
Contents
- Vitamin B5 (Pantothenic Acid) and Biotin (Vitamin B7)
- Pantothenic acid
- Pantothenic Acid function
- Pantothenic acid health benefits
- How much pantothenic acid do you need?
- What foods have Pantothenic acid?
- Pantothenic acid supplement
- What happens if I don’t get enough pantothenic acid?
- Pantothenic acid deficiency
- Pantothenic acid safety
- Biotin
- What does biotin do?
- Biotin health benefits
- How much biotin do I need?
- Biotin supplement
- What foods provide Biotin?
- Biotin deficiency
Vitamin B5 (Pantothenic Acid) and Biotin (Vitamin B7)
Pantothenic acid (also known as vitamin B5) and biotin (also known as vitamin B7 or vitamin H) are types of B vitamins. They are water-soluble, which means that the body can’t store them. If the body can’t use all of the vitamin, the extra vitamins leave the body through the urine. These vitamins must be replaced in the body every day.
Pantothenic acid and biotin are needed for growth. They help the body break down and use food. This is called metabolism. They are both required for making fatty acids.
Pantothenic acid is important for our bodies to properly use carbohydrates, proteins, and lipids and for healthy skin.
Pantothenic acid
Pantothenic acid also known as vitamin B5, is a water-soluble vitamin that is naturally present in some foods (i.e., eggs, milk, vegetables, beef, chicken, and whole grains), added to others, and available as a dietary supplement 1. The main function of pantothenic acid (vitamin B5) is a precursor in the biosynthesis of coenzyme A (CoA) and acyl carrier protein (Figure 1) 2, 3. Coenzyme A (CoA) is essential for fatty acid synthesis and degradation, transfer of acetyl and acyl groups, and a multitude of other anabolic and catabolic processes 4, 5. Acyl carrier protein’s main role is in fatty acid synthesis 3. Coenzyme A (CoA) reacts with acyl groups, giving rise to thioester derivatives, such as acetyl-CoA, succinyl-CoA, malonyl-CoA, and 3-hydroxy-3-methylglutaryl (HMG)-CoA. Coenzyme A (CoA) and its acyl derivatives are required for reactions that generate energy from the degradation of dietary fat, carbohydrates, and proteins 6. In addition, coenzyme A (CoA) in the form of acetyl-CoA and succinyl-CoA is involved in the citric acid cycle, in the synthesis of essential fats, cholesterol, steroid hormones, vitamins A and D, the neurotransmitter acetylcholine, and in the fatty acid beta-oxidation pathway 6. Coenzyme A (CoA) derivatives are also required for the synthesis of the hormone, melatonin, and for a component of hemoglobin called heme. Furthermore, metabolism of a number of drugs and toxins by the liver requires coenzyme A 7.
A wide variety of plant and animal foods contain pantothenic acid (vitamin B5) 2. About 85% of dietary pantothenic acid (vitamin B5) is in the form of coenzyme A (CoA) or phosphopantetheine 3, 5. These forms are converted to pantothenic acid (vitamin B5) by digestive enzymes (nucleosidases, peptidases, and phosphorylases) in the intestinal lumen and intestinal cells. Pantothenic acid (vitamin B5) is absorbed in the intestine and delivered directly into the bloodstream by active transport (and possibly simple diffusion at higher doses) 5, 2. Pantetheine, the dephosphorylated form of phosphopantetheine, however, is first taken up by intestinal cells and converted to pantothenic acid before being delivered into the bloodstream 3. The intestinal flora also produces pantothenic acid, but its contribution to the total amount of pantothenic acid that the body absorbs is not known 5. Red blood cells carry pantothenic acid throughout the body 5. Most pantothenic acid in tissues is in the form of coenzyme A (CoA), but smaller amounts are present as acyl carrier protein or free pantothenic acid 5.
Few data on pantothenic acid (vitamin B5) intakes in the United States are available. However, a typical mixed diet in the United States provides an estimated daily intake of about 6 mg, suggesting that most people in the United States consume adequate amounts 8. Some pantothenic acid (vitamin B5) intake information is available from other Western populations. For example, a 1996–1997 study in New Brunswick, Canada, found average daily pantothenic acid (vitamin B5) intakes of 4.0 mg in women and 5.5 mg in men 9.
Pantothenic acid status is not routinely measured in healthy people 1. Microbiologic growth assays, animal bioassays, and radioimmunoassays can be used to measure pantothenic concentrations in blood, urine, and tissue, but urinary concentrations are the most reliable indicators because of their close relationship with dietary intake 5. With a typical American diet, the urinary excretion rate for pantothenic acid is about 2.6 mg/day 10, 4. Excretion of less than 1 mg pantothenic acid per day suggests deficiency 11, 2. Like urinary concentrations, whole-blood concentrations of pantothenic acid correlate with pantothenic acid intake, but measuring pantothenic acid in whole blood requires enzyme pretreatment to release free pantothenic acid from coenzyme A (CoA) 2. Normal blood concentrations of pantothenic acid range from 1.6 to 2.7 mcmol/L, and blood concentrations below 1 mcmol/L are considered low and suggest deficiency 2. Unlike whole-blood concentrations, plasma levels of pantothenic acid do not correlate well with changes in intake or status 2.
Pantothenic acid (vitamin B5) deficiency is generally rare since the vitamin is present in many foods. However, pantothenic acid (vitamin B5) deficiency can present in people with severe malnutrition 12. An individual with pantothenic acid (vitamin B5) deficiency commonly has deficiencies in other nutrients, which can make it challenging to identify the effects that are specific to pantothenic acid (vitamin B5) deficiency. An experimental pantothenic acid (vitamin B5) deficiency study associated the deficiency with symptoms such as fatigue, headache, malaise, personality changes, numbness, muscle cramps, paresthesia, muscle/ abdominal cramps, nausea, and impaired muscle coordination 11.
Pantothenic acid kinase 2 (PANK2) catalyzes the initial step of phosphorylation of pantothenic acid to 4’-phosphopantothenic acid. Individuals with a mutation in their pantothenate kinase 2 (PANK2) gene are likely to have a pantothenic acid inadequacy as well. Enough PANK2 mutations reduce the activity of pantothenate kinase 2, which can potentially decrease the conversion of pantothenic acid to coenzyme A (CoA) and lead to reduced CoA levels. PANK2 gene mutations also cause pantothenate kinase-associated neurodegeneration (PKAN). A common hallmark of individuals with pantothenate kinase-associated neurodegeneration (PKAN) is an accumulation of iron in the brain that forms a pattern called the “eye of the tiger” sign 13. Pantothenate kinase-associated neurodegeneration (PKAN) disease also presents with a progressive movement disorder, and other symptoms may vary significantly from case to case. Symptoms include dysarthria, dystonia, poor balance, spasticity, and muscle rigidity. Treatment of pantothenate kinase-associated neurodegeneration (PKAN) focuses mainly on reducing symptoms. A few anecdotal reports indicate that vitamin B5 supplements can reduce symptoms, but the benefits of the general use of this supplement in PKAN are not known 14.
Figure 1. Coenzyme A (CoA) synthesis from pantothenic acid (vitamin B5)
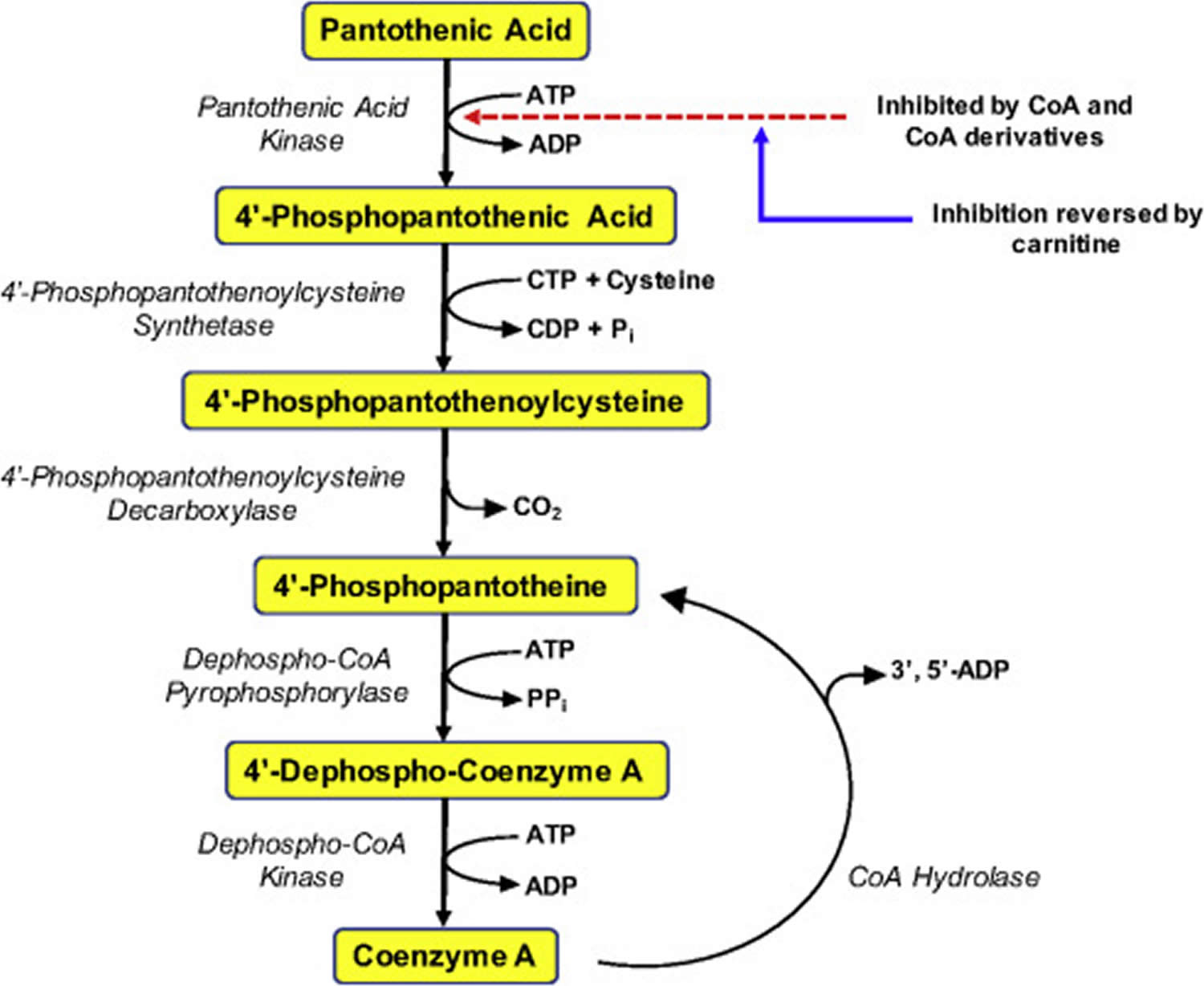
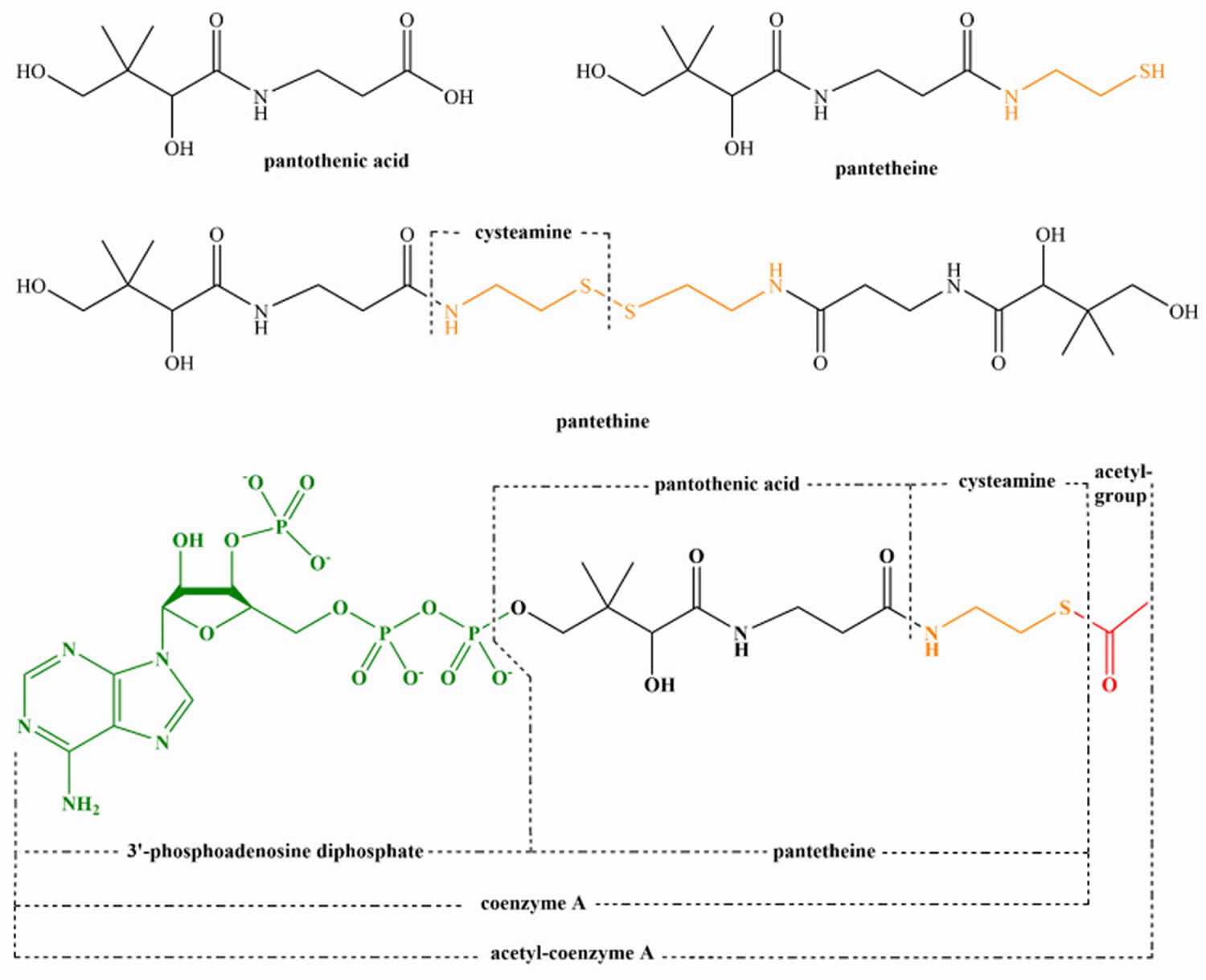
Figure 2. Structure of pantothenic acid and its derivatives (pantetheine, pantethine, and acetyl-CoA).
Pantothenic Acid function
Pantothenic acid or Vitamin B5 is essential for synthesis of coenzyme A (CoA) and acyl-carrier protein (ACP) and phosphopantetheine, which are crucial to fatty acid metabolism 17. Coenzyme A (CoA) plays a vital role in many catabolic and anabolic reactions. It is necessary for synthesis of fatty acids, cholesterol, acetylcholine, bile acids, and others 16. Coenzyme A (CoA) also plays a role in regulation of metabolism and gene expression. Coenzyme A (CoA) is required for processing large organic molecules, such as lipids, carbohydrates, and proteins. These reactions generate energy with formation of acylated forms of CoA, such as acetyl-CoA, succinyl-CoA, propionyl-CoA, isovaleryl-CoA, isobutyryl-CoA, α-methylbutyryl-CoA, and fatty acyl-CoA 18. The structure of pantothenic acid and its derivatives is shown in Figure 2.
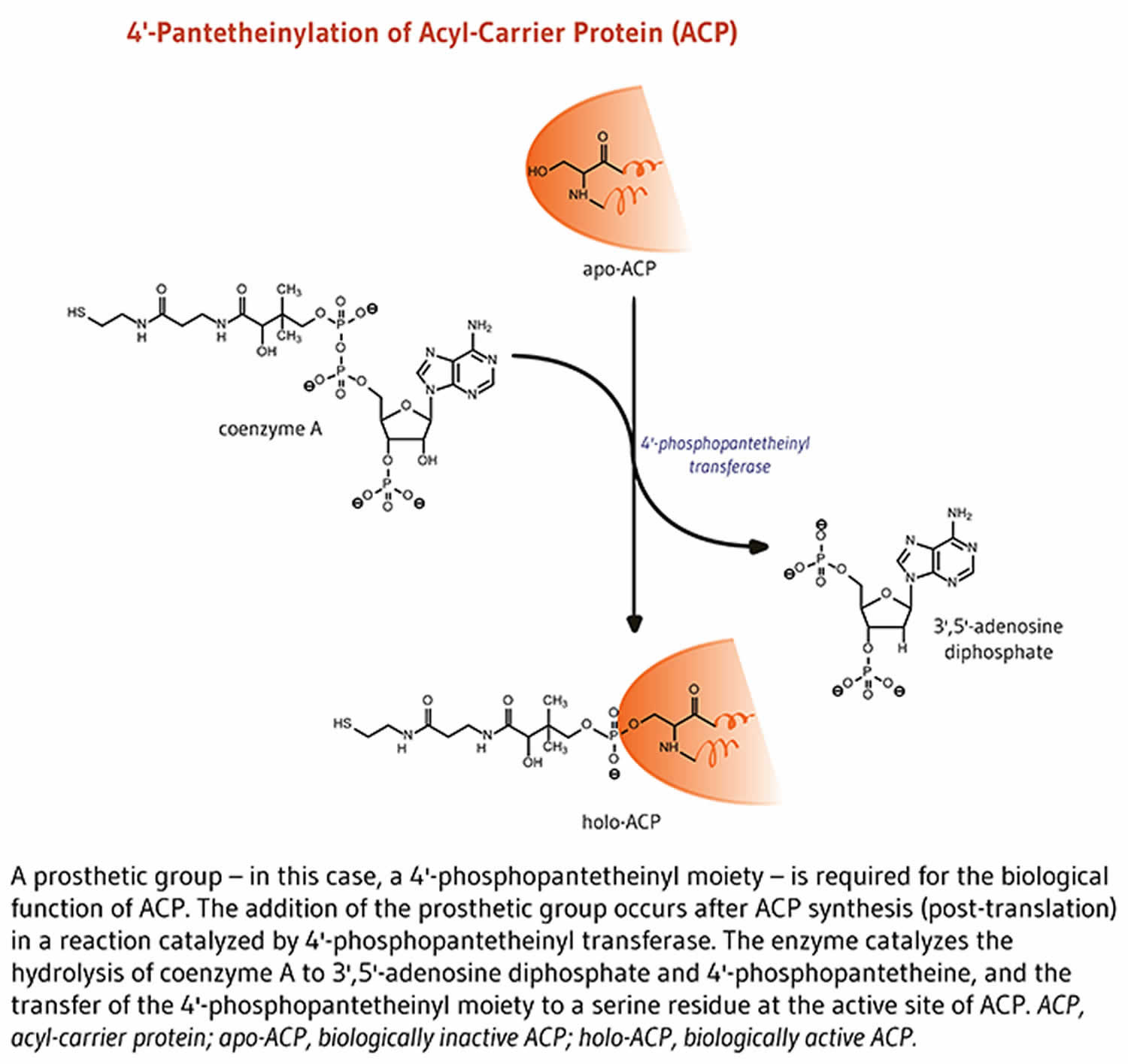
Acyl-carrier protein (ACP) is important for synthesis of fatty acids 16. Acyl-carrier protein (ACP) is expressed in the inactive form, apo-ACP. Its activation to holo-ACP requires the attachment of a prosthetic group (the 4′-phosphopantetheinyl moiety). This happens during the reaction with coenzyme A (CoA) catalyzed by 4′-phosphopantetheinyl transferase 19.
Coenzyme A
Pantothenic acid is a precursor in the biosynthesis of coenzyme A (CoA) (Figure 1), an essential coenzyme in a variety of biochemical reactions that sustain life 6. Pantothenic acid kinase 2 (PANK2) catalyzes the initial step of phosphorylation of pantothenic acid to 4’-phosphopantothenic acid. Coenzyme A (CoA) and its derivatives inhibit the synthesis of 4’-phosphopantothenic acid, but the inhibition can be reversed by carnitine, required for the transport of fatty acids into the mitochondria 20. The subsequent reactions in this biosynthetic pathway include the synthesis of the intermediate 4’-phosphopantetheine, as well as the recycling of coenzyme A to 4’-phosphopantetheine (Figure 1).
Coenzyme A (CoA) reacts with acyl groups, giving rise to thioester derivatives, such as acetyl-CoA, succinyl-CoA, malonyl-CoA, and 3-hydroxy-3-methylglutaryl (HMG)-CoA 6. Coenzyme A (CoA) and its acyl derivatives are required for reactions that generate energy from the degradation of dietary fat, carbohydrates, and proteins. In addition, coenzyme A (CoA) in the form of acetyl-CoA and succinyl-CoA is involved in the citric acid cycle, in the synthesis of essential fats, cholesterol, steroid hormones, vitamins A and D, the neurotransmitter acetylcholine, and in the fatty acid beta-oxidation pathway 6. Coenzyme A (CoA) derivatives are also required for the synthesis of the hormone, melatonin, and for a component of hemoglobin called heme 6. Furthermore, metabolism of a number of drugs and toxins by the liver requires coenzyme A (CoA) 7.
Coenzyme A was named for its role in acetylation reactions 6. Most acetylated proteins in the body have been modified by the addition of an acetate group that was donated by the coenzyme A thioester derivative, acetyl-CoA. Protein acetylation alters the overall charge of proteins, modifying their three-dimensional structure and, potentially, their function 6. For example, acetylation is a mechanism that regulates the activity of peptide hormones, including those produced by the pituitary gland 21. Also, protein acetylation, like other posttranslational modifications, has been shown to regulate the subcellular localization, the function, and the half-life of many signaling molecules, transcription factors, and enzymes. Notably, the acetylation of histones plays a role in the regulation of gene expression by facilitating transcription (i.e., mRNA synthesis), while deacetylated histones are usually associated with chromatin compaction and gene silencing. The acetylation of histones was found to result in structural changes of the chromatin, which affect both DNA-protein and protein-protein interactions. Crosstalk between acetylation marks and other posttranscriptional modifications of the histones also facilitate the recruitment of transcriptional regulators to the promoter of genes that are subsequently transcribed 22.
Finally, a number of signaling molecules are modified by the attachment of long-chain fatty acids donated by coenzyme A (CoA). These modifications are known as protein acylation and have central roles in cell-signaling pathways 7.
Acyl-carrier protein
Lipids are fat molecules essential for normal physiological function and, among other types, include sphingolipids (essential components of the myelin sheath that enhances nerve transmission), phospholipids (important structural components of cell membranes), and fatty acids 6. Fatty acid synthase (FAS) is a multi-enzyme complex that catalyzes the synthesis of fatty acids 6. Within the fatty acid synthase complex, the acyl-carrier protein (ACP) requires pantothenic acid in the form of 4′-phosphopantetheine for its activity as a carrier protein 20. A group, such as the 4’-phosphopantetheinyl moiety for acyl-carrier protein (ACP), is called a prosthetic group; the prosthetic group is not composed of amino acids and is a tightly bound cofactor required for the biological activity of some proteins (Figure 3). Acetyl-CoA, malonyl-CoA, and acyl-carrier protein (ACP) are all required for the synthesis of fatty acids in the cytosol 6. During fatty acid synthesis, the acyl groups of acetyl-CoA and malonyl-CoA are transferred to the sulfhydryl group (-SH) of the 4’-phosphopantetheinyl moiety of acyl-carrier protein (ACP). The prosthetic group is used as a flexible arm to transfer the growing fatty acid chain to each of the enzymatic centers of the type 1 fatty acid synthase complex 23. In the mitochondria, 4′-phosphopantetheine also serves as a prosthetic group for an acyl-carrier protein (ACP) homolog present in mitochondrial type 2 fatty acid synthase complex 23.
Figure 3. Acyl-carrier protein (ACP) function
10-formyltetrahydrofolate dehydrogenase
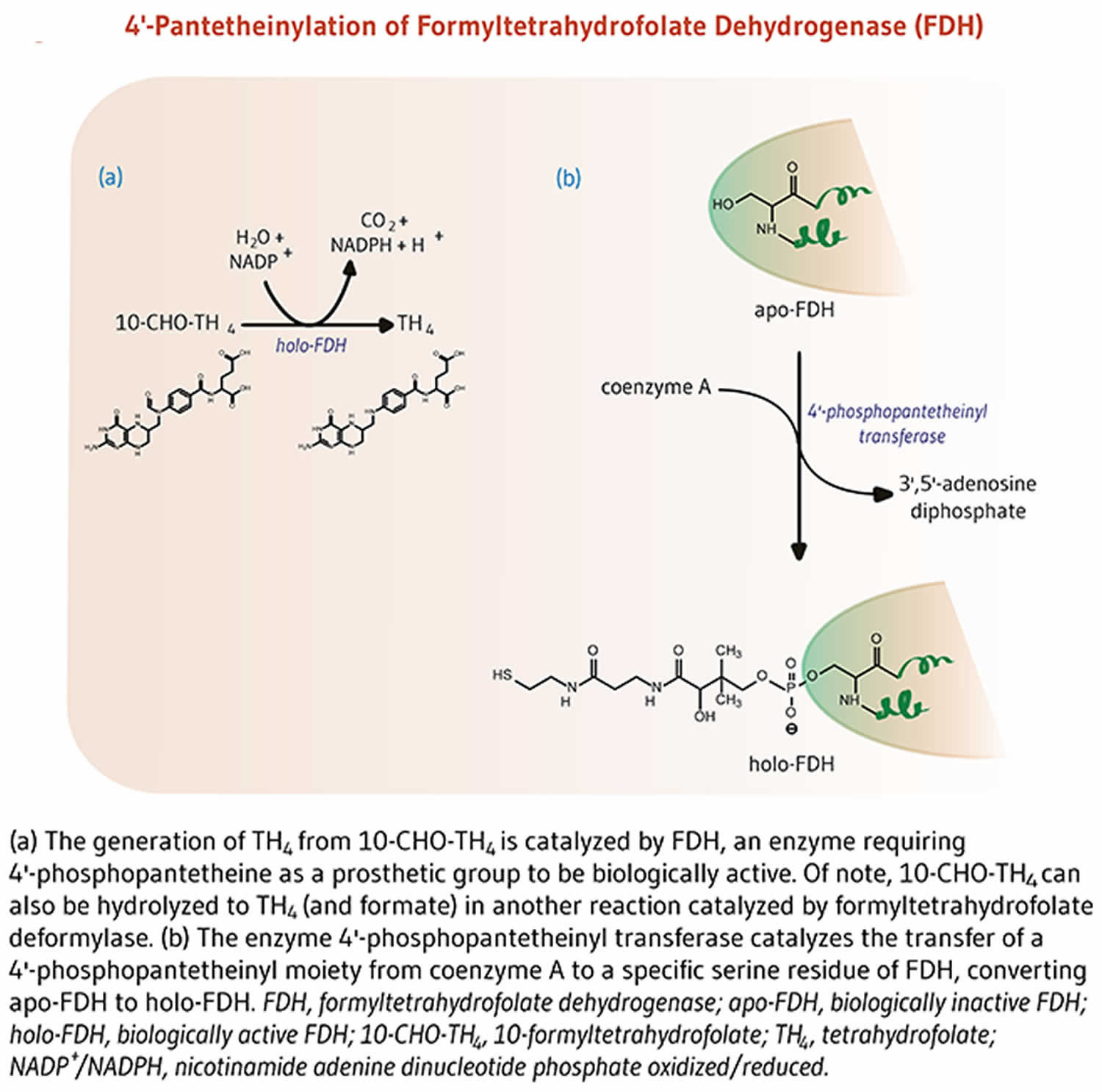
The enzyme 10-formyltetrahydrofolate dehydrogenase (FDH) catalyzes the conversion of 10-formyltetrahydrofolate to tetrahydrofolate, an essential cofactor in the metabolism of nucleic acids and amino acids (Figure 4) 6. Similar to acyl-carrier protein (ACP), 10-formyltetrahydrofolate dehydrogenase requires a 4’-phosphopantetheine prosthetic group for its biological activity 6. The prosthetic group acts as a swinging arm to couple the activities of the two catalytic domains of 10-formyltetrahydrofolate dehydrogenase 24, 25. A homolog of 10-formyltetrahydrofolate dehydrogenase in mitochondria also requires 4’-phosphopantetheinylation to be biologically active 26.
Figure 4. Formyltetrahydrofolate dehydrogenase function
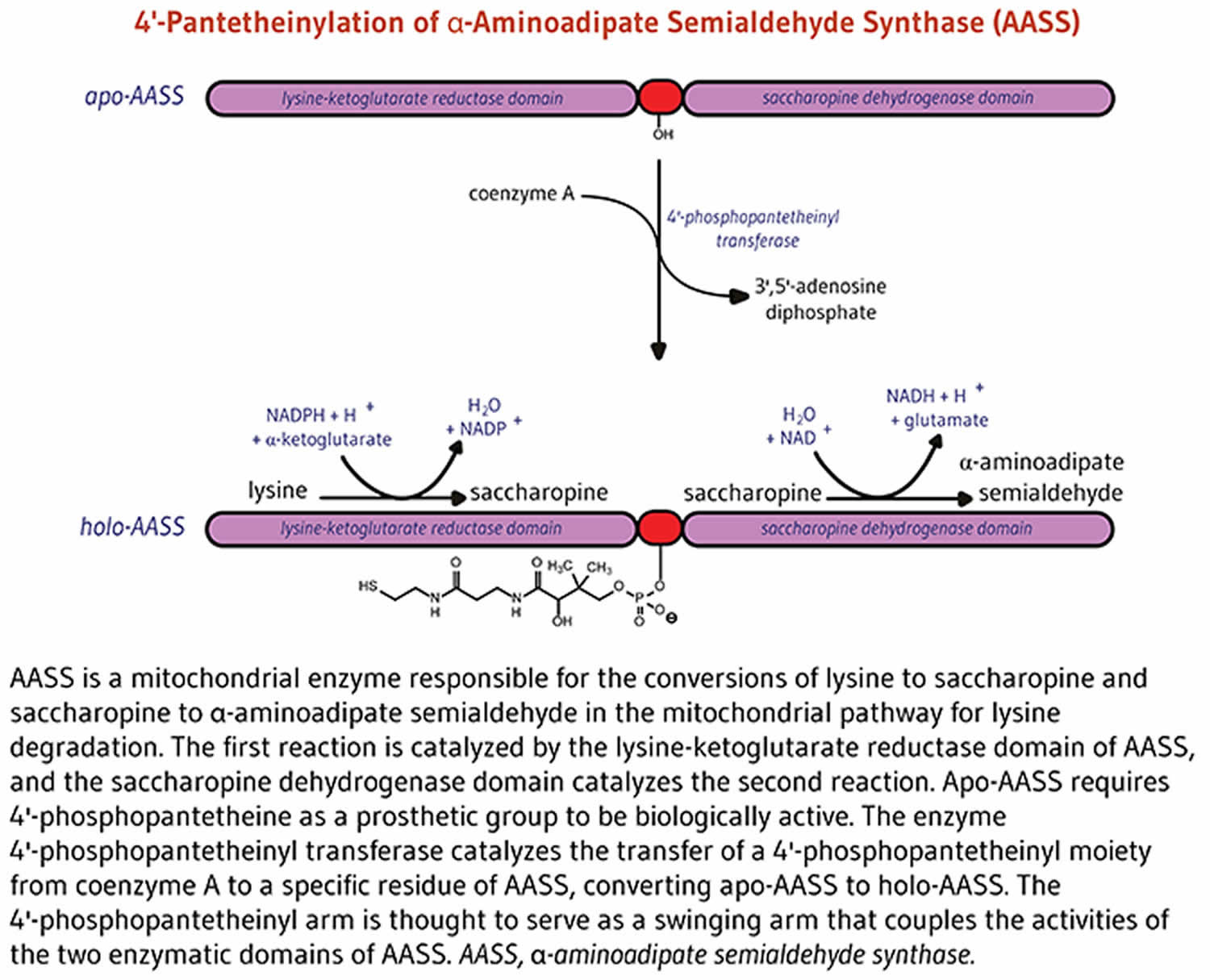
Alpha-aminoadipate semialdehyde synthase
4’-phosphopantetheinylation is required for the biological activity of the apo-enzyme alpha-aminoadipate semialdehyde synthase (AASS) 6. Alpha-aminoadipate semialdehyde synthase (AASS) catalyzes the initial reactions in the mitochondrial pathway for the degradation of lysine — an essential amino acid for humans 6. Alpha-aminoadipate semialdehyde synthase (AASS) is made of two catalytic domains. The lysine-ketoglutarate reductase domain first catalyzes the conversion of lysine to saccharopine. Saccharopine is further converted to α-aminoadipate semialdehyde in a reaction catalyzed by the saccharopine dehydrogenase domain (Figure 5).
Figure 5. Alpha-aminoadipate semialdehyde synthase (AASS) function
Pantothenic acid health benefits
Scientists are studying pantothenic acid or vitamin B5 to understand how it affects health. Here is what research have shown.
High cholesterol and triglyceride levels
Because of pantothenic acid’s role in triglyceride synthesis and lipoprotein metabolism, experts have hypothesized that vitamin B5 or pantothenic acid supplementation might reduce lipid levels in patients with hyperlipidemia 27. The form of pantothenic acid called pantethine is being studied to see if it helps lower total cholesterol, low-density lipoprotein (LDL or “bad”) cholesterol, and triglyceride levels. It’s also being studied to see if it raises levels of high-density lipoprotein (HDL or “good”) cholesterol. The results of these studies so far are promising, but more research is needed to understand the effects of pantethine dietary supplements taken alone or combined with a heart-healthy diet.
Several clinical trials have shown that the form of pantothenic acid known as pantethine reduces lipid levels when taken in large amounts 28, but pantothenic acid itself does not appear to have the same effects 2. A 2005 review included 28 small clinical trials (average sample size of 22 participants) that examined the effect of pantethine supplements (median daily dose of 900 mg for an average of 12.7 weeks) on serum lipid levels in a total of 646 adults with hyperlipidemia 28. On average, the supplements were associated with triglyceride declines of 14.2% at 1 month and 32.9% at 4 months. The corresponding declines in total cholesterol were 8.7% and 15.1%, and for low-density lipoprotein (LDL) cholesterol were 10.4% and 20.1%. The corresponding increases in high-density lipoprotein (HDL) cholesterol were 6.1% and 8.4% 28.
A few additional clinical trials have assessed pantethine’s effects on lipid levels since the publication of the 2005 review. A double-blind trial in China randomly assigned 216 adults with hypertriglyceridemia (204–576 mg/dl) to supplementation with 400 U/day coenzyme A (CoA) or 600 mg/day pantethine 29. All participants also received dietary counseling. Triglyceride levels dropped by a significant 16.5% with pantethine compared with baseline after 8 weeks. Concentrations of total cholesterol and non–HDL cholesterol also declined modestly but significantly from baseline. However, these declines might have been due, at least in part, to the dietary counseling that the participants received 29.
Two randomized, blinded, placebo-controlled studies by the same research group in a total of 152 adults with low to moderate cardiovascular disease risk found that 600 mg/day pantethine for 8 weeks followed by 900 mg/day for 8 weeks plus a therapeutic lifestyle change diet resulted in small but significant reductions in total cholesterol, LDL cholesterol, and non-HDL cholesterol compared with placebo after 16 weeks 27, 30. Increasing the amount of pantethine from 600 to 900 mg/day did not increase the magnitude of reduction in the lipid measures.
Additional studies are needed to determine whether pantethine supplementation has a beneficial effect on hyperlipidemia independently of, and together with, eating a heart-healthy diet. Research is also needed to determine the mechanisms of pantethine’s effects on lipid levels.
Wound healing
The addition of calcium D-pantothenate and/or pantothenol to the medium of cultured skin fibroblasts given an artificial wound was found to increase cell proliferation and migration, thus accelerating wound healing in vitro (test tube studies) 31, 32. Likewise, in vitro (test tube studies) deficiency in pantothenic acid induced the expression of differentiation markers in proliferating skin fibroblasts and inhibited proliferation in human keratinocytes 33. The application of ointments containing either calcium D-pantothenate or pantothenol — also known as D-panthenol or dexpanthenol — to the skin has been shown to accelerate the closure of skin wounds and increase the strength of scar tissue in animals 20.
The effects of dexpanthenol on wound healing are unclear. In a placebo-controlled study that included 12 healthy volunteers, the application of dexpanthenol-containing ointment (every 12 hours for 1 to 6 days) in a model of skin wound healing was associated with an enhanced expression of markers of proliferation, inflammation, and tissue repair 34. However, the study failed to report whether these changes in response to topical dexpanthenol improved the wound-repair process compared to placebo 34. Some studies have shown no effects. Early randomized controlled trials in patients undergoing surgery for tattoo removal found that daily co-supplementation with 1 gram or 3 grams of vitamin C and 200 mg or 900 mg of pantothenic acid (vitamin B5) for 21 days did not significantly improve the wound-healing process 35, 36. Yet, in a recent randomized, double-blind, placebo-controlled study, the use of dexpanthenol pastilles (300 mg/day for up to 14 days post surgery) was found to accelerate mucosal healing after tonsillectomy in children 37.
Facial acne
A randomized, double-blind, placebo-controlled study of adults (average age of 31.8 ± 8.4 years) previously diagnosed with mild to moderate acne vulgaris was performed over over 12-weeks 38, 39. Subjects were randomized to the study agent, a pantothenic acid-based dietary supplement (2.2 g of pantothenic acid twice a day with food), or a placebo for 12 weeks 39. The primary outcome of the study was the difference in total lesion count between the study agent group versus the placebo group from baseline to endpoint. Secondary measurements included differences in mean non-inflammatory and inflammatory lesions, Investigators Global Assessment and Dermatology Life Quality Index (DLQI) scores between the two groups. The results from this study indicate that the administration of a pantothenic acid-based dietary supplement in healthy adults with facial acne lesions is safe, well tolerated and reduced total facial lesion count versus placebo after 12 weeks of administration 39. Secondary analysis shows that the study agent significantly reduced area-specific and inflammatory blemishes 39. Further randomized, placebo-controlled trials are needed to confirm these findings.
Skin conditions
The usage of vitamin B5 is prevalent within the field of dermatology. This interest has led to a study that compares the effectiveness of reduced form of vitamin B5 dexpanthenol (D-panthenol) as an alternative treatment to atopic dermatitis against a standard treatment of hydrocortisone. Overall, the study found that dexpanthenol can potentially treat mild to moderate childhood atopic dermatitis therapy 40. Other research suggests that dexpanthenol cream can be useful in managing mucocutaneous side effects that occur during isotretinoin therapy 41. Isotretinoin therapy is used as a treatment for acne, and its mucocutaneous side effects include dryness of mucous membranes, cheilitis, and xerosis.
The reduced form of vitamin B5 dexpanthenol (D-panthenol) effects are, however, likely not related to the physiological function of vitamin B5 but are mediated by its moisturizing effect, which is based on its hygroscopic property 16. Vitamin B5 dexpanthenol (D-panthenol) could be used topically as a cream, emollient, drops, gel, lotion, oil, ointment, solution, and spray in concentration of 2–5% 42. Dexpanthenol protects epithelium and promotes cellular proliferation. During the wound healing, it helps to recover the epidermal barrier function, has anti-inflammatory activity, and supports wound closure 43.
The healing properties of dexpanthenol-containing cream (5%) were confirmed on superficial skin lesions caused by application of 5% sodium lauryl sulfate solution for 4 hours. One week, twice daily dexpanthenol-containing cream (5%) application led to a significant enhancement of stratum corneum hydration, as well as reduction in skin roughness and inflammation 44. Other studies confirmed the effect of dexpanthenol containing emollient on sodium dodecyl sulfate (0.5%) induced skin barrier dysfunction. Dexpanthenol improved skin hydration and increased ceramide 3, as well as free fatty acid and cholesterol content, in the stratum corneum, and it also supported recolonization of the skin with commensal bacteria 45.
Dexpanthenol in ointment with petroleum jelly led to a significantly faster and pronounced reduction of skin lesions size and better re-epithelialization of ablative CO2 laser photo-damaged skin than the petroleum-jelly cream itself 46. Protective effect of an ointment with dexpanthenol (5%) was also seen in combination with zinc oxide in irritant diaper dermatitis in comparison to the control ointment base 47. Two-week administration of dexpanthenol 5% water-oil formulations 4 to 8 times daily restored the skin barrier of freshly tattooed skin. The disadvantage of this study is that the effect was not compared with a control group 48.
In treatment of atopic dermatitis in children, dexpanthenol (5%) ointment exerted equal effectiveness to hydrocortisone (1%) ointment and, therefore, can be used as alternative to treatment of mild and moderate atopic dermatitis 40.
Use of dexpanthenol cream (5%) on treatment of traumatic nipples of breastfeeding mothers had the same therapeutic effect in comparison with pure lanolin or 0.2% peppermint oil creams administered every 8 hours for 14 days 49.
The application of 2% dexpanthenol drops on corneal epithelial wounds after surface laser ablation only induced little effect on corneal epithelial regeneration, and, in general, the effect was of minimal clinical relevance after 2 months of use 50. However, dexpanthenol has been found to be effective in treatment of dry eye, where it exerted superior improvement in disturbances of corneal epithelium permeability comparing with dexpanthenol-free drops 51.
Dexpanthenol is also added to topical nasal decongestant (sprays and droplets) containing α-sympathomimetics to treat acute allergic or non-allergic rhinitis or after nasal surgery. A combined preparation of oxymethazoline (0.05%) with dexpanthenol (5%) showed a better efficacy than xylomethazoline (0.1%) alone in patients with acute allergic rhinitis or with post-nasal surgery. The relief in nasal congestion was significantly better, recovery time was shorter, and significant improvements in sneezing, nasal discharge, and irritation were also observed 52. Similarly, addition of dexpanthenol to xylometazoline significantly reduced nasal obstruction, rhinorrhea, hyperplasia of nasal concha, and redness of the nasal mucous membrane compared with xylometazoline alone 53, 54.
Graying of hair
Mice that are deficient in pantothenic acid (vitamin B5) developed skin irritation and graying of the fur, which is reversed by pantothenic acid administration. In humans, there is no evidence that taking pantothenic acid (vitamin B5) as supplements or using shampoos containing pantothenic acid can prevent or restore hair color 55.
How much pantothenic acid do you need?
The amount of pantothenic acid or vitamin B5 you need depends on your age and sex. Average daily recommended amounts are listed below in milligrams (mg).
Few data on vitamin B5 or pantothenic acid intakes in the United States are available 1. However, a typical mixed diet in the United States provides an estimated daily intake of about 6 mg, suggesting that most people in the United States consume adequate amounts 8. Some intake information is available from other Western populations. For example, a 1996–1997 study in New Brunswick, Canada, found average daily pantothenic acid intakes of 4 mg in women and 5.5 mg in men 9.
Table 1. Adequate Intakes for Pantothenic Acid (vitamin B5)
| Life Stage | Recommended Amount |
|---|---|
| Birth to 6 months | 1.7 mg |
| Infants 7–12 months | 1.8 mg |
| Children 1–3 years | 2 mg |
| Children 4–8 years | 3 mg |
| Children 9–13 years | 4 mg |
| Teens 14–18 years | 5 mg |
| Adults 19 years and older | 5 mg |
| Pregnant teens and women | 6 mg |
| Breastfeeding teens and women | 7 mg |
Footnote:
- Adequate Intake (AI) = Intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an Recommended Dietary Allowance (RDA).
- Recommended Dietary Allowance (RDA) = Average daily level of intake sufficient to meet the nutrient requirements of nearly all (97%–98%) healthy individuals; often used to plan nutritionally adequate diets for individuals.

What foods have Pantothenic acid?
Pantothenic acid or Vitamin B5 is naturally present in almost all plant- and animal-based foods 5. Vitamin B5 or pantothenic acid is also added to some foods, including some breakfast cereals and beverages (such as energy drinks) 5. Limited data indicate that the body absorbs 40%–61% (or half, on average) of pantothenic acid from foods 10.
The U.S. Department of Agriculture’s (USDA’s) FoodData Central (https://fdc.nal.usda.gov) lists the nutrient content of many foods and provides a comprehensive list of foods containing pantothenic acid arranged by nutrient content (https://www.nal.usda.gov/sites/www.nal.usda.gov/files/pantothenic_acid.pdf).
You can get recommended amounts of vitamin B5 (pantothenic acid) by eating a variety of foods, including the following:
- Beef, poultry, seafood, and organ meats
- Eggs and milk
- Vegetables such as mushrooms (especially shiitakes), avocados, potatoes, and broccoli
- Whole grains, such as whole wheat, brown rice, and oats
- Peanuts, sunflower seeds, and chickpeas.
Food processing may alter the content of vitamin B5 or pantothenic acid 56, 57. The milling of cereals, in which grains, such as wheat, rice, and corn, are dehulled and ground into smaller pieces or flours to improve palatability, reduce cooking time, and create food products, but remove grain parts rich in micronutrients, resulting in considerable losses of vitamin B5 or pantothenic acid 58, 59, 60, 61, 62. Milling reduces vitamin B5 or pantothenic acid contents, in comparison to whole cereals, by 50–55% and 64–88% in wheat and maize, respectively 58, 63, 64. Vitamin B5 or pantothenic acid losses are 50–67% and 18–25% in non-parboiled and parboiled white rice, respectively, compared to brown rice 58, 65, 61, 64.
Vitamin B5 or pantothenic acid is quite stable during thermal processing at pH levels of 5–7; losses of pantothenic acid during the preparation and cooking of foods are normally not very large 66, 56, but substantial losses of pantothenic acid can occur through leaching into the cooking liquids, such as water, soup, gravy, or drippings; when these are consumed along with the cooked food, a great part of the vitamin is retained 67, 68, 69, 70, 71. Vitamin B5 or pantothenic acid content in pork, beef, and chicken is reduced owing to steaming, braising, and, in particular, by boiling, by 15–50% solely in meat due to leaching. In the whole dish, the losses are only 10–20%. Frying decreases the vitamin level by 20%, and it only decreases by 10% when the meat is breaded 67. Similarly, a decrease in pantothenic acid in fish during cooking by different methods comes about 67, 72, 73. Steaming, boiling, baking, and frying of potatoes with the peel bring on pantothenic acid losses of 10% in all cases, but the losses might reach 30% in peeled potatoes when boiled 67, 68. In addition, in vegetables, boiling and steaming usually causes declines of 10% in the total dish, and those of 30–40% and 15%, respectively, in vegetables alone 67, 68, 74, 75. Stewing, frying, and baking lessen pantothenic acid amounts in vegetables by 10% 67, 68. Vitamin B5 or pantothenic acid losses of 24–67% in legumes during boiling are influenced by the pre-soaking method and cooking times 69, 76, 77. Boiling of rice results in a decrease of 59–66% in pantothenic acid content 58. That is why steaming is preferred to boiling, in particular, when cooked vegetables are eaten without cooking liquids 67, 74, 75. Poached, boiled, and fried eggs lose, due to cooking, 4%, 7%, and 9% of their pantothenic acid, respectively 78. In milk, pantothenic acid is stable during pasteurization, since the normal pH of milk is within the optimal pH stability range; milk generally loses less than 10% during processing 69, 56, 79.
In breadmaking, no significant difference of vitamin B5 or pantothenic acid was observed during the kneading phase, while a mild decrease of 12% was documented during baking. This indicates that pantothenic acid is more sensitive to heat than to light and oxygen 80. The roasting of peanuts at 160 °C and 180 °C decreases the amount of pantothenic acid by 24% and 92%, respectively; so, peanuts can be an excellent source if properly processed 81.
Canning leads to various reductions in vitamin B5 or pantothenic acid content: 1–43% in pork luncheon meat, depending on times and temperatures used during thermal processing 82; 20–35%, 46–78%, and 51%, in foods of animal origin (such as meats, fish, and dairy products), vegetables, and fruits and fruit juices, respectively 64. Thermal degradation kinetics of pantothenic acid in extracts of Averrhoa bilimbi fruits showed that increasing the temperature speeds up the decomposition, which was also linearly time-dependent 83. Treatment of food with ionizing radiation used as a method for its preservation has insignificant effects on pantothenic acid content 84, 85. Less vitamin B5 or pantothenic acid is in food products based on nixtamalized (i.e., alkali-treated) maize 86, 87.
Lower contents of vitamin B5 or pantothenic acid in frozen foods, compared to those in raw ones, have been reported; decreases were 18–63% in vegetables, 29–71% in legumes, 7% in fruits and fruit juices, and 4–55% in fish 88, 64, 89. After thawing frozen meat, pantothenic acid, together with other B vitamins, transfer in a drip; amounts of pantothenic acid from defrosted meat found in the drip were 7% and 33% in pork and in beef, respectively. For prevention of the loss of the vitamin, collection and use of the drip is recommended 70, 90, 91.
Regarding fortification of foods using vitamin B5 or pantothenic acid, adult human intake of that vitamin has generally been considered adequate in view of the absence of deficiency in normal populations and the fact that the daily requirement for vitamin B5 or pantothenic acid is easily fulfilled from most natural dietary sources owing to its ubiquitous distribution 58, 92. Pantothenic acid (as calcium pantothenate or sodium pantothenate or dexpanthenol) is added to various foods (such as milk-based products, breakfast cereals, and rice powders) to prevent deficiency due to incorrect nutrition or malnutrition or for certain nutritional requirements (baby foods, e.g., for non-breastfed infants; athletes’ products; low-calorie, reduced-calorie, and vitamin-rich foods) 93, 94, 95, 96, 92, 97.
Table 2. Pantothenic acid (vitamin B5) content of selected foods
| Food | Milligrams (mg) per serving | Percent DV* |
|---|---|---|
| Beef liver, boiled, 3 ounces | 8.3 | 166 |
| Breakfast cereals, fortified with 100% of the DV | 5 | 100 |
| Shitake mushrooms, cooked, ½ cup pieces | 2.6 | 52 |
| Sunflower seeds, ¼ cup | 2.4 | 48 |
| Chicken, breast meat, skinless, roasted, 3 ounces | 1.3 | 26 |
| Tuna, fresh, bluefin, cooked, 3 ounces | 1.2 | 24 |
| Avocados, raw, ½ avocado | 1 | 20 |
| Milk, 2% milkfat, 1 cup | 0.9 | 18 |
| Mushrooms, white, stir fried, ½ cup sliced | 0.8 | 16 |
| Potatoes, russet, flesh and skin, baked, 1 medium | 0.7 | 14 |
| Egg, hard boiled, 1 large | 0.7 | 14 |
| Greek yogurt, vanilla, nonfat, 5.3-ounce container | 0.6 | 12 |
| Ground beef, 85% lean meat, broiled, 3 ounces | 0.6 | 12 |
| Peanuts, roasted in oil, ¼ cup | 0.5 | 10 |
| Broccoli, boiled, ½ cup | 0.5 | 10 |
| Whole-wheat pita, 1 large | 0.5 | 10 |
| Chickpeas, canned, ½ cup | 0.4 | 8 |
| Rice, brown, medium grain, cooked, ½ cup | 0.4 | 8 |
| Oats, regular and quick, cooked with water, ½ cup | 0.4 | 8 |
| Cheese, cheddar, 1.5 ounces | 0.2 | 4 |
| Carrots, chopped, raw, ½ cup | 0.2 | 4 |
| Cabbage, boiled, ½ cup | 0.1 | 2 |
| Clementine, raw, 1 clementine | 0.1 | 2 |
| Tomatoes, raw, chopped or sliced, ½ cup | 0.1 | 2 |
| Cherry tomatoes, raw, ½ cup | 0 | 0 |
| Apple, raw, slices, ½ cup | 0 | 0 |
Footnote: *DV = Daily Value. The Daily Value (DV) for pantothenic acid is 5 mg for adults and children age 4 years and older. The U.S. Food and Drug Administration (FDA) does not require food labels to list pantothenic acid content unless pantothenic acid has been added to the food. Foods providing 20% or more of the DV (Daily Value) are considered to be high sources of a nutrient, but foods providing lower percentages of the DV (Daily Value) also contribute to a healthful diet.
[Source 98 ]Pantothenic acid supplement
Pantothenic acid (vitamin B5) is available in dietary supplements containing only pantothenic acid, in combination with other B-complex dietary supplements and in some multivitamin or multimineral supplements 1. Pantothenic acid (vitamin B5) in dietary supplements is often in the form of calcium pantothenate or pantethine (a dimeric form of pantetheine) 5, 89, 99. Research has not shown that any form of pantothenic acid is better than the others. The amount of pantothenic acid in dietary supplements typically ranges from about 10 mg in multivitamin/multimineral products to up to 1,000 mg in supplements of B-complex vitamins or pantothenic acid alone 1. Pantethine is used as a cholesterol-lowering agent in Japan and is available in the US as a dietary supplement 100.
What happens if I don’t get enough pantothenic acid?
Pantothenic acid or Vitamin B5 deficiency is very rare in the United States because most people in the United States get enough vitamin B5 or pantothenic acid from their diet. However, people with severe malnutrition or people with a rare inherited disorder called pantothenate kinase-associated neurodegeneration mutation (PKAN) can’t use pantothenic acid properly. These disorders can lead to symptoms of pantothenic acid deficiency. Severe vitamin B5 or pantothenic acid deficiency can cause numbness and burning of the hands and feet, headache, extreme tiredness, irritability, restlessness, sleeping problems, stomach pain, heartburn, diarrhea, nausea, vomiting, and loss of appetite.
A common hallmark of individuals with pantothenate kinase-associated neurodegeneration mutation (PKAN) (formerly called Hallervorden-Spatz syndrome) is an accumulation of iron in the brain that forms a pattern called the “eye of the tiger” sign 13, 101. Pantothenate kinase-associated neurodegeneration mutation (PKAN) also presents with a progressive movement disorder, and other symptoms may vary significantly from case to case. Symptoms include dysarthria, dystonia, dysphasia, poor balance, spasticity, and muscle rigidity 13. Dementia, severe mental retardation and severe movement disability may develop at later stages 102. Rare clinical features include rigidity, parkinsonism, choreoathetosis, seizures, optic atrophy, and pigmentary retinopathy 13. Based on age at onset and rate of progression, PKAN can be classified in two major forms. In the classic form of PKAN, onset is usually in the first decade of life. Visual impairment caused by optic atrophy or retinal degeneration have been described in some classical cases 13. Atypical PKAN is presented in the second decade of life with slow progression 13. Neurobehavioral disorders and seizure are common in atypical form 103. All PKAN cases have mutation in pantothenate kinase 2 (PANK2) gene located on the short arm of chromosome 20 (20p13) 104. PANK2 encodes a mitochondrial pantothenate kinase which is the key regulatory enzyme in coenzyme A biosynthesis 105. Treatment of pantothenate kinase-associated neurodegeneration mutation (PKAN) focuses mainly on reducing symptoms. A few anecdotal reports indicate that vitamin B5 supplements can reduce symptoms, but the benefits of the general use of this supplement in PKAN are not known 106.
Pantothenic acid deficiency
Because some pantothenic acid (vitamin B5) is present in almost all foods, pantothenic acid (vitamin B5) deficiency is very rare in the United States except in people with severe malnutrition 5. When someone has a pantothenic acid (vitamin B5) deficiency, it is usually accompanied by deficiencies in other nutrients, making it difficult to identify the effects that are specific to pantothenic acid deficiency 2. The only individuals known to have developed pantothenic acid deficiency were fed diets containing virtually no pantothenic acid or were taking a pantothenic acid metabolic antagonist 4.
On the basis of the experiences of prisoners of war in World War II and studies of diets lacking pantothenic acid in conjunction with administration of an antagonist of pantothenic acid metabolism, a pantothenic acid (vitamin B5) deficiency is associated with numbness and burning of the hands and feet, headache, fatigue, extreme tiredness, irritability, restlessness, disturbed sleep, and gastrointestinal disturbances such as stomach pain, heartburn, diarrhea, nausea, vomiting, and loss of appetite 2, 5, 107, 108, 11.
The following group is most likely to have inadequate pantothenic acid (vitamin B5) status:
Pantothenate kinase-associated neurodegeneration (PKAN)
Pantothenate kinase-associated neurodegeneration (PKAN) also known as Hallervorden-Spatz syndrome is a rare inherited neurological movement disorder characterized by the progressive degeneration of specific regions in the central nervous system (neurodegenerative disorder) and buildup of iron in the brain 109. Pantothenate kinase-associated neurodegeneration (PKAN) is characterized by progressive difficulty with movement, typically beginning in childhood. Movement abnormalities include involuntary muscle spasms, rigidity, and trouble with walking that worsens over time 110, 111, 101. Many people with pantothenate kinase-associated neurodegeneration (PKAN) also develop problems with speech (dysarthria), and some develop vision loss. Additionally, affected individuals may experience a loss of intellectual function (dementia) and psychiatric symptoms such as behavioral problems, personality changes, and depression.
Pantothenate kinase-associated neurodegeneration (PKAN) is the most common type of neurodegeneration with brain iron accumulation (NBIA), a group of clinical disorders marked by progressive abnormal involuntary movements, alterations in muscle tone, and postural disturbances (extrapyramidal) 112, 13, 110, 113, 114, 103. Pantothenate kinase-associated neurodegeneration (PKAN) is also known as neurodegeneration with brain iron accumulation type 1 (NBIA type 1), which accounts for approximately half of the cases of neurodegeneration with brain iron accumulation (NBIA) 13. The neurodegeneration with brain iron accumulation (NBIA) disorders show radiographic evidence of iron accumulation in the brain, called the “eye-of-the-tiger sign”, which is typically seen on magnetic resonance imaging (MRI) scans of the brain in people with pantothenate kinase-associated neurodegeneration (PKAN) (Figure 5). The ‘eye of the tiger’ pattern of iron accumulation in the globus pallidus on T2 weighted magnetic resonance imaging (MRI) which is caused by iron deposition in the periphery (hypointensity) and necrosis on its core (hyperintensity) 115, 116.
Pantothenate kinase-associated neurodegeneration (PKAN) is inherited as an autosomal recessive genetic condition caused by mutations in the pantothenate kinase 2 (PANK2) gene, located on the short arm of chromosome 20 (20p13) 110. The PANK2 gene provides instructions for making an enzyme called pantothenate kinase 2 117. The pantothenate kinase 2 (PANK2) enzyme is active in specialized cellular structures called mitochondria, which are the cell’s energy-producing centers. Within mitochondria, pantothenate kinase 2 regulates the formation of a molecule called coenzyme A (CoA). Coenzyme A (CoA) is found in all living cells, where it is essential for the body’s production of energy from carbohydrates, fats, and some protein building blocks (amino acids).
PANK2 is one of four human genes that provide instructions for making versions of pantothenate kinase. The functions of these different versions probably vary among tissue types and parts of the cell. The version produced by the PANK2 gene is active in cells throughout the body, including nerve cells in the brain.
Vitamin B5 is required for the production of coenzyme A in cells. Disruption of this enzyme affects energy and lipid metabolism and may lead to accumulation of potentially harmful compounds in the brain, including iron. Currently, PANK2 is the only gene known to be associated with pantothenate kinase-associated neurodegeneration (PKAN).
Recessive genetic disorders occur when an individual inherits a non-working gene from each parent 110. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the non-working gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy 110. The chance for a child to receive working genes from both parents is 25% 110. The risk is the same for males and females.
Pantothenate kinase-associated neurodegeneration (PKAN) clinical presentations include dystonia, dysarthria, and dysphasia. Dementia, severe mental retardation and severe movement disability may develop at later stages 102. Rare clinical features include rigidity, parkinsonism, choreoathetosis, seizures, optic atrophy, and pigmentary retinopathy.
Based on age at onset and rate of progression, pantothenate kinase-associated neurodegeneration (PKAN) is usually classified into two major forms: classic and atypical PKAN.
- Classic PKAN causes symptoms in the first 10 years of life, with symptoms that worsen rapidly. Visual impairment caused by optic atrophy or retinal degeneration have been described in some classical cases.
- Atypical PKAN usually occurs after the age of 10 and progresses more slowly. Neurobehavioral disorders and seizure are common in atypical form 103.
- Classic PKAN tends to have onset before 6 years of age, whereas atypical PKAN manifests at a mean age of 14 years 106. Some people have been diagnosed in infancy or adulthood, and some of those affected have characteristics that are between the two categories.
- Signs and symptoms vary, but the atypical PKAN is more likely than the classic PKAN to involve speech defects and psychiatric problems.
- Pantothenate kinase-associated neurodegeneration (PKAN) prevalence is estimated around 1 to 3 per million 106.
All individuals with PKAN have an abnormal buildup of iron in certain areas of the brain. A particular change, called the ‘eye-of-the-tiger sign’, which indicates a buildup of iron, is typically seen on magnetic resonance imaging (MRI) scans of the brain in people with this disorder.
A condition called HARP (hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration) syndrome, which was historically described as a separate syndrome, is now considered part of pantothenate kinase-associated neurodegeneration (PKAN) 101.
The manifestations of pantothenate kinase-associated neurodegeneration (PKAN) can include dystonia (contractions of opposing groups of muscles), spasticity, and pigmentary retinopathy 3, 5, 118. Its progression is rapid and leads to significant disability and loss of function 118. Treatment focuses primarily on reducing symptoms 119.
PKAN is typically diagnosed by molecular genetic testing, most often after a characteristic finding on magnetic resonance imaging (MRI), called the “eye-of-the-tiger” sign, is detected.
There is no specific treatment for individuals with pantothenate kinase-associated neurodegeneration (PKAN) 110. Treatment is directed towards the specific symptoms that appear in each individual and may include medication (such as botulinum toxin), surgery, deep brain stimulation and physical therapy. Whether pantothenate supplementation is beneficial in PKAN is not known, but some anecdotal reports indicate that supplements can reduce symptoms in some patients with atypical PKAN 106. Research is focusing on a better understanding of the underlying cause of this disorder, which may eventually help to find a more effective treatment.
Figure 6. Eye-of-the-tiger sign (MRI scan of the brain in pantothenate kinase-associated neurodegeneration (PKAN))
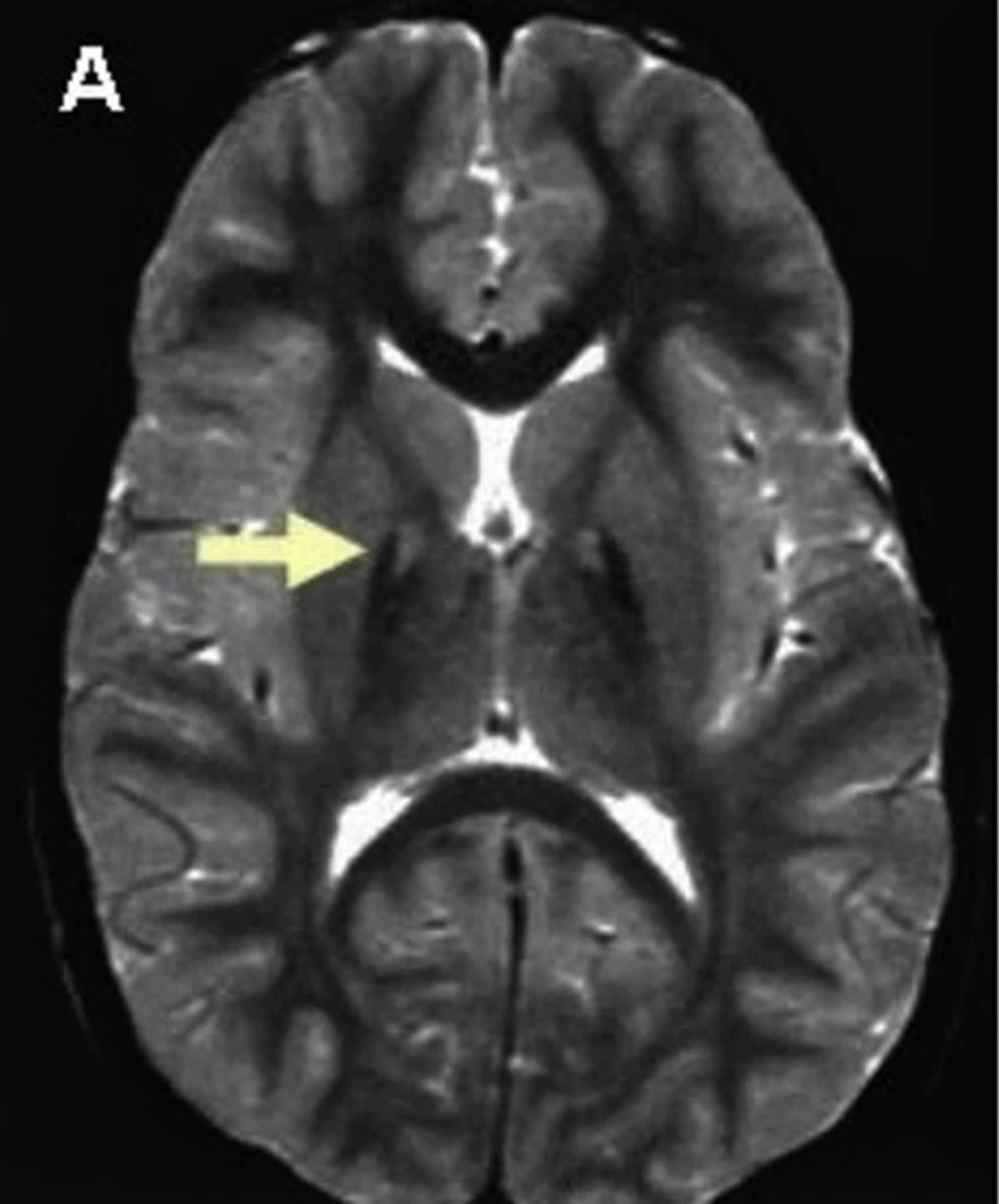
Footnote: T2 weighted brain magnetic resonance imaging (MRI) of a patient with pantothenate kinase-associated neurodegeneration (PKAN) which shows a central hyperintensity (bright spots) within substantia nigra and a surrounding area of hypointensity in globus pallidus (eye-of-the-tiger).
[Source 120 ]Pantothenate kinase-associated neurodegeneration (PKAN) causes
Pantothenate kinase-associated neurodegeneration (PKAN) is an autosomal recessive genetic condition caused by mutations in the pantothenate kinase 2 (PANK2) gene, located on the short arm of chromosome 20 (20p13) 104, 110. The PANK2 gene provides instructions for making an enzyme called pantothenate kinase 2 117. The pantothenate kinase 2 (PANK2) enzyme is active in specialized cellular structures called mitochondria, which are the cell’s energy-producing centers. Within mitochondria, pantothenate kinase 2 regulates the formation of a molecule called coenzyme A (CoA). Coenzyme A (CoA) is found in all living cells, where it is essential for the body’s production of energy from carbohydrates, fats, and some protein building blocks (amino acids).
PANK2 is one of four human genes that provide instructions for making versions of pantothenate kinase. The functions of these different versions probably vary among tissue types and parts of the cell. The version produced by the PANK2 gene is active in cells throughout the body, including nerve cells in the brain.
Individuals with PKAN have abnormal accumulation of iron in certain areas of the brain. This is especially seen in regions of the basal ganglia called the globus pallidus and the substantia nigra 110. The basal ganglia is a collection of structures deep within the base of the brain that assist in regulating movements. The exact relationship between iron accumulation and the symptoms of PKAN is not fully understood 110.
Pantothenate kinase-associated neurodegeneration (PKAN) signs and symptoms
The common feature among all individuals with pantothenate kinase-associated neurodegeneration (PKAN) is iron accumulation in the brain, in a pattern called the ‘eye of the tiger sign,’ along with a progressive movement disorder. Symptoms may vary greatly from case to case. In most cases, progression of the disease extends over several years, leading to death in childhood or early adulthood in classic PKAN cases 110. Some patients experience rapid deterioration and die within 1-2 years 110. Others have a slower progression or can plateau for long periods of time and continue to function into the third decade of life. Atypical individuals often retain a high level of function into later adulthood and some are known to be living in their sixties to seventies 110.
Pantothenate kinase-associated neurodegeneration (PKAN) symptoms include dystonia, (sustained muscle contractions causing repetitive movements), dysarthria (abnormal speech), muscular rigidity, poor balance, and spasticity (sudden involuntary muscle spasms), These features can result in clumsiness, gait (walking) problems, difficulty controlling movement, and speech problems. Another common feature is degeneration of the retina, resulting in progressive night blindness and loss of peripheral (side) vision.
Dystonia is characterized by involuntary muscle contractions that may force certain body parts into unusual, and sometimes painful, movements and positions. In addition, there may be stiffness in the arms and legs because of continuous resistance to muscle relaxing (spasticity) and abnormal tightening of the muscles (muscular rigidity). Spasticity and muscle rigidity usually begin in the legs and later develop in the arms. As affected individuals age, they may eventually lose control of voluntary movements. Muscle spasms combined with decreased bone mass can result in bone fractures (not caused by trauma or accident).
Dystonia affects the muscles in the mouth and throat, which may cause dysarthria and difficulty swallowing (dysphagia). The progression of dystonia in these muscles can result in loss of speech as well as tongue-biting and difficulty with eating.
Specific forms of dystonia that may occur in association with PKAN include blepharospasm and torticollis. Blepharospasm is a condition in which the muscles of the eyelids do not function properly, resulting in excessive blinking and involuntary closing of the eyelids. Torticollis is a condition in which there are involuntary contractions of neck muscles resulting in abnormal movements and positions of the head and neck.
Many of the delays in development pertain to motor skills (movement), although a small subgroup may have intellectual delays. Although intellectual impairment has often been described as a part of the condition in the past, it is unclear if this is a true feature. Intellectual testing may be hampered by the movement disorder; therefore, newer methods of studying intelligence are necessary to determine if there are any cognitive features of this condition.
The symptoms and physical findings associated with PKAN gene mutations can be distinguished between classical and atypical disease. Individuals with classical disease have a more rapid progression of symptoms. In most cases, atypical disease progresses slowly over several years. The symptoms and physical findings vary from case to case.
Classical PKAN develops in the first ten years of life (average age for developing symptoms is three and a half years). These children may initially be perceived as clumsy and later develop more noticeable problems with walking. Speech delay is also common. Eventually, falling becomes a frequent feature. Because of the limited ability to protect themselves during falls, children may have repeated injury to the face and chin. Many individuals with the classic form of PKAN require a wheelchair by their mid-teens (in some cases earlier). Most lose the ability to move/walk independently between 10 and15 years after the beginning of symptoms.
Individuals with classical PKAN are more likely to have specific eye problems. Approximately two-thirds of these patients will have retinal degeneration. This is a progressive degeneration of the nerve-rich membrane lining the eyes (retina), resulting in tunnel vision, night blindness, and loss of peripheral vision. Loss of this peripheral vision may contribute to the more frequent falls and gait disturbances in the early stages. [For more information on this retinopathy (retinitis pigmentosa), choose “retinitis pigmentosa” as your search term in the Rare Disease Database].
The atypical form of PKAN usually occurs after the age of ten years and progresses more slowly. The average age for developing symptoms is 13 years. Loss of independent ambulation (walking) often occurs 15 to 40 years after the initial development of symptoms. The initial presenting symptoms usually involve speech. Common speech problems are repetition of words or phrases (palilalia), rapid speech (tachylalia), and dysarthria. Psychiatric symptoms are more commonly observed and include impulsive behavior, violent outbursts, depression, or a tendency to rapid mood swings. While the movement disorder is a very common feature, it usually develops later. In general, atypical disease is less severe and more slowly progressive than early-onset PKAN.
In cases of neurodegeneration with brain iron accumulation (NBIA) that are not caused by PKAN, the movement-related symptoms (such as dystonia) may be very similar. Nine additional genes causing various subtypes of NBIA have been identified at this time. For those without a specific diagnosis or known cause of NBIA, symptoms are more varied because there are probably several different causes of neurodegeneration in this group. There is a subgroup of patients with moderate to severe intellectual disability. Also, seizure disorders are more common among non-PKAN individuals.
Pantothenate kinase-associated neurodegeneration (PKAN) diagnosis
The diagnosis of pantothenate kinase-associated neurodegeneration (PKAN) is made based upon a detailed patient history, a thorough clinical evaluation, and a variety of specialized tests 110. PKAN is typically suspected when the characteristic brain MRI finding called the “eye-of-the-tiger” sign, which is a dark area indicating accumulation of iron with a bright spot in the center, is observed on T2-weighted MRI. This MRI finding is not seen in other forms of neurodegeneration with brain iron accumulation (NBIA).
Molecular genetic testing for the full gene sequence of the PANK2 gene is the gold standard way to make this diagnosis 110. Approximately 95% of those affected have two identifiable mutations in the PANK2 gene and approximately 5% have only one identifiable mutation. Some PANK2 gene deletions are not detected by sequencing the gene, so for individuals without a detectable mutation or only one detectable mutation, gene deletion/duplication analysis is also recommended 110.
Pantothenate kinase-associated neurodegeneration (PKAN) treatment
There is no specific treatment for individuals with pantothenate kinase-associated neurodegeneration (PKAN) 110. Treatment is directed towards the specific symptoms that appear in each individual. Research is focusing on a better understanding of the underlying cause of this disorder, which may eventually help to find a more comprehensive treatment.
Treatment may require the coordinated efforts of a team of specialists. Physicians that the family may work with include the pediatrician or internist, neurologist, ophthalmologist, physiatrist and geneticist. A team approach to supportive therapy may include physical therapy, exercise physiology, occupation therapy, speech pathology and nutrition/feeding. In addition, many families may benefit from genetic counseling.
The most consistent forms of relief from disabling dystonia are baclofen, trihexyphenidyl, and clonazepam. These medications can be taken orally. Later in disease, a baclofen pump can be used to administer regular doses automatically into the central nervous system. Intramuscular botulinum toxin may also help treat specific regions where dystonia is problematic.
Levodopa/carbidopa does not generally appear to help patients with PKAN, although there may be exceptions. These treatments may have a role in the treatment of other causes of NBIA; however, their overall effectiveness is unknown and the responsiveness in individual cases is unpredictable.
Drugs that reduce the levels of iron in the body (iron chelation) have been attempted to treat individuals with PKAN 110. These early agents were proven ineffective and can cause anemia 110. A clinical trial of the drug deferiprone was completed for PKAN and results were published in 2019. The results suggested a possible modest slowing of disease progression, although the statistical analysis of the data was not able to prove this as significant 121, 122.
Pallidotomy and thalamotomy have been investigational attempts at controlling dystonia. These are both surgical techniques which destroy (ablate) very specific regions of the brain, the globus pallidus and thalamus, respectively. Some families have reported some immediate and temporary relief. However, most patients return to their pre-operative level of dystonia within one year of the operation 110. Deep brain stimulation (DBS) of the globus pallidus has been found to have promising results in some patients with PKAN and NBIA and is now favored over ablative procedures 110.
Individuals experiencing seizures usually benefit from standard anti-convulsive drugs 110. In addition, standard approaches to pain management are generally recommended where there is no identifiable treatment for the underlying cause of pain. Referral to pediatric palliative care specialists can be highly beneficial during later disease stages.
The association between pantothenate kinase and PKAN suggests that supplemental pantothenate (pantothenic acid, calcium pantothenate) taken orally could be beneficial. Pantothenate is another name for vitamin B5, a water soluble vitamin. Theoretically, this is most likely to assist individuals with very low levels of pantothenate kinase activity (atypical PKAN). It is hypothesized that classic PKAN results from complete absence of the enzyme pantothenate kinase, whereas atypical PKAN results from a severe deficiency, although the individuals still may have some level of enzyme activity. Clinical trials are needed to investigate the effectiveness of this treatment 110.
The benefits and limitations of any of the above treatments should be discussed in detail with a physician.
Pantothenic acid deficiency symptoms
Because vitamin B5 or pantothenic acid is widely distributed in nature and deficiency is extremely rare in humans, most information regarding the consequences of vitamin B5 deficiency has been gathered from experimental research in animals 20, 16. The most common symptoms of vitamin B5 or pantothenic acid in animals are growth problems, skin rash, gastrointestinal and nervous symptoms, such as ataxia, loss of coordination, and muscle weakness 16. Similar symptoms appeared also in human studies. Symptoms are described in more detail in Table 3. The diversity of symptoms emphasizes the numerous functions of pantothenic acid in its coenzyme forms.
Humans administered with a vitamin B5 antagonist omega-methyl pantothenic acid developed personality changes with irritability, restlessness, and quarrelsomeness 16. Similar symptoms developed in humans on a diet deficient in vitamin B5 content (8 weeks) 123. An analogous experiment was performed, as well, by Fry et al., in 1976 124, who tested the effect of a diet essentially free from pantothenic acid on human health. In that study, however, no clinical symptoms of deficiency were observed, but some subjects appeared listless and complained of fatigue at the end of diet deficient period (63 days) 124.
Lower levels of pantothenic acid were also detected in some brain regions affected by Alzheimer’s disease compared with controls. It is still unknown whether vitamin B5 depletion participates in the pathophysiology or if this is simply a consequence of the underlying neuropathological process 125.
Vitamin B5 or pantothenic acid deficiency in rats can cause damage to the adrenal glands, breeding problems and failure of embryo implementation with subsequent resorption 126. Vitamin B5 or pantothenic acid deficiency in rats throughout pregnancy has an impact on endocrine function of the placenta, which is linked to a lower production of progesterone and acetylcholine, and underdevelopment of fetuses 127. Among the reported abnormalities in rats include: cerebral and eye defects, digital hemorrhages and edema, interventricular septal defects, anomalies of the aortic arch pattern, hydronephrosis and hydroureter, clubfoot, tail defects, cleft palate, and dermal defects 126.
While vitamin B5 or pantothenic acid-deficient monkeys developed anemia due to decreased synthesis of heme, a component of hemoglobin 6. Dogs with vitamin B5 or pantothenic acid deficiency developed low blood glucose, rapid breathing and heart rates, and convulsions 6. Chickens developed skin irritation, feather abnormalities, and spinal nerve damage associated with the degeneration of the myelin sheath 6. Pantothenic acid-deficient mice showed decreased exercise tolerance and diminished storage of glucose (in the form of glycogen) in muscle and liver 6. Mice also developed skin irritation and graying of the fur, which is reversed by pantothenic acid administration 6.
Table 3. Vitamin B5 or pantothenic acid deficiency symptoms
| Species | Symptoms | Sources |
|---|---|---|
| Humans | Nervous system: headache, irritability, restlessness, quarrelsomeness, excessive fatigue, numbness, paresthesia, muscle cramps, faulty coordination associated with tremor and peculiar gait | 123 |
| Digestive track: abdominal rumbling, diarrhea, epigastric burning, regurgitation | ||
| Glands: loss of eosinophilic response to adrenocorticotropic hormone, increased sensitivity to insulin | ||
| Rodents (rats, mice, guinea pigs) | Growth: retardation, decrease in weight | 128, 129, 130, 131, 132 |
| Skin and mucosa: ruffing and discoloration of the fur, thinning of hair, alopecia, dryness of the skin with scaly desquamation, nasal discharge, watering of the eyes | ||
| Digestive track: diarrhea, duodenal changes (Lieberkühn crypts—enlargement, hyperplasia, increase in space between crypts, atrophy; villi diminution, epithelial changes to cuboid or flat, leading to ulcerations, perforation and chronic lesions), salivation | ||
| Nervous system: muscle weakness of the hind legs, convulsions, coma | ||
| Glands: adrenal lesions | ||
| Birds (ducklings and chicks) | Growth: retardation, decrease in weight | 130, 133, 134 |
| Skin: scaly dermatitis, skin lesions, scabs around beak and eyes, feather depigmentation, dermal edema | ||
| Nervous system: severe ataxia, tendency to fall and inability to rise and laying panting | ||
| Glands: lymphoid cell necrosis in the bursa of Fabricius and the thymus, and a lymphocytic paucity in the spleen | ||
| Pigs | Growth: failure to gain in weight, loss of appetite | 135, 136, 137 |
| Skin: loss of hair, roughness of the coat | ||
| Digestive track: diarrhea, severe colonic lesions | ||
| Nervous system: ataxia, lesions in sensory neurons, sudden lifting one of the limbs from the ground, unusual walk, inability to walk or stand | ||
| Respiratory system: cough and nasal secretion | ||
| Dogs | Growth: retardation Nervous system: sudden weakness, coma, rapid respiratory and heart rate, convulsions, spasticity of the hind legs | 138, 139 |
| Digestive track: decreased appetite, gastrointestinal symptoms, gastritis or enteritis | ||
| Glands: fatty liver, mottled thymusis | ||
| Blood: blood level of glucose and chlorides were lower and non-protein nitrogen was elevated | ||
| Urinary system: hemorrhagic kidney degeneration |
Pantothenic acid safety
Pantothenic acid or vitamin B5 is considered safe, even at high doses 140, 59. However, taking very high doses of vitamin B5 or pantothenic acid supplements (such as 10,000 mg to 20,000 mg/day) can cause an upset stomach and diarrhea, but the mechanism for this effect is not known 2, 59. However, there is one case report of life-threatening eosinophilic pleuropericardial effusion in an elderly woman who took a combination of 10 mg/day of biotin and 300 mg/day of pantothenic acid for two months 141. She was hospitalized with chest pain and breathing problems. Blood tests showed an inflammatory syndrome with a high eosinophil concentration (1200–1500 cells/mm³) 142. Due to the lack of reports of adverse effects when the Dietary Reference Intakes (DRI) for pantothenic acid were established in 1998, the Food and Nutrition Board of the Institute of Medicine did not establish a tolerable upper intake level (UL) for pantothenic acid 143. Pantethine is generally well tolerated in doses up to 1,200 mg/day. However, gastrointestinal side effects, such as nausea and heartburn, have been reported 100. Also, topical formulations containing up to 5% of dexpanthenol (D-panthenol) have been safely used for up to one month. Yet, a few cases of skin irritation, contact dermatitis, and eczema have been reported with the use of dexpanthenol-containing ointments 144, 145.
Pantothenic acid contraindications
Pantothenic acid or vitamin B5 contraindications include patients with hypersensitivity or allergy to the drug or any of its derivatives. A report suggests that pantothenic acid (vitamin B5) intake might correlate with increased cerebral amyloid-beta peptide burden in individuals with cognitive impairment 146. Although further studies are still needed to confirm the findings and discover the molecular mechanisms of this pathway, the current research suggests those with cognitive impairment to be a potential contraindication 146.
Interactions with medications or other supplements
Large doses of pantothenic acid (vitamin B5) have the potential to compete with biotin for intestinal and cellular uptake by the human sodium-dependent multivitamin transporter (hSMVT) 147, 148.
Oral contraceptives (birth control pills) containing estrogen and progestin may increase the requirement for pantothenic acid 140. Use of pantethine in combination with cholesterol-lowering drugs called statins (HMG-CoA reductase inhibitors) or with nicotinic acid (niacin) may produce additive effects on blood lipids 100.
The following drugs have moderate interactions with pantothenic acid (vitamin B5) 12:
- Azithromycin
- Clarithromycin
- Erythromycin base
- Erythromycin ethylsuccinate
- Erythromycin lactobionate
- Erythromycin stearate
- Roxithromycin
Furthermore, there are at least 60 other drugs that have mild interactions with pantothenic acid (vitamin B5).
Biotin
Biotin also known as vitamin B7 or vitamin H, is a water soluble vitamin and is naturally present in liver, soy, beans and egg yolks. Raw egg whites, however, contain the protein avidin that binds to biotin and reduces its availability. Eating 2 or more uncooked egg whites daily for several months has caused biotin deficiency that is serious enough to produce symptoms. Biotin acts as a carrier of carbon dioxide and plays a role in carboxylase enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) that catalyze critical steps fatty acid metabolism, gluconeogenesis (the formation of glucose from sources other than carbohydrates, such as pyruvate, lactate, glycerol, and the glucogenic amino acids), and amino acids 149. Biotin also plays key roles in histone modifications, gene regulation (by modifying the activity of transcription factors), and cell signaling 150.
The recommended daily dietary allowance for biotin has not been formally established, but the amounts needed are small and biotin is found in many foods and is produced by intestinal bacteria. An adequate intake for biotin has been estimated as 30 micrograms (mcg) daily. Thus, most diets provide adequate amounts of biotin and its deficiency is rare. Although there are no nationally representative estimates of biotin intakes in the United States, the average biotin intake from foods in other western populations is about 35–70 mcg/day, indicating that most people in these countries consume adequate amounts of biotin and biotin deficiency is rare 149, 151.
Most biotin in foods is bound to protein, although some dietary biotin is in the free form 152. Gastrointestinal proteases and peptidases break down the protein-bound forms of ingested biotin into biocytin and biotin-oligopeptides, which undergo further processing by biotinidase, an enzyme, in the intestinal lumen to release free biotin 152. The free biotin is then absorbed in the small intestine, and most biotin is stored in the liver 151.
Biotin is available generically in many over-the-counter forms in doses of 5 to 10 mg and is included in most multivitamin preparations, usually in concentrations of 30 to 300 mcg. Biotin is typically added to parenteral nutrition and the doses for deficiencies is in the range of 10 mg daily.
Biotin deficiency has occurred in humans on parenteral nutrition (intravenous administration of nutrition). However, biotin deficiency is very rare in the United States. The signs and symptoms of biotin deficiency typically appear gradually and can include thinning hair with progression to loss of all hair on the body (alopecia); a scaly red rash around the eyes, nose, mouth, and anal area (seborrheic dermatitis); pinkeye (conjunctivitis); lactic acidosis (which occurs when lactate production exceeds lactate clearance) and aciduria (abnormal amounts of acid in urine); skin infection; brittle nails; nervous system disorders (e.g., depression, lethargy, seizures, hallucinations, ataxia and numbness and tingling [paresthesias] of the extremities) in adults; and hypotonia (weak muscle tone), lethargy, sluggishness and developmental delay in infants 149. The characteristic facial rash, together with unusual facial fat distribution in people with biotin deficiency is known as “biotin deficiency facies” 153. Individuals with hereditary disorders of biotin metabolism (inborn metabolic disorders) resulting in functional biotin deficiency often have similar physical findings, as well as seizures and evidence of impaired immune system function and increased susceptibility to bacterial and fungal infections 154, 155.
A limited number of reliable indicators of biotin status is available 156. In healthy adults, the concentration of biotin is 133–329 pmol/L in serum and 18–127 nmol/24 hours in urine 149. Abnormally low urinary excretion of biotin is an indicator of biotin deficiency, as is abnormally high excretion of 3-hydroxyisovaleric acid (higher than 3.3 mmol/mol creatinine) or 3-hydroxyisovalerylcarnitine (higher than 0.06 mmol/mol creatinine) resulting from reduced activity of methylcrotonyl-CoA carboxylase 156. The most reliable individual markers of biotin status, including deficiency and sufficiency, are biotinylated methylcrotonyl-CoA carboxylase and propionyl-CoA carboxylase in white blood cells 156. Oral administration of large doses of biotin increases serum concentrations of biotin and its metabolites 157. However, serum concentrations of biotin and its catabolites are not good indicators of marginal biotin deficiency because they do not decrease sufficiently in people with marginal biotin deficiency for these changes to be detectable with existing tests 151.
What does biotin do?
Biotin helps turn the carbohydrates, fats, and proteins in the food you eat into the energy you need. Biotin functions as a coenzyme; involved in carboxylation, transcarboxylation, and decarboxylation reactions of gluconeogenesis, lipogenesis, fatty acid synthesis, propionate metabolism, and the catabolism of leucine. Biotin acts as a carrier of carbon dioxide and functions as a covalently bound cofactor required for the biological activity of the five known mammalian biotin-dependent carboxylases enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) that catalyze critical steps fatty acid metabolism, gluconeogenesis, and amino acids 149. For acetyl-CoA carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2), biotin serves as a cofactor responsible for transfer of bicarbonate to acetyl-CoA, converting it to malonyl-CoA for fatty acid synthesis. Pyruvate carboxylase (PC) participates in gluconeogenesis. Methylcrotonyl-CoA carboxylase (MCC) catalyzes a step in leucine metabolism. Propionyl-CoA carboxylase (PCC) catalyzes a step in the metabolism of propionyl-CoA 158, 159. Metabolic degradation of the biotinylated carboxylases leads to the formation of biocytin. This compound is further degraded by biotinidase to release biotin, which is then reutilized by holocarboxylase synthetase 159.
Biotin also plays key roles in histone modifications, gene regulation (by modifying the activity of transcription factors), and cell signaling 150.
Enzyme cofactor
Biotin is an essential cofactor to five known mammalian biotin-dependent carboxylases enzymes (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]) in intermediary metabolism and a key regulator of gene expression 160.
Five mammalian carboxylases catalyze essential metabolic reactions 160:
- Both acetyl-Coenzyme A (CoA) carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2) catalyze the conversion of acetyl-CoA to malonyl-CoA using bicarbonate and ATP; however, the two enzymes have different roles in metabolism and different intracellular locations. ACC1 is located in the cytosol, and the malonyl CoA generated by ACC1 is a rate-limiting substrate for the synthesis of fatty acids (Figures 7 and 8). Acetyl-Coenzyme A (CoA) carboxylase 1 (ACC1) is found in all tissues and is particularly active in lipogenic tissues (i.e., liver, white adipose tissue, and mammary gland), heart, and pancreatic islets. Acetyl-CoA carboxylase 2 (ACC2) is located on the outer mitochondrial membrane, and the malonyl CoA generated via ACC2 inhibits CPT1, an enzyme that regulates malonyl-CoA entry into the inner mitochondria, thereby regulating fatty acid oxidation (Figure 9). ACC2 is especially abundant in skeletal muscle and heart 161.
- Pyruvate carboxylase (PC) is a critical enzyme in gluconeogenesis (the formation of glucose from sources other than carbohydrates, such as pyruvate, lactate, glycerol, and the glucogenic amino acids). Pyruvate carboxylase (PC) catalyzes the ATP-dependent incorporation of bicarbonate into pyruvate, producing oxaloacetate; hence, pyruvate carboxylase is anaplerotic for the citric acid cycle (Figure 9). Oxaloacetate can then be converted to phosphoenolpyruvate and eventually to glucose.
- Methylcrotonyl-CoA carboxylase (MCC) catalyzes an essential step in the catabolism of leucine, an essential branched-chain amino acid (BCAA). Methylcrotonyl-CoA carboxylase (MCC) enzyme catalyzes the production of 3-methylglutaconyl-CoA from methylcrotonyl-CoA (Figure 10).
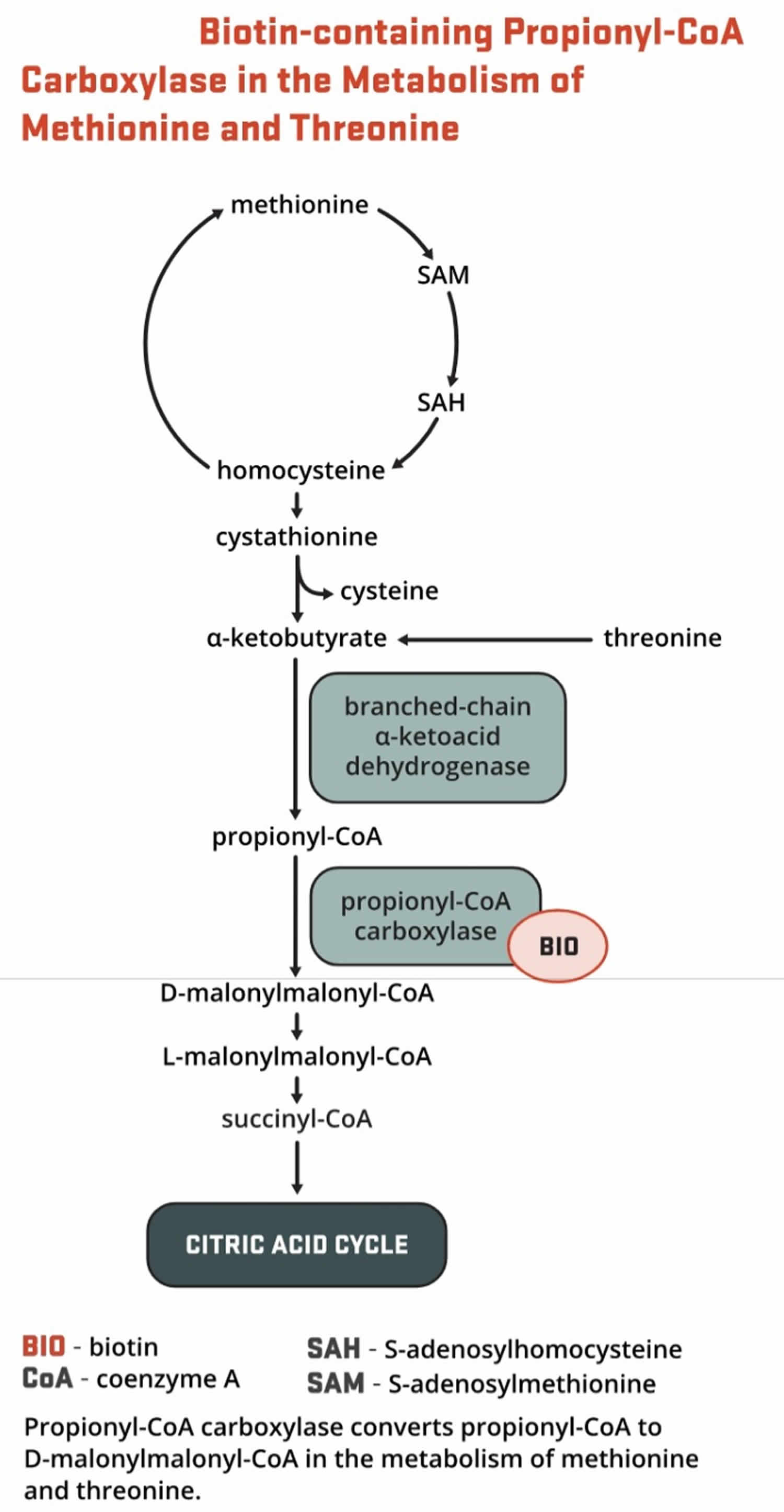
- Propionyl-CoA carboxylase (PCC) produces D-malonylmalonyl-CoA from propionyl-CoA, a by-product in the beta-oxidation of fatty acids with an odd number of carbon atoms (Figure 10). The conversion of propionyl-CoA to D-malonylmalonyl-CoA is also required in the catabolic pathways of two branched-chain amino acids (isoleucine and valine) and the side chain of cholesterol (Figure 10) and of the amino acids methionine and threonine (Figure 11).
Figure 7. Roles of the 5 biotin-dependent carboxylases
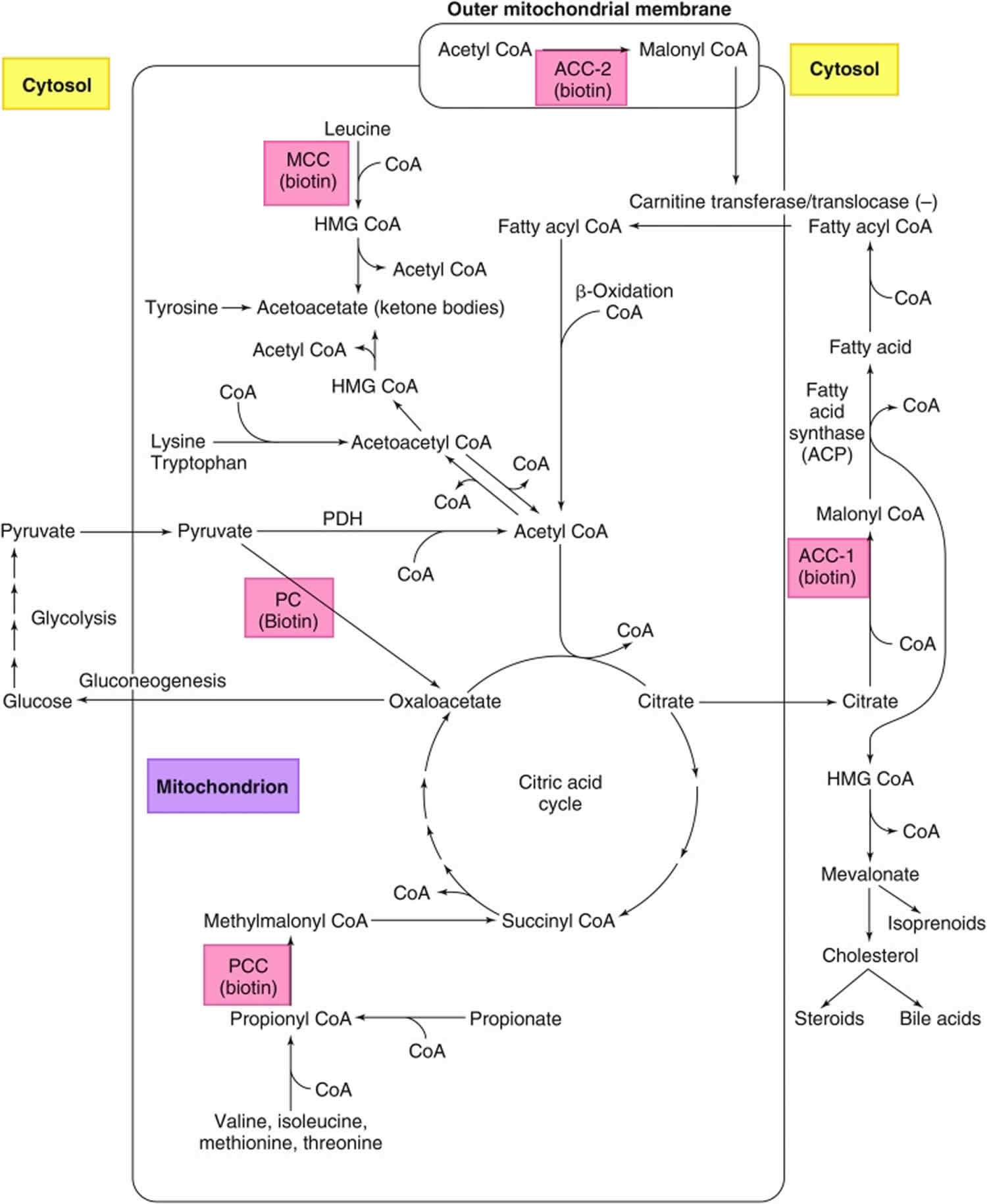
Footnotes: Roles of the 5 biotin-dependent carboxylases of coenzyme A (CoA) and acyl carrier protein (ACP) within the cell. Shown is an overview of the metabolic pathways of acetyl-CoA carboxylase 1 (ACC1) (cytosolic) and acetyl-CoA carboxylase 2 (ACC2) (outer mitochondrial membrane) and the 3 mitochondrial carboxylases propionyl-CoA carboxylase (PCC), methylcrotonyl-CoA carboxylase (MCC) and pyruvate carboxylase (PC).
Abbreviations: ACC1 = acetyl-CoA carboxylase 1; ACC2 = acetyl-CoA carboxylase 2; ACP = acyl carrier protein; HMG = 3-hydroxy-3-methylglutaryl; MCC = methylcrotonyl-CoA carboxylase; PC = pyruvate carboxylase; PCC = propionyl-CoA carboxylase; PDH = pyruvate dehydrogenase.
[Source 162 ]Figure 8. Biotin function as enzyme cofactor
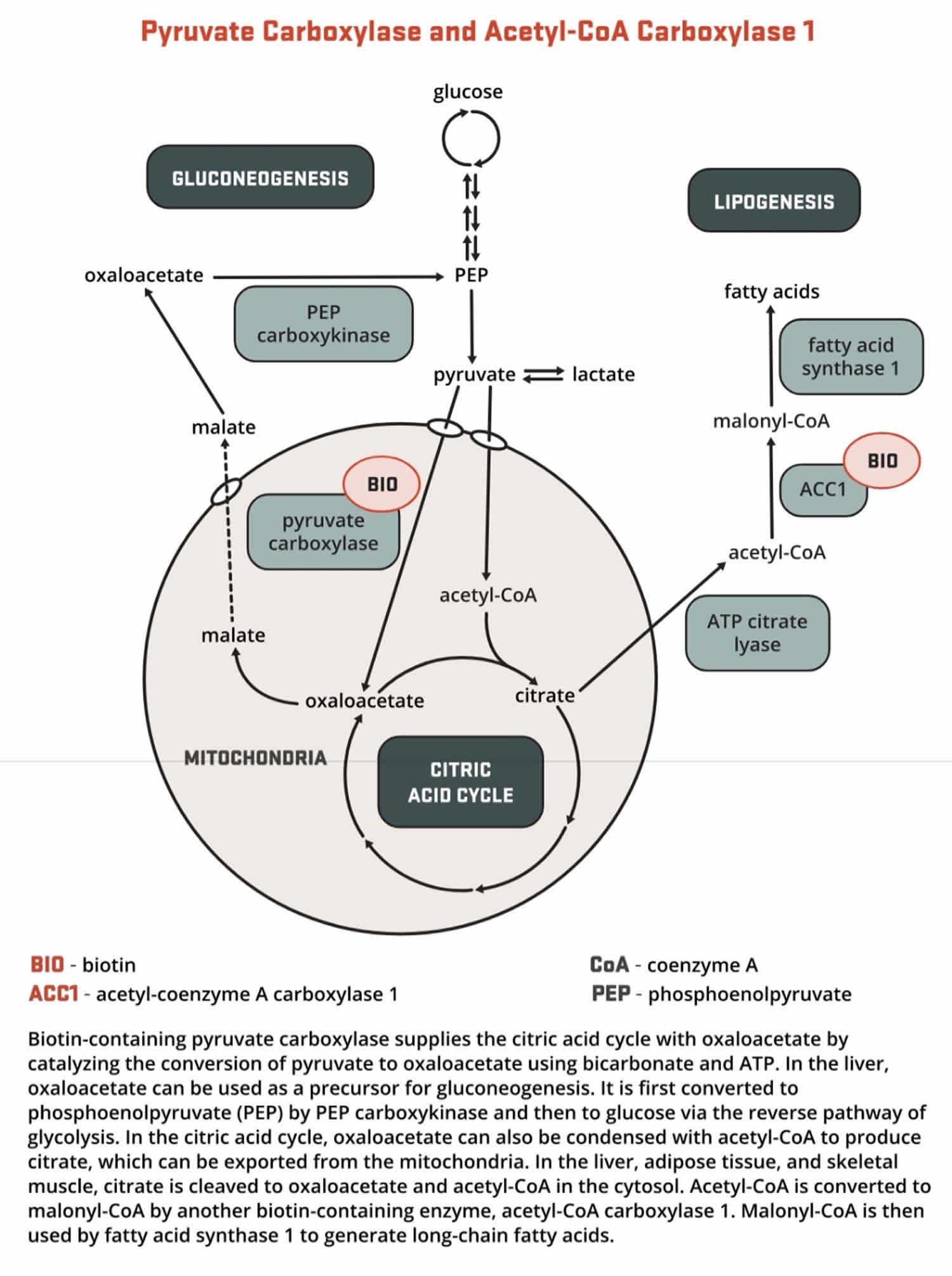
Figure 9. Biotin as enzyme cofactor in gluconeogenesis
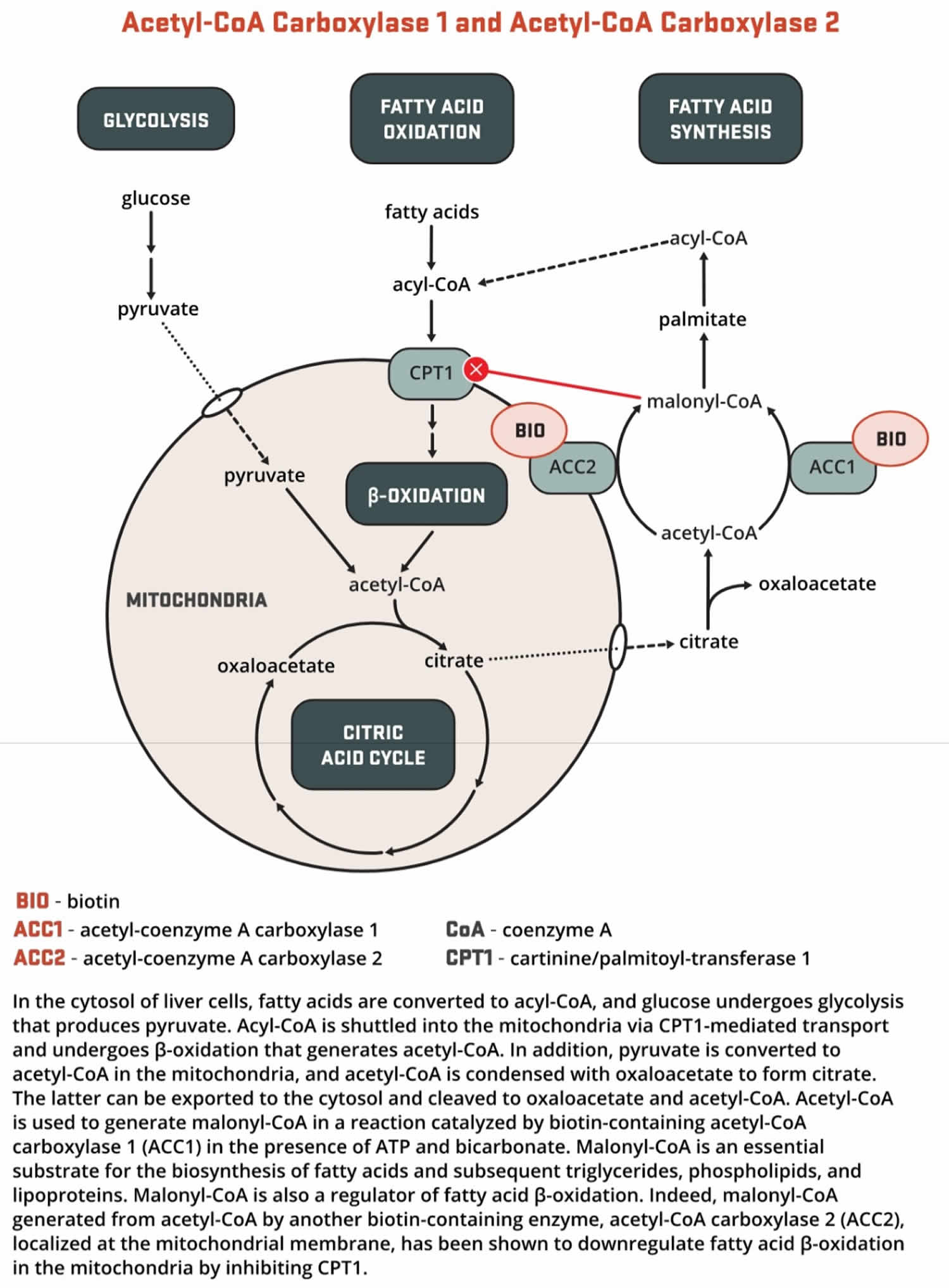
Figure 10. Biotin as enzyme cofactor in fatty acids, amino acids and cholesterol metabolism
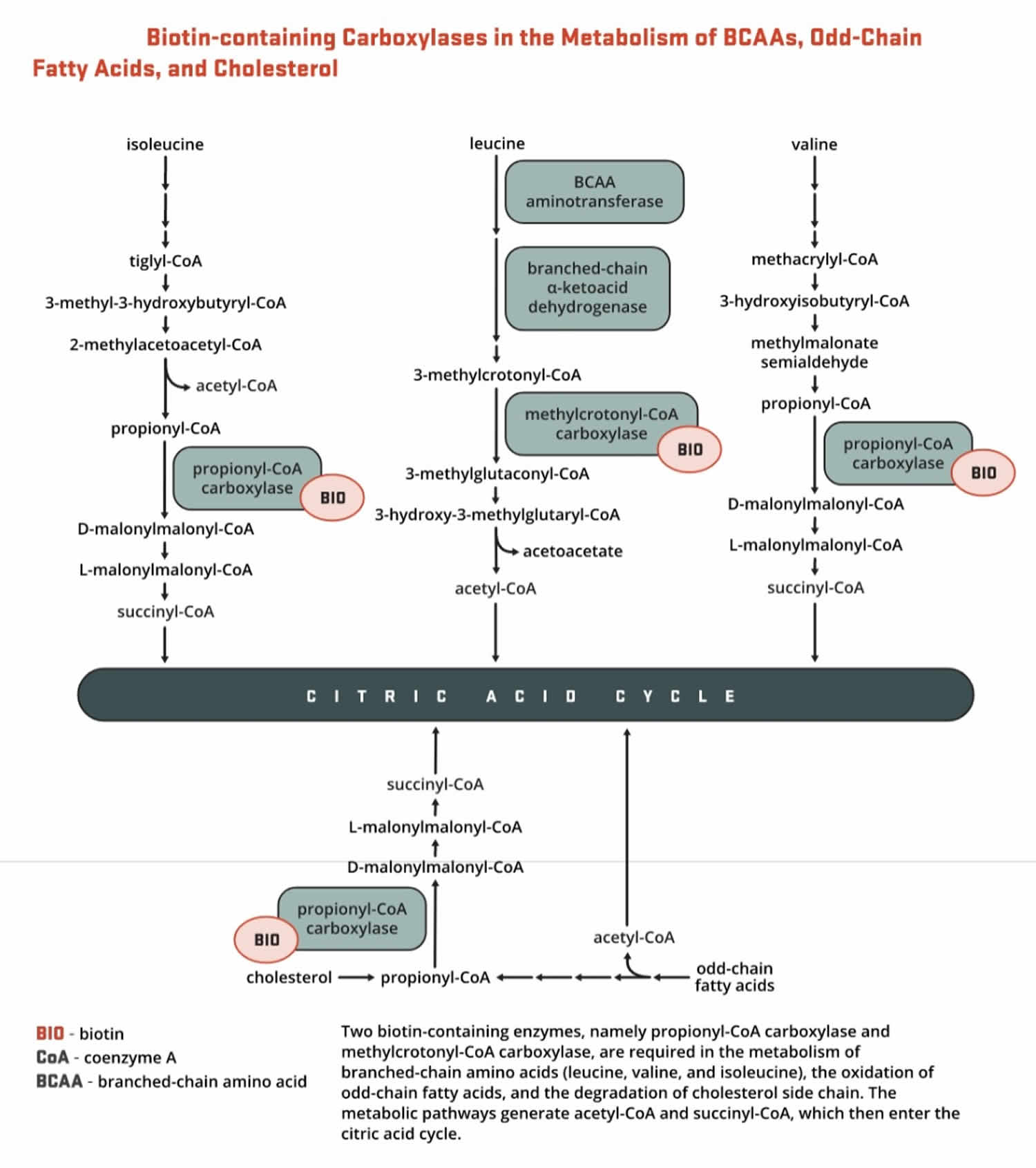
Figure 11. Biotin as enzyme cofactor in amino acids methionine and threonine metabolism
Regulation of chromatin structure and gene expression
In cells that have a nucleus, DNA is packaged into compact structures to form nucleosomes — fundamental units of chromatin. Each nucleosome is composed of 147 base pairs of DNA wrapped around eight histones (paired histones: H2A, H2B, H3, and H4) 160. The H1 linker histone is located at the outer surface of each nucleosome and serves as an anchor to fix the DNA around the histone core. The compact packaging of chromatin must be relaxed for DNA replication and transcription 160. Chemical modifications of DNA and histones affect the folding of chromatin, increasing or reducing DNA accessibility to factors involved in replication and transcription. DNA methylation and a number of chemical modifications within the N-terminal tail of core histones modify their electric charge and structure, thereby changing chromatin conformation and transcriptional activity of genes 160.
The modifications of histone tails (“marks”), including acetylation, methylation, phosphorylation, ubiquitination, SUMOylation, ADP-ribosylation, carbonylation, deimination, hydroxylation, and biotinylation, have various regulatory functions 160. Several sites of biotinylation have been identified in histones H2A, H3, and H4 163. Amongst them, histone H4 biotinylation at lysine (K) 12 (noted H4K12bio) appears to be enriched in heterochromatin, a tightly condensed chromatin associated with repeat regions in (peri)centromeres and telomeres. H4 biotinylation appears to be enriched in transposable elements known as long terminal repeats 164. These biotinylation marks also co-localize with well-known gene repression marks like methylated lysine 9 in histone H3 (H3K9me) in transcriptionally competent chromatin 165. For example, H4K12bio can be found at the promoter of the gene SLC5A6 that codes for the transporter mediating biotin uptake into cells, the human sodium-dependent multivitamin transporter (hSMVT). When biotin is abundant, HCS can biotinylate histones H4 in the SLC5A6 promoter, which down regulates hSMVT synthesis and reduces biotin uptake. Conversely, in biotin-deficient cells, biotinylation marks in the SLC5A6 promoter are removed increasing gene expression and enabling the synthesis of hSMVT and uptake of biotin 166.
Biotinylation
Biotin functions as a covalently bound cofactor required for the biological activity of the five known mammalian biotin-dependent carboxylases (propionyl-CoA carboxylase [PCC], pyruvate carboxylase [PC], methylcrotonyl-CoA carboxylase [MCC], acetyl-CoA carboxylase 1 [ACC1], and acetyl-CoA carboxylase 2 [ACC2]). Such non-protein cofactors are termed “prosthetic groups” and are common in water-soluble vitamins. The covalent attachment of biotin to the apocarboxylase (i.e., the carboxylase protein without the biotin prosthetic group and is catalytically inactive) is catalyzed by the enzyme, holocarboxylase synthetase (HCS). The term “biotinylation” refers to the covalent addition of biotin to any molecule, including the apocarboxylases and histones. Holocarboxylase synthetase (HCS) catalyzes the post-translational biotinylation of the epsilon amino group of a lysine residue at the active site of each apocarboxylase, converting the inactive apocarboxylase into a fully active holocarboxylase (Figure 12). Particular lysine residues within the N-terminal tail of specific histone that help package DNA in eukaryotic nuclei can also be biotinylated 164. Biotinidase is the enzyme that catalyzes the release of biotin from biotinylated histones and from the peptide products of holocarboxylase breakdown (Figure 13).
Figure 12. Biotinylation

Figure 13. Biotin recycling
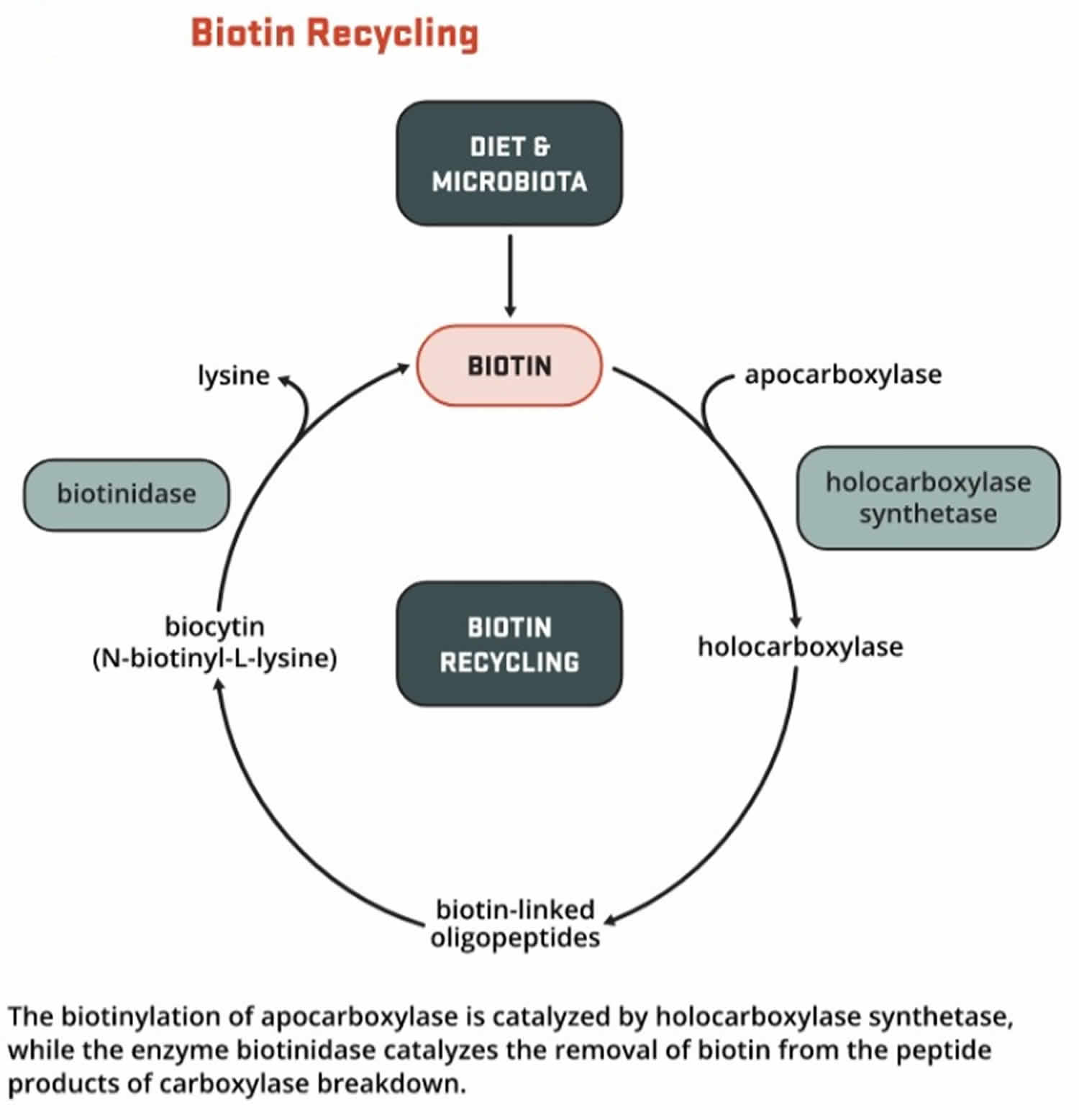
Biotin health benefits
Scientists are studying biotin to understand how it affects health. Biotin is commonly used for hair loss, brittle nails, nerve damage, and many other conditions.
Taking biotin can help treat low blood levels of biotin or biotin deficiency. It can also prevent blood levels of biotin from becoming too low. Low blood levels of biotin can cause thinning of the hair and rash around the eyes, nose, and mouth. Other symptoms include depression, lack of interest, hallucinations, and tingling in the arms and legs. Low biotin levels can occur in people who are pregnant, who have had long-term tube feeding, who are malnourished, who have undergone rapid weight loss, or who have a specific inherited condition. Cigarette smoking might also cause low blood levels of biotin.
Biotin possibly ineffective for:
- Skin rash in infants (seborrheic dermatitis). Taking biotin does not seem to help improve rash in infants.
Insufficient evidence to rate Biotin effectiveness for:
- Hair loss. Taking biotin and zinc by mouth in addition to applying a steroid cream to the skin might help reduce hair loss.
- An inherited disorder called biotin-thiamine-responsive basal ganglia disease. People with this condition experience episodes of altered mental state and muscle problems. Early research shows that taking biotin plus thiamine does not prevent these episodes better than taking thiamine alone. But the combination might shorten how long the episodes last when they do occur.
- Brittle fingernails and toenails. Taking biotin by mouth for up to a year might increase the thickness of fingernails and toenails in people with brittle nails.
- Diabetes. Some early research shows that taking biotin along with chromium might lower blood sugar in people with diabetes. However, taking biotin alone doesn’t seem to improve blood sugar levels in people with diabetes.
- Diabetic nerve pain. Early research shows that taking biotin by mouth or receiving it as a shot might reduce nerve pain in the legs of people with diabetes.
- Muscle cramps related to dialysis. People receiving dialysis tend to have muscle cramps. Early research shows that taking biotin by mouth might reduce muscle cramps in these people.
- Multiple sclerosis. Early research shows that taking high-dose biotin might improve vision and reduce partial paralysis in some people with multiple sclerosis.
- Other conditions.
More evidence is needed to rate biotin for these uses.
Hair, nail, and skin health
Dietary supplements that contain biotin are often promoted to improve the health of your hair, skin, and nails, but there is little scientific evidence to support these claims. In a few small studies, some people with thin and brittle nails who took high doses of biotin had harder nails. Doctors have also reported that in a few cases, high doses of biotin have improved a rare hair disorder in children and skin rash in infants. More research is needed before biotin supplements can be recommended for any of these conditions.
Signs of biotin deficiency include skin rashes, hair loss, and brittle nails 167. Therefore, biotin supplements are often promoted for hair, skin, and nail health 168. However, these claims are supported, at best, by only a few case reports and small studies.
The evidence on biotin supplementation to treat brittle nails includes three small studies that did not include a placebo group, and these reports do not indicate the baseline biotin status of study participants. One of these studies assessed the effects of 2.5 mg/day biotin for 6–15 months in 22 women with brittle, splitting, or soft nails and 10 healthy volunteers 168. In the eight patients with brittle nails whose nail samples were obtained immediately before and after biotin supplementation, nail thickness increased by 25%. In the 14 patients with brittle nails whose nail specimens were obtained 2–4 months after starting treatment and 1–4 months after ending treatment, nail thickness increased by 7%, a difference that was not statistically significant 168. In the second study, 2.5 mg biotin daily for an average of 5.5 months in 45 patients with thin and brittle fingernails resulted in firmer and harder fingernails in 41 of the patients (91%) 169. Finally, the third, retrospective study in 35 patients with brittle nails found that 2.5 mg/day biotin for 6–15 months resulted in clinical improvement in 22 of the 35 patients (63%) 170.
Only case reports are available to support claims that biotin supplements can promote hair health, and these reports were only in children 171, 172. These studies found that 3–5 mg/day biotin in children with uncombable hair syndrome (an inherited condition that is characterized by dry, frizzy hair that cannot be combed flat and is caused by mutations in the PADI3, TGM3, or TCHH gene) significantly improved hair health after 3–4 months 171, 172.
The evidence supporting the use of biotin supplements to support skin health is equally limited to a small number of case reports, all in infants, showing that 100 mcg to 10 mg/day resulted in dramatic improvements in rash or dermatitis as well as alopecia (hair loss) 173, 174.
Future studies are needed to determine whether biotin supplements might improve hair, nail, and skin health, especially among healthy individuals.
Brittle fingernails (onychorrhexis)
The finding that biotin supplements were effective in treating hoof abnormalities in hoofed animals suggested that biotin might also be helpful in strengthening brittle fingernails in humans 175, 176, 177. Three uncontrolled trials examining the effects of biotin supplementation (2.5 mg/day for several months) in women with brittle fingernails have been published 178, 169, 170. In two of the trials, subjective evidence of clinical improvement was reported in 67%-91% of the participants available for follow-up at the end of the treatment period 178, 169. One trial that used scanning electron microscopy to assess fingernail brittleness reported less fingernail splitting and a 25% increase in the thickness of the nail plate in patients supplemented with biotin for 6 to 15 months 170. Biotin supplementation (5 mg/day) was also found to be effective in controlling unruly hair and splitting nails in two toddlers with inherited uncombable hair syndrome 171. Although preliminary evidence suggests that supplemental biotin may help strengthen fragile nails 179, larger placebo-controlled trials are needed to assess the efficacy of high-dose biotin supplementation for the treatment of brittle fingernails.
Hair loss (alopecia)
Biotin administration has been associated with hair loss (alopecia) reversal in children treated with the anticonvulsant valproic acid, as well as with hair regrowth or normal hair growth in some children with inborn errors of biotin metabolism or other genetic disorders (i.e., uncombable hair syndrome) 180. Yet, while hair loss is a symptom of severe biotin deficiency, there are no published scientific studies that support the claim that high-dose biotin supplements are effective in preventing or treating hair loss in men or women 181, 182. Randomized, placebo-controlled trials in healthy individuals would be needed to evaluate this claim.
Diabetes mellitus
Overt biotin deficiency has been shown to impair glucose utilization in mice 183 and cause fatal hypoglycemia in chickens. Overt biotin deficiency likely also causes abnormalities in glucose regulation in humans (see biotin function). One early human study reported lower serum biotin concentrations in 43 patients with type 2 diabetes mellitus compared to 64 control subjects without the disease; an inverse relationship between fasting blood glucose and biotin concentrations was observed as well 184. In a small, randomized, placebo-controlled intervention study in 28 patients with type 2 diabetes, daily supplementation with 9 milligrams (mg) of biotin for one month resulted in a 45% decrease in mean fasting blood glucose concentrations 184. Yet, another small study in 10 patients with type 2 diabetes and 7 controls without diabetes found no effect of biotin supplementation (15 mg/day) for 28 days on fasting blood glucose concentrations in either group 185. A more recent double-blind, placebo-controlled study by the same research group showed that the same biotin regimen lowered plasma triglyceride concentrations in patients with hypertriglyceridemia — independent of whether they had type 2 diabetes 186. In this study, biotin administration did not affect blood glucose concentrations in either patient group. Additionally, a few studies have shown that co-supplementation with biotin and chromium picolinate may be a beneficial adjunct therapy in patients with type 2 diabetes 187, 188, 189, 190.
Potential mechanisms for the glucose and lipid effects have been suggested. As a cofactor of carboxylases required for fatty acid synthesis, biotin may increase the utilization of glucose for fat synthesis. Also, biotin stimulates glucokinase, a liver enzyme that increases synthesis of glycogen, the storage form of glucose. Biotin also triggers the secretion of insulin in the pancreas of rats and improves glucose homeostasis 175. Yet, reduced activity of acetyl-CoA carboxylase 1 (ACC1) and acetyl-CoA carboxylase 2 (ACC2) would be expected to reduce fatty acid synthesis and increase fatty acid oxidation, respectively. Hence, whether pharmacologic doses of biotin benefits the management of hyperglycemia in patients with impaired glucose tolerance remains unclear. Moreover, whether supplemental biotin lowers the risk of cardiovascular complications in patients with diabetes by reducing serum triglycerides and LDL-cholesterol remains to be proven 186, 187, 188.
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease characterized by progressive damage to the myelin sheath surrounding nerve fibers (axons) and neuronal loss in the brain and spinal cord of affected individuals in anatomic locations that vary widely among affected individuals producing variable signs and symptoms. The progression of neurologic disabilities in multiple sclerosis (MS) patients is often assessed by the Expanded Disability Status Scale (EDSS) with scores from 1 to 10, from minimal signs of motor dysfunction (score of 1) to death by multiple sclerosis (score of 10). ATP deficiency due to mitochondrial dysfunction and increased oxidative stress may be partly responsible for the progressive degeneration of neurons in multiple sclerosis 191. Given its role in energy production by intermediary metabolism and fatty acid oxidation and in fatty acid synthesis (required for myelin formation) (see biotin function), high-dose biotin supplementation it has been hypothesized that to exert beneficial effects that would limit or reverse multiple sclerosis-associated functional impairments 191.
The mechanism of action of high-dose biotin has been investigated in a genetic mouse model of chronic axon injury caused by oxidative damage and bioenergetic failure. High-dose biotin restored redox homeostasis, mitochondria biogenesis, and ATP levels, and reversed axonal death and locomotor impairment. Dysregulation of the transcriptional program for lipid synthesis and degradation in the spinal cord was also normalized, possibly as the result of hyperactivation of a nutrient, energy or redox sensor that controls protein synthesis restoring lipid homeostasis.
A nonrandomized, uncontrolled pilot study in 23 patients with progressive multiple sclerosis (MS) found high doses of biotin (100-600 mg/day) to be associated with sustained clinical improvements in five (out of five) patients with progressive visual loss and 16 out of 18 patients with partial paralysis of the limbs after a mean three months following treatment onset 192. Additionally, a multicenter, randomized, placebo-controlled trial in 154 subjects with progressive multiple sclerosis (MS) reported that 13 out of 103 patients supplemented with high-dose, pharmaceutical-grade biotin (300 mg/day) for 12 months achieved MS-related disability reversal — assessed by improved Expanded Disability Status Scale (EDSS) or 25-foot walk time (37). In comparison, none of the 51 patients randomized to the placebo group showed significant clinical improvements 193. However, when this regimen of high-dose biotin supplementation was examined in a larger, international cohort of patients with progressive MS (326 patients receiving biotin and 316 patients receiving placebo), no benefits on EDSS or walk time were seen after 12 months 194. Moreover, a randomized, double-blind, placebo-controlled trial in 93 MS patients with chronic visual loss found that 300 mg/day of pharmaceutical-grade biotin for six months did not improve visual acuity, but an interesting trend favoring the biotin group was observed in the subgroup of patients with progressive optic neuritis 195. Moreover, a meta-analysis of three randomized controlled trials (2 on disability; 3 on adverse effects), involving 889 individuals diagnosed with MS (the preponderance of participants [830] had progressive MS while only 59 had remitting relapsing MS) was conducted 196. Pooling results of two trials found no benefit of high-dose biotin on multiple sclerosis (MS)-related disability, but there was significant heterogeneity between the trials. When the subgroup progressive multiple sclerosis (MS) was analyzed separately, a moderate certainty of evidence suggested a potential benefit in favor of high-dose biotin for the 25-foot minute walk time 196. On balance, studies remain inconclusive but promising.
Biotin-thiamine-responsive basal ganglia disease
Biotin-thiamine-responsive basal ganglia disease also called biotin-responsive basal ganglia disease, thiamine transporter-2 deficiency, and thiamine metabolism dysfunction syndrome-2, is caused by an autosomal recessive mutation in the SLC19A3 gene that codes for thiamin transporter-2 (THTR-2) 197. Biotin-thiamine-responsive basal ganglia disease usually presents around 3 to 10 years of age 197, but an early infantile form of the disease exists with onset as early as one month of age 198. Clinical features include subacute encephalopathy (confusion, drowsiness, altered level of consciousness), ataxia, and seizures.
A retrospective study of 18 affected individuals from the same family or the same tribe in Saudi Arabia showed that biotin monotherapy (5-10 mg/kg/day) efficiently abolished the clinical manifestations of the disease, although one-third of the patients suffered from recurrent acute crises. Often associated with poor outcomes, acute crises were not observed after thiamine (vitamin B1) supplementation started (300-400 mg/day) and during a five-year follow-up period, early diagnosis and immediate treatment with biotin and thiamin led to positive outcomes 199. Although the specific mechanism for therapeutic effects of biotin in biotin-thiamine-responsive basal ganglia disease remains unknown, lifelong high-dose supplementation with a combination of biotin and thiamin is the recommended treatment 197. Early diagnosis and treatment is important to ensure a better prognosis 198, 200.
Birth defects
Current research indicates that at least one-third of women develop marginal biotin deficiency during pregnancy 201. Small observational studies in pregnant women have reported an abnormally high urinary excretion of 3-hydroxyisovaleric acid in both early and late pregnancy, suggesting decreased activity of biotin-dependent methylcrotonyl-CoA carboxylase (MCC) 202, 203. In a randomized, single-blinded intervention study in 26 pregnant women, supplementation with 300 mcg/day of biotin for two weeks limited the excretion of 3-hydroxyisovaleric acid compared to placebo, confirming that increased 3-hydroxyisovaleric acid excretion indeed reflected marginal biotin deficiency in pregnancy 204. A small cross-sectional study in 22 pregnant women reported an incidence of low lymphocyte propionyl-CoA carboxylase (PCC) activity greater than 80% 205. Although these levels of biotin deficiency are not associated with overt signs of deficiency in pregnant women, such observations are sources of concern because subclinical biotin deficiency has been shown to cause cleft palate and limb hypoplasia in several animal species 205. In addition, biotin depletion has been found to suppress the expression of biotin-dependent carboxylases, remove biotin marks from histones, and decrease the proliferation in human embryonic palatal mesenchymal cells in culture 206. Impaired carboxylase activity may result in alterations in lipid metabolism, which have been linked to cleft palate and skeletal abnormalities in animals. Furthermore, biotin deficiency leading to reduced histone biotinylation at specific genomic loci may increase genomic instability and result in chromosome anomalies and fetal malformations 205.
Analogous to pregnant women who are advised to consume supplemental folic acid (vitamin B9) prior to and during pregnancy to prevent neural tube defects, it would also be prudent to ensure adequate biotin intake throughout pregnancy. The current Adequate Intake (intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an Recommended Dietary Allowance [RDA], the average daily level of intake sufficient to meet the nutrient requirements of nearly all (97%–98%) healthy individuals; often used to plan nutritionally adequate diets for individuals) for pregnant women is 30 mcg/day of biotin, and no toxicity has ever been reported at this level of intake.
Lipid metabolism
Consistent with roles of biotin-dependent acetyl-CoA carboxylases 1 (ACC1) and 2 (ACC2), and propionyl-CoA carboxylase (PCC) in lipid metabolism, biotin deficiency causes alterations of the fatty acid profile in liver, skin, and serum of several animal species 207. Biotin deficiency is associated with increased abundance of odd-chain fatty acids, suggesting that odd-chain fatty acid accumulation may be a marker for reduced propionyl-CoA carboxylase (PCC) activity in biotin deficiency. Biotin deficiency does not affect the fatty acid composition in brain tissue to the same extent as in liver 207.
Biotin deficiency also causes abnormalities in fatty acid composition in humans. In patients who developed biotin deficiency during parenteral alimentation, the percentage of odd-chain fatty acids (15:0, 17:0) in serum increased for each of the four major lipid classes, i.e. cholesterol esters, phospholipids, triglycerides, and free fatty acids 207. However, the relative changes in these four classes of lipids have not always been consistent among studies 207.
Immune function and cell stress
Biotin deficiency has adverse effects on cellular and humoral immune functions 208. For example, children with hereditary abnormalities of biotin metabolism developed Candida dermatitis and presented with absent delayed-hypersensitivity skin-tests responses, IgA deficiency, and subnormal percentages of T lymphocytes in peripheral blood 209. Recent studies have shown that biotin also regulates immunological and inflammatory function. Agrawal et al. 210 reported that biotin deficiency facilitates the secretion of tumour necrosis factor (TNF)-α, IL-1, IL-23, and IL-12p40 from dendritic cells, and that biotin-deficient dendritic cells induce significantly higher levels of secretion of IFN-γ, IL-17, and IL-22 from CD4 T cells. These findings suggest that biotin deficiency leads to a shift of Th-cell responses towards Th1/Th17. Among the various skin disorders, the IL-23/Th17 pathway 211 and the IL-1/IL-36 axis 212 have been considered to play a major role in the pathogenesis of psoriasis and pustular psoriasis, respectively.
In rodents, biotin deficiency decreases antibody synthesis 213, decreases the number of spleen cells and the percentage of B lymphocytes in spleen 214, and impairs thymocyte maturation 215. Decreased rates of cell proliferation may cause some of the effects of biotin on immune function 216, 217, 218.
Biotin deficiency is linked also to cell stress, enhancing the nuclear translocation of the transcription factor NF-kappaB in human lymphoid cells 219. NF-kappaB mediates activation of anti-apoptotic genes; this is associated with enhanced survival of biotin-deficient cells in response to cell death signals compared with biotin-sufficient controls 219. Stress-resistant Drosophila can be selected by feeding biotin-deficient diets for multiple generations 220.
How much biotin do I need?
The amount of biotin you need each day depends on your age. Average daily recommended amounts are listed below in micrograms (mcg).
Intake recommendations for biotin and other nutrients are provided in the Dietary Reference Intakes (DRIs) developed by the Food and Nutrition Board at the National Academies of Sciences, Engineering, and Medicine 221. Dietary Reference Intake is the general term for a set of reference values used for planning and assessing nutrient intakes of healthy people. These values, which vary by age and sex, include:
- Recommended Dietary Allowance (RDA): Average daily level of intake sufficient to meet the nutrient requirements of nearly all (97%–98%) healthy individuals; often used to plan nutritionally adequate diets for individuals.
- Adequate Intake (AI): Intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an recommended dietary allowance (RDA).
- Estimated Average Requirement (EAR): Average daily level of intake estimated to meet the requirements of 50% of healthy individuals; usually used to assess the nutrient intakes of groups of people and to plan nutritionally adequate diets for them; can also be used to assess the nutrient intakes of individuals.
- Tolerable Upper Intake Level (UL): Maximum daily intake unlikely to cause adverse health effects.
The Food and Nutrition Board found the available data to be insufficient to derive an Estimated Average Requirement (EAR) and Recommended Dietary Allowance (RDA) for biotin. For this reason, the Food and Nutrition Board established only adequate intakes (AIs) for biotin. The Food and Nutrition Board based its determination of adequate intakes (AIs) for all populations on the amount of biotin in human milk consumed by infants and then used body weight to extrapolate AIs for other groups 222. Table 4 lists the current adequate intakes (AIs) for biotin 221.
Table 4. Adequate Intakes (AIs) for Biotin
| Life Stage | Recommended Amount |
|---|---|
| Birth to 6 months | 5 mcg |
| Infants 7–12 months | 6 mcg |
| Children 1–3 years | 8 mcg |
| Children 4–8 years | 12 mcg |
| Children 9–13 years | 20 mcg |
| Teens 14–18 years | 25 mcg |
| Adults 19+ years | 30 mcg |
| Pregnant teens and women | 30 mcg |
| Breastfeeding teens and women | 35 mcg |
Biotin supplement
Biotin is found in some multivitamin or multimineral supplements, in B-complex supplements, and in supplements containing only biotin. The absorption rate of oral, free biotin is 100%, even when people consume pharmacologic doses of up to 20 mg/day biotin 223.
The following doses have been studied in scientific research:
Adults By mouth
- General: There is no recommended dietary allowance (RDA) established for biotin. The adequate intakes (AI) for biotin are 30 mcg for adults over 18 years and pregnant women, and 35 mcg for breast-feeding women.
- Biotin deficiency: Up to 10 mg daily has been used.
Children By Mouth
- General: There is no recommended dietary allowance (RDA) established for biotin. The adequate intakes (AI) for biotin are 7 mcg for infants 0-12 months, 8 mcg for children 1-3 years, 12 mcg for children 4-8 years, 20 mcg for children 9-13 years, and 25 mcg for adolescents 14-18 years.
- Biotin deficiency: Up to 10 mg daily has been used in infants.
What foods provide Biotin?
Many foods contain some biotin. You can get recommended amounts of biotin by eating a variety of foods, including the following 224:
- Meat, fish, eggs, and organ meats (such as liver)
- Seeds and nuts
- Certain vegetables (such as sweet potatoes, spinach, and broccoli)
Foods that contain the most biotin include organ meats, eggs, fish, meat, seeds, nuts, and certain vegetables (such as sweet potatoes) 149. The biotin content of food can vary; for example, plant variety and season can affect the biotin content of cereal grains, and certain processing techniques (e.g., canning) can reduce the biotin content of foods 225.
Dietary avidin, a glycoprotein in raw egg whites, binds tightly to dietary biotin and prevents biotin’s absorption in the gastrointestinal tract 226. Cooking denatures avidin, making it unable to interfere with biotin absorption 226.
Although there are no nationally representative estimates of biotin intakes in the United States, the average biotin intake from foods in other western populations is about 35–70 mcg/day, indicating that most people in these countries consume adequate amounts of biotin 149.
The U.S. Department of Agriculture’s (USDA’s) FoodData Central (https://fdc.nal.usda.gov) does not list the biotin content of foods or provide lists of foods containing biotin.
Several food sources of biotin are listed in Table 5.
Table 5. Biotin content of selected foods
| Food | Micrograms (mcg) per serving | Percent DV* |
|---|---|---|
| Beef liver, cooked, 3 ounces | 30.8 | 103 |
| Egg, whole, cooked | 10 | 33 |
| Salmon, pink, canned in water, 3 ounces | 5 | 17 |
| Pork chop, cooked, 3 ounces | 3.8 | 13 |
| Hamburger patty, cooked, 3 ounces | 3.8 | 13 |
| Sunflower seeds, roasted, ¼ cup | 2.6 | 9 |
| Sweet potato, cooked, ½ cup | 2.4 | 8 |
| Almonds, roasted, ¼ cup | 1.5 | 5 |
| Tuna, canned in water, 3 ounces | 0.6 | 2 |
| Spinach, boiled, ½ cup | 0.5 | 2 |
| Broccoli, fresh, ½ cup | 0.4 | 1 |
| Cheddar cheese, mild, 1 ounce | 0.4 | 1 |
| Milk, 2%, 1 cup | 0.3 | 1 |
| Plain yogurt, 1 cup | 0.2 | 1 |
| Oatmeal, 1 cup | 0.2 | 1 |
| Banana, ½ cup | 0.2 | 1 |
| Whole wheat bread, 1 slice | 0 | 0 |
| Apple, ½ cup | 0 | 0 |
Footnote: *DV = Daily Value. The U.S. Food and Drug Administration (FDA) developed Daily Values (DVs) to help consumers compare the nutrient contents of foods and dietary supplements within the context of a total diet. The Daily Value (DV) for biotin is 30 mcg for adults and children age 4 years and older 227. FDA does not require food labels to list biotin content unless biotin has been added to the food. Foods providing 20% or more of the DV are considered to be high sources of a nutrient, but foods providing lower percentages of the DV also contribute to a healthful diet.
[Source 228 ]Biotin deficiency
Biotin deficiency also called vitamin B7 deficiency is rare 229, and severe biotin deficiency in healthy individuals eating a normal mixed diet has never been reported 153.
Although clinically overt biotin deficiency or vitamin B7 deficiency is very rare, the human requirement for dietary biotin has been demonstrated in three different situations: prolonged intravenous feeding (parenteral nutrition) without biotin supplementation, infants fed an elemental formula devoid of biotin, and consumption of raw egg white for a prolonged period (many weeks to years) 201. Raw egg white contains avidin; this antimicrobial protein binds biotin with an affinity and specificity that is almost unique as a reversible binding. Because native avidin is resistant to mammalian and microbial digestion, avidin prevents biotin absorption. Cooking egg white denatures avidin, rendering it susceptible to digestion and therefore unable to block the absorption of dietary biotin 163.
The signs and symptoms of biotin deficiency typically appear gradually and can include thinning hair with progression to loss of all hair on the body (alopecia); scaly, red rash around body openings (eyes, nose, mouth, and perineum) also called seborrheic dermatitis; conjunctivitis (pink eye); ketolactic acidosis (which occurs when lactate production exceeds lactate clearance) and aciduria (abnormal amounts of acid in urine); seizures; skin infection; brittle nails; neurological findings (e.g., depression, lethargy, hallucinations, and paresthesias of the extremities) in adults; and hypotonia, lethargy, and developmental delay in infants 149. The rash and unusual distribution of facial fat in people with biotin deficiency is known as “biotin deficiency facies” 153.
The neurological and psychological symptoms can occur with only mild biotin deficiencies while dermatitis, conjunctivitis, and hair loss generally occur only when biotin deficiency becomes more severe 230. Individuals with hereditary disorders of biotin deficiency additionally have evidence of impaired immune system function, including increased susceptibility to bacterial and fungal infections 231.
Regardless of the cause, biotin deficiency or vitamin B7 deficiency can usually be addressed directly with oral biotin supplementation.
Figure 14. Biotin deficiency hair loss
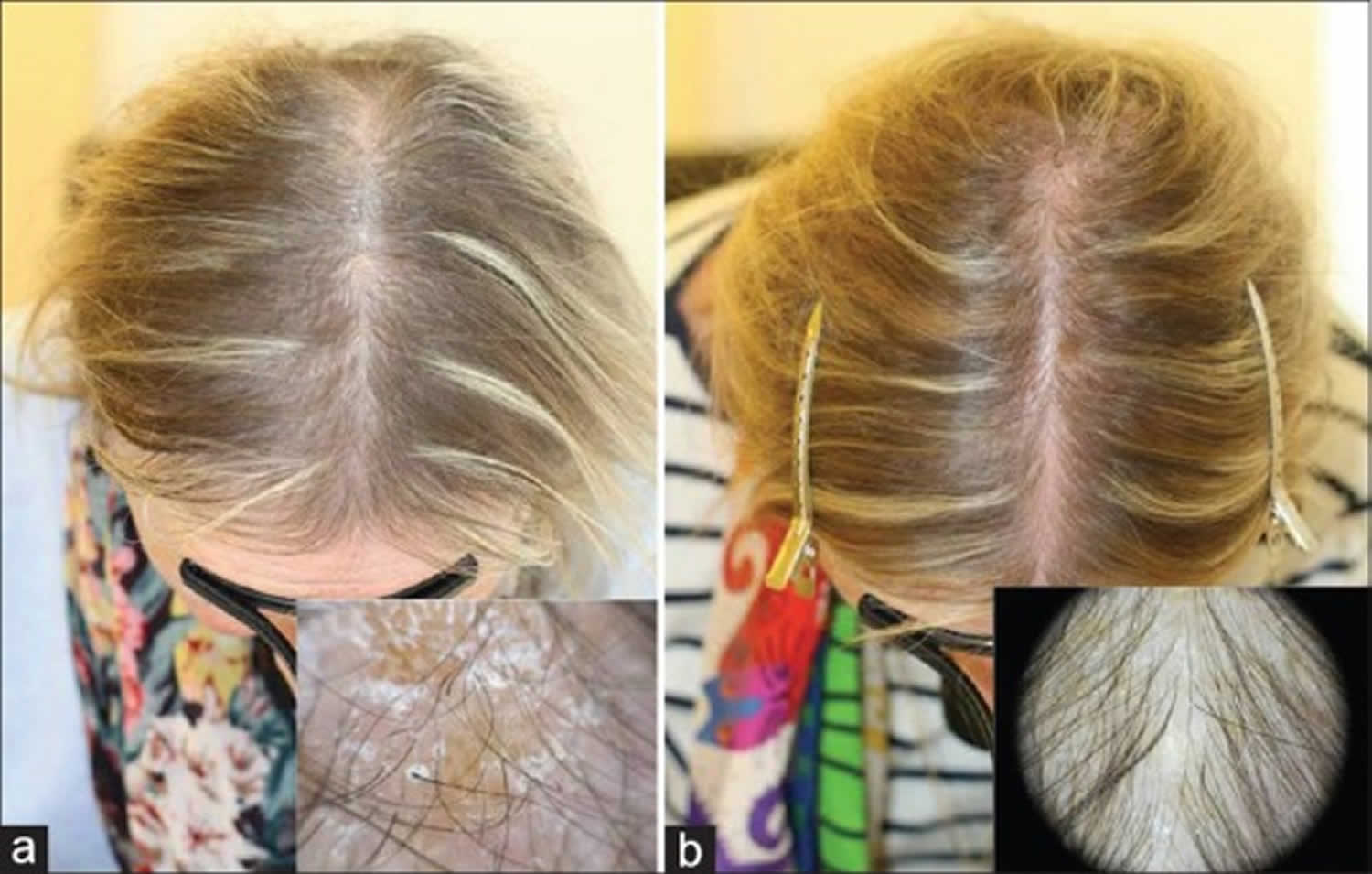
Footnotes: Hair loss (alopecia) and seborrheic-like dermatitis in a patient with biotin deficiency (a) before, and (b) after treatment with 5 mg oral biotin for 3 months.
[Source 230 ]Figure 15. Biotin deficiency in an infant fed with amino acid formula
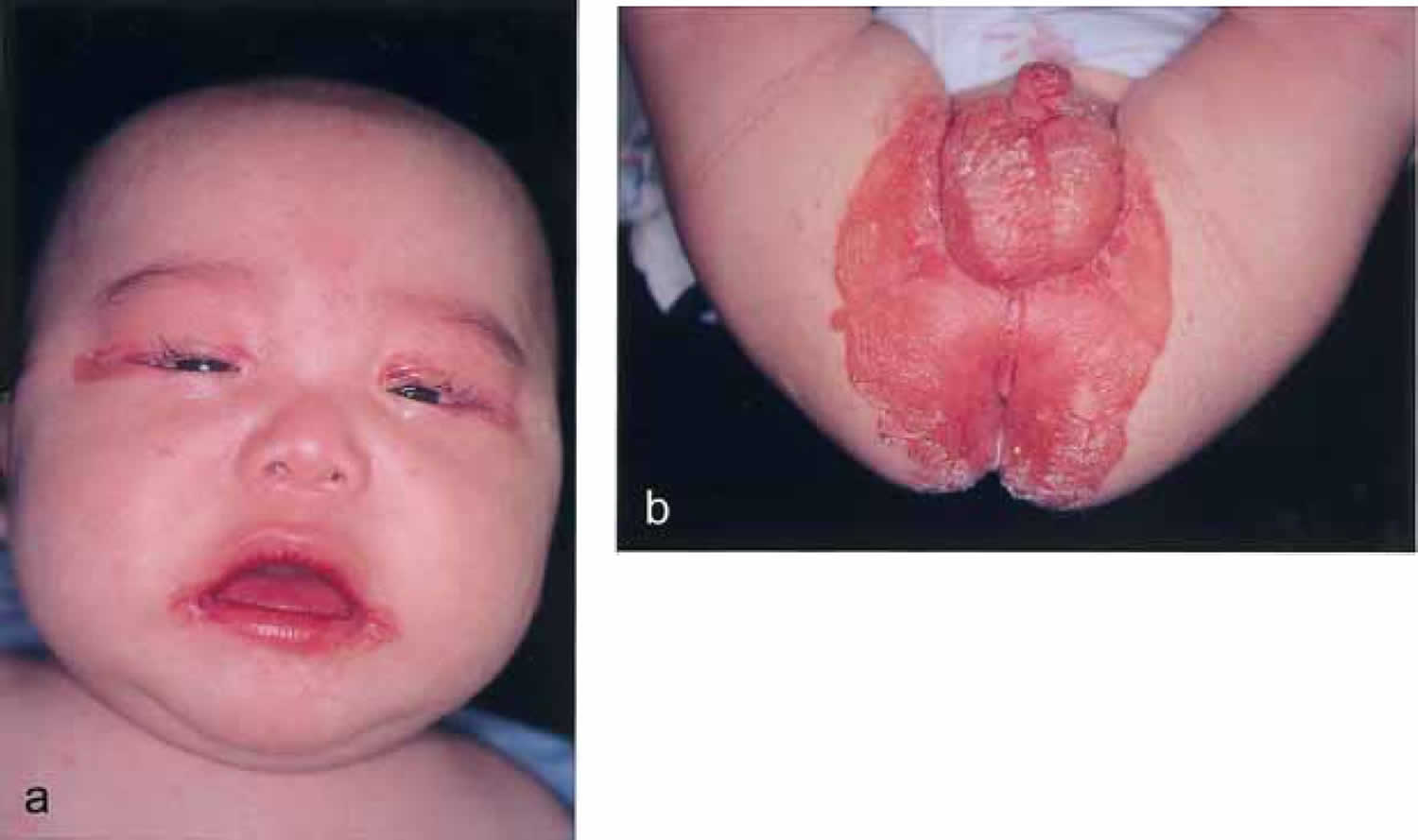
Footnotes: Biotin deficiency is rarely encountered in an infant on weaning from breast and formula feeding. Biotin deficiency is characterized by hair loss (alopecia) and scaly, erythematous dermatitis distributed around the body orifices. A 5-month-old Japanese infant with typical skin lesions who had been diagnosed as a neonate with dyspepsia and fed only an amino acid formula. Serum and urine levels of biotin were below the normal range, but zinc and biotinidase were within normal range. Urinary excretion of 3-methylcrotonylglycine, 3-hydroxyisovaleric acid, and methylcitric acid was significantly elevated. Daily oral supplementation with 1 mg of biotin resulted in dramatic improvement of the periorificial dermatitis and hair growth together with a complete disappearance of the organic aciduria.
[Source 232 ]Figure 16. Biotinidase deficiency
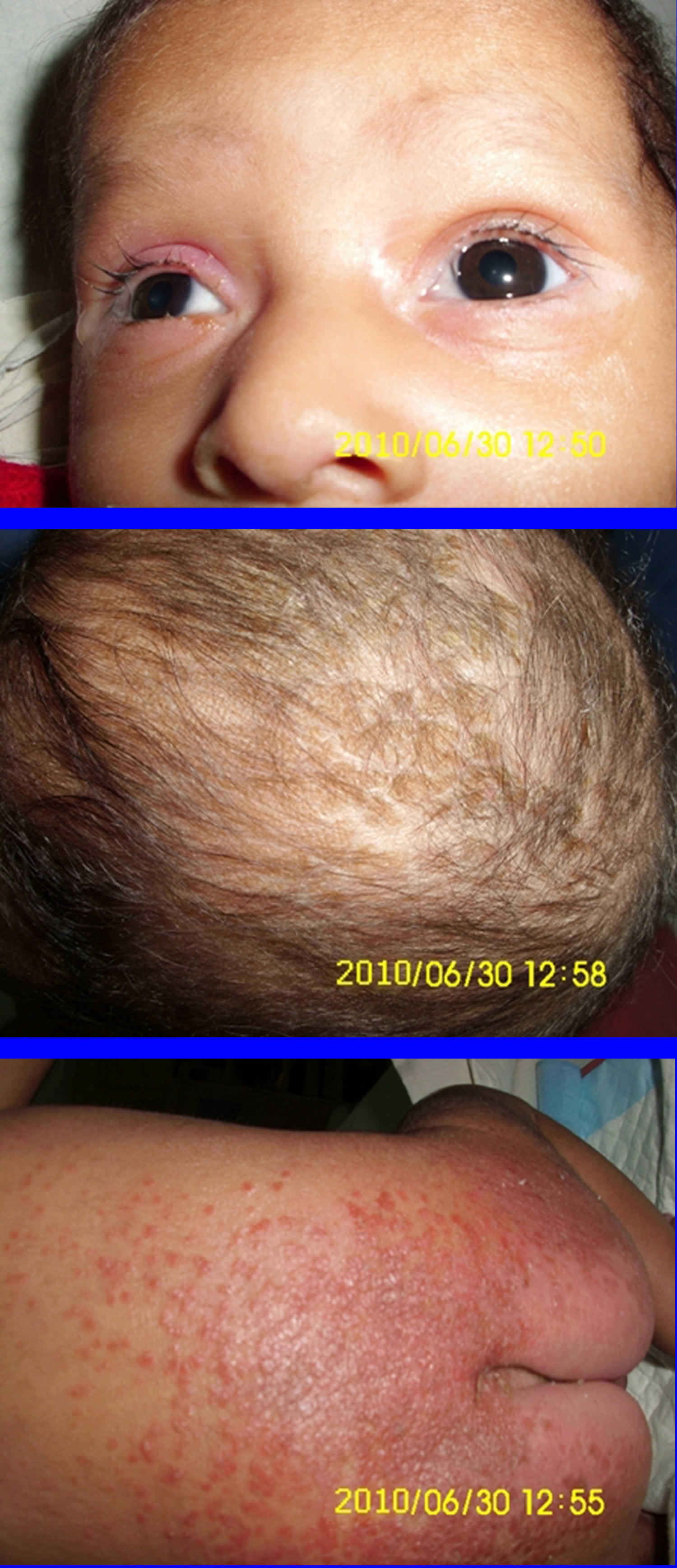
Footnotes: Biotinidase deficiency is a rare autosomal recessive inherited disorder in which the body is unable to recycle biotin, leading to biotin deficiency despite normal biotin intake from foods such as liver, egg yolks, and milk 233. Biotinidase deficiency is caused by genetic changes (mutations) in the BTD gene. The BTD gene provides instructions for making an enzyme called biotinidase 234. The biotinidase enzyme recycles biotin. Biotinidase removes biotin that is bound to proteins in food, leaving the vitamin in its free (unbound) state. Free biotin is needed by enzymes called biotin-dependent carboxylases to break down fats, proteins, and carbohydrates. Because several of these enzymes are impaired in biotinidase deficiency, the condition is considered a form of multiple carboxylase deficiency 233. Biotinidase deficiency in a 58-day-old male infant with seizures for 2 weeks prior to presentation. At admission, the child had multiple episodes of generalized tonic and myoclonic seizures not associated with fever. He also had respiratory distress and stridor. The skin showed erythematous maculopapular rash in the perioral and perianal regions. He had alopecia (hair loss), scanty eyebrows, blepharitis, conjunctivitis, balanitis and seborrheic dermatitis involving the scalp. Hair over the scalp was hypopigmented. On neurological examination, the child was convulsing intermittently. Hypertonia was noted in all the four limbs with exaggerated deep tendon reflexes. Fundus examination revealed papilloedema. Rest of the systemic examination was normal. Within 24 hour of hospitalization, the possibility of biotinidase deficiency was entertained and oral biotin was started empirically at a dose of 10 mg twice a day, after obtaining a blood sample for biotinidase assay. There was total cessation of seizures and considerable improvement in the rash by 48 hour. The hypertonia in the upper limbs resolved within 72 hours though it persisted in the lower limbs, even at discharge, albeit much less than before. The balanitis and blepharitis also resolved by 72 hours. Serum biotinidase levels showed a profound deficiency. The child was then started on regular biotin supplementation and was discharged after 5 days. Anticonvulsants were successfully stopped without recurrence of seizures. The child is currently 10 months old, healthy, with adequate weight gain and normal developmental milestones. At follow-up, there are no sequelae except for a mild persisting lower limb hypertonia.
[Source 235 ]Figure 17. Holocarboxylase synthetase deficiency in baby
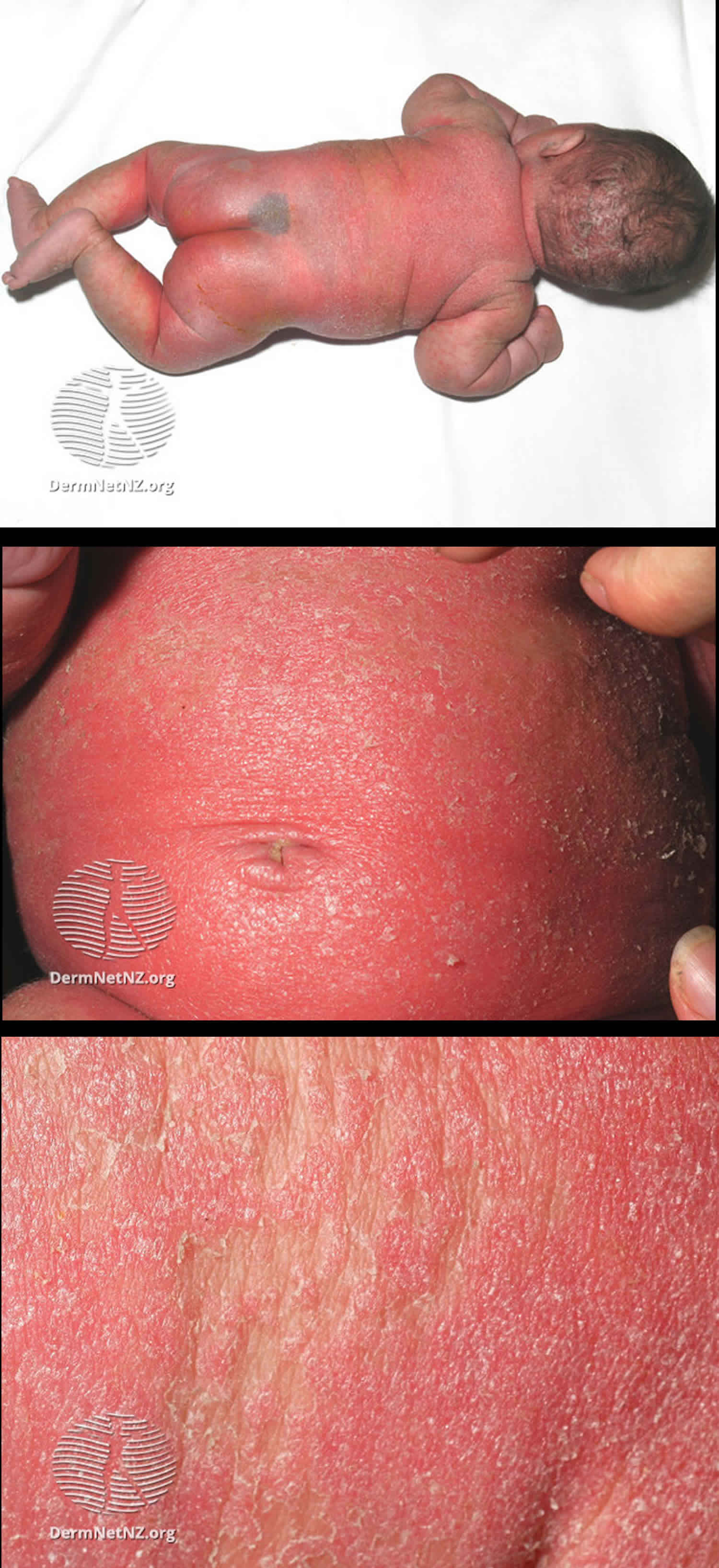
Figure 18. Holocarboxylase synthetase deficiency in adult
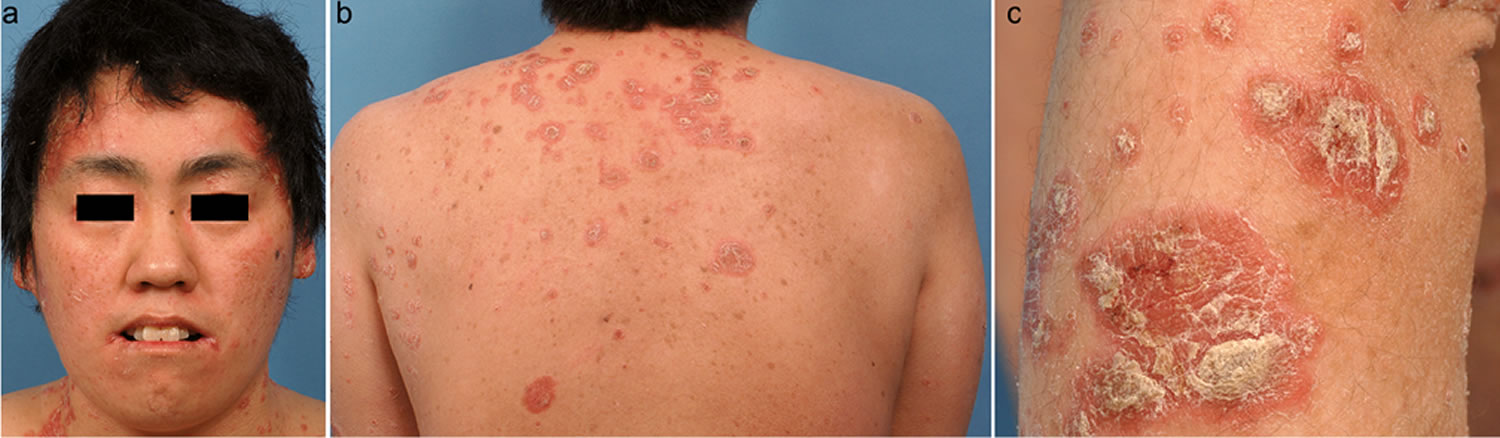
Footnotes: A 34-year-old woman presented with a skin eruption covering the whole body that had persisted since infancy. She had been born to healthy, unrelated Japanese parents. The patient had developed tachypnea and myoclonic seizures from the second day of life. Investigation confirmed severe metabolic acidosis and ketosis. The results of urinary organic acid analysis and fibroblast carboxylase tests were consistent with a diagnosis of holocarboxylase synthetase deficiency. Erythematous plaques with pustules and scales on (a) the face, (b) the abdomen and (c) the back.
[Source 237 ]Biotin deficiency causes
There are many causes of biotin deficiency or vitamin B7 deficiency. It can occur in rare inborn errors of metabolism, namely holocarboxylase synthetase deficiency or biotinidase deficiency 230, 238. Biotinidase deficiency is a rare autosomal recessive inherited disorder in which the body is unable to recycle biotin, leading to biotin deficiency despite normal biotin intake from foods such as liver, egg yolks, and milk 233. Biotinidase deficiency can present as severe biotin deficiency with both neurological and dermatological features. Biotinidase deficiency affects endogenous recycling and failure in releasing biotin from dietary protein. This affects the activity of 5 carboxylases that depends on biotin 239, 240.
Gastrointestinal tract bacterial imbalances resulting from broad-spectrum antibiotics use or inflammatory bowel disease can affect biotin synthesis in the intestine and thus lead to biotin deficiency 241.
Biotin deficiency can also occur in patients on parenteral nutrition without added biotin 230, 210, 242. Therefore, recommended daily dose of biotin must be added to total parental nutrition (TPN), particularly if TPN therapy is likely to be given for more than a week. Currently, all hospital pharmacies add biotin to TPN preparations 210, 243.
Low Biotin levels can occur in patients on antiepileptics (anticonvulsants) such as carbamazepine, phenytoin, and phenobarbital. Possible underlying mechanisms include impaired biotin uptake across the intestinal mucosa, exaggerated biotin catabolism, and inhibition of renal reabsorption. Therefore, patients who are likely to be on anticonvulsants for long periods should receive biotin supplementation 244, 245.
Prolonged use of oral antibiotics may also lead to biotin deficiency. The most likely underlying mechanism is the inhibition of intestinal flora, leading to reduced biotin production. Another possible explanation is the antibiotic-driven overgrowth of biotin-consuming bacteria 246.
Likewise, low biotin levels can occur in patients on isotretinoin (a vitamin-A derivative retinoid) for acne treatment, elderly individuals, people with alcohol use disorder, and smokers (particularly women) 247, 248, 249. Some studies have found biotin deficiency in a large percentage of pregnant and lactating women. Some experts argue that there can be teratogenic effects of decreased biotin levels, and a higher intake of biotin should be advised to pregnant women 250, 251, 205, 252, 253.
Reports exist of biotin deficiency in severely malnourished children in developing countries and through the intake of modified milk without biotin supplementation. Biotin deficiency has been observed in infants consuming hypoallergenic formulas 254.
Consuming large amounts of raw egg whites can lead to acquired biotin deficiency. Raw egg contains the glycoprotein avidin. Avidin binds to biotin in the gastrointestinal tract and prevents biotin absorption, also known as “egg white injury” 210, 238, 230, 208.
The following groups are among those most likely to have inadequate biotin status:
Biotinidase deficiency
Biotinidase deficiency is a rare autosomal recessive inherited disorder in which the body is unable to recycle biotin, leading to biotin deficiency despite normal biotin intake from foods such as liver, egg yolks, and milk 233. Biotinidase deficiency is caused by genetic changes (mutations) in the BTD gene. The BTD gene provides instructions for making an enzyme called biotinidase 234. The biotinidase enzyme recycles biotin. Biotinidase removes biotin that is bound to proteins in food, leaving the vitamin in its free (unbound) state. Free biotin is needed by enzymes called biotin-dependent carboxylases to break down fats, proteins, and carbohydrates. Because several of these enzymes are impaired in biotinidase deficiency, the condition is considered a form of multiple carboxylase deficiency 233.
Biotinidase deficiency can be partial (10 to 30% of enzyme activity) or profound (less than 10% of enzyme activity), significantly impacting the treatment approach.
- Profound biotinidase deficiency results when the activity of biotinidase is reduced to less than 10 percent of normal 233. Profound biotinidase deficiency can lead to coma or death if treatment is not initiated rapidly.
- Partial biotinidase deficiency occurs when biotinidase activity is reduced to between 10 percent and 30 percent of normal 233.
- Without enough of Biotinidase enzyme, biotin cannot be recycled. The resulting shortage of free biotin impairs the activity of biotin-dependent carboxylases, leading to a buildup of potentially toxic compounds in the body. If biotinidase deficiency is not treated promptly, this buildup damages various cells and tissues, causing the signs and symptoms described below.
Infants with biotinidase deficiency may be born without signs of the condition 234. Symptoms of biotinidase deficiency usually appear after the first few weeks or months of life 234. Treating biotinidase deficiency with biotin supplements before symptoms show up can prevent them from happening 234. Below is a list of symptoms that infants and children with profound untreated biotinidase deficiency may have. It is important to know that not every person with biotinidase deficiency will show all of these symptoms.
Many of the symptoms of biotinidase deficiency are neurological, which means they affect the brain and nervous system. About 70% of infants with biotinidase deficiency will experience seizures if they are not treated 234. This is often the first symptom of biotinidase deficiency. Seizures in infants may look different than seizures in adults. Some signs of seizures in infants include:
- Staring spells
- Jerking arm or leg movements
- Stiffening of the body
- Flickering of the eyelids
Because the seizures are caused by the body being unable to recycle biotin, they may not stop with seizure medications (anticonvulsants) 234. However, the biotinidase deficiency seizures do respond to biotin therapy and often should stop within minutes to hours of receiving biotin treatment 234.
Some infants with biotinidase deficiency may have weak muscles and low muscle tone called hypotonia. Infants with hypotonia may look abnormally “floppy.” Hypotonia can affect feeding and motor skills such sitting up without assistance. Biotinidase deficient infants and children may experience delays in reaching developmental milestones, including holding one’s head up or pulling up to stand.
Infants with biotinidase deficiency may also have breathing problems, hearing and vision loss, problems with movement and balance (ataxia) 233. These issues can be prevented if biotin therapy is started early. Some other common features of biotinidase deficiency include eye infections, like pink eye (conjunctivitis), hair loss (alopecia), certain type of skin rash called eczema and a fungal infection called candidiasis. Infants with biotinidase deficiency may have specific molecules in their urine, such as lactic acid (lactic aciduria) or low but noticeable amounts of ammonia.
Some infants with biotinidase deficiency may have other symptoms like:
- Trouble controlling their body’s movements (ataxia)
- Breathing problems
- Drowsiness (lethargy)
- Enlarged liver (hepatomegaly)
- Enlarged spleen (splenomegaly)
- Speech problems.
Partial biotinidase deficiency is a milder form of this condition 233. Without treatment, affected children and adults with partial biotinidase who don’t receive biotin supplements may experience hypotonia (weak muscles and low muscle tone), skin rashes, and hair loss (alopecia), but these problems may appear only during illness, infection, or other times of stress 233.
Profound or partial biotinidase deficiency occurs in approximately 1 in 60,000 newborns 233. All newborns in the United States and many other countries are screened for biotinidase deficiency 255. According to the worldwide neonatal screening survey, the incidence of profound biotinidase deficiency is one in 112,271, and the incidence of partial biotinidase deficiency is one in 129,282 241. The combined incidence of profound and partial biotinidase deficiency is one in 60,089 live births 241. In 2006, the incidence of profound biotinidase deficiency was 1 in 80,000, and the incidence of partial biotinidase deficiency cases was from 1 per 31,000 to 1 per 40,000 in the US. Biotinidase deficiency has been diagnosed more commonly in children of the White race. Research has observed a higher incidence of biotin deficiency in Brazil, Turkey, and Saudi Arabia 256, 239.
The estimated carrier frequency is 1 in 123 individuals. The rates vary from country to country 257, 258, 259.
Early diagnosis and treatment of biotinidase deficiency can prevent symptoms from happening. Nearly all infants with either profound or partial biotinidase deficiency can be detected in the US by newborn screening for metabolic disorders. However, not every country has added biotinidase deficiency to its newborn screening program and the late-onset forms of biotinidase deficiency have been recently described 260, 261, 262. Because the newborn screen is a screening test, a positive result does not mean that an infant definitely has biotinidase deficiency. Often, a repeat test must be done to confirm the diagnosis. A clinical diagnosis is possible after birth by testing for biotinidase activity in the blood 234. Usually, this is performed when signs and symptoms of biotinidase deficiency become clearer. In some infants, a genetic test may be ordered to identify the specific gene changes (mutation) that are causing biotinidase deficiency 234. Prenatal testing of sample fluid from the womb for biotinidase activity is available as early as 12 weeks of pregnancy (this includes chorionic villi sampling and amniocentesis) 234.
Biotinidase activity can be measured in serum, plasma, and also in fibroblasts and leukocytes and other tissue extracts by radioassay 263. The measurement of biotinidase activity in plasma or serum by colorimetric assay is the most frequently used method for the diagnosis of biotinidase deficiency 264. Normal serum biotinidase activity in humans ranges from 4.4 to 10 nmol/min/mL with a mean activity and standard deviation of 7.1 ± 1.2 nmol/min/mL 265.
Serum biotinidase activity of carriers may be similar to those with partial biotinidase deficiency, confounding diagnosis based on enzyme analysis 266. Wolf 267 suggested that evaluation of parental biotinidase activity may be helpful. Mutation in the biotinidase deficiency gene results in deficient levels of enzyme activity 268. In some cases the enzyme activity does not differentiate partial deficiency from heterozygosity for profound deficiency, and genetic analysis is necessary 269.
Biochemically, in untreated patients, metabolic ketoacidosis, lactic acidosis, and/or hyperammonemia can occur 257. Elevation of 3-hydroxyisovaleric, 3-hydroxypropionic, lactic acid, and 3-methylcrotonylglycine can be detected in urine organic acid analysis 240, 270. In previous reports, it showed that urinary excretion of 3-hydroxyisovaleric acid was an indicator of biotin status 271.
Without treatment with biotin, infants with profound biotinidase deficiency can lead to coma or death 272. Because treatment with oral biotin with as much as 5 to 20 milligrams (mg) of biotin daily starting at birth (or before symptoms develop) and continuing for the rest of the person’s life can prevent these symptoms and complications from occurring or improve them if they have already developed 257, 273. However, it takes a few hours to days for seizures and movement disorders to improve and some weeks for skin manifestations to improve. Sometimes, this dose will not be adequate, and the clinical signs may persist. Increasing the oral biotin dose to 40 mg/day is recommended in such scenarios 273. Smaller oral biotin doses may be sufficient, especially later in childhood 274, 257.
In addition to oral biotin therapy, children with residual neurologic deficits may need medical interventions for spasticity, developmental delay, and bulbar dysfunction. Dystonia and spasticity may be treated with intrathecal baclofen and neurotoxins 275.
Biotinidase deficiency prognosis is characteristically good when biotin therapy is introduced in infancy or early childhood and reliably continued for life 154, 274. The prognosis for asymptomatic biotinidase deficiency cases is good if they receive treatment before the symptoms appear. For symptomatic biotinidase deficiency patients, pharmacological biotin therapy improves most clinical features but cannot reverse neurologic damage that has already occurred 276.
Chronic alcohol exposure
Chronic exposure to alcohol inhibits the absorption of biotin 248, 249. Plasma biotin concentrations are low in 15% of people with chronic alcoholism 277. People who excessively consume alcohol have a relatively higher incidence of low biotin levels compared to the general population 230.
Pregnant and breastfeeding women
At least a third of pregnant women develop marginal biotin deficiency in spite of normal biotin intakes 202, 204, 278; plasma and breastmilk concentrations of biotin decrease in lactating women, even when their dietary biotin intakes exceed the adequate intake (AI) 279. Additional research is needed to understand the clinical significance of these findings.
Holocarboxylase synthetase deficiency
Holocarboxylase synthetase deficiency is an inherited autosomal recessive disorder in which the body is unable to use the vitamin biotin effectively 280, 281. Holocarboxylase synthetase deficiency is classified as a multiple carboxylase deficiency, which is a group of disorders characterized by impaired activity of certain enzymes that depend on biotin.
Mutations in the HLCS gene cause holocarboxylase synthetase deficiency 280. The HLCS gene provides instructions for making an enzyme called holocarboxylase synthetase. The holocarboxylase synthetase enzyme is important for the effective use of biotin, a B vitamin found in foods such as liver, egg yolks, and milk. Holocarboxylase synthetase attaches biotin to certain enzymes that are essential for the normal production and breakdown of proteins, fats, and carbohydrates in the body. Mutations in the HLCS gene reduce the holocarboxylase synthetase enzyme’s ability to attach biotin to these enzymes, preventing them from processing nutrients properly and disrupting many cellular functions. These defects lead to the serious medical problems associated with holocarboxylase synthetase deficiency.
The signs and symptoms of holocarboxylase synthetase deficiency typically appear within first few days or first 2 months of life, but the age of onset varies. Affected infants often have difficulty feeding, breathing problems (tachypnea), a skin rash (periorificial and intertriginous dermatitis), hair loss (alopecia), and a lack of energy (lethargy) 280. Seborrheic dermatitis, psoriatic dermatitis and ichthyosis are also seen in some patients 237. The pathogenesis of skin manifestations in holocarboxylase synthetase deficiency has not been fully explained. It has been postulated that a defect of fatty acid synthesis due to reduced activity of acetyl CoA carboxylase is implicated in the skin manifestations of multiple carboxylase deficiency and biotin deficiency 282. Nakajima et al. 283 reported that newborn serine palmitoyltransferase (SPT)-knockout mice showed significantly decreased epidermal levels of ceramide. They then developed alopecia and psoriasis-like skin lesions mediated by interleukin (IL)-23-dependent γδ T cells, which produce IL-17 and IL-22. Therefore, ceramide deficiency resulting from a defect of fatty acid synthesis may lead to psoriasis-like lesions in holocarboxylase synthetase deficiency, although the exact mechanism remains to be determined. These medical problems may be life-threatening in some cases.
Immediate treatment and lifelong management with biotin supplements may prevent many of these complications. If left untreated, holocarboxylase synthetase deficiency can lead to delayed development, seizures, and coma.
Holocarboxylase synthetase deficiency results in decreased formation of all holocarboxylases at physiological blood biotin concentrations; thus, high-dose biotin supplementation (10-80 mg of biotin daily) is required 154. Holocarboxylase synthetase deficiency responds to supplementation with pharmacologic doses of biotin in some cases but not others. The prognosis of holocarboxylase synthetase is usually, but not always, good if biotin therapy is introduced early (even antenatally) and continued for life 284, 154.
Biotin transport deficiency
There has been one case report of a child with biotin transport deficiency who responded to high-dose biotin supplementation 285. Of note, the presence of a defective human sodium-dependent multivitamin transporter (hSMVT) was ruled out as a cause of biotin transport deficiency.
Risk factors for biotin deficiency
Aside from prolonged consumption of raw egg white or total intravenous nutritional support lacking biotin, other conditions may increase the risk of biotin depletion. Smoking has been associated with increased biotin catabolism 247. The rapidly dividing cells of the developing fetus require biotin for synthesis of essential carboxylases and for histone biotinylation; hence, the maternal biotin requirement is likely increased during pregnancy. Research suggests that a substantial number of women develop marginal or subclinical biotin deficiency during normal pregnancy 201, 286, 205. Moreover, certain types of liver disease may decrease biotinidase activity and theoretically increase the requirement for biotin. For example, a study of 62 children with chronic liver disease and 27 healthy controls found serum biotinidase activity to be abnormally low in those with severely impaired liver function due to cirrhosis 287. However, this study did not provide evidence of biotin deficiency. Additionally, anticonvulsant medications used to prevent seizures in individuals with epilepsy increase the risk of biotin depletion 288, 289.
Biotin deficiency signs and symptoms
Signs of overt biotin deficiency include hair loss (alopecia) and a scaly red rash around the eyes, nose, mouth, and genital area (seborrheic dermatitis). Neurologic symptoms in adults have included depression, lethargy, hallucinations, numbness and tingling of the extremities (paresthesia), ataxia, and seizures. The characteristic facial rash, together with unusual facial fat distribution, has been termed the “biotin deficient facies” by some investigators 226. Individuals with hereditary disorders of biotin metabolism (e.g., biotinidase deficiency and holocarboxylase synthetase deficiency) that result in functional biotin deficiency often have similar physical findings, impaired immune system function, and increased susceptibility to bacterial and fungal infections 154, 290.
Biotin deficiency complications
Since biotin plays a crucial role in maintaining cell-mediated and humoral immunity, biotin deficiency due to inborn errors of metabolism can cause candidiasis of the skin in infants and children. There may be IgA deficiency and low percentages of T lymphocytes. They may have absent delayed-hypersensitivity skin-test responses 256, 208.
Biotin deficiency can cause encephalopathies. Patients usually respond well to large doses of biotin. Evidence shows that a lack of biotin is teratogenic in animal models. Strains of mice with biotin deficiency developed fetal malformations, most commonly cleft palate, micrognathia, and micromelia 291, 292.
Biotin deficiency diagnosis
The diagnostic tests for biotin deficiency are urinary 3-hydroxyisovaleric acid and biotin and the status of propionyl-CoA carboxylase (PCC) in lymphocytes 208, 256. Biotin-dependent carboxylases in human lymphocytes are reliable markers for determining biotin status. Decreased beta-methylcrotonyl-CoA carboxylase activity shunts the catabolism to alternative pathways, leading to the elevated formation of 3-hydroxyisovaleric acid. The most reliable marker of biotin deficiency is increased excretion of 3-hydroxyisovaleric acid in the urine (over 195 micromol/24 hours) 271. Evidence shows that serum biotin concentration does not decrease in biotin deficiency patients receiving biotin-free total parenteral nutrition. Therefore, serum biotin levels are not reliable indicators of marginal biotin deficiency 230, 208. If biotin deficiency is suspected, it warrants a thorough neurological examination and other investigations, including vision and hearing testing.
Biotinidase deficiency confirmation is done by DNA analysis, either allele-targeted methods or full-gene sequencing. Currently, all newborn screening programs in the U.S. and more than 30 other countries carry out screening for biotinidase deficiency 239, 272, 240.
Various MRI changes have been reported in individuals with biotinidase deficiency 293, 294. Desai et al. 294 noted MRI findings in four patients and observed:
- Encephalopathy
- Low cerebral volume
- Ventriculomegaly
- Widened extracerebral cerebrospinal fluid spaces
A complete reversal of these findings occurred with biotin use in two patients.
Biotin deficiency treatment
Biotin deficiency treatment essentially means treating the underlying cause 241. Lifelong treatment with biotin supplements is required in patients with genetic disorders disrupting biotin metabolism, such as holocarboxylase synthetase deficiency and biotinidase deficiency 241. If biotin deficiency is related to excess consumption of raw eggs, it should be stopped, and biotin replacement should ensue. Change anticonvulsants if the Biotin deficiency is because of the use of a particular anticonvulsant. Similarly, those on prolonged oral antibiotic therapy may benefit from biotin supplementation.
Oral biotin supplements have high bioavailability. Usually, a dose of 5 mg/day is given regardless of the cause of biotin deficiency 230. The Food and Nutrition Board of the National Research Council recommends a range of 5 mcg/day in newborn infants to 35 mcg/day in lactating women.
Healthcare professionals should be aware that biotin requirements may increase during anticonvulsant therapy 208. In biotinidase deficiency, patient therapy typically consists of lifelong doses of biotin. Biotin doses in the range of 5 to 20 mg can treat and prevent clinical signs and biotinidase deficiency symptoms 240, 256.
In the cases of holocarboxylase synthetase deficiency detected antenatally, antenatal biotin treatment has been found to be very useful 284.
Biotin deficiency prognosis
Biotin deficiency is rare and has a relatively good prognosis 241. Children diagnosed with biotinidase deficiency require early intervention and life-long biotin treatment. Children who quit therapy develop symptoms again within weeks to months. When neonates diagnosed by neonatal screening receive biotin, they develop normally without having any symptoms, and those with symptoms respond quickly to biotin treatment. Failure to evaluate and manage biotinidase deficiency at an early stage can cause irreversible neurodevelopmental abnormalities and lead to developmental delay and autistic behavior 239, 240, 256.
- Pantothenic Acid. https://ods.od.nih.gov/factsheets/PantothenicAcid-HealthProfessional[↩][↩][↩][↩][↩]
- Miller JW, Rucker RB. Pantothenic acid. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:375-90.[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Sweetman L. Pantothenic acid. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:604-11.[↩][↩][↩][↩][↩]
- Institute of Medicine. Food and Nutrition Board. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press; 1998.[↩][↩][↩][↩]
- Trumbo PR. Pantothenic acid. In: Ross AC, Caballero B, Cousins RJ, et al., eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:351-7.[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Pantothenic Acid. https://lpi.oregonstate.edu/mic/vitamins/pantothenic-acid[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bauerly K, Rucker RB. Pantothenic acid. In: Zempleni J, Rucker RB, McCormick DB, Suttie JW, eds. Handbook of vitamins. 4th ed. Boca Raton: CRC Press; 2007:289-314.[↩][↩][↩]
- Iyenga GV, Wolfe WR, Tanner JT, Morris ER. Content of minor and trace elements, and organic nutrients in representative mixed total diet composites from the USA. Sci Total Environ. 2000 Jul 10;256(2-3):215-26. doi: 10.1016/s0048-9697(00)00494-0[↩][↩]
- Provincial Epidemiology Service, New Brunswick Department of Health and Wellness. New Brunswick nutrition survey; 1997.[↩][↩]
- Tarr JB, Tamura T, Stokstad EL. Availability of vitamin B6 and pantothenate in an average American diet in man. Am J Clin Nutr. 1981 Jul;34(7):1328-37. doi: 10.1093/ajcn/34.7.1328[↩][↩]
- Hodges RE, Ohlson MA, Bean WB. Pantothenic acid deficiency in man. J Clin Invest. 1958 Nov;37(11):1642-57. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1062846/pdf/jcinvest00347-0188.pdf[↩][↩][↩]
- Sanvictores T, Chauhan S. Vitamin B5 (Pantothenic Acid) [Updated 2022 Oct 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK563233[↩][↩]
- Dezfouli MA, Jaberi E, Alavi A, Rezvani M, Shahidi G, Elahi E, Rohani M. Pantothenate kinase 2 mutation with eye-of-the-tiger sign on magnetic resonance imaging in three siblings. Iran J Neurol. 2012;11(4):155-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3829266[↩][↩][↩][↩][↩][↩][↩][↩]
- Kurian MA, Hayflick SJ. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int Rev Neurobiol. 2013;110:49-71. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6059649[↩]
- Chapter 16 – Pantothenic acid. Present Knowledge in Nutrition (Eleventh Edition). Volume 1: Basic Nutrition and Metabolism. 2020, Pages 273-287. https://doi.org/10.1016/B978-0-323-66162-1.00016-0[↩]
- Hrubša M, Siatka T, Nejmanová I, Vopršalová M, Kujovská Krčmová L, Matoušová K, Javorská L, Macáková K, Mercolini L, Remião F, Máťuš M, Mladěnka P, On Behalf Of The Oemonom. Biological Properties of Vitamins of the B-Complex, Part 1: Vitamins B1, B2, B3, and B5. Nutrients. 2022 Jan 22;14(3):484. doi: 10.3390/nu14030484[↩][↩][↩][↩][↩][↩][↩][↩]
- Rucker R.B. Pantothenic Acid. Academic Press; Cambridge, MA, USA: 2016.[↩]
- Czumaj A., Szrok-Jurga S., Hebanowska A., Turyn J., Swierczynski J., Sledzinski T., Stelmanska E. The Pathophysiological Role of CoA. Int. J. Mol. Sci. 2020;21:9057. doi: 10.3390/ijms21239057[↩]
- Mindrebo J.T., Patel A., Misson L.E., Kim W.E., Davis T.D., Ni Q.Z., La Clair J.J., Burkart M.D. 1.04-Structural Basis of Acyl-Carrier Protein Interactions in Fatty Acid and Polyketide Biosynthesis. In: Hung-Wen L., Begley T.P., editors. Comprehensive Natural Products III. Elsevier; Amsterdam, The Netherlands: 2020.[↩]
- Miller JW, Rucker RB. Pantothenic acid. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Ames: Wiley-Blackwell; 2012:375-390.[↩][↩][↩][↩]
- Takahashi A, Mizusawa K. Posttranslational modifications of proopiomelanocortin in vertebrates and their biological significance. Front Endocrinol (Lausanne). 2013 Oct 17;4:143. doi: 10.3389/fendo.2013.00143[↩]
- Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014 Aug;15(8):536-50. doi: 10.1038/nrm3841[↩]
- Bunkoczi G, Pasta S, Joshi A, Wu X, Kavanagh KL, Smith S, Oppermann U. Mechanism and substrate recognition of human holo ACP synthase. Chem Biol. 2007 Nov;14(11):1243-53. doi: 10.1016/j.chembiol.2007.10.013[↩][↩]
- Donato H, Krupenko NI, Tsybovsky Y, Krupenko SA. 10-formyltetrahydrofolate dehydrogenase requires a 4′-phosphopantetheine prosthetic group for catalysis. J Biol Chem. 2007 Nov 23;282(47):34159-66. doi: 10.1074/jbc.M707627200[↩]
- Strickland KC, Hoeferlin LA, Oleinik NV, Krupenko NI, Krupenko SA. Acyl carrier protein-specific 4′-phosphopantetheinyl transferase activates 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 2010 Jan 15;285(3):1627-33. doi: 10.1074/jbc.M109.080556[↩]
- Strickland KC, Krupenko NI, Dubard ME, Hu CJ, Tsybovsky Y, Krupenko SA. Enzymatic properties of ALDH1L2, a mitochondrial 10-formyltetrahydrofolate dehydrogenase. Chem Biol Interact. 2011 May 30;191(1-3):129-36. doi: 10.1016/j.cbi.2011.01.008[↩]
- Rumberger JA, Napolitano J, Azumano I, Kamiya T, Evans M. Pantethine, a derivative of vitamin B(5) used as a nutritional supplement, favorably alters low-density lipoprotein cholesterol metabolism in low- to moderate-cardiovascular risk North American subjects: a triple-blinded placebo and diet-controlled investigation. Nutr Res. 2011 Aug;31(8):608-15. doi: 10.1016/j.nutres.2011.08.001[↩][↩]
- McRae MP. Treatment of hyperlipoproteinemia with pantethine: a review and analysis of efficacy and tolerability. 2005. In: Database of Abstracts of Reviews of Effects (DARE): Quality-assessed Reviews [Internet]. York (UK): Centre for Reviews and Dissemination (UK); 1995-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK71836[↩][↩][↩]
- Chen YQ, Zhao SP, Zhao YH. Efficacy and tolerability of coenzyme A vs pantethine for the treatment of patients with hyperlipidemia: A randomized, double-blind, multicenter study. J Clin Lipidol. 2015 Sep-Oct;9(5):692-7. doi: 10.1016/j.jacl.2015.07.003[↩][↩]
- Evans M, Rumberger JA, Azumano I, Napolitano JJ, Citrolo D, Kamiya T. Pantethine, a derivative of vitamin B5, favorably alters total, LDL and non-HDL cholesterol in low to moderate cardiovascular risk subjects eligible for statin therapy: a triple-blinded placebo and diet-controlled investigation. Vasc Health Risk Manag. 2014 Feb 27;10:89-100. doi: 10.2147/VHRM.S57116[↩]
- Weimann BI, Hermann D. Studies on wound healing: effects of calcium D-pantothenate on the migration, proliferation and protein synthesis of human dermal fibroblasts in culture. Int J Vitam Nutr Res. 1999 Mar;69(2):113-9. doi: 10.1024/0300-9831.69.2.113[↩]
- Wiederholt, T., Heise, R., Skazik, C., Marquardt, Y., Joussen, S., Erdmann, K., Schröder, H., Merk, H.F. and Baron, J.M. (2009), Calcium pantothenate modulates gene expression in proliferating human dermal fibroblasts. Experimental Dermatology, 18: 969-978. https://doi.org/10.1111/j.1600-0625.2009.00884.x[↩]
- Kobayashi D, Kusama M, Onda M, Nakahata N. The effect of pantothenic acid deficiency on keratinocyte proliferation and the synthesis of keratinocyte growth factor and collagen in fibroblasts. J Pharmacol Sci. 2011;115(2):230-4. doi: 10.1254/jphs.10224sc[↩]
- Heise R, Skazik C, Marquardt Y, Czaja K, Sebastian K, Kurschat P, Gan L, Denecke B, Ekanayake-Bohlig S, Wilhelm KP, Merk HF, Baron JM. Dexpanthenol modulates gene expression in skin wound healing in vivo. Skin Pharmacol Physiol. 2012;25(5):241-8. doi: 10.1159/000341144[↩][↩]
- Vaxman F, Olender S, Lambert A, Nisand G, Aprahamian M, Bruch JF, Didier E, Volkmar P, Grenier JF. Effect of pantothenic acid and ascorbic acid supplementation on human skin wound healing process. A double-blind, prospective and randomized trial. Eur Surg Res. 1995;27(3):158-66. doi: 10.1159/000129395[↩]
- Vaxman F, Olender S, Lambert A, Nisand G, Grenier JF. Can the wound healing process be improved by vitamin supplementation? Experimental study on humans. Eur Surg Res. 1996 Jul-Aug;28(4):306-14. doi: 10.1159/000129471[↩]
- Celebi S, Tepe C, Yelken K, Celik O. Efficacy of dexpanthenol for pediatric post-tonsillectomy pain and wound healing. Ann Otol Rhinol Laryngol. 2013 Jul;122(7):464-7. doi: 10.1177/000348941312200710[↩]
- Capodice J.L. Feasibility, Tolerability, Safety and Efficacy of a Pantothenic Acid Based Dietary Supplement in Subjects with Mild to Moderate Facial Acne Blemishes. J. Cosmet. Dermatol. Sci. Appl. 2012;2:132–135. doi: 10.4236/jcdsa.2012.23026[↩]
- Yang M, Moclair B, Hatcher V, Kaminetsky J, Mekas M, Chapas A, Capodice J. A randomized, double-blind, placebo-controlled study of a novel pantothenic Acid-based dietary supplement in subjects with mild to moderate facial acne. Dermatol Ther (Heidelb). 2014 Jun;4(1):93-101. doi: 10.1007/s13555-014-0052-3[↩][↩][↩][↩]
- Udompataikul M, Limpa-o-vart D. Comparative trial of 5% dexpanthenol in water-in-oil formulation with 1% hydrocortisone ointment in the treatment of childhood atopic dermatitis: a pilot study. J Drugs Dermatol. 2012 Mar;11(3):366-74.[↩][↩]
- Kuwahara, R.T. and Skinner, R.B. (2002), Granuloma Annulare Resolved With Topical Application Of Imiquimod. Pediatric Dermatology, 19: 368-371. https://doi.org/10.1046/j.1525-1470.2002.00105.x[↩]
- Proksch E., de Bony R., Trapp S., Boudon S. Topical use of dexpanthenol: A 70th anniversary article. J. Dermatol. Treat. 2017;28:766–773. doi: 10.1080/09546634.2017.1325310[↩]
- Wollina U., Kubicki J. Dexpanthenol supports healing of superficial wounds and injuries. Kosm. Med. 2006;27:240–249.[↩]
- Proksch E., Nissen H.P. Dexpanthenol enhances skin barrier repair and reduces inflammation after sodium lauryl sulphate-induced irritation. J. Dermatol. Treat. 2002;13:173–178. doi: 10.1080/09546630212345674[↩]
- Stettler H., Kurka P., Lunau N., Manger C., Bohling A., Bielfeldt S., Wilhelm K.P., Dahnhardt-Pfeiffer S., Dahnhardt D., Brill F.H., et al. A new topical panthenol-containing emollient: Results from two randomized controlled studies assessing its skin moisturization and barrier restoration potential, and the effect on skin microflora. J. Dermatol. Treat. 2017;28:173–180. doi: 10.1080/09546634.2016.1214235[↩]
- Heise R., Schmitt L., Huth L., Krings L., Kluwig D., Katsoulari K.V., Steiner T., Holzle F., Baron J.M., Huth S. Accelerated wound healing with a dexpanthenol-containing ointment after fractional ablative CO2 laser resurfacing of photo-damaged skin in a randomized prospective clinical trial. Cutan. Ocul. Toxicol. 2019;38:274–278. doi: 10.1080/15569527.2019.1597879[↩]
- Wananukul S., Limpongsanuruk W., Singalavanija S., Wisuthsarewong W. Comparison of dexpanthenol and zinc oxide ointment with ointment base in the treatment of irritant diaper dermatitis from diarrhea: A multicenter study. J. Med. Assoc. Thai. 2006;89:1654–1658.[↩]
- Olsavszky R., Nanu E.A., Macura-Biegun A., Kurka P., Trapp S. Skin barrier restoration upon topical use of two 5% dexpanthenol water-in-oil formulations on freshly tattooed skin: Results from a single-blind prospective study. Wounds Int. 2019;10:33–39.[↩]
- Shanazi M., Farshbaf Khalili A., Kamalifard M., Asghari Jafarabadi M., Masoudin K., Esmaeli F. Comparison of the Effects of Lanolin, Peppermint, and Dexpanthenol Creams on Treatment of Traumatic Nipples in Breastfeeding Mothers. J. Caring Sci. 2015;4:297–307. doi: 10.15171/jcs.2015.030[↩]
- Hamdi I.M. Effect of D-Panthenol on Corneal Epithelial Healing after Surface Laser Ablation. J. Ophthalmol. 2018;2018:6537413. doi: 10.1155/2018/6537413[↩]
- Gobbels M., Gross D. Clinical study of the effectiveness of a dexpanthenol containing artificial tears solution (Siccaprotect) in treatment of dry eyes. Klin. Monbl. Augenheilkd. 1996;209:84–88. doi: 10.1055/s-2008-1035283[↩]
- Jagade M.V., Langade D.G., Pophale R.R., Prabhu A. Oxymetazoline plus dexpanthenol in nasal congestion. Indian J. Otolaryngol. Head Neck Surg. 2008;60:393–397. doi: 10.1007/s12070-008-0125-7[↩]
- Kehrl W., Sonnemann U., Dethlefsen U. Advance in therapy of acute rhinitis—Comparison of efficacy and safety of xylometazoline in combination xylometazoline-dexpanthenol in patients with acute rhinitis. Laryngo-Rhino-Otologie. 2003;82:266–271. doi: 10.1055/s-2003-38941[↩]
- Kehrl W., Sonnemann U. Improving wound healing after nose surgery by combined administration of xylometazoline and dexpanthenol. Laryngo-Rhino-Otologie. 2000;79:151–154. doi: 10.1055/s-2000-295[↩]
- Pantothenic Acid. https://lpi.oregonstate.edu/mic/vitamins/pantothenic-acid#disease-treatment[↩]
- Berry Ottaway P. Stability of vitamins during food processing and storage. In: Skibsted L.H., Risbo J., Andersen M.L., editors. Chemical Deterioration and Physical Instability of Food and Beverages. Woodhead Publishing; Cambridge, UK: 2010.[↩][↩][↩]
- Walsh J.H., Wyse B.W., Hansen R.G. Pantothenic acid content of 75 processed and cooked foods. J. Am. Diet. Assoc. 1981;78:140–144. doi: 10.1016/S0002-8223(21)04766-0[↩]
- Garg M., Sharma A., Vats S., Tiwari V., Kumari A., Mishra V., Krishania M. Vitamins in Cereals: A Critical Review of Content, Health Effects, Processing Losses, Bioaccessibility, Fortification, and Biofortification Strategies for Their Improvement. Front. Nutr. 2021;8:586815. doi: 10.3389/fnut.2021.586815[↩][↩][↩][↩][↩]
- Chawla J, Kvarnberg D. Hydrosoluble vitamins. Handb Clin Neurol. 2014;120:891-914. doi: 10.1016/B978-0-7020-4087-0.00059-0[↩][↩][↩]
- MacDonald R., Reitmeier C., editors. Understanding Food Systems. Academic Press; Cambridge, MA, USA: 2017. Food Processing; pp. 179–225.[↩]
- Kyritsi A., Tzia C., Karathanos V.T. Vitamin fortified rice grain using spraying and soaking methods. LWT Food Sci. Technol. 2011;44:312–320. doi: 10.1016/j.lwt.2010.06.001[↩][↩]
- Miller J.W., Rucker R.B. Present Knowledge in Nutrition. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2012. Pantothenic Acid; pp. 375–390.[↩]
- Gwirtz J.A., Garcia-Casal M.N. Processing maize flour and corn meal food products. Ann. N. Y. Acad. Sci. 2014;1312:66–75. doi: 10.1111/nyas.12299[↩]
- Schroeder H.A. Losses of vitamins and trace minerals resulting from processing and preservation of foods. Am. J. Clin. Nutr. 1971;24:562–573. doi: 10.1093/ajcn/24.5.562[↩][↩][↩][↩]
- Tiozon R.N., Fernie A.R., Sreenivasulu N. Meeting human dietary vitamin requirements in the staple rice via strategies of biofortification and post-harvest fortification. Trends Food Sci. Technol. 2021;109:65–82. doi: 10.1016/j.tifs.2021.01.023[↩]
- Godoy H.T., Amaya-Farfan J., Rodriguez-Amaya D.B. Degradation of vitamins. In: Rodriguez-Amaya D.B., Amaya-Farfan J., editors. Chemical Changes During Processing and Storage of Foods. Academic Press; Cambridge, MA, USA: 2021. pp. 329–383.[↩]
- Bognár A. Tables on Weight Yield of Food and Retention Factors of Food Constituents for the Calculation of Nutrient Composition of Cooked Foods (Dishes) Bundesforschungsanstalt für Ernährung; Karlsruhe, Germany: 2002.[↩][↩][↩][↩][↩][↩][↩]
- Bell S., Becker W., Vásquez-Caicedo A., Hartmann B., Møller A., Butriss J. Report on Nutrient Losses and Gains Factors Used in European Food Composition Databases. Federal Research Centre for Nutrition and Food; Karlsruhe, Germany: 2006.[↩][↩][↩][↩]
- Lešková E., Kubíková J., Kováčiková E., Košická M., Porubská J., Holčíková K. Vitamin losses: Retention during heat treatment and continual changes expressed by mathematical models. J. Food Compos. Anal. 2006;19:252–276. doi: 10.1016/j.jfca.2005.04.014[↩][↩][↩]
- Engler P.P., Bowers J.A. B-vitamin retention in meat during storage and preparation. A review. J. Am. Diet. Assoc. 1976;69:253–257. doi: 10.1016/S0002-8223(21)06708-0[↩][↩]
- Meyer B.H., Mysinger M.A., Wodarski L.A. Pantothenic acid and vitamin B6 in beef. J. Am. Diet Assoc. 1969;54:122–125. doi: 10.1016/S0002-8223(21)12596-9[↩]
- Roe M., Church S., Pinchen H., Finglas P. Nutrient Analysis of Fish and Fish Products. Institute of Food Research; Norwich, UK: 2013. pp. 14–69. Analytical Report.[↩]
- Bodwell C., Anderson B. Nutritional Composition and Value of Meat and Meat Products. Academic Press; Cambridge, MA, USA: 1986. pp. 321–369.[↩]
- Roe M., Church S., Pinchen H., Finglas P. Nutrient Analysis of Fruit and Vegetables. Institute of Food Research; Norwich, UK: 2013. pp. 17–76. Analytical Report.[↩][↩]
- Cheng T.S., Eitenmiller R.R. Effects of Processing and Storage on the Pantothenic-Acid Content of Spinach and Broccoli. J. Food Process. Preserv. 1988;12:115–123. doi: 10.1111/j.1745-4549.1988.tb00071.x[↩][↩]
- Hoppner K., Lampi B. Pantothenic-Acid and Biotin Retention in Cooked Legumes. J. Food Sci. 1993;58:1084–1085. doi: 10.1111/j.1365-2621.1993.tb06119.x[↩]
- Khalil A.H., Mansour E.H. The Effect of Cooking, Autoclaving and Germination on the Nutritional Quality of Faba Beans. Food Chem. 1995;54:177–182. doi: 10.1016/0308-8146(95)00024-D[↩]
- Roe M., Church S., Pinchen H., Finglas P. Nutrient Analysis of Eggs. Institute of Food Research; Norwich, UK: 2013. pp. 1–44. Analytical Report.[↩]
- Rolls B.A., Porter J.W.G. Some effects of processing and storage on the nutritive value of milk and milk products. Proc. Nutr Soc. 1973;32:9–15. doi: 10.1079/PNS19730003[↩]
- Nurit E., Lyan B., Pujos-Guillot E., Branlard G., Piquet A. Change in B and E vitamin and lutein, β-sitosterol contents in industrial milling fractions and during toasted bread production. J. Cereal Sci. 2016;69:290–296. doi: 10.1016/j.jcs.2016.04.005[↩]
- Dunn K.R., Goddard V.R. Effect of heat upon the nutritive values of peanuts; riboflavin and pantothenic acid content. Food Res. 1948;13:512–517. doi: 10.1111/j.1365-2621.1948.tb16652.x[↩]
- Greenwood D.A., Kraybill H.R., Feaster J.F., Jackson J.M. Vitamin Retention in Processed Meat. Ind. Eng. Chem. 1944;36:922–927. doi: 10.1021/ie50418a012[↩]
- Muhamad N., Yusoff M.M., Gimbun J. Thermal degradation kinetics of nicotinic acid, pantothenic acid and catechin derived from Averrhoa bilimbi fruits. RSC Adv. 2015;5:74132–74137. doi: 10.1039/C5RA11950B[↩]
- Woodside J. Nutritional aspects of irradiated food. Stewart Postharvest Rev. 2015;11:1–6. doi: 10.2212/spr.2015.3.2[↩]
- Kilcast D. Effect of Irradiation on Vitamins. Food Chem. 1994;49:157–164. doi: 10.1016/0308-8146(94)90152-X[↩]
- Suri D.J., Tanumihardjo S.A. Effects of Different Processing Methods on the Micronutrient and Phytochemical Contents of Maize: From A to Z. Compr. Rev. Food Sci. Food Saf. 2016;15:912–926. doi: 10.1111/1541-4337.12216[↩]
- Escalante-Aburto A., Mariscal-Moreno R.M., Santiago-Ramos D., Ponce-García N. An Update of Different Nixtamalization Technologies, and Its Effects on Chemical Composition and Nutritional Value of Corn Tortillas. Food Rev. Int. 2020;36:456–498. doi: 10.1080/87559129.2019.1649693[↩]
- Gonzalez-Lopez J., Aliaga L., Gonzalez-Martinez A., Martinez-Toledo M.V. Industrial Biotechnology of Vitamins, Biopigments, and Antioxidants. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2016. Pantothenic Acid; pp. 67–101.[↩]
- Kelly GS. Pantothenic acid. Monograph. Altern Med Rev. 2011 Sep;16(3):263-74.[↩][↩]
- Pearson A., West R., Luecke R. The vitamin and amino acid content of drip obtained upon defrosting frozen pork. Food Res. 1959;24:515–519. doi: 10.1111/j.1365-2621.1959.tb17302.x[↩]
- Pearson A.M., Burnside J.E., Edwards H.M., Glasscock R.S., Cunha T.J., Novak A.F. Vitamin losses in drip obtained upon defrosting frozen meat. Food Res. 1951;16:85–87. doi: 10.1111/j.1365-2621.1951.tb17354.x[↩]
- Woollard D.C., Indyk H.E., Christiansen S.K. The analysis of pantothenic acid in milk and infant formulas by HPLC. Food Chem. 2000;69:201–208. doi: 10.1016/S0308-8146(99)00255-1[↩][↩]
- Liberato S.C., Pinheiro-Sant’Ana H.M. Fortification of industrialized foods with vitamins. Rev. Nutr. 2006;19:215–231. doi: 10.1590/S1415-52732006000200009[↩]
- EU Commision E. Commission Directive 2006/125/EC of 5 December 2006 on Processed Cereal-Based Foods and Baby Foods for Infants and Young Children. https://eur-lex.europa.eu/eli/dir/2006/125/oj[↩]
- Müller M.A., Medlock J., Prágai Z., Warnke I., Litta G., Kleefeldt A., Kaiser K., De Potzolli B. Ullmann’s Encyclopedia of Industrial Chemistry. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2019. Vitamins, 9. Vitamin B5; pp. 1–16.[↩]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA) Scientific Opinion on Dietary Reference Values for pantothenic acid. EFSA J. 2014;12:3581. doi: 10.2903/j.efsa.2014.3581[↩]
- Lu B., Ren Y., Huang B., Liao W., Cai Z., Tie X. Simultaneous determination of four water-soluble vitamins in fortified infant foods by ultra-performance liquid chromatography coupled with triple quadrupole mass spectrometry. J. Chromatogr. Sci. 2008;46:225–232. doi: 10.1093/chromsci/46.3.225[↩]
- U.S. Department of Agriculture, Agricultural Research Service. FoodData Central. https://fdc.nal.usda.gov[↩]
- Horváth Z, Vécsei L. Current medical aspects of pantethine. Ideggyogy Sz. 2009 Jul 30;62(7-8):220-9.[↩]
- Hendler SS, Rorvik DR, eds. PDR for Nutritional Supplements. 2nd ed. Montvale: Thomson Reuters; 2008.[↩][↩][↩]
- Pantothenate kinase-associated neurodegeneration. https://medlineplus.gov/genetics/condition/pantothenate-kinase-associated-neurodegeneration[↩][↩][↩]
- Hartig MB, Hörtnagel K, Garavaglia B, Zorzi G, Kmiec T, Klopstock T, Rostasy K, Svetel M, Kostic VS, Schuelke M, Botz E, Weindl A, Novakovic I, Nardocci N, Prokisch H, Meitinger T. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol. 2006 Feb;59(2):248-56. doi: 10.1002/ana.20771[↩][↩]
- Schneider SA, Hardy J, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord. 2012 Jan;27(1):42-53. doi: 10.1002/mds.23971[↩][↩][↩]
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001 Aug;28(4):345-9. doi: 10.1038/ng572[↩][↩]
- Hörtnagel K, Prokisch H, Meitinger T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum Mol Genet. 2003 Feb 1;12(3):321-7. doi: 10.1093/hmg/ddg026[↩]
- Kurian MA, Hayflick SJ. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int Rev Neurobiol. 2013;110:49-71. doi: 10.1016/B978-0-12-410502-7.00003-X[↩][↩][↩][↩]
- Glusman M. The syndrome of burning feet (nutritional melalgia) as a manifestation of nutritional deficiency. Am J Med 1947;3:211-23. doi: 10.1016/0002-9343(47)90151-4[↩]
- Hodges RE, Bean WB, Ohlson MA, et al. Human pantothenic acid deficiency produced by omega-methyl pantothenic acid. J Clin Invest 1959;38:1421-5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC442097/pdf/jcinvest00451-0153.pdf[↩]
- Johnson M.A., Kuo Y.M., Westaway S.K., Parker S.M., Ching K.H., Gitschier J., Hayflick S.J. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann. N. Y. Acad. Sci. 2004;1012:282–298. doi: 10.1196/annals.1306.023[↩]
- Pantothenate Kinase-Associated Neurodegeneration. https://rarediseases.org/rare-diseases/pantothenate-kinase-associated-neurodegeneration[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Pantothenate kinase-associated neurodegeneration. https://rarediseases.info.nih.gov/diseases/6564/pantothenate-kinase-associated-neurodegeneration[↩]
- Sanvictores T, Chauhan S. Vitamin B5 (Pantothenic Acid) [Updated 2022 Oct 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK563233[↩]
- Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009 Feb;46(2):73-80. doi: 10.1136/jmg.2008.061929[↩]
- Gregory A, Hayflick SJ. Pantothenate Kinase-Associated Neurodegeneration. Orphanet Encyclopedia. 2004.[↩]
- Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, Gitschier J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003 Jan 2;348(1):33-40. doi: 10.1056/NEJMoa020817[↩]
- Hayflick SJ, Hartman M, Coryell J, Gitschier J, Rowley H. Brain MRI in neurodegeneration with brain iron accumulation with and without PANK2 mutations. AJNR Am J Neuroradiol. 2006 Jun-Jul;27(6):1230-3. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2099458[↩]
- PANK2 gene. https://medlineplus.gov/genetics/gene/pank2[↩][↩]
- Hayflick SJ. Defective pantothenate metabolism and neurodegeneration. Biochem Soc Trans. 2014 Aug;42(4):1063-8. doi: 10.1042/BST20140098[↩][↩]
- Gregory A, Hayflick SJ. Pantothenate Kinase-Associated Neurodegeneration. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2017.[↩]
- Pantothenate kinase-associated neurodegeneration. https://medlineplus.gov/images/PX000366_PRESENTATION.jpeg[↩]
- Olivieri NF, Sabouhanian A, Gallie BL. Single-center retrospective study of the effectiveness and toxicity of the oral iron chelating drugs deferiprone and deferasirox. PLoS One. 2019 Feb 27;14(2):e0211942. doi: 10.1371/journal.pone.0211942. Erratum in: PLoS One. 2019 Mar 13;14(3):e0214005[↩]
- Munshi MI, Yao SJ, Ben Mamoun C. Redesigning therapies for pantothenate kinase-associated neurodegeneration. J Biol Chem. 2022 Mar;298(3):101577. doi: 10.1016/j.jbc.2022.101577[↩]
- Hodges R.E., Ohlson M.A., Bean W.B. Pantothenic acid deficiency in man. J. Clin. Investig. 1958;37:1642–1657. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1062846/pdf/jcinvest00347-0188.pdf[↩][↩]
- Fry P.C., Fox H.M., Tao H.G. Metabolic response to a pantothenic acid deficient diet in humans. J. Nutr. Sci. Vitam. 1976;22:339–346. doi: 10.3177/jnsv.22.339[↩][↩]
- Xu J., Patassini S., Begley P., Church S., Waldvogel H.J., Faull R.L.M., Unwin R.D., Cooper G.J.S. Cerebral deficiency of vitamin B5 (d-pantothenic acid; pantothenate) as a potentially-reversible cause of neurodegeneration and dementia in sporadic Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2020;527:676–681. doi: 10.1016/j.bbrc.2020.05.015[↩]
- Nelson M.M., Evans H.M. Pantothenic acid deficiency and reproduction in the rat. J. Nutr. 1946;31:497–507. doi: 10.1093/jn/31.4.497[↩][↩]
- Chen M.-C., Song Y., Song W.O. Fetal growth retardation and death in pantothenic acid-deficient rats is due to imparired placental function. J. Nutr. Biochem. 1996;7:451–456. doi: 10.1016/0955-2863(96)00078-2[↩]
- Drell W., Dunn M.S. Production of pantothenic acid deficiency syndrome in mice with-methylpantothenic acid. Arch. Biochem. Biophys. 1951;33:110–119. doi: 10.1016/0003-9861(51)90085-9[↩]
- Pudelkewicz C., Roderuck C. Pantothenic acid deficiency in the young guinea pig. J. Nutr. 1960;70:348–352. doi: 10.1093/jn/70.3.348[↩]
- Olson R.E., Kaplan N.O. The effect of pantothenic acid deficiency upon the coenzyme A content and pyruvate utilization of rat and duck tissues. J. Biol. Chem. 1948;175:515–529. doi: 10.1016/S0021-9258(18)57172-6[↩][↩]
- Hatano M. Pantothenic acid deficiency in rats. J. Vitam. 1962;8:143–159. doi: 10.5925/jnsv1954.8.143[↩]
- BERG BN. Duodenitis and duodenal ulcers produced in rats by pantothenic acid deficiency. Br J Exp Pathol. 1959 Aug;40(4):371-4.[↩]
- Groody T.C., Groody M.E. Feather Depigmentation and Pantothenic Acid Deficiency in Chicks. Science. 1942;95:655–656. doi: 10.1126/science.95.2478.655[↩]
- Gries C.L., Scott M.L. The pathology of thiamin, riboflavin, pantothenic acid and niacin deficiencies in the chick. J. Nutr. 1972;102:1269–1285. doi: 10.1093/jn/102.10.1269[↩]
- Wintrobe M.M., Follis R.H., Jr., Alcayaga R., Paulson M., Humphreys S. Pantothenic acid deficiency in swine with particular reference to the effects on growth and on the alimentary tract. Bul. Johns Hopkins Hosp. 1943;73:313–341.[↩]
- Follis R.H., Wintrobe M.M. A Comparison of the Effects of Pyridoxine and Pantothenic Acid Deficiencies on the Nervous Tissues of Swine. J. Exp. Med. 1945;81:539–552. doi: 10.1084/jem.81.6.539[↩]
- Nelson R.A. Intestinal transport, coenzyme A, and colitis in pantothenic acid deficiency. Am. J. Clin. Nutr. 1968;21:495–501. doi: 10.1093/ajcn/21.5.495[↩]
- Schaefer A.E., McKibbin J.M., Elvehjem C.A. Pantothenic acid deficiency studies in the dog. J. Biol. Chem. 1942;143:321–330. doi: 10.1016/S0021-9258(18)72619-7[↩]
- Silber R.H. Studies of Pantothenic Acid Deficiency in Dogs: Three Figures. J. Nutr. 1944;27:425–433. doi: 10.1093/jn/27.5.25[↩]
- Flodin N. Pharmacology of micronutrients. New York: Alan R. Liss, Inc.; 1988.[↩][↩]
- Debourdeau PM, Djezzar S, Estival JL, Zammit CM, Richard RC, Castot AC. Life-threatening eosinophilic pleuropericardial effusion related to vitamins B5 and H. Ann Pharmacother. 2001 Apr;35(4):424-6. doi: 10.1345/aph.10213[↩]
- Debourdeau P.M., Djezzar S., Estival J.L., Zammit C.M., Richard R.C., Castot A.C. Life-threatening eosinophilic pleuropericardial effusion related to vitamins B5 and H. Ann. Pharmacother. 2001;35:424–426. doi: 10.1345/aph.10213[↩]
- Food and Nutrition Board, Institute of Medicine. Pantothenic acid. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, D.C.: National Academy Press; 1998:357-373.[↩]
- Herbst, R.A., Uter, W., Pirker, C., Geier, J. and Frosch, P.J. (2004), Allergic and non-allergic periorbital dermatitis: patch test results of the Information Network of the Departments of Dermatology during a 5-year period. Contact Dermatitis, 51: 13-19. https://doi.org/10.1111/j.0105-1873.2004.00334.x[↩]
- M. Schmuth, M.A. Wimmer, S. Hofer, A. Sztankay, G. Weinlich, D.M. Linder, P.M. Elias, P.O. Fritsch, E. Fritsch, Topical corticosteroid therapy for acute radiation dermatitis: a prospective, randomized, double‐blind study, British Journal of Dermatology, Volume 146, Issue 6, 1 June 2002, Pages 983–991, https://doi.org/10.1046/j.1365-2133.2002.04751.x[↩]
- Lee JH, Ahn SY, Lee HA, Won KS, Chang HW, Oh JS, Kim HW. Dietary intake of pantothenic acid is associated with cerebral amyloid burden in patients with cognitive impairment. Food Nutr Res. 2018 Dec 10;62. doi: 10.29219/fnr.v62.1415[↩][↩]
- Chirapu SR, Rotter CJ, Miller EL, Varma MV, Dow RL, Finn MG. High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid. Curr Top Med Chem. 2013;13(7):837-42. doi: 10.2174/1568026611313070006[↩]
- Said HM, Ortiz A, McCloud E, Dyer D, Moyer MP, Rubin S. Biotin uptake by human colonic epithelial NCM460 cells: a carrier-mediated process shared with pantothenic acid. Am J Physiol. 1998 Nov;275(5):C1365-71. doi: 10.1152/ajpcell.1998.275.5.C1365[↩]
- Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:390-8.[↩][↩][↩][↩][↩][↩][↩][↩]
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:359-74[↩][↩]
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:359-74.[↩][↩][↩]
- Said HM. Biotin: biochemical, physiological and clinical aspects. Subcell Biochem 2012;56:1-19.[↩][↩]
- Mock DM. Biotin. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:43-51[↩][↩][↩]
- Elrefai S, Wolf B. Disorders of biotin metabolism. In: Rosenberg RN, Pascual JM, eds. Rosenberg’s Molecular and Genetic basis of Neurological and Psychiatric Disease. 5th ed. United States of America: Elsevier; 2015:531-539.[↩][↩][↩][↩][↩]
- Regula Baumgartner, E. and Suormala, T. (1999), Inherited defects of biotin metabolism. BioFactors, 10: 287-290. https://doi.org/10.1002/biof.5520100229[↩]
- Eng WK, Giraud D, Schlegel VL, Wang D, Lee BH, Zempleni J. Identification and assessment of markers of biotin status in healthy adults. Br J Nutr 2013;110:321-9[↩][↩][↩]
- Li D, Radulescu A, Shrestha RT, Root M, Karger AB, Killeen AA, et al. Association of biotin ingestion with performance of hormone and nonhormone assays in healthy adults. JAMA 2017;318:1150-60[↩]
- Zempleni J, Mock DM. Biotin biochemistry and human requirements. J Nutr Biochem. 1999 Mar;10(3):128-38. doi: 10.1016/s0955-2863(98)00095-3[↩]
- Present Knowledge in Nutrition. Basic Nutrition and Metabolism 11th Edition pp. 289–304, July 17, 2020. ISBN: 9780323661621[↩][↩]
- Biotin. https://lpi.oregonstate.edu/mic/vitamins/biotin[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253-72. doi: 10.1146/annurev.nutr.28.061807.155434[↩]
- Mock D, Matthews N. Biotin and pantothenic acid In: Stipanuk MH, Caudill MA, editors. Biochemical, physiological and molecular aspects of human nutrition. 3rd ed Amsterdam (Netherlands): Elsevier; 2012. p. 610–25.[↩]
- Zempleni J, Wijeratne SSK, Kuroishi T. Biotin. In: Erdman JWJ, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed: John Wiley & Sons, Inc.; 2012:359-374.[↩][↩]
- Zempleni J, Teixeira DC, Kuroishi T, Cordonier EL, Baier S. Biotin requirements for DNA damage prevention. Mutat Res. 2012 May 1;733(1-2):58-60. doi: 10.1016/j.mrfmmm.2011.08.001[↩][↩]
- Zempleni J, Li Y, Xue J, Cordonier EL. The role of holocarboxylase synthetase in genome stability is mediated partly by epigenomic synergies between methylation and biotinylation events. Epigenetics. 2011 Jul;6(7):892-4. doi: 10.4161/epi.6.7.15544[↩]
- Zempleni J, Gralla M, Camporeale G, Hassan YI. Sodium-dependent multivitamin transporter gene is regulated at the chromatin level by histone biotinylation in human Jurkat lymphoblastoma cells. J Nutr. 2009 Jan;139(1):163-6. doi: 10.3945/jn.108.091967[↩]
- Zempleni J, Wijeratne SSK, Hassan YI. Biotin. Biofactors 2009;35:36-46.[↩]
- Colombo VE, Gerber F, Bronhofer M, Floersheim GL. Treatment of brittle fingernails and onychoschizia with biotin: scanning electron microscopy. J Am Acad Dermatol. 1990 Dec;23(6 Pt 1):1127-32. doi: 10.1016/0190-9622(90)70345-i[↩][↩][↩]
- Floersheim GL. Behandlung brüchiger Fingernägel mit Biotin [Treatment of brittle fingernails with biotin]. Z Hautkr. 1989 Jan 15;64(1):41-8. German.[↩][↩][↩]
- Hochman LG, Scher RK, Meyerson MS. Brittle nails: response to daily biotin supplementation. Cutis. 1993 Apr;51(4):303-5.[↩][↩][↩]
- Boccaletti V, Zendri E, Giordano G, Gnetti L, De Panfilis G. Familial Uncombable Hair Syndrome: Ultrastructural Hair Study and Response to Biotin. Pediatr Dermatol. 2007 May-Jun;24(3):E14-6. doi: 10.1111/j.1525-1470.2007.00385.x[↩][↩][↩]
- Shelley WB, Shelley ED. Uncombable hair syndrome: observations on response to biotin and occurrence in siblings with ectodermal dysplasia. J Am Acad Dermatol. 1985 Jul;13(1):97-102. doi: 10.1016/s0190-9622(85)70150-8[↩][↩]
- Mock DM, Baswell DL, Baker H, Holman RT, Sweetman L. Biotin deficiency complicating parenteral alimentation: diagnosis, metabolic repercussions, and treatment. J Pediatr. 1985 May;106(5):762-9. doi: 10.1016/s0022-3476(85)80350-4[↩]
- Fujimoto W, Inaoki M, Fukui T, Inoue Y, Kuhara T. Biotin deficiency in an infant fed with amino acid formula. J Dermatol. 2005 Apr;32(4):256-61. doi: 10.1111/j.1346-8138.2005.tb00758.x[↩]
- Randhawa SS, Dua K, Randhawa CS, Randhawa SS, Munshi SK. Effect of biotin supplementation on hoof health and ceramide composition in dairy cattle. Vet Res Commun. 2008 Dec;32(8):599-608. doi: 10.1007/s11259-008-9060-z[↩][↩]
- Reilly JD, Cottrell DF, Martin RJ, Cuddeford DJ. Effect of supplementary dietary biotin on hoof growth and hoof growth rate in ponies: a controlled trial. Equine Vet J Suppl. 1998 Sep;(26):51-7. doi: 10.1111/j.2042-3306.1998.tb05122.x[↩]
- Zenker W, Josseck H, Geyer H. Histological and physical assessment of poor hoof horn quality in Lipizzaner horses and a therapeutic trial with biotin and a placebo. Equine Vet J. 1995 May;27(3):183-91. doi: 10.1111/j.2042-3306.1995.tb03061.x[↩]
- Romero-Navarro G, Cabrera-Valladares G, German MS, Matschinsky FM, Velazquez A, Wang J, Fernandez-Mejia C. Biotin regulation of pancreatic glucokinase and insulin in primary cultured rat islets and in biotin-deficient rats. Endocrinology. 1999 Oct;140(10):4595-600. doi: 10.1210/endo.140.10.7084[↩][↩]
- Lipner SR, Scher RK. Biotin for the treatment of nail disease: what is the evidence? J Dermatolog Treat. 2018 Jun;29(4):411-414. doi: 10.1080/09546634.2017.1395799[↩]
- Walth CB, Wessman LL, Wipf A, Carina A, Hordinsky MK, Farah RS. Response to: “Rethinking biotin therapy for hair, nail, and skin disorders”. J Am Acad Dermatol. 2018 Dec;79(6):e121-e124. doi: 10.1016/j.jaad.2018.07.055[↩]
- Famenini S, Goh C. Evidence for supplemental treatments in androgenetic alopecia. J Drugs Dermatol. 2014 Jul;13(7):809-12. https://jddonline.com/articles/evidence-for-supplemental-treatments-in-androgenetic-alopecia-S1545961614P0809X[↩]
- Patel DP, Swink SM, Castelo-Soccio L. A Review of the Use of Biotin for Hair Loss. Skin Appendage Disord. 2017 Aug;3(3):166-169. doi: 10.1159/000462981 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5582478[↩]
- Larrieta E, Vega-Monroy ML, Vital P, Aguilera A, German MS, Hafidi ME, Fernandez-Mejia C. Effects of biotin deficiency on pancreatic islet morphology, insulin sensitivity and glucose homeostasis. J Nutr Biochem. 2012 Apr;23(4):392-9. doi: 10.1016/j.jnutbio.2011.01.003[↩]
- Maebashi M, Makino Y, Furukawa Y, Ohinata K, Kimura S, Sato T. Therapeutic evaluation of the effect of biotin on hyperglycemia in pateints with non-insulin dependent diabetes mellitus. J Clin Biochem Nutr.1993;14:211-218.[↩][↩]
- Báez-Saldaña A, Zendejas-Ruiz I, Revilla-Monsalve C, Islas-Andrade S, Cárdenas A, Rojas-Ochoa A, Vilches A, Fernandez-Mejia C. Effects of biotin on pyruvate carboxylase, acetyl-CoA carboxylase, propionyl-CoA carboxylase, and markers for glucose and lipid homeostasis in type 2 diabetic patients and nondiabetic subjects. Am J Clin Nutr. 2004 Feb;79(2):238-43. doi: 10.1093/ajcn/79.2.238[↩]
- Revilla-Monsalve C, Zendejas-Ruiz I, Islas-Andrade S, Báez-Saldaña A, Palomino-Garibay MA, Hernández-Quiróz PM, Fernandez-Mejia C. Biotin supplementation reduces plasma triacylglycerol and VLDL in type 2 diabetic patients and in nondiabetic subjects with hypertriglyceridemia. Biomed Pharmacother. 2006 May;60(4):182-5. doi: 10.1016/j.biopha.2006.03.005[↩][↩]
- Geohas J, Daly A, Juturu V, Finch M, Komorowski JR. Chromium picolinate and biotin combination reduces atherogenic index of plasma in patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized clinical trial. Am J Med Sci. 2007 Mar;333(3):145-53. doi: 10.1097/MAJ.0b013e318031b3c9[↩][↩]
- Albarracin C, Fuqua B, Geohas J, Juturu V, Finch MR, Komorowski JR. Combination of chromium and biotin improves coronary risk factors in hypercholesterolemic type 2 diabetes mellitus: a placebo-controlled, double-blind randomized clinical trial. J Cardiometab Syndr. 2007 Spring;2(2):91-7. doi: 10.1111/j.1559-4564.2007.06366.x[↩][↩]
- Singer GM, Geohas J. The effect of chromium picolinate and biotin supplementation on glycemic control in poorly controlled patients with type 2 diabetes mellitus: a placebo-controlled, double-blinded, randomized trial. Diabetes Technol Ther. 2006 Dec;8(6):636-43. doi: 10.1089/dia.2006.8.636[↩]
- Albarracin CA, Fuqua BC, Evans JL, Goldfine ID. Chromium picolinate and biotin combination improves glucose metabolism in treated, uncontrolled overweight to obese patients with type 2 diabetes. Diabetes Metab Res Rev. 2008 Jan-Feb;24(1):41-51. doi: 10.1002/dmrr.755[↩]
- Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2016 Nov;110(Pt B):644-653. doi: 10.1016/j.neuropharm.2015.08.028[↩][↩]
- Sedel F, Papeix C, Bellanger A, Touitou V, Lebrun-Frenay C, Galanaud D, Gout O, Lyon-Caen O, Tourbah A. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015 Mar;4(2):159-69. doi: 10.1016/j.msard.2015.01.005[↩]
- Tourbah A, Lebrun-Frenay C, Edan G, Clanet M, Papeix C, Vukusic S, De Sèze J, Debouverie M, Gout O, Clavelou P, Defer G, Laplaud DA, Moreau T, Labauge P, Brochet B, Sedel F, Pelletier J; MS-SPI study group. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult Scler. 2016 Nov;22(13):1719-1731. doi: 10.1177/1352458516667568[↩]
- Cree BAC, Cutter G, Wolinsky JS, Freedman MS, Comi G, Giovannoni G, Hartung HP, Arnold D, Kuhle J, Block V, Munschauer FE, Sedel F, Lublin FD; SPI2 investigative teams. Safety and efficacy of MD1003 (high-dose biotin) in patients with progressive multiple sclerosis (SPI2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2020 Dec;19(12):988-997. doi: 10.1016/S1474-4422(20)30347-1[↩]
- Tourbah A, Gout O, Vighetto A, Deburghgraeve V, Pelletier J, Papeix C, Lebrun-Frenay C, Labauge P, Brassat D, Toosy A, Laplaud DA, Outteryck O, Moreau T, Debouverie M, Clavelou P, Heinzlef O, De Sèze J, Defer G, Sedel F, Arndt C. MD1003 (High-Dose Pharmaceutical-Grade Biotin) for the Treatment of Chronic Visual Loss Related to Optic Neuritis in Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Study. CNS Drugs. 2018 Jul;32(7):661-672. doi: 10.1007/s40263-018-0528-2[↩]
- Espiritu AI, Remalante-Rayco PPM. High-dose biotin for multiple sclerosis: A systematic review and meta-analyses of randomized controlled trials. Mult Scler Relat Disord. 2021 Oct;55:103159. doi: 10.1016/j.msard.2021.103159[↩][↩]
- Tabarki B, Al-Hashem A, Alfadhel M. Biotin-Thiamine-Responsive Basal Ganglia Disease. 2013 Nov 21 [Updated 2020 Aug 20]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK169615[↩][↩][↩]
- Kılıç B, Topçu Y, Dursun Ş, Erol İ, Dolu MH, Taşdemir HA, Aydın K. Single gene, two diseases, and multiple clinical presentations: Biotin-thiamine-responsive basal ganglia disease. Brain Dev. 2020 Sep;42(8):572-580. doi: 10.1016/j.braindev.2020.05.008[↩][↩]
- Alfadhel M, Almuntashri M, Jadah RH, Bashiri FA, Al Rifai MT, Al Shalaan H, Al Balwi M, Al Rumayan A, Eyaid W, Al-Twaijri W. Biotin-responsive basal ganglia disease should be renamed biotin-thiamine-responsive basal ganglia disease: a retrospective review of the clinical, radiological and molecular findings of 18 new cases. Orphanet J Rare Dis. 2013 Jun 6;8:83. doi: 10.1186/1750-1172-8-83[↩]
- Algahtani H, Ghamdi S, Shirah B, Alharbi B, Algahtani R, Bazaid A. Biotin-thiamine-responsive basal ganglia disease: catastrophic consequences of delay in diagnosis and treatment. Neurol Res. 2017 Feb;39(2):117-125. doi: 10.1080/01616412.2016.1263176[↩]
- Mock DM. Biotin. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed: Lippincott Williams & Wilkins; 2014:390-398.[↩][↩][↩]
- Mock DM, Stadler DD. Conflicting indicators of biotin status from a cross-sectional study of normal pregnancy. J Am Coll Nutr. 1997 Jun;16(3):252-7. doi: 10.1080/07315724.1997.10718682[↩][↩]
- Mock DM, Stadler DD, Stratton SL, Mock NI. Biotin status assessed longitudinally in pregnant women. J Nutr. 1997 May;127(5):710-6. doi: 10.1093/jn/127.5.710[↩]
- Mock DM, Quirk JG, Mock NI. Marginal biotin deficiency during normal pregnancy. Am J Clin Nutr. 2002 Feb;75(2):295-9. doi: 10.1093/ajcn/75.2.295[↩][↩]
- Mock DM. Marginal biotin deficiency is common in normal human pregnancy and is highly teratogenic in mice. J Nutr. 2009 Jan;139(1):154-7. doi: 10.3945/jn.108.095273[↩][↩][↩][↩][↩]
- Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr. 2008 Apr;138(4):680-4. doi: 10.1093/jn/138.4.680[↩]
- Zempleni J, Mock DM. Marginal biotin deficiency is teratogenic. Proc Soc Exp Biol Med. 2000 Jan;223(1):14-21. doi: 10.1046/j.1525-1373.2000.22303.x[↩][↩][↩][↩]
- Zempleni J, Wijeratne SS, Hassan YI. Biotin. Biofactors. 2009 Jan-Feb;35(1):36-46. doi: 10.1002/biof.8[↩][↩][↩][↩][↩][↩]
- Cowan MJ, Wara DW, Packman S, Ammann AJ, Yoshino M, Sweetman L, Nyhan W. Multiple biotin-dependent carboxylase deficiencies associated with defects in T-cell and B-cell immunity. Lancet. 1979 Jul 21;2(8134):115-8. doi: 10.1016/s0140-6736(79)90002-3[↩]
- Agrawal S, Agrawal A, Said HM. Biotin deficiency enhances the inflammatory response of human dendritic cells. Am J Physiol Cell Physiol. 2016 Sep 1;311(3):C386-91. doi: 10.1152/ajpcell.00141.2016[↩][↩][↩][↩]
- Boehncke WH, Schön MP. Psoriasis. Lancet. 2015 Sep 5;386(9997):983-94. doi: 10.1016/S0140-6736(14)61909-7[↩]
- Johnston A, Xing X, Wolterink L, Barnes DH, Yin Z, Reingold L, Kahlenberg JM, Harms PW, Gudjonsson JE. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017 Jul;140(1):109-120. doi: 10.1016/j.jaci.2016.08.056[↩]
- Kumar M, Axelrod AE. Cellular antibody synthesis in thiamin, riboflavin, biotin and folic acid-deficient rats. Proc Soc Exp Biol Med. 1978 Mar;157(3):421-3. doi: 10.3181/00379727-157-40068[↩]
- Báez-Saldaña A, Díaz G, Espinoza B, Ortega E. Biotin deficiency induces changes in subpopulations of spleen lymphocytes in mice. Am J Clin Nutr. 1998 Mar;67(3):431-7. doi: 10.1093/ajcn/67.3.431[↩]
- Báez-Saldaña A, Ortega E. Biotin deficiency blocks thymocyte maturation, accelerates thymus involution, and decreases nose-rump length in mice. J Nutr. 2004 Aug;134(8):1970-7. doi: 10.1093/jn/134.8.1970[↩]
- Manthey KC, Griffin JB, Zempleni J. Biotin supply affects expression of biotin transporters, biotinylation of carboxylases and metabolism of interleukin-2 in Jurkat cells. J Nutr. 2002 May;132(5):887-92. doi: 10.1093/jn/132.5.887. Erratum in: J Nutr 2002 Aug;132(8):2326[↩]
- Crisp SE, Griffin JB, White BR, Toombs CF, Camporeale G, Said HM, Zempleni J. Biotin supply affects rates of cell proliferation, biotinylation of carboxylases and histones, and expression of the gene encoding the sodium-dependent multivitamin transporter in JAr choriocarcinoma cells. Eur J Nutr. 2004 Feb;43(1):23-31. doi: 10.1007/s00394-004-0435-9[↩]
- Dakshinamurti K, Chalifour L, Bhullar RP. Requirement for biotin and the function of biotin in cells in culture. Ann N Y Acad Sci. 1985;447:38-55. doi: 10.1111/j.1749-6632.1985.tb18424.x[↩]
- Rodriguez-Melendez R, Schwab LD, Zempleni J. Jurkat cells respond to biotin deficiency with increased nuclear translocation of NF-kappaB, mediating cell survival. Int J Vitam Nutr Res. 2004 May;74(3):209-16. doi: 10.1024/0300-9831.74.3.209[↩][↩]
- Smith EM, Hoi JT, Eissenberg JC, Shoemaker JD, Neckameyer WS, Ilvarsonn AM, Harshman LG, Schlegel VL, Zempleni J. Feeding Drosophila a biotin-deficient diet for multiple generations increases stress resistance and lifespan and alters gene expression and histone biotinylation patterns. J Nutr. 2007 Sep;137(9):2006-12. doi: 10.1093/jn/137.9.2006[↩]
- Institute of Medicine. Food and Nutrition Board. Dietary Reference Intakes: Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press; 1998. https://www.ncbi.nlm.nih.gov/books/NBK114310/pdf/Bookshelf_NBK114310.pdf[↩][↩][↩]
- Institute of Medicine. Dietary reference intakes: the essential guide to nutrient requirements. Washington, DC: National Academies Press; 2006. https://www.nap.edu/catalog/11537/dietary-reference-intakes-the-essential-guide-to-nutrient-requirements[↩]
- Zempleni J, Mock DM. Bioavailability of biotin given orally to humans in pharmacologic doses. Am J Clin Nutr. 1999 Mar;69(3):504-8. doi: 10.1093/ajcn/69.3.504[↩]
- Biotin. https://ods.od.nih.gov/factsheets/Biotin-Consumer[↩]
- Combs GF, Jr. Biotin. In: Combs GF, Jr., ed. The vitamins: fundamental aspects in nutrition and health. Third ed. Burlington, MA: Elsevier Academic Press; 2008:331-44.[↩]
- Mock DM. Biotin. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:43-51.[↩][↩][↩]
- Food Labeling: Revision of the Nutrition and Supplement Facts Labels. https://www.federalregister.gov/documents/2016/05/27/2016-11867/food-labeling-revision-of-the-nutrition-and-supplement-facts-labels[↩]
- Staggs CG, Sealey WM, McCabe BJ, Teague AM, Mock DM. Determination of the biotin content of select foods using accurate and sensitive HPLC/avidin binding. J Food Compost Anal. 2004 Dec;17(6):767-776. doi: 10.1016/j.jfca.2003.09.015[↩]
- Perry CA, West AA, Gayle A, Lucas LK, Yan J, Jiang X, et al. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr 2014;144:1977-84.[↩]
- Trüeb RM. Serum Biotin Levels in Women Complaining of Hair Loss. Int J Trichology. 2016 Apr-Jun;8(2):73-7. doi: 10.4103/0974-7753.188040[↩][↩][↩][↩][↩][↩][↩][↩]
- Seymons K, De Moor A, De Raeve H, Lambert J. Dermatologic signs of biotin deficiency leading to the diagnosis of multiple carboxylase deficiency. Pediatr Dermatol. 2004 May-Jun;21(3):231-5. doi: 10.1111/j.0736-8046.2004.21308.x[↩]
- Fujimoto, W., Inaoki, M., Fukui, T., Inoue, Y. and Kuhara, T. (2005), Biotin Deficiency in an Infant Fed with Amino Acid Formula. The Journal of Dermatology, 32: 256-261. https://doi.org/10.1111/j.1346-8138.2005.tb00758.x[↩]
- Biotinidase deficiency. https://medlineplus.gov/genetics/condition/biotinidase-deficiency[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Biotinidase Deficiency. https://rarediseases.org/rare-diseases/biotinidase-deficiency[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Rajendiran A, Sampath S. Biotinidase deficiency–clinching the diagnosis rapidly can make all the difference! BMJ Case Rep. 2011 Sep 28;2011:bcr0720114494. doi: 10.1136/bcr.07.2011.4494[↩]
- Biotin responsive dermatoses. https://dermnetnz.org/topics/biotin-responsive-dermatoses[↩]
- Watabe D, Watanabe A, Akasaka T, Sakamoto O, Amano H. Psoriasis-like Dermatitis in Adulthood: A Skin Manifestation of Holocarboxylase Synthetase Deficiency. Acta Derm Venereol. 2018 Aug 29;98(8):805-806. doi: 10.2340/00015555-2954[↩][↩]
- Ogawa Y, Kinoshita M, Sato T, Shimada S, Kawamura T. Biotin Is Required for the Zinc Homeostasis in the Skin. Nutrients. 2019 Apr 24;11(4):919. doi: 10.3390/nu11040919[↩][↩]
- Hsu RH, Chien YH, Hwu WL, Chang IF, Ho HC, Chou SP, Huang TM, Lee NC. Genotypic and phenotypic correlations of biotinidase deficiency in the Chinese population. Orphanet J Rare Dis. 2019 Jan 7;14(1):6. doi: 10.1186/s13023-018-0992-2[↩][↩][↩][↩]
- Strovel ET, Cowan TM, Scott AI, Wolf B. ERRATUM: Laboratory diagnosis of biotinidase deficiency, 2017 update: a technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med. 2018 Feb;20(2):282. https://doi.org/10.1038/gim.2017.201 Erratum Genet Med online publication 6 July 2017; doi:https://doi.org/10.1038/gim.2017.84[↩][↩][↩][↩][↩]
- Saleem F, Soos MP. Biotin Deficiency. [Updated 2023 Feb 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547751[↩][↩][↩][↩][↩][↩]
- Innis SM, Allardyce DB. Possible biotin deficiency in adults receiving long-term total parenteral nutrition. Am J Clin Nutr. 1983 Feb;37(2):185-7. doi: 10.1093/ajcn/37.2.185[↩]
- Khalidi N, Wesley JR, Thoene JG, Whitehouse WM Jr, Baker WL. Biotin deficiency in a patient with short bowel syndrome during home parenteral nutrition. JPEN J Parenter Enteral Nutr. 1984 May-Jun;8(3):311-4. doi: 10.1177/0148607184008003311[↩]
- Krause KH, Bonjour JP, Berlit P, Kochen W. Biotin status of epileptics. Ann N Y Acad Sci. 1985;447:297-313. doi: 10.1111/j.1749-6632.1985.tb18447.x[↩]
- Said HM. Biotin: biochemical, physiological and clinical aspects. Subcell Biochem. 2012;56:1-19. doi: 10.1007/978-94-007-2199-9_1[↩]
- Hayashi A, Mikami Y, Miyamoto K, Kamada N, Sato T, Mizuno S, Naganuma M, Teratani T, Aoki R, Fukuda S, Suda W, Hattori M, Amagai M, Ohyama M, Kanai T. Intestinal Dysbiosis and Biotin Deprivation Induce Alopecia through Overgrowth of Lactobacillus murinus in Mice. Cell Rep. 2017 Aug 15;20(7):1513-1524. doi: 10.1016/j.celrep.2017.07.057[↩]
- Sealey WM, Teague AM, Stratton SL, Mock DM. Smoking accelerates biotin catabolism in women. Am J Clin Nutr. 2004 Oct;80(4):932-5. doi: 10.1093/ajcn/80.4.932[↩][↩]
- Srinivasan P, Kapadia R, Biswas A, Said HM. Chronic alcohol exposure inhibits biotin uptake by pancreatic acinar cells: possible involvement of epigenetic mechanisms. Am J Physiol Gastrointest Liver Physiol. 2014 Nov 1;307(9):G941-9. doi: 10.1152/ajpgi.00278.2014[↩][↩]
- Subramanya SB, Subramanian VS, Kumar JS, Hoiness R, Said HM. Inhibition of intestinal biotin absorption by chronic alcohol feeding: cellular and molecular mechanisms. Am J Physiol Gastrointest Liver Physiol. 2011 Mar;300(3):G494-501. doi: 10.1152/ajpgi.00465.2010[↩][↩]
- Mock DM. Adequate intake of biotin in pregnancy: why bother? J Nutr. 2014 Dec;144(12):1885-6. doi: 10.3945/jn.114.203356[↩]
- Mock DM. Biotin: From Nutrition to Therapeutics. J Nutr. 2017 Aug;147(8):1487-1492. doi: 10.3945/jn.116.238956[↩]
- Czeizel AE, Dudás I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992 Dec 24;327(26):1832-5. doi: 10.1056/NEJM199212243272602[↩]
- Mock DM, Mock NI, Stewart CW, LaBorde JB, Hansen DK. Marginal biotin deficiency is teratogenic in ICR mice. J Nutr. 2003 Aug;133(8):2519-25. doi: 10.1093/jn/133.8.2519[↩]
- Hayashi H, Tokuriki S, Okuno T, Shigematsu Y, Yasushi A, Matsuyama G, Sawada K, Ohshima Y. Biotin and carnitine deficiency due to hypoallergenic formula nutrition in infants with milk allergy. Pediatr Int. 2014 Apr;56(2):286-8. doi: 10.1111/ped.12319[↩]
- Küry S, Ramaekers V, Bézieau S, Wolf B. Clinical utility gene card for: Biotinidase deficiency-update 2015. Eur J Hum Genet. 2016 Jul;24(7). doi: 10.1038/ejhg.2015.246[↩]
- Zempleni J, Hassan YI, Wijeratne SS. Biotin and biotinidase deficiency. Expert Rev Endocrinol Metab. 2008 Nov 1;3(6):715-724. doi: 10.1586/17446651.3.6.715[↩][↩][↩][↩][↩]
- Canda E, Kalkan Uçar S, Çoker M. Biotinidase Deficiency: Prevalence, Impact And Management Strategies. Pediatric Health Med Ther. 2020 May 4;11:127-133. doi: 10.2147/PHMT.S198656[↩][↩][↩][↩]
- Pindolia K, Chen J, Cardwell C, Cui X, Chopp M, Wolf B. Neurological deficits in mice with profound biotinidase deficiency are associated with demylination and axonal degeneration. Neurobiol Dis. 2012 Sep;47(3):428-35. doi: 10.1016/j.nbd.2012.04.016[↩]
- Wolf B. Biotinidase Deficiency. 2000 Mar 24 [Updated 2023 May 25]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1322[↩]
- Yang Y, Yang JY, Chen XJ. Biotinidase deficiency characterized by skin and hair findings. Clin Dermatol. 2020 Jul-Aug;38(4):477-483. doi: 10.1016/j.clindermatol.2020.03.004[↩]
- Mohite K, Nair KV, Sapare A, Bhat V, Shukla A, Kekatpure M, Patil SJ. Late Onset Subacute Profound Biotinidase Deficiency Caused by a Novel Homozygous Variant c.466-3T>G in the BTD Gene. Indian J Pediatr. 2022 Jun;89(6):594-596. doi: 10.1007/s12098-021-04000-3[↩]
- Radelfahr F, Riedhammer KM, Keidel LF, Gramer G, Meitinger T, Klopstock T, Wagner M. Biotinidase deficiency: A treatable cause of hereditary spastic paraparesis. Neurol Genet. 2020 Oct 13;6(6):e525. doi: 10.1212/NXG.0000000000000525[↩]
- Wolf B, Secor McVoy J. A sensitive radioassay for biotinidase activity: deficient activity in tissues of serum biotinidase-deficient individuals. Clin Chim Acta. 1983;135(3):275–281. doi: 10.1016/0009-8981(83)90286-3[↩]
- Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131(3):273–281. doi: 10.1016/0009-8981(83)90096-7[↩]
- Wolf B, Grier RE, Secor McVoy JR, Heard GS. Biotinidase deficiency: a novel vitamin recycling defect. J Inherit Metab Dis. 1985;8(Suppl 1):53–58. doi: 10.1007/BF01800660[↩]
- Gannavarapu S, Prasad C, DiRaimo J, et al. Biotinidase deficiency: spectrum of molecular, enzymatic and clinical information from newborn screening Ontario, Canada (2007–2014). Mol Genet Metab. 2015;116(3):146–151. doi: 10.1016/j.ymgme.2015.08.010[↩]
- Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med. 2012;14(6):565–575. doi: 10.1038/gim.2011.6[↩]
- Hymes J, Stanley CM, Wolf B. Mutations in BTD causing biotinidase deficiency. Hum Mutat. 2001;18(5):375–381. doi: 10.1002/(ISSN)1098-1004[↩]
- Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100(1):6–13. doi: 10.1016/j.ymgme.2010.01.003[↩]
- Schubiger G, Caflisch U, Baumgartner R, Suormala T, Bachmann C. Biotinidase deficiency: clinical course and biochemical findings. J Inherit Metab Dis. 1984;7(3):129-30. doi: 10.1007/BF01801771[↩]
- Horvath TD, Matthews NI, Stratton SL, Mock DM, Boysen G. Measurement of 3-hydroxyisovaleric acid in urine from marginally biotin-deficient humans by UPLC-MS/MS. Anal Bioanal Chem. 2011;401(9):2805–2810. doi: 10.1007/s00216-011-5356-x[↩][↩]
- Wolf B. Biotinidase deficiency and our champagne legacy. Gene. 2016 Sep 10;589(2):142-50. doi: 10.1016/j.gene.2015.10.010[↩][↩]
- Saleem H, Simpson B. Biotinidase Deficiency. [Updated 2023 Feb 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560607[↩][↩]
- Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med. 2012 Jun;14(6):565-75. doi: 10.1038/gim.2011.6[↩][↩]
- Armstrong RW, Steinbok P, Cochrane DD, Kube SD, Fife SE, Farrell K. Intrathecally administered baclofen for treatment of children with spasticity of cerebral origin. J Neurosurg. 1997 Sep;87(3):409-14. doi: 10.3171/jns.1997.87.3.0409[↩]
- Szymańska E, Średzińska M, Ługowska A, Pajdowska M, Rokicki D, Tylki-Szymańska A. Outcomes of oral biotin treatment in patients with biotinidase deficiency – Twenty years follow-up. Mol Genet Metab Rep. 2015 Oct 6;5:33-35. doi: 10.1016/j.ymgmr.2015.09.004[↩]
- Combs GF, Jr. Biotin. In: Combs GF, Jr., ed. The vitamins: fundamental aspects in nutrition and health. Third ed. Burlington, MA: Elsevier Academic Press; 2008:331-44[↩]
- Stratton SL, Bogusiewicz A, Mock MM, Mock NI, Wells AM, Mock DM. Lymphocyte propionyl-CoA carboxylase and its activation by biotin are sensitive indicators of marginal biotin deficiency in humans. Am J Clin Nutr. 2006 Aug;84(2):384-8. doi: 10.1093/ajcn/84.1.384[↩]
- Perry CA, West AA, Gayle A, Lucas LK, Yan J, Jiang X, et al. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr 2014;144:1977-84[↩]
- Holocarboxylase synthetase deficiency. https://medlineplus.gov/genetics/condition/holocarboxylase-synthetase-deficiency[↩][↩][↩]
- Kalter, H. (1972), The metabolic basis of inherited disease. 3rd Ed. J. B. Stanbury, J. B. Wyngaarden, and D. S. Fredrickson, eds. McGraw-Hill, New York. 1778 pp. 1972. Teratology, 6: 362-362. https://doi.org/10.1002/tera.1420060320[↩]
- Mock, Donald M.. Evidence for a Pathogenic Role of ω6 Polyunsaturated Fatty Acid in the Cutaneous Manifestations of Biotin Deficiency. Journal of Pediatric Gastroenterology and Nutrition 10(2):p 222-229, February 1990. doi: 10.1097/00005176-199002000-00013[↩]
- Nakajima K, Terao M, Takaishi M, Kataoka S, Goto-Inoue N, Setou M, Horie K, Sakamoto F, Ito M, Azukizawa H, Kitaba S, Murota H, Itami S, Katayama I, Takeda J, Sano S. Barrier abnormality due to ceramide deficiency leads to psoriasiform inflammation in a mouse model. J Invest Dermatol. 2013 Nov;133(11):2555-2565. doi: 10.1038/jid.2013.199[↩]
- Bandaralage SP, Farnaghi S, Dulhunty JM, Kothari A. Antenatal and postnatal radiologic diagnosis of holocarboxylase synthetase deficiency: a systematic review. Pediatr Radiol. 2016 Mar;46(3):357-64. doi: 10.1007/s00247-015-3492-8[↩][↩]
- Mardach R, Zempleni J, Wolf B, Cannon MJ, Jennings ML, Cress S, Boylan J, Roth S, Cederbaum S, Mock DM. Biotin dependency due to a defect in biotin transport. J Clin Invest. 2002 Jun;109(12):1617-23. doi: 10.1172/JCI13138[↩]
- Perry CA, West AA, Gayle A, Lucas LK, Yan J, Jiang X, Malysheva O, Caudill MA. Pregnancy and lactation alter biomarkers of biotin metabolism in women consuming a controlled diet. J Nutr. 2014 Dec;144(12):1977-84. doi: 10.3945/jn.114.194472[↩]
- Pabuçcuoğlu A, Aydoğdu S, Baş M. Serum biotinidase activity in children with chronic liver disease and its clinical significance. J Pediatr Gastroenterol Nutr. 2002 Jan;34(1):59-62. doi: 10.1097/00005176-200201000-00014[↩]
- Mock DM. Biotin status: which are valid indicators and how do we know? J Nutr. 1999 Feb;129(2S Suppl):498S-503S[↩]
- Schulpis KH, Karikas GA, Tjamouranis J, Regoutas S, Tsakiris S. Low serum biotinidase activity in children with valproic acid monotherapy. Epilepsia. 2001 Oct;42(10):1359-62. doi: 10.1046/j.1528-1157.2001.47000.x[↩]
- Baumgartner ER, Suormala T. Inherited defects of biotin metabolism. Biofactors. 1999;10(2-3):287-90. doi: 10.1002/biof.5520100229[↩]
- Misir R, Blair R, Doige CE. Development of a system for clinical evaluation of the biotin status of sows. Can Vet J. 1986 Jan;27(1):6-12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1680218/pdf/canvetj00589-0012.pdf[↩]
- BROWN A. The effect of egg white and crystalline biotin methyl ester on a skin lesion in three infants. Glasgow Med J. 1948 Sep;29(9):309-16. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5966187/pdf/glasgowmedj76301-0017.pdf[↩]
- Bunch M, Singh A. Peculiar neuroimaging and electrophysiological findings in a patient with biotinidase deficiency. Seizure. 2011 Jan;20(1):83-6. doi: 10.1016/j.seizure.2010.10.001[↩]
- Desai S, Ganesan K, Hegde A. Biotinidase deficiency: a reversible metabolic encephalopathy. Neuroimaging and MR spectroscopic findings in a series of four patients. Pediatr Radiol. 2008 Aug;38(8):848-56. doi: 10.1007/s00247-008-0904-z[↩][↩]





{kind=link}