Contents
What is acetylcholine
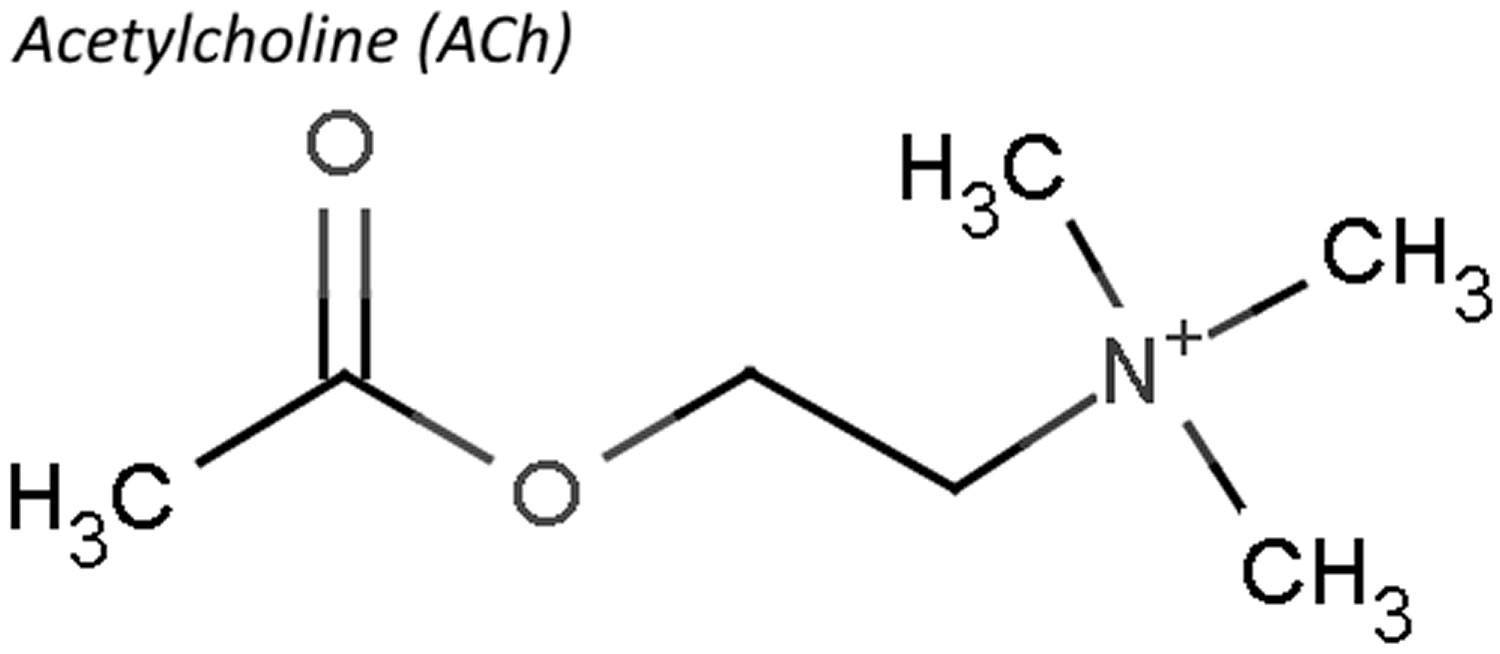
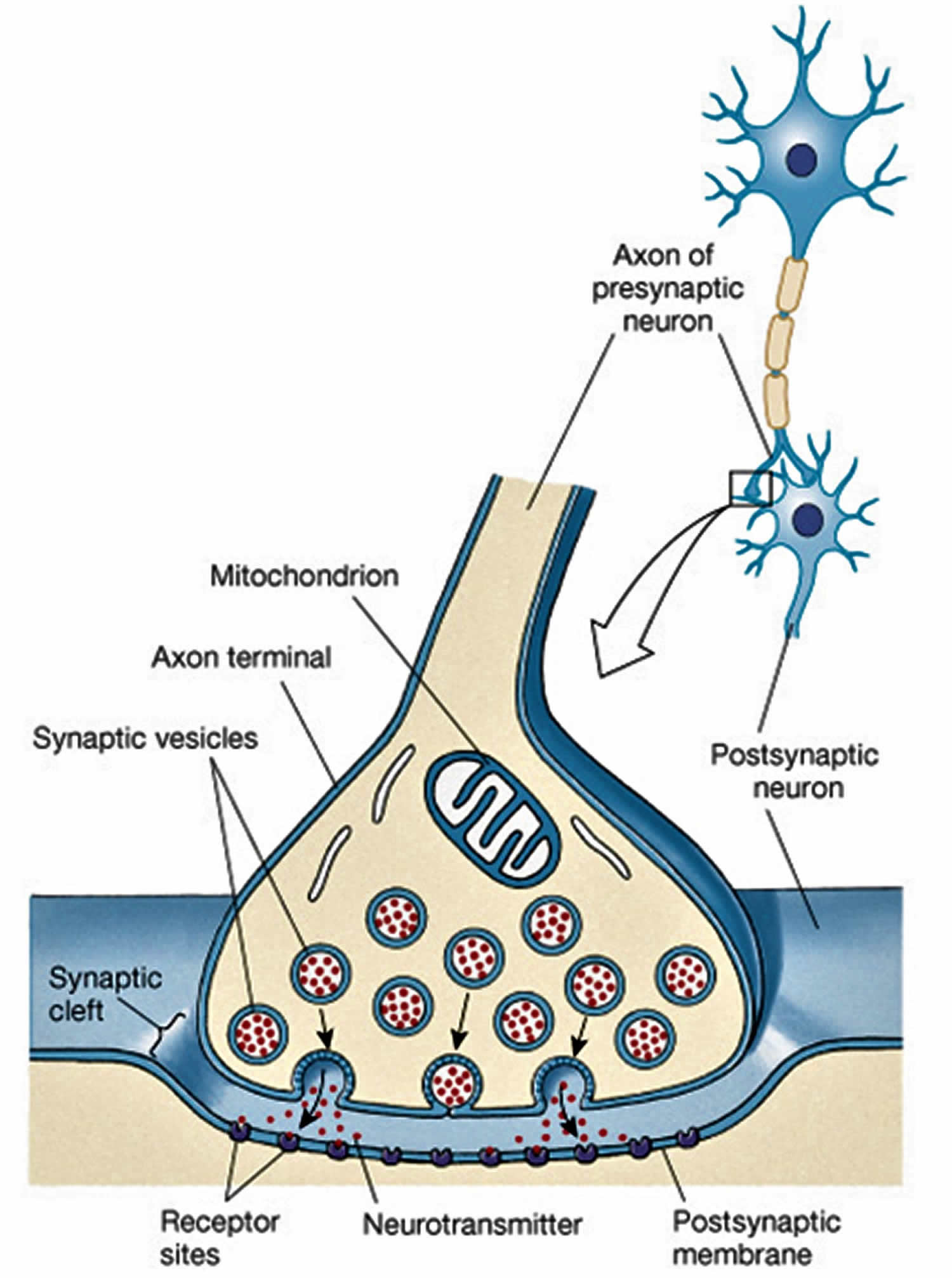
Acetylcholine (ACh) is a neurotransmitter found at neuromuscular junctions, autonomic ganglia, parasympathetic effector junctions, a subset of sympathetic effector junctions, and at many sites in the central nervous system. Neurotransmitters are chemicals that neurons, or brain cells, use to communicate information. Normally when electrical signals or impulses travel down a motor nerve, the nerve endings release a neurotransmitter called acetylcholine. Acetylcholine travels from the nerve ending and binds to acetylcholine receptors on the muscle. The binding of acetylcholine to its receptor activates the muscle and causes a muscle contraction. Nerve fibers that secrete acetylcholine are called cholinergic fibers.
Acetylcholine is used during eye surgery to make the eye muscles contract and constrict the pupil.
Despite a great deal is known about the function of cholinergic transmission at the neuromuscular junction and at ganglionic synapses, the actions of acetylcholine in the central nervous system are not as well understood 1. Acetylcholine (ACh) has been considered an important excitatory neurotransmitter in the carotid body 2. Various nicotinic and muscarinic acetylcholine receptors are present in both afferent nerve endings and glomus cells. Therefore, acetylcholine can depolarize or hyperpolarize the cell membrane depending on the available receptor type in the vicinity. Binding of Acetylcholine (ACh) to its receptor can create a wide variety of cellular responses including opening cation channels (nicotinic acetylcholine receptor activation), releasing Ca2+ from intracellular storage sites (via muscarinic acetylcholine receptors), and modulating activities of K+ and Ca2+ channels. Interactions between acetylcholine and other neurotransmitters (dopamine, adenosine, nitric oxide) have been known, and they may induce complicated responses.
Acetylcholine is synthesized in nerve terminals from acetyl coenzyme A (acetyl CoA, which is synthesized from glucose) and choline (Ch), in a reaction catalyzed by choline acetyltransferase (CAT) (Figure 2). The presence of choline acetyltransferase (CAT) in a neuron is thus a strong indication that acetylcholine (ACh) is used as one of its transmitters. Choline is present in plasma at a concentration of about 10 mM, and is taken up into cholinergic neurons by a high-affinity Na+/choline transporter. About 10,000 molecules of acetylcholine (ACh) are packaged into each vesicle by a vesicular acetylcholine (ACh) transporter.
Acetylcholine release
The cholinergic nerve fiber, exemplified by its endings at the neuromuscular junction, also became the archetypical nerve terminal for studying neurotransmitter release, especially through the work of Katz and Fatt 3. It was here that quantal release was first demonstrated and the essential role of calcium elucidated 4. These concepts proved generally applicable at other synapses, though with variations in the quantal content of the evoked synaptic response or release efficiency. It was also at the neuromuscular junction that the pharmacological concept of the ‘false transmitter’ was first mooted, to explain the inhibitory action of triethylcholine 5, even though this pharmacological modus operandi was later applied to aminergic fibers, with the introduction of α-methyldopa in antihypertensive therapy. More recent pharmacological approaches to modifying acetylcholine release have been directed at selective blockade of presynaptic muscarinic autoreceptors (see below).
Figure 1. Cell to cell conduction at the neuronal synapse
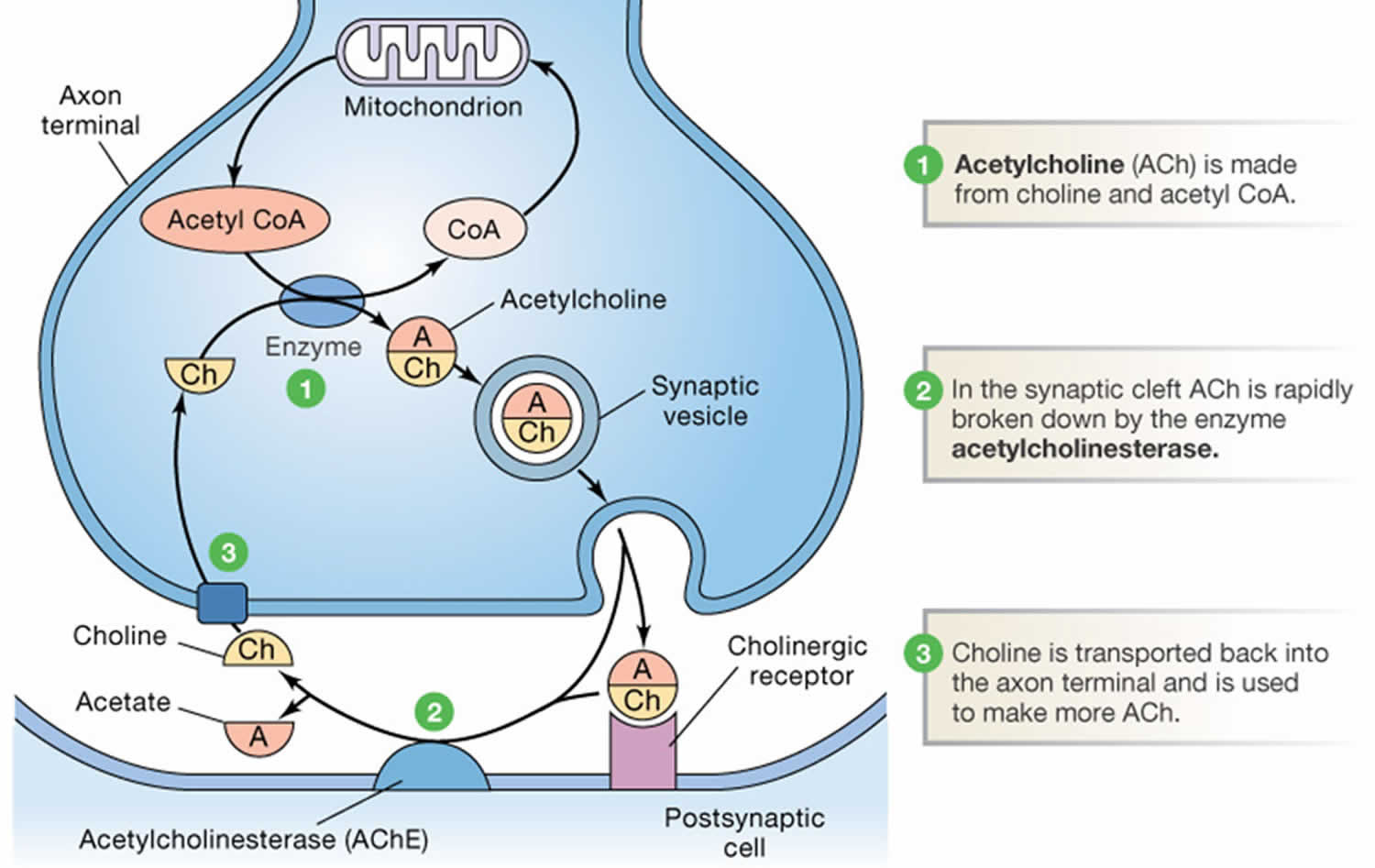
Footnotes: Acetylcholine (ACh) metabolism in cholinergic nerve terminals. The synthesis of acetylcholine (ACh) from choline (Ch) and acetyl CoA requires choline acetyltransferase (CAT) (#1). Acetyl CoA is derived from pyruvate generated by glycolysis, while choline (Ch) is transported into the terminals via a Na+ dependent transporter. After release, acetylcholine (ACh) is rapidly metabolized by acetylcholinesterase (AChE) (#2) and choline (Ch) is transported back into the terminal by the Na+-choline transporter (#3).
[Source 6]In contrast to most other small-molecule neurotransmitters, the postsynaptic action of acetylcholine (ACh) at many cholinergic synapses (the neuromuscular junction in particular) are not terminated by reuptake, but by a powerful hydrolytic enzyme, acetylcholinesterase (AChE) (see Figure 2). This enzyme is concentrated in the synaptic cleft, ensuring a rapid decrease in acetylcholine concentration after its release from the presynaptic terminal. Acetylcholinesterase (AChE) has a very high catalytic activity (about 5000 molecules of acetylcholine per acetylcholinesterase (AChE) molecule per second) and hydrolyzes acetylcholine into acetate and choline (Ch). As already mentioned, cholinergic nerve terminals typically contain a high-affinity, Na+-choline transporter that takes up the choline produced by acetylcholine hydrolysis.
Among the many interesting drugs that interact with cholinergic enzymes are the organophosphates. Compounds such as diphenyl trichloroethane (DTT) and the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) were originally developed as insecticides. This group also includes some potent chemical warfare agents. One such compound is the nerve gas “Sarin”. Organophosphates can be lethal to humans (and insects) because they inhibit acetylcholinesterase (AChE), causing acetylcholine to accumulate at cholinergic synapses. This build-up of acetylcholine depolarizes the postsynaptic cell and renders it refractory to subsequent acetylcholine release, causing, among other effects, neuromuscular paralysis.
Acetylcholine receptors
Actions of acetylcholine in the periphery are the result of activation of either the ionotropic nicotinic receptor or the metabotropic muscarinic receptor 7. In the mammalian central nervous system (CNS), both nicotinic and muscarinic receptor subtypes are present on neurons, although there is as yet very limited evidence for a physiological role for nicotinic receptors in synaptic function in the mammalian brain 8. In the periphery, among other effects, muscarinic receptors mediate smooth muscle contraction, glandular secretion, and modulation of cardiac rate and force. In the central nervous system (CNS), there is evidence that muscarinic receptors are involved in motor control, temperature regulation, cardiovascular regulation, and memory. Interest in the classification of muscarinic receptors involved in functions at different locations has been heightened by the potential therapeutic application of selective agents in areas such as Alzheimer’s disease, Parkinson’s disease, asthma, analgesia, and disorders of intestinal motility and cardiac and urinary bladder function.
At the molecular level, scientists await the crystal structures of the complete nicotinic and muscarinic receptors. At the functional level, scientists anticipate an increasingly detailed description of the precise role of individual nicotinic receptor subunits in defined pathways and systems from targeted and timed knockouts and from siRNA knockdowns (which are not restricted to mice), and, hopefully, the circumstances under which they are activated. This will also take us outside the nervous system, since nicotinic receptors (especially α7 receptors) are present and functional in epithelial cells, various tumor cells and in elements of the immune system, such as B-lymphocytes 9. From these two approaches, and others, scientists might optimistically look forward to some potentially beneficial advances in pharmacotherapy of disorders such as schizophrenia, neuropathic pain, Alzheimer’s disease, and some forms of cancer and immune disorders – or at least the replacement of nicotine with less addictive substitutes.
As for muscarinic actions, some advances have already emerged from the identification of muscarinic receptor subtypes in the treatment of bladder incontinence 10 and bronchoconstriction 11, and there may be scope for some additional treatment of Parkinson’s disease via an M4 muscarinic receptor antagonist 12. However, the initial optimism that M1 muscarinic receptor agonists might be beneficial in Alzheimer’s disease has not yet been fully rewarded.
Overall, given the limited number, and overlapping distributions and functions, of muscarinic receptor subtypes, further ‘quantum leaps’ in therapeutic drug development based on subtype-specific agonists, antagonists or inverse agonists seem somewhat remote; perhaps allosteric enhancers and inhibitors offer more scope, since they modify ongoing functions, rather than imitating or suppressing them. Of course, this is also what anticholinesterase drugs do, and they certainly have some valuable uses, but their limitations in the treatment of Alzheimer’s disease are equally apparent.
Muscarinic acetylcholine receptor
There are five subtypes of muscarinic receptor (M1 to M5) emerged from cloning experiments 13, with differential coupling to G-proteins (M1, M3, M5 to the Gq/G11 family, and M2, M4 to the Gi/Go family), and, to some degree, differential sensitivities to some antagonists 14. Cloning of complementary deoxyribonucleic acids for muscarinic receptor genes was spearheaded by the work of Numa and colleagues, who cloned the M1 and M2 genes 15, and was extended by the discovery of the M3, M4, and M5 genes 13. These five genes encode muscarinic receptor proteins (actually glycoproteins) which have the structural features of the seven transmembrane helix G-protein-coupled receptor family. Muscarinic receptor sequences have significant homologies with other members of this receptor superfamily 16. The chromosomal localization of the human M1 to M5 genes are reported to be 11q12–13, 7q35–36, 1q43–44, 11p12–11.2, and 15q26, respectively 17.
However, the paucity of highly selective antagonists, and the lack of any selective agonists has impeded the unambiguous identification of muscarinic receptor subtypes mediating many important responses 7. It is hoped that the discovery of compounds (and toxins) with greater receptor subtype selectivity will aid this process 7.
Structurally and functionally, the muscarinic receptor appears to show a close homology to rhodopsin 18. Like other G-protein-coupled receptors, muscarinic receptors can form dimers or oligomers, but these are apparently subtype-specific, that is, homodimers, not heterodimers 19; so dimerization is unlikely to have pharmacological consequences.
Nicotinic acetylcholine receptor
One of the most important post-1930 advances in nicotinic receptor pharmacology was the synthesis of the polymethylene bis-trimethylammonium (‘methonium’) compounds, and the consequent demonstration of a clear difference between neural (ganglionic) and muscle (neuromuscular junction of striated muscle) receptors: the former were selectively blocked by hexamethonium and pentamethonium, whereas the neuromuscular junction of striated muscle receptor was first excited and then blocked by decamethonium 20. This difference between nicotinic receptors was, apart from its scientific significance, also of immense clinical significance, since the fact that reducing the blood pressure with hexamethonium was actually life-saving in malignant hypertension was instrumental in persuading clinicians that the hypertension was indeed the cause of death, rather than an essential adaptation. This analysis provided the clinical impetus for all subsequent (and extensive) developments in antihypertensive pharmacotherapy. The selective ganglion-blocking action of tetraethylammonium was already known, but tetraethylammonium was too short-acting to be of much clinical use. Hexamethonium was a very imperfect drug from a practical viewpoint, with poor absorption and numerous side effects. These defects induced an avalanche of novel ganglion-blocking agents developed by U.K. and U.S. pharmaceutical companies, such as pentolinium 21, pempidine 22, mecamylamine, trimetaphan and chlorisondamine, before alternative approaches to blood pressure control were developed (though some ganglion-blocking agents are still in use for selected purposes).
Notwithstanding the exact mechanism of block, the distinction between muscle and neural nicotinic receptors has been extended to the central nervous system (CNS), where virtually all of the nicotinic effects of acetylcholine are blocked by hexamethonium or, more conveniently, by the lipophilic mecamylamine. This distinction was emphasized by the cloning of the nicotinic receptor subunits. Thus, while the (adult) muscle receptor has a constant composition of two α1 subunits with one each of β1, ɛ and δ subunits, neural receptors are composed of quite distinct α and β subunits (or of homomeric α subunits), of which, collectively, there are 12 known types – α2–α10 and β2–β4 18. Since different subunit combinations show different agonist rank orders and some antagonist selectivity (at least, for toxins), much effort has been, and is being, devoted to finding out which subunit composition represents the physiologically relevant receptor in different neurons and at different synapses 23.
As with muscarinic receptors, information regarding the more global functions of different nicotinic receptor subunits has been derived from ‘knock-outs’ 24. Thus, though largely restricted to the autonomic nervous system, an α3 knock-out causes early postnatal death – possibly due to an intestinal disorder since it replicates a known human genetic intestinal defect with a short (3.6 months) survival time. At the other end of the spectrum, a β2 knock-out accelerates the reduction in cortical thickness with aging, as well as impairing passive avoidance learning. They also affect responses to nicotine – for example, both α4 and β2 knock-outs reduce the antinociceptive effect of nicotine, while β2 knock-outs also reduce nicotine self-administration.
Overall, more is now known about the nicotinic acetylcholine receptor than any other neurotransmitter receptor 25. Single-channel analysis has revealed much about the likely conformational changes of a single receptor molecule on a sub-millisecond time-scale; the overall physical structure has been determined from cryo-electron microscopy at 4 Å resolution 26, and the likely arrangement of atoms in the external domain of the receptor determined from X-ray crystallography of the homologous snail acetylcholine-binding protein. Much of this, plus the results of extensive mutational analysis concerning ligand-binding, channel-gating and channel conductance, are summarized by Karlin 27. Thus, ‘nicotine action’ of acetylcholine can now be explained in molecular terms.
Acetylcholine function
Acetylcholine role in learning and memory
Pharmacological studies in human subjects conclusively demonstrate that blockade of muscarinic cholinergic receptors by drugs such as scopolamine impairs the encoding of new memories, but not the retrieval of previously stored memories 28, and impairs working memory for some stimuli 29. The neurochemical acetylcholine has been implicated in a range of cognitive processes, including episodic memory, which is impaired in Alzheimer’s disease, in which there is a decrease in acetylcholine innervation 30. However, systemic administration of acetylcholinesterase inhibitors produces only small acute improvements in memory 31. Despite the lack of evidence that procholinergic therapies in Alzheimer’s disease lead to lasting improvements in memory, they do slow cognitive decline 32. Moreover, drugs which activate nicotinic receptors enhance the encoding of new information 33.
Localized infusions of acetylcholine antagonists into specific anatomical structures demonstrate the importance of cholinergic receptors for particular aspects of memory tasks 34. Localized infusions of scopolamine into parahippocampal structures demonstrate a role of cholinergic receptors in these structures for the encoding of information for subsequent recognition in both monkeys 35 and rats 36. These studies used tasks in which animals are exposed to one or multiple sample stimuli during encoding, and are subsequently tested on their delayed recognition of these sample stimuli and rejection of other stimuli which were not presented during the sample phase. Local infusions into perirhinal cortex in monkeys impair encoding for subsequent recognition, whereas infusions into dentate gyrus or inferotemporal cortex do not 35. Local infusions into perirhinal cortex in rats impair object recognition, as measured by exploration time, but do not impair spatial alternation, suggesting task specificity 36.
Local application of cholinergic antagonists into other regions also cause selective impairments. Infusions of scopolamine into the hippocampus impair spatial encoding 37 and infusions into the medial septum impair spatial learning and reduce acetylcholine release in the hippocampus 38. Infusions of carbachol into the medial septum, which increase levels of hippocampal acetylcholine, also impair memory 38, possibly by interfering with consolidation 39. Infusions of scopolamine into region CA3 cause selective impairments of encoding but not retrieval in the Hebb-Williams maze 40.
Acetylcholine and Alzheimer’s disease
Alzheimer’s disease is a major form of senile dementia, characterized by progressive memory and neuronal loss combined with cognitive impairment 41. Alzheimer’s disease is the most common neurodegenerative disease worldwide, affecting one-fifth of those aged over 85 years 41. Recent therapeutic approaches have been strongly influenced by five neuropathological hallmarks of Alzheimer’s disease: acetylcholine deficiency, glutamate excitotoxicity, extracellular deposition of amyloid-β (Aβ plague), formation of intraneuronal neurofibrillary tangles (NTFs), and neuroinflammation 41. The lowered concentrations of acetylcholine (ACh) in Alzheimer’s disease result in a progressive and significant loss of cognitive and behavioral function. Current Alzheimer’s disease medications, memantine and acetylcholinesterase inhibitors (AChEIs) alleviate some of these symptoms by enhancing cholinergic signaling, but they are not curative 41. Since 2003, no new drugs have been approved for the treatment of Alzheimer’s disease.
Alzheimer’s disease is characterized by neuronal death, which usually correlates with the appearance of key neuropathological changes, including acetylcholine deficiency, glutamate excitotoxicity, extracellular deposition of β-amyloid (Aβ plaques), intracellular neurofibrillary tangles by hyperphosphorylated tau protein deposits, neuroinflammation, and widespread neuronal loss 42. The role of β-amyloid (Aβ plaques) and tau proteins in the pathophysiology of Alzheimer’s disease remains unclear. Different theories suggest that inflammation, accumulation of reactive oxygen species (ROS), mitochondrial damage, genetic factors, cerebrovascular disease, traumatic brain injury, age-related loss of sex steroid hormones in both women and man, are some of the established risk factors considered to be promising targets for drug discovery in the treatment of Alzheimer’s disease 43. Hung and Yu 41 have classified therapeutic drugs and targets in the treatment of Alzheimer’s disease according to the neuropathological hallmarks of Alzheimer’s disease (Figure 3).
Current drug treatment for Alzheimer’s disease patients, essentially symptomatic, is based on three cholinesterase inhibitors (rivastigmine, donepezil and galantamine) and memantine, affecting the glutamatergic system. These drugs do not represent a cure, as they do not arrest the progression of dementia, but rather, they lead to a temporary slowdown in the loss of cognitive function by decreasing cholinesterase activity, resulting in higher acetylcholine (ACh) levels and improved brain function. Over two hundred compounds have reached Phase II clinical trials since 2003, but no new drugs have been approved for the treatment of Alzheimer’s disease 44. Most Phase II clinical trials ending with a positive outcome do not succeed in Phase III, often due to serious adverse effects or lack of therapeutic efficacy 45. Other challenges facing drug development in Alzheimer’s disease include a lack of validated objective diagnostic criteria and robust biological markers of Alzheimer’s disease that might be useful as clinical endpoints and efficacy standards. Despite decades of study efforts to develop therapies, there is no effective therapy available to cure Alzheimer’s disease or significantly inhibits the progression of Alzheimer’s disease symptoms 41.
Figure 3. Classification of therapeutic drugs or targets in the treatment of Alzheimer’s disease according to neuropathological hallmarks

Lack of acetylcholine in Alzheimer’s disease
In Alzheimer’s disease, the loss of cholinergic tone and acetylcholine levels in the brain is hypothesized to be responsible for the gradual cognitive decline.
Enhancement of the acetylcholine response by acetylcholinesterase inhibitors
In 1976, Davies and Maloney were the first to hypothesize that selective loss of central cholinergic neurons in Alzheimer’s disease plays a key role in its pathophysiology 46. The release in 1993 of tacrine, the first reversible acetylcholinesterase inhibitor (AChEI) to reach the market for the treatment of Alzheimer’s disease, was withdrawn soon after because of reports of liver toxicity. Three other cholinesterase inhibitors- donepezil, galantamine, and rivastigmine are currently used in the treatment of Alzheimer’s disease to reduce the activity of acetylcholinesterase (AChE). These agents do not delay the progression of dementia but temporarily slow the loss of cognitive function.
Enhancement of the acetylcholine response using 5-HT6 receptor antagonists
The serotonergic neurotransmitter system is impaired as Alzheimer’s disease develops and progresses; modulation of this pathway is therefore considered to be of therapeutic value 47. Serotonin (5-HT) activates specific 5-HT receptors, consisting of seven distinct classes (5-HT1 to 5-HT7) based on their structural and function characteristics. The 5-HT6 receptor is expressed primarily in brain areas involved in learning and memory processes – the cortex and hippocampus. 5-HT6 receptor antagonists are thought to enhance cholinergic neurotransmission 47. Idalopirdine (Lu AE58054) is an orally available 5-HT6 antagonist, that showed promising efficacy and safety data in Phase II trials (Clinical Trial Identifier: NCT01019421). Although idalopirdine is safe and well tolerated as an adjunctive therapy to donepezil (acetylcholinesterase inhibitor) in patients with mild-to-moderate Alzheimer’s disease, however, idalopirdine did not meet its primary efficacy endpoint versus placebo in recent two phase III trials 48 (Clinical Trial Identifier: NCT02006641 and NCT02006654) (Table 1). Intepirdine (RVT-101) is another 5-HT6 antagonist that is currently in Phase III clinical trials in patients with mild-to-moderate Alzheimer’s disease already on donepezil therapy (Clinical Trial Identifier: NCT02585934 and NCT02586909) (Table 1). Analysis of data from Phase II evaluation of treatment with intepirdine indicates that addition of this treatment to donepezil may improve the cognition and function of patients with mild-to-moderate Alzheimer’s disease 49 (Clinical Trial Identifier: NCT02910102).
Enhancement of the acetylcholine response using H3 receptor antagonists
Histamine H3 receptors are widely distributed throughout the CNS. Blockade of this receptor augments the presynaptic release of both histamine and other neurotransmitters including acetylcholine. Several histamine H3 antagonists have entered therapeutic programs for cognition disorders 50, including ABT-288, GSK239512, and SUVN-G3031 (Table 1). A Phase II trial of ABT-288 in patients with mild-to-moderate Alzheimer’s disease as adjunct treatment on stable donepezil was ended due to lack of clinical efficacy 51 (Clinical Trial Identifier: NCT01018875). Similarly, GSK239512 was discontinued in a Phase II study because of lack of improvement in memory testing in patients with mild-to-moderate Alzheimer’s disease (Clinical Trial Identifier: NCT01009255), suggesting that H3 receptor antagonists are not effective in treating cognitive dysfunction in Alzheimer’s disease 52. SUVN-G3031 is an orally active H3 receptor antagonist, which is currently undergoing Phase I investigation evaluating the safety, tolerability, and pharmacokinetics study in healthy volunteers (Clinical Trial Identifier: NCT02342041).
Enhancement of the acetylcholine response by α7 nicotinic acetylcholine receptor (α7nAChR) agonists
It is well established that acetylcholine neurotransmission plays a crucial role in learning and memory. Current medication is aimed at enhancing cholinergic signaling for treating cognitive deficits and memory impairment in neurodegenerative disorders, including Alzheimer’s disease. The nicotinic acetylcholine receptor family (nAChR) and the muscarinic acetylcholine receptor family (mAChR) are targeted by acetylcholine in the brain 53. The alkaloid galantamine, used for the treatment of mild-to-severe dementia, has shown activity in modulating the nicotinic cholinergic receptors on cholinergic neurons to increase acetylcholine release 53. α7 nicotinic acetylcholine receptor (α7nAChR) belongs to the family of ligand-gated ion channels and is expressed in key brain regions (e.g. prefrontal and frontal cortices, hippocampus) 54. α7 nicotinic acetylcholine receptor (α7nAChR) is involved in essential cognitive functions such as memory, thinking, comprehension, the capacity to learn, calculate, orientate, language abilities, and judgment 54. Notably, α7 nicotinic acetylcholine receptor (α7nAChR) acts as a carrier to bind with extracellular Aβ, which further inhibits Aβ-induced neurotoxicity via autophagic degradation, an important step in Aβ detoxification 55. Encenicline (EVP-6124, MT-4666), a partial selective agonist of α7 nicotinic acetylcholine receptor (α7nAChR), has been developed for the treatment of cognitive deficits in Alzheimer’s disease and schizophrenia and Alzheimer’s disease (Table 1). Clinical study findings show that encenicline also can act as a co-agonist with acetylcholine to enhance cognition 56. In 2015, the U.S. FDA imposed a clinical hold on encenicline following reports of gastrointestinal side effects in two Phase III Alzheimer studies 57 (Clinical Trial Identifier: NCT01969136 and NCT01969123).
Table 1. Update of selected anti-Alzheimer’s disease drugs in clinical trials (updated in June 2017)
| Target | Drug name | Therapy type | Trial status | Reasons for Discontinuation | Company | Clinical Trial Identifier |
| Serotoninergic (5-HT6 receptor antagonist) | Idalopirdine | Small molecule | Phase III Discontinued in 2017 | No clinical efficacy | H. Lundbeck, Otsuka Pharmaceutical Co., Ltd. | NCT01019421 NCT02006641 NCT02006654 |
| Intepirdine | Small molecule | Phase II/III | Not applicable | Axovant Sciences Ltd. | NCT02910102 NCT02585934 NCT02586909 | |
| Histaminergic (H3 receptor antagonist) | ABT-288 | Small molecule | Phase II Discontinued in 2011 | No clinical efficacy | AbbVie | NCT01018875 |
| GSK239512 | Small molecule | Phase II Discontinued in 2012 | No improvements in memory test | GlaxoSmithKline (GSK) | NCT01009255 | |
| SUVN-G3031 | Small molecule | Phase I | Not applicable | Suven Life Sciences Ltd | NCT02342041 | |
| Acetylcholine response ↑ (α7nAChR agonist) | Encenicline | Small molecule | Phase III Discontinued in 2015 | Adverse effects: gastrointestinal side effect | FORUM Pharmaceuticals Inc., Mitsubishi Tanabe Pharma | NCT01969136 NCT01969123 |
| Glutaminergic | Riluzole | Small molecule | Phase II | Not applicable | Sanofi | NCT01703117 |
| BACE inhibitor | BI 1181181 | Small molecule | Phase I Discontinued in 2015 | Low oral bioavailability and low blood-brain barrier penetration | Boehringer Ingelheim, Vitae Pharmaceuticals | NCT02044406 NCT02106247 NCT02254161 |
| RG7129 | Small molecule | Phase I Discontinued in 2013 | Liver toxicity | Roche | NCT01664143 NCT01592331 | |
| LY2811376 | Small molecule | Phase I Discontinued In 2008 | Liver toxicity | Eli Lilly & Co. | NCT00838084 | |
| LY2886721 | Small molecule | Phase II Discontinued In 2013 | Liver toxicity | Eli Lilly & Co. | NCT01561430 | |
| E2609 | Small molecule | Phase III | Not applicable | Biogen, Eisai Co., Ltd. | NCT03036280 NCT02956486 | |
| AZD3293 | Small molecule | Phase III | Not applicable | AstraZeneca, Eli Lilly & Co. | NCT02783573 | |
| CNP520 | Small molecule | Phase II/III | Not applicable | Amgen, Inc., Novartis Pharmaceuticals Corporation | NCT02576639 NCT02565511 | |
| JNJ-54861911 | Small molecule | Phase II/III | Not applicable | Janssen, Shionogi Pharma | NCT02406027 NCT02569398 | |
| Verubecestat | Small molecule | Phase III | Not applicable | Merck | NCT01953601 | |
| γ-Secretase inhibitor | Semagacestat | Small molecule | Phase III Discontinued in 2012 | No clinical efficacy and adverse effects: skin cancer and infections | Eli Lilly & Co. | NCT00762411 NCT00594568 NCT01035138 |
| Avagacestat | Small molecule | Phase II Discontinued in 2012 | Adverse effects: cerebral microbleeds, glycosuria, and nonmelanoma skin cancer | Bristol-Myers Squibb | NCT00890890 | |
| EVP-0962 | Small molecule | Phase II Discontinued in 2016 | Not applicable | FORUM Pharmaceuticals Inc. | NCT01661673 | |
| NIC5-15 | Small molecule | Phase II | Not applicable | Humanetics Pharmaceuticals Corporation | NCT00470418 NCT01928420 | |
| Aβ clearance | AN-1792 | Active immunotherapy (Aβ1-42 peptides) | Phase II Discontinued in 2002 | Adverse effects: meningoencephalitis | Janssen, Pfizer | NCT00021723 |
| CAD106 | Active immunotherapy (Aβ1-6 peptides) | Phase II/III | Not applicable | Novartis Pharmaceuticals Corporation | NCT01097096 NCT02565511 | |
| ACC-001 | Active immunotherapy (Aβ1-4 peptides) | Phase II Discontinued in 2013 | Adverse effects: strong autoimmune response | Janssen | NCT01238991 NCT00479557 NCT00498602 | |
| Affitope AD02 | Active immunotherapy (6 a.a. peptides mimic Aβ1-42 N-terminus) | Phase II-discontinued in 2014 | Not applicable | AFFiRiS AG | NCT01117818 | |
| Bapineuzumab | Passive immunotherapy (against Aβ N-terminal) | Phase III- discontinued in 2012 | No clinical efficacy | Janssen, Pfizer | NCT00667810 NCT00676143 | |
| AAB-003 | Passive immunotherapy | Phase I | Not applicable | Janssen, Pfizer | NCT01193608 NCT01369225 | |
| GSK933776 | Passive immunotherapy (against Aβ N-terminal) | Phase I Discontinued in 2012 | No clinical benefit | GlaxoSmithKline (GSK) | NCT00459550 NCT01424436 | |
| Solanezumab | Passive immunotherapy (against Aβ16-24) | Phase III Discontinued in 2016 | Missed primary endpoint | Eli Lilly & Co. | NCT01127633 NCT01900665 | |
| Crenezumab | Passive immunotherapy against Aβ) | Phase III | Not applicable | AC Immune SA, Genentech, Hoffmann-La Roche | NCT02670083 | |
| Gantenerumab | Passive immunotherapy (against Aβ3-12 & Aβ18-27) | Phase III | Not applicable | Chugai Pharmaceutical Co., Ltd., Hoffmann-La Roche | NCT02051608 NCT01900665 | |
| BAN2401 | Passive immunotherapy (against large soluble Aβ protofibrils) | Phase II | Not applicable | Biogen, Eisai Co., Ltd. | NCT01767311 | |
| Aducanumab | Passive immunotherapy (against aggregated Aβ) | Phase III | Not applicable | Biogen | NCT02477800 NCT02484547 | |
| Tau stabilization | Epothilone D | Small molecule | Phase I Discontinued in 2013 | Not applicable | Bristol-Myers Squibb | Not available |
| TPI 287 | Small molecule | Phase I | Not applicable | Cortice Biosciences | NCT01966666 | |
| Tau aggregation inhibitor | Rember™ | Small molecule | Phase II Discontinued in 2007 | Adverse effects: diarrhea, urinary urgency, and painful urination, etc. | TauRx Therapeutics Ltd | NCT00515333 NCT00684944 |
| TRx0237 | Small molecule | Phase III | Not applicable | TauRx Therapeutics Ltd | NCT01689233 NCT01689246 NCT01626378 | |
| p-Tau clearance | AADvac-1 | Active immunotherapy (synthetic peptide truncated and misfolded tau) | Phase II | Not applicable | Axon Neuroscience SE | NCT01850238 NCT02031198 NCT02579252 |
| ACI-35 | Active immunotherapy (Human protein tau sequence 393 to 408 of longest tau isoform phosphorylated at S396 and S404) | Phase I | Not applicable | AC Immune SA, Janssen | Main ID in the WHO International Clinical Trials Registry Platform: ISRCTN13033912 | |
| Microglial activation inhibitor | Alzhemed™ | Small molecule | Phase III Discontinued in 2007 | No clinical efficacy | Neurochem, Inc. | Not available |
| Azeliragon | Small molecule | Phase III | Not applicable | Pfizer, TransTech Pharma, Inc., vTv Therapeutics LLC | NCT02080364 | |
| Ibuprofen | Small molecule | Phase IV Discontinued in 2005 | No clinical efficacy | Not applicable | Not available | |
| Flurizan™ | Small molecule | Phase III Discontinued in 2008 | No clinical efficacy | Myriad Genetics & Laboratories | NCT00322036 | |
| CHF 5074 | Small molecule | Phase II | Not applicable | CereSpir™ Incorporated, Chiesi Pharmaceuticals Inc. | NCT01421056 |
Acetylcholine receptor antibody
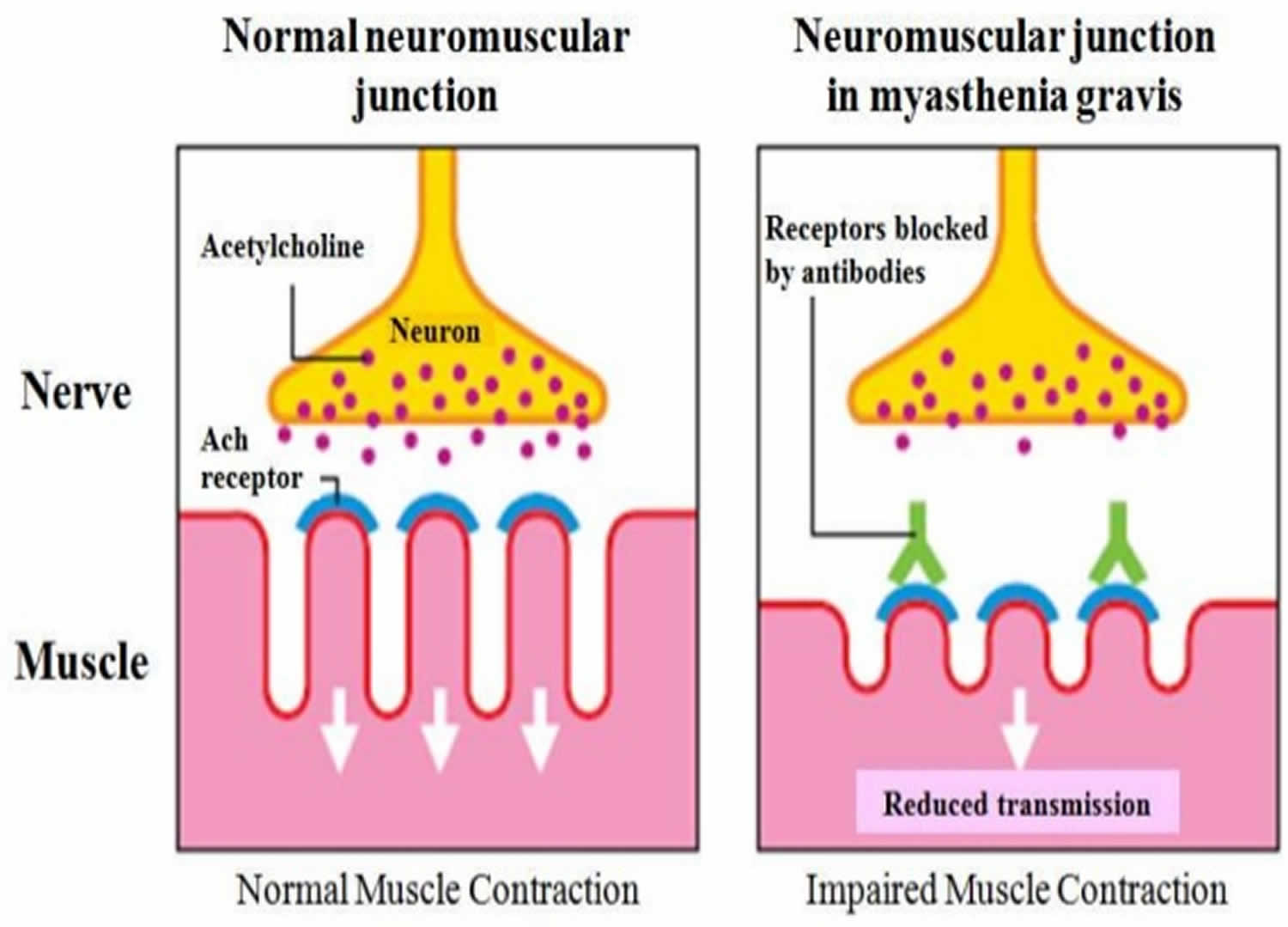
Acetylcholine receptor antibody is a protein found in the blood of most people with myasthenia gravis. The acetylcholine receptor antibody affects a chemical that sends signals from nerves to muscles and between nerves in the brain. Myasthenia gravis is a chronic autoimmune disease in which autoantibodies against the acetylcholine receptor on skeletal muscle cells cause a block in neuromuscular junctions, leading to progressive weakness in the skeletal muscles, which are responsible for breathing and moving parts of the body, including the arms and legs and eventually death. The name myasthenia gravis, which is Latin and Greek in origin, means “grave, or serious, muscle weakness.” People who have myasthenia gravis are more likely to also have other autoimmune disorders, such as rheumatoid arthritis or lupus.
Myasthenia gravis most commonly affects the muscles that control the eyes and eyelids, facial expressions, chewing, swallowing and speaking. But it can affect most parts of the body.
In around one in five people, the condition only affects the eye muscles. This is known as “ocular myasthenia”. But for most people, the weakness spreads to other parts of the body over a few weeks, months or years. If you’ve only had symptoms affecting your eyes for two years or more, it’s unusual for other parts of your body to be affected later on.
Acetylcholine receptor antibodies impede communication between nerves and skeletal muscles, inhibit muscle contraction, and cause rapid muscle fatigue by preventing activation of the acetylcholine receptors. They do this in three major ways:
- Binding antibodies attach to the receptors on nerve cells and may initiate an inflammatory reaction that destroys the receptors.
- Blocking antibodies may sit on the receptors, preventing acetylcholine from binding.
- Modulating antibodies may cross-link the receptors, causing them to be taken up into the muscle cell and removed from the neuromuscular junction.
Figure 4. Acetylcholine receptor antibody
Three different types of tests are available to determine which of these may be the problem in a particular individual. However, the test that measures “binding” antibodies is most commonly used because it is generally rare for the other two tests to be positive without the “binding” test being positive as well. These other tests may be used when a healthcare practitioner strongly suspects myasthenia gravis and the “binding” test is negative.
The hallmark of myasthenia gravis is muscle weakness that worsens after periods of activity and improves after periods of rest. Certain muscles such as those that control eye and eyelid movement, facial expression, chewing, talking, and swallowing are often (but not always) involved in the disorder. The muscles that control breathing and neck and limb movements may also be affected.
There is no known cure but with current therapies most cases of myasthenia gravis are not as “grave” as the name implies. Available treatments can control symptoms and often allow people to have a relatively high quality of life. Most individuals with the condition have a normal life expectancy.
What causes myasthenia gravis?
Myasthenia gravis is caused by an error in the transmission of nerve impulses to muscles. It occurs when normal communication between the nerve and muscle is interrupted at the neuromuscular junction—the place where nerve cells connect with the muscles they control. Use of drugs such as succinylcholine can increase acetylcholine receptor antibodiess.
Neurotransmitters are chemicals that neurons, or brain cells, use to communicate information. Normally when electrical signals or impulses travel down a motor nerve, the nerve endings release a neurotransmitter called acetylcholine. Acetylcholine travels from the nerve ending and binds to acetylcholine receptors on the muscle. The binding of acetylcholine to its receptor activates the muscle and causes a muscle contraction.
In myasthenia gravis, antibodies (immune proteins) block, alter, or destroy the receptors for acetylcholine at the neuromuscular junction, which prevents the muscle from contracting. In most individuals with myasthenia gravis, this is caused by antibodies to the acetylcholine receptor itself. However, antibodies to other proteins, such as MuSK (Muscle-Specific Kinase) protein, can also lead to impaired transmission at the neuromuscular junction.
These antibodies are produced by the body’s own immune system. Myasthenia gravis is an autoimmune disease because the immune system—which normally protects the body from foreign organisms—mistakenly attacks itself.
The thymus is a gland that controls immune function and maybe associated with myasthenia gravis. Located in the chest behind the breast bone, the gland is largest in children. It grows gradually until puberty, and then gets smaller and is replaced by fat. Throughout childhood, the thymus plays an important role in the development of the immune system because it is responsible for producing T-lymphocytes or T cells, a specific type of white blood cell that protects the body from viruses and infections.
In many adults with myasthenia gravis, the thymus gland remains large. People with the disease typically have clusters of immune cells in their thymus gland similar to lymphoid hyperplasia—a condition that usually only happens in the spleen and lymph nodes during an active immune response. Some individuals with myasthenia gravis develop thymomas (tumors of the thymus gland). Thymomas are most often harmless, but they can become cancerous.
The thymus gland plays a role in myasthenia gravis, but its function is not fully understood. Scientists believe that the thymus gland may give incorrect instructions to developing immune cells, ultimately causing the immune system to attack its own cells and tissues and produce acetylcholine receptor antibodies—setting the stage for the attack on neuromuscular transmission.
Who gets myasthenia gravis?
Myasthenia gravis affects both men and women and occurs across all racial and ethnic groups. It most commonly impacts young adult women (under 40) and older men (over 60), but it can occur at any age, including childhood. Myasthenia gravis is not inherited nor is it contagious. Occasionally, the disease may occur in more than one member of the same family.
Although myasthenia gravis is rarely seen in infants, the fetus may acquire antibodies from a mother affected with myasthenia gravis—a condition called neonatal myasthenia. Generally, neonatal myasthenia gravis is temporary and the child’s symptoms usually disappear within two to three months after birth. Rarely, children of a healthy mother may develop congenital myasthenia. This is not an autoimmune disorder (it is caused by defective genes that produce abnormal proteins in the neuromuscular junction) and can cause similar symptoms to myasthenia gravis.
Can myasthenia gravis be inherited?
As an autoimmune process, no. Some people may inherit a genetic defect that causes congenital myasthenic syndrome, a condition with similar symptoms.
Is there anything I can do to prevent getting acetylcholine receptor antibodies?
No, the cause of myasthenia gravis is not known and the condition is not preventable.
Can myasthenia gravis be passed from one person to another?
Myasthenia gravis is not contagious, but a pregnant woman with myasthenia gravis can pass some of her acetylcholine receptor antibodies to her fetus. This can cause a newborn to have myasthenia gravis symptoms for several weeks after birth.
What are the symptoms of myasthenia gravis?
Although myasthenia gravis may affect any skeletal muscle, muscles that control eye and eyelid movement, facial expression, and swallowing are most frequently affected. The onset of the disorder may be sudden and symptoms often are not immediately recognized as myasthenia gravis.
In most cases, the first noticeable symptom is weakness of the eye muscles. In others, difficulty swallowing and slurred speech may be the first signs. The degree of muscle weakness involved in myasthenia gravis varies greatly among individuals, ranging from a localized form limited to eye muscles (ocular myasthenia), to a severe or generalized form in which many muscles—sometimes including those that control breathing—are affected.
Myasthenia gravis symptoms may include:
- drooping of one or both eyelids (ptosis)
- blurred or double vision (diplopia) due to weakness of the muscles that control eye movements
- a change in facial expression or difficulty making facial expressions
- problems with chewing and difficulty swallowing
- shortness of breath and occasionally serious breathing difficulties
- impaired speech or slurred speech (dysarthria)
- weakness in the arms, hands, fingers, legs, and neck.
The symptoms tend to get worse when you’re tired. Many people find they are worse towards the end of the day, and better the next morning after getting some sleep or rest.
In some people, the symptoms can also have a number of other triggers, such as stress, infections and certain medicines.
Can myasthenia gravis affect my heart?
No, the receptors for heart and smooth (digestive) muscles are different from skeletal muscles so they are not affected by the formation of acetylcholine receptor binding antibodies.
What is a myasthenic crisis?
A myasthenic crisis is a medical emergency that occurs when the muscles that control breathing weaken to the point where individuals require a ventilator to help them breathe.
Approximately 15 to 20 percent of people with myasthenia gravis experience at least one myasthenic crisis. This condition usually requires immediate medical attention and may be triggered by infection, stress, surgery, or an adverse reaction to medication. However, up to one-half of people may have no obvious cause for their myasthenic crisis. Certain medications have been shown to cause myasthenia gravis. However, sometimes these medications may still be used if it is more important to treat an underlying condition.
How is myasthenia gravis diagnosed?
A doctor may perform or order several tests to confirm the diagnosis, including:
- A physical and neurological examination. A physician will first review an individual’s medical history and conduct a physical examination. In a neurological examination, the physician will check muscle strength and tone, coordination, sense of touch, and look for impairment of eye movements.
- An edrophonium test. This test uses injections of edrophonium chloride to briefly relieve weakness in people with myasthenia gravis. The drug blocks the breakdown of acetylcholine and temporarily increases the levels of acetylcholine at the neuromuscular junction. It is usually used to test ocular muscle weakness. If you have a sudden but temporary improvement in muscle strength after the injection, it’s likely you have myasthenia gravis. But edrophonium test is rarely done these days because there’s a risk it could cause potentially serious side effects, such as a slow heartbeat and breathing problems. It’s only done if absolutely necessary and in a hospital setting where treatment for any side effects is readily available.
- A blood test. Most individuals with myasthenia gravis have abnormally elevated levels of acetylcholine receptor antibodies. A second antibody—called the anti-MuSK (muscle-specific kinase) antibody—has been found in about half of individuals with myasthenia gravis who do not have acetylcholine receptor antibodies. A blood test can also detect this antibody. However, in some individuals with myasthenia gravis, neither of these antibodies is present. These individuals are said to have seronegative (negative antibody) myasthenia.
- Electrodiagnostics. Diagnostic tests include repetitive nerve stimulation, which repeatedly stimulates a person’s nerves with small pulses of electricity to tire specific muscles. Muscle fibers in myasthenia gravis, as well as other neuromuscular disorders, do not respond as well to repeated electrical stimulation compared to muscles from normal individuals. Single fiber electromyography (EMG), considered the most sensitive test for myasthenia gravis, detects impaired nerve-to-muscle transmission. EMG can be very helpful in diagnosing mild cases of myasthenia gravis when other tests fail to demonstrate abnormalities.
- Diagnostic imaging. Diagnostic imaging of the chest using computed tomography (CT) or magnetic resonance imaging (MRI) may identify the presence of a thymoma.
- Pulmonary function testing. Measuring breathing strength can help predict if respiration may fail and lead to a myasthenic crisis.
Because weakness is a common symptom of many other disorders, the diagnosis of myasthenia gravis is often missed or delayed (sometimes up to two years) in people who experience mild weakness or in those individuals whose weakness is restricted to only a few muscles.
Myasthenia gravis treatment
Today, myasthenia gravis can generally be controlled. There are several therapies available to help reduce and improve muscle weakness.
- Thymectomy. This operation to remove the thymus gland (which often is abnormal in individuals with myasthenia gravis) can reduce symptoms and may cure some people, possibly by rebalancing the immune system. A recent study found that thymectomy is beneficial both for people with thymoma and those with no evidence of the tumors. The clinical trial followed 126 people with myasthenia gravis and no visible thymoma and found that the surgery reduced muscle weakness and the need for immunosuppressive drugs.
- Anticholinesterase medications. Medications to treat the disorder include anticholinesterase agents such as mestinon or pyridostigmine, which slow the breakdown of acetylcholine at the neuromuscular junction and thereby improve neuromuscular transmission and increase muscle strength.
- Immunosuppressive drugs. These drugs improve muscle strength by suppressing the production of abnormal antibodies. They include prednisone, azathioprine, mycophenolate mofetil, tacrolimus, and rituximab. The drugs can cause significant side effects and must be carefully monitored by a physician.
- Plasmapheresis and intravenous immunoglobulin. These therapies may be options in severe cases of myasthenia gravis. Individuals can have antibodies in their plasma (a liquid component in blood) that attack the neuromuscular junction. These treatments remove the destructive antibodies, although their effectiveness usually only lasts for a few weeks to months.
- Plasmapheresis is a procedure using a machine to remove harmful antibodies in plasma and replace them with good plasma or a plasma substitute.
- Intravenous immunoglobulin is a highly concentrated injection of antibodies pooled from many healthy donors that temporarily changes the way the immune system operates. It works by binding to the antibodies that cause myasthenia gravis and removing them from circulation.
- Avoiding triggers. The symptoms of myasthenia gravis can sometimes have a specific trigger. Doing what you can to avoid your triggers may help. It’s also a good idea to avoid activities that could be dangerous if you experience sudden weakness, such as swimming alone.
- Common triggers include:
- tiredness and exhaustion – getting plenty of rest and not over-exerting yourself may help
- stress – read some tips to help reduce your stress levels
- infections – you may be advised to have an annual flu jab and the one-off pneumococcal vaccine, but get advice before having a “live” vaccine, such as
- the shingles vaccine
- medicines – make sure your doctor is aware of your condition and get advice before taking anything on the list of medicines that can trigger
- myasthenia gravis symptoms
- surgery – make sure your surgeon is aware of your condition before having any operation.
Myasthenia gravis prognosis
With treatment, most individuals with myasthenia can significantly improve their muscle weakness and lead normal or nearly normal lives. Sometimes the severe weakness of myasthenia gravis may cause respiratory failure, which requires immediate emergency medical care.
Some cases of myasthenia gravis may go into remission—either temporarily or permanently—and muscle weakness may disappear completely so that medications can be discontinued. Stable, long-lasting complete remissions are the goal of thymectomy and may occur in about 50 percent of individuals who undergo this procedure.
- Purves D, Augustine GJ, Fitzpatrick D, et al., editors. Neuroscience. 2nd edition. Sunderland (MA): Sinauer Associates; 2001. Acetylcholine. Available from: https://www.ncbi.nlm.nih.gov/books/NBK11143[↩][↩]
- Role of acetylcholine in neurotransmission of the carotid body. Respir Physiol Neurobiol. 2007 Jul 1;157(1):93-105. Epub 2007 Jan 11. https://doi.org/10.1016/j.resp.2006.12.010[↩]
- Spontaneous subthreshold activity at motor nerve endings. FATT P, KATZ B. J Physiol. 1952 May; 117(1):109-28.[↩]
- The nature of the antagonism between calcium and magnesium ions at the neuromuscular junction. JENKINSON DH. J Physiol. 1957 Oct 30; 138(3):434-44[↩]
- Actions of triethylcholine on neuromuscular transmission. BOWMAN WC, RAND MJ. Br J Pharmacol Chemother. 1961 Oct; 17():176-95.[↩]
- Acetylcholine synthesis and recycling. http://faculty.pasadena.edu/dkwon/chap%208_files/textmostly/slide58.html[↩]
- International Union of Pharmacology. XVII. Classification of Muscarinic Acetylcholine Receptors. Pharmacological Reviews June 1, 1998, 50 (2) 279-290. http://pharmrev.aspetjournals.org/content/50/2/279.long[↩][↩][↩]
- Role LW, Berg DK. (1996) Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16:1077–1085.[↩]
- Functional nicotinic acetylcholine receptors are expressed in B lymphocyte-derived cell lines. Skok MV, Kalashnik EN, Koval LN, Tsetlin VI, Utkin YN, Changeux JP, Grailhe R. Mol Pharmacol. 2003 Oct; 64(4):885-9.[↩]
- Role of muscarinic receptor antagonists in urgency and nocturia. Michel MC, de la Rosette JJ. BJU Int. 2005 Sep; 96 Suppl 1:37-42.[↩]
- Tiotropium bromide: a novel once-daily anticholinergic bronchodilator for the treatment of COPD. Hansel TT, Barnes PJ. Drugs Today (Barc). 2002 Sep; 38(9):585-600.[↩]
- WESS J. Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu. Rev. Pharmacol. Toxicol. 2004;44:423–450.[↩]
- Identification of a family of muscarinic acetylcholine receptor genes. Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987 Jul 31; 237(4814):527-32. https://www.ncbi.nlm.nih.gov/pubmed/3037705/[↩][↩]
- ALEXANDER S.P.H., MATHIE A., PETERS J.A. Acetylcholine (muscarinic) receptors: guide to receptors and channels. Br. J. Pharmacol. 2004;141:S6.[↩]
- Kubo T, Fukuda K, Mikami A, Maeda A, Takahashi H, Mishina M, Haga T, Haga K, Ichiyama A, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. (1986a) Cloning, sequencing and expression of complementary DNA encoding the muscarinic acetylcholine receptor. Nature 323:411–416.[↩]
- Hulme EC, Birdsall NJM, Buckley NJ. (1990) Muscarinic receptor subtypes. Ann Rev Pharmacol Toxicol 30:633–673.[↩]
- Bonner TI, Modi WS, Seuanez HN, O’Brien SJ. (1991) Chromosomal mapping of the five human genes encoding muscarinic acetylcholine receptors. Cytogenet Cell Genet 58:1850–1851.[↩]
- Brown DA. Acetylcholine. British Journal of Pharmacology. 2006;147(Suppl 1):S120-S126. doi:10.1038/sj.bjp.0706474. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1760728/[↩][↩]
- Molecular aspects of muscarinic receptor dimerization. Zeng F, Wess J. Neuropsychopharmacology. 2000 Oct; 23(4 Suppl):S19-31. https://www.ncbi.nlm.nih.gov/pubmed/11008064/[↩]
- The pharmacological actions of polymethylene bistrimethyl-ammonium salts. PATON WD, ZAIMIS EJ. Br J Pharmacol Chemother. 1949 Dec; 4(4):381-400. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1509928/pdf/bripharmchem00112-0068.pdf[↩]
- The actions of heterocyclic bisquaternary compounds, especially of a pyrrolidinium series. MASON DF, WIEN R. Br J Pharmacol Chemother. 1955 Mar; 10(1):124-32[↩]
- Pharmacological properties of pempidine (1:2:2:6:6-pentamethylpiperidine), a new ganglion-blocking compound. CORNE SJ, EDGE ND. Br J Pharmacol Chemother. 1958 Sep; 13(3):339-49.[↩]
- Nicotinic receptors in the brain: correlating physiology with function. Jones S, Sudweeks S, Yakel JL. Trends Neurosci. 1999 Dec; 22(12):555-61. https://www.ncbi.nlm.nih.gov/pubmed/10542436/[↩]
- Nicotinic receptor function: new perspectives from knockout mice. Cordero-Erausquin M, Marubio LM, Klink R, Changeux JP. Trends Pharmacol Sci. 2000 Jun; 21(6):211-7.[↩]
- COLQUHOUN D., UNWIN N., SHELLEY C., BATTON C., SIVILOTTI L.G. 2003 Nicotinic acetylcholine receptors Drug Discovery and Drug Developmented. Abrahams, D, pp. 357–405.New York: John Wiley;[↩]
- Refined structure of the nicotinic acetylcholine receptor at 4A resolution. Unwin N. J Mol Biol. 2005 Mar 4; 346(4):967-89.[↩]
- Emerging structure of the nicotinic acetylcholine receptors. Karlin A. Nat Rev Neurosci. 2002 Feb; 3(2):102-14.[↩]
- Atri A, Sherman S, Norman KA, Kirchhoff BA, Nicolas MM, Greicius MD, Cramer SC, Breiter HC, Hasselmo ME, Stern CE. Blockade of central cholinergic receptors impairs new learning and increases proactive interference in a word paired-associate memory task. Behav Neurosci. 2004;118:223–236.[↩]
- Green A, Ellis KA, Ellis J, Bartholomeusz CF, Ilic S, Croft RJ, Luan Phan K, Nathan PJ. Muscarinic and nicotinic receptor modulation of object and spatial n-back working memory in humans. Pharmacol Biochem Behav. 2005;81:575–584[↩]
- On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Bartus RT. Exp Neurol. 2000 Jun; 163(2):495-529.[↩]
- Further analysis of the cognitive effects of tetrahydroaminoacridine (THA) in Alzheimer’s disease: assessment of attentional and mnemonic function using CANTAB. Sahakian BJ, Owen AM, Morant NJ, Eagger SA, Boddington S, Crayton L, Crockford HA, Crooks M, Hill K, Levy R. Psychopharmacology (Berl). 1993; 110(4):395-401.[↩]
- Cognitive performance in Alzheimer’s disease patients receiving rivastigmine for up to 5 years. Small GW, Kaufer D, Mendiondo MS, Quarg P, Spiegel R. Int J Clin Pract. 2005 Apr; 59(4):473-7.[↩]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–539[↩]
- Hasselmo ME. The Role of Acetylcholine in Learning and Memory. Current opinion in neurobiology. 2006;16(6):710-715. doi:10.1016/j.conb.2006.09.002. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2659740/[↩]
- Tang Y, Mishkin M, Aigner TG. Effects of muscarinic blockade in perirhinal cortex during visual recognition. Proc. Natl. Acad. Sci. USA. 1997;94:12667–12669[↩][↩]
- Winters BD, Bussey TJ. Removal of cholinergic input to perirhinal cortex disrupts object recognition but not spatial working memory in the rat. Eur J Neurosci. 2005;21:2263–2270[↩][↩]
- Blokland A, Honig W, Raaijmakers WGM. Effects of intra-hippocampal scopolamine injections in a repeated spatial acquisition task in the rat. Psychopharmacology. 1992;109:373–376.[↩]
- Elvander E, Schott PA, Sandin J, Bjelke B, Kehr J, Yoshitake T, Ogren SO. Intraseptal muscarinic ligands and galanin: influence on hippocampal acetylcholine and cognition. Neuroscience. 2004;126:541–557.[↩][↩]
- Bunce JG, Sabolek HR, Chrobak JJ. Intraseptal infusion of the cholinergic agonist carbachol impairs delayed-non-match-to-sample radial arm maze performance in the rat. Hippocampus. 2004;14:450–459.[↩]
- Rogers JL, Kesner RP. Cholinergic modulation of the hippocampus during encoding and retrieval. Neurobiol Learn Mem. 2003;80:332–342.[↩]
- Hung S-Y, Fu W-M. Drug candidates in clinical trials for Alzheimer’s disease. Journal of Biomedical Science. 2017;24:47. doi:10.1186/s12929-017-0355-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516350/[↩][↩][↩][↩][↩][↩][↩][↩]
- Graham WV, Bonito-Oliva A, Sakmar TP. Update on Alzheimer’s disease Therapy and Prevention Strategies. Annu Rev Med. 2017;68:413–430. doi: 10.1146/annurev-med-042915-103753[↩]
- Grimm A, Mensah-Nyagan AG, Eckert A. Alzheimer, mitochondria and gender. Neurosci Biobehav Rev. 2016;67:89–101. doi: 10.1016/j.neubiorev.2016.04.012[↩]
- Godyn J, Jonczyk J, Panek D, Malawska B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep. 2016;68:127–138. doi: 10.1016/j.pharep.2015.07.006[↩]
- Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6:37. doi: 10.1186/alzrt269[↩]
- Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403. doi: 10.1016/S0140-6736(76)91936-X[↩]
- Upton N, Chuang TT, Hunter AJ, Virley DJ. 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics. 2008;5:458–469. doi: 10.1016/j.nurt.2008.05.008[↩][↩]
- Brauser D: Two more phase 3 trials of Alzheimer’s drug idalopirdine fail. Medscape; 2017[↩]
- Axovant Unveils New Data Analysis Showing Addition of Intepirdine to Standard Therapy May Help People with Alzheimer’s Disease Maintain Independence Longer. PR Newswire; 2016. https://www.prnewswire.com/news-releases/axovant-unveils-new-data-analysis-showing-addition-of-intepirdine-to-standard-therapy-may-help-people-with-alzheimers-disease-maintain-independence-longer-300376136.html[↩]
- Passani MB, Blandina P. Cognitive implications for H3 and 5-HT3 receptor modulation of cortical cholinergic function: a parallel story. Methods Find Exp Clin Pharmacol. 1998;20:725–733. doi: 10.1358/mf.1998.20.8.487510[↩]
- Haig GM, Pritchett Y, Meier A, Othman AA, Hall C, Gault LM, Lenz RA. A randomized study of H3 antagonist ABT-288 in mild-to-moderate Alzheimer’s dementia. J Alzheimers Dis. 2014;42:959–971.[↩]
- Kubo M, Kishi T, Matsunaga S, Iwata N. Histamine H3 receptor antagonists for Alzheimer’s disease: a systematic review and meta-analysis of randomized placebo-controlled trials. J Alzheimers Dis. 2015;48:667–671. doi: 10.3233/JAD-150393[↩]
- Woodruff-Pak DS, Vogel RW, 3rd, Wenk GL. Galantamine: effect on nicotinic receptor binding, acetylcholinesterase inhibition, and learning. Proc Natl Acad Sci U S A. 2001;98:2089–2094. doi: 10.1073/pnas.98.4.2089[↩][↩]
- Russo P, Del Bufalo A, Frustaci A, Fini M, Cesario A. Beyond acetylcholinesterase inhibitors for treating Alzheimer’s disease: alpha7-nAChR agonists in human clinical trials. Curr Pharm Des. 2014;20:6014–6021. doi: 10.2174/1381612820666140316130720[↩][↩]
- Hung SY, Huang WP, Liou HC, Fu WM. LC3 overexpression reduces Abeta neurotoxicity through increasing alpha7nAchR expression and autophagic activity in neurons and mice. Neuropharmacology. 2015;93:243–251. doi: 10.1016/j.neuropharm.2015.02.003[↩]
- Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, Reneerkens OA, Flood DG, Hilt D, Gawryl M, et al. EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology. 2012;62:1099–1110. doi: 10.1016/j.neuropharm.2011.10.024[↩]
- Rare but Severe Side Effects Sideline Some Phase 3 Encenicline Trials. https://www.alzforum.org/news/research-news/rare-severe-side-effects-sideline-some-phase-3-encenicline-trials[↩]





{kind=link}