Contents
What is alkaptonuria
Alkaptonuria is a rare genetic metabolic disorder characterized by the accumulation of homogentisic acid in the body. Affected individuals lack enough functional levels of an enzyme required to breakdown homogentisic acid. Affected individuals may have dark urine or urine that turns black when exposed to air. However, this change may not occur for several hours after urination and often goes unnoticed. Aside from dark urine that is present from infancy, affected individuals generally do not develop symptoms (asymptomatic) during infancy or childhood and often remain unaware of their alkaptonuria until adulthood. Affected individuals eventually develop ochronosis, which is a buildup of dark pigment (bluish-black discoloration) in connective tissues such as cartilage and skin and other tissue within the body. Affected individuals may develop discoloration of the skin overlying cartilage within the body such as over part of the outer ear. This blue-black pigmentation usually appears after age 30. In some cases, the whites of the eyes (sclera) may also become discolored. Beginning in early adulthood, affected individuals also develop progressive arthritis of the spine and large joints. Other features of alkaptonuria can include heart problems, kidney stones, and prostate stones.
Alkaptonuria is rare, affecting 1 in 250,000 to 1 million people worldwide. Alkaptonuria is more common in certain areas of Slovakia (where it has an incidence of about 1 in 19,000 people) and in the Dominican Republic.
Alkaptonuria is caused by mutations in the HGD gene. The HGD gene codes for the enzyme required for the breakdown of homogentisic acid. Alkaptonuria is inherited in an autosomal recessive fashion 1.
Alkaptonuria does not cause developmental delays or cognitive impairment and does not appear to affect life span. However, chronic pain and mobility issues can develop.
There is still no cure for alkaptonuria. Treatment includes the management of joint pain, physical and occupational therapy, joint replacements and surgery when needed.
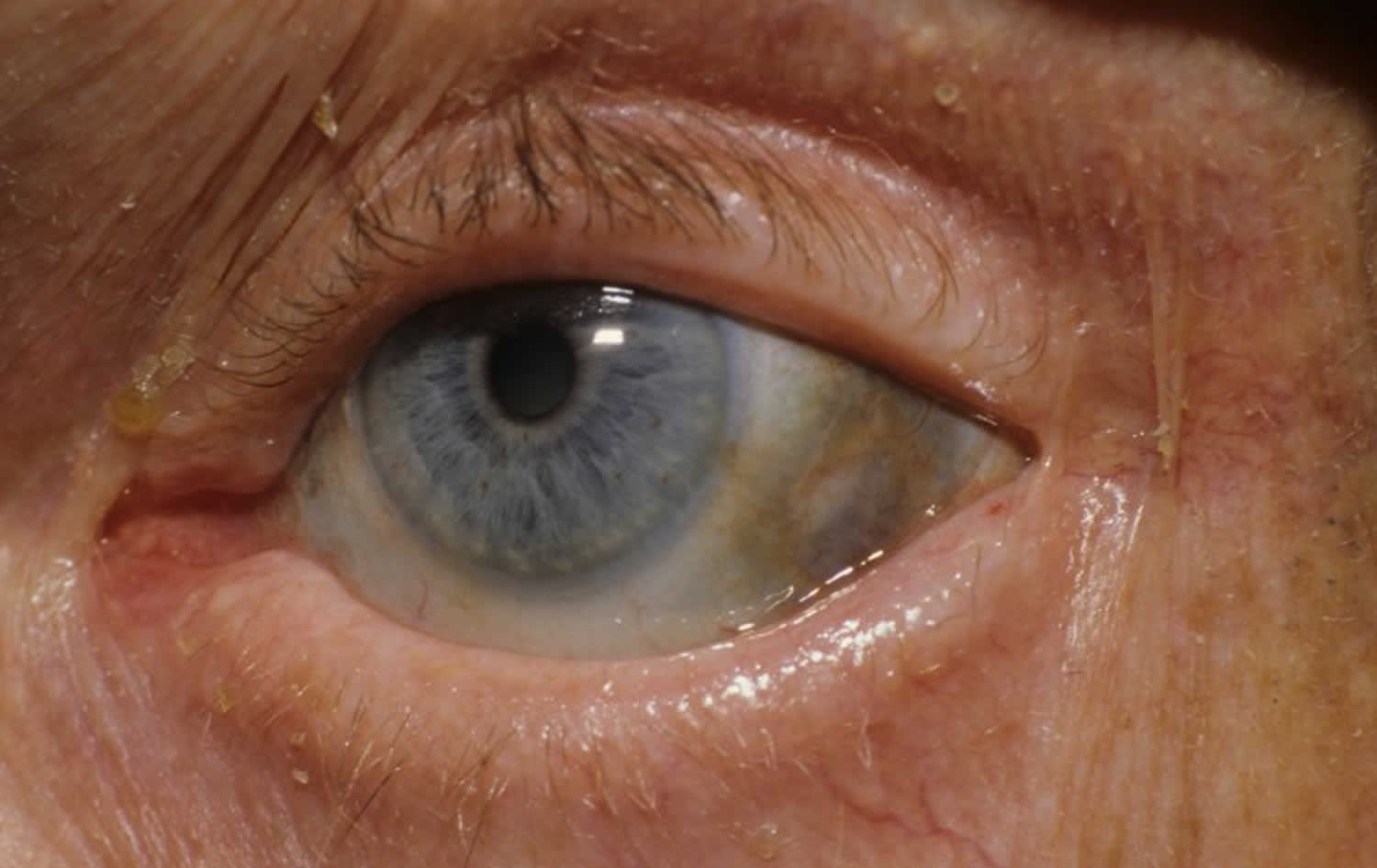
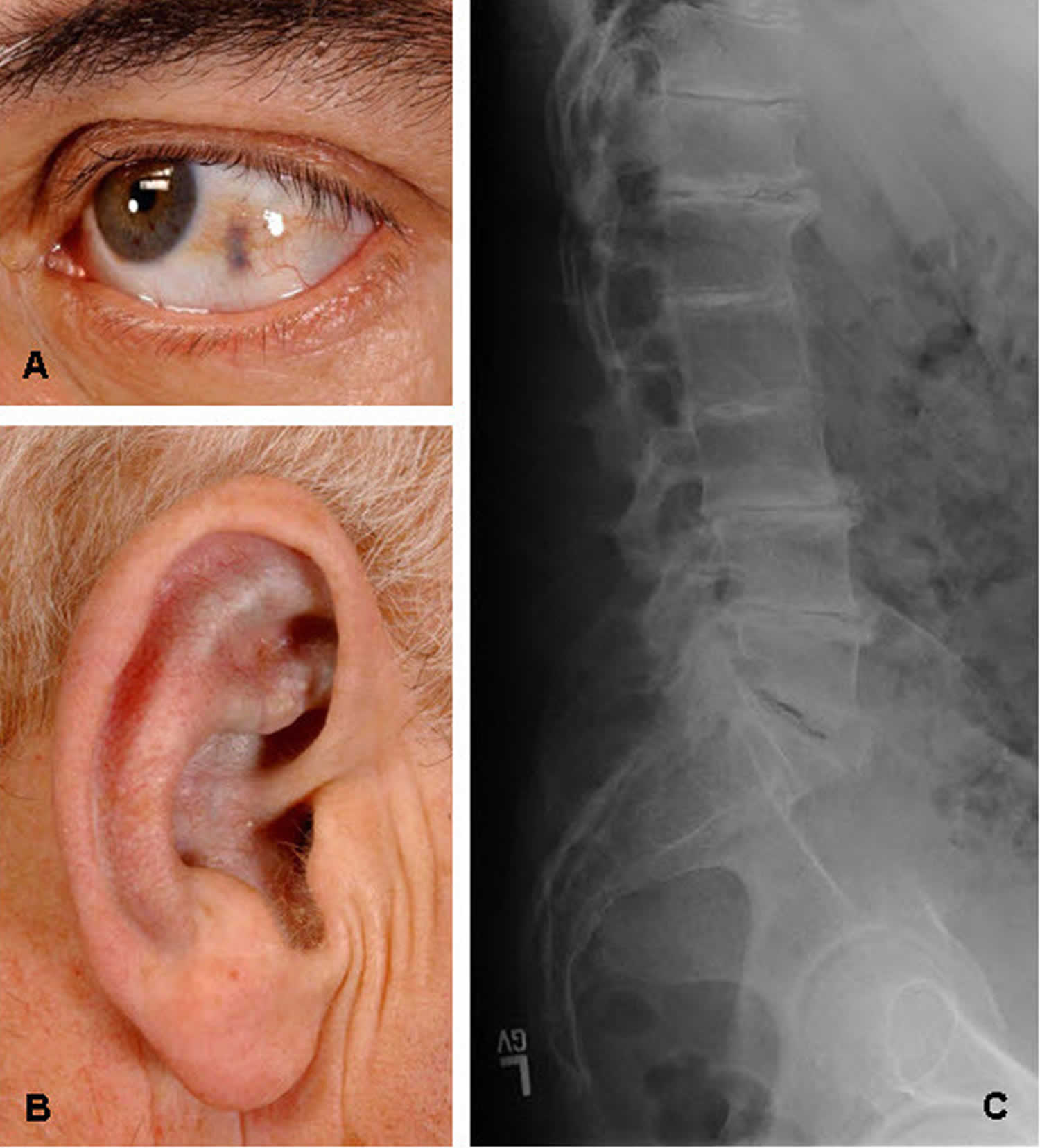
Figure 1. Ochronosis
Footnote:
A. Ochronosis of the sclera of the eye
B. Ochronosis of the antihelix and concha
C. Classic radiographic findings of the lumbar spine with disc flattening, calcification, and osteophyte formation
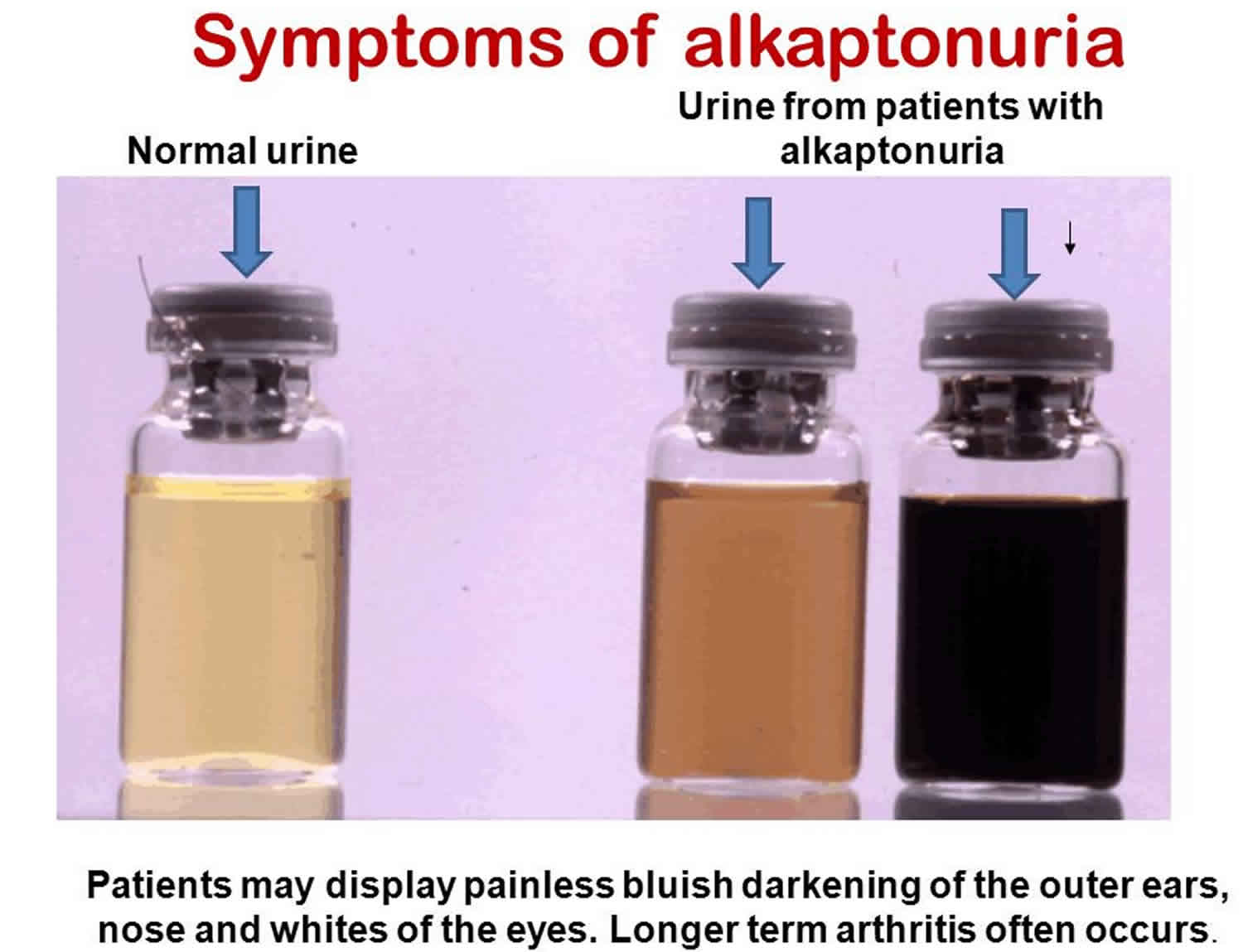
[Source 2 ]Figure 2. Alkaptonuria urine
Alkaptonuria causes
Mutations in the homogentisate 1,2-dioxygenase (HGD) gene cause alkaptonuria. The HGD (homogentisate 1,2-dioxygenase) gene provides instructions for making an enzyme called homogentisate 1,2-dioxygenase. This enzyme helps break down the amino acids phenylalanine and tyrosine, which are important building blocks of proteins. Mutations in the HGD gene impair the enzyme’s role in this process. As a result, a substance called homogentisic acid, which is produced as phenylalanine and tyrosine are broken down, accumulates in the body. Excess homogentisic acid and related compounds are deposited in connective tissues, which causes cartilage and skin to darken. Over time, a buildup of this substance in the joints leads to arthritis. Homogentisic acid is also excreted in urine, making the urine turn dark when exposed to air.
Alkaptonuria genetics
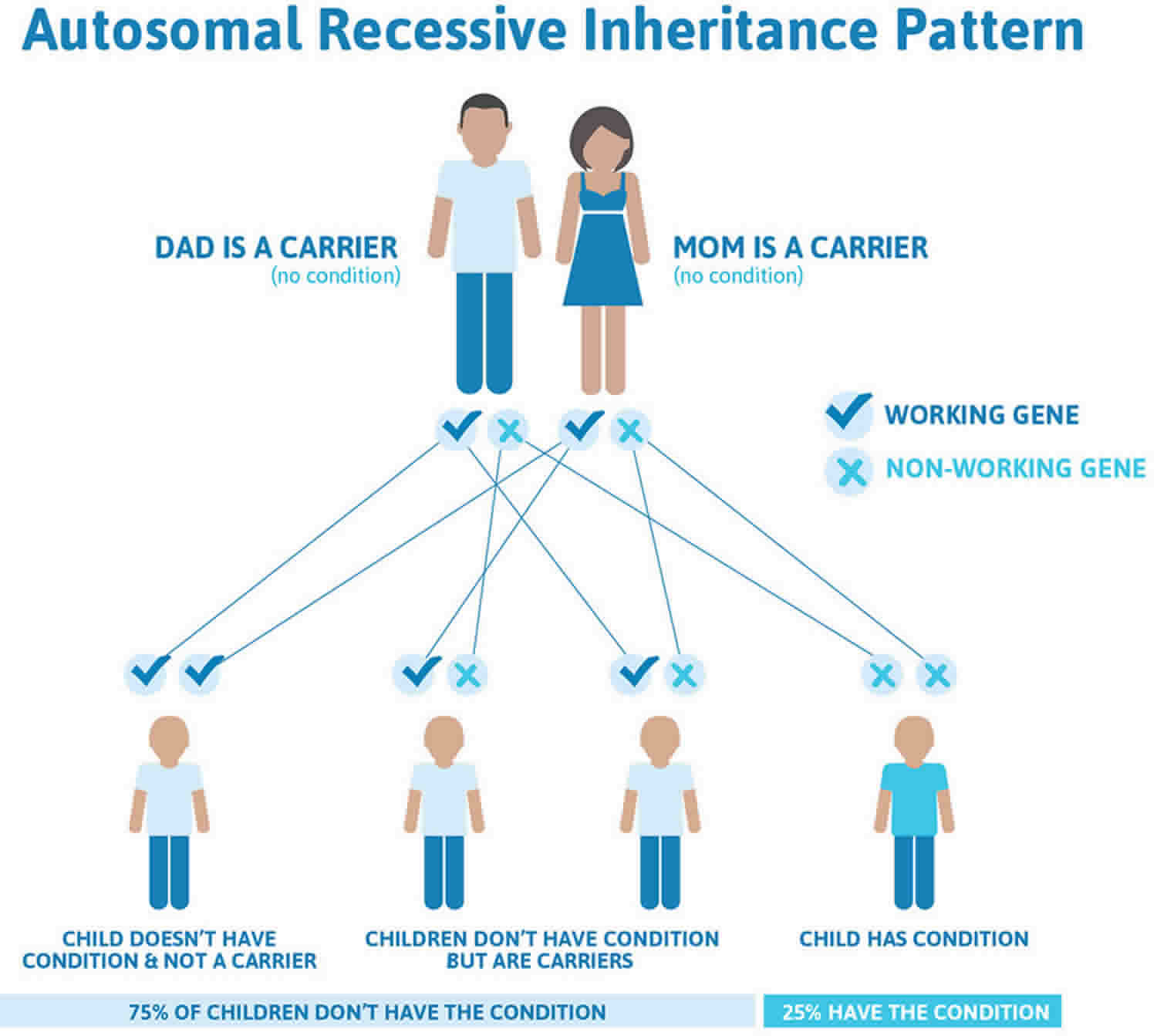
Alkaptonuria is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Key points to remember
- A person must inherit two copies of a abnormal gene, one from each parent, in order to be affected by the condition (25% chance). If a person inherits only one abnormal gene then they will be a carrier (50% chance). These outcomes occur randomly. They remain the same in every pregnancy and are the same for boys and girls.
- If both parents are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
- A abnormal gene cannot be corrected – it is present for life.
- A abnormal gene is not something that can be caught from other people. They can still be a blood donor, for example.
- People often feel guilty about a genetic condition which runs in the family. It is important to remember that it is no-one’s fault and no-one has done anything to cause it to happen.
Figure 3. Alkaptonuria autosomal recessive inheritance pattern
Alkaptonuria symptoms
Symptoms are generally slowly progressive. The urine of individuals with alkaptonuria may be abnormally dark or it may turn black upon long-standing exposure to the air. However, since this change often takes several hours, it often goes unnoticed. During infancy, diapers may be stained black (from urine exposure to air), although this is often missed or ignored.
The first noticeable signs and symptoms of alkaptonuria usually do not develop until approximately 30 years of age and are due to chronic accumulation of homogentisic acid in connective tissue, especially cartilage. Affected individuals develop a condition called ochronosis, in which connective tissue such as cartilage turns blue, grey or black due to the chronic accumulation of homogentisic acid. In many individuals, cartilage within the ear may become thickened, irregular and discolored blue, grey or black. Eventually, this discoloration may be apparent on the skin overlying the cartilage. In many cases, the whites of the eyes (sclera) also become discolored. However, this pigmentation does not interfere with vision.
In addition to cartilage, homogentisic acid accumulates in other connective tissue including tendons and ligaments and even bone. Over time, affected tissue becomes discolored, brittle and weak. Affected individuals may develop abnormalities affecting the tendons including thickened Achilles tendons and inflammation of the tendons (tendonitis). Affected tendons and ligaments may be particularly susceptible to rupturing. Eventually, discoloration of tendons may become visible on the overlying skin.
Long-standing alkaptonuria leads to chronic joint pain and inflammation (arthritis), especially in the spine and large joints (ochronotic arthropathy). Arthritis can be severe and disabling. Low back pain and stiffness are common symptoms and are sometimes seen before the age of 30. Discs between the vertebrae flatten and calcify. Eventually, vertebrae or other bones may fuse causing stiffening or immobility of affected joints (ankylosis). Spinal involvement may lead to abnormal outward curvature of the spine causing hunching of the back (kyphosis) and loss of height. The hip, knees and shoulders are commonly affected as well. Joint mobility is usually diminished and fluid buildup in affected joints (effusions) may also occur. Joint abnormalities are progressive and may eventually necessitate a joint replacement. Joint disease in alkaptonuria tends to begin earlier and progress more rapidly in males than females.
Less often, additional symptoms may occur in alkaptonuria. Although these symptoms occur less often than the main symptoms of alkaptonuria, they occur with greater frequency than would be expected in the general population. Such symptoms include kidney stones, which develop in 50 percent of affected individuals over 64 years of age. Men with alkaptonuria may also develop prostate stones. Passage of these black stones can be extremely painful.
In some individuals, heart disease may develop due to the accumulation of homogentisic acid within the aortic or mitral valves. This accumulation causes thickening of the valves and narrowing (stenosis) of the openings of the valves. Occasionally, the narrowing is severe enough that the aortic valve needs to be replaced. The aortic valve connects the lower left chamber (main pumping chamber) of the heart with the aorta (the main artery of the body). The mitral valve is located between the left upper and left lower chambers of the heart. Affected individuals may develop calcification of the valves and/or backflow of blood back through the affected valves (regurgitation), which can lead to reduced blood flow throughout the body. Widening (dilation) of the aorta may also occur. In some cases, calcification of the small blood vessels that supply blood and oxygen to the heart (coronary blood vessels) may also occur.
Alkaptonuria diagnosis
Alkaptonuria should be suspected in individuals with dark urine. However, since some individuals with alkaptonuria do not have dark urine, it may be advisable to rule out the disorder for all individuals with osteoarthritis, especially those with an early onset of symptoms. The diagnosis of alkaptonuria is based on the detection of a significant amount of homogentisic acid (HGA) in the urine by gas chromatography-mass spectrometry analysis. A normal 24-hour urine sample contains 20-30 mg of homogentisic acid (HGA). The amount of homogentisic acid (HGA) excreted per day in individuals with alkaptonuria is usually between one and eight grams (1000 to 8000 mg). Identification of biallelic pathogenic variants in HGD on molecular genetic testing confirms the diagnosis and allows family studies. However, this testing is not required to confirm the diagnosis.
In individuals over 40, echocardiography may be recommended to detect potential cardiac complications such as aortic dilation or calcification or regurgitation of the aortic or mitral valves. With echocardiography, sound waves are bounced off the heart (echoes), enabling physicians to study cardiac function and motion.
Computed tomography (CT) scan may be recommended to detect coronary artery calcification.
Alkaptonuria treatment
There is no cure for alkaptonuria, but there is treatment for some individual signs and symptoms of the condition. Joint pain may be substantial in individuals with alkaptonuria, and close attention to pain control is usually necessary. Individuals with alkaptonuria often receive anti-inflammatory medications to treat joint pain. In severe cases, stronger medications such as narcotics may be recommended. Pain management is tailored to each individual’s specific case and requires long-term follow up and adjustment.
Physical and occupational therapy can be important to promote muscle strength and flexibility. Knee, hip, and shoulder replacement surgeries may be options for managing significant arthritis, often by 50-60 years of age. In general, however, the goal of joint replacement is pain relief rather than increased range of motion. Maintaining joint range of motion through moderate non-weight-bearing exercise such as swimming may have beneficial effects. Infrequently, individuals require spinal surgery, including fusion and/or removal of the lumber discs. Surgery to replace the aortic or mitral valves may also be necessary. In some cases, chronic and painful kidney or prostate stones may require surgical intervention or preventive (prophylactic) therapy 2.
No therapy has proven to prevent or correct the pigmentary changes of ochronosis 2.
Dietary restriction of phenylalanine and tyrosine has been proposed to reduce the production of homogentisic acid, but severe restriction of these amino acids is not practical in the long term and may be dangerous 2. In addition, long-term, severe restriction of protein intake can be associated with complications.
In older children and adults, high-doses of vitamin C have also been used to treat alkaptonuria because it hinders the accumulation and deposition of homogentisic acid. However, long-term use of vitamin C has generally proven ineffective and definite clinical studies on its efficacy are lacking.
Activities that place significant physical stress to the spine and joints such as high impact sports or heavy manual labor should be avoided.
- Alkaptonuria. https://ghr.nlm.nih.gov/condition/alkaptonuria[↩]
- Introne WJ, Gahl WA. Alkaptonuria. 2003 May 9 [Updated 2016 May 12]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1454[↩][↩][↩][↩]




{kind=link}