
Contents
- What is Batten disease
- What causes Batten disease?
- How are Batten Diseases inherited?
- Types of Batten Disease
- Batten Disease Infantile onset and others
- Late-infantile Batten disease
- Juvenile Batten disease
- Variant Late Infantile Batten disease
- Variant late-infantile onset Batten disease
- Epilepsy with Progressive Mental Retardation and Late Infantile Variant Batten Disease
- Congenital, neonatal and late infantile Batten disease
- Adult Onset Batten Disease
- Batten disease life expectancy
- Batten disease symptoms
- Batten Disease Diagnosis
- Batten disease treatment
What is Batten disease
Batten disease known as Spielmeyer-Vogt-Sjogren-Batten Disease, it is the most common form of a group of disorders called neuronal ceroid lipofuscinoses (or NCLs). Batten disease is named after the British pediatrician who first described it in 1903 and is the name for a group of inherited nervous system disorders that most often begin in childhood and interfere with a cell’s ability to recycle a cellular residue called lipofuscin 1. Affected children suffer increasing mental impairment, worsening seizures, and progressive loss of sight and motor skills. Eventually, children with Batten disease become blind, bedridden and demented. Batten disease is often fatal by the late teens or twenties.
Batten disease is commonly being used to describe the many forms of the disease, called neuronal ceroid lipofuscinosis (NCL). The many forms of the disease are classified by the gene that causes the disorder, with each gene being called CLN (ceroid lipofucinosis, neuronal) and given a different number as its subtype. Because of the different gene mutations, signs and symptoms range in severity and progress at different rates. Symptoms generally include:
- progressive vision loss leading to blindness,
- seizures,
- movement disorder, and
- dementia.
Developmental skills such as standing, walking, and talking may now be achieved or are gradually lost. Other symptoms that continue to worsen over time include learning difficulties, poor concentration, and progressive loss of language skills and speech. Most children become bedridden and unable to communicate. Some children develop problems sleeping. Currently, most diagnoses of Batten disease are made by genetic testing. Each gene is called CLN (ceroid lipofuscinosis, neuronal) and given a different number designation as its subtype. There are 14 known forms of Batten disease and you will often hear them referred to as CLN1-CLN14.
Although Batten Disease is usually regarded as the juvenile form of Neuronal Ceroid Lipofuscinosis (NCL), it has now become the term to describe all forms of Neuronal Ceroid Lipofuscinosis (NCL). It is estimated that 2-4 births per 100,000 in the U.S. are affected by Batten disease, though some researchers in the field suggest these numbers are low 2. The basic cause, progression, and the outcome are the same. The forms of Neuronal Ceroid Lipofuscinosis (NCL) are classified by age of onset, and eight different genes [CLN1 – CLN8] have been identified as causing the disorder.
The first sign of the disease is often loss of vision, and may first be suspected during an eye examination. Other early signs are subtle, but may include personality and behaviour changes, slow learning, clumsiness or stumbling. Over time, affected children suffer mental impairment, worsening seizures, and progressive loss of sight and motor skills. Eventually, children with Juvenile Batten Disease become blind, bedridden, and unable to communicate. Juvenile Batten Disease is always fatal by the late teens or twenties.
There are four main types of Batten disease, including two forms that begin earlier in childhood and a very rare form that strikes adults. The symptoms are similar but they become apparent at different ages and progress at different rates.
- Infantile Batten disease (Santavuori-Haltia disease): begins between about 6 months and 2 years of age and progresses rapidly. Affected children fail to thrive and have abnormally small heads (microcephaly). Also typical are short, sharp muscle contractions called myoclonic jerks. Initial signs of this disorder include delayed psychomotor development with progressive deterioration, other motor disorders, or seizures. The infantile form has the most rapid progression and children live into their mid childhood years.
- Late Infantile Batten disease (Jansky-Bielschowsky disease) begins between ages 2 and 4. The typical early signs are loss of muscle coordination (ataxia) and seizures along with progressive mental deterioration. This form progresses rapidly and ends in death between ages 8 and 12.
- Juvenile Batten disease (Batten Disease) begins between the ages of 5 and 8. The typical early signs are progressive vision loss, seizures, ataxia or clumsiness. This form progresses less rapidly and ends in death in the late teens or early 20s, although some may live into their 30s.
- Adult Batten disease (Kufs Disease or Parry’s Disease) generally begins before the age of 40, causes milder symptoms that progress slowly, and does not cause blindness. Although age of death is variable among affected individuals, this form does shorten life expectancy.
A related disorder, Northern Epilepsy [(NE) or Progressive Epilepsy with Mental Retardation (PEMR)], so far identified only in patients of Finnish origin, generally begins between 5 and 10 years of age, and is characterised by tonic-clonic or complex partial seizures, and mental and motor deterioration. Although the frequency of seizures declines after puberty, cognitive function continues to decline. Some affected individuals have lived past the age of 60.
Batten Disease and other forms of NCL are relatively rare, occurring in an estimated 2 to 4 of every 100,000 births in the United States. It is not known how many people have Batten disease, but by some estimates it can be as frequent as in 1 in 12,500 people in some populations. These disorders appear to be more common in Finland, Sweden, other parts of northern Europe, and Newfoundland, Canada, although the disease has been identified worldwide. Although NCLs are classified as rare diseases, they often strike more than one person in families that carry the defective genes.
In those families where the disease-causing mutation has been identified, prenatal and carrier testing is available for family members willing to be tested, and genetic counselling can assist them to make informed decisions according to their personal circumstances.
Siblings of an affected individual have a 25 % chance of also being affected, a 50% chance of being unaffected but being a carrier of the defective genes, and a 25% chance of being both unaffected and a non-carrier. Unaffected siblings of parents with an affected child also have a 50% chance of being carriers.
In families known to be carriers of the defective genes, prenatal testing can be performed at either 10-12 weeks [by chorionic villus sampling or CVS], or 16-18 weeks [amniocentesis] to discover if the fetus is affected.
What causes Batten disease?
Batten disease is an inherited genetic disorder that appears to affect the function of tiny bodies within cells called lysosomes. Lysosomes are the “recycle bin” of the cell and regularly break down waste, proteins, and naturally occurring fatty compounds called lipids into smaller components that can be discarded out of the cell or recycled. Lipids include fatty acids, oils, waxes, and sterols. In Batten disease/NCLs, the mutated genes do not produce the proper amounts of proteins important for lysosomal function. Each gene (representing a form of the disease) provides information for a specific protein that is in turn, defective and not produced. These proteins are needed for brain cells (neurons) and other cells to work efficiently. The lack of a functional protein causes the abnormal buildup of “junk” material in the lysosomes—as well as the abnormal buildup of the residue called lipofuscin that occurs naturally as part of the lysosomal breakdown of lipids. It is not known whether the lipofuscin itself is toxic or if the buildup is a marker of impaired lysosomal function.
How are Batten Diseases inherited?
People normally have two copies of the same gene in their cells, one comes from the father and one from the mother. This means that, in some cases, the cells have a “back up” system if only one copy is needed for the cell to function properly. Batten disease is caused when both copies (one from each parent) of the specific gene causing the disease are defective. This is known as autosomal recessive disease. People who only have one defective copy (carriers) will not develop symptoms and are usually unaware of their carrier condition. The rare exception may be for Adult NCL (see below).
If both parents carry one defective gene that causes NCL, there is a 1 in 4 chance during each pregnancy of having a child with the disease. At the same time, during each pregnancy there is 50 percent chance for the baby to inherit only one copy of the defective gene, which would make the child a “carrier” like the parent, as a normal copy will be inherited from the other parent. Carriers most often are not affected by the disease but can pass the abnormal gene to their children in a similar fashion to which they inherited it from their own parents. Finally, there is a 1 in 4 chance for the baby to inherit two completely normal genes.
At risk for any form of Batten disease are children whose parents have Batten disease, and children whose parents are carriers of an NCL gene that causes the disorder but aren’t severely affected by the disorder, if at all.
Adult NCL/Kufs disease B may be inherited as an autosomal recessive or, less often, as an autosomal dominant disorder. In autosomal dominant inheritance, everyone who inherits a defective of the disease gene develops the disease, even when they may have inherited a normal copy.
Types of Batten Disease
Batten disease or Neuronal Ceroid Lipofuscinosis (NCL), has 14 known forms. Progressions of the different forms of the disease vary (the phenotype) based on the type of genetic mutation (the genotype) and other factors. Each form is classified by the gene that causes the disorder. Each gene is called CLN (ceroid lipofuscinosis, neuronal) and given a different number designation as its subtype. The disorders generally include a combination of vision loss, epilepsy, and dementia.
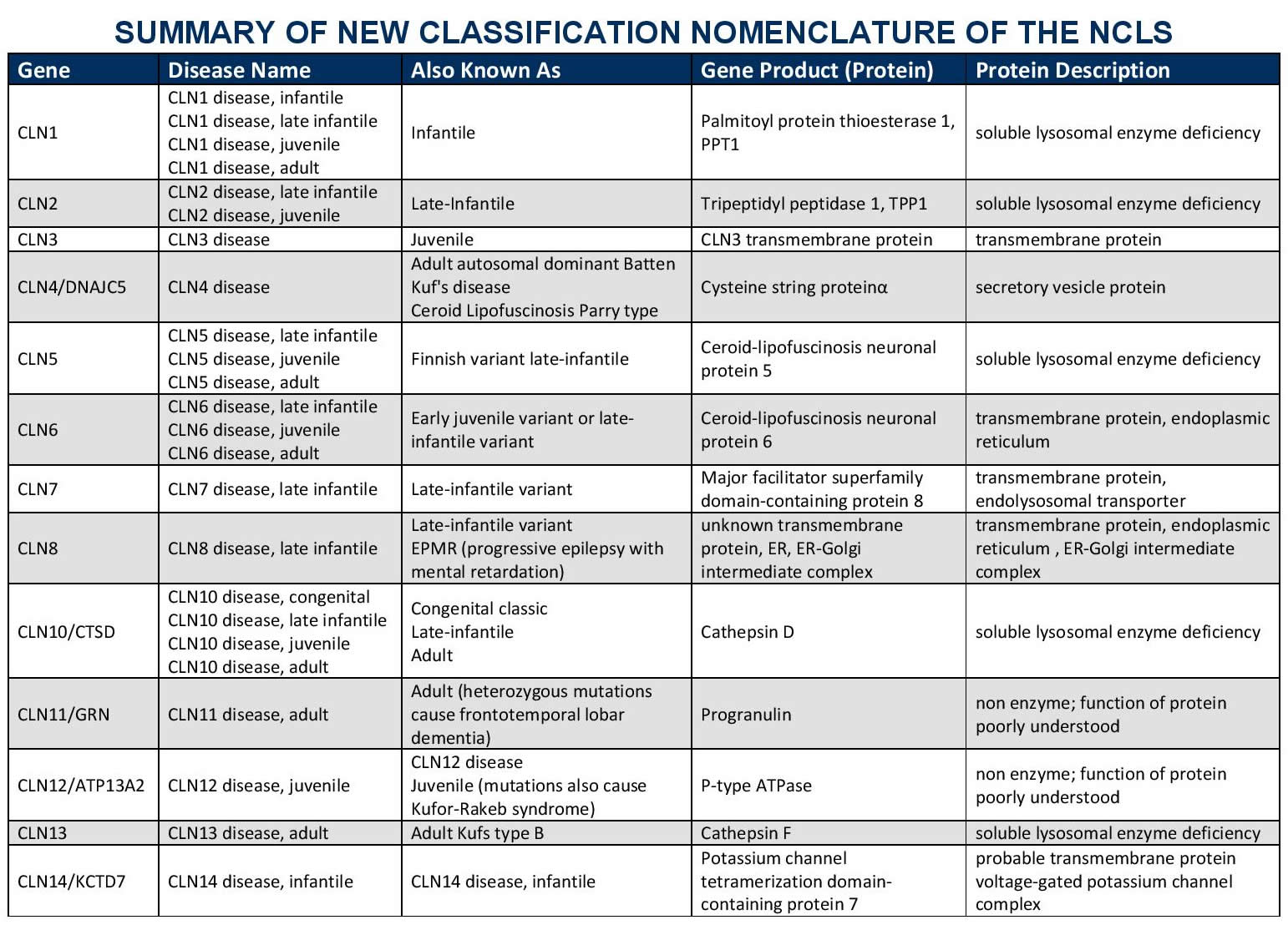
Batten Disease Infantile onset and others
CLN1 disease was first described in the 1970s in Finland and is sometimes called Haltia-Santavuori Disease, Infantile neuronal ceroid lipofuscinoses, or INCL.
What is the cause?
The gene called CLN1 lies on chromosome 1. CLN1 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry mutations in the CLN1 gene, and both parents are unaffected carriers. The gene was discovered in 1995. CLN1 normally directs production of a lysosomal enzyme called Palmitoyl protein thioesterase 1 or PPT1. A deficiency of PPT1 results in abnormal storage of proteins and lipids in neurons and other cells and impaired cellular function. The cells cannot function as they should and symptoms develop.
How is it diagnosed?
The diagnosis is usually made by enzyme (PPT1) and genetic (CLN1) tests on blood samples. Occasionally a skin biopsy may be necessary. Granular osmiophilic deposits (GRODSs) are the characteristic storage body at the electron microscope level.
How does the disease progress?
Classical CLN1 disease, infantile
Babies are healthy and develop normally for the first few months of life. Towards the end of the first year, developmental progress starts to slow down. Infants may have difficulty sleeping through the night and may become more restless and irritable during the day. Some infants develop repetitive hand movements and fiddling. They often become floppy and developmental skills such as walking, standing and speech are lost.
Children become less able and increasingly dependent during the toddler years. By the age of 2 years, most will have epileptic seizures and jerks. Vision gets worse until they are no longer able to see. From about the age of three years, children are completely dependent, unable to play, feed themselves, sit independently or communicate. They may need a feeding tube and their arms and legs usually become stiff. Some children get frequent chest infections. Most affected children die in early to mid-childhood.
CLN1 disease, juvenile onset
Some children with CLN1 abnormalities develop the disease after infancy—around age 5 or 6—and have slower disease progression. Affected children may live into their teenage years. Others may not develop symptoms until adolescence and may live into adulthood. CLN1 disease, variant late infantile and adult types. A wide variety of age at symptom onset and disease progression is seen with mutations in CLN1.
Late-infantile Batten disease
CLN2 late infantile disease is sometimes called Jansky-Bielschowsky Disease or late infantile NCL (LINCL).
What is the cause?
The gene called CLN2 lies on chromosome 11. CLN2 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry mutations in the CLN2 gene, and both parents are unaffected carriers. The gene was discovered in 1998. CLN2 normally directs production of a lysosomal enzyme called tripeptidyl peptidase1 or TPP1. A deficiency of TPP1 results in abnormal storage of proteins and lipids in neurons and other cells and impaired cellular function. The cells cannot function as they should and symptoms develop.
How is it diagnosed?
The diagnosis is usually made by enzyme (TPP1) and genetic (CLN2) tests on blood samples. Occasionally a skin biopsy may be necessary. Curvilinear bodies (CVB) are the characteristic storage body at the electron microscope level.
How does the disease progress?
Children are healthy and develop normally for the first few years of life. Towards the end of the second year, developmental progress may start to slow down. Some children are slow to talk. The first definite sign of the disease is usually epilepsy. Seizures may be drops, vacant spells or motor seizures with violent jerking of the limbs and loss of consciousness. Seizures may be controlled by medicines for several months but always recur, becoming difficult to control. Children tend to become unsteady on their feet with frequent falls and gradually skills such as walking, playing and speech are lost. Children become less able, and increasingly dependent. By 4-5 years the children usually have myoclonic jerks of their limbs and head nods. They may have difficulties sleeping and become distressed around this time, often for no obvious reason. Vision is gradually lost. By the age of 6 years, most will be completely dependent on families and careers for all of their daily needs. They may need a feeding tube and their arms and legs may become stiff. Some children get frequent chest infections. Death usually occurs between the ages of 6 and 12 years (but occasionally later).
CLN2 disease, later onset
Some children with CLN2 abnormalities develop the disease later in childhood —around age 6 or 7—and have slower disease progression. In later-onset CLN2 disease, loss of coordination (ataxia) may be the initial symptom. Affected children may live into their teenage years.
The TTP1 deficiency in atypical CLN2 can present as symptoms of ataxia and cerebellar atrophy but the individual does not necessarily develop seizures or vision loss. This example of atypical CLN2 is referred to as SCAR7, or spinocerebellar ataxia autosomal recessive 7.
Treatment
CLN2 is currently the only form of Batten disease that has an FDA approved treatment. The enzyme replacement therapy, Brineura® (cerliponase alfa), was created by BioMarin and was approved by the FDA in April 2017. It was approved to slow loss of ability to walk or crawl (ambulation) in symptomatic pediatric CLN2 Batten disease patients 3 years of age and older.
Juvenile Batten disease
At the beginning of the 20th century Dr. Frederick Batten described a group of disorders that now bear his name. Over time it was discovered that there were several types of the disease with similar but distinct features and ages at onset of symptoms: infantile, late infantile, juvenile, and adult. CLN3 disease is often called Batten disease, or Spielmeyer- Sjogren-Vogt disease.
What is the cause?
The gene called CLN3 lies on chromosome 16 and was discovered in 1995. CLN3 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry mutations in the CLN3 gene, and both parents are unaffected carriers. This gene codes for a transmembrane protein. The nerve cells cannot function as they should and symptoms develop.
How is it diagnosed?
The diagnosis is usually made by genetic (CLN3) tests on blood samples. Occasionally a skin biopsy may be necessary.
How does the disease progress?
Children are healthy and develop normally for the first few years of life. The first sign of the disease is usually a gradual loss of vision between 4 and 7 years of age. This may be noticed first at nursery or at school. Vision changes rapidly over 6 to 12 months initially but children retain some awareness of color and light/dark until later. By the end of primary school, children are beginning to show some difficulties with concentration, short-term memory and learning. Many are still able to attend mainstream school but may need extra learning support in the classroom. The next stage of the disease starts with the onset of epileptic seizures (average age of onset of seizures is 10 years). Often the first seizures are motor seizures with violent jerking of the limbs and loss of consciousness. Seizures may be controlled by medicines for several months or years, but always recur, eventually becoming difficult to control completely. The pattern of seizures may change over time and other seizure types may evolve, such as vacant spells and episodes of partial awareness with fiddling and muddled speech.
During the teenage years children tend to slowly become more unsteady on their feet. At around the same time speech may become repetitive and gradually more difficult to understand. Not uncommonly children become anxious and tend to worry. Some feel things, hear voices or see things that are unreal. Teenagers become less able and increasingly dependent. The course of the disease is extremely variable even for children from the same family. The teenagers and young adults are much more able some days than others, especially in terms of mobility, communication and feeding skills. The disease progresses with periods of stability which may last months or years alternating with periods of deterioration lasting several months which may be triggered by intercurrent illness. Death usually occurs between the ages of 15 and 35 years (but occasionally later).
Variant Late Infantile Batten disease
CLN5 disease, variant late-infantile may also be referred to as variant late-infantile CLN5 disease. It has previously been described as Finnish, Turkish, Indian, or Mediterranean Variant NCL, alongside Variant Late Infantile Neuronal Ceroid Lipofuscinosis (LINCL); though was more commonly known as Variant Late-Infantile Batten Disease.
What is the cause?
This disease is caused by problems with lysosomal protein called CLN5, whose function is unknown. The CLN5 gene is located on chromosome 13.
How is it diagnosed?
The diagnosis is usually made by histological and genetic tests on blood samples. A skin biopsy may be necessary and the abnormal storage material takes on a mixed appearance with granular osmiophilic deposits (GRODS), curvilinear bodies (CVB), rectilinear profiles (RLP), and/or fingerprint profiles (FPP). The appearance of the storage material can guide the genetic diagnostic tests in some cases.
Genetic testing is recommended to look for the exact mutation or mistake in the CLN5 gene. A blood or saliva sample will be taken to extract DNA from the cells for the test.
How does the disease progress?
Children progress normally for the first few years of life before they start losing skills and develop behavior problems. Seizures and myoclonic jerks begin usually between ages 6 and 13. Vision deteriorates and is eventually lost. Children have learning disabilities and problems with concentration and memory. Some may need a feeding tube. Most children with CLN5 live into their late childhood or teenage years.
Variant late-infantile onset and adult onset Batten disease
CLN6 disease, variant late-infantile was one of the first variant types of Neuronal Ceroid Lipofuscinosis (NCLs) to be identified and was originally termed early Juvenile Batten disease.
What is the cause?
This disease is caused by problems with lysosomal protein called CLN6, whose function is unknown. The CLN6 gene is located on chromosome 15 directs the production of the protein CLN6, also called linclin. The protein is found in the membranes of the cell (most predominantly in a structure called the endoplasmic reticulum). Its function has not been identified.
How is it diagnosed?
The diagnosis is usually made by histological and genetic tests on blood samples. A skin biopsy may be necessary and the abnormal storage material takes on a mixed appearance with granular osmiophilic deposits (GRODS), curvilinear bodies (CVB), rectilinear profiles (RLP), and/or fingerprint profiles (FPP). The appearance of the storage material can guide the genetic diagnostic tests in some cases.
Genetic testing is recommended to look for the exact mutation or mistake in the CLN6 gene. A blood or saliva sample will be taken to extract DNA from the cells for the test.
How does the disease progress?
CLN6 disease, Variant Late-Infantile
Symptoms vary among children, but typically start after the first few years of life and include developmental delay, changes in behavior, and seizures. Children eventually lose skills for walking, playing, and speech. The also develop myoclonic jerks, problems sleeping, and vision loss. Most children with CNL6 die during late childhood or in their early teenage years.
CLN6, Adult Onset
Also known as Kufs’ disease Type A, this form of CLN6 disease shows signs in early adulthood that include epilepsy, inability to control muscles in the arms and legs (resulting in a lack of balance or coordination, or problems with walking), and slow but progressive cognitive decline.
Variant late-infantile onset Batten disease
CLN7 disease, variant late-infantile may also be referred to as variant late-infantile CLN7 disease, alongside Variant Late Infantile Neuronal Ceroid Lipofuscinosis; though was more commonly known as Variant Late-Infantile Batten Disease.
What is the cause?
This disease is caused mutations in the CLN7 gene, is located on chromosome 4, which produced the protein MFSD8. MFSD8 a member of a protein family called the major facilitator superfamily. This superfamily is involved with transporting substances across the cell membranes.
How is it diagnosed?
The diagnosis is usually made by histological and genetic tests on blood samples. A skin biopsy may be necessary and the abnormal storage material takes on a mixed appearance with granular osmiophilic deposits (GRODS), curvilinear bodies (CVB), rectilinear profiles (RLP), and/or fingerprint profiles (FPP). The appearance of the storage material can guide the genetic diagnostic tests in some cases.
Genetic testing is recommended to look for the exact mutation or mistake in the CLN7 gene. A blood or saliva sample will be taken to extract DNA from the cells for the test.
How does the disease progress?
Developmental delays begin after a few years of normal development. Children usually develop epilepsy between the ages of 3 and 7 years, along with problems sleeping and myoclonic jerks. Children begin to lose the ability to walk, play, and speak. A rapid advancement of symptoms is seen between the ages of 9 and 11. Most children with the disorder live until their late childhood or teenage years.
Epilepsy with Progressive Mental Retardation and Late Infantile Variant Batten Disease
What is the cause?
The gene called CLN8 lies on chromosome 8. CLN8 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry mutations in the CLN8 gene, and both parents are unaffected carriers. The gene was discovered in 1999. CLN8 normally directs production of a protein that is embedded in internal cell membranes. The cells cannot function as they should and symptoms develop.
How is it diagnosed?
The diagnosis is usually made by histological and genetic (CLN8) tests on blood samples. A skin biopsy may be necessary. The characteristic storage bodies at the electron microscope level often show a mixture of fingerprint profiles (FPP) and curvilinear bodies (CVB).
How does the disease progress?
CLN8 disease, Epilepsy with Progressive Mental Retardation (EPMR) or Northern Epilepsy
This disease is caused by mutations in CLN8 but has seldom been described outside Scandinavian countries. Symptoms usually start between the ages of 5 and 10 years, with seizures. Cognitive decline occurs at around the same time. Seizure frequency increases until puberty. Cognitive deterioration is more rapid during puberty. Behavioral disturbances can occur, eg: irritability, restlessness, inactivity and these features may continue into adulthood. Epilepsy is partially responsive to treatment. The number of seizures decreases spontaneously after puberty, even with no change in treatment, and by the second-third decade they become relatively sporadic. Cognitive decline continues and in some cases loss of speech has been reported. Motor function is also impaired. In a number of cases, visual acuity is reduced (without evidence of retinal degeneration). The disease has a chronic course and survival to the sixth or seventh decade has been reported. Epilepsy with Progressive Mental Retardation is very unusual amongst the NCLs of childhood onset in this respect.
CLN8 disease, variant late infantile
All children have developmental delay before the onset of symptoms at 2 -7 years of age: myoclonic seizures and an unsteady gait are commonly the initial symptoms; other seizures follow soon after. Cognitive decline and visual impairment usually occur. Behavioral abnormalities are frequent. Rapid disease progression with loss of cognitive skills is observed over two years from the time of diagnosis. By the age of 8-10 years severe deterioration of neurological and cognitive skills is apparent together with medication resistant epilepsy. Spasticity, dystonic posturing, tremors, and other extrapyramidal signs are also observed commonly. In the second decade of life children are unable to walk or stand without support. The life-expectancy of children affected by this disease is not yet known. The eldest patients known are now in their second decade, and their general health remains good.
Congenital, neonatal and late infantile Batten disease
What is the cause?
This is a very rare form of NCL and only a small number of cases have been written about. There may be undiagnosed cases. This disease is caused by mutations in a gene called Cathepsin D (CTSD gene) also called CLN10, which lies on chromosome 11. CLN10 disease is inherited as an autosomal recessive disorder, which means that both chromosomes carry mutations in the CLN10 gene, and both parents are unaffected carriers. The gene was discovered in 2006. CLN10 normally directs production of a lysosomal enzyme called cathepsin D. If no enzyme is produced, symptoms start very early in life, or even before birth. If some enzyme is working, symptoms develop later and disease progression is slower.
How is it diagnosed?
The diagnosis is usually made by enzyme (CTSD) and genetic (CLN10) tests on blood samples. Occasionally a skin biopsy may be necessary. Granular deposits are the characteristic storage body at the electron microscope level.
How does the disease progress?
CLN10 disease, congenital
Seizures may occur before birth but are hard to differentiate from normal baby movements. Following birth, babies may have seizures that do not respond to treatment, problems with breathing that can progress to respiratory failure, and obstructive sleep apnea. Babies may die shortly after birth or within the first weeks of life.
CLN10 disease, late infantile
Some children with mutations in CLN10 have a later onset of symptoms and slower disease progression, like variant late infantile NCL. As children age, they develop seizures and progressive problems with vision, balance, and intellectual skills. Affected individuals also may have problems coordinating muscle movement and trouble with walking (called ataxia) as well as very stiff muscles (spasticity). Children with the disease often die in early childhood.
Adult Onset Batten Disease
Adult Batten Disease is very rare although affected families have been described from several different countries.
Adult Batten Disease has often been called Kufs disease and doctors recognize two main types – called type A and type B. Unlike the childhood NCLs, vision is not affected in either type. In some families inheritance is recessive but in others a dominant inheritance pattern is seen. Until the genetic basis for the adult NCLs is fully understood, diagnosis is usually dependent on a brain biopsy. It is emerging that several genes can cause adult onset NCL diseases, some of which have been presented at this meeting. Type A presents in early adulthood with a progressive myoclonic epilepsy, ataxia and slow cognitive deterioration over many years. Type B usually presents with an early dementia or evolving movement disorder. Mild mutations in childhood NCL genes may also cause NCL disease with delayed age of onset and slow disease progression but vision is generally affected and abnormal storage is seen more reliably in peripheral tissues.
Batten disease life expectancy
Children with Batten disease have a greatly shortened life expectancy. Children with infantile Batten disease often die in early childhood. Children with later onset forms of the disease may live into their teens to thirties, while those who develop the disease in adulthood may have a normal life expectancy.
Over time, affected children suffer cognitive impairment, worsening seizures, and progressive loss of sight and motor skills. Eventually, children with Batten disease become blind, bedridden, and have dementia.
- CLN1 disease, infantile onset: Most affected children die in early to mid-childhood.
- CLN1 disease, juvenile onset: Affected children may live into their teenage years. Others may not develop symptoms until adolescence and may live into adulthood.
- CLN2 disease, late-infantile onset: By age 6 most children are completely dependent on their caregivers, and many will require a feeding tube. Most children with CLN2 disease die between the ages of 6–12 years.
- CLN2 disease, later-onset: Affected children may live into their teenage years.
- CLN3 disease, juvenile onset (ages 4-7): Most children with the disease die between the ages of 15 and 30.
- CLN4 disease, adult onset: The age of death varies among affected individuals.
- CLN5 disease, variant late-infantile onset: Most children with CLN5 live into their late childhood or teenage years.
- CLN6, variant late-infantile onset: Most children with CNL6 die during late childhood or in their early teenage years.
- CLN6, adult onset: no data available.
- CLN7, variant late-infantile onset: Most children with the disorder live until their late childhood or teenage years.
- CLN8 disease with Epilepsy with Progressive Mental Retardation: Affected individuals can live into adulthood.
- CLN8 disease, late-variant onset: Life expectancy is uncertain; some children have lived into their second decade of life.
- CLN10 disease:
- In the congenital form babies may die shortly after birth or within the first weeks of life.
- A late-infantile form children with the disease often die in early childhood.
Batten disease symptoms
The symptoms of Batten disease stem from its classification as a lysosomal storage disorder, interfering with cells’ ability to break down wastes. The build-up of waste or lipofuscin, causes cell death and leads to the early death of children and some adults. During the progression of the disease, which varies by type, the affected child or adult can experience a combination of some of the below characteristics. It is important to note, each presentation is unique and not all symptoms are present in every phenotype.
- Seizures
- Visual impairment/blindness
- Personality and behavior changes
- Ataxia
- Myoclonus
- Dementia
- Cognitive decline
- Psychiatric symptoms (i.e. aggression)
- Extrapyramidal symptoms (i.e. spasms, restlessness, rigidity, tremors, jerky movements)
- Loss of motor skills and the ability to walk, talk and communicate
Batten Disease Diagnosis
Families tell of very long diagnostic journeys, often years after the first signs of seizures or loss of sight. In a recent needs assessment completed by BDSRA, more than 30 different diagnoses were reported by families before the final Batten diagnostic determination.
Autism, seizure disorder, epilepsy, pervasive developmental disorder and others are common early diagnoses. There have been recent improvements in genetic testing that have made diagnosing Batten disease much quicker and more reliable than in years past. Ultimately, we would like to see batten on newborn screening panels and are collaborating in efforts to do so.
Following a review of the person’s individual and family medical history and a neurological exam, several tests can be used to diagnose Batten disease and other neuronal ceroid lipofuscinoses. Currently, most diagnoses of Batten disease are made by genetic testing. Possible diagnostic tests include:
- DNA analysis/genetic testing. DNA analysis can confirm the presence of one of the mutated genes that cause an NCL disease, as well as be used in prenatal diagnosis of the disease. Increasingly, the NCL genes are being included on commercially available epilepsy gene panels that test several genes at the same time.
- Measurement of enzyme activity. Various enzymes are either deficient or missing in NCL disorders. Measuring enzyme levels can be used to confirm or rule out CLN1 and CLN2 disease.
- Blood or urine tests. These tests can detect abnormalities that may indicate Batten disease. For example, elevated levels of a chemical called dolichol are found in the urine of many individuals with NCL. And the presence of abnormal white blood cells that contain holes or cavities—called vacuolated lymphocytes—is common to certain disease mutations.
- Skin or tissue sampling. The accumulated lipofuscins form distinctive shapes—some look like half-moons while others look like fingerprints—when viewed under a microscope. The lipofuscins also take on a greenish-yellow color when viewed under an ultraviolet light microscope.
- Electroencephalogram (EEG). An EEG monitors brain activity through the skull, using electrodes that are placed on the scalp. Telltale patterns in the brain’s electrical activity suggest an individual has seizures.
- Electrical studies of the eyes. These tests, which include visual-evoked responses (which measure electrical activity in the brain generated by sight) and electroretinograms (used to detect abnormalities with the retina), can identify various eye problems common in several NCLs. And the greenish-yellow color of lipofuscins can sometimes be detected by examining the back of the eye. These are less frequently performed now, as most diagnosis can be made with DNA testing.
- Diagnostic imaging. Computed tomography (CT) and magnetic resonance imaging (MRI) create scans that can help doctors look for changes in the brain’s appearance.
Batten disease treatment
As yet, no specific treatment is known that can halt or reverse the symptoms of Batten Disease or other NCLs. However, seizures can sometimes be reduced or controlled with anticonvulsant drugs, and other medical problems can be treated appropriately as they arise. Other medicines are available to treat anxiety, depression, parkinsonism, and spasticity. Additional medical problems can be treated appropriately as they arise. At the same time, physical and occupational therapy may help patients retain function as long as possible. Support groups can help affected children, adults, and families to share common concerns and experiences, and to cope with the severe symptoms of the disease (visit Batten Disease Support and Research Association at https://bdsra.org).
Some reports have described a slowing of the disease in children with Batten Disease who were treated with vitamins C and E and with diets low in vitamin A. However, these treatments did not prevent the fatal outcome of the disease.
In 2017, the U.S. Food and Drug Administration has approved the use of Brineura® (cerliponase alfa) to slow the progression of symptoms in children with a late infantile form of the disorder called CLN2 (TTP1 deficiency) or Jansky-Bielschowsky Disease. Brineura® (cerliponase alfa) is available commercially through several hospitals across the U.S. For more information about this treatment visit https://www.brineura.com.
There are ongoing efforts for CLN2 treatments in both gene therapy and small molecule therapy. The work in scientific research for further treatments and a cure for all forms of Batten disease continues.





{kind=link}