic A. Ovarian differentiation and gonadal failure. Am J Med Genet. 1999 Dec 29;89(4):186-200. doi: 10.1002/(sici)1096-8628(19991229)89:4<186::aid-ajmg3>3.0.co;2-5[↩]
- Marozzi A, Manfredini E, Tibiletti MG, Furlan D, Villa N, Vegetti W, Crosignani PG, Ginelli E, Meneveri R, Dalprà L. Molecular definition of Xq common-deleted region in patients affected by premature ovarian failure. Hum Genet. 2000 Oct;107(4):304-11. doi: 10.1007/s004390000364[↩]
- Ledig S, Röpke A, Haeusler G, Hinney B, Wieacker P. BMP15 mutations in XX gonadal dysgenesis and premature ovarian failure. Am J Obstet Gynecol. 2008 Jan;198(1):84.e1-5. doi: 10.1016/j.ajog.2007.05.029[↩][↩][↩]
- BONE MORPHOGENETIC PROTEIN 15; BMP15. https://www.omim.org/entry/300247[↩]
- TRANSFORMING GROWTH FACTOR, BETA-1; TGFB1. https://www.omim.org/entry/190180[↩]
- Bashamboo A, Eozenou C, Rojo S, McElreavey K. Anomalies in human sex determination provide unique insights into the complex genetic interactions of early gonad development. Clin Genet. 2017 Feb;91(2):143-156. doi: 10.1111/cge.12932[↩][↩][↩]
- CYP21A2 gene. https://medlineplus.gov/genetics/gene/cyp21a2/[↩][↩]
- Elliott, G. A., Sandler, A., Rabinowitz, D. Gonadal dysgenesis in three sisters. J. Clin. Endocr. 19: 995-1003. doi: 10.1210/jcem-19-8-995[↩][↩]
- KLOTZ HP, MERGER R, AVRIL J. Syndrome de Turner chez deux soeurs issues de cousins germains; considérations pathogéniques [Turner syndrome in two sisters born of first cousins; pathogenetic considerations]. Ann Endocrinol (Paris). 1956 May;17(1):43-6. French.[↩]
- Christakos AC, Simpson JL, Younger JB, Christian CD. Gonadal dysgenesis as an autosomal recessive condition. Am J Obstet Gynecol. 1969 Aug 1;104(7):1027-30. doi: 10.1016/0002-9378(69)90697-8[↩]
- Simpson JL, Christakos AC, Horwith M, Silverman FS. Gonadal dysgenesis in individuals with apparently normal chromosomal complements: tabulation of cases and compilation of genetic data. Birth Defects Orig Artic Ser. 1971 May;7(6):215-28.[↩]
- Vesely DL, Bower RH, Kohler PO, Char F. Familial ovarian dysgenesis in 46,XX females. Am J Med Sci. 1980 Nov-Dec;280(3):157-66. doi: 10.1097/00000441-198011000-00004[↩]
- Aleem FA. Familial 46,XX gonadal dysgenesis. Fertil Steril. 1981 Mar;35(3):317-20. doi: 10.1016/s0015-0282(16)45378-1[↩]
- Aittomäki K. The genetics of XX gonadal dysgenesis. Am J Hum Genet. 1994 May;54(5):844-51. https://pmc.ncbi.nlm.nih.gov/articles/instance/1918251/pdf/ajhg00050-0116.pdf[↩][↩]
- de la Chapelle A. Disease gene mapping in isolated human populations: the example of Finland. J Med Genet. 1993 Oct;30(10):857-65. https://pmc.ncbi.nlm.nih.gov/articles/instance/1016570/pdf/jmedgene00012-0059.pdf[↩]
- Aittomäki K, Herva R, Stenman UH, Juntunen K, Ylöstalo P, Hovatta O, de la Chapelle A. Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene. J Clin Endocrinol Metab. 1996 Oct;81(10):3722-6. doi: 10.1210/jcem.81.10.8855829[↩][↩]
- 46,XX gonadal dysgenesis. https://www.orpha.net/en/disease/detail/243[↩]
- Azidah AK, Nik Hazlina NH, Aishah MN. Swyer syndrome in a woman with pure 46, XY gonadal dysgenesis and a hypoplastic uterus. Malays Fam Physician. 2013; 8(2):58-61. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4170479[↩]
- Swyer syndrome. https://rarediseases.org/rare-diseases/swyer-syndrome/[↩]
- Salete Da Silva Rios, et. al. A Case of Swyer Syndrome Associated with Advanced Gonadal Dysgerminoma Involving Long Survival. Case Rep Oncol. January-April, 2015; 8(1):179-184.[↩]
- Swyer syndrome. https://ghr.nlm.nih.gov/condition/swyer-syndrome[↩]
- Mohnach L, Fechner PY, Keegan CE. Nonsyndromic Disorders of Testicular Development. 2008 May 21 [Updated 2016 Jun 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1547[↩][↩]
- McCann-Crosby B, Mansouri R, Dietrich JE, McCullough LB, Sutton VR, Austin EG, Schlomer B, Roth DR, Karaviti L, Gunn S, Hicks MJ, Macias CG. State of the art review in gonadal dysgenesis: challenges in diagnosis and management. Int J Pediatr Endocrinol. 2014;2014:4. doi: 10.1186/1687-9856-2014-4[↩]
- Wolffenbuttel KP, Hersmus R, Stoop H, Biermann K, Hoebeke P, Cools M, Looijenga LH. Gonadal dysgenesis in disorders of sex development: Diagnosis and surgical management. J Pediatr Urol. 2016;12:411–416. doi: 10.1016/j.jpurol.2016.08.015[↩]
- Baxter RM, Arboleda VA, Lee H, Barseghyan H, Adam MP, Fechner PY, Bargman R, Keegan C, Travers S, Schelley S, Hudgins L, Mathew RP, Stalker HJ, Zori R, Gordon OK, Ramos-Platt L, Pawlikowska-Haddal A, Eskin A, Nelson SF, Delot E, Vilain E. Exome sequencing for the diagnosis of 46,XY disorders of sex development. J Clin Endocrinol Metab. 2015;100:E333–344. doi: 10.1210/jc.2014-2605[↩][↩]
- Eggers S, Sadedin S, van den Bergen JA, Robevska G, et al. Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016;17:243. doi: 10.1186/s13059-016-1105-y[↩]
- Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol. 2013;9:79–91. doi: 10.1038/nrendo.2012.235[↩][↩]
- Nistal M, Paniagua R, González-Peramato P, Reyes-Múgica M. Perspectives in Pediatric Pathology, Chapter 5. Gonadal Dysgenesis. Pediatr Dev Pathol. 2015 Jul-Aug;18(4):259-78. doi: 10.2350/14-04-1471-PB.1[↩]
- Granados A, Alaniz VI, Mohnach L, Barseghyan H, Vilain E, Ostrer H, Quint EH, Chen M, Keegan CE. MAP3K1-related gonadal dysgenesis: Six new cases and review of the literature. Am J Med Genet C Semin Med Genet. 2017 Jun;175(2):253-259. doi: 10.1002/ajmg.c.31559[↩]
- Riccabona M, Darge K, Lobo ML, Ording-Muller LS, Augdal TA, Avni FE, Blickman J, Damasio BM, Ntoulia A, Papadopoulou F, Vivier PH, Willi U. ESPR Uroradiology Taskforce–imaging recommendations in paediatric uroradiology, part VIII: retrograde urethrography, imaging disorder of sexual development and imaging childhood testicular torsion. Pediatr Radiol. 2015 Dec;45(13):2023-8. doi: 10.1007/s00247-015-3452-3[↩]
- Kopac M, Riccabona M, Haim M. Contrast-enhanced voiding urosonography and genitography in a baby with ambiguous genitalia and urogenital sinus. Ultraschall Med. 2009 Jun;30(3):299-300. doi: 10.1055/s-0028-1109353[↩]
- Chavhan GB, Parra DA, Oudjhane K, Miller SF, Babyn PS, Pippi Salle FL. Imaging of ambiguous genitalia: classification and diagnostic approach. Radiographics. 2008 Nov-Dec;28(7):1891-904. doi: 10.1148/rg.287085034[↩]
- Morgan T. Turner syndrome: diagnosis and management. Am Fam Physician. 2007 Aug 1;76(3):405-10. https://www.aafp.org/pubs/afp/issues/2007/0801/p405.html[↩]
- Finlayson C, Fritsch MK, Johnson EK, Rosoklija I, Gosiengfiao Y, Yerkes E, Madonna MB, Woodruff TK, Cheng E. Presence of Germ Cells in Disorders of Sex Development: Implications for Fertility Potential and Preservation. J Urol. 2017 Mar;197(3 Pt 2):937-943. doi: 10.1016/j.juro.2016.08.108[↩]
- Vidal I, Gorduza DB, Haraux E, Gay CL, Chatelain P, Nicolino M, Mure PY, Mouriquand P. Surgical options in disorders of sex development (dsd) with ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010 Apr;24(2):311-24. doi: 10.1016/j.beem.2009.10.004[↩][↩]
- Wolffenbuttel KP, Crouch NS. Timing of feminising surgery in disorders of sex development. Endocr Dev. 2014;27:210-21. doi: 10.1159/000363665[↩]
- Eckoldt-Wolke F. Timing of surgery for feminizing genitoplasty in patients suffering from congenital adrenal hyperplasia. Endocr Dev. 2014;27:203-9. doi: 10.1159/000363664[↩]
- Springer A, Baskin LS. Timing of hypospadias repair in patients with disorders of sex development. Endocr Dev. 2014;27:197-202. doi: 10.1159/000363662[↩]





{kind=link}