Contents
- Congenital adrenal hyperplasia
- What are adrenal glands?
- Congenital adrenal hyperplasia types
- Congenital adrenal hyperplasia causes
- Congenital adrenal hyperplasia pathophysiology
- Congenital adrenal hyperplasia prevention
- Congenital adrenal hyperplasia symptoms
- Congenital adrenal hyperplasia complications
- Congenital adrenal hyperplasia diagnosis
- Congenital adrenal hyperplasia treatment
- Congenital adrenal hyperplasia prognosis
Congenital adrenal hyperplasia
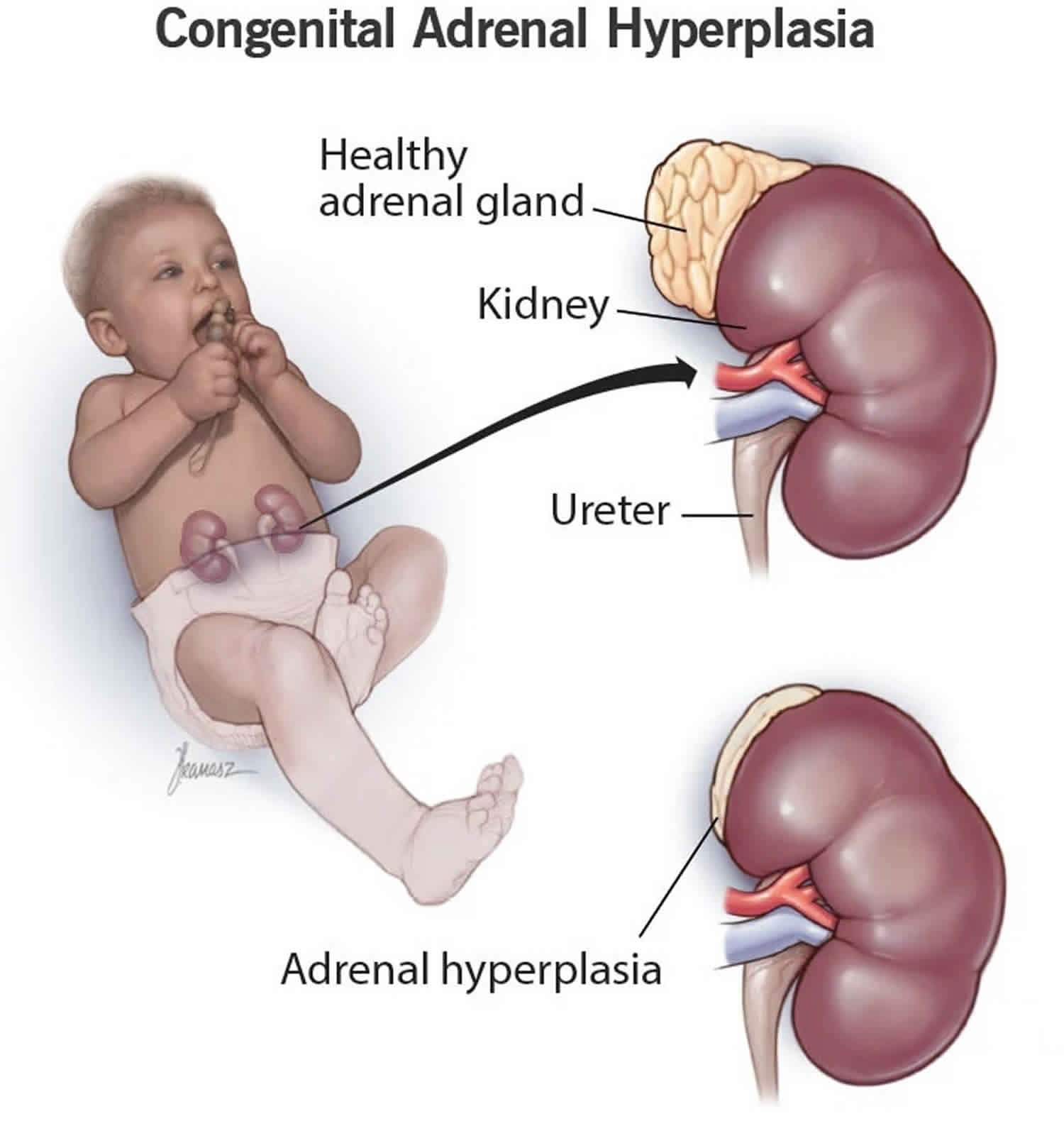
Congenital adrenal hyperplasia also called “CAH” or 21-hydroxylase deficiency (21OHD), is a group of rare inherited autosomal recessive disorders that affect the adrenal glands and it’s characterized by a deficiency of one of the enzymes needed to make specific hormones 1. Normally, the adrenal glands are responsible for producing three different hormones: corticosteroids (cortisol), which regulates energy, blood pressure and blood sugar and helps you recover from a sudden illness; mineralocorticoids (aldosterone), which regulate salt and water levels; and androgens, a group of hormones which control the development of male characteristics. People with CAH cannot properly make cortisol and in some cases, are unable to produce aldosterone. They also produce too much of some androgens, such as testosterone and 17-hydroxyprogesterone. These hormone imbalances (too little cortisol, too little aldosterone and excess androgens) can lead to serious illness, atypical genitalia, early puberty, growth concerns and other problems in infants, children, and adults. CAH can also cause imbalances in the hormone adrenaline, which affects blood sugar levels, blood pressure, and the body’s response to stress 2.
Approximately 90% to 99% of cases of congenital adrenal hyperplasia (CAH) are caused by 21-hydroxylase deficiency (21OHD) due to mutations in CYP21A2 gene 3, 4. Different mutations in the CYP21A2 gene responsible for 21-hydroxylase result in different levels of the enzyme, producing a spectrum of effects. Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency is broken down further into two subcategories: classical congenital adrenal hyperplasia, which can be sub-divided into the salt-losing form or the simple-virilizing form, and non-classical congenital adrenal hyperplasia.
There are two forms of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency (21OHD).
- Classical CAH. This is the most severe form of the disease and is less common. Classic CAH is usually detected at birth or in early infancy. In people with classical CAH, the body fails to produce any cortisol, which is needed to regulate blood pressure, blood sugar, and help your body respond to illness or injury. Many children with classical CAH are also unable to secrete aldosterone or able to maintain an adequate amount of salt in their bodies, a situation that can lead to severe dehydration and even death if left untreated. Classic CAH is usually detected at birth through routine newborn screening or when babies have atypical genitalia. CAH may also be identified when male or female babies show signs of severe illness due to low levels of cortisol, aldosterone or both. Classic CAH can be caused by either 21-hydroxylase or 11-hydroxylase deficiency.
- Nonclassical CAH also called late-onset CAH. This is the most common form of CAH and is mild in presentation. The body makes enough cortisol, but too much of certain androgens, like testosterone and 17-hydroxyprogesterone. Aldosterone secretion is normal in people with nonclassical CAH. Nonclassic CAH may not be identified until childhood or early adulthood. In children who have nonclassic CAH, signs and symptoms of early puberty may appear. If you have concerns about your child’s growth or development, make an appointment with your child’s doctor.
Since the absence of 21-hydroxylase makes these individuals unable to make the hormone cortisol and, in the case of salt-losing CAH, aldosterone, the body produces more androgens which cause a variety of symptoms such as abnormal genital development in infant girls. There are other much rarer forms of CAH as well, including 11-beta hydroxylase deficiency (11B-HD), 17a-hydroxylase deficiency (17a-HD), 3-beta-hydroxysteroid dehydrogenase deficiency (3B-HSD), congenital lipoid adrenal hyperplasia and p450 oxidoreductase deficiency which all present different symptoms.
Congenital adrenal hyperplasia can affect both boys and girls. About 1 in 10,000 to 18,000 children are born with congenital adrenal hyperplasia 5.
Congenital adrenal hyperplasia signs and symptoms present in each person depend on many factors including the type of congenital adrenal hyperplasia someone has (depending on which gene is affected and the level of enzyme deficiency), their age when the disorder is diagnosed, and the sex of the affected person.
Classical congenital adrenal hyperplasia. Children with classical congenital adrenal hyperplasia may develop an “adrenal crisis” which produces symptoms including:
- Vomiting
- Severe dehydration
- Low blood pressure
- Life-threatening shock
In classical CAH, the body also overproduces androgens, which can cause:
- Newborn girls may have atypical genitalia (genitals that look more male than female)
- Other signs and symptoms of classic CAH in infants include:
- an enlarged penis for boys
- poor weight gain or weight loss
- dehydration
- vomiting
- Older children to have early puberty, rapid growth, short adult height and difficulties with fertility
Nonclassical congenital adrenal hyperplasia. Children with nonclassical congenital adrenal hyperplasia may have:
- Early puberty
- Oily skin
- Acne
- Rapid growth during adolescence
- Short adult height
- Irregular periods (in women)
- Fertility problems
Children and adults with either type of CAH can have:
- rapid growth and early puberty, followed by shorter than average final height 6
- irregular menstrual cycles
- infertility
- excessive facial or body hair, and a deep voice in females
- acne, which can be severe
At any age, people with CAH are vulnerable to an ‘adrenal crisis‘, in which they feel very ill, weak and tired, and start vomiting. An adrenal crisis usually occurs during times of physical stress.
Adrenal crisis can be prevented with early treatment, so it is important to be able to recognize the signs and see your doctor immediately.
Although CAH is not curable, as long as patients receive adequate care and treatment, they can go on to lead normal lives.
Your child’s doctor will likely refer your child to a specialist in childhood hormonal issues (pediatric endocrinologist) for treatment of CAH. The health care team may also include other specialists, such as a urologist, psychologist, gynecologist and geneticist. Treatment may include medications, reconstructive surgery and psychological support.
The goal of congenital adrenal hyperplasia treatment is to return hormone levels to normal, or near normal. This is done by taking a form of cortisol, most often hydrocortisone. People may need additional doses of medicine during times of stress, such as severe illness or surgery. Steroids used to treat congenital adrenal hyperplasia do not usually cause side effects such as obesity or weak bones, because the doses replace the hormones that the child’s body cannot make. It is important for parents to report signs of infection and stress to their child’s provider because the child may need more medicine. Steroids cannot be stopped suddenly because doing so may lead to adrenal insufficiency.
Your doctor will also determine the genetic sex of the baby with abnormal genitalia by checking the chromosomes (karyotyping).
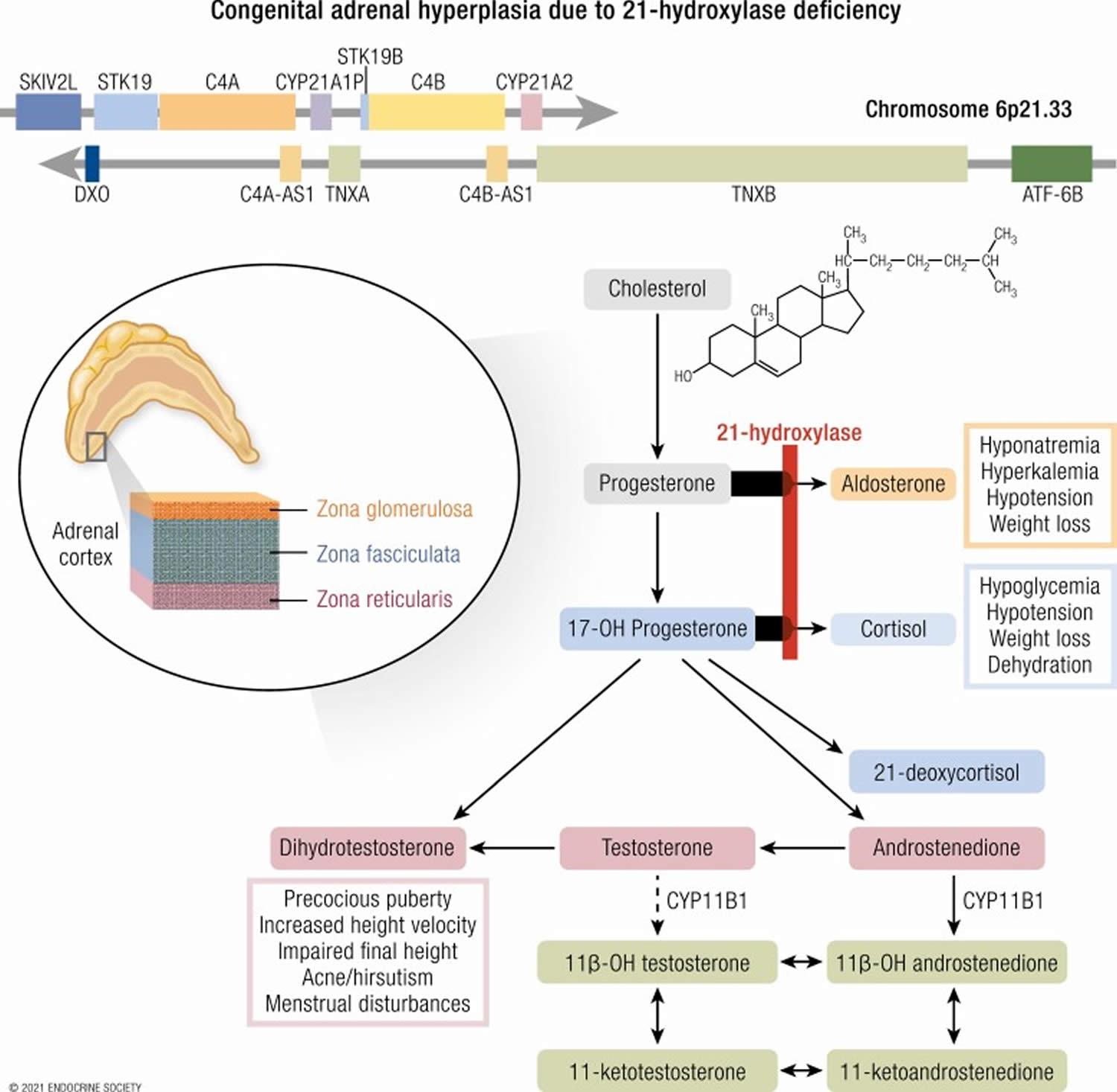
Figure 1. Congenital adrenal hyperplasia 21 hydroxylase deficiency
If my child has congenital adrenal hyperplasia can he/she go to day care and school?
Yes, children with CAH can attend day care and school. Before enrolling children in day care, parents should explain that the child has adrenal insufficiency, which might require the administration of emergency medication. Parents should discuss the day care provider’s policy on giving medications to children. They should also provide a set of written instructions, as well as a list of emergency contact names and numbers.
Before a child starts school, parents should consider meeting with the teacher, principal, and school nurse to explain the child’s condition. Parents can also discuss precautions to take if the child becomes ill.
I have CAH and want to start a family. What should I be thinking about?
Anyone with CAH, or from a family in which CAH has been diagnosed, should consider genetic counseling. Genetic counselors discuss all options for having a child. They explain the risks and benefits of each option.
The genes for CAH are passed down from parents to their children. In general, people have two copies of every gene in their bodies. They receive one copy from each parent. For an infant to have CAH, both copies must have an error that affects an adrenal-gland enzyme.
CAH is an example of an autosomal recessive disorder:
- Autosomal means the gene is not on the X chromosome or Y chromosome.
- Recessive means that both copies of the gene must have the error for the disease or disorder to occur.
If both parents have CAH, all of their children also will have it. If each parent carries one affected gene and one normal gene, there is a one in four chance of a child having CAH.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://findageneticcounselor.nsgc.org) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://www.acmg.net/ACMG/Directories.aspx) has a searchable database of medical genetics clinic services in the United States.
For women
Women with CAH can get pregnant. Women with CAH who become pregnant should continue taking their medications.
In some of the women, high levels of androgens disrupt the regular release of the egg from the ovary, a process known as ovulation. Some women also have irregular menstrual cycles. These problems can make it more difficult to get pregnant. These women often can be helped with medicines. Women with CAH who want to become pregnant can meet with a reproductive endocrinologist. This is a doctor who specializes in fertility issues.
For men
Men with CAH can father children. The main challenges for these men are low testosterone (a hormone important for male fertility and sexual function), and growths in the testicles called testicular adrenal rests tumors (TART). These problems can cause reduced sperm production. These issues tend to occur when hormone imbalances are not well controlled with medicines. Men who wish to father children should take all medicines as directed. A health care provider may recommend that males with CAH who have gone through puberty get an ultrasound of the testicles. The ultrasound provides a picture of the inside of the testicle and can help a health care provider detect abnormal growths. Future ultrasounds can be compared with the original to quickly identify any problems.
What are adrenal glands?
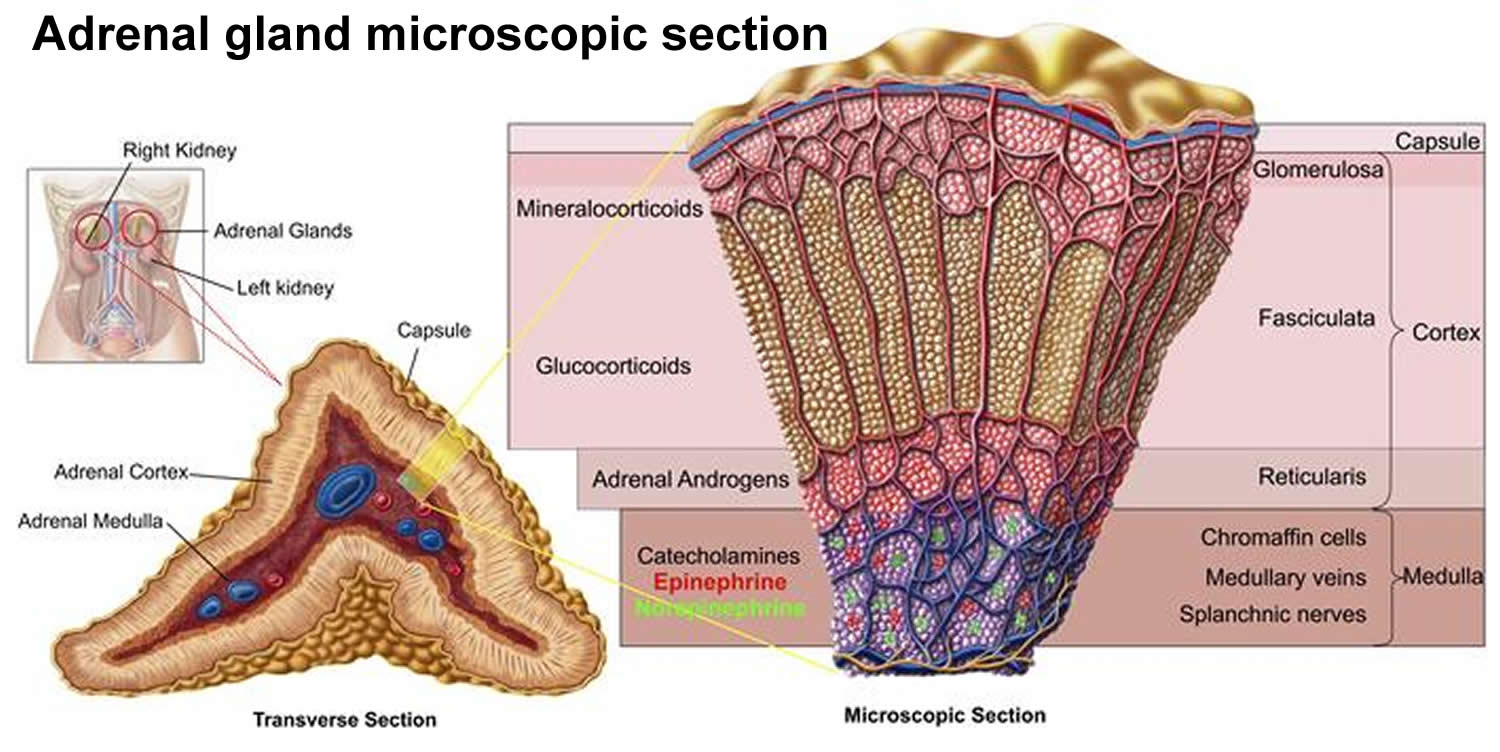
The adrenal glands are two two triangle-shaped glands that measure about 1.5 inches in height and 3 inches in length. They are located on top of each kidney like a cap and is embedded in the mass of fatty tissue that encloses the kidney (Figure 1 and 2). Their name directly relates to their location (ad—near or at; renes—kidneys) 7.
The adrenal glands, located on the top of each kidney, are responsible for releasing different classes of hormones.
The outer part of the adrenal gland, called the adrenal cortex, produces the hormones cortisol, aldosterone, and hormones that can be changed into testosterone 7. The inner part of the gland, called the adrenal medulla, produces the hormones adrenaline and noradrenaline. These hormones are also called epinephrine and norepinephrine 7.
These hormones—cortisol, aldosterone, adrenaline, and noradrenaline—control many important functions in the body, including:
- Maintaining metabolic processes, such as managing blood sugar levels and regulating inflammation.
- Regulating the balance of salt and water.
- Controlling the “fight or flight” response to stress.
- Maintaining pregnancy.
- Initiating and controlling sexual maturation during childhood and puberty.
The adrenal glands are also an important source of sex steroids, such as estrogen and testosterone.
When the adrenal glands produce more or less hormones than normal, you can become sick. This might happen at birth or later in life 8.
The adrenal glands can be affected by many diseases, such as autoimmune disorders, infections, tumors, and bleeding 8.
Conditions related to adrenal gland problems include:
- Addison disease (also called adrenal insufficiency)
- Congenital adrenal hyperplasia
- Cushing syndrome
- Diabetes – caused by another medical problem
- Glucocorticoid medicines
- Excessive or unwanted hair in women (hirsutism)
- Hump behind shoulders (dorsocervical fat pad)
- Hypoglycemia
- Primary aldosteronism (Conn syndrome)
- Waterhouse-Friderichsen syndrome
Location of adrenal glands
Figure 2. Location of the adrenal glands on top of each kidneys
Structure of the adrenal glands
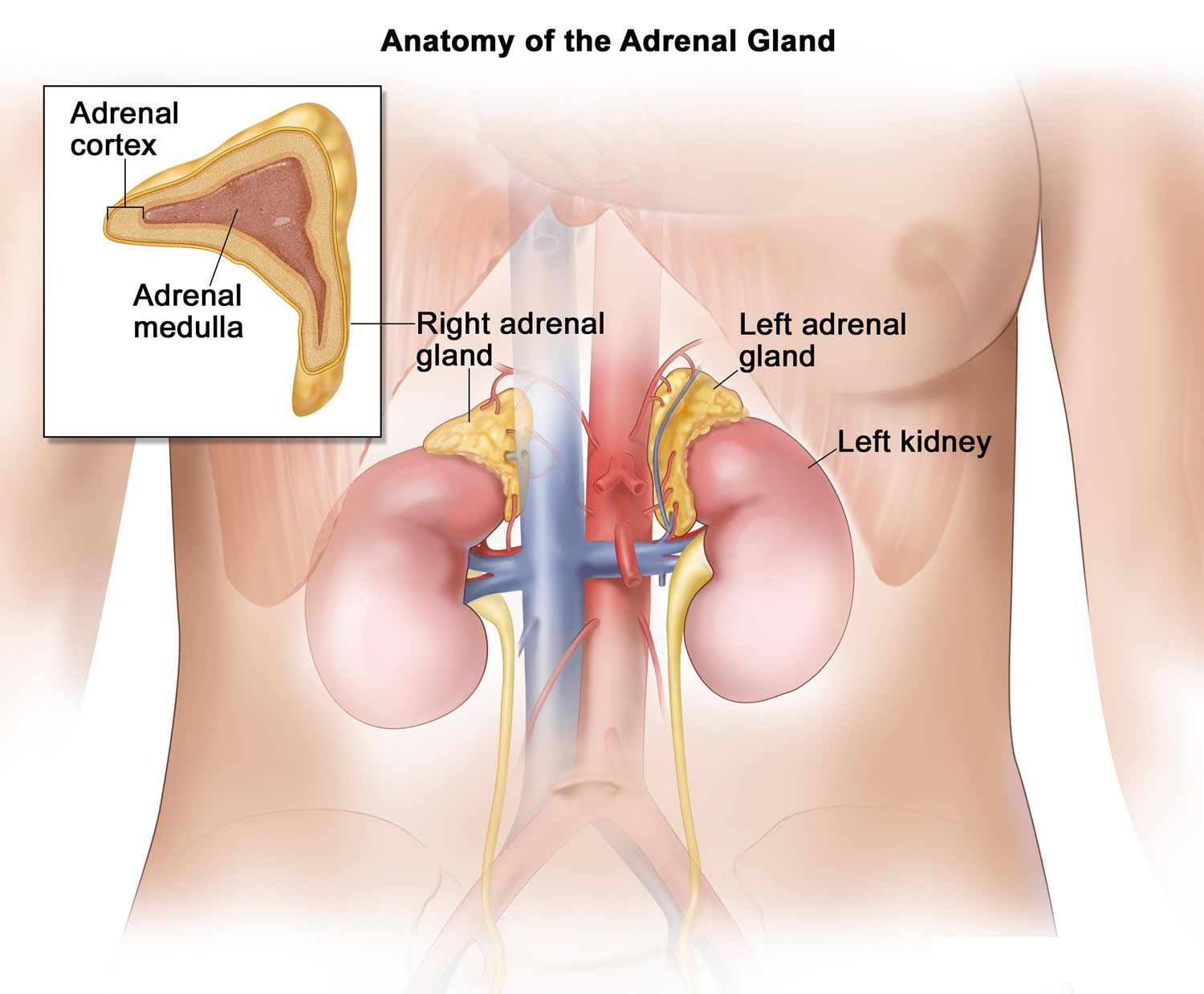
Each adrenal gland is about the size of the top part of the thumb. Each adrenal gland is very vascular and consists of two parts:
- The outer part is the adrenal cortex.
- The central portion is the adrenal medulla.
These regions are not sharply divided, but they are functionally distinct structures that secrete different hormones.
The adrenal cortex and the adrenal medulla have very different functions. One of the main distinctions between them is that the hormones released by the adrenal cortex are necessary for life; those secreted by the adrenal medulla are not.
The adrenal medulla consists of irregularly shaped cells organized in groups around blood vessels. These cells are intimately connected with the sympathetic division of the autonomic nervous system. Adrenal medullary cells are actually modified postganglionic neurons. Preganglionic autonomic nerve fibers control their secretions. The adrenal medulla produces epinephrine and norepinephrine. These hormones are also called adrenaline and noradrenaline.
The adrenal cortex, which makes up the bulk of the adrenal gland, is composed of closely packed masses of epithelial cells, organized in layers. These layers form the outer (glomerulosa), middle (fasciculata), and inner (reticularis) zones of the cortex (Figure 3). As in the adrenal medulla, the cells of the adrenal cortex are well supplied with blood vessels. The adrenal cortex produces steroid hormones such as cortisol, aldosterone, and hormones that can be changed into testosterone.
Figure 3. Adrenal gland microscopic section
Adrenal Cortex Hormones
The adrenal cortex produces two main groups of corticosteroid hormones—glucocorticoids and mineralcorticoids. The release of glucocorticoids is triggered by the hypothalamus and pituitary gland. Mineralcorticoids are mediated by signals triggered by the kidney.
The principle mineralcorticoid is aldosterone, which maintains the right balance of salt and water while helping control blood pressure. Aldosterone is a hormone produced by the adrenal glands and helps to control the amount of fluid in the body by affecting how much salt and water the kidney retains or excretes. The adrenal glands produce aldosterone in response to another hormone called renin. Renin is produced by specialised cells in the kidney that detect when the body lacks salt; renin released by the kidney signals the adrenal glands to release aldosterone. The kidney detects an increase in aldosterone in the bloodstream and responds by retaining extra salt rather than excreting it in the urine. As the body regains the salt it needs, the level of renin in the bloodstream drops and therefore the amount of aldosterone in the blood also falls, meaning more water is excreted in the urine. This is an example of a feedback system.
When the hypothalamus produces corticotrophin-releasing hormone (CRH), it stimulates the pituitary gland to release adrenal corticotrophic hormone (ACTH). These hormones, in turn, alert the adrenal glands to produce corticosteroid hormones.
Glucocorticoids released by the adrenal cortex include:
- 1) Cortisol: Also known as hydrocortisone, it regulates how the body converts fats, proteins, and carbohydrates to energy. It also helps regulate blood pressure and cardiovascular function.
- 2) Corticosterone: This hormone works with hydrocortisone to regulate immune response and suppress inflammatory reactions.
There is a third class of hormone released by the adrenal cortex, known as sex steroids or adrenal sex hormones. The adrenal cortex releases small amounts of male and female sex hormones. These hormones are male types (adrenal androgens), but some are converted to female hormones (estrogens) in the skin, liver, and adipose tissue. The amounts of adrenal sex hormones are very small compared to the supply of sex hormones from the gonads, but they may contribute to
early development of reproductive organs. However, their impact is usually overshadowed by the greater amounts of hormones (such as estrogen and testosterone) released by the ovaries or testes. Cells in the inner zone of the adrenal cortex produce sex hormones.
The more important actions of cortisol include:
- Inhibition of protein synthesis in tissues, increasing the blood concentration of amino acids.
- Promotion of fatty acid release from adipose tissue, increasing the utilization of fatty acids and decreasing the use of glucose as energy sources.
- Stimulation of liver cells to synthesize glucose from noncarbohydrates, such as circulating amino acids and glycerol, increasing the blood glucose concentration.
These actions of cortisol help keep blood glucose concentration within the normal range between meals. This control is important, because a few hours without food can exhaust the supply of liver glycogen, a major source of glucose.
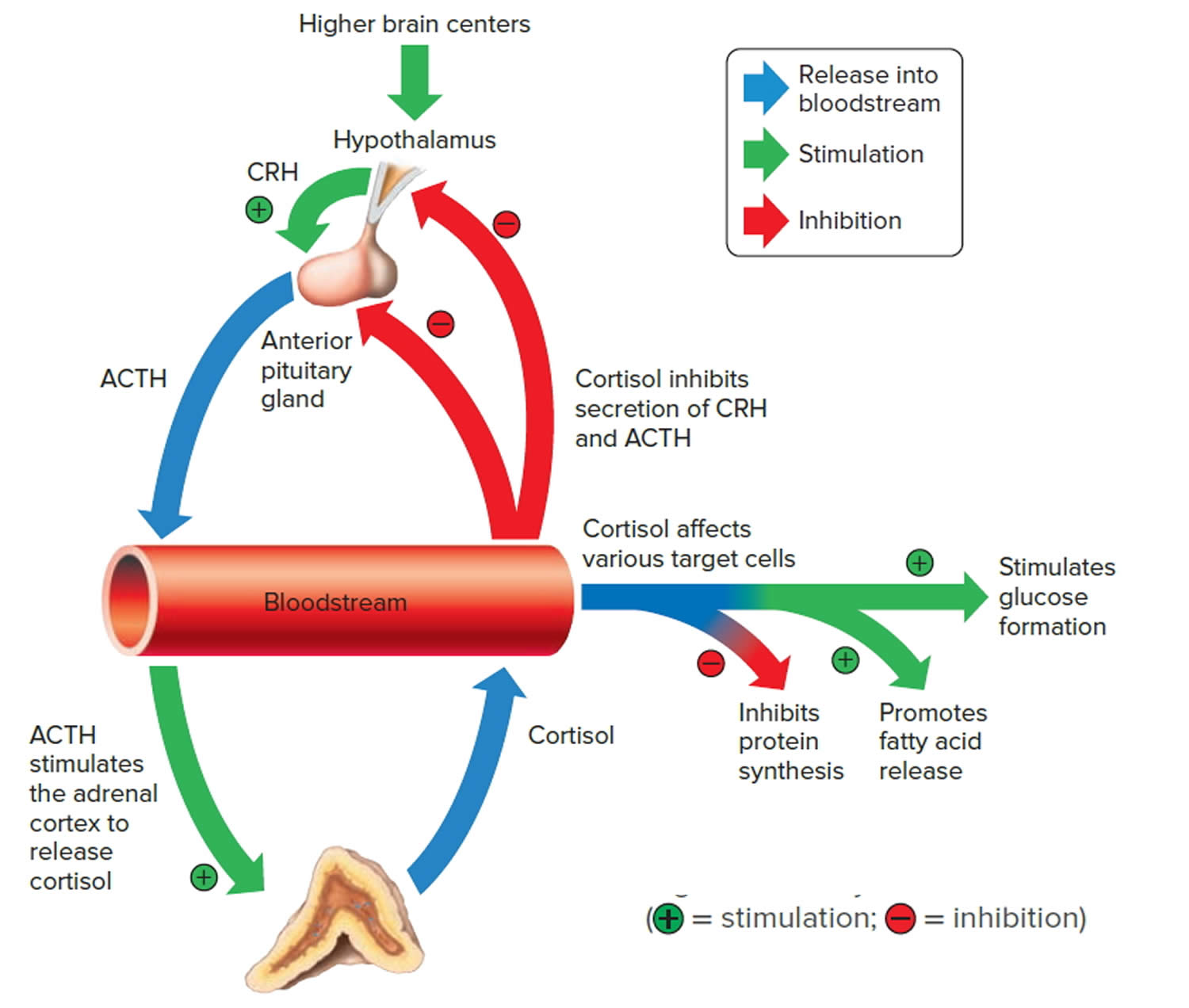
Negative feedback controls cortisol release (see Figure 4). This is much like control of thyroid hormones, involving the hypothalamus, anterior pituitary gland, and adrenal cortex. The hypothalamus secretes corticotropin-releasing hormone (CRH) into the pituitary gland portal veins, which carry CRH to the anterior pituitary, stimulating it to secrete adrenal corticotrophic hormone (ACTH). In turn, ACTH stimulates the adrenal cortex to release cortisol. Cortisol inhibits the release of CRH and ACTH, and as concentrations of these fall, cortisol production drops.
The set point of the feedback mechanism controlling cortisol secretion may change to meet the demands of changing conditions. For example, under stress—as from injury, disease, or emotional upset—information concerning the stressful condition reaches the brain. In response, brain centers signal the hypothalamus to release more corticotropin-releasing hormone (CRH), elevating the blood cortisol concentration until the stress subsides.
Figure 4. Negative feedback regulates cortisol secretion
Adrenal Medulla Hormones
Unlike the adrenal cortex, the adrenal medulla does not perform any vital functions. That is, you don’t need it to live 9. But that hardly means the adrenal medulla is useless. The hormones of the adrenal medulla are released after the sympathetic nervous system is stimulated, which occurs when you’re stressed. As such, the adrenal medulla helps you deal with physical and emotional stress.
You may be familiar with the fight-or-flight response—a process initiated by the sympathetic nervous system when your body encounters a threatening (stressful) situation. The hormones of the adrenal medulla contribute to this response.
Hormones secreted by the adrenal medulla are:
- Epinephrine: Most people know epinephrine by its other name—adrenaline. This hormone rapidly responds to stress by increasing your heart rate and rushing blood to the muscles and brain. It also spikes your blood sugar level by helping convert glycogen to glucose in the liver. (Glycogen is the liver’s storage form of glucose.)
- Norepinephrine: Also known as noradrenaline, this hormone works with epinephrine in responding to stress. However, it can cause vasoconstriction (the narrowing of blood vessels). This results in high blood pressure.
These adrenal medullary hormones have similar molecular structures and physiological functions. In fact, adrenaline (epinephrine), which makes up 80 percent
of the adrenal medullary secretion, is synthesized from noradrenaline (norepinephrine).
The effects of the adrenal medullary hormones resemble those of sympathetic neurons stimulating their effectors. The hormonal effects, however, last up to ten times longer than nervous stimulation because hormones are broken down more slowly than are neurotransmitters. Adrenaline (epinephrine) and noradrenaline (norepinephrine) increase heart rate, the force of cardiac muscle contraction, and blood glucose level. They also dilate airways, which makes breathing easier, elevate blood pressure, and decrease digestive activity.
Impulses arriving on sympathetic preganglionic nerve fibers stimulate the adrenal medulla to release its hormones at the same time that sympathetic impulses are stimulating other effectors. These sympathetic impulses originate in the hypothalamus in response to stress. In this way, adrenal medullary secretions function with the sympathetic division of the autonomic nervous system in preparing the body for energy-expending action, also called “fight-or-flight responses.”
Congenital adrenal hyperplasia types
In children with CAH, the adrenal glands lack certain enzymes needed to process cortisol and aldosterone. In close to 90% to 99% of cases , the missing enzyme is called 21-hydroxylase. CAH caused by this missing enzyme is called 21-hydroxylase deficiency (21OHD).
- Classical congenital adrenal hyperplasia is by far the more severe form with impaired cortisol production and can result in adrenal crisis and death if not detected and treated. The classic congenital adrenal hyperplasia occurs in 1 in 14,000 to 18,000 children based on newborn screening.
- Classic congenital adrenal hyperplasia can be sub-divided into the salt-losing form (most severe form due to little or no residual enzymatic activity, resulting in cortisol and aldosterone deficiency) or the simple-virilizing form.
- Salt-wasting form of classic CAH if not promptly treated, infants with this form of CAH quickly develop potentially fatal “salt-wasting crises” with hyponatremia (low blood sodium), hyperkalemia (low blood potassium), acidosis, and shock.
- Non-classical congenital adrenal hyperplasia sometimes called late-onset CAH is milder, and may or may not present symptoms.
Table 1. Types of Congenital Adrenal Hyperplasia
| Condition Onset Abnormality | Genitalia Mineralocorticoid Effect | Gene Chromosomal Location Typical Features |
|---|---|---|
| Lipoid CAH Congenital StAR Protein | Female, with no sexual development Salt wasting | StAR 8p11.2 All steroid products low |
| Lipoid CAH Congenital P450scc | Female, with no sexual development Salt wasting | CYP11A 15q23-24 All steroid products low |
| 3-beta-hydroxysteroid dehydrogenase deficiency (3B-HSD) Congenital 3-beta-hydroxysteroid dehydrogenase deficiency (3B-HSD) | Females virilized, males hypovirilized Salt wasting | HSD3B2 1p13.1 Elevated DHEA, 17-pregnenolone, low androstenedione, testosterone, elevated K, low Na, CO2 |
| 17α-OH deficiency Congenital P450c17 | Males hypovirilized, Hyperkalemic low-renin hypertension | CYP17 CYP17 10q24.3 Decreased androgens and estrogen, elevated DOC, corticosterone |
| Classic CAH, salt wasting Congenital P450c21 | Females prenatally virilized, males unchanged Salt wasting occurs in ¾ of 21OHD patients | CYP21 6p21.3 Elevated 17-hydroxyprogesterone (17-OHP), DHEA, and androstenedione, elevated K, low Na, CO2, low aldosterone, high plasma renin |
| Classic CAH, simple virilizing Congenital P450c21 | Females prenatally virilized, males unchanged No salt wasting | CYP21 6p21.3 Elevated 17-hydroxyprogesterone (17-OHP), DHEA, and androstenedione, normal electrolytes |
| Non-classic CAH Postnatal P450c21 | All with normal genitalia at birth, hyperandrogenism postnatally No salt wasting | CYP21 6p21.3 Elevated 17-hydroxyprogesterone (17-OHP), DHEA, and androstenedione on ACTH stimulation |
| 11β-OH deficiency Congenital P450c11B1 | Females virilized, males unchanged Low-renin hypertension | CYP11B1 8q24.3 Elevated DOC, 11-deoxycortisol (S); androgens, low K, elevated Na, CO2 |
| P450 Oxidoreductase deficiency (POR), Congenital P450 oxidoreductase | Males undervirilized, females unchanged Variable degree of mineralocorticoid deficiency | P450 Oxidoreductase gene (POR) 7q11.2 Combined and variable enzymatic defects of P450c21, P450c17 and P450aro Wide range of phenotypes: normal to genital ambiguity +/- skeletal abnormalities (Antley Bixler type) |
Classical congenital adrenal hyperplasia
Classical congenital adrenal hyperplasia is by far the more severe form with impaired cortisol production and can result in adrenal crisis and death if not detected and treated. Classic CAH is usually detected at birth through routine newborn screening or when babies have atypical genitalia. CAH may also be identified when male or female babies show signs of severe illness due to low levels of cortisol, aldosterone or both. Classic CAH can be caused by either 21-hydroxylase or 11-hydroxylase deficiency. The classic congenital adrenal hyperplasia occurs in 1 in 14,000 to 18,000 children based on newborn screening.
- Classic congenital adrenal hyperplasia can be sub-divided into the salt-losing form (most severe form due to little or no residual enzymatic activity, resulting in cortisol and aldosterone deficiency) or the simple-virilizing form.
- Salt-wasting form of classic CAH if not promptly treated, infants with this form of CAH quickly develop potentially fatal “salt-wasting crises” with hyponatremia (low blood sodium), hyperkalemia (low blood potassium), acidosis (metabolic acidosis), and shock.
- Symptoms of classic CAH due to 11-hydroxylase deficiency are similar to those of simple virilizing CAH 11. About two-thirds of people with classic 11-hydroxylase deficiency also have high blood pressure (hypertension) 12.
Signs and symptoms of classic CAH may include:
- Insufficient cortisol. Classic CAH causes the body to produce an insufficient amount of cortisol. This can cause problems maintaining normal blood pressure, blood sugar and energy levels, and cause problems during physical stress such as illness.
- Adrenal crisis. People with classic CAH can be seriously affected by a lack of cortisol, aldosterone or both. This is known as an adrenal crisis, and it can be life-threatening.
- Atypical genitalia. Female infants may have atypical genitalia appearance, such as an enlarged clitoris that may resemble a penis, and a partially closed labia resembling a scrotum. The urinary opening (urethra) and the vagina may be only one opening instead of two separate openings. The uterus, fallopian tubes and ovaries usually develop typically. Male infants usually have typical-appearing genitals.
- Excess androgen. An excess of the male sex hormone androgen can result in short height and early puberty for both males and females. Pubic hair and other signs of puberty may appear at a very early age. Severe acne also may occur. Excess androgen hormones in females may result in facial hair, excessive body hair and a deepening voice.
- Altered growth. Rapid growth may occur during childhood with an advanced bone age. Final height may be shorter than average.
- Fertility issues. These can include irregular menstrual periods, or not having any at all, and having infertility problems in females. Fertility issues can sometimes occur in males.
Salt-wasting CAH
Salt-wasting CAH is the severe form of classic 21-hydroxylase deficiency. In this type of CAH, the adrenal glands make too little aldosterone, causing the body to be unable to retain enough sodium (salt). Too much sodium is lost in urine (thus the name, “salt-wasting”). If undiagnosed, symptoms of classic salt-wasting CAH appear within days or weeks of birth and, in some cases, death occurs.
Salt-wasting CAH symptoms may include:
- Dehydration
- Poor feeding
- Diarrhea
- Vomiting
- Heart rhythm problems (arrhythmias)
- Low blood pressure (hypotension)
- Very low blood sodium levels (hyponatremia)
- Low blood glucose (hypoglycemia)
- Too much acid in the blood, called metabolic acidosis
- Weight loss
- Shock, a condition where not enough blood gets to the brain and other organs. Shock in infants with salt-wasting is called adrenal crisis. Signs include confusion, irritability, rapid heart rate, and/or coma.
Even when carefully treated, children with salt-wasting CAH are still at risk for adrenal crises when they become ill or are under physical stress. The body needs more than the usual amount of adrenal hormones during illness, injury, or physical stress. This means a child with CAH must be given more medication during these times to prevent an adrenal crisis.
Salt-wasting CAH also involves symptoms caused by low cortisol and high androgens. These symptoms may include:
- In female newborns, external genitalia can be ambiguous, i.e., not typical female appearing , with normal internal reproductive organs (ovaries, uterus, and fallopian tubes)
- Enlarged genitalia in male newborns
- Development of certain qualities called virilization in boys or girls before the normal age of puberty, sometimes as early as age 2 or 3. This is a condition characterized by:
- Rapid growth
- Appearance of pubic and armpit hair
- Deep voice
- Failure to menstruate, or abnormal or irregular menstrual periods (females)
- Well-developed muscles
- Enlarged penis (males)
- Unusually tall height as children, but being shorter than normal as adults
- Possible difficulties getting pregnant (females)
- Excess facial hair (females)
- Early beard (males)
- Severe acne
- Benign testicular tumors and infertility (males)
Simple virilizing (non-salt wasting) CAH
Simple virilizing CAH is the moderate form of classic 21-hydroxylase deficiency. This type of CAH involves less severe aldosterone deficiency. Therefore, there are no severe or life-threatening sodium-deficiency symptoms in newborns. Like salt-wasting CAH, simple virilizing CAH involves too little cortisol and too much androgen. Female newborns typically have ambiguous genitalia, and young children typically display virilization.
Non-classical congenital adrenal hyperplasia
Non-classical congenital adrenal hyperplasia sometimes called late-onset CAH is milder, and may or may not present symptoms. Often there are no symptoms of nonclassic CAH when a baby is born. Some people with nonclassic CAH never have symptoms. Some people have nonclassic CAH and never know it because the symptoms are so mild 13.
Nonclassic CAH is almost always caused by 21-hydroxylase deficiency. The condition is not identified on routine infant blood screening and usually becomes evident in late childhood or early adulthood. Cortisol may be the only hormone that’s deficient.
Females who have nonclassic CAH may have typical-appearing genitals at birth. Later in life, they may experience:
- Irregular menstrual periods, or not having any at all, and problems getting pregnant
- Masculine characteristics such as facial hair, excessive body hair and a deepening voice
- Fertility problems (in about 10% to 15% of women)
In both females and males, signs of nonclassic CAH may also include:
- Early appearance of pubic hair and other signs of early puberty
- Severe acne
- Rapid growth during childhood with an advanced bone age and shorter than expected final height
- Male-pattern baldness (hair loss near the temples)
- Enlarged penis (males)
- Small testicles (males)
Congenital adrenal hyperplasia causes
Congenital adrenal hyperplasia is a genetic disorder, which means it’s inherited from parents and is present at birth. The most common cause of CAH is the lack of the enzyme known as 21-hydroxylase due to mutations in CYP21A2 gene 3, 4. In CAH due to 21-hydroxylase deficiency, the adrenal glands cannot make enough cortisol or aldosterone. In addition, the glands make too much androgen. People with 21-hydroxylase deficiency also may not produce enough adrenaline 14.
CAH caused by 21-hydroxylasse deficiency can affect both boys and girls equally. Different mutations in the CYP21A2 gene responsible for 21-hydroxylase result in different levels of the enzyme, producing a spectrum of effects. One in 10,000 to 18,000 children are born with classical CAH, while the nonclassical form is much more common.
Children who have congenital adrenal hyperplasia (CAH) have two parents who either have CAH themselves or who are both carriers of the genetic change that causes the condition. This is known as the autosomal recessive inheritance pattern. In order for a child to be born with CAH, both parents must be carriers of the mutated gene and pass it on to their baby (Figure 5).
Factors that increase the risk of having CAH include:
- Parents who both have CAH or are both carriers of the genetic change for the disorder
- Being of Ashkenazi Jewish, Latino, Mediterranean, Yugoslav or Yup’ik ancestry
There are other much rarer enzyme deficiencies that also cause CAH including 11-beta hydroxylase deficiency (11B-HD), 17a-hydroxylase deficiency (17a-HD), 3-beta-hydroxysteroid dehydrogenase deficiency (3B-HSD), congenital lipoid adrenal hyperplasia and p450 oxidoreductase deficiency which all present different symptoms.
Congenital adrenal hyperplasia inheritance
The genes for CAH are passed down from parents to their children. In general, people have two copies of every gene in their bodies. They receive one copy from each parent. For an infant to have CAH, both copies must have an error that affects an adrenal-gland enzyme.
Congenital adrenal hyperplasia (CAH) is an example of an autosomal recessive disorder:
- Autosomal means the gene is not on the X chromosome or Y chromosome.
- Recessive means that both copies of the gene must have the error for the disease or disorder to occur.
If both parents have CAH, all of their children will also have it. If each parent carries one affected gene and one normal gene (called a “carrier”), there is a one-in-four chance of their child having CAH.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 5 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 5. Congenital adrenal hyperplasia inheritance
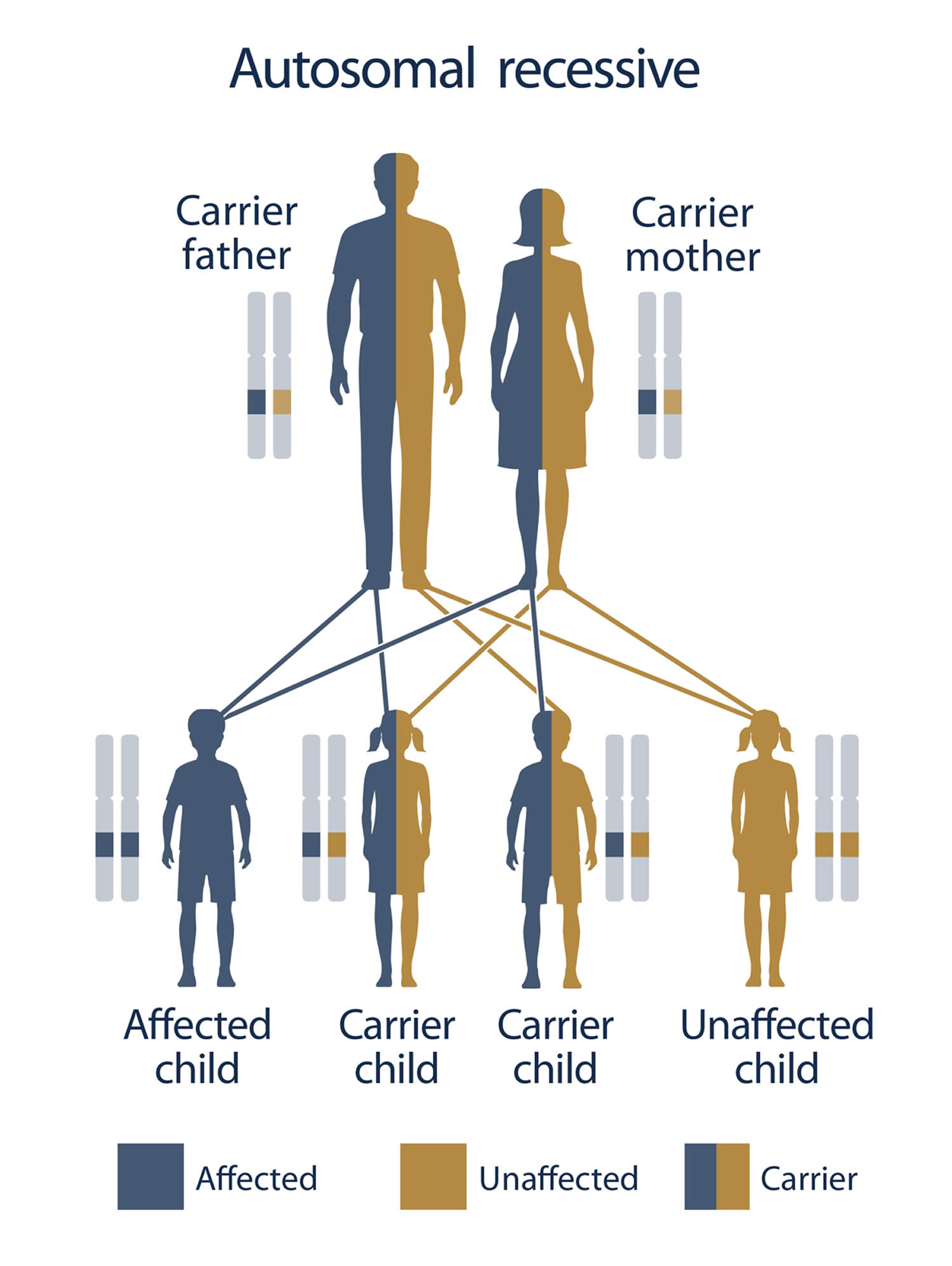
Footnotes: To have an autosomal recessive disorder, you inherit two mutated genes, one from each parent (who are carriers of the mutated gene). These disorders are usually passed on by two carriers. Carriers of the mutated gene is rarely affected, but they have one mutated gene (recessive gene) and one normal gene (dominant gene) for the condition. Two carriers have a 25% chance of having an unaffected child with two normal genes (left), a 50% chance of having an unaffected child who also is a carrier (middle), and a 25% chance of having an affected child with two recessive genes (right).
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Congenital adrenal hyperplasia pathophysiology
Steroid synthesis in the adrenal cortex takes place in 3 concentric zones: the outermost zona glomerulosa (mineralocorticoid biosynthesis), the zona fasciculata (glucocorticoid biosynthesis), and the innermost zona reticularis (sex steroid precursor biosynthesis) (see Figure 3). It entails conversion of cholesterol to active steroid hormones, and involves many enzymes, cofactors, and accessory proteins (Figures 1 and 6). Glucocorticoids (particularly cortisol), androgens, and estrogens are synthesized in the zona fasciculata and reticularis; and aldosterone in the zona glomerulosa.
The hypothalamus-pituitary-adrenal (HPA) feedback system is mediated through the circulating level of plasma cortisol by negative feedback of cortisol on corticotropin-releasing hormone (CRH) and adrenal corticotrophic hormone (ACTH) secretion. Therefore, a decrease in cortisol secretion leads to increased adrenal corticotrophic hormone (ACTH) production, which in turn stimulates (1) excessive synthesis of adrenal products in those pathways unimpaired by the enzyme deficiency and (2) an increase of precursor molecules in pathways blocked by the enzyme deficiency.
The clinical symptoms of the five different forms of CAH result from the particular hormones that are deficient and those that are produced in excess as outlined in Table 1 above. In 21-hydroxylase deficiency-CAH, there is an accumulation of 17-hydroxyprogesterone (17-OHP), a precursor to the 21-hydroxylation step, which is then shunted into the intact androgen pathway, where the 17,20-lyase enzyme converts the 17-hydroxyprogesterone (17-OHP) to D4-androstenedione, which is converted into androgens. Mineralocorticoid deficiency is a feature of classic salt wasting-CAH, the most severe form of CAH. The enzyme defect in non-classic-CAH is only partial and salt wasting in this mild form of the disease does not occur. The analogy of all other enzyme deficiencies in terms of precursor retention and product deficiencies are shown in Table 1 above.
The clinical manifestations of each form of congenital adrenal hyperplasia are related to the degree of cortisol deficiency and/or the degree of aldosterone deficiency. In some cases, these manifestations reflect the accumulation of precursor adrenocortical hormones. When present in supraphysiologic concentrations, these precursors lead to excess androgen production with resultant virilization, or because of mineralocorticoid properties, cause sodium retention and hypertension.
Figure 6. Adrenal steroidogenesis
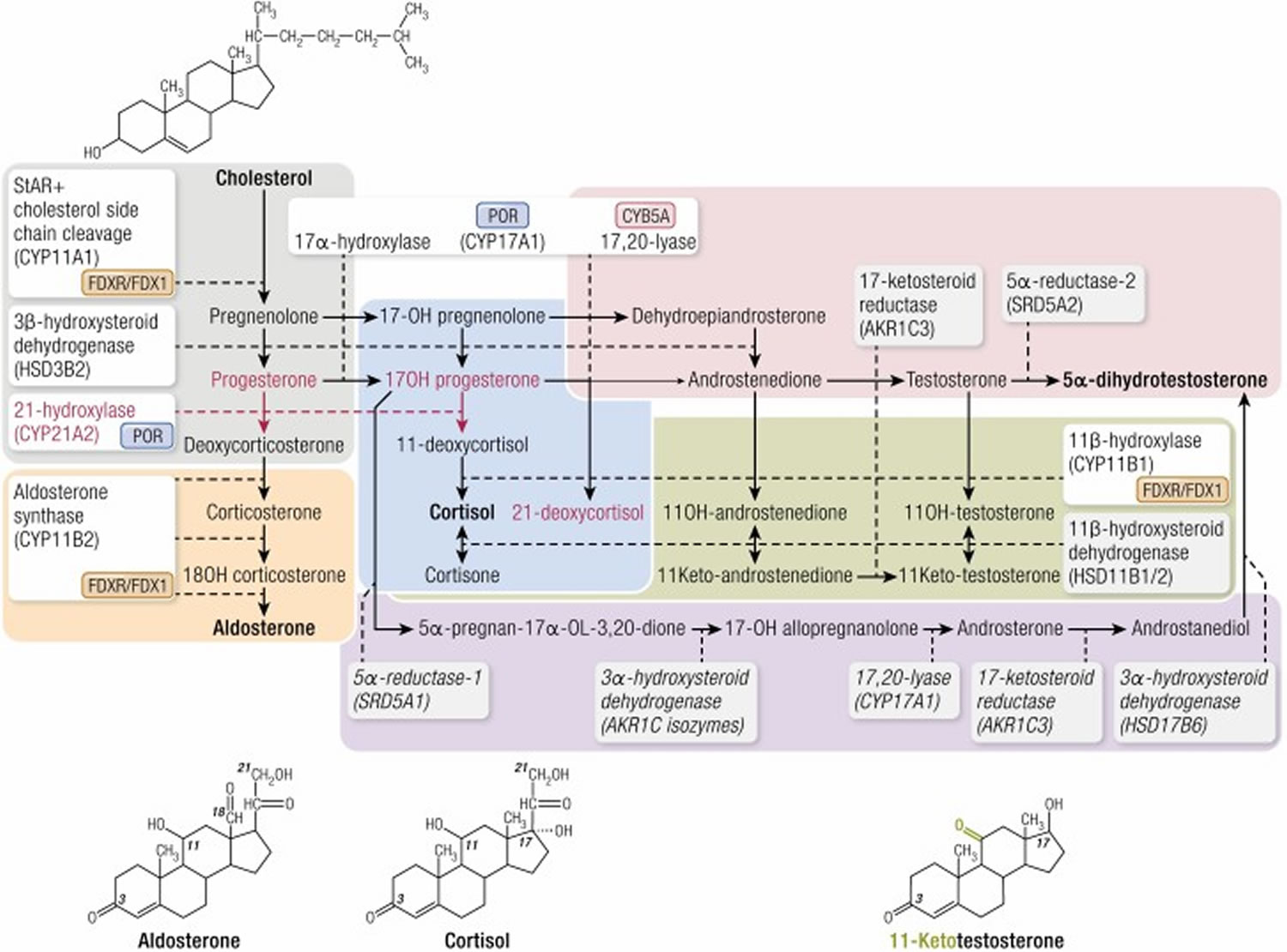
Footnotes: Adrenal steroidogenesis occurs in three major pathways: glucocorticoids, mineralocorticoids, and sex steroids. Enzymes are boxed with dotted lines extending to arrows denoting each enzymatic conversion; 2 enzymes, CYP11B2 and CYP17, catalyze several successive enzymatic conversions. Accessory proteins required for activity of cytochrome P450 enzymes are shown next to each such enzyme: POR, P450 oxidoreductase, required by CYP enzymes in the endoplasmic reticulum; FDXR/FDX1, ferredoxin reductase and ferredoxin, required by mitochondrial CYP enzymes. Cytochrome B5 (CYP5A) is required for full 17,20-lyase activity of CYP17A1. There are 2 11β-hydroxysteroid dehydrogenase isozymes; HSD11B1, expressed mainly in the liver, catalyzes reduction (eg, cortisone to cortisol), whereas HSD11B2, expressed mainly in the kidney, catalyzes oxidation (eg, cortisol to cortisone). The steps affected by 21-hydroxylase deficiency (21OHD), including steroids secreted in increased amounts in this disease, are denoted by red lines and red lettering. Steps taking place only in the adrenal glands are in unshaded boxes; steps taking place partly or predominantly outside the adrenal cortex are denoted by shaded boxes. Planar structures of cholesterol, aldosterone, cortisol, and testosterone are illustrated; the position of the 11-oxo (11-keto) group in 11-ketotestosterone is illustrated in green. Colored rectangles indicate the following: gray, early steps of steroidogenesis common to all zones of the cortex; orange, steps in the zona glomerulosa leading to aldosterone; blue, steps in the zona fasciculata leading to cortisol; magenta; steps in the zona reticularis and extra-adrenal tissues leading to androgens; purple, the “backdoor” or alternate pathway from 17-hydroxyprogesterone (17-OH progesterone) to dihydrotestosterone (for clarity, the alternative pathway from progesterone is not shown); green, conversions leading to 11-oxo androgens.
[Source 1 ]Congenital adrenal hyperplasia prevention
There is no known way to prevent CAH. If you’re thinking of starting a family and you’re at risk of having a child with CAH, you should consider genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Prenatal diagnosis is available for some forms of congenital adrenal hyperplasia. Diagnosis is made in the first trimester by chorionic villus sampling. Diagnosis in the second trimester is made by measuring hormones such as 17-hydroxyprogesterone in the amniotic fluid.
A newborn screening test is available for the most common form of congenital adrenal hyperplasia. It can be done on heel stick blood (as part of the routine screenings done on newborns). This test is currently performed in most states.
Congenital adrenal hyperplasia symptoms
Congenital adrenal hyperplasia signs and symptoms present in each person depend on many factors including the type of congenital adrenal hyperplasia someone has, their age when the disorder is diagnosed, and the sex of the affected person.
- Children with milder forms may not have signs or symptoms of congenital adrenal hyperplasia and may not be diagnosed until as late as adolescence.
- Girls with a more severe form often have masculinized genitals at birth and may be diagnosed before symptoms appear.
- Boys will appear normal at birth, even if they have a more severe form.
In children with the more severe form of the disorder, symptoms often develop within 2 or 3 weeks after birth.
- Poor feeding or vomiting
- Dehydration
- Electrolyte changes (abnormal levels of sodium and potassium in the blood)
- Abnormal heart rhythm
Girls with the milder form will usually have normal female reproductive organs (ovaries, uterus, and fallopian tubes). They may also have the following changes:
- Abnormal menstrual periods or failure to menstruate
- Early appearance of pubic or armpit hair
- Excessive hair growth or facial hair
- Some enlargement of the clitoris
Boys with the milder form often appear normal at birth. However, they may appear to enter puberty early. Symptoms may include:
- Deepening voice
- Early appearance of pubic or armpit hair
- Enlarged penis but normal testes
- Well-developed muscles
Both boys and girls will be tall as children, but much shorter than normal as adults.
Congenital adrenal hyperplasia complications
Congenital adrenal hyperplasia complications may include:
- High blood pressure (hypertension)
- Low blood sugar (hypoglycemia)
- Low sodium (hyponatremia)
People who have classic CAH are at risk of adrenal crisis. This is a life-threatening medical emergency that requires immediate treatment. Adrenal crisis can occur within the first few days after birth. It can also be triggered at any age by infectious illness or physical stress such as surgery.
Very low levels of cortisol in the blood can cause diarrhea, vomiting, dehydration, low blood sugar levels, seizures and shock. Aldosterone also may be low, which leads to dehydration and low sodium and high potassium levels. The nonclassic form of CAH doesn’t cause adrenal crisis.
People who have either classic or nonclassic CAH may experience fertility problems.
Congenital adrenal hyperplasia diagnosis
All infants born in the United States are screened for congenital adrenal hyperplasia through a blood test. It is performed by measuring the 17-hydroxyprogesterone level in a tiny blood spot obtained on a filter paper by heel prick of newborns usually at 24 to 48 hours of life (the time of the collection varies in each state). The screening test is performed by the laboratory of Department of Public Health in most state’s regional programs. Check with your regional Department of Public Health on newborn screening for CAH for current status.
If your child is diagnosed with CAH, his physician may order additional blood tests, as well as other tests, including ultrasounds or X-rays of the abdomen and pelvis, to view the adrenal glands and the structure of your child’s internal genitalia. Common blood tests include:
- Serum electrolytes
- Aldosterone
- Renin
- Cortisol
X-ray of the left hand and wrist may show that the child’s bones appear to be those of someone older than their actual age.
Genetic tests can help diagnose or confirm the diagnosis, but they are rarely needed.
Prenatal testing during pregnancy
If a woman already has a child with CAH and becomes pregnant with the same partner, her fetus has a one in four chance of having CAH (see Figure 5). For this reason, prenatal testing can be done for some forms of CAH. A health care provider checks for the disorder by using techniques called amniocentesis or chorionic villus sampling.
Tests used to identify CAH in fetuses who are at risk for the disorder include:
- Amniocentesis. This procedure involves inserting a needle into the womb, through the abdomen, to withdraw a small amount of amniotic fluid from the womb and then examining the cells. The procedure is usually done between the 15th and 20th week of pregnancy.
- Chorionic villus sampling. This test involves withdrawing cells from the placenta for examination. A health care provider inserts a needle into the womb, either through the abdomen or the cervix, and extracts a small piece of tissue from the chorionic villi (the tissue that will later become the placenta). This procedure is usually done between the 10th and 12th week of pregnancy.
After a health care provider takes a sample using one of these techniques, he or she will perform a genetic test on the sample. This test will reveal whether the fetus has a gene change that causes CAH.
Tests to confirm the diagnosis of CAH are done after the baby is born.
Parents may also choose to wait until birth to have the newborn tested. Talking to your doctor may help you identify the option that is right for you.
Newborns and infants
In the United States and many other countries, routine screening of all newborns for genetic 21-hydroxylase deficiency is recommended during the first few days of life. This test identifies the classic form of CAH but doesn’t identify the nonclassic form.
In female infants who have severe atypical genitalia, tests can be done to analyze chromosomes to identify genetic sex. Also, pelvic ultrasound can identify the presence of female reproductive structures such as the uterus and ovaries.
Children and young adults
Diagnosis of CAH in children and young adults includes:
- Physical exam. Your health care provider will do a physical exam, check your child’s blood pressure and heart rate, and review symptoms to identify possible CAH. The next step is to confirm the diagnosis with blood and urine tests.
- Blood and urine tests. These tests look for hormones produced by the adrenal glands at levels outside the standard ranges. The tests also check the levels of electrolytes. These are minerals such as sodium that balance the amount of water in the body.
- Genetic testing. Genetic testing may be needed to diagnose CAH.
An X-ray can help to diagnose CAH in children. Because some children with CAH grow too quickly, their bones will be more developed than normal for their age.
Congenital adrenal hyperplasia treatment
Treatments for CAH include medication and reconstructive surgery as well as psychological support 15.
Medications
The goal of treating CAH with medications is to reduce excess androgen production and replace deficient hormones. People who have the classic form of CAH can successfully manage the condition by taking hormone replacement medications throughout their lives. If a patient stops taking his or her medication, symptoms will return.
People who have nonclassic CAH may not require treatment or may need only small doses of corticosteroids. Symptoms of nonclassic CAH that signal that the patient may need treatment are:
- Early puberty
- Excess body hair
- Irregular menstrual periods (females)
- Infertility
It may be possible for patients with nonclassic CAH to stop medication as adults if their symptoms go away.
Medications for CAH are taken daily. During periods of illness or significant stress, such as surgery, additional medications or higher doses may be needed.
Medications may include:
- Corticosteroids to replace cortisol. CAH requires lifelong treatment with cortisol. Your child will typically begin taking a synthetic cortisol or glucocorticoid (such as hydrocortisone, prednisone or dexamethasone) as a pill given by mouth every day. This pill replaces the cortisol your child cannot produce and also decreases the levels of androgens in the blood stream. When a child with CAH gets sick, is injured or needs surgery, she will need high doses of cortisol, called “stress doses.”
- Mineralocorticoids to replace aldosterone to help retain salt and get rid of excess potassium. If your child is unable to make aldosterone, her doctor may also prescribe an aldosterone replacement, which is also given as a pill once or twice a day.
- Salt supplements to help retain salt
Monitoring the effectiveness of medication includes regularly scheduled:
- Physical exams. The health care provider will check your child’s growth and development, including monitoring changes in height, weight, blood pressure and bone growth. Assessing health at regular visits is lifelong.
- Monitoring for side effects. The health care provider will monitor for medication side effects, such as the loss of bone mass and impaired growth, particularly if steroid-type replacement medication doses are high and used long term.
- Blood tests to check hormone levels. It’s critical to have regular blood tests to ensure that hormone levels are balanced. Children who haven’t yet reached puberty need enough cortisone to suppress androgens to grow to a typical height. For females who have CAH, it’s important to suppress androgens to minimize unwanted masculine characteristics. On the other hand, too much cortisone can cause Cushing syndrome. Cushing’s syndrome symptoms can include:
- Weight gain
- Slowed growth
- Stretch marks on the skin
- Rounded face
- High blood pressure
- Bone loss
- High blood sugar
People who have classic CAH need to wear a medical alert identification bracelet or necklace. To alert medical professionals in case of an emergency, the bracelet or necklace should read: “adrenal insufficiency, requires hydrocortisone.”
Adults or parents also need to learn how to give an injection of hydrocortisone if there is an emergency.
Reconstructive surgery
Some infant girls who have severe atypical genitalia because of classic CAH may require reconstructive surgery to improve genital function and provide a more typical appearance. In this situation, your child will be evaluated by a genitourinary surgeon. The genitourinary surgeon will meet with your family to counsel you on the different options available, discuss what surgery can do for your child, explain the risks and benefits of the procedure, and develop a treatment plan to meet your child’s individual needs.
Reconstructive surgery may involve reducing the size of the clitoris and reconstructing the vaginal opening. The surgery is typically performed between 2 and 6 months of age. Females who have reconstructive genital surgery may need more cosmetic surgery later in life.
Genital surgery is easier to perform when a child is very young. However, some parents choose to wait for surgery until their child is old enough to understand the risks and choose the gender assignment.
Before making decisions about the best treatment approach for your child, talk with your health care provider about these issues. Working together, you and your provider can make informed choices that will help your child thrive.
CAH girls with nonclassic CAH have normal genitals, so they do not need surgery.
Psychological support
Psychological support is important to the emotional health and social adjustment of children and adults who have CAH.
Early and steady support from family and health care professionals can help your child have healthy self-esteem and a satisfying social life. These approaches may help:
- Include psychological counseling in your child’s treatment plan as needed
- Seek help from a mental health professional if you’re having trouble coping and to help you develop healthy parenting strategies
Ongoing monitoring
Treatment of CAH can be challenging. Excessive treatment with glucocorticoids can lead to stunted growth, excessive weight gain and other long-term problems such as osteoporosis. Overtreatment with either glucocorticoids or fludrocortisone can result in hypertension.
On the other hand, under treatment usually results in elevated androgen levels. As a result, children may experience early puberty and a short window for growth with short adult height. In adolescent girls, undertreating may cause irregular periods and skin problems such as acne and excessive, male-pattern hair growth (hirsutism). In adolescent boys, poor CAH control can cause growth of testicular masses that resemble adrenal tissue, called testicular adrenal rests tumors (TART).
To mediate the risks of over and under treatment, a team of doctors will monitor children and teens with CAH for all potential complications associated with the disorder, including:
- Slow or rapid of growth
- Advanced bone age leading to shorter adult height
- Early or delayed puberty
- Increased body weight, insulin resistance and increased risk for type 2 diabetes
- High blood pressure
- Irregular menstruation in young girls
- Increased acne and facial hair growth in young girls
- Testicular adrenal rests tumors (TART) and decreased sperm count in young men
- Genetic counseling for affected families considering having a child
- Osteoporosis
Follow-up and transitioning to adult care
Babies and children with classical CAH are seen frequently depending on age and response to treatment. Follow-up care continues throughout childhood as doctots track your child’s height, weight, bone age and sexual development.
Long-term care may also include appointments with nutritionists, psychologists and counselors. Having all of these specialists work together on one team helps to deliver the best-coordinated care for your child.
Once your child reaches adulthood, your child will work closely with adult endocrinologists and adult infertility and reproductive endocrinologists to help make a seamless transition from pediatric to adult care.
Congenital adrenal hyperplasia prognosis
People with congenital adrenal hyperplasia must take medicine their entire life. With proper treatment, children with congenital adrenal hyperplasia can live normal lives and participate fully in school and other activities. However, they may be shorter than normal adults, even with treatment.
In some cases, congenital adrenal hyperplasia can affect fertility.
Girls with CAH may also grow and develop normally, have regular menstrual cycles and have children.
- Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, Flück CE, Guasti L, Huebner A, Kortmann BBM, Krone N, Merke DP, Miller WL, Nordenström A, Reisch N, Sandberg DE, Stikkelbroeck NMML, Touraine P, Utari A, Wudy SA, White PC. Congenital Adrenal Hyperplasia-Current Insights in Pathophysiology, Diagnostics, and Management. Endocr Rev. 2022 Jan 12;43(1):91-159. doi: 10.1210/endrev/bnab016[↩][↩][↩]
- Speiser, P.W. and White, P.C. (2003). Congenital Adrenal Hyperplasia. New England Journal of Medicine, 349, 776-788. https://www.nejm.org/doi/full/10.1056/NEJMra021561[↩]
- White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000 Jun;21(3):245-91. doi: 10.1210/edrv.21.3.0398. Erratum in: Endocr Rev 2000 Oct;21(5):550.[↩][↩]
- Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, Nordenström A. One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet Diabetes Endocrinol. 2013 Sep;1(1):35-42. doi: 10.1016/S2213-8587(13)70007-X Erratum in: Lancet Diabetes Endocrinol. 2013 Aug;1 Suppl 1:s22.[↩][↩]
- Congenital adrenal hyperplasia. https://medlineplus.gov/ency/article/000411.htm[↩]
- Muthusamy K, Elamin MB, Smushkin G, Murad MH, Lampropulos JF, Elamin KB, Abu Elnour NO, Gallegos-Orozco JF, Fatourechi MM, Agrwal N, Lane MA, Albuquerque FN, Erwin PJ, Montori VM. Clinical review: Adult height in patients with congenital adrenal hyperplasia: a systematic review and metaanalysis. J Clin Endocrinol Metab. 2010 Sep;95(9):4161-72. doi: 10.1210/jc.2009-2616[↩]
- US Department of Health and Human Services. National Institutes of Health. About Adrenal Gland Disorders. https://www.nichd.nih.gov/health/topics/adrenalgland/conditioninfo/Pages/default.aspx[↩][↩][↩]
- U.S. National Library of Medicine. Medline Plus. Adrenal glands. https://medlineplus.gov/ency/article/002219.htm[↩][↩]
- Vertical Health Endocrine Web. An Overview of the Adrenal Glands. https://www.endocrineweb.com/endocrinology/overview-adrenal-glands[↩]
- Yau M, Gujral J, New MI. Congenital Adrenal Hyperplasia: Diagnosis and Emergency Treatment. [Updated 2019 Apr 16]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279085[↩]
- Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency. https://medlineplus.gov/genetics/condition/congenital-adrenal-hyperplasia-due-to-11-beta-hydroxylase-deficiency[↩]
- C-11 Hydroxylase Deficiency. https://emedicine.medscape.com/article/117012-overview#showall[↩]
- Congenital Adrenal Hyperplasia (CAH). https://www.nadf.us/congenital-adrenal-hyperplasia-cah.html[↩]
- About Congenital Adrenal Hyperplasia. https://www.nichd.nih.gov/health/topics/cah/conditioninfo[↩]
- What are the treatments for congenital adrenal hyperplasia (CAH)? https://www.nichd.nih.gov/health/topics/cah/conditioninfo/treatments[↩]




{kind=link}