Contents
Andersen Tawil syndrome
Andersen-Tawil syndrome also called Andersen syndrome, long QT syndrome 7 or LQTS7, is a rare genetic disorder that causes episodes of muscle weakness (periodic paralysis), abnormalities affecting the electrical system of the heart that can cause abnormal life-threatening heart rhythms (arrhythmias), and a variety of distinctive facial and skeletal features, such as short stature, clinodactyly (an inward curvature of the 5th fingers), short index fingers, fused or webbed second and third toes (syndactyly), scoliosis (crooked spine), widely spaced eyes (ocular hypertelorism), low-set ears, broad nose, a broad forehead, and a small jaw (micrognathia) 1, 2, 3, 4, 5, 6. The specific signs and severity can vary greatly from one person to another, even among members of the same family or they may exist in other family members who do not experience muscle weakness or paralysis. Some individuals will not develop all of the facial and skeletal features. Distinctive facial features may be so mild as to go unnoticed 7. Periodic paralysis begins early in life, and episodes last from hours to days. These episodes may occur after exercise or long periods of rest, but they often have no obvious trigger. Muscle strength usually returns to normal between episodes. However, mild muscle weakness may eventually become permanent.
The relationship between Andersen-Tawil syndrome and potassium is inconsistent, and it varies between patients 8. It’s also possible for one patient to have different experiences at different times. Some attacks are caused by a rise in blood potassium levels (hyperkalemia) or fall in blood potassium levels (hypokalemia) but can also happen with normal blood potassium levels (normokalemia). The levels of rise or fall in potassium levels in Andersen Tawil syndrome are not dramatic as in other forms of periodic paralysis and so treatment does not usually include lowering or raising the blood levels of potassium.
Andersen-Tawil syndrome is sometimes referred to as long QT syndrome 7 because some individuals in early reports of the disorder had a prolonged QT interval, which is measured on an electrocardiogram (ECG) and indicates that the heart muscle is taking longer than usual to recharge between beats. Long QT means that there is a longer than normal period of time between the start of the Q wave and the end of the T wave as seen on an ECG. The prolongation of this period tends to trigger irregular heart rhythms. In people with Andersen-Tawil syndrome, the most common changes affecting the heart are ventricular arrhythmia, which is a disruption in the rhythm of the heart’s lower chambers (the ventricles), and long QT syndrome. However, subsequent clinical reports have shown the QT interval is not prolonged or only mildly prolonged in most cases. Instead, the Q-U interval is markedly prolonged. In addition, unlike most forms of long QT syndrome, Andersen-Tawil syndrome is associated with symptoms in addition to disturbances of the electrical system of the heart. Although still sub-classified as a form of long QT syndrome, Andersen-Tawil syndrome is recognized as separate from traditional long QT syndromes. Long QT syndrome is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. The irregular heartbeats can lead to discomfort, such as the feeling that the heart is skipping beats (palpitations). Uncommonly, the irregular heartbeats can cause fainting (syncope), and even more rarely, sudden death due to cardiac arrest 9.
Physical abnormalities associated with Andersen-Tawil syndrome typically affect the face, other parts of the head, and the limbs. These features often include a very small lower jaw (micrognathia), dental abnormalities (such as crowded teeth), low-set ears, widely spaced eyes, fusion (syndactyly) of the second and third toes, and unusual curving of the fingers or toes (clinodactyly). Some affected people also have short stature and an abnormal side-to-side curvature of the spine (scoliosis).
The signs and symptoms of Andersen-Tawil syndrome vary widely, and they can be different even among affected members of the same family. About 60 percent of affected individuals have all three major features (periodic paralysis, cardiac arrhythmia, and physical abnormalities) and up to 80% of the cases express two of the three major features 10.
About 60% of cases of Andersen-Tawil syndrome are caused by mutations in the KCNJ2 gene, a gene encoding the inward rectifier potassium channel 2 protein (Kir2.1) 11, 12. The cause of the remaining 40% of Andersen Tawil syndrome cases remains unknown. Andersen-Tawil syndrome is inherited in an autosomal dominant pattern 13.
The terms Andersen-Tawil syndrome type 1 or type 2 are also used in the medical literature. Andersen-Tawil syndrome type 1 (ATS1) refers to cases caused by a known KCNJ2 gene mutation; Andersen-Tawil syndrome type 2 (ATS2) refers to cases without an identified KCNJ2 mutation 14.
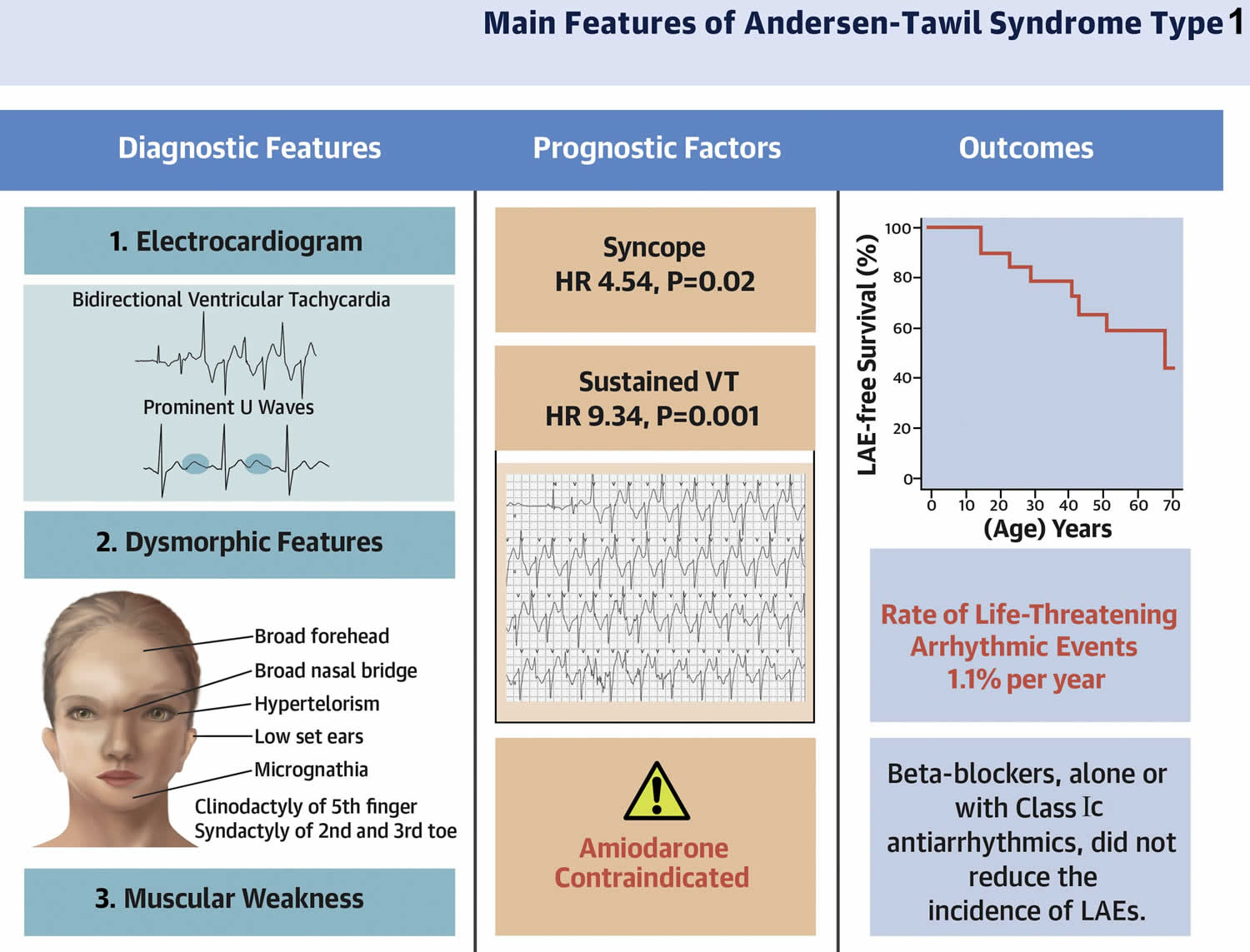
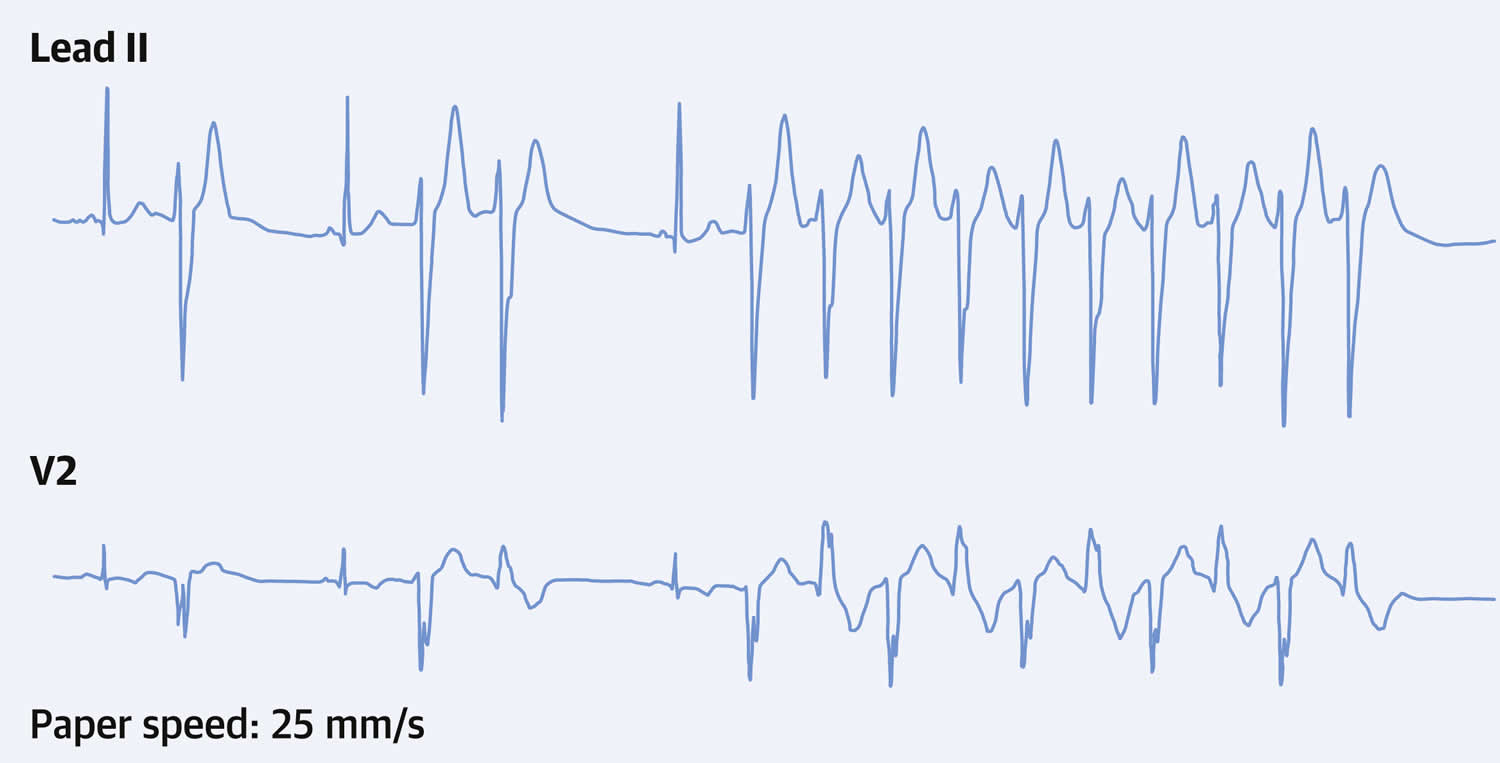
In a study involving 57 Andersen-Tawil syndrome affected individuals (82% female; age at first visit 20 ± 15 years, range 1.5 years to 51 years) and their 61 affected relatives (54% female; age 24 ± 21 years, range 1 month to 69 years) 15. Overall, 97% of patients with Andersen-Tawil Syndrome type 1 (ATS1) exhibited heart symptoms (more frequently a large U-wave and ventricular bigeminy), and 75% of patients presented dysmorphic features (more often a small mandible) (see Figure 1) 15. A total of 77 patients with Andersen-Tawil Syndrome type 1 (ATS1) had the documentation of ventricular tachycardia (VT) and/or nonsustained VT during their clinical evaluation, and in 58 (75%) of 77 of them episodes of bidirectional VT (BidVT). In most cases, the episodes of bidirectional VT (BidVT) were observed as brief “interludes” that interrupted prolonged episodes of polymorphic VT (Figure 3). A family history of sudden death was present in 11 (19%) of 57 kindreds, with a total of 19 individuals (68% female), who died suddenly at 29 ± 18 years of age 15. All sudden death cases occurred in families in which the KCNJ2 gene mutation segregated with the Andersen-Tawil syndrome type 1 (ATS1) phenotype 15. The highest incidence of life-threatening arrhythmic events in that population occurred between the third and the fifth decades 15. Four cases, the arrhythmic events occurred within the first decade of life, suggesting that Andersen-Tawil syndrome may manifest with life-threatening arrhythmic event during childhood 15.
Andersen-Tawil syndrome exact prevalence is unknown, although it is estimated to affect 1 in 1 million people worldwide 6, 5. About 200 affected individuals have been described in the medical literature. Researchers believe that Andersen-Tawil syndrome accounts for less than 10 percent of all cases of periodic paralysis. Consequently information on the clinical course derive from case series that are either small 16 or devoid of outcome data 17.
Andersen-Tawil syndrome needs to be distinguished from other forms of periodic paralysis including hypokalemic periodic paralysis (HypoPP), hyperkalemic period paralysis (HyperPP), and thyrotoxic periodic paralysis (TPP). Other conditions that cause a prolonged QT interval or syncope should be ruled out as well including vasovagal syncope, orthostatic hypotension, hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and catecholamingergic polymorphic ventricular tachycardia (CPVT).
Treatment for Andersen Tawil syndrome is complicated because different patients have different symptoms, and what helps one patient might hurt another 8. For this reason, it is crucial that doctors come up with an individualized treatment plan for each patient. It is also essential that the patient is followed by a neurologist experienced with managing patients with periodic paralysis as well as a cardiologist for regular follow-up to determine if and when they may need to be on heart medications to prevent irregularities in the heart rhythm 8. Medications which regulate the heart are often prescribed, other treatment depends on the individual and their reaction to potassium. Patients with severe heart rhythm problems may require a pacemaker-like device.
Beta-blockers such as propranolol, anti-arrhythmics such as flecainide, and calcium-channel blockers such as verapamil are possible options for heart rhythm problems, but some of these heart medications could make muscle weakness worse, so caution is needed. Also, people with Andersen Tawil syndrome should avoid medications that can make their heart’s long QT interval even longer. See CredibleMeds (https://crediblemeds.org) for a complete and updated list (free registration required). Salbutamol inhalers, which may be used in the treatment of primary hyperkalemic periodic paralysis, should be avoided because of the potential for exacerbation of cardiac arrhythmias. Thiazide and other potassium-wasting diuretics may provoke drug-induced hypokalemia and could aggravate the QT interval prolongation.
Carbonic anhydrase inhibitors drug such as acetazolamide is likely to help with muscle weakness attacks, as well as dichlorphenamide (brand name KEVEYIS, the only FDA-approved periodic paralysis treatment). Taking potassium supplements can help patients whose attacks are triggered by not having enough potassium in their blood. On the other hand, patients whose attacks are triggered by high potassium (hyperkalemia) may be able to shorten the length of attacks by having a high-carbohydrate food or beverage (sugary foods and drinks) during an attack.
Lifestyle changes are important, too. Andersen Tawil syndrome patients should try to avoid being in cold environments, and they should also be careful when exercising so that they are not overexerting themselves. Stress management also plays a big role in reducing the chances of an attack. It’s helpful to have healthy ways to cope with stressful situations. Changes in diet may be necessary, because certain foods may trigger attacks.
Figure 1. Andersen Tawil syndrome
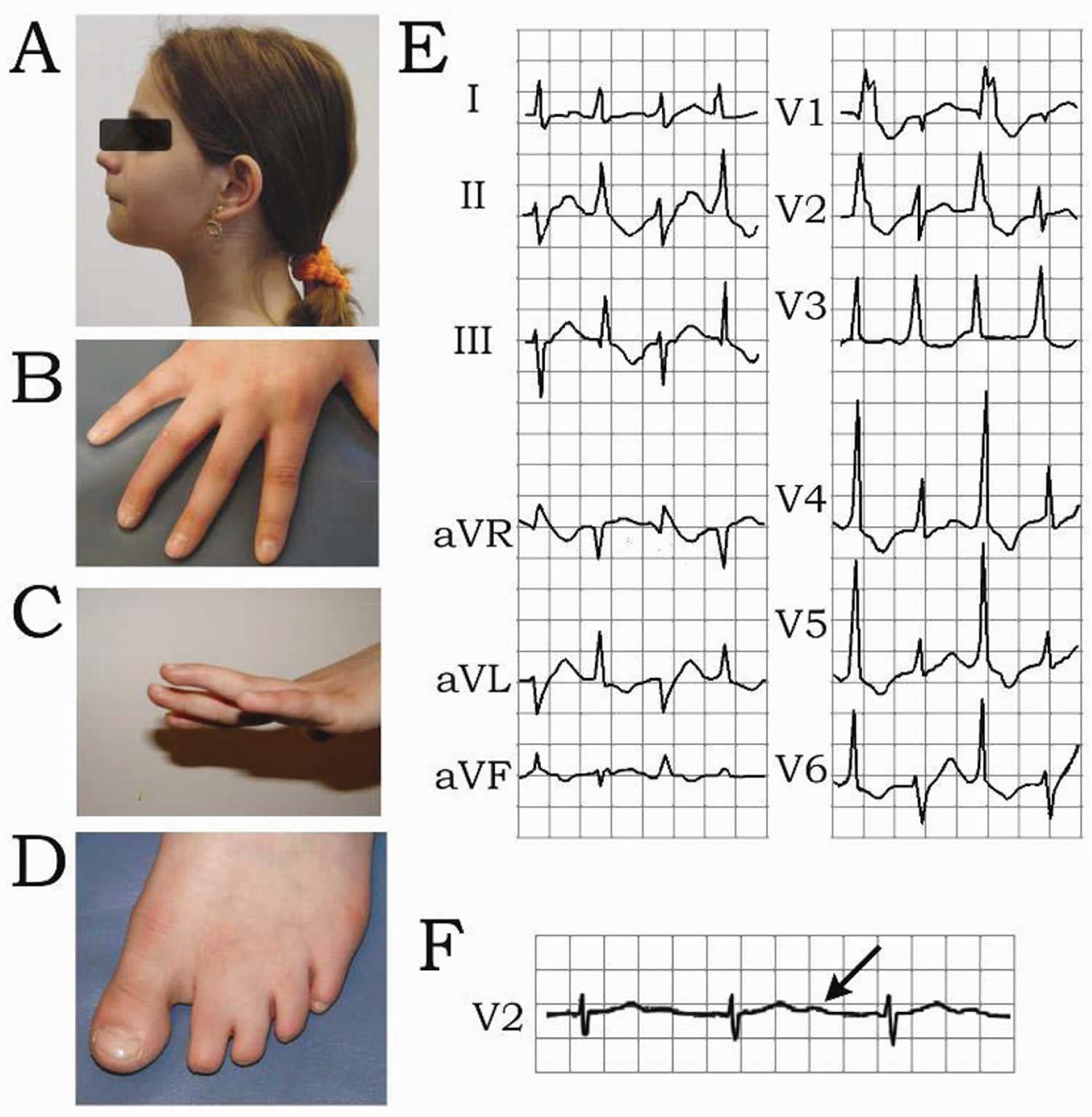
Footnote: Andersen-Tawil syndrome is characterized by dysmorphic features and cardiac arrhythmias. Patient exhibited micrognathia (small chin), retrognathia and hypertelorism (A), clinodactyly (B and C) and syndactyly (D). Electrocardiogram rhythm strip shows bidirectional ventricular tachycardia (BiVT) (E) and prolonged U wave (QUc=626 ms) (F).
[Source 18 ]Figure 2. Andersen Tawil syndrome symptoms
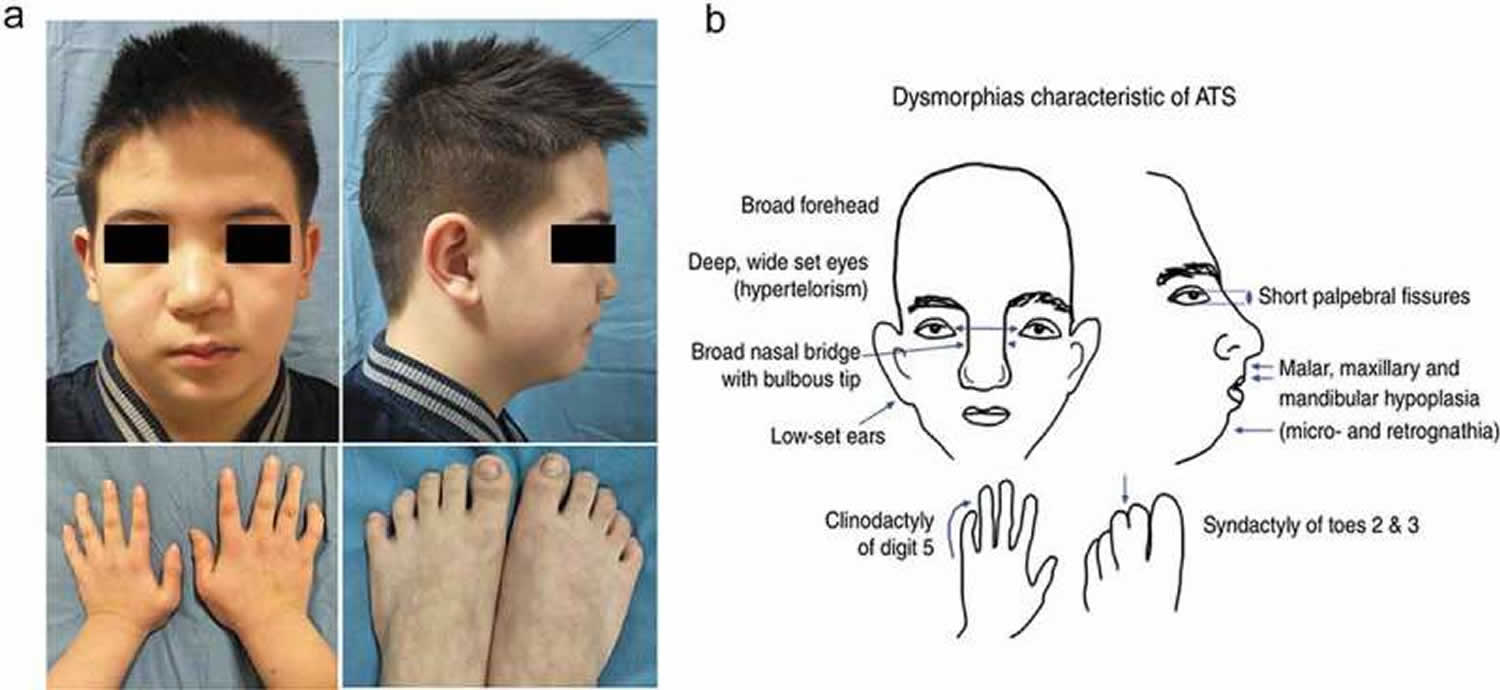
Footnotes: (a) Widely spaced eyes, small mandible, low-set ears, and fifth-digit clinodactyly. (b) Craniofacial anomalies characteristic of Andersen‐Tawil syndrome, one of the three features used for diagnosis of Andersen‐Tawil syndrome, craniofacial anomalies are seen in a majority of patients. These features are variably penetrant, however, with different patients showing different degrees of severity and different subsets of those shown 19, 20.
Abbreviation: LAEs = life-threatening arrhythmic events
[Sources 15, 21 ]Figure 3. Bidirectional ventricular tachycardia in Andersen Tawil syndrome
Will I pass Andersen Tawil syndrome on to my children?
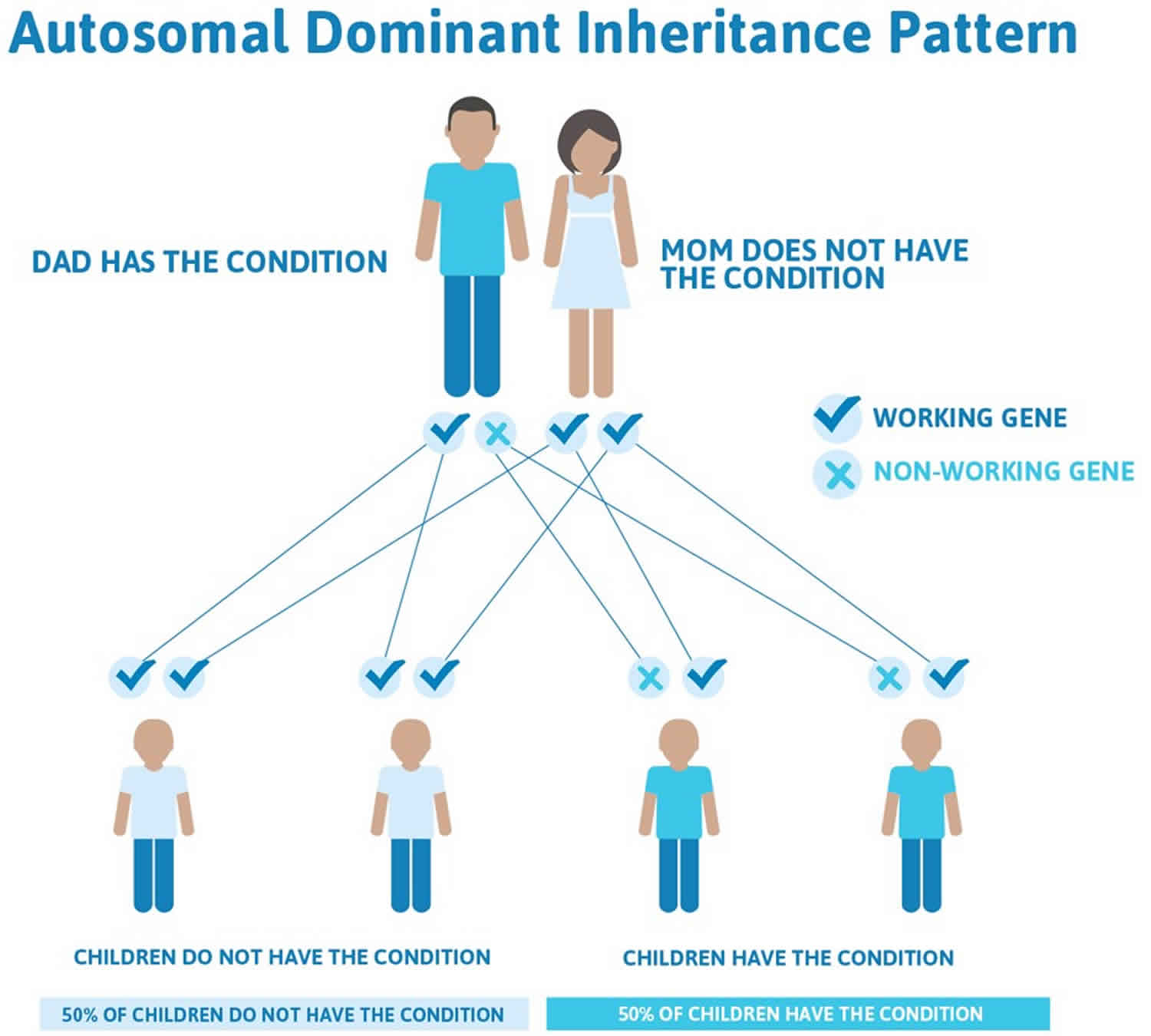
Andersen Tawil syndrome is inherited in an autosomal dominant pattern (see Figure 5 below). The chances that a child of an affected person will inherit the defective KCNJ2 gene is ‘statistically’ 50%, but not all who inherit the gene will have symptoms. The degree to which children are affected may vary from one child to the next. One child may be mildly affected, the next seriously affected and the next completely unaffected.
However, in approximately 50% of cases, Andersen Tawil syndrome results from a new (de novo) mutation in the KCNJ2 gene and not inherited from either parent, which means that in those specific cases the gene mutation has occurred at the time of the formation of the egg or sperm for that child only and no other family member will be affected. These cases occur in people with no history of Andersen Tawil syndrome in their family and the disorder is usually not inherited from or “carried” by a healthy parent. Speak with a doctor or genetic counselor if you or someone in your family has been diagnosed with Andersen Tawil syndrome.
Andersen Tawil syndrome causes
Mutations in the potassium inwardly rectifying channel subfamily J member 2 gene or KCNJ2 gene cause about 60 percent of all cases of Andersen-Tawil syndrome and more than 60 mutations in the KCNJ2 gene have been found to cause Andersen-Tawil syndrome 22. When the disorder is caused by mutations in KCNJ2 gene, it is classified as Andersen-Tawil syndrome type 1 (ATS1). In the other 40% of Andersen Tawil syndrome cases (Andersen-Tawil syndrome type 2), the underlying genetic mutation is unknown, suggesting that additional as-yet-unidentified genes also cause the disorder.
The KCNJ2 gene provides instructions for making the inward rectifier potassium (Kir2.1) channels that transport positively charged potassium ions across the membrane of skeletal muscle and cardiac muscle cells (Figure 4) 23, 24, 25, 26. Most of the KCNJ2 gene mutations change a single protein building block (amino acid) in the KCNJ2 protein. The movement of potassium ions through these channels is critical for maintaining the normal function of muscles used for movement (skeletal muscles) and cardiac muscle. Mutations in the KCNJ2 gene alter the usual structure and function of these potassium channels. These changes disrupt the flow of potassium ions in skeletal and cardiac muscle, leading to the periodic paralysis and irregular heart rhythm characteristic of Andersen-Tawil syndrome.
Many KCNJ2 mutations prevent phosphatidylinositol 4,5-bisphosphate (PIP2) from effectively binding to and activating potassium channels 1, 2. If the KCNJ2 protein is unable to bind to phosphatidylinositol 4,5-bisphosphate (PIP2), the channels remain closed and potassium ions are unable to flow across the cell membrane. Researchers believe that problems with PIP2 binding are a major cause of Andersen-Tawil syndrome.
A loss of inward rectifier potassium (Kir2.1) channel’s function in skeletal and cardiac muscle cells disrupts the normal flow of potassium ions out of these cells, resulting in periodic paralysis and an irregular heart rhythm. It is not known how mutations in the KCNJ2 gene contribute to the skeletal changes and other physical abnormalities often found in Andersen-Tawil syndrome 22.
In the 40 percent of cases not caused by KCNJ2 gene mutations, the cause of Andersen-Tawil syndrome is usually unknown. These cases are classified as Andersen-Tawil syndrome type 2 (ATS2). Studies suggest that variations in at least one other potassium channel gene may underlie the disorder in some of these affected individuals.
Figure 4. Structure of Kir2.1 Channel in Andersen Tawil Syndrome type 1 (ATS1)
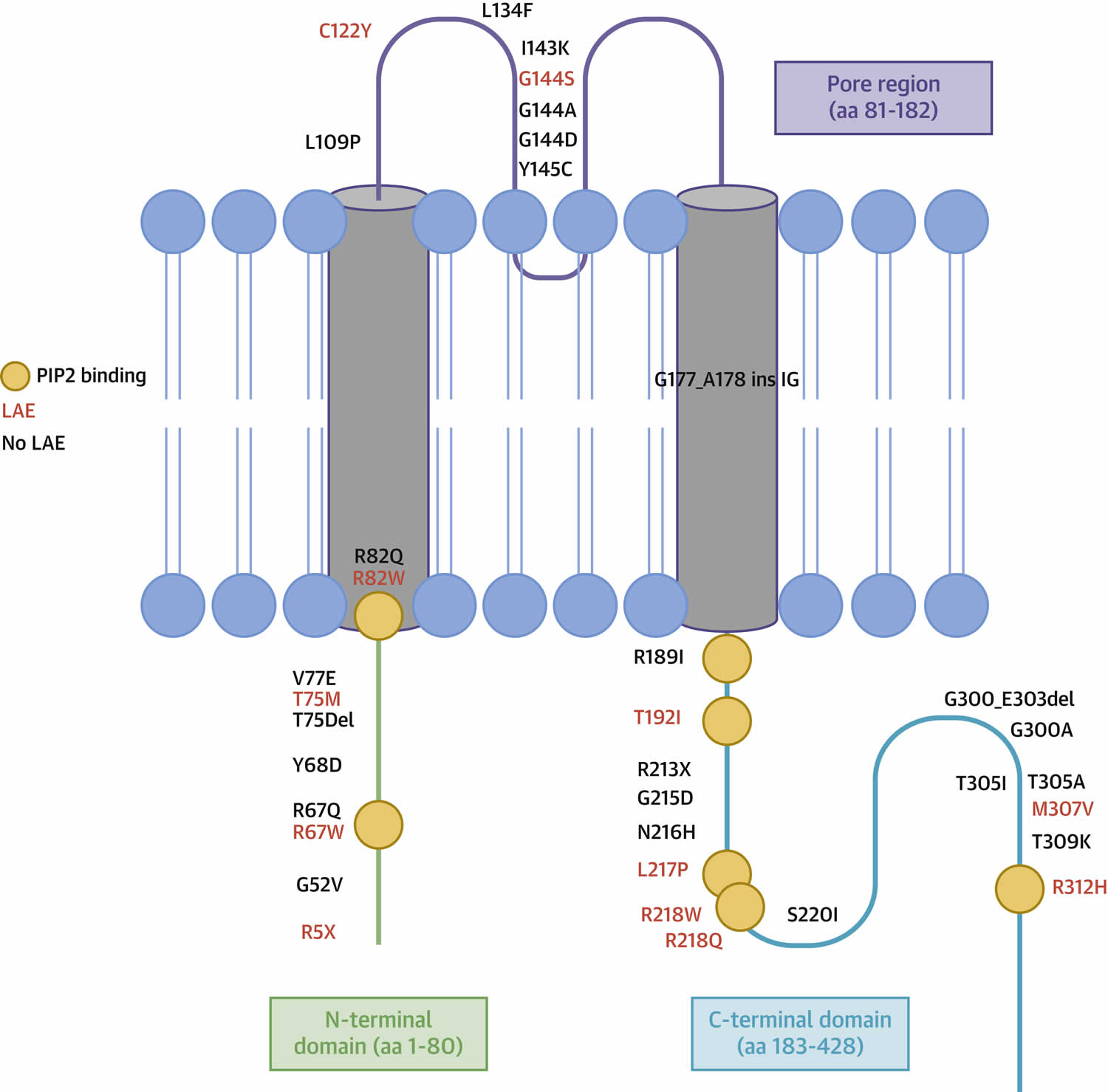
Footnote: Amino acid location of the 35 different KCNJ2 mutations. N-terminal domain is highlighted in green, transmembrane and pore domains in purple, and C-terminal domain in light blue. Mutations associated with life-threatening arrhythmic events (LAEs) are reported in red. Orange circles indicate phosphatidylinositol (4, 5)-bisphosphate (PIP2) binding residues. Blue circles and tails represent the phospholipid bilayer of the cell membrane.
Abbreviation: LAE = life-threatening arrhythmic event
[Source 15 ]Andersen Tawil syndrome triggers
Triggers may vary from person to person. For example food triggers for Andersen Tawil syndrome are not as easily recognized as they are in other types of periodic paralysis, where it is possible to say potassium-rich foods or carbohydrate-rich foods trigger episodes. Attacks can be triggered by high blood potassium levels (hyperkalemia) or low blood potassium levels (hypokalemia), and during attacks, potassium may rise, fall, or remain normal (normokalemia). Since the person’s potassium level may vary from episode to episode it may take real attention to one’s body’s signals to recognize food triggers. In some Andersen Tawil syndrome patients food does not seem to be a trigger at all. Because Andersen Tawil syndrome causes attacks that vary in severity and triggers, it is recommended that patients keep a diary to help them determine what triggers their attacks of weakness. Here’s how: After you have an attack, you should write down what happened, how you felt, what you were doing before the attack, and how you were able to recover from it. After doing this for a while, you’ll be able to look back on your previous diary entries and possibly notice certain patterns. Even if there are no clear patterns, the diary will be useful to discuss with your doctor.
Common triggers are rest after activity and periods of inactivity (for example; sitting through a long class or church service, especially in a cool room). Sleep is a potent trigger. Going too long without eating is another trigger, as is eating a large meal. Getting chilled is a trigger. Emotional events may trigger attacks. Gasoline or paint fumes and car exhaust have been reported as triggers by some patients, and are probably best avoided when possible.
Andersen Tawil syndrome inheritance pattern
Andersen Tawil syndrome is inherited (passed through families) in an autosomal dominant pattern, which means one copy of an altered KCNJ2 gene in each cell is sufficient to cause the condition (see Figure 5). When Andersen Tawil syndrome results from a mutation in the KCNJ2 gene, an affected individual may inherit the mutation from one affected parent. If an individual has Andersen Tawil syndrome, there is a one in two (50%) chance that they will pass it on to each of their children. However, in approximately 50% of cases, Andersen Tawil syndrome results from a new (de novo) mutation in the KCNJ2 gene and not inherited from either parent, which means that in those specific cases the gene mutation has occurred at the time of the formation of the egg or sperm for that child only and no other family member will be affected. These cases occur in people with no history of Andersen Tawil syndrome in their family and the disorder is usually not inherited from or “carried” by a healthy parent. Speak with a doctor or genetic counselor if you or someone in your family has been diagnosed with Andersen Tawil syndrome.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 5 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 5. Andersen Tawil syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Andersen Tawil syndrome symptoms
Andersen-Tawil syndrome is defined by three main features (i.e. a clinical triad), specifically:
- Episodic flaccid muscle weakness (periodic paralysis): Intermittent weakness occurs spontaneously, or alternatively may be triggered by prolonged rest or rest following exertion. The attack frequency, duration, and severity are variable between and within affected individuals. Mild permanent weakness is common 27. Affected individuals can develop fixed proximal weakness over time.
- Arrhythmias and heart abnormalities (ventricular arrhythmias, prolonged QTc or QUc intervals, and prominent U waves):
- Ventricular arrhythmias including bidirectional ventricular tachycardia (VT), polymorphic VT, and multifocal premature ventricular contractions may be asymptomatic, or may manifest (most commonly) as palpitations. Less common symptomatic presentations include syncope, cardiac arrest, or sudden death 28. While the ECG may reveal a long QTc (LQT) interval, characteristic T-U patterns including enlarged U waves, a wide T-U junction, and prolonged terminal T-wave downslope distinguish Andersen Tawil syndrome from other LQT syndromes 29. A large case series found no significant difference in the incidence of ventricular tachyarrhythmias between individuals with typical and atypical presentations of Andersen Tawil syndrome 30. A retrospective multicenter French study of 36 individuals with Andersen Tawil syndrome followed for an average of 9.5 years reported no deaths in follow up; four individuals experienced syncope and one individual had a non-fatal cardiac arrest 31.
- Dilated cardiomyopathy was observed in two of three affected individuals in a single kindred with pathogenic variant p.Arg218Trp 32. Additionally, cardiomyopathy was documented and reversed by treatment with flecainide in an individual with pathogenic variant p.Arg67Trp 33 and reversed by bisoprolol in another individual with typical clinical features of Andersen Tawil syndrome and novel variant p.Leu222Ser 34. These reports suggest that dilated cardiomyopathy is a secondary phenotype as a consequence of chronic tachycardia rather than a primary phenotypic manifestation. Additional study is needed to further delineate this clinical feature.
- Distinctive physical features. Distinctive physical features recognized initially included low-set ears, widely spaced eyes, small mandible, fifth-digit clinodactyly, second and third toe syndactyly, short stature, broad nasal root, and scoliosis 28. Dental enamel discoloration was noted in two kindreds with the p.Gly300Asp and p.Arg218Trp pathogenic variants 35. Detailed, prospectively collected data in ten individuals with confirmed KCNJ2 pathogenic variants have expanded the phenotype to include a characteristic facies and dental and skeletal anomalies 36. Characteristic facies include broad forehead, short palpebral fissures, wide nasal bridge with bulbous nose, hypoplasia of maxilla and mandible, thin upper lip, and a triangular face.
- Dental findings include (among others) persistent primary dentition, multiple missing teeth (oligodontia), and dental crowding.
- Skeletal findings include mild syndactyly of toes two and three as well as fifth-digit clinodactyly.
- Novel findings include small hands and feet (<10th centile for age) and joint laxity.
However, the disorder is highly variable and not all affected individuals will develop all three of these characteristic symptoms. Andersen-Tawil syndrome can vary greatly in expression and severity from one person to another, even among members of the same family.
Most people who have Andersen-Tawil syndrome start experiencing symptoms before they turn 20. Some will begin to have heart palpitations and fainting spells, while others may start having episodes of transient muscle weakness. Severity and frequency can vary greatly, and there are many possible triggers, so patients may not be able to pinpoint what triggers the episodes of weakness.
Although researchers have established a clear syndrome with characteristic or “core” symptoms, much about the disorder is not fully understood. Several factors including the small number of identified cases, the lack of large clinical studies, and the possibility of other genes influencing the disorder prevent physicians from developing a complete picture of associated symptoms and prognosis. Therefore, it is important to note that affected individuals may not have all of the symptoms discussed below. Parents should talk to their children’s physician and medical team about their specific case, associated symptoms and overall prognosis.
Affected individuals may experience temporary episodes of flaccid, muscle weakness or paralysis, known as periodic paralysis. The legs are most often affected and the severity of muscle weakness can range from mild weakness to an inability to walk unassisted. The arms, hands, legs and feet are also commonly affected. The frequency and duration of episodes varies from one person to another and from one episode to the next for the same person. Some episodes may last only minutes to hours; others can go on for days. Episodes can occur without warning (spontaneously), but can also occur following prolonged exercise, prolonged rest (e.g. upon awaking in the morning), rest after exercise, going too long without eating, eating a large meal, or emotional stress. Episodes can range in frequency from once per day to once per year. In some cases, a mild, but permanent weakness, present even between episodes, can develop with age and progress slowly over time.
In most cases, periodic paralysis may be associated with low levels of potassium in the blood (hypokalemia), a common finding with other forms of periodic paralysis. However, some individuals who experience periodic paralysis have had normal potassium levels or even elevated levels (hyperkalemia). Low potassium levels can also impact the function of heart muscle cells.
Affected individuals may experience disturbances of the normal rhythm of the heartbeat (arrhythmias), which can include abnormally fast heartbeats that originate in the lower chamber of the heart (ventricular tachycardia). Generally, this may not cause any symptoms (asymptomatic) or may cause shortness of breath or palpitations. In some cases, these arrhythmias may cause episodes of fainting or loss of consciousness (syncope). In severe cases, the possibility of cardiac arrest and sudden death exists. Although sudden death due to the cardiac abnormalities has occurred in Andersen-Tawil syndrome, it is extremely rare.
Some affected individuals also have characteristic physical features including distinctive facial features, which are often mild in expression. Such features include a broad forehead, low-set ears, eyes that are spaced apart wider than usual (hypertelorism), and a small jaw (micrognathia). Additional facial features include a round (bulbous) nose, a thin upper lip, a triangular-shaped face, highly-arched roof of the mouth (palate), a cleft palate, and underdevelopment of the cheek bones (malar hypoplasia). Common physical features include webbing (syndactyly) of the second and/or third toes, pinkies that are fixed in a bent or crooked position (clinodactyly), and disproportionately small fingers and toes (brachydactyly). Additional findings include small hands and feet, loose joints, and abnormal sideways curvature of the spine (scoliosis). Dental anomalies have also been reported including delayed loss of primary or ‘baby’ teeth (persistent primary dentition), multiple missing teeth (oligodontia), and teeth that are abnormally crowded together.
As affected children grow into adulthood, short stature may become evident. Short stature refers to an individual whose height is much shorter than would otherwise be expected based upon age and gender.
Some individuals with Andersen-Tawil syndrome have experienced neuropsychiatric abnormalities including mild learning disabilities, depression, and deficits in executive functioning and abstract reasoning. Some infants experience seizures without fever (afebrile seizures). Afebrile seizures occurring only in infancy were reported in 4/23 (17%) of a Japanese cohort with molecularly confirmed Andersen-Tawil syndrome 29.
Isolated reports of renal anomalies include unilateral hypoplastic kidney 37 and renal tubular defect 35.
Mild learning difficulties have been described 35. A distinct neurocognitive phenotype (i.e., deficits in executive function and abstract reasoning) has been recognized in individuals with a KCNJ2 pathogenic variant despite IQ levels similar to those of their unaffected sibs 38. Growth restriction and developmental delay have been described as well 39.
Andersen Tawil syndrome diagnosis
A diagnosis of Andersen-Tawil syndrome is based upon identification of characteristic symptoms (e.g. periodic paralysis, symptomatic arrhythmias, and/or distinctive facial and skeletal features), a detailed family and patient history, a thorough clinical evaluation and a variety of specialized tests.
The diagnosis of Andersen-Tawil syndrome might be suspected in individuals with either A or B 40:
- A. Presence of two of the following three criteria:
- Periodic paralysis
- Symptomatic cardiac arrhythmias or evidence of enlarged U-waves, ventricular ectopy, or a prolonged QTc or QUc interval on electrocardiogram (ECG)
- Characteristic facial features, dental abnormalities, small hands and feet, AND at least two of the following:
- Low-set ears
- Widely spaced eyes
- Small lower jaw (mandible)
- Fifth-digit clinodactyly (curved pinky finger)
- Syndactyly of toes 2 and 3
OR
- B. One of the above three criteria in addition to at least one other family member who meets two of the three criteria 41.
The presence of a mutation in the KCNJ2 gene confirms the diagnosis of Andersen-Tawil syndrome 13.
Table 1. Recommended evaluations following initial diagnosis in individuals with Andersen Tawil syndrome
| Organ System | Evaluation | Comment |
|---|---|---|
| Cardiovascular | Baseline assessment | Performed by cardiologist familiar with long QT management |
| 12-lead EKG & 24-hour Holter monitor | ||
| Serum potassium concentrations | Performed at baseline & during attacks of weakness | |
| Neurologic | Baseline assessment | Performed by neurologist familiar w/periodic paralysis |
| Electrophysiologic studies incl long exercise protocol | ||
| Dental | Baseline assessment for dental abnormalities assoc with Andersen-Tawil syndrome | Follow up as needed |
| Musculoskeletal | Baseline assessment & to establish care with orthopedist or spine surgeon if scoliosis identified | Follow up as needed |
| Miscellaneous/ Other | Serum TSH concentration | Verification that serum TSH concentration is w/in normal limits |
| Consultation with clinical geneticist and/or genetic counselor |
Abbreviation: TSH = thyroid-stimulating hormone
[Source 40 ]Clinical testing and workup
Because potassium levels may be reduced during an episode of periodic paralysis, a blood test to determine the serum potassium levels during an episode can be helpful in diagnosing the disorder in some cases.
Long exercise nerve conduction studies have been used to help diagnose individuals with Andersen-Tawil syndrome. During this test, an affected individual will perform voluntary muscle contractions of a small muscle on the ulnar side of the palm of the hand for approximately 2-5 minutes. This test allows physician to evaluate muscle function and specific results can be indicative of periodic paralysis.
An electrocardiogram or EKG records the heart’s electrical impulses and may reveal abnormal electrical patterns or activity commonly associated with Andersen-Tawil syndrome including prominent U waves, prolonged QU intervals, prolonged QT intervals, premature ventricular contractions, or polymorphic ventricular tachycardia.
Some individuals may undergo 24-Holter monitoring, during which an affected individual wears a small device for 24 hours. Through electrodes attached to the chest, this device continuously records the rhythm of the heart in order to detect the presence, frequency and duration of ventricular tachycardia and other symptoms.
Molecular genetic testing can confirm a diagnosis of Andersen-Tawil syndrome in some cases. Molecular genetic testing can detect mutations in the KCNJ2 gene known to cause the disorder, but is available only as a diagnostic service at specialized laboratories.
Supportive findings
Individuals with either episodic weakness or cardiac symptoms require careful evaluation by a neurologist and/or cardiologist as well as measurement of serum potassium concentration (baseline and during attacks of flaccid paralysis), a 12-lead ECG, a 24-hour Holter monitor, and possibly the long exercise protocol.
- Serum potassium concentration during episodes of weakness may be elevated, normal, or, most commonly, reduced (<3.5 mmol/L) 41, 26.
- Routine nerve conduction electrophysiology is normal between episodes. A more sensitive electrophysiologic study, the long exercise protocol, may reveal an immediate post-exercise increment followed by an abnormal decrement in the compound motor action potential (CMAP) amplitude (>40%) 42 or area (>50%) 20-40 minutes post exercise 43, 44. In a study of 11 individuals with Andersen-Tawil syndrome, 82% met long-exercise amplitude decrement criteria for abnormal testing 45.
- Electrocardiogram (ECG) may reveal characteristic abnormalities including prominent U waves, prolonged Q-U intervals, premature ventricular contractions, polymorphic ventricular tachycardia, and bidirectional ventricular tachycardia 41, 17, 16, 46.
- 24-hour Holter monitoring is important to document the presence, frequency, and duration of ventricular tachycardia (VT) and the presence or absence of associated symptoms.
Andersen Tawil syndrome treatment
The treatment of Andersen-Tawil syndrome is directed toward the specific symptoms that are apparent in each individual. Management of individuals with Andersen Tawil syndrome requires the coordinated efforts of a team of specialists. Pediatricians, neurologists experienced in the treatment of periodic paralysis, cardiologists experienced in the treatment of long QT syndrome, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment.
There are no standardized treatment protocols or guidelines for affected individuals. Due to the rarity of Andersen Tawil syndrome, there are no randomized clinical therapeutic trials that have been tested on a large group of patients. Various treatments have been reported in the medical literature as part of single case reports or small series of patients. Treatment trials would be very helpful to determine the long-term safety and effectiveness of specific medications and treatments for individuals with Andersen-Tawil syndrome.
Affected individuals are encouraged to avoid potential triggers of periodic paralysis (e.g. rest following exercise or prolonged exercise). Avoidance of drugs that can prolong the QT interval is also recommended.
When periodic paralysis is associated with low potassium levels, treatment with oral supplemental potassium can be beneficial. In individuals prone to low potassium levels, daily potassium supplementation can be considered. If serum potassium concentration is low (<3.0 mmol/L), administration of oral potassium (20-30 mEq/L) every 15-30 minutes (not to exceed 200 mEq in a 12-hour period) until the serum concentration normalizes; if a relative drop in serum potassium within the normal range causes episodic paralysis, an individual potassium replacement regimen with a goal of maintaining serum potassium levels in the high range of normal can be considered; if serum potassium concentration is high, ingesting carbohydrates may lower serum potassium levels. Mild exercise may shorten or reduce the severity of the attack. Potassium supplementation may also shorten the QT interval, which can be of benefit for individuals who also experience a long QT interval.
A periodic paralysis episode that occurs when potassium levels are high usually resolve on their own within 60 minutes. However, eating carbohydrates or continuing mild exercise can shorten the duration of the episode.
Specific drugs known as carbonic anhydrase inhibitors, such as acetazolamide and dichlorpenamide, are used to treat periodic paralysis in individuals with Andersen-Tawil syndrome. Clinical trials in other forms of periodic paralysis showed that dichlorphenamide reduces the frequency and severity of attacks of periodic paralysis and is now an FDA approved for the treatment of periodic paralysis.
Despite a high frequency of ventricular arrhythmias in some individuals with Andersen-Tawil syndrome, they rarely degenerate into life-threatening arrhythmias. Many arrhythmias do not cause symptoms and go away on their own without problems (self-terminate). Various different drugs have been used, but no standard, effective therapy has been established. Beta-adrenergic blocking drugs (beta blockers), drugs that suppress abnormal heart rhythms (anti-arrhythmics) such as flecainide or amiodarone, or calcium-channel blocking drugs such as verapamil have all shown some effect. Beta blockers are commonly used to treat abnormal heart rhythms. These drugs, which include propranolol, atenolol, metroprolol, and nadolol, reduce the workload of the heart by decreasing the electrical stimulation of the heart, thereby slowing the heartbeat and preventing symptoms. Beta blockers have been used in conjunction with flecainide. Some anti-arrhythmic drugs can worsen neuromuscular symptoms and should be used with caution in individuals with Andersen-Tawil syndrome.
Treatment with an implantable automatic cardioverter-defibrillator (ICD) is necessary in rare cases. Implantable automatic cardioverter-defibrillators (ICDs) are considered for individuals in whom cardiac arrhythmias are severe and symptomatic. These small devices are implanted under the skin of the chest. The device detects the abnormal heartbeat automatically and selectively delivers an electrical impulse to restore the proper heartbeat. Opting for an ICD is a lifelong therapy that carries significant implications including the potential for complications, especially in younger individuals, and should be undertaken only after consultation with appropriate medical personnel and a careful risk vs. benefit evaluation.
Genetic counseling is recommended for affected individuals and their families.
Episodic weakness or paralysis treatment
Management of attacks of episodic weakness depends on the associated serum potassium concentration 40:
- If the serum potassium concentration is low (<3.0 mmol/L), administer oral potassium (20-30 mEq/L) every 15-30 minutes (not to exceed 200 mEq in a 12-hour period) until the serum concentration normalizes often shortens the attack. As dysphagia is almost never a problem during attacks of paralysis, oral potassium replacement is the safest route. If intravenous potassium replacement is needed, a 5% mannitol solution instead of a saline or glucose solution (both of which may exacerbate weakness) is recommended. Close monitoring of serum potassium concentrations and ECG is necessary during potassium replacement therapy in an emergency setting to avoid secondary hyperkalemia.
- Whether a relative drop in serum potassium within the normal range causes episodic paralysis is not clear. If such cases are suspected, affected individuals can work with their physician to devise an individual potassium replacement regimen, with a goal of maintaining serum potassium levels in the high range of normal.
- Attacks of weakness when serum potassium concentration is high usually resolve within 60 minutes. Episodes may be shortened by ingesting carbohydrates or continuing mild exercise. Intravenous calcium gluconate is rarely necessary for management in an individual seen in an emergency setting.
Vasovagal syncope in individuals with Andersen Tawil syndrome mandates a careful cardiology assessment 47.
Prevention of Andersen Tawil syndrome attacks
Prophylactic treatment aimed at reduction of attack frequency and severity can be achieved, as in other forms of periodic paralysis, with the following 40:
- Lifestyle and dietary modification to avoid known triggers
- Use of carbonic anhydrase inhibitors (acetazolamide: adults 125-1,000 mg daily and children 5-10 mg/kg/day divided 1-2x/day or dichlorphenamide 50-200 mg/1-2x/day). Use of potassium-sparing diuretic should be individualized based on patient needs.
- Daily use of slow-release potassium supplements, which may also be helpful in controlling attack rates in individuals prone to hypokalemia. Elevating the serum potassium concentration (>4 mEq/L) has the added benefit of narrowing the QT interval, thus reducing the risk of long QT-associated arrhythmias.
- An implantable cardioverter-defibrillator in individuals with tachycardia-induced syncope 48
- Empiric treatment with flecainide should be considered for significant, frequent ventricular arrhythmias in the setting of reduced left ventricular function 49, 50, 51, 52. A prospective open label study in ten individuals with Andersen Tawil syndrome and a confirmed KCNJ2 pathogenic variant tested the effect of flecainide, a type 1c antiarrhythmic, for the prevention of cardiac arrhythmias 53. Outcomes included a 24-hour Holter monitor before and after treatment and a treadmill exercise test. Flecainide was found to significantly reduce the number of ventricular arrhythmias seen on Holter monitor and to suppress exercise-induced ventricular arrhythmias. Individuals were then followed for a mean of 23 months and no syncope or cardiac arrest was documented 53. Other case studies have reported beneficial effects with flecainide 41. A recent study showed that fleicainide suppresses arrhythmogenicity through Na+/Ca2+ exchanger flux in induced pluripotent stem cells derived from patients with Andersen Tawil syndrome 54. Threfore, flecainide may reduce cardiac arrhythmias in Andersen Tawil syndrome.
Prevention of secondary complications
Cardiologists should be aware that some antiarrhythmic drugs (e.g., lidocaine, mexiletine, propafenone, quinidine), particularly Class 1 drugs, may paradoxically exacerbate the neuromuscular symptoms and should be used cautiously in individuals with Andersen Tawil syndrome 40.
Although malignant hyperthermia has not been reported in Andersen Tawil syndrome, appropriate anesthetic precautions should be undertaken, as with individuals with other forms of periodic paralysis 40.
Pregnancy management
The rarity of Andersen Tawil syndrome and the paucity of reports pertaining to pregnancy in women with Andersen Tawil syndrome make an evidence-based approach to pregnancy management difficult to formulate 40. One case study reported an uneventful pregnancy, with increased episodes of weakness but reduced ventricular ectopy compared to the pre-pregnancy period 55. However, as data are limited, a multidisciplinary approach to individual care and anticipation of increased risk (as can be seen in those with long QT syndrome) seems reasonable 40.
- Moreno-Manuel AI, Gutiérrez LK, Vera-Pedrosa ML, Cruz FM, Bermúdez-Jiménez FJ, Martínez-Carrascoso I, Sánchez-Pérez P, Macías Á, Jalife J. Molecular stratification of arrhythmogenic mechanisms in the Andersen Tawil syndrome. Cardiovasc Res. 2023 May 2;119(4):919-932. doi: 10.1093/cvr/cvac118[↩][↩]
- Yim J, Kim KB, Kim M, Lee GD, Kim M. Andersen-Tawil Syndrome With Novel Mutation in KCNJ2: Case Report. Front Pediatr. 2022 Jan 31;9:790075. doi: 10.3389/fped.2021.790075[↩][↩]
- Andersen ED, Krasilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr Scand. 1971 Sep;60(5):559-64. doi: 10.1111/j.1651-2227.1971.tb06990.x[↩]
- Tawil R, Ptacek LJ, Pavlakis SG, DeVivo DC, Penn AS, Ozdemir C, Griggs RC. Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol. 1994 Mar;35(3):326-30. doi: 10.1002/ana.410350313[↩]
- Stunnenberg BC, Raaphorst J, Deenen JCW, Links TP, Wilde AA, Verbove DJ, Kamsteeg EJ, van den Wijngaard A, Faber CG, van der Wilt GJ, van Engelen BGM, Drost G, Ginjaar HB. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul Disord. 2018 May;28(5):402-407. doi: 10.1016/j.nmd.2018.03.006[↩][↩]
- Andersen-Tawil syndrome. https://medlineplus.gov/genetics/condition/andersen-tawil-syndrome[↩][↩]
- Andersen-Tawil Syndrome. https://rarediseases.org/rare-diseases/andersen-tawil-syndrome[↩]
- Andersen-Tawil Syndrome. https://periodicparalysis.org/andersen-tawil-syndrome[↩][↩][↩]
- Mazzanti A, Guz D, Trancuccio A, et al. Natural History and Risk Stratification in Andersen-Tawil Syndrome Type 1. J Am Coll Cardiol. 2020 Apr 21;75(15):1772-1784. doi: 10.1016/j.jacc.2020.02.033[↩]
- van der Werf-‘t Lam AS, van Haeringen A, Rinnen T, Robles de Medina RM, Wilde AAM, Hennekam RC, Barge-Schaapveld DQCM. Andersen-Tawil syndrome: Overlapping clinical features with Noonan syndrome? Eur J Med Genet. 2022 Jan;65(1):104382. doi: 10.1016/j.ejmg.2021.104382[↩]
- Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptácek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001 May 18;105(4):511-9. doi: 10.1016/s0092-8674(01)00342-7[↩]
- Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J Physiol. 1984 Feb;347:641-57. doi: 10.1113/jphysiol.1984.sp015088[↩]
- Veerapandiyan A, Statland JM, Tawil R. Andersen-Tawil Syndrome. 2004 Nov 22 [Updated 2018 Jun 7]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1264[↩][↩]
- Donaldson MR, Yoon G, Fu YH, Ptacek LJ. Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med. 2004;36 Suppl 1:92-7. doi: 10.1080/17431380410032490[↩]
- Mazzanti A, Guz D, Trancuccio A, et al. Natural History and Risk Stratification in Andersen-Tawil Syndrome Type 1. J Am Coll Cardiol. 2020 Apr 21;75(15):1772-1784. https://www.jacc.org/doi/10.1016/j.jacc.2020.02.033[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Delannoy E, Sacher F, Maury P, Mabo P, Mansourati J, Magnin I, Camous JP, Tournant G, Rendu E, Kyndt F, Haïssaguerre M, Bézieau S, Guyomarch B, Le Marec H, Fressart V, Denjoy I, Probst V. Cardiac characteristics and long-term outcome in Andersen-Tawil syndrome patients related to KCNJ2 mutation. Europace. 2013 Dec;15(12):1805-11. doi: 10.1093/europace/eut160[↩][↩]
- Zhang L, Benson DW, Tristani-Firouzi M, Ptacek LJ, Tawil R, Schwartz PJ, George AL, Horie M, Andelfinger G, Snow GL, Fu YH, Ackerman MJ, Vincent GM. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation. 2005 May 31;111(21):2720-6. doi: 10.1161/CIRCULATIONAHA.104.472498[↩][↩]
- Barajas-Martinez H, Hu D, Ontiveros G, et al. Biophysical and molecular characterization of a novel de novo KCNJ2 mutation associated with Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia mimicry. Circ Cardiovasc Genet. 2011;4(1):51-57. doi:10.1161/CIRCGENETICS.110.957696 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3041844[↩]
- Adams DS, Uzel SG, Akagi J, Wlodkowic D, Andreeva V, Yelick PC, Devitt-Lee A, Pare JF, Levin M. Bioelectric signalling via potassium channels: a mechanism for craniofacial dysmorphogenesis in KCNJ2-associated Andersen-Tawil Syndrome. J Physiol. 2016 Jun 15;594(12):3245-70. doi: 10.1113/JP271930[↩]
- Yoon G, Oberoi S, Tristani-Firouzi M, Etheridge SP, Quitania L, Kramer JH, Miller BL, Fu YH, Ptácek LJ. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am J Med Genet A. 2006 Feb 15;140(4):312-21. doi: 10.1002/ajmg.a.31092[↩]
- Wang Q, Zhao Z, Shen H, Bing Q, Li N, Hu J. The clinical and genetic heterogeneity analysis of five families with primary periodic paralysis. Channels (Austin). 2021 Dec;15(1):20-30. doi: 10.1080/19336950.2020.1857980[↩]
- KCNJ2 gene. https://medlineplus.gov/genetics/gene/kcnj2[↩][↩]
- Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 2002 Jun 13;34(6):933-44. doi: 10.1016/s0896-6273(02)00725-0[↩]
- Ai T, Fujiwara Y, Tsuji K, Otani H, Nakano S, Kubo Y, Horie M. Novel KCNJ2 mutation in familial periodic paralysis with ventricular dysrhythmia. Circulation. 2002 Jun 4;105(22):2592-4. doi: 10.1161/01.cir.0000019906.35135.a3[↩]
- Davies NP, Imbrici P, Fialho D, Herd C, Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman BR, Giunti P, Parsons LM, Thomas M, Manzur AY, Jurkat-Rott K, Lehmann-Horn F, Chinnery PF, Rose M, Kullmann DM, Hanna MG. Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology. 2005 Oct 11;65(7):1083-9. doi: 10.1212/01.wnl.0000178888.03767.74[↩]
- Sansone V, Tawil R. Management and treatment of Andersen-Tawil syndrome (ATS). Neurotherapeutics. 2007 Apr;4(2):233-7. doi: 10.1016/j.nurt.2007.01.005[↩][↩]
- Tristani-Firouzi M, Jensen JL, Donaldson MR, Sansone V, Meola G, Hahn A, Bendahhou S, Kwiecinski H, Fidzianska A, Plaster N, Fu YH, Ptacek LJ, Tawil R. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J Clin Invest. 2002;110:381–8.[↩]
- Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, Szepetowski P, Pereira S, Mozaffar T, Escolar DM, Fu YH, Ptácek LJ. PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology. 2003;60:1811–6.[↩][↩]
- Haruna Y, Kobori A, Makiyama T, Yoshida H, Akao M, Doi T, Tsuji K, Ono S, Nishio Y, Shimizu W, Inoue T, Murakami T, Tsuboi N, Yamanouchi H, Ushinohama H, Nakamura Y, Yoshinaga M, Horigome H, Aizawa Y, Kita T, Horie M. Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum Mutat. 2007;28:208.[↩][↩]
- Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, Wu J, Ohno S, Makiyama T, Miyamoto A, Naiki N, Wang Q, Xie Y, Suzuki T, Tateno S, Nakamura Y, Zang WJ, Ito M, Matsuura H, Horie M. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012;5:344–53.[↩]
- Delannoy E, Sacher F, Maury P, Mabo P, Mansourati J, Magnin I, Camous JP, Tournant G, Rendu E, Kyndt F, Haïssaguerre M, Bézieau S, Guyomarch B, Le Marec H, Fressart V, Denjoy I, Probst V. Cardiac characteristics and long-term outcome in Andersen-Tawil syndrome patients related to KCNJ2 mutation. Europace. 2013;15:1805–11.[↩]
- Schoonderwoerd BA, Wiesfeld AC, Wilde AA, van den Heuvel F, Van Tintelen JP, van den Berg MP, Van Veldhuisen DJ, Van Gelder IC. A family with Andersen-Tawil syndrome and dilated cardiomyopathy. Heart Rhythm. 2006;3:1346–50.[↩]
- Pellizzón OA, Kalaizich L, Ptácek LJ, Tristani-Firouzi M, Gonzalez MD. Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J Cardiovasc Electrophysiol. 2008;19:95–7.[↩]
- Rezazadeh S, Guo J, Duff HJ, Ferrier RA, Gerull B. Reversible dilated cardiomyopathy caused by a high burden of ventricular arrhythmias in Andersen-Tawil syndrome. Can J Cardiol. 2016;32:1576.e15–e18.[↩]
- Davies NP, Imbrici P, Fialho D, Herd C, Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman BR, Giunti P, Parsons LM, Thomas M, Manzur AY, Jurkat-Rott K, Lehmann-Horn F, Chinnery PF, Rose M, Kullmann DM, Hanna MG. Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology. 2005;65:1083–9.[↩][↩][↩]
- Yoon G, Oberoi S, Tristani-Firouzi M, Etheridge SP, Quitania L, Kramer JH, Miller BL, Fu YH, Ptácek LJ. Andersen-Tawil syndrome: prospective cohort analysis and expansion of the phenotype. Am J Med Genet A. 2006a;140:312–21.[↩]
- Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL Jr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71:663–8.[↩]
- Yoon G, Quitania L, Kramer JH, Fu YH, Miller BL, Ptácek LJ. Andersen-Tawil syndrome: definition of a neurocognitive phenotype. Neurology. 2006b;66:1703–10.[↩]
- Kim NR, Jang J, Jean GW, Cho E, Sin JB. Identification of the KCNJ2 mutation in a Korean family with Andersen-Tawil syndrome and developmental delay. Ann Clin Lab Sci. 2016;46:110–3.[↩]
- Veerapandiyan A, Statland JM, Tawil R. Andersen-Tawil Syndrome. 2004 Nov 22 [Updated 2018 Jun 7]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1264[↩][↩][↩][↩][↩][↩][↩][↩]
- Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, Sansone VA, Shieh PB, Tawil RN, Trivedi J, Cannon SC, Griggs RC. Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve. 2018 Apr;57(4):522-530. doi: 10.1002/mus.26009[↩][↩][↩][↩]
- Katz JS, Wolfe GI, Iannaccone S, Bryan WW, Barohn RJ. The exercise test in Andersen syndrome. Arch Neurol. 1999 Mar;56(3):352-6. doi: 10.1001/archneur.56.3.352[↩]
- Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B, Willer JC, Tabti N, Fontaine B. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. 2004 Nov;56(5):650-61. doi: 10.1002/ana.20241[↩]
- Kuntzer T, Flocard F, Vial C, Kohler A, Magistris M, Labarre-Vila A, Gonnaud PM, Ochsner F, Soichot P, Chan V, Monnier G. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000 Jul;23(7):1089-94. doi: 10.1002/1097-4598(200007)23:7<1089::aid-mus12>3.0.co;2-q[↩]
- Tan SV, Matthews E, Barber M, Burge JA, Rajakulendran S, Fialho D, Sud R, Haworth A, Koltzenburg M, Hanna MG. Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies. Ann Neurol. 2011 Feb;69(2):328-40. doi: 10.1002/ana.22238[↩]
- Koppikar S, Barbosa-Barros R, Baranchuk A. A Practical Approach to the Investigation of an rSr’ Pattern in Leads V1-V2. Can J Cardiol. 2015 Dec;31(12):1493-6. doi: 10.1016/j.cjca.2015.04.008[↩]
- Airey KJ, Etheridge SP, Tawil R, Tristani-Firouzi M. Resuscitated sudden cardiac death in Andersen-Tawil syndrome. Heart Rhythm. 2009 Dec;6(12):1814-7. doi: 10.1016/j.hrthm.2009.08.032[↩]
- Chun TU, Epstein MR, Dick M 2nd, Andelfinger G, Ballester L, Vanoye CG, George AL Jr, Benson DW. Polymorphic ventricular tachycardia and KCNJ2 mutations. Heart Rhythm. 2004 Jul;1(2):235-41. doi: 10.1016/j.hrthm.2004.02.017[↩]
- Bökenkamp R, Wilde AA, Schalij MJ, Blom NA. Flecainide for recurrent malignant ventricular arrhythmias in two siblings with Andersen-Tawil syndrome. Heart Rhythm. 2007 Apr;4(4):508-11. doi: 10.1016/j.hrthm.2006.12.031[↩]
- Fox DJ, Klein GJ, Hahn A, Skanes AC, Gula LJ, Yee RK, Subbiah RN, Krahn AD. Reduction of complex ventricular ectopy and improvement in exercise capacity with flecainide therapy in Andersen-Tawil syndrome. Europace. 2008 Aug;10(8):1006-8. doi: 10.1093/europace/eun180[↩]
- Pellizzón OA, Kalaizich L, Ptácek LJ, Tristani-Firouzi M, Gonzalez MD. Flecainide suppresses bidirectional ventricular tachycardia and reverses tachycardia-induced cardiomyopathy in Andersen-Tawil syndrome. J Cardiovasc Electrophysiol. 2008 Jan;19(1):95-7. doi: 10.1111/j.1540-8167.2007.00910.x[↩]
- Tristani-Firouzi M, Etheridge SP. Kir 2.1 channelopathies: the Andersen-Tawil syndrome. Pflugers Arch. 2010 Jul;460(2):289-94. doi: 10.1007/s00424-010-0820-6[↩]
- Miyamoto K, Aiba T, Kimura H, Hayashi H, Ohno S, Yasuoka C, Tanioka Y, Tsuchiya T, Yoshida Y, Hayashi H, Tsuboi I, Nakajima I, Ishibashi K, Okamura H, Noda T, Ishihara M, Anzai T, Yasuda S, Miyamoto Y, Kamakura S, Kusano K, Ogawa H, Horie M, Shimizu W. Efficacy and safety of flecainide for ventricular arrhythmias in patients with Andersen-Tawil syndrome with KCNJ2 mutations. Heart Rhythm. 2015 Mar;12(3):596-603. doi: 10.1016/j.hrthm.2014.12.009[↩][↩]
- Kuroda Y, Yuasa S, Watanabe Y, Ito S, Egashira T, Seki T, Hattori T, Ohno S, Kodaira M, Suzuki T, Hashimoto H, Okata S, Tanaka A, Aizawa Y, Murata M, Aiba T, Makita N, Furukawa T, Shimizu W, Kodama I, Ogawa S, Kokubun N, Horigome H, Horie M, Kamiya K, Fukuda K. Flecainide ameliorates arrhythmogenicity through NCX flux in Andersen-Tawil syndrome-iPS cell-derived cardiomyocytes. Biochem Biophys Rep. 2017 Jan 11;9:245-256. doi: 10.1016/j.bbrep.2017.01.002[↩]
- Subbiah RN, Gula LJ, Skanes AC, Krahn AD. Andersen-Tawil syndrome: management challenges during pregnancy, labor, and delivery. J Cardiovasc Electrophysiol. 2008 Sep;19(9):987-9. doi: 10.1111/j.1540-8167.2008.01216.x[↩]





{kind=link}