Contents
What is aldosterone
Aldosterone also called mineralocorticoid, is a hormone that is crucial for sodium conservation in the kidney, salivary glands, sweat glands and colon. Aldosterone stimulates sodium transport across cell membranes, particularly in the distal renal tubule where sodium is exchanged for hydrogen and potassium. Secondarily, aldosterone is important in the maintenance of blood pressure and blood volume. Renin is an enzyme that controls aldosterone production (see Figures 3 and 4, the renin angiotensin aldosterone system).
Aldosterone is produced by the adrenal glands located at the top of each kidney, in their outer portion (called the adrenal cortex) (see Figures 1 and 2). Aldosterone stimulates the retention of sodium (salt) and the elimination of potassium by the kidneys. Renin is produced by the kidneys and controls the activation of the hormone angiotensin, which stimulates the adrenal glands to produce aldosterone.
The renin-angiotensin system is the primary regulator of the synthesis and secretion of aldosterone. Likewise, increased concentrations of potassium in the plasma may directly stimulate adrenal production of the hormone. Under physiologic conditions, pituitary adrenocorticotropic hormone (ACTH) is not a major factor in regulating aldosterone secretion.
The kidneys release renin when there is a drop in blood pressure or a decrease in sodium chloride concentration in the tubules in the kidney. Renin cleaves the blood protein angiotensinogen to form angiotensin 1, which is then converted by a second enzyme to angiotensin 2. Angiotensin 2 causes blood vessels to constrict, and it stimulates aldosterone production. Overall, this raises blood pressure and keeps sodium and potassium at normal levels.
A variety of conditions can lead to aldosterone overproduction (hyperaldosteronism, usually just called aldosteronism) or underproduction (hypoaldosteronism). Since renin and aldosterone are so closely related, both substances are often tested together to identify the cause of an abnormal aldosterone.
Figure 1. Location of the adrenal glands on top of each kidneys
Figure 2. Adrenal gland microscopic section
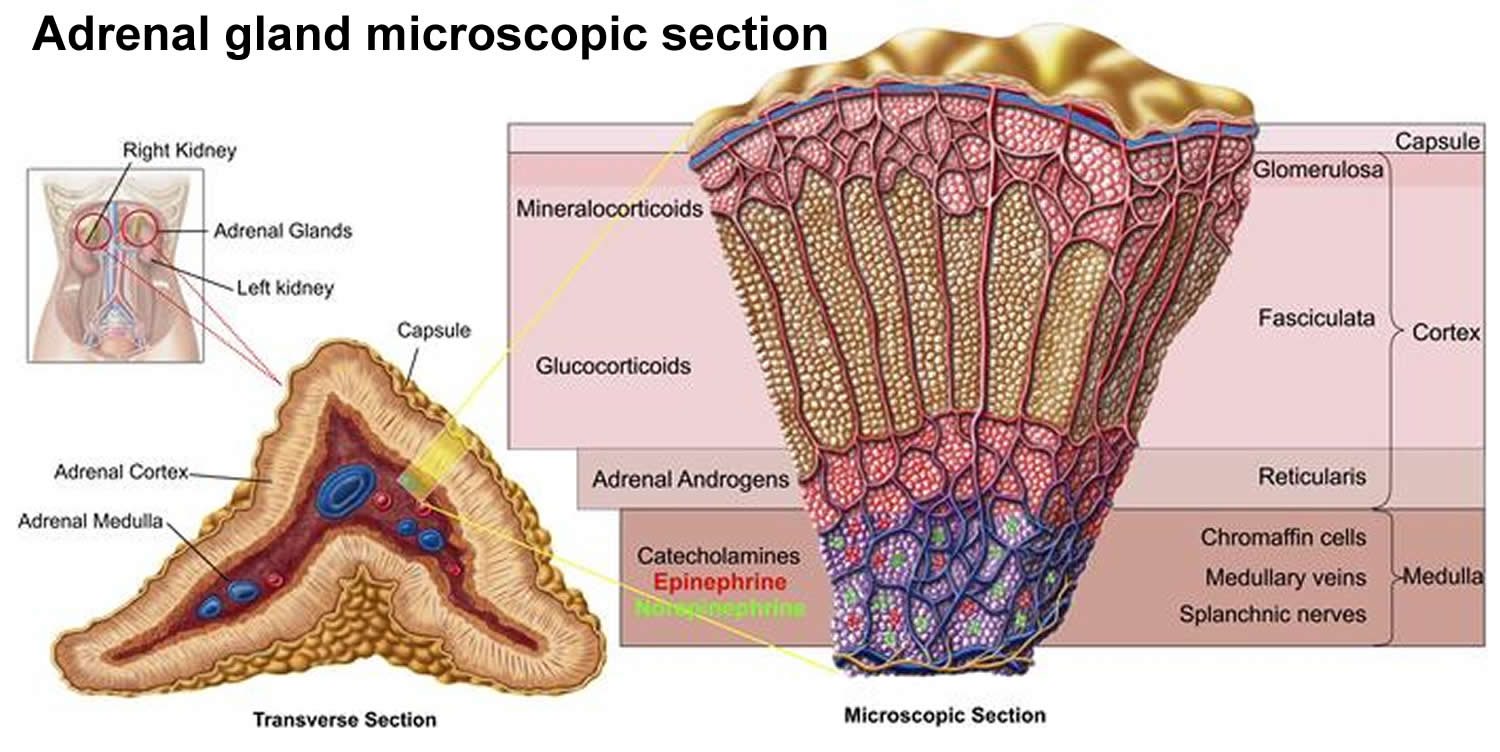
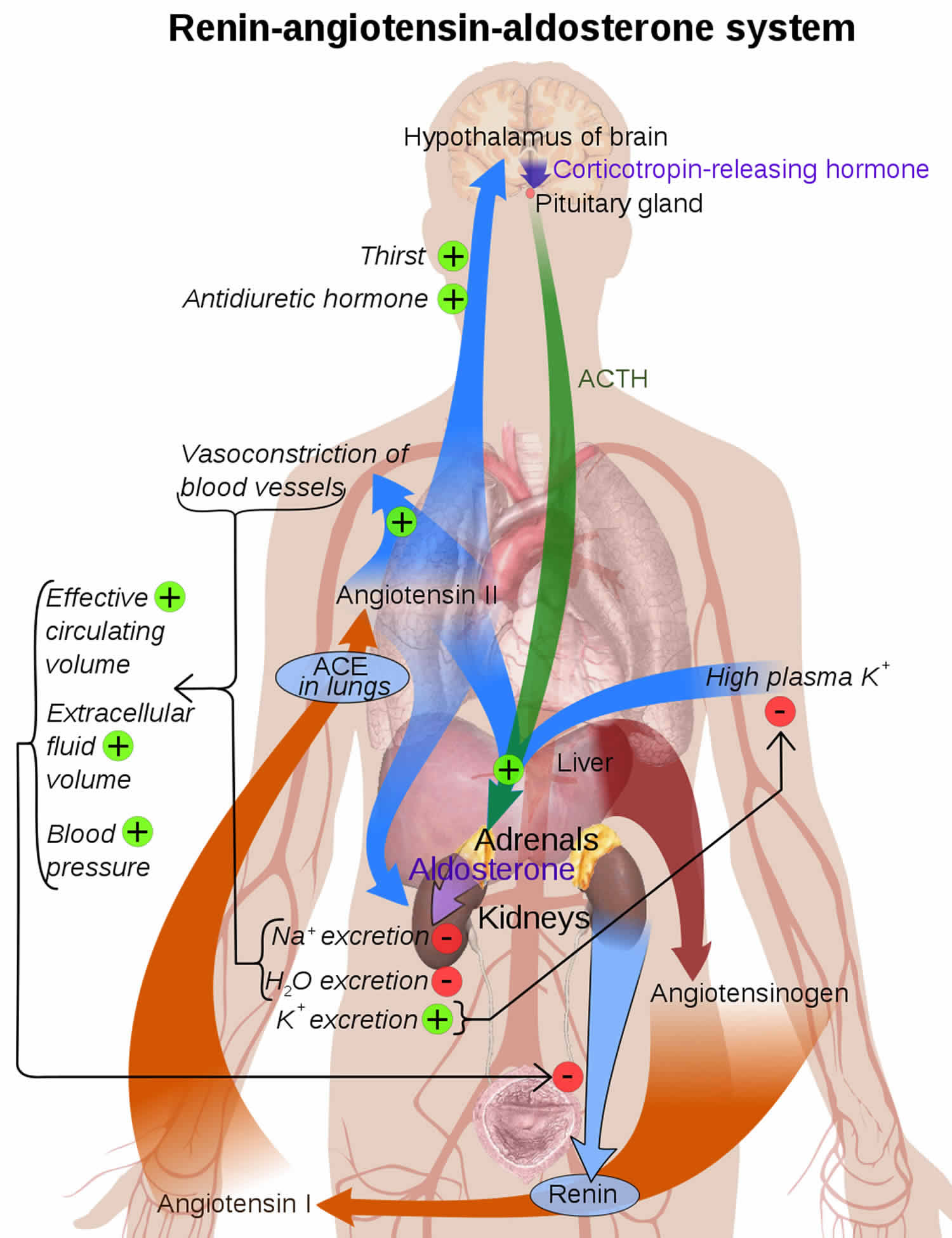
Figure 3. Renin angiotensin aldosterone system
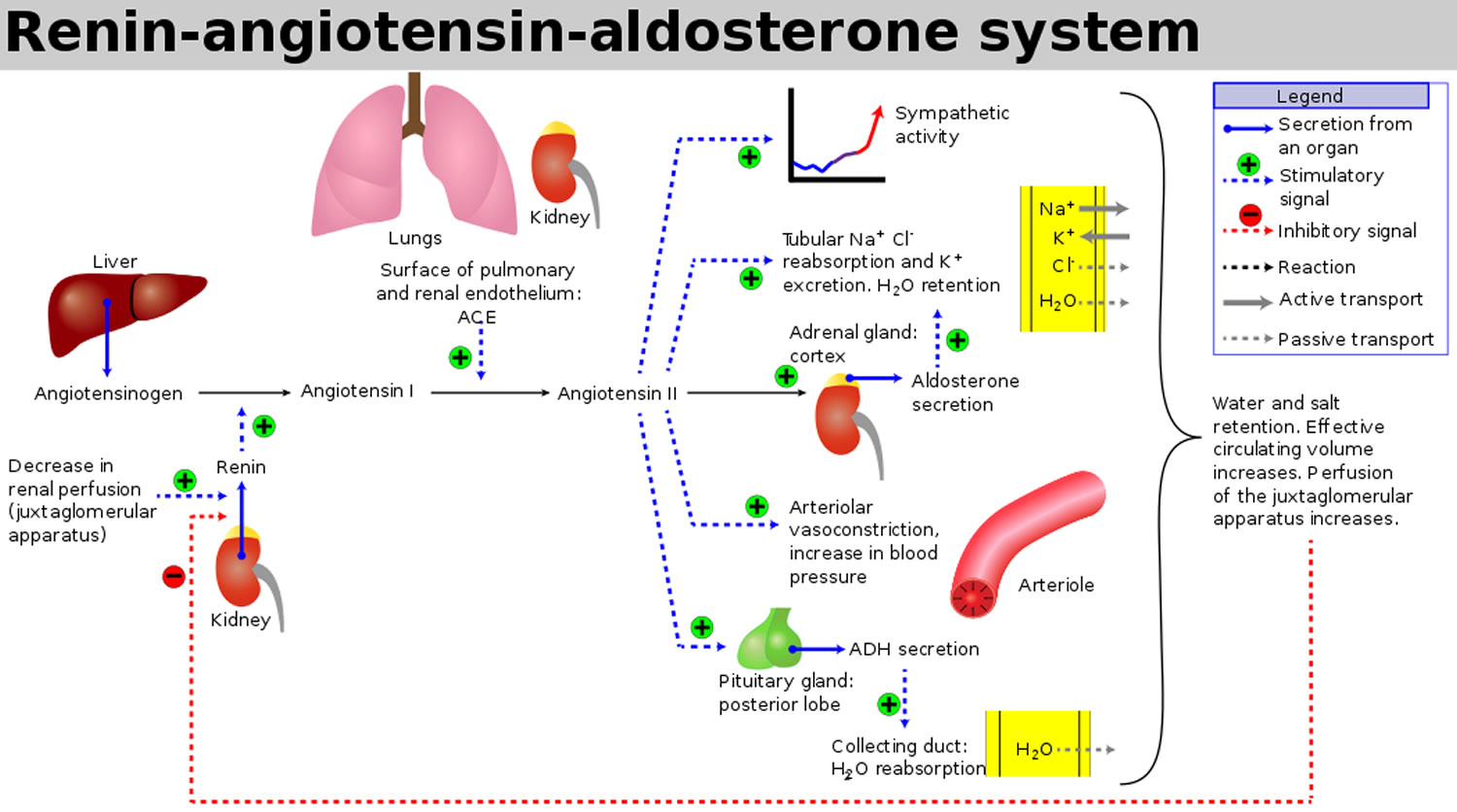
Figure 4. Renin angiotensin aldosterone system mechanism of action
Where is aldosterone produced?
Aldosterone is synthesized exclusively in the zona glomerulosa of the adrenal gland (see Figure 2 above).
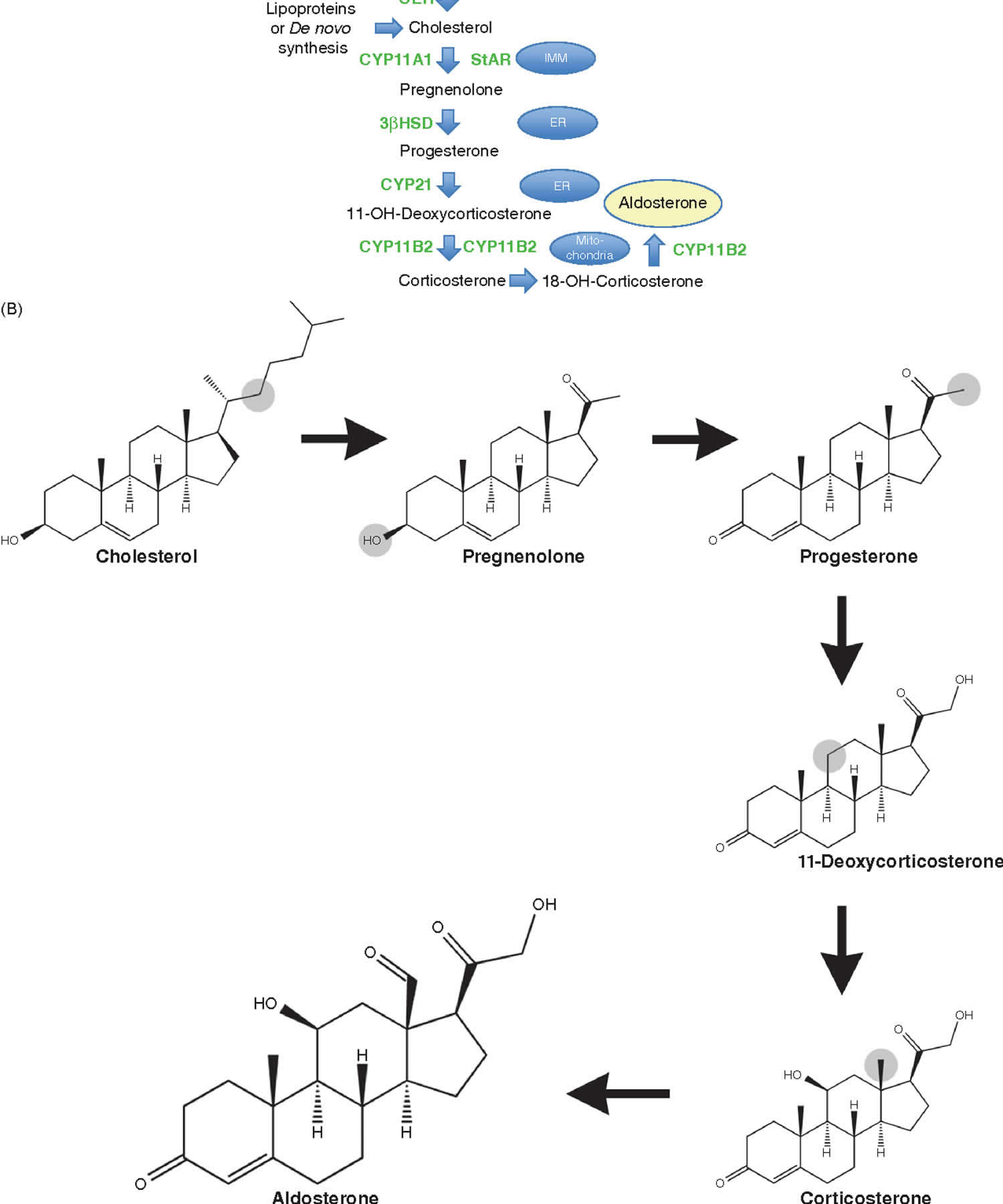
Aldosterone biosynthesis
All human steroid hormones are derived from cholesterol. Aldosterone is synthesized in the zona glomerulosa of the adrenal cortex through four enzymes, cholesterol desmolase (CYP11A1), 21-hydroxylase (CYP21A2), aldosterone synthase (CYP11B2) and 3β-hydroxysteroid dehydrogenase (3β-HSD) (Figure 5).
Figure 5. Aldosterone biosynthesis
 Regulation of aldosterone secretion
Regulation of aldosterone secretion
Aldosterone secretion is regulated by multiple factors. The renin-angiotensin system and potassium ion are the major regulators, whereas adrenocorticotropic hormone (ACTH) and other pro-opiomelanocortin (POMC) peptides, sodium ion, vasopressin (antidiuretic hormone or ADH), dopamine, atrial natriuretic peptide (ANP), β-adrenergic agents, serotonin and somatostatin are minor modulators.
The Renin angiotensin aldosterone system
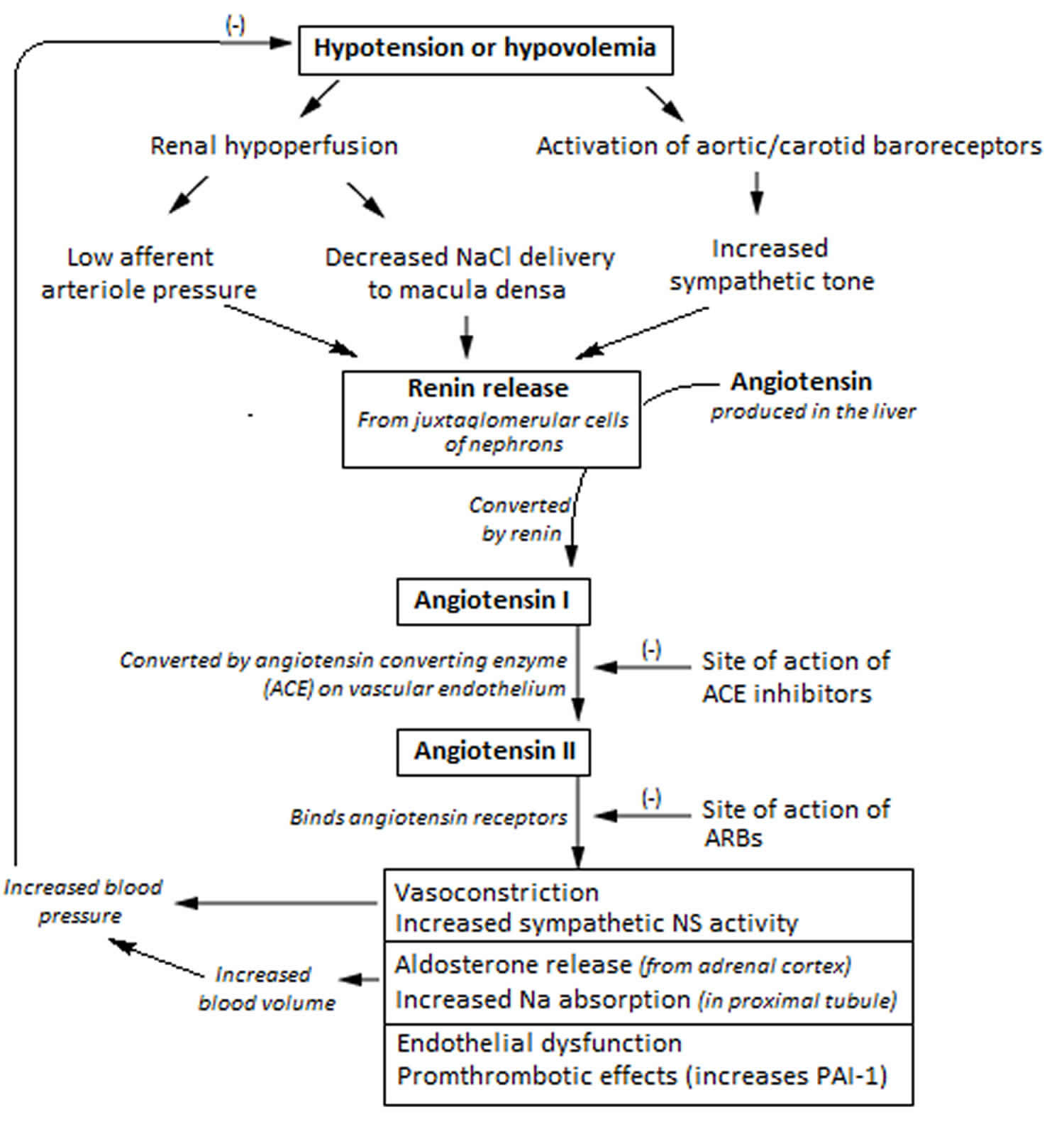
Within the afferent arterioles of the kidney, specialized cells called juxtaglomerular cells contain prorenin. While prorenin is secreted constitutively in its inactive form, activation of juxtaglomerular cells causes the cleavage of prorenin to renin. Activation of these cells occurs in response to decreased blood pressure, beta-activation, or activation by macula densa cells in response to a decreased sodium load in the distal convoluted tubule.
Once renin has been released into the blood, it can act on its target, angiotensinogen. Angiotensinogen is produced in the liver and is found continuously circulating in the plasma. Renin then acts to cleave angiotensinogen into angiotensin 1. Angiotensin 1 is physiologically inactive, but acts as a precursor for angiotensin 2.
The conversion of angiotensin 1 to angiotensin 2 is catalyzed by an enzyme called angiotensin converting enzyme (ACE). Angiotensin converting enzyme (ACE) is found primarily in the vascular endothelium of the lungs and kidneys. After angiotensin 1 is converted to angiotensin 2, it has effects on the kidney, adrenal cortex, arterioles, and brain by binding to angiotensin 2 type I and type II receptors. The effects discussed below are a result of binding to angiotensin receptors. The role of angiotensin receptors is still being investigated, but pertinently, they have been shown to cause vasodilation by nitric oxide generation. In the plasma, angiotensin 2 has a half-life of 1-2 minutes, at which point peptidases degrade it into angiotensin 3 and 4. Angiotensin 3 has been shown to have 100% of the aldosterone stimulating effect of angiotensin 2, but 40% of the pressor effects, while angiotensin 4 has some lesser pressor effect 1. Angiotensin 4 has cognitive-enhancing effects in the central nervous system 2. The exact identity of angiotensin 4 receptors has not been established. There is evidence that the angiotensin 4 receptor is insulin-regulated aminopeptidase 3 receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem. 2001 Dec 28;276(52):48623-6. Epub 2001 Nov 13. http://www.jbc.org/content/276/52/48623.long)). There is also evidence that angiotensin 4 interacts with the hepatocyte growth factor (HGF) through the c-Met receptor 4.
In the proximal convoluted tubule of the kidney, angiotensin 2 acts to increase Na-H exchange, increasing sodium reabsorption. Increased levels of Na in the body acts to increase the osmolarity of the blood, leading to a shift of fluid into the blood volume and extracellular space (ECF). This increases the arterial pressure of the patient.
Angiotensin 2 also acts on the adrenal cortex, specifically the zona glomerulosa. Here, it stimulates the release of aldosterone. Aldosterone is a steroid hormone that causes an increase in sodium reabsorption and potassium excretion at the distal tubule and collecting duct of the nephron. Aldosterone works by stimulating the insertion of luminal Na channels and basolateral Na-K ATPase proteins. The net effect is an increased level of sodium reabsorption. This has the same effect as mentioned previously: the increased total body sodium leads to an increase in osmolarity and subsequent increase in blood and extracellular space (ECF) volume. In contrast to angiotensin II, aldosterone is a steroid hormone. As a result, it enacts change by binding to nuclear receptors and altering gene transcription. Thus, the effects of aldosterone may take hours to days to begin, while the effects of angiotensin 2 are rapid.
The effect of angiotensin 2 on vasoconstriction takes place in systemic arterioles. Here, angiotensin 2 binds to G protein-coupled receptors, leading to a secondary messenger cascade that results in potent arteriolar vasoconstriction. This acts to increase total peripheral resistance, causing an increase in blood pressure.
Finally, angiotensin 2 acts on the brain. Here, it has three effects. First, it binds to the hypothalamus, stimulating thirst and increased water intake. Second, it stimulates the release of antidiuretic hormone (ADH) by the posterior pituitary. Antidiuretic hormone (ADH), or vasopressin, acts to increase water reabsorption in the kidney by inserting aquaporin channels at the collecting duct. Finally, angiotensin 2 decreases the sensitivity of the baroreceptor reflex. This diminishes baroreceptor response to an increase in blood pressure, which would be counterproductive to the goal of the renin angiotensin aldosterone system.
The net effect of these interactions is an increase in total body sodium, total body water, and vascular tone.
The renin–angiotensin–aldosterone system (RAAS) is a critical regulator of blood volume and systemic vascular resistance. The renin–angiotensin–aldosterone system functions to elevate blood volume and arterial tone in a prolonged manner. The renin–angiotensin–aldosterone system does this by increasing sodium reabsorption, water reabsorption, and vascular tone. While the baroreceptor reflex responds in a short-term manner to decreased arterial pressure, the renin angiotensin aldosterone system is responsible for more chronic alterations 1. The renin–angiotensin–aldosterone system (RAAS) is composed of three major compounds: renin, angiotensin 2 and aldosterone. These three act to elevate arterial blood pressure in response to decreased renal blood pressure, decreased salt delivery to the distal convoluted tubule, and/or beta-agonism. Through these mechanisms, the body can elevate the blood pressure in a prolonged manner.
The renin angiotensin aldosterone system acts to manage blood volume and arteriolar tone on a long-term basis. While minor and rapid shifts are typically managed via the baroreceptor reflex, the renin angiotensin aldosterone system can alter blood volume chronically. Though the renin angiotensin aldosterone system serves a critical function, it can be activated inappropriately in several conditions that may then lead to the development of hypertension. For example, renal artery stenosis results in a decreased volume of blood reaching one (or both) kidneys. As a result, the juxtaglomerular cells will sense a decrease in blood volume, activating the renin angiotensin aldosterone system. This can lead to an inappropriate elevation of circulating blood volume and arteriolar tone due to poor renal perfusion.
Pharmacologically, the renin angiotensin aldosterone system (RAAS) is a frequently manipulated system in the management of heart failure, hypertension, diabetes mellitus, and acute myocardial infarction. Angiotensin converting enzyme inhibitors (e.g., enalapril), angiotensin receptor blockers (ARBs, e.g., losartan), and aldosterone antagonists (e.g., spironolactone) all act to decrease the effect of the renin angiotensin aldosterone system.
Figure 6. The renin angiotensin aldosterone system involves the kidneys, lungs, systemic vasculature, and the brain.
Potassium
Potassium directly increases aldosterone secretion by the adrenal cortex and aldosterone then lowers serum potassium by stimulating its excretion by the kidney. High dietary potassium intake increases plasma aldosterone and enhances the aldosterone response to a subsequent potassium or angiotensin 2 infusion 5. The primary action of potassium for stimulating aldosterone secretion is to depolarize the plasma membrane, which activates voltage-dependent calcium channels, that permit influx or exflux of extracellular calcium 5, leading to the activation of calmodulin and calmodulin-dependent kinase, subsequently.The activated kinase phospholylates both activating transcription factor and members of CRE-binding protein family, which bind to 5’ flanking promotor regions of the CYP11B2 gene and trigger gene transcription in the zona glomerulosa, then increase aldosterone biosynthesis as angiotensin 2 does 6.
Pituitary Factors
Adrenocorticotropic hormone (ACTH) and possibly other pro-opiomelanocortin (POMC)-derived peptides, including α-MSH, α-MSH, β-LPH and β-END, influence aldosterone secretion, however, the role of ACTH in aldosterone secretion is minor 5. ACTH increases aldosterone secretion by binding to glomerulosa cell-surface melanocortin-2 receptor, activating adenylate cyclase, and increasing intracellular cAMP 7. Like other agents, ACTH stimulates the same two early and late steps of aldosterone biosynthesis.
Vasopressin has a modest and transient stimulatory effect on aldosterone secretion from zona granulosa cells in vitro. This effect is probably mediated via V2 receptors and phospholipase C generating IP3 and diacylglycerol 8.
Sodium
Sodium intake influences aldosterone secretion by an indirect effect through renin and to a minor extent by direct effects on zona glomerulosa responsiveness to angiotensin 2. High sodium intake increases vascular volume, which suppresses renin secretion and angiotensin 2 generation and decreases the sensitivity of aldosterone response to angiotensin 2 9.
Inhibitory agents
Dopamine inhibits aldosterone secretion in humans by a mechanism that is independent of the effects of prolactin, ACTH, electrolytes and the renin-angiotensin system 10. This inhibitory effect may involve binding to D2 receptors on glomerulosa cells 11. Atrial natriuretic peptide (ANP) directly inhibits aldosterone secretion and blocks the stimulatory effects of angiotensin 2, potassium and ACTH, at least in part, by interfering with extracellular calcium influx 12.
What does aldosterone do
Aldosterone plays an important role in maintaining normal sodium and potassium concentrations in blood and in controlling blood volume and blood pressure.
Aldosterone stimulates sodium transport across cell membranes, particularly in the distal renal tubule where sodium is exchanged for hydrogen and potassium. Secondarily, aldosterone is important in the maintenance of blood pressure and blood volume.
Aldosterone promotes active sodium transport and excretion of potassium via the mineralocorticoid receptor and the resultant activation of specific amiloride-sensitive sodium channels and the Na-K ATPase pump in the target tissues 13. Defective aldosterone action results in various symptoms and laboratory characteristics such as hyponatremia (low blood sodium), hyperkalemia (low blood potassium), acidosis, and so on.
Aldosterone actions on the cardiovascular system
Hypertrophy and fibrosis
Aldosterone has been shown to induce ventricular hypertrophy and fibrosis in cardiac interstitial and perivascular tissues. Myocardial fibrosis does not occur in cardiac hypertrophy unless there is concurrent activation of the renin-angiotensin-aldosterone system 14. Thus chronic anemia, arteriovenous fistula, and atrial septal defect lead to hypertrophy without fibrosis. This observation led Brilla et al. 15 to explore the role of aldosterone in the genesis of cardiac fibrosis. They compared different experimental hypertensive rat models: elevated angiotensin 2 and aldosterone induced by a renovascular lesion, normal angiotensin 2 and aldosterone in infrarenal aorta binding, isolated elevated aldosterone by minipumps plus high salt intake, and control. The results showed ventricular hypertrophy with myocardial fibrosis in the models with elevated aldosterone levels and with high, normal, and suppressed angiotensin 2 levels, suggesting that elevation in circulating angiotensin 2 alone was not the decisive mediator of the fibrous tissue response. They concluded that elevated aldosterone was important in the regulation of collagen synthesis within the cardiac interstitium and in the adventitia of intramyocardial coronary arteries 14. Thus chronic anemia, arteriovenous fistula, and atrial septal defect lead to hypertrophy without fibrosis. This observation led Brilla et al. 15. Subsequently, the same laboratory demonstrated that administration of an aldosterone antagonist, spironolactone, at a dose that had no antihypertensive effect, could prevent cardiac fibrosis 16. Recently, Schlaich et al. 17 confirmed by use of M-mode echocardiography a correlation between plasma levels of aldosterone and cardiac left ventricular mass in human subjects. This correlation was not related to differences in blood pressure. A recent prospective clinical trial has shown that treatment with spironolactone significantly reduced morbidity and mortality among patients with severe heart failure. Blood pressure was not affected by spironolactone treatment, suggesting that endogenous aldosterone has direct deleterious effects on the cardiovascular system in these patients 18.
The cellular mechanisms by which aldosterone induces fibrosis are still under investigation. Initial studies from Weber’s laboratory reported that aldosterone stimulated transcription of collagen I mRNA and increased levels of collagen I synthesis in rat cardiac fibroblasts 19. Robert et al. 20 also demonstrated an increase in cardiac type I and type III collagen mRNA in rats made hypertensive with aldosterone and salt treatment. However, other laboratories 21 could not confirm these findings. They proposed that aldosterone does not influence fibroblast collagen synthesis directly but increases endothelin receptor number, which leads to increased collagen synthesis 21. These studies are difficult to interpret, because the effect of aldosterone to increase cardiac fibrosis occurs over a long period of time and may not be detectable in an in vitro culture system.
Plasminogen activator inhibitor-1
Aldosterone may have a role in the regulation of plasminogen activator inhibitor-1 (PAI-1) 22. Although aldosterone had no direct effect on plasminogen activator inhibitor-1 activity in vitro, it did enhance the stimulatory effect of angiotensin 2 on plasminogen activator inhibitor-1 activity. In a rat model of radiation-induced glomerulosclerosis, spironolactone decreased the degree of sclerosis and the level of plasminogen activator inhibitor-1 mRNA expression 23.
Effects on vascular tone
Aldosterone also modulates vascular tone, although the exact mechanisms remain to be elucidated. Possible mechanisms include increased vasoconstrictive effects of catecholamines through decreased tissue catecholamine uptake 24, impaired vasodilatation in response to acetylcholine 25, upregulation of β-adrenergic 26 and angiotensin 2 27 receptors, and a direct aldosterone effect, possibly through nongenomic mechanisms. There is evidence that aldosterone also affects vascular smooth muscle cells via mineralocorticoid receptors, causing transmembrane influx of sodium and potentiating angiotensin 2-induced leucine incorporation into smooth muscle cells in vitro 28. These mechanisms may be involved in hypertrophy of smooth muscle cells. Aldosterone administration, in nonpressor doses, augmented neointimal thickening in the rabbit iliac artery after balloon-induced vascular injury. Conversely, spironolactone administration reduced the thickening compared with both aldosterone and control groups 29. Wang et al. 24 demonstrated that perfusion of the carotid sinus with aldosterone reduced baroreceptor discharge in the dog, and a similar finding has been reported in humans 30.
Cerebrovascular effects
Studies in stroke-prone spontaneously hypertensive rats have indicated a possible role for aldosterone in vascular remodeling and stroke. Spironolactone treatment in these animals did not reduce blood pressure but did prevent spontaneous cerebral infarction 31. Captopril, an angiotensin-converting enzyme inhibitor, also prevented cerebral infarction, but coadministration of aldosterone reversed the captopril effect 32. Dorrance et al. 33 reported that spironolactone treatment reduced the size of cerebral infarcts induced by middle cerebral artery ligation. These authors also found that mineralocorticoid treatment increased vascular expression of receptors for epidermal growth factor. They proposed that aldosterone increases vascular remodeling by increasing vascular epidermal growth factor receptor activity 33.
Local biosynthesis
In addition to effects of circulating mineralocorticoids on the heart, local biosynthesis of aldosterone has recently been reported and may have physiological or pathophysiological importance. In the isolated rat heart, Silvestre et al. 34 used quantitative reverse transcription-polymerase chain reaction analysis to document mRNA expression of both aldosterone synthase (CYP11B2) and 11β-hydroxylase (CYP11B1), crucial enzymes in the pathway of mineralocorticoid synthesis. They used celite column chromatography coupled to radioimmunoassay to measure tissue levels of aldosterone. The cardiac aldosterone synthesis pathway is responsive to a low-sodium/high-potassium diet and to angiotensin 2 in a manner similar to that of the adrenal cortex. Silvestre et al. 34 demonstrated that cardiac aldosterone levels are many times higher than those in plasma, suggesting that locally synthesized aldosterone within the heart may be important for local paracrine or autocrine function. The physiological role of this local system remains to be explored.
In summary, aldosterone appears to play an important role in the pathogenesis of cardiovascular disease. Possible mechanisms of these effects include promotion of fibrosis, modulation of vascular tone and vascular remodeling, and enhancement of angiotensin 2-mediated plasminogen activator inhibitor-1 (PAI-1) stimulation.
Aldosterone actions on the central nervous system
The classic glucocorticoid receptor is widely distributed in brain in both neurons and glial cells, with a high density in the hippocampus and other parts of the limbic system 35. Mineralocorticoid receptors are less prevalent in the central nervous system (CNS), and their location is predominantly limited to neurons of the hippocampus and lateral septum 36. Unlike mineralocorticoid receptors targets in epithelial tissue or the cardiovascular system, the brain lacks significant 11βHSD2 activity; therefore, brain mineralocorticoid receptors are not protected from glucocorticoid binding, and mineralocorticoid receptors can bind to both mineralocorticoids and glucocorticoids. Thus, it appears likely that glucocorticoids are the primary ligand for most brain mineralocorticoid receptors. Reul and de Kloet 36 showed that >80% of mineralocorticoid receptors in the brain are occupied by endogenous hormones under basal resting conditions. In contrast, brain glucocorticoid receptor are significantly occupied only when circulating glucocorticoid levels are high. De Kloet and colleagues 37 postulated that mineralocorticoid receptors mediate tonic actions of corticosteroids involved in maintenance of homeostasis. In contrast, the glucocorticoid receptor is likely to be important in dynamic responses and the restoration of homeostasis. Corticosteroids in brain appear to be involved in the regulation of several physiological systems: learning and memory 37, neuroendocrine hypothalmic-pituitary-adrenal axis regulation 38, autonomic function, control of sodium homeostasis, and blood pressure regulation. Most of these effects are mediated through activation of mineralocorticoid receptors, possibly by interaction between mineralocorticoid receptors and either glucocorticoids or mineralocorticoids.
Despite almost negligible levels of 11βHSD2 activity in the brain, there is evidence that certain areas contain mineralocorticoid receptors that bind preferentially to mineralocorticoids. This has been demonstrated in the anterior hypothalamus, hippocampus, anterior pituitary, and some nuclei in the brain stem 39. The mineralocorticoid receptors in the hypothalamus and circumventricular organs are involved in the regulation of blood pressure and sodium homeostasis.
Aldosterone acts directly in the CNS to increase blood pressure. This action is separate from its systemic effects and is not associated with changes in fluid and electrolyte balance, salt appetite, or vascular reactivity. Gomez-Sanchez and colleagues 40 have extensively explored this issue in a series of experiments comparing the effects of subcutaneous and intracerebroventricular administration of corticosteroid agonists and antagonists in rats. Continuous intracerebroventricular infusion of aldosterone produced a significant increase in blood pressure that was blocked by concomitant intracerebroventricular infusion of mineralocorticoid receptors antagonists, either prorenone 41 or RU-28318 42. The hypertension induced by the continuous intracerebroventricular infusion of aldosterone was dose responsive and independent of changes in renal sodium handling 43; blood pressure increased in sodium-deplete as well as sodium-replete dogs and rats 44. The doses of steroids given intracerebroventricularly were small and did not cause changes in blood pressure when given subcutaneously. Similar findings were also reported in dogs receiving centrally administered aldosterone 45. Gomez-Sanchez et al. 40 also demonstrated that corticosterone antagonized the pressor effect of centrally administered aldosterone in a dose-dependent fashion if it was coinfused intracerebroventricularly with aldosterone. Bilateral adrenalectomy in rats prevented the pressor effect of intracerebroventricular aldosterone, and the effect was restored by administration of physiological replacement doses of corticosterone 40. Central infusion of a selective mineralocorticoid receptors antagonist, RU-28318, diminished the hypertensive effect of subcutaneous aldosterone infusion 42, and this amount of intracerebroventricular RU-28318 was less than that required to inhibit the increased appetite for saline associated with excess systemic mineralocorticoids 46.
Deoxycorticosterone acetate (DOCA) in combination with dietary salt is commonly used to induce mineralocorticoid hypertension. In this experimental model, attenuation of the baroreflex response can usually be detected before elevation of blood pressure occurs. In deoxycorticosterone acetate-salt-treated rats, intracerebroventricular infusion of RU-28318 normalized the baroreflex, reduced sympathetic tone, and prevented hypertension 47. The prehypertensive stage of intracerebroventricular aldosterone-induced hypertension, in contrast to that of systemic mineralocorticoid excess states, is not associated with a decrease in baroreceptor reactivity, nor is there an increase in vascular reactivity to the intravenous infusion of angiotensin 2, norepinephrine, or arginine vasopressin 47.
These results, taken together, indicate that direct CNS effects of aldosterone play a significant role in the pressor effects that occur when steroids are administered systemically.
A recent study by Gomez-Sanchez et al. 48 suggested that the brain has the capability to produce aldosterone. Southern blot hybridization demonstrated aldosterone synthase in the hypothalamus, hippocampus, amygdala, cerebrum, and cerebellum of rat brain. Incubation of [3H]DOCA, a substrate for aldosterone synthase, with minced brain from hippocampus, hypothalamus, and cerebellum, yielded [3H]aldosterone. In contrast, incubation of minced brain with aldosterone synthesis inhibitors, either cortisol or metyrapone, inhibited aldosterone production.
In conclusion, it is clear that aldosterone has important physiological and pathophysiological effects on nonepithelial tissues. In the cardiovascular system it promotes ventricular hypertrophy and interstitial cardiac and perivascular fibrosis. It may also increase PAI-1 activity. It probably plays an important role in the pathophysiology of heart failure. In the CNS, aldosterone acts to increase blood pressure and sympathetic activity. The importance of locally produced aldosterone in these tissues and the role of nongenomic actions of aldosterone remain to be determined.
Normal aldosterone level
Aldosterone and renin tests are used to evaluate whether the adrenal glands are producing appropriate amounts of aldosterone and to distinguish between the potential causes of excess or deficiency. Aldosterone may be measured in the blood or in a 24-hour urine sample, which measures the amount of aldosterone removed in the urine in a day. Renin is always measured in blood.
Both aldosterone and renin levels are highest in the morning and vary throughout the day. They are affected by the body’s position, by stress, and by a variety of prescribed medications. Late p.m. levels can be up to 30% lower than early a.m. levels. Supine values are on average 50% lower than upright collections. Sodium deplete subjects have significantly elevated serum aldosterone levels, potentially exceeding the upper limit of the salt replete upright reference range by several fold. To account for these variables, at least in part, it is recommended that plasma renin activity is measured concomitantly. In situations of physiological variability, Pplasma renin activity should be altered in the same direction as aldosterone.
- Age 0-30 days: 17-154 ng/dL
- Age 31 days-11 months: 6.5-86 ng/dL
- Age 1-10 years:
- Age < or =40 ng/dL (supine)
- Age < or =124 ng/dL (upright)
- Age > or =11 years: < or =21 ng/dL (a.m. peripheral vein specimen)
When is aldosterone test ordered?
A blood aldosterone test and a renin test are usually ordered together when someone has high blood pressure, especially if the person also has low potassium. Even if potassium is normal, testing may be done if typical medications do not control the high blood pressure or if hypertension develops at an early age. Primary aldosteronism (Conn syndrome) is a potentially curable form of hypertension, so it is important to detect and treat it properly.
Aldosterone levels are occasionally ordered, along with other tests, when a healthcare practitioner suspects that someone has adrenal insufficiency or Addison disease. One of those tests, the aldosterone stimulation test, also called ACTH stimulation, tests aldosterone and cortisol to determine if someone has Addison disease, low pituitary function, or a pituitary tumor. A normal result is a cortisol increase and an increase in aldosterone after stimulation by ACTH.
What does abnormal aldosterone test result mean?
The table below indicates the changes in renin, aldosterone, and cortisol that occur with different disorders.
| Disease | Aldosterone | Cortisol | Renin |
|---|---|---|---|
| Primary aldosteronism (Conn syndrome) | High | Normal | Low |
| Secondary aldosteronism | High | Normal | High |
| Adrenal insufficiency (Addison disease) | Low | Low | High |
| Cushing syndrome | Low | High | Low |
A high ratio of serum aldosterone in ng/dL to plasma renin activity in ng/mL per hour, is a positive screening test result, a finding that warrants further testing. An serum aldosterone/renin ratio > or =20 is only interpretable with an serum aldosterone > or =15 ng/dL and indicates probable primary aldosteronism (Conn syndrome).
Renal disease, such as unilateral renal artery stenosis, results in elevated renin and aldosterone levels. Renal venous catheterization may be helpful. A positive test is a renal venous renin ratio (affected/normal) >1.5.
The plasma renin activity cannot be interpreted if the patient is being treated with spironolactone (Aldactone). Spironolactone should be discontinued for 4 to 6 weeks before testing.
Angiotensin converting enzyme (ACE) inhibitors have the potential to falsely elevate plasma renin activity. Therefore, in a patient treated with an ACE inhibitor, the findings of a detectable plasma renin activity level or a low serum aldosterone/plasma renin activity ratio do not exclude the diagnosis of primary aldosteronism (Conn syndrome). In addition, a strong predictor for primary aldosteronism is a plasma renin activity level undetectably low in a patient taking an ACE inhibitor.
The amount of salt in your diet and medications, such as over-the-counter pain relievers of the non-steroidal anti-inflammatory drugs (NSAIDs), diuretics, beta blockers, steroids, angiotensin-converting enzyme (ACE) inhibitors, and oral contraceptives can affect the test results. Some of these drugs are used to treat high blood pressure. Stress, exercise, and pregnancy can also affect the test results. Your healthcare provider will tell you if you should change the amount of sodium (salt) you ingest in your diet, your use of diuretics or other medications, or your exercise routine before aldosterone testing.
Licorice may mimic aldosterone properties and should be avoided for at least two weeks before the test because it can decrease aldosterone results. This refers only to the actual products of the licorice plant (hard licorice); most soft licorice and other forms of licorice sold in North America do not actually contain licorice. Check the package label if you are uncertain, or bring a package with you to ask the healthcare practitioner.
Aldosterone levels become very low with severe illness, so testing should not be done at times when someone is very ill.
What is an aldosterone/renin ratio?
An aldosterone/renin ratio is a screening test to detect primary aldosteronism (Conn syndrome) in high-risk, hypertensive individuals. To determine the ratio, blood levels of aldosterone and renin are measured and a calculation is done by dividing the aldosterone result by the renin result. The aldosterone/renin ratio is considered the most reliable screening for primary aldosteronism (Conn syndrome), though it is not straightforward to interpret. Anything that could interfere with the test, such as medications, posture, sodium intake, and plasma potassium, needs to be taken into account before the test to avoid false positives or false negatives. Other tests, like suppression tests, are used to confirm the diagnosis after screening.
What are aldosterone stimulation and suppression tests?
Aldosterone suppression tests are used to confirm a diagnosis of primary aldosteronism. There are a few different types of suppression tests:
- You may be instructed to follow a high-salt diet for three days, then have your aldosterone and sodium in your urine measured.
- You may have a saline (salt) solution administered through a vein (intravenous, IV) and then have your aldosterone level measured.
- You may follow a high-salt diet and be administered a synthetic corticosteroid called fludrocortisone, then have your aldosterone level measured.
In healthy people who are on a high-salt diet or who are administered saline or fludrocortisone, their aldosterone level will be suppressed.
The aldosterone stimulation test, also called adrenocorticotropic hormone (ACTH) stimulation, tests aldosterone and cortisol to determine if someone has Addison disease, low pituitary function, or a pituitary tumor. This test involves measuring aldosterone and cortisol before and after an injection of synthetic adrenocorticotropic hormone (ACTH). A normal result is an increase in aldosterone and cortisol after stimulation by adrenocorticotropic hormone (ACTH).
Low aldosterone
Low aldosterone (hypoaldosteronism) usually occurs as part of adrenal insufficiency. It causes dehydration, low blood pressure, a low blood sodium level, and a high potassium level. When infants lack an enzyme needed to make cortisol, a condition called congenital adrenal hyperplasia, this can decrease production of aldosterone in some cases.
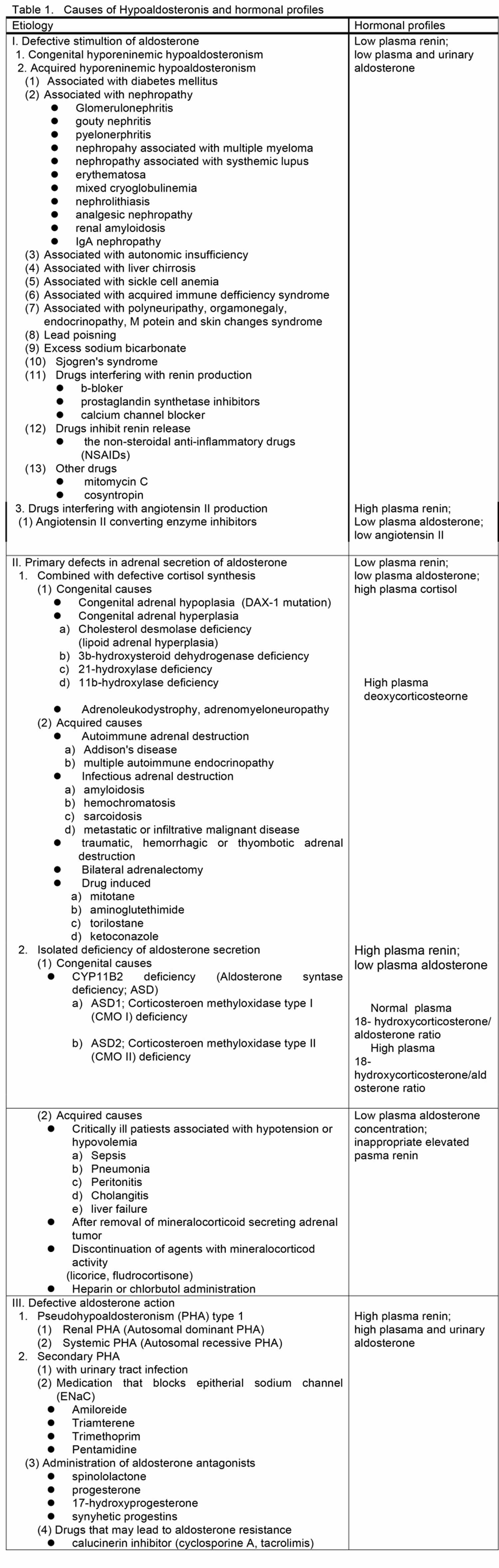
Various syndromes are characterized by or associated with hypoaldosteronism. Hypoaldosteronism is classified in three large categories, defective stimulation of aldosterone secretion, primary defects in adrenal synthesis or secretion of aldosterone and aldosterone resistance, according to their pathophysiology and summarized in Table 1.
 [Source 49 ]
[Source 49 ]
Defective Stimulation of Aldosterone Secretion
The first category of conditions, which is characterized by defective stimulation of aldosterone secretion, includes the syndromes of congenital and acquired hyporeninemic hypoaldosteronism. One of these conditions is due to a defect of renin secretion such as hyporeninemia resulting from β-blockers, prostaglandin synthetase inhibitors, and calcium channel blockers. Another condition is due to decrease in the conversion of angiotensin 1 to angiotensin 2 mediated by converting enzyme inhibitor medications and is associated with hyperreninemia.
Primary Defects in Adrenal Biosynthesis or Secretion of Aldosterone
The second category of conditions, which are characterized by primary defects in adrenal synthesis or secretion of aldosterone, includes all causes of primary adrenal insufficiency and primary hypoaldosteronism caused by aldosterone synthase (CYP11B2) deficiency or as an acquired state. Primary adrenal insufficiency causes include congenital adrenal hypoplasia, congenital adrenal hyperplasia, adrenoleukodystrophy/adrenomyeloneuropathy, acquired adrenal insufficiency due to autoimmune, infectious and infiltrative disease, bilateral adrenalectomy and use of adrenolytic agents and enzyme inhibitors that block cortisol and aldosterone biosynthesis. These conditions are usually combined with defective cortisol synthesis. Aldosterone synthase (CYP11B2) deficiency leads to reduced aldosterone production associated with low or high levels of 18-hydroxycorticosterone, referred to as CMO I or CMO II deficiency, respectively. Several conditions may be associated with aldosterone biosynthetic activity. Heparin suppresses aldosterone synthesis. Critically ill patients with persistent hypovolemia and hypotension also have inappropriately low plasma aldosterone concentrations in relation to the activity of the renin-angiotensin system. Isolated primary hypoaldosteronism in occasionally associated with metastatic cancer of the adrenal gland.
Defective Aldosterone Actions
The third category which is characterized by defective aldosterone action includes syndromes of aldosterone resistance such as pseudohypoaldosteronism type 1 and sodium-wasting states resulting from excessive amounts of circulating mineralocorticoid antagonists, such as spinololactone and its analogues, and synthetic progestin or natural agonists, such as progesterone or 17-hydroxyprogesterone. These mineralocorticoid antagonists may antagonize aldosterone at the levels of mineralocorticoid receptor 50 and frequently, these states are compensated for by elevated concentrations of plasma aldosterone.
High aldosterone
Primary aldosteronism (Conn syndrome) is caused by the overproduction of aldosterone by the adrenal glands, usually by a benign tumor of one of the glands. The high aldosterone level increases reabsorption of sodium (salt) and loss of potassium by the kidneys, often resulting in an electrolyte imbalance. The excess sodium in turn holds on to water, increasing your blood volume and blood pressure. Diagnosis and treatment of primary aldosteronism are important because people with this form of high blood pressure have a higher risk of heart disease and stroke. Also, the high blood pressure associated with primary aldosteronism may be curable. Options for people with primary aldosteronism include medications, lifestyle modifications and surgery.
Lower than normal blood potassium (hypokalemia) in someone with hypertension suggests the need to look for aldosteronism. Sometimes, to determine whether only one or both adrenal glands are affected, blood may be taken from both of the adrenal veins and testing is done to determine whether there is a difference in the amount of aldosterone (and sometimes cortisol) produced by each of the adrenal glands.
Common conditions causing the overproduction of aldosterone include:
- A benign growth in an adrenal gland (aldosterone-producing adenoma) — a condition also known as Conn’s syndrome
- Overactivity of both adrenal glands (idiopathic hyperaldosteronism)
In rare cases, primary aldosteronism may be caused by:
- A cancerous (malignant) growth of the outer layer (cortex) of the adrenal gland (adrenal cortical carcinoma)
- A rare type of primary aldosteronism called glucocorticoid-remediable aldosteronism that runs in families and causes high blood pressure in children and young adults
Secondary aldosteronism, which is more common than primary aldosteronism, is caused by anything that leads to excess aldosterone, other than a disorder of the adrenal glands. It could be caused by any condition that decreases blood flow to the kidneys, decreases blood pressure, or lowers sodium levels. Secondary aldosteronism may be seen with congestive heart failure, cirrhosis of the liver, kidney disease, and toxemia of pregnancy (pre-eclampsia). It is also common in dehydration. In these conditions, the cause of aldosteronism is usually obvious.
The most important cause of secondary aldosteronism is narrowing of the blood vessels that supply the kidney, termed renal artery stenosis. This causes high blood pressure due to high renin and aldosterone and may be cured by surgery or angioplasty. Sometimes, to see if only one kidney is affected, a catheter is inserted through the groin and blood is collected directly from the veins draining the kidney (renal vein renin levels). If the value is significantly higher in one side, this indicates where the narrowing of the artery is present.
What is Bartter syndrome?
Bartter syndrome is a group of rare congenital disorders that affect the kidney’s ability to reabsorb sodium. People with Bartter syndrome lose too much sodium through the urine. This causes a rise in the level of the aldosterone and makes the kidneys remove too much potassium from the body. The syndrome is therefore associated with high levels of renin and aldosterone in the blood, increased blood pH (alkalosis), and high levels of potassium, calcium, and chloride in the urine.
The syndrome, which is usually diagnosed in early childhood, can be caused by mutations in at least one of five genes, and genetic testing can confirm a diagnosis. There are different types of Bartter syndrome, defined based upon which gene is the cause of the condition.
Signs and symptoms will vary depending on the type of Bartter syndrome. The antenatal form (appears before birth) can be life-threatening. The classical form found in infants and young children usually causes failure to thrive, constipation, muscle cramping and weakness as well as dehydration, increased urine production, and weakened bones.
The condition cannot be cured, but a few treatments are available, such as keeping an affected person’s blood potassium from being abnormally low through a potassium-rich diet or by taking supplements. While, with treatment, prognosis is good, those affected must be careful to maintain fluid and electrolyte balance. Kidney failure is a possible complication of Bartter syndrome.
High aldosterone symptoms
Signs and symptoms of high aldosterone due to primary aldosteronism (Conn syndrome) include high blood pressure, headache, and muscle weakness, especially if potassium levels are very low.
Primary and secondary hyperaldosteronism have common symptoms, including:
- High blood pressure
- Low level of potassium in the blood
- Feeling tired all the time
- Headache
- Muscle weakness
- Numbness
Primary hyperaldosteronism can cause very high blood pressure, which can damage many organs, including the eyes, kidneys, heart and brain.
Erection problems and gynecomastia (enlarged breasts in men) may occur with long-term use of medicines to block the effect of hyperaldosteronism.
High aldosterone complications
Primary aldosteronism can lead to high blood pressure and low potassium levels. These complications in turn can lead to other problems.
Problems related to high blood pressure
Persistently elevated blood pressure can lead to problems with your heart and kidneys, including:
- Heart attack
- Heart failure
- Left ventricular hypertrophy — enlargement of the muscle that makes up the wall of the left ventricle, one of your heart’s pumping chambers
- Stroke
- Kidney disease or kidney failure
- Premature death
High blood pressure caused by primary aldosteronism carries a higher risk of cardiovascular complications than do other types of high blood pressure. This excess risk is due to the high aldosterone levels, which can cause heart and blood vessel damage independent of complications related to high blood pressure.
Problems related to low potassium levels
Some, but not all, people with primary aldosteronism have low potassium levels (hypokalemia). Mild hypokalemia may not cause any symptoms, but very low levels of potassium can lead to:
- Weakness
- Cardiac arrhythmias
- Muscle cramps
- Excess thirst or urination
High aldosterone diagnosis
The health care provider will perform a physical exam and ask about your symptoms to help diagnose primary aldosteronism.
Screening test
Initially, your doctor is likely to measure the levels of aldosterone and renin in your blood. Renin is an enzyme released by your kidneys that helps regulate blood pressure. The combination of a very low renin level with a high aldosterone level suggests that primary aldosteronism may be the cause of your high blood pressure.
Confirmation tests
If the aldosterone-renin test suggests that you might have primary aldosteronism, you’ll need another test to confirm the diagnosis, such as one of the following:
- Oral salt loading. You’ll follow a high-sodium diet for three days before your doctor measures aldosterone and sodium levels in your urine.
- Saline infusion test. Your aldosterone levels are tested after sodium mixed with water (saline) is infused into your bloodstream for several hours.
- Fludrocortisone suppression test. After you’ve followed a high-sodium diet and taken fludrocortisone — which mimics the action of aldosterone — for several days, aldosterone levels in your blood are measured.
Additional tests
If you receive a diagnosis of primary aldosteronism, your doctor will run additional tests to determine whether the underlying cause is an aldosterone-producing adenoma or overactivity of both adrenal glands. Tests may include:
- Abdominal computerized tomography (CT) scan. A CT scan can help identify a tumor on your adrenal gland or an enlargement that suggests overactivity. You may still need additional testing after a CT scan because this imaging test may miss small but important abnormalities or find tumors that don’t produce aldosterone.
- Adrenal vein sampling. This test involves placing a tube in a vein in your groin and threading it up to the adrenal veins. A radiologist then draws blood from both your right and left adrenal veins and compares the two samples. Aldosterone levels that are significantly higher on one side indicate the presence of an aldosteronoma on that side. Similar aldosterone levels on both sides point to overactivity in both glands. Though essential for determining the appropriate treatment, this test carries the risk of bleeding or a blood clot in the vein.
Primary hyperaldosteronism treatment
Treatment for primary aldosteronism depends on the underlying cause, but its basic goal is to normalize or block the effect of high aldosterone levels and prevent the potential complications of high blood pressure and low potassium levels.
Treatment for an adrenal gland tumor
An adrenal gland tumor may be treated with surgery or medications and lifestyle changes.
- Surgical removal of the gland. Surgical removal of the adrenal gland containing the tumor (adrenalectomy) is usually recommended because it may permanently resolve high blood pressure and potassium deficiency, and it can bring aldosterone levels back to normal. Blood pressure usually drops gradually after a unilateral adrenalectomy. Your doctor will follow you closely after surgery and progressively adjust or eliminate your high blood pressure medications. An adrenalectomy carries the usual risks of abdominal surgery, including bleeding and infection. However, adrenal hormone replacement is not necessary after a unilateral adrenalectomy because the other adrenal gland is able to produce adequate amounts of all the hormones on its own.
- Aldosterone-blocking drugs. If you’re unable to have surgery or prefer not to, primary aldosteronism caused by a benign tumor can also be treated with aldosterone-blocking drugs (mineralocorticoid receptor antagonists) and lifestyle changes. But high blood pressure and low potassium will return if you stop taking your medications.
Treatment for overactivity of both adrenal glands
A combination of medications and lifestyle modifications can effectively treat primary aldosteronism caused by overactivity of both adrenal glands (bilateral adrenal hyperplasia).
- Medications. Mineralocorticoid receptor antagonists block the action of aldosterone in your body. Your doctor may first prescribe spironolactone. This medication helps correct high blood pressure and low potassium, but it may cause problems. In addition to blocking aldosterone receptors, spironolactone blocks androgen and progesterone receptors and may inhibit the action of these hormones. Side effects can include male breast enlargement (gynecomastia), decreased sexual desire, impotence, menstrual irregularities and gastrointestinal distress. A newer, more expensive mineralocorticoid receptor antagonist called eplerenone acts just on aldosterone receptors, but eliminates the sex hormone side effects associated with spironolactone. Your doctor may recommend eplerenone if you have serious side effects with spironolactone. You may also need other medications for high blood pressure.
- Lifestyle changes. High blood pressure medications are more effective when combined with a healthy diet and lifestyle. Work with your doctor to create a plan to reduce the sodium in your diet and maintain a healthy body weight. Getting regular exercise, limiting the amount of alcohol you drink and stopping smoking also may improve your response to medications.
Limiting salt intake and taking medicine may control the symptoms without surgery.
A healthy lifestyle is essential for keeping blood pressure low and maintaining long-term heart health. Here are some healthy lifestyle suggestions:
- Follow a healthy diet. Limit the sodium in your diet by focusing on fresh foods and reduced-sodium products, avoiding condiments, and removing salt from recipes. Diets that also emphasize a healthy variety of foods — including grains, fruits, vegetables and low-fat dairy products — can promote weight loss and help lower blood pressure. Try the Dietary Approaches to Stop Hypertension (DASH) diet — it has proven benefits for your heart.
- Achieve a healthy weight. If your body mass index (BMI) is 25 or more, losing as few as 10 pounds (4.5 kilograms) may reduce your blood pressure.
- Exercise. Regular aerobic exercise can help lower blood pressure. You don’t have to hit the gym — taking vigorous walks most days of the week can significantly improve your health. Try walking with a friend at lunch instead of dining out.
- Don’t smoke. Quitting smoking will improve your overall cardiovascular health. Nicotine in tobacco makes your heart work harder by constricting your blood vessels and increasing your heart rate and blood pressure. Talk to your doctor about medications that can help you stop smoking.
- Limit alcohol and caffeine. Both substances can raise your blood pressure, and alcohol can interfere with the effectiveness of some blood pressure medications. Ask your doctor whether moderate alcohol consumption is safe for you.
Secondary hyperaldosteronism is treated with medicines (as described above) and limiting salt intake. Surgery is usually not used.
Primary hyperaldosteronism prognosis
The outlook for primary hyperaldosteronism is good with early diagnosis and treatment.
The outlook for secondary hyperaldosteronism depends on the cause of the condition.
- Fountain JH, Lappin SL. Physiology, Renin Angiotensin System. [Updated 2017 Dec 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470410/[↩][↩]
- Cognitive-enhancing effects of angiotensin IV. BMC Neurosci. 2008;9 Suppl 2(Suppl 2):S15. Published 2008 Dec 3. doi:10.1186/1471-2202-9-S2-S15 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2604899/[↩]
- Evidence that the angiotensin IV (AT(4[↩]
- The Brain Hepatocyte Growth Factor/c-Met Receptor System: A New Target for the Treatment of Alzheimer’s Disease. J Alzheimers Dis. 2015;45(4):985-1000. doi: 10.3233/JAD-142814. https://www.ncbi.nlm.nih.gov/pubmed/25649658[↩]
- Quinn SJ, Williams GH. Regulation of aldosterone secretion. Ann Rev Physiol 50:409-429, 1988.[↩][↩][↩]
- Nogueria EF, Olala LO, Bollag WB, et al. Regulation of aldosterone synthase by activator transcription factor/cAMP response element-binding protein family members. Endocrinology 151: 1060-1070, 2010.[↩]
- Kojima I, Kojima K, Rasmussen H. Role of calcium and cAMP in the action of adrenocorticotropin on aldosterone secretion. J Biol Chem 269:4248-4256, 1985.[↩]
- Woodcock EA, McLeod JK, Johnston CI. Vasopressin stimulates phosphatidylinositol turnover and aldosterone synthesis in rat adrenal glomerulosa cells: comparison with angiotensin II. Endocrinology 118:2432-2436, 1986.[↩]
- Hollenberg NK, Chenitz WR, Adams DF, et al. Reciprocal influence of salt intake on adrenal glomerulosa and vascular response to angiotensin II in normal man. J Cin Invest 54:34-42, 1974.[↩]
- Carey RM. Acute dopaminergic inhibition of aldosterone secretion is independent of angiotensin II and adrenocorticotropin. J Clin Endocrinol Metab 54:463-469, 1982.[↩]
- Missale C, Liberini P, Memo M, et al. Characterization of dopamine receptors associated with aldosterone secretion in rat adrenal glomerulosa. Endocrinology 119:2227-2232, 1986.[↩]
- Chartier L, Schiffrin EL. Role of calcium in effects of atrial natriuretic peptide on aldosterone production in adrenal glomerulosa cells. Am J Physiol 252:E485-E491, 1987.[↩]
- Jorgensen PL. Structure, function and regulation of Na, K-ATPase in the kidney. Kidney Intern 20:10-20, 1986.[↩]
- Nontraditional aspects of aldosterone physiology. Am J Physiol Endocrinol Metab 281: E1122–E1127, 2001. https://www.physiology.org/doi/pdf/10.1152/ajpendo.2001.281.6.E1122[↩][↩]
- Brilla CG, Pick R, Tan LB, Janicki JS, and Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 67: 1355–1364, 1990.[↩][↩]
- Brilla CG, Matsubara LS, and Weber KT. Antifibrotic effects of spironolactone in preventing myocardial fibrosis in systemic arterial hypertension. Am J Cardiol 71: 12A–16A, 1993.[↩]
- Schlaich MP, Schobel HP, Hilgers K, and Schmieder RE. Impact of aldosterone on left ventricular structure and function in young normotensive and mildly hypertensive subjects. Am J Cardiol 85: 1199–1206, 2000.[↩]
- Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, and Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. New Engl J Med 341: 709–717, 1999.[↩]
- Chapman D, Weber KT, and Eghbali M. Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circ Res 67: 787–794, 1990.[↩]
- Robert V, Silvestre JS, Charlemagne D, Sabri A, Trouve P, Wassef M, Swynghedauw B, and Delcayre C. Biological determinants of aldosterone-induced cardiac fibrosis in rats. Hypertension 26: 971–978, 1995.[↩]
- Fullerton MJ and Funder JW. Aldosterone and cardiac fibrosis: in vitro studies. Cardiovasc Res 28: 1863–1867, 1994.[↩][↩]
- Brown NJ, Kim KS, Chen YQ, Blevins LS, Nadeau JH, Meranze SG, and Vaughan DE. Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production. J Clin Endocrinol Metab 85: 336–344, 2000.[↩]
- Brown NJ, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M, Vaughan DE, and Fogo AB. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int 58: 1219–1227, 2000.[↩]
- Wang W, McClain JM, and Zucker IH. Aldosterone reduces baroreceptor discharge in the dog. Hypertension 19: 270–277, 1992.[↩][↩]
- Taddei S, Virdis A, Mattei P, and Salvetti A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 21: 929–933, 1993.[↩]
- Jazayeri A and Meyer WJ III. Mineralocorticoid-induced increase in beta-adrenergic receptors of cultured rat arterial smooth muscle cells. J Steroid Biochem 33: 987–991, 1989.[↩]
- Slight SH, Joseph J, Ganjam VK, and Weber KT. Extraadrenal mineralocorticoids and cardiovascular tissue. J Mol Cell Cardiol 31: 1175–1184, 1999.[↩]
- Hatakeyama H, Miyamori I, Fujita T, Takeda Y, Takeda R, and Yamamoto H. Vascular aldosterone. Biosynthesis and a link to angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem 269: 24316–24320, 1994.[↩]
- Van Belle E, Bauters C, Wernert N, Hamon M, McFadden EP, Racadot A, Dupuis B, Lablanche JM, and Bertrand ME. Neointimal thickening after balloon denudation is enhanced by aldosterone and inhibited by spironolactone, an aldosterone antagonist. Cardiovas Res 29: 27–32, 1995.[↩]
- Struthers AD. Aldosterone escape during ACE inhibitor therapy in chronic heart failure. Eur Heart J 16: 103–106, 1995.[↩]
- Rocha R, Chander PN, Khanna K, Zuckerman A, and Stier CT Jr. Mineralocorticoid blockade reduces vascular injury in stroke-prone hypertensive rats. Hypertension 31: 451–458, 1998.[↩]
- MacLeod AB, Vasdev S, and Smeda JS. The role of blood pressure and aldosterone in the production of hemorrhagic stroke in captopril-treated hypertensive rats. Stroke 28: 1821–1829, 1997.[↩]
- Dorrance AM, Grekin R, and Webb R. Aldosterone antagonism reduces cerebral infarct size in normotensive rats. J Hypertens 18: S84, 2000.[↩][↩]
- Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B, Mouas C, Moalic JM, Swynghedauw B, and Delcayre C. Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation. J Biol Chem 273: 4883–4891, 1998[↩][↩]
- Van Steensel B, van Binnendijk EP, Hornsby CD, van der Voort HT, Krozowski ZS, de Kloet ER, and van Driel R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J Cell Sci 109: 787–792, 1996.[↩]
- Reul JM and de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117: 2505–2511, 1985.[↩][↩]
- De Kloet ER, Van Acker SA, Sibug RM, Oitzl MS, Meijer OC, Rahmouni K, and de Jong W. Brain mineralocorticoid receptors and centrally regulated functions. Kidney Int 57: 1329–1336, 2000.[↩][↩]
- De Kloet ER, Vreugdenhil E, Oitzl MS, and Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev 19: 269–301, 1998.[↩]
- De Nicola AF, Tornello S, Weisenberg L, Fridman O, and Birmingham MK. Uptake and binding of [3H]aldosterone by the anterior pituitary and brain regions in adrenalectomized rats. Horm Metab Res 13: 103–106, 1981.[↩]
- Gomez-Sanchez EP, Venkataraman MT, Thwaites D, and Fort C. ICV infusion of corticosterone antagonizes ICV-aldosterone hypertension. Am J Physiol Endocrinol Metab 258: E649–E653, 1990.[↩][↩][↩]
- Gomez-Sanchez EP. Intracerebroventricular infusion of aldosterone induces hypertension in rats. Endocrinology 118: 819–823, 1986.[↩]
- Gomez-Sanchez EP, Fort CM, and Gomez-Sanchez CE. Intracerebroventricular infusion of RU28318 blocks aldosteronesalt hypertension. Am J Physiol Endocrinol Metab 258: E482–E484, 1990.[↩][↩]
- Peysner K, Henry CA, and Malvin RL. Central infusion of aldosterone increases blood pressure by mechanisms independent of Na retention. Clin Exp Hypertens 12: 399–414, 1990.[↩]
- Chen M, Lee J, and Malvin RL. Central administration of aldosterone increases blood pressure in rats. Clin Exp Hypertens 11: 459–472, 1989.[↩]
- Kageyama Y and Bravo EL. Hypertensive mechanisms associated with centrally administered aldosterone in dogs. Hypertension 11: 750–753, 1988.[↩]
- Gomez-Sanchez EP. Intracerebroventricular infusion of aldosterone induces hypertension in rats. Endocrinology 118: 819– 823, 1986.[↩]
- Janiak PC, Lewis SJ, and Brody MJ. Role of central mineralocorticoid binding sites in development of hypertension. Am J Physiol Regulatory Integrative Comp Physiol 259: R1025–R1034, 1990.[↩][↩]
- Gomez-Sanchez CE, Zhou MY, Cozza EN, Morita H, Foecking MF, and Gomez-Sanchez EP. Aldosterone biosynthesis in the rat brain. Endocrinology 138: 3369–3373, 1997.[↩]
- Arai K, Chrousos GP. Aldosterone Deficiency and Resistance. [Updated 2016 May 11]. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279079[↩]
- Landau RL, Lugibihl K. Inhibition of the sodium-retaining influence of aldosterone by progesterone. J Clin Endocrunol Metab 18:1237-1245, 1958.[↩]





{kind=link}