Contents
- What is chelation therapy
- Chelation therapy benefits
- Calcium Disodium Ethylenediamine Tetraacetic Acid (CaNa2EDTA)
- Calcium Trisodium DTPA
- D-Penicillamine
- British Anti Lewisite (BAL)
- DMSA (Meso-2,3-Dimercaptosuccinic Acid)
- DMPS (Sodium 2,3 Dimercaptopropane-l-Sulphonate)
- Deferoxamine (DFO)
- Deferiprone (L1)
- TETA (Tetraethylenetetraamine)
- Nitrilotriacetic Acid (NTA)
- Chelation therapy autism
- Chelation therapy for heart disease
- Iron chelation therapy
- Chelation therapy for lead
- Chelation therapy side effects
- Chelation therapy benefits
What is chelation therapy
Chelation therapy is a treatment that involves administering a drug called a chelator, which has a magnetically charged pocket that can “grab” a metal and hang on to it, allowing it to be excreted in the urine- kind of like a baseball mitt with a magnet in its pocket 1. Chelating agents are capable of binding to toxic metal ions to form complex structures which are easily excreted from the body removing them from intracellular or extracellular spaces 2. One chelator, calcium EDTA (ethylenediaminetetraacetic acid), is FDA approved to treat lead poisoning.
Chelating agents are organic or inorganic compounds capable of binding metal ions to form complex ring-like structure called ‘chelates’ 2. Chelating agents possess “ligand” binding atoms that form either two covalent linkages or one covalent and one co-ordinate or two co-ordinate linkages in the case of bidentate chelates. Mainly atoms like S, N and O function as ligand atoms in the form of chemical groups like –SH, –S-S, –NH2, =NH, –OH, –OPO3H, or >C=O. Bidenate or multidentate ligands form ring structures that include the metal ion and the two-ligand atoms attached to the metal 3. Many donors act as bidentate ligands. Five-membered chelate rings are specially stable and they are often formed by ligands with YCCY skeltons such as Y-CH2-CH2-Y, Y-CO-CH2-Y etc. where Y is OR, NR2, O, S, NR, etc. There are also examples of inorganic chelate ligands which form five-membered ring with metal ions. Other types of chelating ligands are possible, like EDTA4−, which is a hexadentate ligand. In the simplest case a proton (H+) that can absorb the lone pair of electrons of ligand-binding atom(s) of the chelator may be involved in the coordination complex formation. However, the positive charge on proton remains since there is no loss or gain of electrons in the process.
The latter may also be known as the ‘net ionic charge’ of the complex, which plays a crucial role in governing the pharmacokinetic fate and ultimately the toxicological behavior of such complexes in vivo. In the biological environment metal cations viz. Na+, Mg+, Cu+, Cu2+, and Zn2+ and specially the transition metals like Mn, Fe and Co may be involved in such complex formation 2. Although the stability of such complexes varies, the deciding factors are based on the properties of both the chelating agent and the chelated metal. The stability constant of a complex can be quantitatively expressed in equilibrium equation values, which depend on the atomic structure of the chelated metals. For example, the stability constants for different metals with EDTA are on the scale shown in Table 1, where a metal with higher k constant competes for the chelating agent with a metal of lower stability value and ultimately removes the latter 2.
Table 1. EDTA-metal complex stability constants
| Metal | Na | Li | Ba | Sr | Mg | Ca | Mn | Fe | Co | Zn | Cd | Pb | Ni |
| K (log) | 1.7 | 2.8 | 7.8 | 8.6 | 8.7 | 10.6 | 13.4 | 14.4 | 16.1 | 16.1 | 16.4 | 18.3 | 18.4 |
An ideal chelator should have high solubility in water, resistance to biotransformation, ability to reach the sites of metal storage, retain chelating ability at the pH of body fluids and the property of forming metal complexes that are less toxic than the free metal ion.
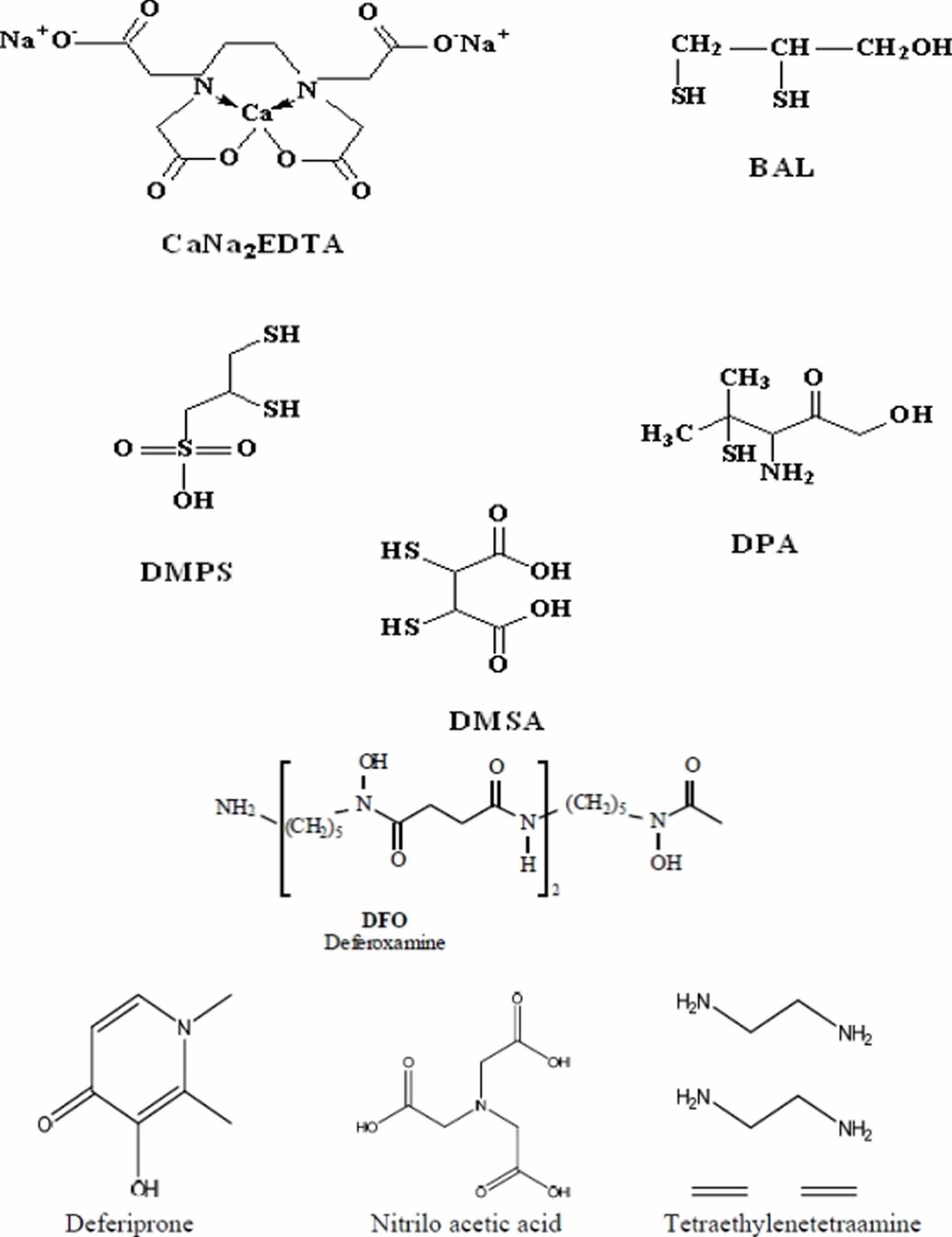
During Second World War, dimercaprol (also named British Anti-Lewisite or BAL), an organic dithiol compound, was developed as an experimental antidote against the arsenic-based poison gas Lewisite. After World War II, mass lead poisoning was observed in large number of navy personnel, later identified as a result of their jobs repainting the hulls of ships. This introduced the medical use of EDTA as a lead chelating agent. BAL (British Anti-Lewisite) has dominated medical prescriptions for general metal intoxication due to its high efficacy for human arsenic and mercury poisoning. In the 1960s, BAL (British Anti-Lewisite) was modified into meso 2,3-dimercaptosuccinic acid (DMSA), a related dithiol with far fewer side effects. Another dithiol, sodium 2,3-dimercaptopropane 1-sulfonate (DMPS), was introduced as a mercury-chelating agent by researchers in the former Soviet Union. Chelation therapy has historically been used in attempts to reduce the body burden of toxic metals in highly symptomatic patients with elevated biological markers 4. Chelating agents can affect metal toxicity by mobilizing the toxic metal mainly into urine. A chelating agent forming a stable complex with a toxic metal may shield biological targets from the metal ion, thereby reducing the local toxicity 5. Desferrioxamine (DFOA), an iron chelator, completely covers the surface of Fe3+ during complex formation, thereby preventing iron-catalyzed free radical reactions 6. However, sometimes a chelator may expose the metal to the biological environment and thus increase the toxicity of the metal. Ethylenediamine-tetraacetic acid (EDTA) is not able to shield the surface of the Fe3+ ion, but forms an open complex (basket complex), thereby increasing the catalytic capacity of Fe3+ for generating oxidative stress 7. Structures of various chelating agents are presented in Figure 1.
Figure 1. Chelation therapy agents (structures of various chelating agents used to treat cases of heavy metal poisoning)
Chelation therapy benefits
Calcium Disodium Ethylenediamine Tetraacetic Acid (CaNa2EDTA)
Calcium disodium ethylenediamine tetraacetic acid (CaNa2EDTA) is the most commonly used chelating agent. It is a derivative of ethylenediamine tetraacetic acid (EDTA); a synthetic polyamino-polycarboxylic acid and since 1950s has been one of the mainstays for the treatment of childhood lead poisoning 8. The drug has been claimed beneficial in vascular disease since 1955. It is believed that chelation therapy may alter plaque morphology and volume, or improve endothelial function, and suggest that it might replace coronary artery bypass draft surgery 9. Several theories have been proposed to support latter mainly focusing on calcium chelation. EDTA is said to work in vascular conditions either by removing calcium found in fatty plaques either directly via chelation effect or alternately by stimulating release of hormones that in turn cause calcium removal or lower cholesterol levels. Another theory suggests that EDTA therapy may reduce the oxidative stress injury and inflammation in blood vessel walls 10.
Although a number of trials conducted indicate the utility of CaNa2EDTA in coronary heart disease, the evidence in the absence of the controlled trial is not as convincing. In view of reports where symptomatic improvements have been comparable with placebo effects and the risks associated with the therapy, it has been facing criticism 11. The American Heart Association has stated that “there have been no adequate, controlled, published scientific studies using currently approved scientific methodology to support this therapy for coronary heart disease. Using this form of unproven treatment for coronary heart disease may deprive patients of the well-established benefits from the many other valuable methods of treating these diseases” 12 for the program to assess alternative treatment strategies to achieve cardiac health (PATCH) investigators also concluded after a randomized control trial with 84 patient that there is no evidence to support a beneficial effect of calcium chelation therapy with EDTA in patients with ischemic heart disease, stable angina and a positive treadmill test for ischemia. Another sub-study under PATCH rejects the proposed benefits of EDTA in combination with vitamins to improve impaired endothelium-dependent brachial artery flow-mediated vasodilation in patients with coronary artery disease 13. But recently all randomized clinical trials have been underpowered and the NCCAM and the National Heart, Lung, and Blood Institute launched the Trial to Assess Chelation Therapy (TACT). TACT is the first largest, multicenter study to assess safety and efficacy of EDTA chelation therapy for patients with coronary artery disease. This placebo-controlled, double-blind trial has started recruiting participants, aiming at 2,372 sample size, aged ≥ 50 years with prior myocardial infarction to test whether EDTA chelation therapy, high dose vitamin therapy, or both are effective in secondary prevention. The study started in 2003 and was expected to complete in 5 years 14. However, trials are ongoing and conclusive results are not expected before June 2012.
CaNa2EDTA can be valuable for the treatment of poisoning by metals that have higher affinity for chelating agent than does Ca2+. The successful use of CaNa2EDTA in the treatment of lead poisoning is due, in part, to the capacity of lead to displace calcium from the chelate. Initially EDTA was introduced as its sodium salt (NaEDTA) which when administered in vivo resulted in the urinary excretion of calcium leading to hypocalcemia with the risk of tetany due to the formation of its calcium complex. To overcome this hazard, CaNa2EDTA was introduced for the treatment of lead poisoning. The Pb-EDTA complex has high stability constant thus CaNa2EDTA was found to chelate lead from the body fluids, excreting PbNa2EDTA leaving Ca behind.
Pharmacological profile
CaNa2EDTA is poorly absorbed in the gastrointestinal tract (<5%) thus can only be administered by parenteral route. Intravenous administration of this drug results in good absorption but is very painful at the injection site. Hence intravenous injection could be given either by diluting in 5% dextrose or saline (iv infusion) 8. CaNa2EDTA is distributed mainly in the extracellular fluids, which limits its capacity to chelate out metals from inside the cells. The latter also contributes to one of its major drawbacks that of redistributing lead from other tissues to the brain. Reporting the same, Flora et al. 15 recommended avoiding the use of CaNa2EDTA for lead mobilization tests in children. It was suggested that CaNa2EDTA when administered in animals chronically exposed to lead, mobilizes the metal deposited in hard tissue for chelation. This then allows mobilized circulating lead to redistribute in soft tissues like the brain and liver in order to achieve equilibrium. CaNa2EDTA is not significantly metabolized and is excreted rapidly by glomerular filtration, entirely unchanged in urine, 50% of which appears within one hour 16. The drug shows an elimination half life of 1.4 to 3 hours in adult and is entirely excreted within 24 hours.
The risks associated with CaNa2EDTA therapy are substantial, including renal failures, arrhythmias, tetany, hypocalcaemia, hypotension, bone marrow depression, prolonged bleeding time, convulsions, respiratory arrest, etc. 12. Although the nephrotoxicity by CaNa2EDTA is dose dependent and reversible after cessation of therapy yet exceeding maximal daily dose of 75 mg/kg could be fatal. However, there also are reports that highlight the efficacy of EDTA in chronic renal artery diseases 17. Other adverse effects may include fatigue, headache, fever, nasal congestion, lacrimation, mucocutaneous lesions, glycosuria, myalgia, hepatotoxicity, increased urinary frequency, hypotension, abnormal changes in ECG and gastrointestinal symptoms 18. Prolonged treatment with CaNa2EDTA results in depletion of essential metal, especially Zn, Cu and Mn 19. It has been reported that zinc supplementation during and after chelation is also beneficial 20. Although zinc depletion by CaNa2EDTA therapy is rapidly reversible by zinc supplementation, it is considered to be key mechanism for teratogenic effects of the drug, especially when administered between days 11 to 14 at doses comparable to humans. This allows the use of EDTA as a research tool to study mechanisms and role of zinc dependent molecules at critical periods of pregnancy. CaNa2EDTA has the LD50 value of 16.4 mmol/kg in mouse 8. The drug is contraindicated in pregnancy, active renal diseases or anuria, hepatitis, and hypersensitivity to edentate products 18.
Calcium Trisodium DTPA
Calcium or zinc trisodium diethylenetriaminepentaacetate (CaNa3DTPA or ZnNa3DTPA respectively) have been used against plutonium and other transuranic elements like californium, americium, and curium 21. The drug is effective against cobalt and zinc poisoning in experimental models 22. Its efficacy against acute cadmium poisoning is promising however is less effective as compare to carbodithioates 23.
Pharmacological profile
Like CaNa2EDTA, CaNa3DTPA/ZnNa3DTPA is poorly absorbed in the gastrointestinal tract, and thus they are parenterally administered and are distributed extracellularly. They may be administered by intravenous or intramuscular routes or by inhalation through a nebulizer 24, where the former are painful. CaNa3DTPA shows an expected disadvantage of depleting Zn from the system that may be overcome by supplementation or using the zinc salt of the drug. Despite this disadvantage, although CaNa3DTPA is still preferred over the Zn form due to its higher efficacy, it is not recommended for prolonged therapy. Although CaNa3DTPA is well tolerated, some adverse effects may be observed, including nausea, vomiting, diarrhea, chills, fever and muscle cramps during the first 24 hrs 24. CaNa3DTPA is teratogenic like CaNa2EDTA due to its Zn and Mn depletion effect that is confirmed by the teratogenic effects of ZnNa3DTPA at a dose 16 times lower than CaNa3DTPA. Thus, although the drug is contraindicated during pregnancy ZnNa3DTPA may be carefully administered if unavoidable. Other contraindications include prescription to children, in patients with renal insufficiency and bone marrow depression.
D-Penicillamine
d-Penicillamine (DPA; β-β-dimethylcysteine or 3-mercapto-d-valine) is a sulfhydryl containing amino acid and a degradation product of penicillin. Only the d-isomer is used because the l-isomer causes optic neuritis. d-Penicillamine is used mainly as a chelating agent in heavy metal toxicity viz. lead, mercury and copper poisoning (Wilson’s disease) 25.
Pharmacological profile
d-Penicillamine is well absorbed via the gastrointestinal tract and can be administered orally or by iv route. It is approximately 50% absorbed orally and mainly follows extracellular distribution. Peak plasma concentration is reached between 1 and 4 hours after oral administration. A minimal drug fraction under-goes hepatic metabolism to disulfides, and most of the drug is excreted unchanged in urine. Elimination half life ranges from 1 to more than 7 hours. Although low adverse reactions have been reported, some serious ones including thrombocytopenia and leukocytopenia (incidence 5–15%), and rarely aplastic anemia may occur. Prolonged d-Penicillamine treatment may lead to anorexia, nausea and vomiting. The ulcerogenic activity of d-Penicillamine in rats, which may involve the stimulation of histaminergic (H1 and H2) receptors, has been reported 26. Other toxic effects of d-Penicillamine include gastrointestinal disturbances (10–30%), changes/loss of taste (5–30%), hair loss (1–2%), and partly proteinuria (5–20%) 27. Severe adverse effects are autoimmune phenomena such as pemphigus, d-Penicillamine-induced lupus erythematosus, polymyositis/dermatomyositis, membranous glomerulopathy and hypersensitivity pneumonitis 27. d-Penicillamine is a well recognized teratogen and lathyrogen that causes skeletal, cutaneous and pulmonary abnormalities 26. The most important contraindication is in patients allergic to penicillin and in cases of renal insufficiency. Co-administration of d-Penicillamine to patients receiving gold therapy, antimalarial or cytotoxic drugs, phenyl-butazone, or oxyphenbutazone must be contraindicated since it can result in more serious adverse reactions.
British Anti Lewisite (BAL)
2,3-Dimercaprol (BAL) is a traditional chelating agent developed by British biochemists at Oxford University during World War II 28. BAL has a 3-carbon backbone with two sulfhydryl (–SH) groups and a hydroxyl group. It has been used clinically since 1949 in arsenic, cadmium and mercury poisoning. It is an oily, clear, colorless liquid with a pungent, unpleasant typical mercaptan smell. It detoxifies lewisite by forming a five membered stable complex with arsenic. In humans and experimental models, the antidotal efficacy of BAL has been shown to be most effective when administered immediately after the exposure. Besides rapid mobilization of arsenic and mercury from the body, it causes a significant increase in brain deposition of arsenic and mercury compounds 29.
Pharmacological profile
Due to its oily nature, the drug is not absorbed orally and administration of BAL requires deep intra-muscular injection that is extremely painful and allergenic. It was found to mobilize and relocate lead to the brain, increasing its neurotoxic effects 30. Although treatment with 2,3-dimercaprol (BAL) increases the excretion of cadmium, there is a concomitant increase in renal cadmium concentration, so its use should be avoided in cases of cadmium toxicity. The drug has a short half life. Thus, the major drawbacks of BAL include:
- Low therapeutic index (small margin of safety)
- Tendency to redistribute arsenic to brain and testes
- Need for (painful) intramuscular injection
- Unpleasant odor (rotten eggs)
Other common adverse effects include fever, conjunctivitis (eye inflammation), lacrimation (tearing), constricted feeling (chest, limbs, jaw, abdomen), headache, paresthesias (tingling sensation), tremor, nausea, and pain at the injection site 31. More serious complications may be infections (abscesses) at the injection site, liver damage, elevated blood pressure and heart rate, and hemolysis (destruction of red blood cells) in patients with glucose-6-phophate deficiency (G6PD) 31.
DMSA (Meso-2,3-Dimercaptosuccinic Acid)
A chemical derivative of dimercaprol which has gained more and more attention, is meso-2,3-dimercaptosuccinic acid (DMSA). DMSA is a dithiol compound (containing two sulfhydryl, or –SH, groups) and an analogue of dimercaprol (BAL). It was 40 years ago when initial studies identified DMSA as an effective antidote to heavy metal poisoning. Subsequently DMSA was studied for more than twenty years in the People’s Republic of China, Japan, and Russia before scientists in Europe and the United States “discovered” the substance and its potential usefulness in the mid-1970s 32.
DMSA has been found to have fewer side effects with greater efficacy. The heavy metal chelating property of DMSA is due to the presence of vicinal dithiol groups that may also act as an oxygen radical scavenger and thus inhibit lipid peroxidation, giving an added advantage of fighting heavy metal induced oxidative stress. However, its hydrophilic properties do not allow it to cross the cell membrane. It was recently observed that esters of DMSA might be more effective antidotes for heavy metal toxicity. A large number of esters of DMSA have been synthesized aiming to for achieve optimal chelation and transport effects, compared to DMSA. These esters are mainly the mono- and diesters of DMSA that have been studied experimentally with the aim of enhancing tissue uptake of chelating agents. In order to make the compounds more lipophilic, the carbon chain length of the parent DMSA was increased by controlled esterification with the corresponding alcohol (methyl, ethyl, propyl, isopropyl, butyl, isobutyl, pentyl, isopentyl and hexyl). It has also been reported that these mono and diesters have a better potential in mobilizing heavy metal from the tissues in mice 33. Walker et al. 33 studied the effects of seven different monoalkyl esters of DMSA on the mobilization of lead in mice and observed that after a single parenteral dose of the chelator DMSA there was a 52% reduction in the lead concentrations while with the monoesters the reduction varied from 54% to 75%. In most of these published reports, it has been observed that the analogues of DMSA were capable of crossing the biomembranes and were more effective in reducing arsenic burden in acute and sub-chronic intoxication. These studies have also suggested that the monoesters may be preferred over DMSA diesters owing to their higher efficacy against arsenic intoxication and lower toxicity.
Pharmacological profile
The hydrophilic nature of DMSA causes considerable absorption in gastro intestinal tract thus possible oral route of administration creates its distinct advantage over BAL. The drug is 95 % plasma protein bound, most likely by virtue of binding on one of its sulfhydryl groups to a cysteine residue on albumin, leaving the other –SH available to chelate metals 34. DMSA has a large therapeutic window and is the least toxic amongst the dithiol compounds 35. No significant loss of essential metals like zinc, iron, calcium and magnesium are observed. However, metabolism of copper may be altered in humans after administration of DMSA 36. A major drawback associated with DMSA is its extracellular distribution, since it is unable to cross the cell membrane. Adverse reaction of DMSA includes gastrointestinal discomfort, skin reaction, mild neutropenia and elevated liver enzymes. The LD50 value of sodium salt of DMSA is about 3% greater than that of DMPS 37, in mice it is: 2.4 for i.v., 3.8 for i.m., 4.4 for i.p., and 8.5 gm/kg for oral route.
Numerous animal and human studies have shown that DMSA administration increases urinary mercury excretion and reduces blood and tissue mercury concentration 38. Chelation therapy with DMSA must be initiated shortly after exposure to prevent accumulation and avoid toxicity. Studies in animal models have shown that neither DMSA nor any other chelating or mobilizing agents were able to ameliorate the brain burden of mercury 39. The treatment with DMSA after exposure to inorganic mercury caused an increased elevation of mercury into motor axons presumably owing to redistribution of mercury, which was mobilized from non-neural tissues (e.g., kidneys and liver) 39.
Monoisoamyl DMSA (MiADMSA)
Monoisoamyl DMSA was synthesized by controlled esterification of DMSA with isoamyl alcohol. Controlled esterification made MiADMSA, a C5 branched chain alkyl monoester of DMSA which was lipophilic in nature as compared to the parent DMSA. Chelation studies with MiADMSA found it to be highly effective in reducing heavy metal burden from various organs in heavy metal exposed animals 40.
Pharmacological profile
MiADMSA is a potential drug candidate that is still in its developmental phase thus, its entire pharmacological profile has not been established yet.
Monomethyl DMSA (MmDMSA) and Monocyclohexyl DMSA (MchDMSA)
MmDMSA has a straight and branched chain methyl group, while MchDMSA has a cyclic carbon chain. Thus, they both have better lipophilic characteristics and might penetrate cells more readily that extracellularly acting chelating agent like DMSA. Both these chelating agents are orally active. Jones et al. 41 in their in vivo study on male albino mice exposed to cadmium for seven days, observed that administration of MmDMSA and MchDMSA produced significant reductions in whole body cadmium levels. Further, no redistribution of cadmium in brain was observed. The in vivo evaluation of these monoesters derived from higher alcohols (C3–C6 monoesters) proved to have better efficacy as compared to the monoesters derived from lower ones (C1–C2 monoesters) 41. Their ability to be administered orally suggests that they may possess considerable advantages in the clinical treatment of lead toxicity however; extensive studies are further required to reach a final conclusion.
DMPS (Sodium 2,3 Dimercaptopropane-l-Sulphonate)
Sodium 2,3-dimercaptopropane sulfonate (DMPS) is another analogue of BAL. DMPS is not considered as an appropriate drug against lead toxicity. A pilot study of DMPS in lead poisoned children by Gersl and co workers indicates less efficiency than CaNa2EDTA and DMSA 42. DMPS, although known for its antidotal efficacy against mercury, it has also been reported to have limited efficacy for treating lead and arsenic poisoning 43. The drug is registered in Germany for treatment of mercury intoxication, but it is not approved in the United States, so unless special permission is given by the U.S. Food and Drug Administration, it is unlawful for physicians to use it in the United States, nor is it lawful for pharmacies to compound it. Still, DMPS is being illegally used by members of the alternative health industry to treat people allegedly suffering from mercury intoxication, most often claimed to be due to amalgam dental fillings.
Pharmacological profile
DMPS, being hydrophilic in nature, is mainly distributed in the extracellular space but may enter cells by specific transport mechanisms. Thus, DMPS is known to be distributed both in the extracellular and to small extent in intracellular spaces 16. DMPS appears to be bio-transformed in humans to acyclic and cyclic disulphides. The drug and its metabolites are rapidly eliminated from the body through the kidneys. It is important to note that this drug does not redistribute arsenic, lead, or inorganic mercury to the brain 44. No major adverse effects following DMPS administration in humans or animals have been reported 44. Adverse reactions during treatment with DMPS include gastrointestinal discomfort, skin reactions, mild neutropenia, and elevated liver enzymes. Some patients, especially those with allergic asthma symptoms, may develop hypersensitivity to DMPS 45. Further, oral administration of DMPS did not adversely affects late gestation, parturition or lactation in mature mice and fetal and neonatal development do not appear to be adversely affected.
Deferoxamine (DFO)
Deferoxamine is a trihydroxamic acid, siderphore secreted by Streptomyces pilosus, a fungus. This chelating agent is known for its strong binding affinity for trivalent iron and less affinity for other metals making it a specific chelating agent for iron related diseases such as thalassaemia major as well as aluminium poisoning associated with chronic renal dialysis.
Pharmacological profile
The absorption of deferoxamine (DFO) in the gastrointestinal tract is low and thus it is administered mainly through intravenous injection or infusion. Deferoxamine is mainly distributed extracellularly and the protein binding in plasma is low (<10%). It complexes with iron and is excreted rapidly as ferrioxamine, mainly through kidney and one third into bile through faces. Thus, its efficacy also is determined by adequate urine output and may be facilitated by dialysis in case of dysfunctioning. This drug is generally well tolerated with few cases where side effects include opthalamic and auditory toxicity, bacterial and fungal infections, alterations in blood histology, allergic and skin reaction besides few reported adverse effect on lungs, kidney and nervous system 46. The total intravenous dose of deferoxamine should generally not exceed 80 mg/kg/24 hours. The intramuscular route of administration is not recommended in circulatory shock states.
Deferiprone (L1)
Deferiprone (L1; CP20; 1,2-dimethyl-3-hydroxypyrid-4-one) is an iron chelator and is considered a suitable alternative to deferoxamine in the trasfusional iron overload 47. It is comparatively less expensive than deferoxamine. Clinical studies suggest that combined administration of deferoxamine and deferiprone for three days per week is very effective in depleting iron overload accompanied by improvement in the cardiac function in transfusion dependent thalassemia patients. It may be noted that deferoxamine is known for reversal of cardiac dysfunction, and deferiprone has also shown cardio-protective effects. These may be attributed to reduction in iron overload however, the specific mechanism is not clear. Besides, due to higher incidences of agranulocyrosis, treatment must be closely monitored 48.
Pharmacological profile
The major advantages include oral administration and rapid absorption through gastrointestinal tract. Deferiprone (L1) is mainly excreted via renal route and has a half life of 47–134 minutes. Dosages of 75–100 mg/kg body weight/day of deferiprone have been considered effective to maintain stable iron balance and to reduce serum ferritin levels within one year of treatment in iron-overloaded thalassemic patients. Deferiprone (L1) is rapidly absorbed mainly from the stomach and reaches the circulation quickly. However, they might be the possibility of food–drug interaction or other gastric factors that delays the appearance of the drug in blood following oral administration. Deferiprone (L1) is mainly metabolized as glucoronide conjugates and excreted from urine. Wide variation in the metabolism and clearance of L1 amongst patients have been observed, which mainly depend on iron overload and availability of chelatable iron 49. The main reported side effects during a deferiprone therapy are arthropathy, gastrointestinal symptoms, headache, and moderate zinc deficiency. These adverse reactions are usually reversed on reducing the dose or discontinuing the drug. Except for severe joint symptoms in few patients, most of the subjects in different clinical trials have been able to continue with L1 therapy for a long term. The most severe, but rare complication following administration of deferiprone is agranulocytosis or neutropenia.
TETA (Tetraethylenetetraamine)
Tetraethylenetetraamine or trientine is a drug of choice for acute copper intoxication. Although nowadays there is less use of copper utensils, in ancient times the use of copper utensils could lead to extensive exposure to copper. Increased urinary copper excretion has been reported after administration of TETA.
Pharmacological profile
Normally TETA is administered through the oral route but its absorption is relatively poor, as indicated by the as evident from the recovery observed in urine and caracass after administration of an oral dose of C14-labeled TETA. Two major metabolites of TETA have been identified, i.e., N1-acetyltriethylenetetramine (MAT) and N1,N10-diacetyltriethylenetetramine (DAT). The former plays a significant role in the molecular mechanism by which TETA extracts copper from the system. The 5–18% of TETA that is systemically absorbed is said to be extensively metabolized, with the majority being excreted in urine as metabolite(s) 50. Wilson’s disease is characterized by disturbance in copper homeostasis which is inherited and leads to progressive increase in accumulation of copper and may even be fatal if not cured. Wilson’s disease was originally treated with DPA but TETA was a better chelator and found to be potentially free of side effects like those of DPA. The recommended dosage to treat Wilson’s disease patients is 0.75–2 g/day. The oral LD50 for rats for TETA is 2.5 g/kg body weight and is very close to the recommended dose for treatment of Wilson’s disease.
Nitrilotriacetic Acid (NTA)
Nitrilotriacetic acid (NTA, C6H9NO6), is a polyamino carboxylic acid and is also used as a chelating agent which forms coordination compounds (chelates) with metal ions such as Ca2+, Cu2+ or Fe3+. The uses of NTA are quite similar to EDTA. However, in contrast to EDTA, NTA is easily biodegradable. NTA is available in two forms, one that is a sodium derivative (Na3NTA) and the other iron (FeNTA). Both of these forms have been used as chelating agents 51.
NTA has been used shown to possess the ability to mobilize nickel from brain, heart, kidney and liver of nickel poisoned rats. In a comparative study with six metal binding agents, NTA was highly effective in dialyzing out nickel from the subcellular fractions of liver, kidney and blood corpuscles in rats that were exposed to nickel sulphate 52. Apart from nickel mobilization, a single dose of NTA has been tried in the removal of manganese from various organs and plasma in rats. Results from these studies have indicated that NTA binds rapidly to Mn and forms stable and diffusible complexes that result in faster excretion of Mn from the rats 53. Since NTA is considered to be non-mutagenic in vitro 54 an epigenetic mechanism is assumed, based on the fact that there is sustained cytotoxicity of zinc ions transfer to the urinary tract. Vacuolated cells of the proximal tubules represent the earliest lesions 54, followed by sloughing, necrosis/apoptosis, and proliferation. With chronic application of a high dose level, these alterations lead as a continuum to hyperplastic foci, adenomas, and adenocarcinomas 55. This ability of both NTA to form tumors cannot be ruled out. Studies however have indicated that tumor formation ability of both NTA is highly route and dose dependented. While FeNTA causes an iron overload and lipid peroxidation in cells and is genotoxic 56, Na3NTA predominantly binds to zinc and calcium, thereby exerting its toxic effects 54.
Chelation therapy autism
Chelation therapy is not an effective autism treatment, and it may be dangerous.
Some doctors and parents have considered chelation therapy as a potential autism treatment. Proponents believe that autism is caused by mercury exposure, such as from childhood vaccines. Chelation therapy supposedly removes mercury from the body, which chelation supporters say cures autism — but there’s no evidence of a link between mercury exposure and autism. In addition, chelation therapy can be associated with serious side effects, including potentially deadly kidney damage.
The subject of exposure to environmental toxic agents — such as mercury, lead and a host of other toxins — and links to autism spectrum disorder is complex and the quality of studies varies considerably. That’s in part because of all the many variables, such as geography, genetic factors, metabolism differences in individuals and sampling sources. Studies show conflicting results with no reproducible proof.
There’s no cure for autism — now called autism spectrum disorder (ASD) in the newest Diagnostic and Statistical Manual of Mental Disorders (DSM-5), published by the American Psychiatric Association. As a result, many unproven alternative therapies are often suggested. However, these alternative therapies are usually found to be ineffective and sometimes harmful.
Consult your primary doctor if you’re considering an alternative treatment for autism spectrum disorder. Your doctor may help you identify treatment options and local resources that provide support or refer you to a health professional who can do so.
Chelation therapy for heart disease
It’s unclear whether chelation therapy can treat heart disease. Chelation therapy has been used for many years as a treatment for mercury and lead poisoning, but it isn’t a proven treatment for heart disease. Chelation therapy can potentially cause serious side effects when used as a heart disease treatment. Even so, some doctors and complementary health practitioners have used chelation therapy to treat heart disease and stroke.
In chelation therapy, a dose of a medication called ethylenediaminetetraacetic acid (EDTA) is delivered into your bloodstream through an intravenous (IV) line. This medication seeks out and binds to minerals in your bloodstream. Once the medication binds to the minerals, it creates a compound that leaves your body in your urine.
The theory behind using chelation therapy for heart disease is that the medicine used in the treatment binds to the calcium that’s in fatty deposits (plaques) in your arteries. Once the medicine (EDTA) binds to the calcium, the plaques are swept away as the medicine moves through your bloodstream.
The safety and effectiveness of chelation therapy for heart disease can’t be determined, even after a large-scale study was conducted to determine just that. Results of the Trial to Assess Chelation Therapy (TACT) 57, sponsored by the National Center for Complementary and Alternative Medicine and the National Heart Lung and Blood Institute, a clinical trial of chelation therapy in patients age 50 or older with a prior heart attack and good kidney function to finally understand whether EDTA chelation for coronary disease was safe and effective. The Trial to Assess Chelation Therapy (TACT) 58 enrolled 1,708 patients who were at least 50 years old and had a prior heart attack. The proposed treatment was intensive – 40 intravenous infusions, three hours each, all given over a little more than a year. Half of the patients received EDTA chelation. The other half received a saltwater placebo. Overall, patients received 55,222 IV infusions in 134 offices and hospitals across the US and Canada.
Overall, there was an 18% reduction in heart events (death, another heart attack, stroke, stenting or bypass, and hospitalization for heart pains) by EDTA infusions above and beyond that provided by our effective treatments including statins and aspirin 58. When the group that took the EDTA infusions plus oral vitamins was analyzed, the reduction was by 26% compared with placebo 57. The effect was even more striking in patients with diabetes, where there was a 41% reduction in clinical events, including a 43% reduction in deaths over 5 years 59. However, the Trial to Assess Chelation Therapy (TACT) didn’t provide enough evidence to support routine use of chelation therapy for heart disease.
Figure 2. Chelation therapy for heart disease (risk of death, heart attack, stroke, stenting, bypass, or hospitalization for angina in patients with, or without chelation therapy.)
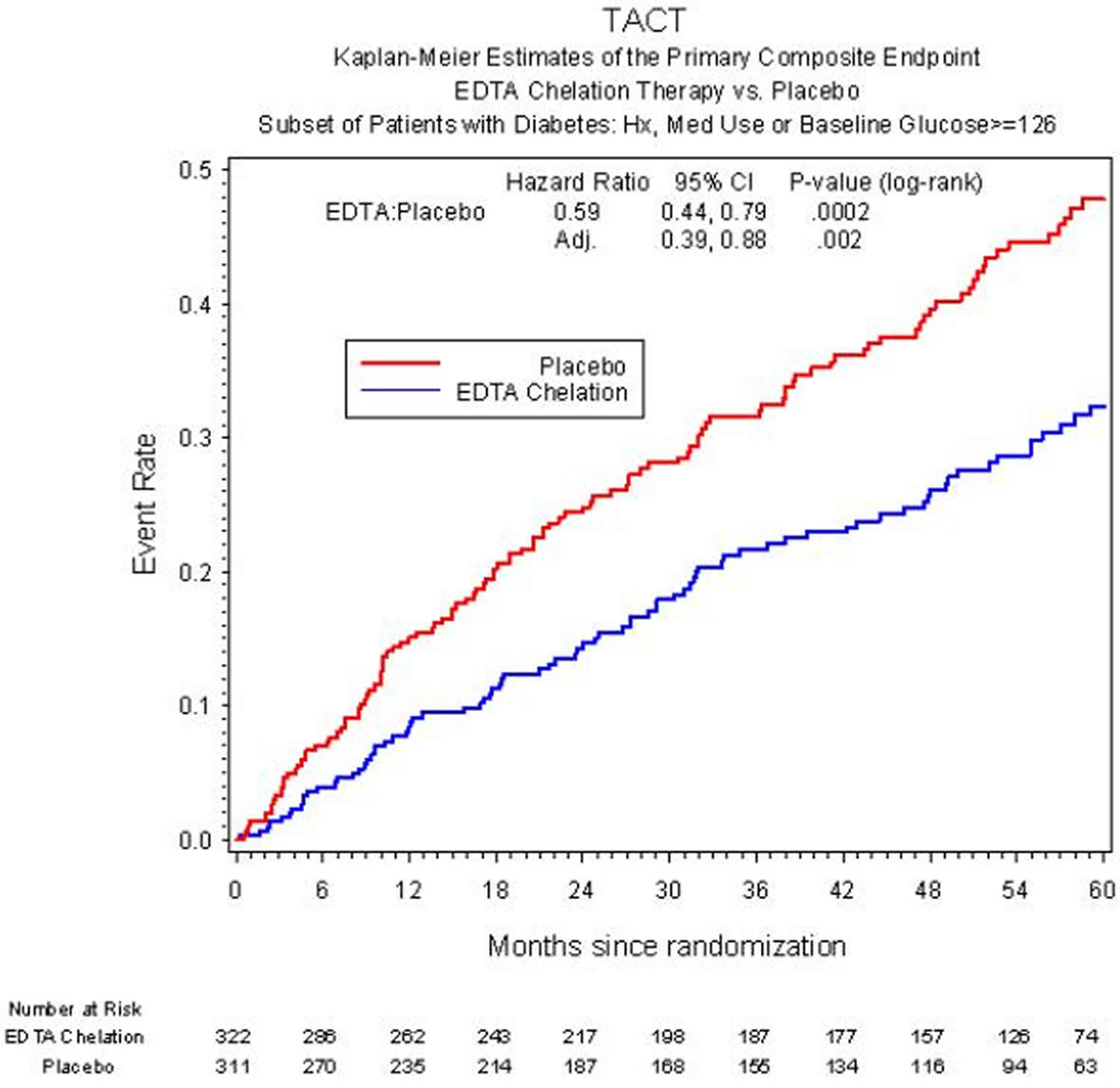
The American Heart Association and the American College of Cardiology have determined that it’s uncertain whether chelation therapy is useful as a treatment for heart disease, and the US Food and Drug Administration hasn’t approved chelation therapy for use as a heart disease treatment. The US Food and Drug Administration encouraged the researchers to carry out another study to confirm these results (TACT2 is being planned).
Some doctors are concerned about the safety of chelation therapy as a treatment for heart disease. A burning sensation at the IV site is the most common side effect. Less common side effects include fever, headache, nausea or vomiting. Other rare but serious complications that have been reported include abnormally low blood-calcium levels (hypocalcemia), permanent kidney damage or death.
Because of the known risks of chelation therapy, discuss this and other options with your doctor before trying chelation therapy as a heart disease treatment.
Bottom line
- Overall, TACT showed that infusions of disodium EDTA chelation therapy produced a modest reduction in cardiovascular events. However, further examination of the data showed that chelation therapy benefitted only the patients with diabetes.
- Patients with diabetes, who made up approximately one-third of the 1,708 TACT participants, had a 41 percent overall reduction in the risk of any cardiovascular event; a 40 percent reduction in the risk of death from heart disease, nonfatal stroke, or nonfatal heart attack; a 52 percent reduction in recurrent heart attacks; and a 43 percent reduction in death from any cause. In contrast, there was no significant benefit of EDTA treatment in participants who didn’t have diabetes.
- The TACT study team also looked at the impact of taking high-dose vitamins and minerals in addition to chelation therapy. They found that chelation plus high-dose vitamins and minerals produced the greatest reduction in risk of cardiovascular events versus placebo.
- In the TACT study, which had extensive safety monitoring, 16 percent of people receiving chelation and 15 percent of people receiving the placebo stopped their infusions because of an adverse event. Four of those events were serious; two were in the chelation group (one death) and two were in the placebo group (one death).
- The most common side effect of EDTA chelation is a burning sensation where the solution is placed the vein. Rare side effects can include fever, headache, nausea, and vomiting. Even more rare are serious and potentially fatal side effects that can include heart failure, a sudden drop in blood pressure, abnormally low calcium levels in the blood (hypocalcemia), permanent kidney damage, and bone marrow depression (blood cell counts fall). Hypocalcemia and death may occur particularly if disodium EDTA is infused too rapidly. Reversible injury to the kidneys, although infrequent, has been reported with EDTA chelation therapy. Other serious side effects can occur if EDTA is not administered by a trained health professional.
- If you’re considering chelation therapy, discuss it first with your cardiologist or other health care provider for your heart care. Seek out and consider information available from scientific studies on the therapy.
- Further research is needed to fully understand the TACT results. Since this is the first clinical trial to show a benefit, these results are not, by themselves, sufficient to support the routine use of chelation as a post-heart attack therapy in people with diabetes.
- A new study, called the Trial To Assess Chelation Therapy 2 (TACT2), is now in its early stages (https://tact2.org/). Its purpose is to repeat the first TACT study—but only in patients with diabetes and a prior heart attack—to see if the apparent benefit can be confirmed. The results of TACT2 will help the FDA determine whether disodium EDTA chelation therapy should be approved to reduce the risk of further cardiovascular events in patients who have both coronary artery disease and diabetes.
Iron chelation therapy
Iron chelation therapy is used when you have a condition called iron overload or hemochromatosis. Iron overload means you have too much iron in your body. Hemochromatosis is a disease in which too much iron builds up in your body (iron overload). Iron is a mineral found in many foods.
Too much iron is toxic to your body. It can poison your organs and cause organ failure. In hemochromatosis, iron can build up in most of your body’s organs, but especially in the liver, heart, and pancreas. Too much iron in the liver can cause an enlarged liver, liver failure, liver cancer, or cirrhosis. Cirrhosis is scarring of the liver, which causes the organ to not work well.
Too much iron in the heart can cause irregular heartbeats called arrhythmias and heart failure. Too much iron in the pancreas can lead to diabetes.
If hemochromatosis isn’t treated, it may even cause death. However, only a doctor can decide if you have iron overload or hemochromatosis and need treatment.
Iron overload can also be a problem for people who get lots of red blood cell transfusions. Red blood cells contain iron. Each time you get a red blood cell transfusion you are putting more iron in your body. Your body has no good way to get rid of the extra iron. This iron can build up in your vital organs and may injure them over time.
Extra iron that is not immediately needed to make new red blood cells is normally stored in the liver, spleen, and bone marrow. Excess iron may accumulate in these 3 organs and in other organs that don’t normally store iron, such as the pancreas, heart, joints and skin. This excess iron can injure of these organs.
Only your doctor can decide if iron overload is a problem for you. To decide, your doctor will look at how many blood transfusions you have had and do blood tests to check how much iron is in your blood and body. The blood tests might include serum ferritin level, iron concentration, and transferrin saturation. It is possible your doctor may do other tests like a liver biopsy or an MRI (Magnetic Resonance Imaging) to understand if iron is building up in organs.
Early on, iron overload can cause no symptoms, or it can cause non-specific symptoms that are also seen in other conditions. Some of these symptoms include:
- tiredness or weakness
- loss of sex drive
- weight loss
- abdominal pain
- joint aches or pain
Young people with iron overload might not grow or go through puberty normally.
Women who have iron overload might stop getting their periods.
With severe iron overload, you may experience:
- gray-colored or bronze-colored skin
- shortness of breath
- arthritis
- liver disease, including cirrhosis or liver cancer
- enlarged spleen that may cause abdominal pain or difficulty eating a normal-sized meal
- diabetes
- shrunken testicles
- heart problems, including both heart failure and heart rhythm problems
High levels of iron can be detected through two simple blood tests. These tests tell doctors how much iron is stored in your body.
Treatments for hemochromatosis include therapeutic phlebotomy, iron chelation therapy, dietary changes, and treatment for complications.
The goals of treating hemochromatosis include:
- Reducing the amount of iron in your body to normal levels
- Preventing or delaying organ damage from iron overload
- Treating complications of the disease
- Maintaining a normal amount of iron in your body for the rest of your life
Iron chelation therapy uses medicine to remove excess iron from your body. This treatment is a good option for people who can’t have routine blood removal.
The medicine used in iron chelation therapy is either injected or taken orally (by mouth). Injected iron chelation therapy is done at a doctor’s office. Oral iron chelation therapy can be done at home.
There are two iron chelators approved by the U.S. Food and Drug Administration (FDA) for use in the U.S.
- Deferoxamine (Desferal®) is usually administered by subcutaneous (under the skin) infusion using a small portable pump about the size of a CD player. The pump is worn for 8-12 hours a day, usually at night while sleeping. Many patients find taking deferoxamine difficult because of the need to carry around a pump.
- Deferasirox is a newer iron chelator that now comes in two forms (see below). Deferasirox is not recommended for people with high-risk Myelodysplastic Syndromes.
- Exjade® is a tablet form that must be dissolved in juice or water and taken (by mouth) once a day. Most patients tolerate it very well.
- Jadenu® is new tablet formulation of deferasirox approved for use in 2015. It is taken on an empty stomach or with a light meal once a day with water or other liquids.
One additional iron chelator is currently being used in Europe, Asia, and Canada, but is not yet approved by the FDA for use in the United States:
- Deferiprone or L1 (Ferriprox™) comes in a pill form and is taken 3 times a day. It is generally well tolerated by patients, but it can cause a drop in white blood cell counts, so patients need to have their blood checked weekly while taking this drug (white blood cells protect you against infections).
Iron chelators side effects
Some common side effects of iron chelators include:
- Nausea and dizziness
- Diarrhea
- Rash
- Vomiting
- Vision and hearing problems
More serious effects such as kidney or liver injury can occur. Once the body gets used to the drug, side effects usually go away. Your doctor should monitor your liver and kidneys for potentially serious side effects while you are taking deferasirox.
When taking either deferoxamine or deferasirox, you should:
- Have your vision and hearing tested prior to starting therapy, with re-testing every 6-12 months. Both deferoxamine and deferasirox can cause damage to the eyes and ears.
- Avoid taking Vitamin C unless it is prescribed by your doctor. Under your doctor’s specific orders, Vitamin C can be added at a later time to iron chelation therapy and may improve results for some patients. Vitamin C should only be taken in a moderate dose, such as 100 mg daily.
Chelation therapy for lead
Lead is a naturally occurring element found in small amounts in the earth’s crust. While it has some beneficial uses, it can be toxic to humans and animals causing of health effects.
Lead can be found in all parts of our environment – the air, the soil, the water, and even inside our homes. Much of our exposure comes from human activities including the use of fossil fuels including past use of leaded gasoline, some types of industrial facilities, and past use of lead-based paint in homes. Lead and lead compounds have been used in a wide variety of products found in and around our homes, including paint, ceramics, pipes and plumbing materials, solders, gasoline, batteries, ammunition, and cosmetics.
Lead is particularly dangerous to children because their growing bodies absorb more lead than adults do and their brains and nervous systems are more sensitive to the damaging effects of lead. Babies and young children can also be more highly exposed to lead because they often put their hands and other objects that can have lead from dust or soil on them into their mouths. Children may also be exposed to lead by eating and drinking food or water containing lead or from dishes or glasses that contain lead, inhaling lead dust from lead-based paint or lead-contaminated soil or from playing with toys with lead paint.
Adults may be exposed to lead by eating and drinking food or water containing lead or from dishes or glasses that contain lead. They may also breath lead dust by spending time in areas where lead-based paint is deteriorating, and during renovation or repair work that disturbs painted surfaces in older homes and buildings. Working in a job or engaging in hobbies where lead is used, such as making stained glass, can increase exposure as can certain folk remedies containing lead. A pregnant woman’s exposure to lead from these sources is of particular concern because it can result in exposure to her developing baby.
Health effects of lead
Lead can affect almost every organ and system in your body. Children six years old and younger are most susceptible to the effects of lead.
Children
Even low levels of lead in the blood of children can result in:
- Behavior and learning problems
- Lower IQ and Hyperactivity
- Slowed growth
- Hearing Problems
- Anemia
In rare cases, ingestion of lead can cause seizures, coma and even death.
When kids do develop symptoms of lead poisoning, they usually appear as:
- irritability or behavioral problems
- difficulty concentrating
- headaches
- loss of appetite
- weight loss
- sluggishness or fatigue
- abdominal pain
- vomiting or nausea
- constipation
- pallor (pale skin) from anemia
- metallic taste in mouth
- muscle and joint weakness or pain
- seizures
Pregnant Women
Lead can accumulate in our bodies over time, where it is stored in bones along with calcium. During pregnancy, lead is released from the mother’s bones along with calcium and can pass from the mother exposing the fetus or the breastfeeding infant to lead. This can result in serious effects to the developing fetus and infant, including:
- Cause the baby to be born too early or too small;
- Hurt the baby’s brain, kidney’s, and nervous system;
- Increase the likelihood of learning or behavioral problems; and
- Put the mother at risk for miscarriage.
Adults
Lead is also harmful to other adults. Adults exposed to lead can suffer from:
- Cardiovascular effects, increased blood pressure and incidence of hypertension
- Decreased kidney function
- Reproductive problems (in both men and women)
Chelation agents for lead
The majority of chelating agents bind to heavy metals in extracellular fluid and cannot cross the cellular membrane 60. Because they cause essential metal loss and may cause adverse drug effects such as hepatotoxicity or nephrotoxicity, while there are benefits to their use in cases of acute poisoning, they are not recommended for cases of chronic poisoning 61. Due to the risk caused by the adverse side effects of the medicine and the redistribution of lead, chelation therapy is generally not recommended in cases where blood lead levels are below 45 μg/dL in adults 62. Additionally, as rebounding commonly occurs after chelation therapy, blood lead levels should be measured before and after the treatment.
The drugs used as chelating agents for lead include dimercaptosuccinic acid (DMSA), dimercaptopropane sulfonate (DMPS), dimercaprol (British Anti-Lewisite, BAL), penicillamine, and CaNa2EDTA (Table 2).
An analogue of dimercaprol, dimercaptosuccinic acid (DMSA) is a dithiol compound which contains two sulfhydryl (–SH) groups. Dimercaptosuccinic acid (DMSA) has a large therapeutic window, and is the least toxic dithiol compound 63. While chelation therapy can result in a reduction in essential metals, DMSA has been found to not cause a reduction in essential metals such as zinc, iron, calcium, and magnesium 64. DMSA is the most efficient and safe chelating agent for lead exposure, so the chelators being used the most are DMSA recently 65.
Dimercaptopropane sulfonate (DMPS) is another analogue of dimercaprol. Less effective than CaNa2EDTA or DMSA, dimercaptopropane sulfonate (DMPS) is not commonly used in lead chelation therapy 66. Dimercaptopropane sulfonate (DMPS) is mainly used in cases of arsenic or mercury poisoning.
The drawbacks of CaNa2EDTA are that it contributes to a greater loss in essential minerals compared to DMSA or DMPS and that it redistributes lead to the brain 65. Generally, one gram of EDTA is mixed with 250 mL of a 5 % dextrose solution, gradually settling over one or two hours, and treatment is administered over a five day period. During the treatment period, the patient should be hydrated intravenously, and tests should be conducted daily on kidney function, electrolyte levels, and urine.
Penicillamine was the first chelating agent used in the treatment of lead poisoning. It reaches its greatest capacity one to four hours after oral administration. Food, antioxidants, and iron supplements can reduce absorption of penicillamine. In the past, the daily dose used was 500–1000 mg, but recent findings show that using a lower dosage had equivalent lead removal effects while reducing adverse side effects 67. Due to the narrow therapeutic window and harsh side effects of dimercaprol (British Anti-Lewisite, BAL), it is rarely used these days.
In general, chelation therapy is recommended for children only in cases where blood lead levels are 45 μg/dL or greater. In cases where blood lead levels are 45–69 μg/dL, chelation therapy can be attempted with DMSA. McKay 68 tried chelation therapy on children with blood lead levels of 20–44 μg/dL. While the blood lead levels of the children who received chelation therapy decreased temporarily compared with the control group, when a follow-up study was conducted three years later, the blood lead levels of the therapy group had risen again, and no significant difference was observed in cognitive or behavioral function nor in blood lead levels between the therapy group and the control group.
While removing the source of lead exposure is most important for reducing the health impact of lead and similar harmful heavy metals, naturally occurring essential minerals (calcium, magnesium, selenium, etc.) and other related nutrient replacement therapies can be useful in decreasing the absorption of harmful heavy metals or promoting their excretion. New strategies in heavy metal chelation therapy include the use of structurally different chelating agents (combination therapy such as DMSA and EDTA) and the co-administration of antioxidants such as DMSA and taurine 65.
Table 2. Overview of chelation drugs
| Chemical name (common names, abbreviations) | Dose | Adverse effects | Elements chelated |
|---|---|---|---|
| 2,3-bis(sulfanyl)butanedioic acid (Dimercaptosuccinic acid; DMSA; Succimer) | 10 mg/kg (or 350 mg/m2) per 8 h for 5 days, then 10 mg/kg per 12 h for 14 days (a total of 19 days), POa. | Gastrointestinal disturbances, mild increase in serum transaminase | Lead, arsenic, mercury, cadmium, silver, tin, copper |
| Sodium 2,3-bis(sulfanyl)propane-1-sulfonate (Dimercaptopropanesulfonate; DMPS; Dimaval) | Adult: 5 mg/kg per 6–8 h, PO, IMb, IVc, or SQd. | Low back (kidney) pain, gastrointestinal disturbances, skin rash, fatigue, hypersensitivity reactions | Mercury, arsenic, lead, cadmium, tin, silver, copper, selenium, zinc, magnesium |
| Children: 5-day course of 200 or 400 mg/m2/day | |||
| 2-[2-[bis(carboxymethyl)amino]ethyl-(carboxymethyl)amino]acetic acid (Ethylenediaminetetraacetic acid; EDTA; CaNa2EDTA) | 1000–1500 mg/m2/day (1–2 g/24 h for a 70-kg adult) as an IV infusion for 5 days | Renal toxicity | Lead, cadmium, zinc |
| (2S)-2-amino-3-methyl-3-sulfanylbutanoic acid (3-Sulfanyl-D-valine; Penicillamine; D- Penicillamine) | 10 mg/kg/day for 7 days with a possibility of a prolonged treatment during 2 to 3 weeks, PO. | Interstitial nephritis, hypersensitivity reactions, gastrointestinal disturbances, leukopenia and thrombocytopenia | Copper, arsenic, zinc, mercury, lead |
| 2,3-bis(sulfanyl)propan-1-ol (Dimercaprol; British Anti-Lewisite; BAL) | 50–75 mg/m2 per 4 h for 5 days, deep IM. | Allergy, gastrointestinal symptoms, tachycardia, fever, elevation of liver function tests | Arsenic, gold, mercury, lead (BAL in combination with CaNa2EDTA) |
Footnotes:
a PO = oral ingestion
b IM = intramuscular injection
c IV = intravenous injection
d SQ = subcutaneous injection
[Source 63 ]Chelation therapy side effects
The risks associated with CaNa2EDTA therapy are substantial, including renal failures, arrhythmias, tetany, hypocalcaemia, hypotension, bone marrow depression, prolonged bleeding time, convulsions, respiratory arrest, etc. 12. Although the nephrotoxicity by CaNa2EDTA is dose dependent and reversible after cessation of therapy yet exceeding maximal daily dose of 75 mg/kg could be fatal. However, there also are reports that highlight the efficacy of EDTA in chronic renal artery diseases 17. Other adverse effects may include fatigue, headache, fever, nasal congestion, lacrimation, mucocutaneous lesions, glycosuria, myalgia, hepatotoxicity, increased urinary frequency, hypotension, abnormal changes in ECG and gastrointestinal symptoms 18. Prolonged treatment with CaNa2EDTA results in depletion of essential metal, especially Zn, Cu and Mn 19. It has been reported that zinc supplementation during and after chelation is also beneficial 20. Although zinc depletion by CaNa2EDTA therapy is rapidly reversible by zinc supplementation, it is considered to be key mechanism for teratogenic effects of the drug, especially when administered between days 11 to 14 at doses comparable to humans.
CaNa3DTPA adverse effects may include nausea, vomiting, diarrhea, chills, fever and muscle cramps during the first 24 hrs 24. CaNa3DTPA is teratogenic like CaNa2EDTA due to its Zn and Mn depletion effect that is confirmed by the teratogenic effects of ZnNa3DTPA at a dose 16 times lower than CaNa3DTPA. Thus, although the drug is contraindicated during pregnancy ZnNa3DTPA may be carefully administered if unavoidable. Other contraindications include prescription to children, in patients with renal insufficiency and bone marrow depression.
D-Penicillamine adverse reactions have been reported, some serious ones including thrombocytopenia and leukocytopenia (incidence 5–15%), and rarely aplastic anemia may occur. Prolonged d-Penicillamine treatment may lead to anorexia, nausea and vomiting. The ulcerogenic activity of d-Penicillamine in rats, which may involve the stimulation of histaminergic (H1 and H2) receptors, has been reported 26. Other toxic effects of d-Penicillamine include gastrointestinal disturbances (10–30%), changes/loss of taste (5–30%), hair loss (1–2%), and partly proteinuria (5–20%) 27. Severe adverse effects are autoimmune phenomena such as pemphigus, d-Penicillamine-induced lupus erythematosus, polymyositis/dermatomyositis, membranous glomerulopathy and hypersensitivity pneumonitis 27. d-Penicillamine is a well recognized teratogen and lathyrogen that causes skeletal, cutaneous and pulmonary abnormalities 26. The most important contraindication is in patients allergic to penicillin and in cases of renal insufficiency. Co-administration of d-Penicillamine to patients receiving gold therapy, antimalarial or cytotoxic drugs, phenyl-butazone, or oxyphenbutazone must be contraindicated since it can result in more serious adverse reactions.
BAL was found to mobilize and relocate lead to the brain, increasing its neurotoxic effects 30. Other common adverse effects include fever, conjunctivitis (eye inflammation), lacrimation (tearing), constricted feeling (chest, limbs, jaw, abdomen), headache, paresthesias (tingling sensation), tremor, nausea, and pain at the injection site 31. More serious complications may be infections (abscesses) at the injection site, liver damage, elevated blood pressure and heart rate, and hemolysis (destruction of red blood cells) in patients with glucose-6-phophate deficiency (G6PD) 31.
Adverse reaction of DMSA includes gastrointestinal discomfort, skin reaction, mild neutropenia and elevated liver enzymes. The LD50 value of sodium salt of DMSA is about 3% greater than that of DMPS 37, in mice it is: 2.4 for i.v., 3.8 for i.m., 4.4 for i.p., and 8.5 gm/kg for oral route.
Adverse reactions during treatment with DMPS include gastrointestinal discomfort, skin reactions, mild neutropenia, and elevated liver enzymes. Some patients, especially those with allergic asthma symptoms, may develop hypersensitivity to DMPS 45. Further, oral administration of DMPS did not adversely affects late gestation, parturition or lactation in mature mice and fetal and neonatal development do not appear to be adversely affected.
Deferoxamine (DFO) is generally well tolerated with few cases where side effects include opthalamic and auditory toxicity, bacterial and fungal infections, alterations in blood histology, allergic and skin reaction besides few reported adverse effect on lungs, kidney and nervous system 46.
The main reported side effects during a Deferiprone (L1; CP20; 1,2-dimethyl-3-hydroxypyrid-4-one) therapy are arthropathy, gastrointestinal symptoms, headache, and moderate zinc deficiency. These adverse reactions are usually reversed on reducing the dose or discontinuing the drug. Except for severe joint symptoms in few patients, most of the subjects in different clinical trials have been able to continue with L1 therapy for a long term. The most severe, but rare complication following administration of deferiprone is agranulocytosis or neutropenia.
- Lamas GA. Cardiology Patient Page. Chelation therapy: a new look at an old treatment for heart disease, particularly in diabetics. Circulation. 2015;131(21):e505-6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4448121[↩]
- Flora SJ, Pachauri V. Chelation in metal intoxication. Int J Environ Res Public Health. 2010;7(7):2745-88. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2922724/[↩][↩][↩][↩][↩][↩]
- Principles and recent developments in chelation treatment of metal intoxication. Andersen O. Chem Rev. 1999 Sep 8; 99(9):2683-710.[↩]
- Treatment of mercury intoxication. Baum CR. Curr Opin Pediatr. 1999 Jun; 11(3):265-8.[↩]
- Oral cadmium exposure in mice: toxicokinetics and efficiency of chelating agents. Andersen O. Crit Rev Toxicol. 1989; 20(2):83-112.[↩]
- Iron chelators for clinical use. Tilbrook GS, Hider RC. Met Ions Biol Syst. 1998; 35():691-730.[↩]
- Therapeutic iron chelators and their potential side-effects. Singh S, Khodr H, Taylor MI, Hider RC. Biochem Soc Symp. 1995; 61():127-37.[↩]
- Klaassen CD. Heavy metals and heavy metal antagonists. In: Goodman L, Gilman A, editors. The Pharmacological Basis of Therapeutics. McGraw Hill, Medical Publishing Division; New York, NY, USA: 2006. pp. 1825–1872.[↩][↩][↩]
- Quan H, Ghali WA, Verhoef MJ, Norris CN, Galbraith PD, Knudtson ML. Use of chelation therapy after coronary angiography. Am. J. Med. 2001;111:686–691[↩]
- Miller KL, Liebowitz RS, Newby LK. Complementary and alternative medicine in cardiovascular diseases: A review of biologically based approaches. Am. Heart J. 2004;147:401–411[↩]
- Ernst E. Chelation therapy for coronary heart disease: An overview of all clinical investigations. Am. Heart J. 2000;140:139–141[↩]
- Knudtson ML, Wyse KG, Galbraith PD. Chelation therapy for ischemic heart disease. A randomized controlled trail. JAMA. 2002;287:481–486.[↩][↩][↩]
- Anderson TJ, Hubacek J, Wyse DG, Knudtson ML. Effect of chelation therapy on endothelial function in patients with coronary artery disease: PATCH substudy. J. Amer. Coll. Cardiol. 2003;41:420–425.[↩]
- Miller KL, Liebowitz RS, Newby LK. Complementary and alternative medicine in cardiovascular diseases: A review of biologically based approaches. Am. Heart J. 2004;147:401–411.[↩]
- Flora SJS, Bhattacharya R, Vijayaraghavan R. Combined therapeutic potential of meso 2,3-dimercaptosuccinic acid and calcium disodium edetate in the mobilization and distribution of lead in experimental lead intoxication in rats. Fund. Appl. Toxicol. 1995;25:233–240[↩]
- Andersen O. Principles and recent developments in chelation treatment of metal intoxication. Chem. Rev. 1999;99:2683–2710.[↩][↩]
- Lin-Tan DT, Lin JL, Yen TH, Chen KH, Huang YL. Long-term outcome of repeated lead chelation therapy in progressive non-diabetic chronic kidney diseases. Nephrol. Dial. Transplant. 2007;22:2924–2931.[↩][↩]
- DRUGDEX Drug evaluation. 2004;122 Thomson MICROMEDEX Healthcare Series[↩][↩][↩]
- Ibim SE, Trotman J, Musey PI, Semafuko WE. Depletion of essential elements by calcium disodium EDTA treatment in the dog. Toxicology. 1992;73:229–237[↩][↩]
- Flora SJS, Tandon SK. Beneficial effects of zinc supplementation during chelation treatment of lead intoxication in rats. Toxicology. 1990;64:129–139[↩][↩]
- Spoor NL. The use of EDTA and DTPA for accelerating the removal of deposited transuranic elements from human. Harwell; Didcot, UK: 1977[↩]
- Llobet JM, Domingo JL, Corbella J. Comparison of the effectiveness of several chelators after single administration on the toxicity, excretion and distribution of cobalt. Arch. Toxicol. 1986;58:278–281.[↩]
- Walker EM, Jr, Gale GR, Fody EP, Atkins LM, Smith AB, Jones MM. Comparative antidotal effects of diethyldithicarbamate, dimercaptosuccinate and diethylene triamine pentaacetate against cadmium induced testicular toxicity in mice. Res. Commun. Chem. Pathol. Pharmacol. 1986;51:231–244.[↩]
- Oak Ridge Institute for Science and Education; Oak Ridge, TN, USA: 2002. Radiation emergency assistance center, Training site (REAC/TS). Ca-DTPA[↩][↩][↩]
- Roussaeux CG, MacNabb LG. Oral administration of D-pencillamine causes neonatal mortality without morphological defects in CD-1 mice. J. Appl. Toxicol. 1992;12:35–38.[↩]
- Gupta B, Srivastava RK, Saxena KK, Prasad DN. A study on the penicillamine induced gastric ulceration in the rat. Ind. J. Pharmacol. 1980;12:247–252.[↩][↩][↩][↩]
- Grasedyck K. D-penicillamine—side effects, pathogenesis and decreasing the risks. Z. Rheumatol. 1988;47:17–19.[↩][↩][↩][↩]
- Peters R, Stocken L, Thompson R. British Anti-Lewisite (BAL) Nature. 1945;156:616–619[↩]
- Hoover TD, Aposhian HV. BAL increases the arsenic-74 content of rabbit brain. Toxicol. Appl. Pharmacol. 1983;70:160–162.[↩]
- Andersen O. Chemical and biological considerations in the treatment of metal intoxications by chelating agents. Mini Rev. Med. Chem. 2004;4:11–21[↩][↩]
- Janakiraman N. Hemolysis during BAL chelation therapy for high blood lead levels in two G6PD deficient children. Clin. Pediatr. 1978;17:485–487[↩][↩][↩][↩]
- Aposhian HV. DMSA and DMPS—Water soluble antidotes for heavy metal poisoning. Ann. Rev. Pharmacol. Toxicol. 1983;23:193–215.[↩]
- Walker EM, Stone A, Milligan LB, Gale GR, Atkins LM, Smith AB, Jones MM, Singh PK, Basinger MA. Mobilization of lead in mice by administration of monoalkyl esters of meso-2,3-dimercaptosuccinic acid. Toxicology. 1992;76:79–87.[↩][↩]
- Alan L, Miller ND. Dimercaptosuccinic Acid (DMSA), a non-toxic, water-soluble treatment for heavy metal toxicity. Altern. Med. Rev. 1998;3:199–207[↩]
- Graziano JH. Role of 2,3-dimercaptosuccinic acid in the treatment of heavy metal poisoning. Med. Tox. 1986;1:155–162.[↩]
- Aposhian HV, Maiorino RM, Dart RC, Perry DF. Urinary excretion of meso-2,3-dimercaptosuccinic acid in human subjects. Clin. Pharmacol. Ther. 1989;45:520–526.[↩]
- Aposhian HV. DMSA and DMPS—Water soluble antidotes for heavy metal poisoning. Ann. Rev. Pharmacol. Toxicol. 1983;23:193–215[↩][↩]
- Aaseth J, Alexander J, Raknerud N. Treatment of mercuric chloride poisoning with dimercaptosuccinic acid and diuretics: preliminary studies. J. Toxicol. Clin. Toxicol. 1982;19:173–186.[↩]
- Ewan KB, Pamphlett R. Increased inorganic mercury in spinal motor neurons following chelating agents. Neurotoxicology. 1996;17:343–349.[↩][↩]
- Flora SJS, Pande M, Kannan GM, Mehta A. Lead induced oxidative stress and its recovery following co-administration of melatonin or n-acetylcysteine during chelation with succimer in male rats. Cell. Mol. Biol. 2004;50:543–551[↩]
- Jones MM, Singh PK, Gale GR, Smith AB, Atkins LM. Cadmium mobilization in vivo by intraperitoneal or oral administration of mono alkyl esters of meso-2,3-dimercaptosuccinic acid. Pharmacol. Toxicol. 1992;70:336–343[↩][↩]
- Gersl V, Hrdina R, Vavrova J, Holeckova M, Palicka V, Vogkova J, Mazurova Y, Bajgar J. Effects of repeated administration of dithiol chelating agent- sodium 2,3-dimercapto 1-propanesulphonate (DMPS)- on biochemical and hematological parameters in rabbits. Acta. Medica. 1997;40:3–8[↩]
- Kalia K, Flora SJS. Strategies for Safe and Effective Treatment for Chronic Arsenic and Lead Poisoning. J. Occup. Hlth. 2005;47:1–21.[↩]
- Aposhian MM, Maiorino RM, Xu Z. Sodium 2,3-dimercapto-1-propanesulfonate (DMPS) treatment does not redistribute lead or mercury to the brain of rats. Toxicology. 1996;109:49–55.[↩][↩]
- McNeill Consumer Products Co . Chemet Product Information. McNeill Consumer Products Co; Fort Washington, PA, USA: 1994.[↩][↩]
- Winship KA. Toxicity of aluminium: a historical review, Part 2. Adverse Drug React Toxicol. Rev. 1993;12:177–211[↩][↩]
- Hoffbrand AV, Cohen A, Hershko C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood. 2003;102:17–24[↩]
- Kattamis A, Ladis V, Berdousi H, Kelekis NL, Alexopoulou E, Papasotiriou I, Drakaki K, Kaloumenou I, Galani A, Kattamis C. Iron chelation treatment with combination therapy with deferiprone and deferioxamine: A 12-month trial. Blood Cell. Mol. Dis. 2006;36:21–25[↩]
- Cappellini MD, Pattoneri P. Oral iron chelators. Annu. Rev. Med. 2009;60:25–38[↩]
- Kodama H, Murata Y, Iitsuka T, Abe T. Metabolism of administered triethylene tetramine dihydrochloride in humans. Life Sci. 1997;61:899–907[↩]
- Bahnemann R, Leibold E, Kittel B, Mellert W, Jackh R. Different patterns of kidney toxicity after sub-acute administration of Na-nitrilotriacetic acid and Fe-nitrilotriacetic acid to Wistar rats. Toxicol. Sci. 1998;46:166–175.[↩]
- Tandon SK, Mathur AK. Chelation in Metal Intoxication. III. Lowering of Nickel Content in Poisoned Rat Organs. Acta Pharmacol. Toxicol. 1976;38:401–408[↩]
- Kaur G, Hasan SK, Srivastava RC. Effect of nitrilotriacetic acid (NTA) on the distribution of manganese-54 in rats. Arch. Toxicol. 1980;45:203–206.[↩]
- Anderson RL. The role of zinc in nitrilotriacetate (NTA)-associated renal tubular cell toxicity. Fd Cosmet. Toxicol. 1981;19:639–650[↩][↩][↩]
- Dietrich DR, Swenberg JA. Preneoplastic lesions in rodent kidney induced spontaneously or by non-genotoxic agents: Predictive nature and comparison to lesion induced by genotoxic carcinogens. Mutat. Res. 1991;248:239–260.[↩]
- Umemura T, Hasegawa R, Sai-Kato K, Nishikawa A, Furukawa F, Toyokum S, Uchida K, Inouc T, Kurokawa Y. Prevention by 2-mercaptoethane sulfonate and N-acetylcysteine of renal oxidative damage in rats treated with ferric nitriltriacetate. Jpn. J. Cancer Res. 1996;87:882–886[↩]
- EDTA chelation therapy alone and in combination with oral high-dose multivitamins and minerals for coronary disease: The factorial group results of the Trial to Assess Chelation Therapy. Lamas GA, Boineau R, Goertz C, Mark DB, Rosenberg Y, Stylianou M, Rozema T, Nahin RL, Terry Chappell L, Lindblad L, Lewis EF, Drisko J, Lee KL. Am Heart J. 2014 Jul; 168(1):37-44.e5.[↩][↩]
- Effect of disodium EDTA chelation regimen on cardiovascular events in patients with previous myocardial infarction: the TACT randomized trial. Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Lindblad L, Lewis EF, Drisko J, Lee KL, TACT Investigators. JAMA. 2013 Mar 27; 309(12):1241-50.[↩][↩]
- The effect of an EDTA-based chelation regimen on patients with diabetes mellitus and prior myocardial infarction in the Trial to Assess Chelation Therapy (TACT). Escolar E, Lamas GA, Mark DB, Boineau R, Goertz C, Rosenberg Y, Nahin RL, Ouyang P, Rozema T, Magaziner A, Nahas R, Lewis EF, Lindblad L, Lee KL. Circ Cardiovasc Qual Outcomes. 2014 Jan; 7(1):15-24.[↩][↩]
- Kim HC, Jang TW, Chae HJ, et al. Evaluation and management of lead exposure. Ann Occup Environ Med. 2015;27:30. Published 2015 Dec 15. doi:10.1186/s40557-015-0085-9 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4681084/[↩]
- Chelation in metal intoxication. Flora SJ, Pachauri V. Int J Environ Res Public Health. 2010 Jul; 7(7):2745-88.[↩]
- Lead toxicity and chelation therapy. Gracia RC, Snodgrass WR. Am J Health Syst Pharm. 2007 Jan 1; 64(1):45-53.[↩]
- Kim HC, Jang TW, Chae HJ, et al. Evaluation and management of lead exposure. Ann Occup Environ Med. 2015;27:30. Published 2015 Dec 15. doi:10.1186/s40557-015-0085-9[↩][↩]
- Bradberry S, Vale A. Dimercaptosuccinic acid (succimer; DMSA) in inorganic lead poisoning. Clin Toxicol (Phila) 2009;47:617–31. doi: 10.1080/15563650903174828.[↩]
- Sears ME. Chelation: harnessing and enhancing heavy metal detoxification–a review. ScientificWorldJournal. 2013;2013:219840. doi: 10.1155/2013/219840[↩][↩][↩]
- Flora SJ, Pachauri V. Chelation in metal intoxication. Int J Environ Res Public Health. 2010;7:2745–88. doi: 10.3390/ijerph7072745[↩]
- Shannon MW, Townsend MK. Adverse effects of reduced-dose d-penicillamine in children with mild-to-moderate lead poisoning. Ann Pharmacother. 2000;34:15–8. doi: 10.1345/aph.19084[↩]
- McKay CA., Jr Role of chelation in the treatment of lead poisoning: discussion of the Treatment of Lead-Exposed Children Trial (TLC) J Med Toxicol. 2013;9:339–43. doi: 10.1007/s13181-013-0341-8.[↩]




{kind=link}