Contents
- ALA dehydratase deficiency porphyria
- ALA dehydratase deficiency porphyria cause
- ALA dehydratase deficiency porphyria signs and symptoms
- ALA dehydratase deficiency porphyria complications
- ALA dehydratase deficiency porphyria diagnosis
- ALA dehydratase deficiency porphyria differential diagnosis
- ALA dehydratase deficiency porphyria treatment
- ALA dehydratase deficiency porphyria prognosis
ALA dehydratase deficiency porphyria
ALA dehydratase deficiency porphyria (ADP) also called ALAD deficiency porphyria, delta-aminolevulinate dehydratase deficiency or Doss porphyria is an extremely rare genetic disorder that involve defects in heme or ‘haem’ biosynthetic pathway caused by mutations (changes) in the ALAD gene which results in deficiency of the enzyme delta-aminolevulinate dehydratase (ALAD) (also known as porphobilinogen synthase [PBG synthase]), the second enzyme in the heme biosynthetic pathway that produces porphyrins and heme 1, 2, 3, 4, 5, 6, 7, 8. Deficiency of delta-aminolevulinate dehydratase (ALAD) enzyme is crucial for the synthesis of heme, a component of hemoglobin in red blood cells, which leads to a buildup of porphyrin precursors, primarily delta-aminolevulinic acid (ALA), in the body, causing a variety of symptoms. ALA dehydratase deficiency porphyria (ADP) is a type of acute hepatic porphyria that is characterized by severe episodic neurovisceral symptoms resembling acute intermittent porphyria (AIP) and no skin signs and symptoms. ALA dehydratase deficiency porphyria (ADP) symptoms vary from one person to another, but usually come from the neurological and gastrointestinal systems.
ALAD deficiency porphyria is caused by mutation is in the ALAD gene localized at chromosome 9q34 causing a deficiency of the enzyme delta-aminolevulinic acid dehydratase (ALAD). ALA dehydratase deficiency porphyria (ADP) is inherited as an autosomal recessive disorder, meaning one copy of the abnormal ALAD gene is inherited from each parent, whereas the other three acute porphyrias are autosomal dominant (i.e., acute intermittent porphyria [AIP], hereditary coproporphyria [HCP] and variegate porphyria [VP]). Each parent of an affected individual must have a mutation in one of their delta-aminolevulinic acid dehydratase (ALAD) genes and both must pass their mutation on to their child.
ALA dehydratase deficiency porphyria (ADP) is the rarest type of porphyria, with only 10 documented cases reported worldwide 9, 10, 11, 12. To date, all documented ALA dehydratase deficiency porphyria cases have been in male patients in contrast to the other acute porphyrias, with symptoms manifestating at age 14 4, 5. ALA dehydratase deficiency porphyria (ADP) is the least common of all the porphyrias with less than 10 cases documented to date. Of these cases, 2 of them were identified at birth, one of them at age 7, four patients were diagnosed at age 12 to 15, and one patient in Belgium was diagnosed at age 63 who had symptoms when he developed a myeloproliferative neoplasm (polycythemia vera) 1, 13, 10, 5.
A variety of different triggers have been identified that can precipitate an acute attack in individuals with ALA dehydratase deficiency porphyria (ADP). These triggers include alcohol, certain drugs, physical and psychological stress, infection, fasting (reduced caloric intake) and dehydration. The use of estrogen or progesterone is also suspected of triggering an acute attack.
Individuals with ALA dehydratase deficiency porphyria (ADP) may have bouts where symptoms are intense, which are referred to as “neurovisceral symptoms” or “acute attacks“. An attack may last for several weeks. During an attack, affected individuals may experience severe abdominal cramping or pain accompanied by nausea, vomiting, constipation, bloating, and rarely diarrhea. During infancy, gastrointestinal abnormalities may cause an affected child to fail to grow and gain weight as expected. Since the abdominal pain is neurological, abdominal tenderness or rebound, leukocytosis (an elevated white blood cell count in the blood), and fever are either minimal or absent. No skin findings were noted.
Several other neurological symptoms can occur during an acute attack due to problems with the nerves outside the central nervous system (peripheral neuropathy), resulting in numbness or tingling in the hands and feet, burning pain, sensitivity to touch, and a lack of coordination (sensory neuropathy). In severe cases, the motor nerves are involved (motor neuropathy), resulting in loss or partial impairment of the ability to use voluntary muscles. Motor dysfunction and muscle weakness were seen in 6 out of the 8 patients.
ALA dehydratase deficiency porphyria (ADP) can also be associated with neuropsychiatric changes during an acute attack. Neuropsychiatric symptoms can include disorientation, agitation, anxiety, restlessness, hysteria, hallucinations, delirium, altered consciousness, apathy, depression, and phobias. In severe cases, loss of contact from reality (psychosis) has been reported.
Additional symptoms that occur during acute attacks include a rapid heartbeat (tachycardia), high blood pressure (hypertension), seizures, and breathing (respiratory) impairment.
There are many laboratory tests available for the porphyrias, and it is often difficult to decide which should be chosen. Many of these tests are expensive and the results are often difficult to interpret. When the classic triad of abdominal pain, peripheral neuropathy, and neuropsychiatric symptoms suggest an acute porphyria, the best screening tests are urinary aminolevulinic acid (ALA) and urinary porphobilinogen (PBG), obtained preferably during an acute porphyria attack (e.g. when someone is having abdominal pain, etc). The total urine porphyrin level will be elevated. Urinary porphobilinogen (PBG) is not significantly increased in ALA dehydratase porphyria (ADP) 14.
If these initials tests are normal in a patient with classic symptoms of ALA dehydratase deficiency porphyria (ADP), then it is suggested to repeat the first-line tests rather than proceeding to second-line testing. Second-line testing in a patient is performed if it is found that total urine porphyrins are elevated without porphobilinogen (PBG) elevation (findings suggestive of ALA dehydratase deficiency porphyria), and clinical suspicion of acute porphyria is very high.
Note the most frequent errors lie in (1) not considering the diagnosis in a timely fashion, leading to the average 15‐year delay in the United States from time of first symptoms to eventual correct diagnosis, and (2) ordering a random urine screening for porphyrins rather than for ALA and PBG 15, 16.
Second-line testing includes urine aminolevulinic acid (ALA) level, fractionation of porphyrins, and measurement of red blood cell (erythrocyte) protoporphyrin. Aminolevulinic acid (ALA) and red blood cell (erythrocyte) protoporphyrin is markedly elevated, and there is a predominance of coproporphyrin III noted.
Diagnosis is confirmed by measuring ALA levels in erythrocytes and DNA testing to identify the specific ALAD genetic mutation, which reveals heterozygous or homozygous ALAD mutations 17. Patients are usually compound heterozygotes for ALAD mutations. Heterozygotes may be more susceptible to lead toxicity, which inhibits ALAD enzyme by displacing zinc 18.
ALA dehydratase deficiency porphyria (ADP) treatment is the same as in the other acute porphyrias and is directed toward the specific symptoms that are present in each individual. Because there have been so few cases of ALA dehydratase deficiency porphyria (ADP), there is only limited information on treatment for the disorder 1.
Acute attacks are managed by withdrawing and avoiding the offending agents that can precipitate acute attacks and supportive care for nausea, vomiting, pain, seizures, and electrolyte imbalances. Offending agents can include alcohol, oral contraceptives, antibiotics (such as nitrofurantoin, rifampin, trimethoprim-sulfamethoxazole), anti-seizure medications (such as topiramate, phenytoin, phenobarbital, ethosuximide, valproic acid), and many more medications. Physical, psychological, and emotional stress may also precipitate attacks and must be minimized 15.
Attacks can be prevented in many cases by avoiding harmful drugs and adverse dietary practices such as fasting, and low carbohydrate diets is recommended for affected individuals.
The specific drugs that may need to be avoided in one person can differ from the drugs that need to be avoided in another.
- The American Porphyria Foundation offers a drug database with safety information about the interaction of specific drugs in patients with porphyria (https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/) or https://porphyriafoundation.org/drugdatabase/drug-safety-database-search/.
- The EPNET/NAPOS Database should also be consulted. The Norwegian Porphyria Centre (NAPOS), with the European Porphyria Network (EPNET), has created a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/). These drugs include ketamine, thiopental, chloramphenicol, erythromycin, nitrofurantoin, rifampicin, trimethoprim/sulfamethoxazole, spironolactone, methyldopa, valproic acid, carbamazepine, phenytoin, phenobarbital, primidone, and risperidone 25. For information on prescribing medication in the context of certain conditions (e.g., HIV, epilepsy, malaria), see https://porphyria.uct.ac.za/porphyria-professionals/prescribing-porphyria-treatment-specific-disorders-poprhyria/therapy-epilepsy.
Hospitalization is often necessary for acute attacks. Medications for pain, nausea and vomiting, and close observation are generally required with monitoring of salt and water balance.
Two standard treatments for acute porphyrias in general are intravenous infusions of hemin (Panhematin®) and supplementation with glucose. However, these therapies have not been universally effective in treating individuals with ALA dehydratase deficiency porphyria (ADP).
Hemin is an orphan drug (a pharmaceutical agent developed to treat a rare medical condition, one that affects a small number of people) that has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of acute porphyria. The drug known as Panhematin® (hemin for injection) is usually given to treat an acute attack. Panhematin® (hemin for injection) is an enzyme inhibitor derived from red blood cells that is potent in suppressing acute attacks of porphyria. Giving IV hemin causes negative feedback to heme synthesis by inhibiting the synthesis of the ALA synthase enzyme (ALAS, rate-limiting step), which limits liver and bone marrow porphyrin synthesis. Suppression of ALA synthase (ALAS) enzyme leads to decreased accumulation of heme precursors and their byproducts and rapid and dramatic reductions in plasma and urinary porphobilinogen (PBG) and aminolevulinic acid (ALA). Panhematin almost always returns porphyrin and porphyrin precursor levels to normal values. The U.S. Food and Drug Administration (FDA) approved Panhematin for the treatment of recurrent attacks of acute intermittent porphyria (AIP) related to the menstrual cycle in susceptible women, after a trial of glucose therapy and should be administered only by physicians experienced in the management of porphyrias in a hospital setting. Based on much experience, it is used for treating and even preventing acute attacks, often without an initial trial of glucose, and has been found to be safe during pregnancy 19, 20, 21.
Normosang (heme arginate) is another heme preparation that can be used to treat individuals with acute intermittent porphyria (AIP). Normosang is not available in the United States, but is used in many other countries where Panhematin is not available.
In 2019, the FDA approved Givlaari (givosiran) for the treatment of adult patients with acute hepatic porphyria. Givlaari (givosiran) aims to reduce the number of attacks patients experience.
Wearing a Medic Alert bracelet or the use of a wallet card is advisable in individuals who have ALA dehydratase deficiency porphyria (ADP).
Figure 1. ALA dehydratase deficiency porphyria
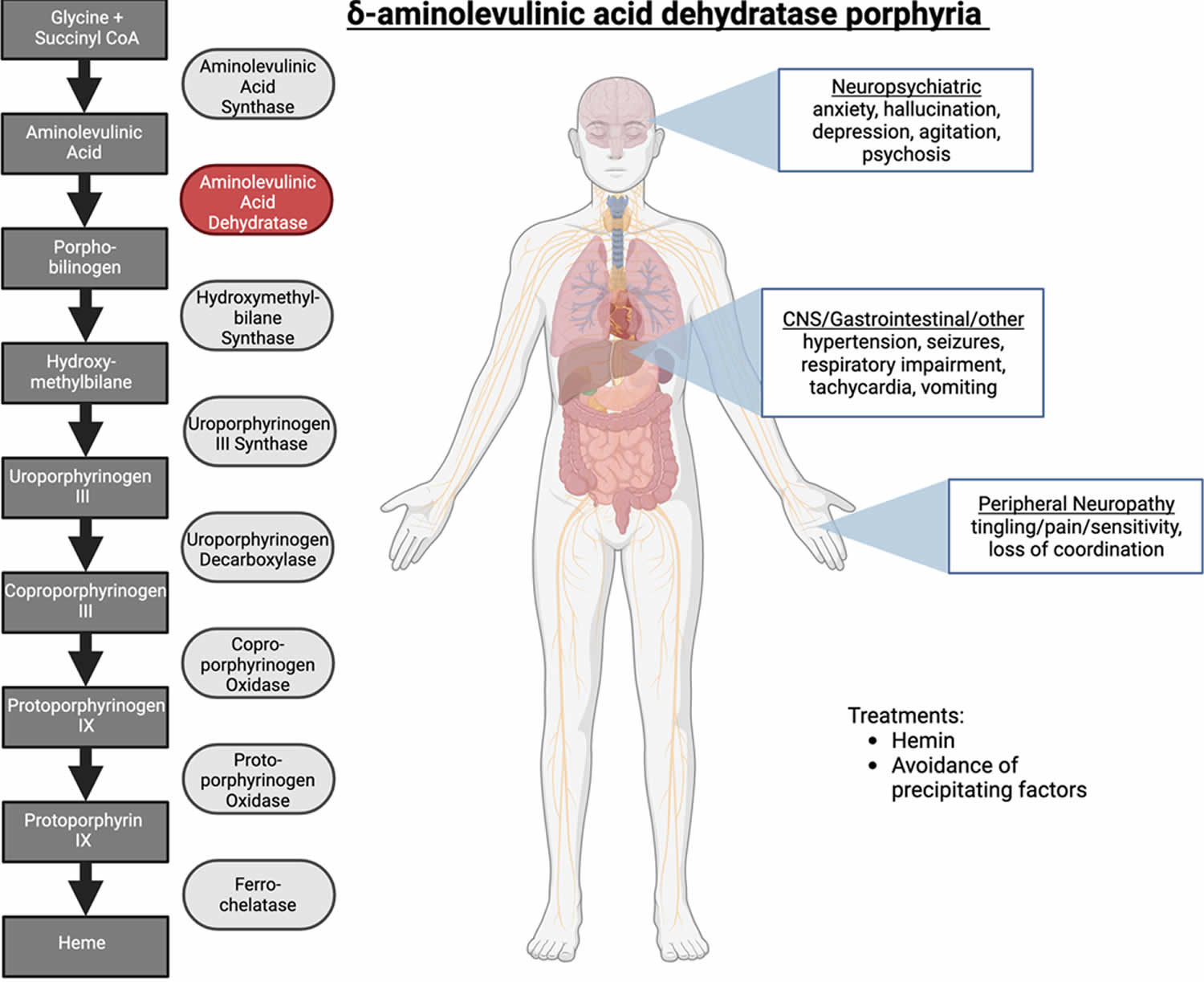
Footnotes: Delta-aminolevulinic acid dehydratase porphyria (ADP). Patients with ALA dehydratase deficiency porphyria (ADP) have a deficiency in the enzyme delta-aminolevulinate dehydratase (ALAD). Symptoms attributed to the accumulation of delta-aminolevulinic acid (ALA) include acute attacks and neurological and gastrointestinal manifestations of disease. Hemin is an effective treatment in some cases, and avoidance of precipitating factors such as alcohol are strongly recommended.
[Source 3 ]Figure 2. Porphyrin molecular structure
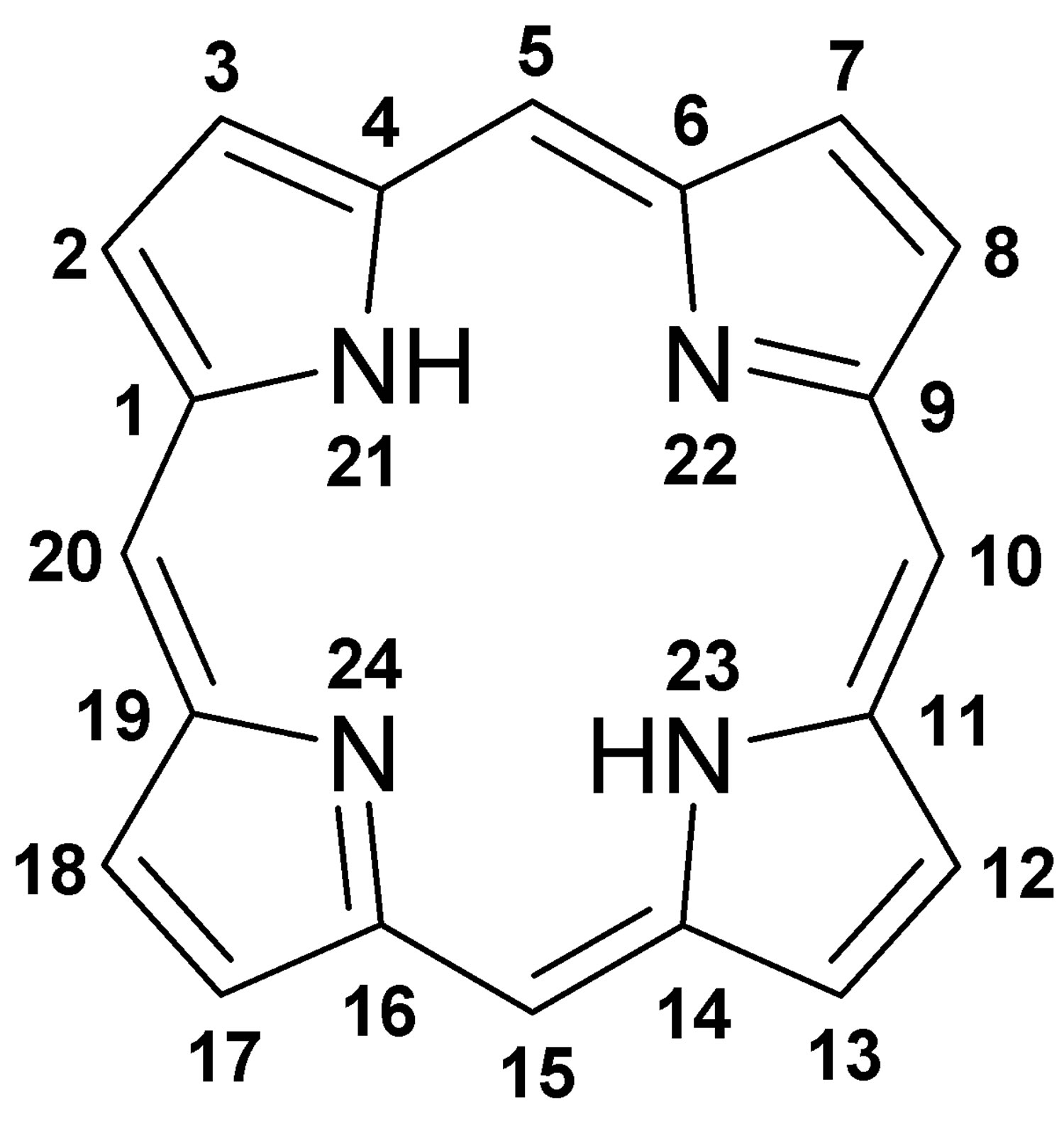
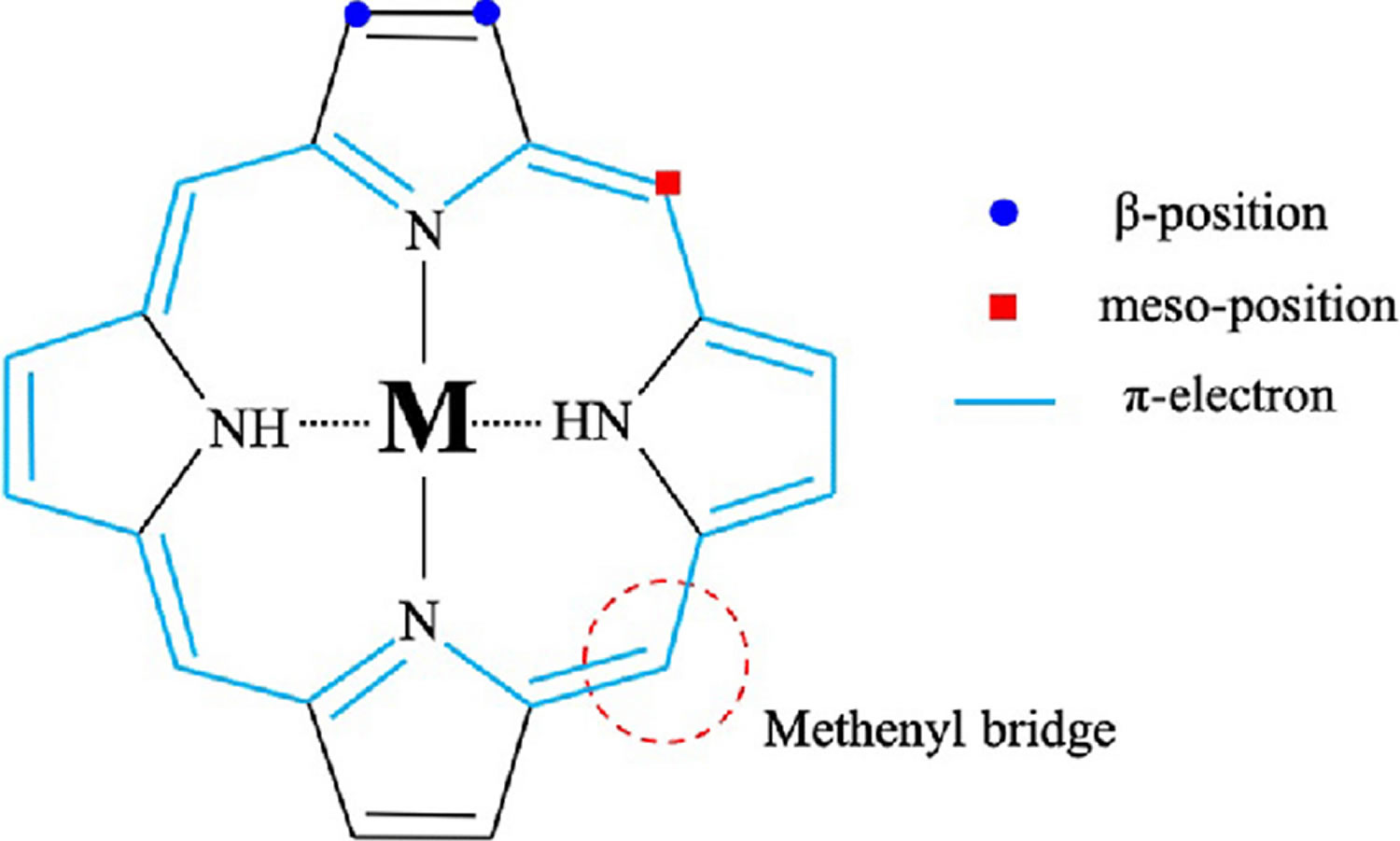
Footnote: Molecular structure of porphyrin (M represent metal ions, such as Mg, Cu, Fe, Zn, etc.).
[Source 22 ]Figure 3. Hemoglobin molecular structure
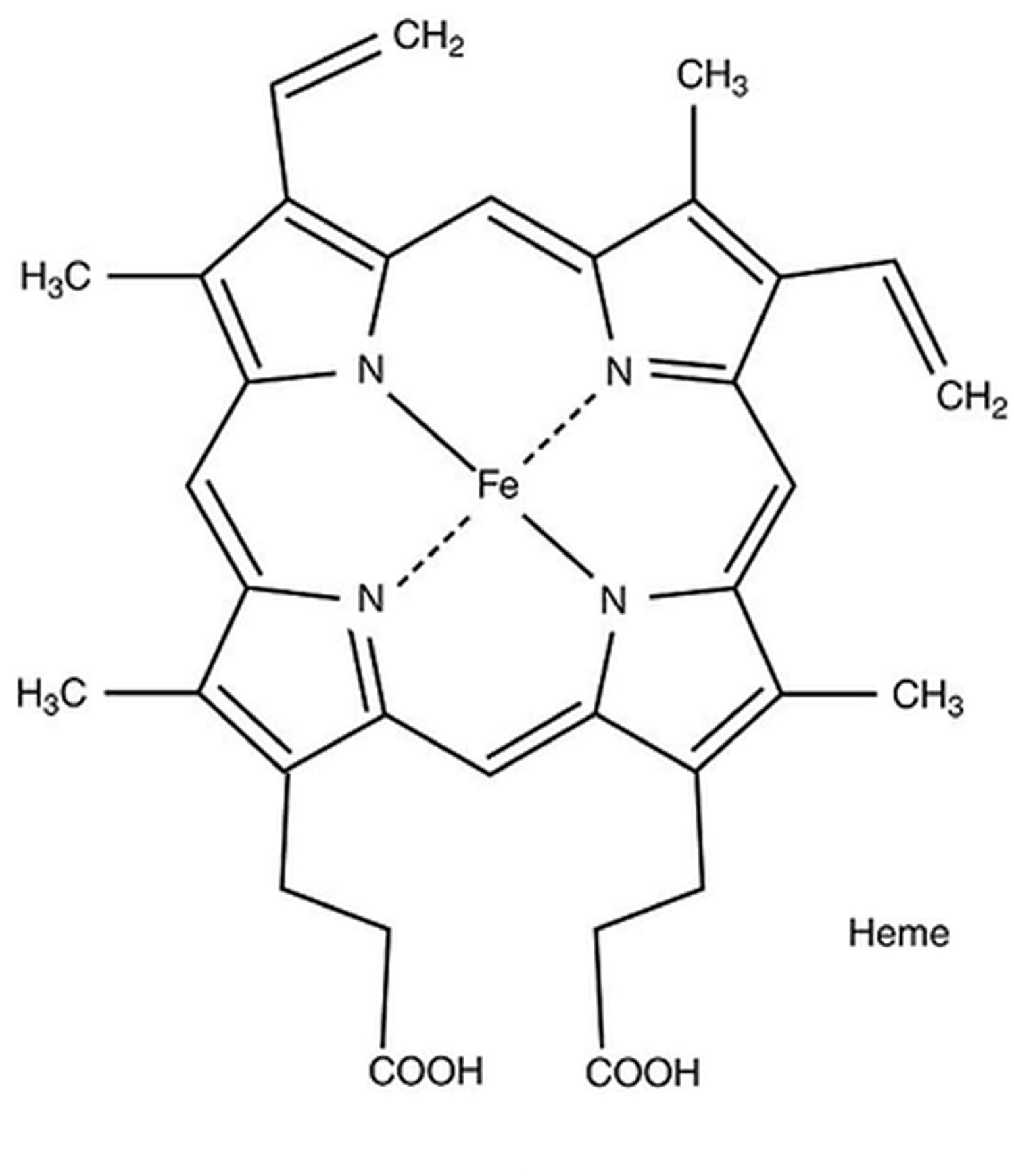
Figure 4. Heme biosynthesis pathway
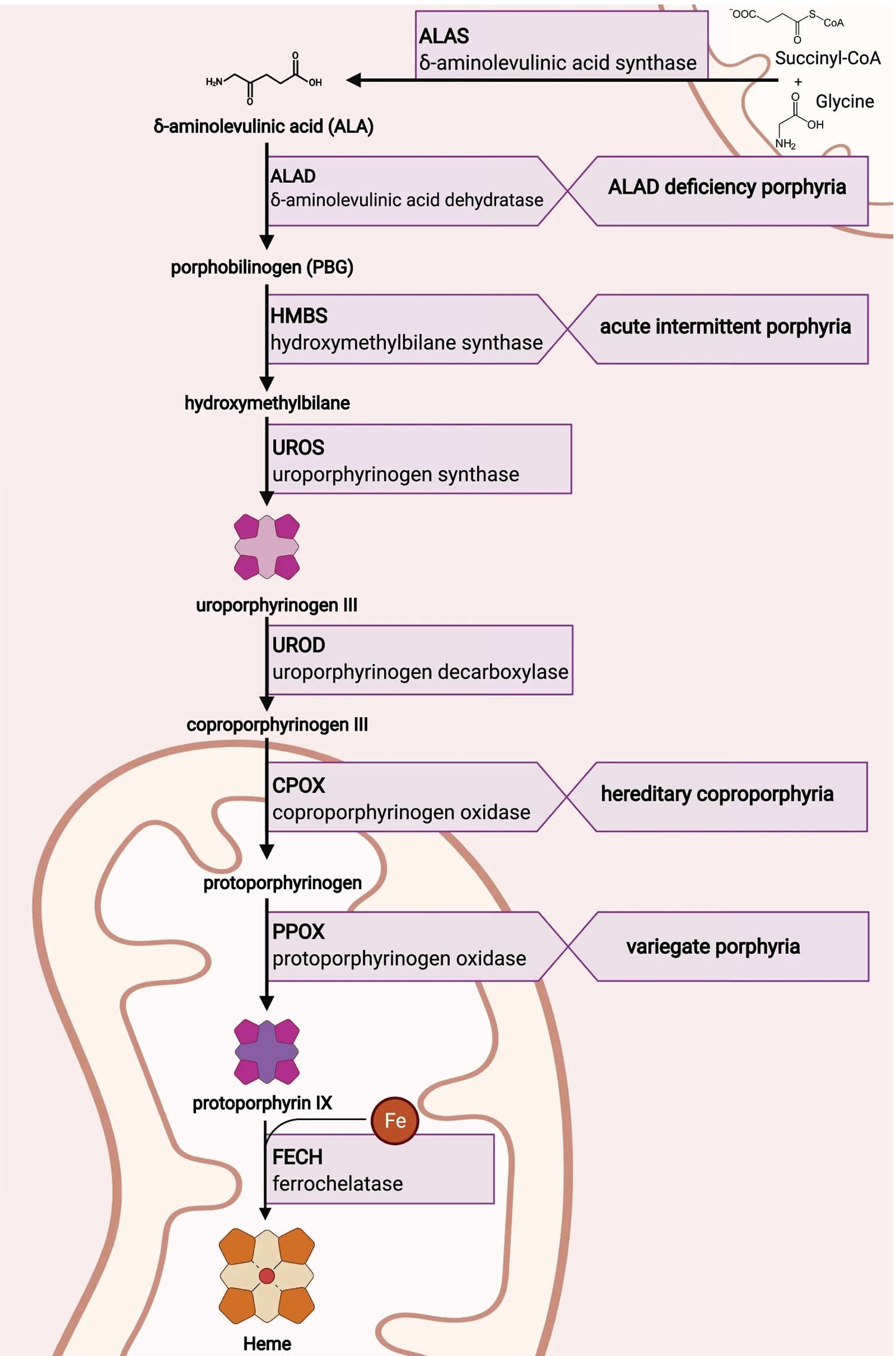
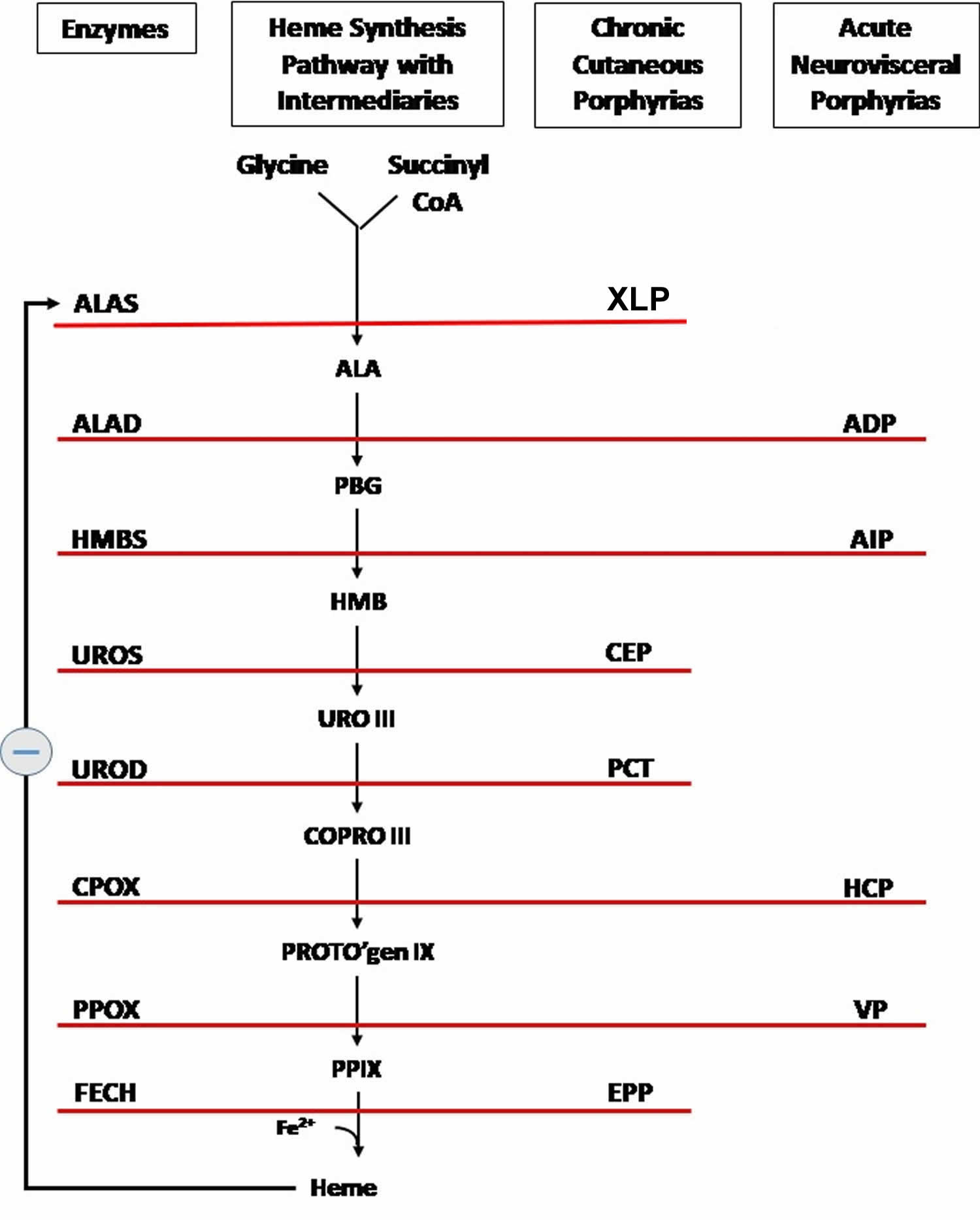
Footnotes: The heme biosynthetic pathway requires 8 enzymatic steps. Heme synthesis pathway showing the enzymes involved in the heme synthesis pathway and the associated porphyrias with the disruption of each specific enzyme. Gain-of-function variants in ALAS2 result in X-linked protoporphyria (XLP), and loss-of-functions variants in FECH result in erythropoietic protoporphyria (EPP). In both X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP), metal-free protoporphyrin IX (PPIX) accumulates in erythroblasts, erythrocytes, the plasma, and the biliary system. Metal-free protoporphyrin IX (PPIX) is photosensitive, particularly to visible light in the blue range, and the light-mediated activation of metal-free protoporphyrin IX (PPIX) produces free radicals that damage the surrounding tissues.
Enzymes, encoded by genes, catalyze each of the steps. Gene mutations cause deficient enzyme production. Disruptions are indicated by red lines connecting enzymes with the resultant porphyrias. ALAS (ALAS2) = aminolevulinate synthase (aminolevulinate synthase 2); ALAD = delta-aminolevulinic acid dehydratase; PBGD = porphobilinogen dehydratase; HMBS = hydroxymethylbilane synthase; UROS = uroporphyrinogen-III synthase; UROD = uroporphyrinogen III decarboxylase; CPOX = coproporphyrinogen-III oxidase; PPOX = protoporphyrinogen oxidase; FECH = ferrochelatase.
Porphyrias resulting from disruption of enzyme production. XLP (X-linked protoporphyria); ADP (aminolevulinic acid dehydratase porphyria); AIP (acute intermittent porphyria); CEP (congenital erythropoietic porphyria); PCT (porphyria cutanea tarda); HCP (hereditary coproporphyria); VP (variegate porphyria); EPP (erythropoietic protoporphyria).
Abbreviations: ALA = aminolevulinic acid; PBG = porphobilinogen; HMB = hydroxymethylbilane; URO III = uroporphyrinogen III; COPRO III = coproporphyrinogen III; PROTO’gen IX protoporphyrinogen IX; PPIX = protoporphyrin IX; Fe2+ = iron.
[Source 14 ]ALA dehydratase deficiency porphyria cause
ALA dehydratase deficiency porphyria (ADP) is caused by mutation in the aminolevulinate dehydratase (ALAD) gene localized at chromosome 9q34 causing a deficiency of the enzyme delta-aminolevulinic acid dehydratase (ALAD) 24, 25. The ALAD gene provides instructions for making an enzyme known as delta-aminolevulinate dehydratase (ALAD) also known as porphobilinogen synthase (PBG synthase), the second enzyme in the heme biosynthetic pathway that produces porphyrins and heme 1, 2, 3. The delta-aminolevulinate dehydratase (ALAD) enzyme is involved in the production of a molecule called heme (haem). Heme is vital for all of the body’s organs, although it is found mostly in the blood, bone marrow, and liver. Heme is an essential component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood).
The production of heme (haem) is a multi-step process that requires 8 different enzymes. Delta-aminolevulinate dehydratase (ALAD) enzyme is responsible for the second step in this process, which combines two molecules of ALA (delta-aminolevulinic acid, the product of the first step) to form a compound called porphobilinogen (PBG) in the cytosol of cells in the liver and red blood cells and play a chief role in the biosynthetic pathway of heme. In subsequent steps, four molecules of porphobilinogen (PBG) are combined and then modified to produce heme.
Mutations of the ALAD (ALA dehydratase deficiency) gene result in deficient levels of porphobilinogen (PBG) in your body, with accumulation of delta-aminolevulinic acid (ALA, the product of the first step) within the red blood cells (erythrocytes) and liver cells (hepatocytes), which can subsequently leak into the plasma and can cause toxicity to various tissues causing the symptoms associated with ALA dehydratase deficiency porphyria (ADP). ALA (delta-aminolevulinic acid) is further metabolized to other heme precursors, and as a result coproporphyrin III is excreted in urine in large amounts and protoporphyrin IX (PPIX) is increased in red blood cells (erythrocytes).
ALAD is markedly deficient and ALA substantially elevated in plasma and urine in patients with ALA dehydratase deficiency porphyria (ADP). As in other acute porphyrias, the mechanism of neurological damage is not well understood.
Aminolevulinate dehydratase (ALAD) enzyme is present in vast abundance in mammalian cells; for example in liver its activity is 80 to 100-fold greater than that of aminolevulinate synthase (ALAS), the first and rate-limiting enzyme for liver heme biosynthesis 26. Under normal conditions, the activity of aminolevulinate dehydratase (ALAD) enzyme is present in excess and a partial deficiency of the enzyme (~50%) is not associated with disturbances of heme synthesis. The normal abundance of ALAD enzyme offers an explanation as to why heterozygotes with ~50% of normal ALAD activity remain asymptomatic 27, 4.
ALA dehydratase deficiency porphyria (ADP) cases usually have compound heterozygotes for ALAD mutations. Heterozygotes may be more susceptible to lead toxicity, which inhibits ALAD enzyme by displacing zinc 18. Heterozygous subjects have approximately 50 percent of normal delta-aminolevulinate dehydratase (ALAD) enzyme activity and are clinically asymptomatic 28.
On the basis of an analysis of a small population, it has been estimated that the prevalence of individuals with 50% of normal aminolevulinate dehydratase (ALAD) activity, putatively caused by one aberrant ALAD gene, is 2% in the normal asymptomatic population 29. This study suggested that most instances of compound heterozygosity in ALAD gene result in spontaneous abortions. The frequency of genetic carriers for ALAD deficiency (i.e., heterozygotes) in the normal population in Sweden was estimated to be approximately 2 percent. Although subjects with heterozygosity for ALAD deficiency are asymptomatic, they may be considered to be at risk for developing ALA dehydratase deficiency porphyria (ADP) when exposed to environmental chemicals or toxins, which further inhibit ALAD activity 29.
A variety of different triggers have been identified that can precipitatean acute attack in individuals with ALA dehydratase deficiency porphyria (ADP). These triggers include alcohol, certain drugs, physical and psychological stress, infection, fasting (reduced caloric intake) and dehydration. The use of estrogen or progesterone is also suspect of triggering an acute attack.
ALA dehydratase deficiency porphyria inheritance pattern
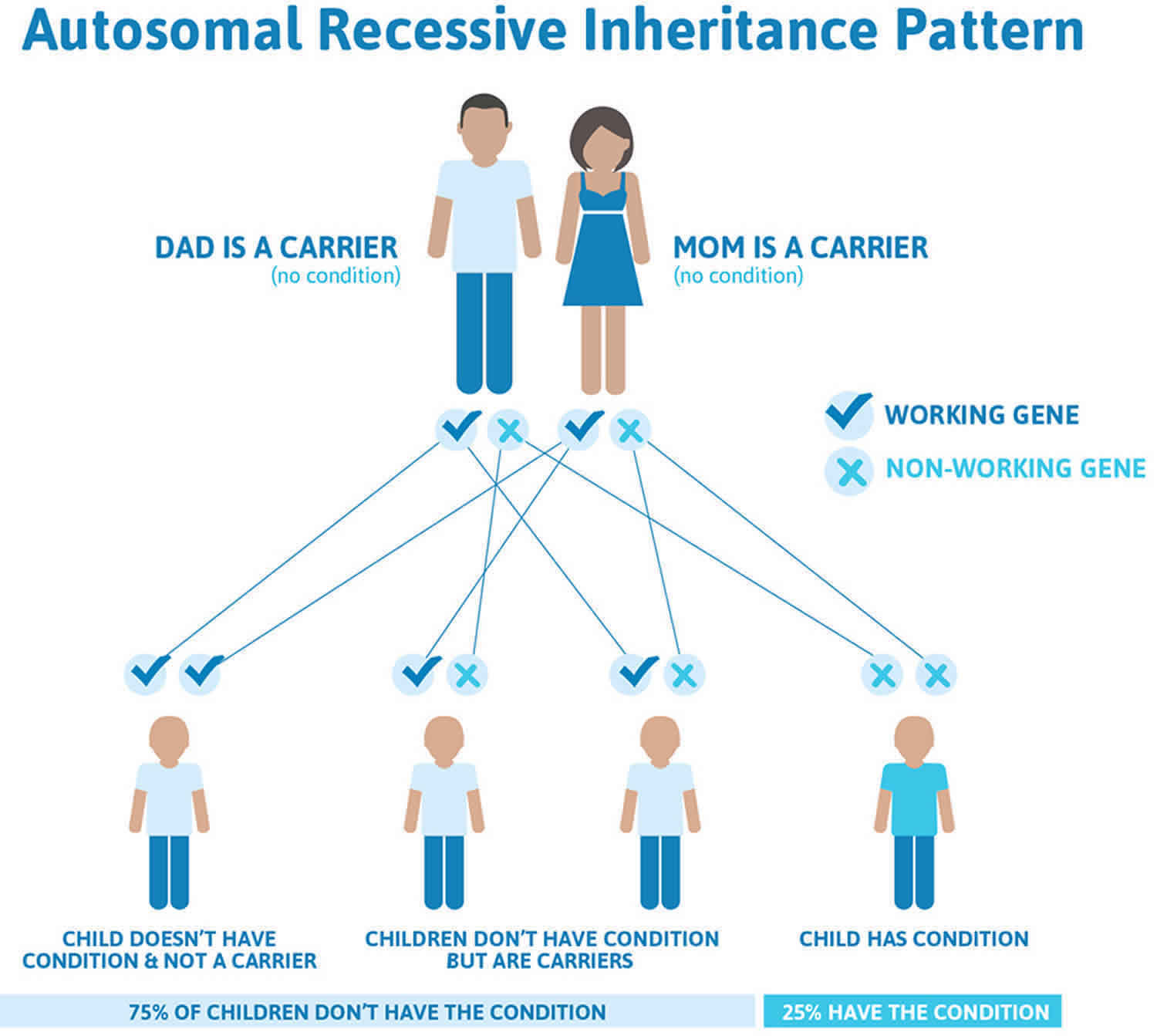
ALA dehydratase deficiency porphyria (ADP) is inherited as an autosomal recessive disorder 6, 1. This means that both copies of the ALAD (ALA dehydratase deficiency) gene have a mutation. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 5. ALA dehydratase deficiency porphyria (ADP) autosomal recessive inheritance pattern
ALA dehydratase deficiency porphyria signs and symptoms
ALA dehydratase deficiency porphyria (ADP) is more severe than the other acute porphyrias 1. Individuals with ALA dehydratase deficiency porphyria (ADP) may have bouts where symptoms are intense, which are referred to as “neurovisceral symptoms” or “acute attacks“. An attack may last for several weeks. During an attack, affected individuals may experience severe abdominal cramping or pain accompanied by nausea, vomiting, constipation, bloating, and rarely diarrhea. During infancy, gastrointestinal abnormalities may cause an affected child to fail to grow and gain weight as expected. Since the abdominal pain is neurological, abdominal tenderness or rebound, leukocytosis (an elevated white blood cell count in the blood), and fever are either minimal or absent. No skin findings were noted.
Several other neurological symptoms can occur during an acute attack due to problems with the nerves outside the central nervous system (peripheral neuropathy), resulting in numbness or tingling in the hands and feet, burning pain, sensitivity to touch, and a lack of coordination (sensory neuropathy). In severe cases, the motor nerves are involved (motor neuropathy), resulting in loss or partial impairment of the ability to use voluntary muscles. Motor dysfunction and muscle weakness were seen in 6 out of the 8 patients.
ALA dehydratase deficiency porphyria (ADP) can also be associated with neuropsychiatric changes during an acute attack. Neuropsychiatric symptoms can include disorientation, agitation, anxiety, restlessness, hysteria, hallucinations, delirium, altered consciousness, apathy, depression, and phobias. In severe cases, loss of contact from reality (psychosis) has been reported.
Additional symptoms that occur during acute attacks include a rapid heartbeat (tachycardia), high blood pressure (hypertension), seizures, and breathing (respiratory) impairment.
An infant diagnosed in Sweden had severe symptoms that progressed to pain, polyneuropathy (multiple peripheral nerves are damaged, often leading to a range of symptoms like numbness, tingling, pain, and muscle weakness, particularly in the extremities), low blood sodium level (hyponatremia), vomiting, and respiratory dysfunction 30, 31, 32, 33. The male from Belgium developed symptoms of acute porphyria along with myeloproliferative disorder (polycythemia vera) at 63 years of age. His motor neuropathy gradually progressed to cause the loss of strength in both his arms 34, 35, 36, 37. Many times, the symptoms are exacerbated by drugs, which can induce CYP enzymes. CYP enzymes lead to the induction of ALA synthase 1 (ALAS1) in the liver. Induction of ALA synthase 1 (ALAS1) enzyme causes exacerbation or precipitation of symptoms.
ALA dehydratase deficiency porphyria complications
Progressive disease and recurrent attacks lead to severe motor weakness involving all limbs (quadriparesis), slurred speech (dysarthria) and may even progress to severe respiratory insufficiency requiring mechanical ventilation 34, 38. Early-onset ALA dehydratase deficiency porphyria (ADP) in the Swedish infant was complicated by mild intellectual disability, autism, impaired hearing, and walking difficulties due to bilateral ankle contractures.
Myeloproliferative disorder (polycythemia vera) was noted in the Belgian male 38. Myeloproliferative disorder is unlikely to be associated with ALA dehydratase deficiency porphyria (ADP) but could have contributed to disease severity since myeloproliferative disorder is associated with increased production of hemoglobin and red blood cells (erythrocytes), thus unmasking his underlying pre-existing ALA dehydratase deficiency porphyria (ADP).
Complications related to hemin infusion include infusion site phlebitis and iron overload after multiple hemin transfusions (monitor serum ferritin).
ALA dehydratase deficiency porphyria diagnosis
There are many laboratory tests available for the porphyrias, and it is often difficult to decide which should be chosen. Many of these tests are expensive and the results are often difficult to interpret. When the classic triad of abdominal pain, peripheral neuropathy, and neuropsychiatric symptoms suggest an acute porphyria, the best screening tests are urinary aminolevulinic acid (ALA) and urinary porphobilinogen (PBG), obtained preferably during an acute porphyria attack (e.g. when someone is having abdominal pain, etc). The total urine porphyrin level will be elevated. Urinary porphobilinogen (PBG) is not significantly increased in ALA dehydratase porphyria (ADP) 14.
If these initials tests are normal in a patient with classic symptoms of ALA dehydratase deficiency porphyria (ADP), then it is suggested to repeat the first-line tests rather than proceeding to second-line testing. Second-line testing in a patient is performed if it is found that total urine porphyrins are elevated without porphobilinogen (PBG) elevation (findings suggestive of ALA dehydratase deficiency porphyria), and clinical suspicion of acute porphyria is very high.
Note the most frequent errors lie in (1) not considering the diagnosis in a timely fashion, leading to the average 15‐year delay in the United States from time of first symptoms to eventual correct diagnosis, and (2) ordering a random urine screening for porphyrins rather than for ALA and PBG 15, 16.
Second-line testing includes urine aminolevulinic acid (ALA) level, fractionation of porphyrins, and measurement of red blood cell (erythrocyte) protoporphyrin. Aminolevulinic acid (ALA) and red blood cell (erythrocyte) protoporphyrin is markedly elevated, and there is a predominance of coproporphyrin III noted.
DNA testing to identify the specific ALAD genetic mutation, which reveals heterozygous or homozygous ALAD mutations is the most specific and sensitive test to confirm the diagnosis of ALA dehydratase deficiency porphyria. Before requesting DNA testing, it is recommended that patients have biochemical testing (urinary, stool and/or plasma porphyrins and porphyrin precursors (ALA and PBG) and/or enzyme assays) 17.
ALA dehydratase deficiency porphyria differential diagnosis
Diseases with similar to the non-specific findings of ALA dehydratase deficiency porphyria (ADP) include other acute porphyrias that have neurovisceral manifestations. These include acute intermittent porphyria (AIP), variegate porphyria (VP), and hereditary coproporphyria (HCP). Each of them has a characteristic elevation of different urinary biomarkers.
Acute intermittent porphyria (AIP) is characterized by elevated urinary porphobilinogen (PBG) on an initial urine sample, unlike ALA dehydratase deficiency porphyria (ADP). Variegate porphyria (VP) presents with neurovisceral symptoms and skin signs and symptoms such as blistering skin lesions. Variegate porphyria (VP) is characterized by elevated urinary PBG (porphobilinogen) and elevated plasma and fecal porphyrins, unlike ALA dehydratase deficiency porphyria (ADP). Hereditary coproporphyria (HCP) presents with acute neurovisceral symptoms similar to ALA dehydratase deficiency porphyria (ADP) but may also have blistering skin lesions. Hereditary coproporphyria (HCP) is characterized by elevated urinary PBG (porphobilinogen) and fecal porphyrins on the initial spot urine sample, unlike ALA dehydratase deficiency porphyria (ADP). Other differentials include diseases where ALAD is inhibited.
The ALAD enzyme activity is reduced in individuals with diabetes mellitus 39, in hemodialysis patients with chronic kidney failure 40 and in smokers and alcoholics 41. An acquired form of ALAD porphyria has also been described in which six diabetic patients with advanced kidney disease developed a syndrome similar to acute intermittent porphyria (AIP) after initiation of treatment with erythropoietin (EPO) 42. The symptoms varied but resolved in all patients when erythropoietin was stopped, and reappeared in four patients when erythropoietin was restarted. In all of the patients, the enzyme ALA dehydratase (ALAD) was low 42.
Lead poisoning can also mimic ALA dehydratase deficiency porphyria (ADP). Lead is a potent inhibitor of ALAD enzyme. Lead metal inhibits ALAD enzyme activity by displacing zinc from the ALAD enzyme 43, 44. Individuals with heterozygous ALAD deficiency may be more susceptible to lead poisoning 45. Lead poisoning (plumbism) can produce the same symptoms and biochemical abnormalities as ALA dehydratase deficiency porphyria (ADP), including increased urinary aminolevulinic acid (ALA) and coproporphyrin III, and erythrocyte zinc protoporphyrin. However, lead levels are normal in ALA dehydratase deficiency porphyria (ADP) but elevated in lead poisoning 46.
The ALAD enzyme activity has also been shown to be reduced when rats were exposed to trichloroethylene 47, styrene 48, lead 49, and iron 50.
Succinylacetone, a structural analogue of ALA and a potent inhibitor of ALAD, is found in high quantities in the blood and urine of patients with hereditary tyrosinemia type 1 (HT1) 51, 52. Approximately 40% of children with hereditary tyrosinemia type 1 (HT1) develop signs and symptoms of ALA dehydratase deficiency porphyria (ADP) 52. Children with hereditary tyrosinemia type 1 (HT1) also have symptoms related to other metabolic effects, including progressive liver disease and renal tubular dysfunction. Those with HT1 have elevated urinary organic acids and FAH gene mutations.
ALA dehydratase deficiency porphyria treatment
ALA dehydratase deficiency porphyria (ADP) treatment is the same as in the other acute porphyrias and is directed toward the specific symptoms that are present in each individual. Because there have been so few cases of ALA dehydratase deficiency porphyria (ADP), there is only limited information on treatment for the disorder 1.
Acute attacks are managed by withdrawing and avoiding the offending agents that can precipitate acute attacks and supportive care for nausea, vomiting, pain, seizures, and electrolyte imbalances. Offending agents can include alcohol, oral contraceptives, antibiotics (such as nitrofurantoin, rifampin, trimethoprim-sulfamethoxazole), anti-seizure medications (such as topiramate, phenytoin, phenobarbital, ethosuximide, valproic acid), and many more medications. Physical, psychological, and emotional stress may also precipitate attacks and must be minimized 15.
Attacks can be prevented in many cases by avoiding harmful drugs and adverse dietary practices such as fasting, and low carbohydrate diets is recommended for affected individuals.
The specific drugs that may need to be avoided in one person can differ from the drugs that need to be avoided in another.
- The American Porphyria Foundation offers a drug database with safety information about the interaction of specific drugs in patients with porphyria (https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/) or https://porphyriafoundation.org/drugdatabase/drug-safety-database-search/.
- The EPNET/NAPOS Database should also be consulted. The Norwegian Porphyria Centre (NAPOS), with the European Porphyria Network (EPNET), has created a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/). These drugs include ketamine, thiopental, chloramphenicol, erythromycin, nitrofurantoin, rifampicin, trimethoprim/sulfamethoxazole, spironolactone, methyldopa, valproic acid, carbamazepine, phenytoin, phenobarbital, primidone, and risperidone 25. For information on prescribing medication in the context of certain conditions (e.g., HIV, epilepsy, malaria), see https://porphyria.uct.ac.za/porphyria-professionals/prescribing-porphyria-treatment-specific-disorders-poprhyria/therapy-epilepsy.
Hospitalization is often necessary for acute attacks. Medications for pain, nausea and vomiting, and close observation are generally required with monitoring of salt and water balance.
Two standard treatments for acute porphyrias in general are intravenous infusions of hemin (Panhematin®) and supplementation with glucose. However, these therapies have not been universally effective in treating individuals with ALA dehydratase deficiency porphyria (ADP).
Hemin is an orphan drug that has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of acute porphyria. The drug known as Panhematin® (hemin for injection) is usually given to treat an acute attack. Panhematin® (hemin for injection) is an enzyme inhibitor derived from red blood cells that is potent in suppressing acute attacks of porphyria. Giving IV hemin causes negative feedback to heme synthesis by inhibiting the synthesis of the ALA synthase enzyme (ALAS, rate-limiting step), which limits liver and bone marrow porphyrin synthesis. Suppression of ALA synthase (ALAS) enzyme leads to decreased accumulation of heme precursors and their byproducts and rapid and dramatic reductions in plasma and urinary porphobilinogen (PBG) and aminolevulinic acid (ALA). Panhematin almost always returns porphyrin and porphyrin precursor levels to normal values. The U.S. Food and Drug Administration (FDA) approved Panhematin for the treatment of recurrent attacks of acute intermittent porphyria (AIP) related to the menstrual cycle in susceptible women, after a trial of glucose therapy and should be administered only by physicians experienced in the management of porphyrias in a hospital setting. Based on much experience, it is used for treating and even preventing acute attacks, often without an initial trial of glucose, and has been found to be safe during pregnancy 19, 20, 21.
Normosang (heme arginate) is another heme preparation that can be used to treat individuals with acute intermittent porphyria (AIP). Normosang is not available in the United States, but is used in many other countries where Panhematin is not available.
Indications for the use of hemin include attacks severe enough to require hospitalization, opioid analgesia, or other intravenous medication; or accompanied by nausea and vomiting, motor neuropathy, paresis, seizures, agitation, delirium, psychosis, ileus that prevents oral intake, or hyponatremia 1. Hemin should be administered at a dose of 3 to 4 mg/kg body weight intravenously as a single daily dose for four days (dosing more than once per day is not recommended as it is unlikely to improve efficacy) 1. Hemin is mixed with 25 percent human albumin for infusion (it was previously mixed with sterile water but resulted in adverse reactions) 1. Treatment duration can be extended beyond four days in patients with advanced motor neuropathy where resolution is not seen.
The use of hemin decreased hospital stay by 3 days and reduced opioid usage 53. All four adolescent cases with onset of symptoms at ages 12 to 15 years showed effective resolution of attacks with hemin, and in two of these cases, prophylactic hemin infusions prevented recurrent attacks 11, 54. However, a Swedish child with early-onset ALA dehydratase deficiency porphyria (ADP) did not respond to hemin 32, 33.
Glucose loading may be tried for mild symptoms if hemin is not readily available, but there is not sufficient evidence to support this 33.
Blood transfusions and hydroxyurea (hydroxycarbamide) to suppress erythropoiesis showed beneficial results in one case 38. This deserves further study.
In 2019, the FDA approved Givlaari (givosiran), an RNA interference (RNAi) drug, for the treatment of adult patients with acute hepatic porphyria. Givlaari (givosiran) aims to reduce the number of attacks patients experience 55.
It is unclear if a liver transplant may be of benefit in less severe refractory cases, but it failed to benefit a child with severe ALA dehydratase deficiency porphyria (ADP) 33.
Wearing a Medic Alert bracelet or the use of a wallet card is advisable in individuals who have ALA dehydratase deficiency porphyria (ADP).
ALA dehydratase deficiency porphyria prognosis
ALA dehydratase deficiency porphyria (ADP) is the least common of all the porphyrias with less than 10 cases documented to date. Only 2 patients have had complete follow-up. One was a Swedish infant who eventually required liver transplantation but died 3 years after transplantation at 9 years 1. The other was a Belgian male diagnosed at 63 years old; he went on to develop a myeloproliferative disorder (polycythemia vera) and eventually succumbed to his blood cancer, not the porphyria 54.
An infant diagnosed in Sweden had severe symptoms that progressed to pain, polyneuropathy (multiple peripheral nerves are damaged, often leading to a range of symptoms like numbness, tingling, pain, and muscle weakness, particularly in the extremities), low blood sodium level (hyponatremia), vomiting, and respiratory dysfunction 30, 31, 32, 33. The male from Belgium developed symptoms of acute porphyria along with myeloproliferative disorder (polycythemia vera) at 63 years of age. His motor neuropathy gradually progressed to cause the loss of strength in both his arms 34, 35, 36, 37. Many times, the symptoms are exacerbated by drugs, which can induce CYP enzymes. CYP enzymes lead to the induction of ALA synthase 1 (ALAS1) in the liver. Induction of ALA synthase 1 (ALAS1) enzyme causes exacerbation or precipitation of symptoms.
- Mohan G, Madan A. ALA Dehydratase Deficiency Porphyria. [Updated 2023 Jul 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560836[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Doss M, Schmidt A. Zwei Suchteste für Porphyrien [Two screening tests for porphyrias (author’s transl)]. Z Klin Chem Klin Biochem. 1972 May;10(5):230-1. German.[↩][↩]
- Balogun O, Nejak-Bowen K. Understanding Hepatic Porphyrias: Symptoms, Treatments, and Unmet Needs. Semin Liver Dis. 2024 May;44(2):209-225. doi: 10.1055/s-0044-1787076[↩][↩][↩]
- Ramanujam VS, Anderson KE. Porphyria Diagnostics-Part 1: A Brief Overview of the Porphyrias. Curr Protoc Hum Genet. 2015 Jul 1;86:17.20.1-17.20.26. doi: 10.1002/0471142905.hg1720s86[↩][↩][↩]
- Maruno M, Furuyama K, Akagi R, Horie Y, Meguro K, Garbaczewski L, Chiorazzi N, Doss MO, Hassoun A, Mercelis R, Verstraeten L, Harper P, Floderus Y, Thunell S, Sassa S. Highly heterogeneous nature of delta-aminolevulinate dehydratase (ALAD) deficiencies in ALAD porphyria. Blood. 2001 May 15;97(10):2972-8. doi: 10.1182/blood.v97.10.2972[↩][↩][↩]
- Besur S, Schmeltzer P, Bonkovsky HL. Acute Porphyrias. J Emerg Med. 2015 Sep;49(3):305-12. doi: 10.1016/j.jemermed.2015.04.034[↩][↩]
- Chakraborty A, Muranjan M, Karande S, Kharkar V. Porphyrias: Uncommon disorders masquerading as common childhood diseases. J Postgrad Med. 2023 Jul-Sep;69(3):164-171. doi: 10.4103/jpgm.jpgm_698_22[↩]
- Doss M, Look D, Henning H, Nawrocki P, Schmidt A, Dölle W, Korb G, Lüders CJ, Strohmeyer G. Hepatic porphyrins and urinary porphyrins and porphyrin precursors in liver cirrhosis. Klin Wochenschr. 1972 Nov 15;50(22):1025-32. doi: 10.1007/BF01486762[↩]
- ALAD-Deficiency Porphyria (ADP). https://porphyriafoundation.org/for-patients/types-of-porphyria/adp/[↩]
- Sassa S. ALAD porphyria. Semin Liver Dis. 1998;18(1):95-101. doi: 10.1055/s-2007-1007145[↩][↩]
- Akagi R, Kato N, Inoue R, Anderson KE, Jaffe EK, Sassa S. delta-Aminolevulinate dehydratase (ALAD) porphyria: the first case in North America with two novel ALAD mutations. Mol Genet Metab. 2006 Apr;87(4):329-36. doi: 10.1016/j.ymgme.2005.10.011[↩][↩]
- Balwani M, Desnick RJ. The porphyrias: advances in diagnosis and treatment. Blood. 2012 Nov 29;120(23):4496-504. doi: 10.1182/blood-2012-05-423186. Epub 2012 Jul 12. Erratum in: Blood. 2013 Oct 24;122(17):3090.[↩]
- Doss MO, Stauch T, Gross U, Renz M, Akagi R, Doss-Frank M, Seelig HP, Sassa S. The third case of Doss porphyria (delta-amino-levulinic acid dehydratase deficiency) in Germany. J Inherit Metab Dis. 2004;27(4):529–536. doi: 10.1023/B:BOLI.0000037341.21975.9d[↩]
- Edel Y, Mamet R. Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. 2018 Apr 19;9(2):e0013. doi: 10.5041/RMMJ.10333[↩][↩][↩]
- Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute Hepatic Porphyrias: Review and Recent Progress. Hepatol Commun. 2018 Dec 20;3(2):193-206. doi: 10.1002/hep4.1297[↩][↩][↩][↩]
- Bonkovsky HL, Maddukuri VC, Yazici C, Anderson KE, Bissell DM, Bloomer JR, Phillips JD, Naik H, Peter I, Baillargeon G, Bossi K, Gandolfo L, Light C, Bishop D, Desnick RJ. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med. 2014 Dec;127(12):1233-41. doi: 10.1016/j.amjmed.2014.06.036[↩][↩]
- Phillips JD. Heme biosynthesis and the porphyrias. Mol Genet Metab. 2019 Nov;128(3):164-177. doi: 10.1016/j.ymgme.2019.04.008[↩][↩]
- Scinicariello F, Murray HE, Moffett DB, Abadin HG, Sexton MJ, Fowler BA. Lead and delta-aminolevulinic acid dehydratase polymorphism: where does it lead? A meta-analysis. Environ Health Perspect. 2007 Jan;115(1):35-41. doi: 10.1289/ehp.9448[↩][↩]
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005 Mar 15;142(6):439-50. doi: 10.7326/0003-4819-142-6-200503150-00010. Erratum in: Ann Intern Med. 2005 Aug 16;143(4):316.[↩][↩]
- Tenhunen R, Mustajoki P. Acute porphyria: treatment with heme. Semin Liver Dis. 1998;18(1):53-5. doi: 10.1055/s-2007-1007140[↩][↩]
- Mustajoki P, Nordmann Y. Early Administration of Heme Arginate for Acute Porphyric Attacks. Arch Intern Med. 1993;153(17):2004–2008. doi:10.1001/archinte.1993.00410170078008[↩][↩]
- Lin, Jou & Shi, Donglu. (2021). Photothermal and photovoltaic properties of transparent thin films of porphyrin compounds for energy applications. Applied Physics Reviews. 8. 011302. https://doi.org/10.1063/5.0036961[↩]
- Panawala, Lakna. (2017). What is the Function of Hemoglobin in the Human Body. https://www.researchgate.net/publication/313841668_What_is_the_Function_of_Hemoglobin_in_the_Human_Body[↩]
- Wetmur JG, Bishop DF, Ostasiewicz L, Desnick RJ. Molecular cloning of a cDNA for human delta-aminolevulinate dehydratase. Gene. 1986;43(1–2):123–130. doi: 10.1016/0378-1119(86)90015-6[↩]
- Potluri VR, Astrin KH, Wetmur JG, Bishop DF, Desnick RJ. Human delta-aminolevulinate dehydratase: chromosomal localization to 9q34 by in situ hybridization. Hum Genet. 1987;6(3):236–239. doi: 10.1007/BF00283614[↩]
- Kappas A, Sassa S, Galbraith RA, Nordmann Y. The porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Metabolic and Molecular Bases of Inherited Disease. 7. Vol. 2. McGraw-Hill; New York: 1995. pp. 2103–2159.[↩]
- Kappas A The porphyrias. In, The Metabolic and Molecular Basis of Inherited Disease, 7th Edition; 1995: 2103–2159.[↩]
- Bird TD, Hamernyik P, Nutter JY, Labbe RF. Inherited deficiency of delta-aminolevulinic acid dehydratase. Am J Hum Genet. 1979 Nov;31(6):662-8. https://pmc.ncbi.nlm.nih.gov/articles/instance/1686033/pdf/ajhg00198-0010.pdf[↩]
- Thunell S, Holmberg L, Lundgren J. Aminolaevulinate dehydratase porphyria in infancy. A clinical and biochemical study. J Clin Chem Clin Biochem. 1987;25(1):5–14. doi: 10.1515/cclm.1987.25.1.5[↩][↩]
- Thunell S, Holmberg L, Lundgren J. Aminolaevulinate dehydratase porphyria in infancy. A clinical and biochemical study. J Clin Chem Clin Biochem. 1987 Jan;25(1):5-14. doi: 10.1515/cclm.1987.25.1.5[↩][↩]
- Fujita H, Sassa S, Lundgren J, Holmberg L, Thunell S, Kappas A. Enzymatic defect in a child with hereditary hepatic porphyria due to homozygous delta-aminolevulinic acid dehydratase deficiency: immunochemical studies. Pediatrics. 1987 Dec;80(6):880-5.[↩][↩]
- Plewinska M, Thunell S, Holmberg L, Wetmur JG, Desnick RJ. delta-Aminolevulinate dehydratase deficient porphyria: identification of the molecular lesions in a severely affected homozygote. Am J Hum Genet. 1991 Jul;49(1):167-74. https://pmc.ncbi.nlm.nih.gov/articles/instance/1683193/pdf/ajhg00078-0172.pdf[↩][↩][↩]
- Thunell S, Henrichson A, Floderus Y, Groth CG, Eriksson BG, Barkholt L, Nemeth A, Strandvik B, Eleborg L, Holmberg L, et al. Liver transplantation in a boy with acute porphyria due to aminolaevulinate dehydratase deficiency. Eur J Clin Chem Clin Biochem. 1992 Oct;30(10):599-606. doi: 10.1515/cclm.1992.30.10.599[↩][↩][↩][↩][↩]
- Mercelis R, Hassoun A, Verstraeten L, De Bock R, Martin JJ. Porphyric neuropathy and hereditary delta-aminolevulinic acid dehydratase deficiency in an adult. J Neurol Sci. 1990 Jan;95(1):39-47. doi: 10.1016/0022-510x(90)90115-4[↩][↩][↩]
- Akagi R, Nishitani C, Harigae H, Horie Y, Garbaczewski L, Hassoun A, Mercelis R, Verstraeten L, Sassa S. Molecular analysis of delta-aminolevulinate dehydratase deficiency in a patient with an unusual late-onset porphyria. Blood. 2000 Nov 15;96(10):3618-23. https://doi.org/10.1182/blood.V96.10.3618[↩][↩]
- Hassoun A, Verstraeten L, Mercelis R, Martin JJ. Biochemical diagnosis of an hereditary aminolaevulinate dehydratase deficiency in a 63-year-old man. J Clin Chem Clin Biochem. 1989 Oct;27(10):781-6. doi: 10.1515/cclm.1989.27.10.781[↩][↩]
- Sassa S, Fujita H, Doss M, Hassoun A, Verstraeten L, Mercelis R, Kappas A. Hereditary hepatic porphyria due to homozygous delta-aminolevulinic acid dehydratase deficiency: studies in lymphocytes and erythrocytes. Eur J Clin Invest. 1991 Apr;21(2):244-8. doi: 10.1111/j.1365-2362.1991.tb01817.x[↩][↩]
- Neeleman RA, van Beers EJ, Friesema EC, Koole-Lesuis R, van der Pol WL, Wilson JHP, Langendonk JG. Clinical Remission of Delta-Aminolevulinic Acid Dehydratase Deficiency Through Suppression of Erythroid Heme Synthesis. Hepatology. 2019 Jul;70(1):434-436. doi: 10.1002/hep.30543[↩][↩][↩]
- Fernández-Cuartero B, Rebollar JL, Batlle A, Enriquez de Salamanca R. Delta aminolevulinate dehydratase (ALA-D) activity in human and experimental diabetes mellitus. Int J Biochem Cell Biol. 1999;31(3–4):479–488. doi: 10.1016/s1357-2725(98)00145-9[↩]
- Guolo M, Stella AM, Melito V, Parera V, Del C Batlle AM. Altered 5-aminolevulinic acid metabolism leading to pseudoporphyria in hemodialysed patients. Int J Biochem Cell Biol. 1996 Mar;28(3):311-7. doi: 10.1016/1357-2725(95)00133-6[↩]
- McColl KE, Moore MR, Thompson GG, Goldberg A. Abnormal haem biosynthesis in chronic alcoholics. Eur J Clin Invest. 1981;11:461–468. doi: 10.1111/j.1365-2362.1981.tb02014.x[↩]
- Hedger RW, Wehrmacher WH, French AV. Porphyria syndrome associated with diabetic nephrosclerosis and erythropoietin. Compr Ther. 2006 Fall;32(3):163-71. doi: 10.1007/s12019-006-0007-4[↩][↩]
- Bergdahl IA, Grubb A, Schütz A, Schutz A, Desnick RJ, Wetmur JG, Sassa S, Skerfving S. Lead binding to delta-aminolevulinic acid dehydratase (ALAD) in human erythrocytes. Pharmacol Toxicol. 1997;81(4):153–158. doi: 10.1111/j.1600-0773.1997.tb02061.x[↩]
- Granick JL, Sassa S, Kappas A. Some biochemical and clinical aspects of lead intoxication. Adv Clin Chem. 1978;20:287-339. doi: 10.1016/s0065-2423(08)60025-6[↩]
- Doss M, Laubenthal F, Stoeppler M. Lead poisoning in inherited delta-aminolevulinic acid dehydratase deficiency. Int Arch Occup Environ Health. 1984;54(1):55–63. doi: 10.1007/BF00378728[↩]
- Anderson KE, Fischbein A, Kestenbaum D, Sassa S, Alvares AP, Kappas A. Plumbism from airborne lead in a firing range. An unusual exposure to a toxic heavy metal. Am J Med. 1977 Aug;63(2):306-12. doi: 10.1016/0002-9343(77)90246-7[↩]
- Fujita H, Koizumi A, Yamamoto M, Kumai M, Sadamoto T, Ikeda M. Inhibition of delta-aminolevulinate dehydratase in trichloroethylene-exposed rats, and the effects on heme regulation. Biochim Biophys Acta. 1984;800(1):1–10. doi: 10.1016/0304-4165(84)90087-4[↩]
- Fujita H, Koizumi A, Hayashi N, Ikeda M. Reduced synthesis of 5-aminolevulinate dehydratase in styrene-treated rats. Biochim Biophys Acta. 1986;867(3):89–96. doi: 10.1016/0167-4781(86)90068-0[↩]
- Sassa S, Granick S, Kappas A. Effect of lead and genetic factors on heme biosynthesis in the human red cell. Ann N Y Acad Sci. 1975;244:419–440. doi: 10.1111/j.1749-6632.1975.tb41546.x[↩]
- Bonkovsky HL, Healey JF, Lincoln B, Bacon BR, Bishop DF, Elder GH. Hepatic heme synthesis in a new model of experimental hemochromatosis: studies in rats fed finely divided elemental iron. Hepatology. 1987;7(6):1195–1203. doi: 10.1002/hep.1840070605[↩]
- Sassa S, Kappas A. Hereditary tyrosinemia and the heme biosynthetic pathway: Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J Clin Invest. 1983;71(3):625–634. doi: 10.1172/JCI110809[↩]
- Lindblad B, Lindstedt S, Steen G. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci U S A. 1977 Oct;74(10):4641-5. https://pmc.ncbi.nlm.nih.gov/articles/instance/432003/pdf/pnas00032-0537.pdf[↩][↩]
- Herrick AL, McColl KE, Moore MR, Cook A, Goldberg A. Controlled trial of haem arginate in acute hepatic porphyria. Lancet. 1989 Jun 10;1(8650):1295-7. doi: 10.1016/s0140-6736(89)92688-3[↩]
- Doss MO, Stauch T, Gross U, Renz M, Akagi R, Doss-Frank M, Seelig HP, Sassa S. The third case of Doss porphyria (delta-amino-levulinic acid dehydratase deficiency) in Germany. J Inherit Metab Dis. 2004;27(4):529-36. doi: 10.1023/B:BOLI.0000037341.21975.9d[↩][↩]
- Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017 Aug 31;377(9):862-872. doi: 10.1056/NEJMra1608634[↩]





{kind=link}