Contents
- Congenital erythropoietic porphyria
Congenital erythropoietic porphyria
Congenital erythropoietic porphyria (CEP) also called Gunther’s disease is an extremely rare inherited autosomal recessive disorder that involve defects in heme or ‘haem’ biosynthetic pathway caused by mutations (changes) in the UROS gene which results in low levels of the enzyme uroporphyrinogen III cosynthase or uroporphyrinogen-III synthase (UROS), the fourth enzyme in the heme biosynthetic pathway 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20. The UROS (uroporphyrinogen III synthase or uroporphyrinogen III cosynthase) enzyme is involved in the production of a molecule called heme (haem). Heme is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). The production of heme is a multi-step process that requires 8 different enzymes. In the presence of insufficient uroporphyrinogen III synthase (UROS) activity, hydroxymethylbilane (HMB), the product of the third step spontaneously closes and forms the non-physiological compound uroporphyrinogen I, which is then acted upon by uroporphyrinogen decarboxylase (UROD) the fifth enzyme in the heme synthetic pathway to form coproporphyrinogen I 21. Coproporphyrinogen I then oxidizes spontaneously to form coproporphyrin I and uroporphyrin I, two harmful and non-physiological compounds associated with abnormalities involving various organs 1. In congenital erythropoietic porphyria (CEP), these toxic porphyrins mostly accumulate in red blood cell precursors (erythroblasts) in your bone marrow, as well as your teeth, bones, urine, and feces. These toxic porphyrins are not significantly elevated in your liver. Your bone marrow, the spongy tissue inside your bones, is essential for producing blood cells and supporting the immune system. Your bone marrow acts as a factory for red blood cells, white blood cells, and platelets, which are vital for oxygen transport, fighting infection, and blood clotting, respectively. Your bone marrow also plays a role in storing fat and supporting bone and cartilage health. As these toxic porphyrins accumulate in your bone marrow, they may be released into your blood and subsequently found in the plasma and mature red blood cells. They then may travel to your skin, and upon exposure to light, generate free radicals that can damage cells and tissues. This leads to disease characterized by severe skin photosensitivity and breakdown of red blood cells (hemolysis) 22. The major symptom of congenital erythropoietic porphyria (CEP) is hypersensitivity of your skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the photo-activated porphyrins in your skin cause skin friability and blistering (bullae) and the fluid-filled sacs may rupture and the lesions often get infected. These infected lesions can lead to scarring, bone loss, and in severe instances, this can lead to skin mutilation and deformities 23. Your face, ears, neck, arms, forearms and hands are the most commonly affected areas.
In most cases, congenital erythropoietic porphyria (CEP) is caused by UROS gene mutations located on chromosome 10q26.2 and follows an autosomal recessive inheritance pattern 24, 25. Typically, there is no family history of the disease. Neither parent has symptoms of congenital erythropoietic porphyria (CEP), but each carries a defective UROS gene that they can pass to their children. Affected offspring have two copies of the defective UROS gene, one inherited from each parent.
Rarely congenital erythropoietic porphyria (CEP) is caused by GATA1 gene mutation located on the X chromosome (chromosome Xp11.23) that affect males 26, 15, 27. Females who are heterozygous for GATA1 gene will be either asymptomatic or have a milder disease with predominantly hematologic abnormalities due to skewed X-chromosome inactivation 15, 27.
In a small percentage of congenital erythropoietic porphyria patients (>10%), a disease-causing mutation has not been detected in the UROS or GATA1 genes, raising the possibility that additional gene(s) may play a role in congenital erythropoietic porphyria pathogenesis 28.
Disease-causing mutations in either UROS gene or GATA1 gene result in absent or markedly reduced UROS (uroporphyrinogen III synthase) enzymatic activity. UROS (uroporphyrinogen III synthase) enzyme catalyzes the cyclization of hydroxymethylbilane (HMB) with concomitant inversion of ring D to yield uroporphyrinogen III (URO III) 29. Deficiency of UROS (uroporphyrinogen III synthase) enzyme results in accumulation of HMB (hydroxymethylbilane), most of which is converted nonenzymatically to non-physiological compound uroporphyrinogen I, which is then acted upon by uroporphyrinogen decarboxylase (UROD) the fifth enzyme in the heme synthetic pathway to form coproporphyrinogen I 21. Coproporphyrinogen I then oxidizes spontaneously to form the porphyrins coproporphyrin I and uroporphyrin I. In congenital erythropoietic porphyria (CEP), these toxic porphyrins mostly accumulate in red blood cell precursors in your bone marrow, as well as your teeth, bones, urine, and feces. These toxic porphyrins are not significantly elevated in your liver. Since these toxic porphyrins are photocatalytic compounds, exposure of the skin to sunlight and other sources of long-wave ultraviolet light elicits a phototoxic reaction, resulting in blistering and vesicle formation as well as increased friability of the skin and breakdown of red blood cells (hemolysis) 22, 30, 31.
Congenital erythropoietic porphyria (CEP) is a very rare genetic disorder affecting less than 1 in 1,000,000 children and it affects males and females in equal numbers. So far, about 280 congenital erythropoietic porphyria patients have been described in the literature, primarily in Western countries 28, 32, 33 and their clinical manifestations are markedly heterogeneous, ranging from non-immune hydrops fetalis to milder, later-onset forms characterized by mild cutaneous involvement without hematologic symptoms in adult life 31, 34. Severely affected patients are transfusion-dependent throughout life, have secondary hypersplenism and significant cutaneous involvement. Severe complications such as secondary skin infections with subsequent bone resorption and photomutilation, leading to loss of digits and facial features are common 22. Chronic infections have caused osteomyelitis in some congenital erythropoietic porphyria (CEP) patients 35, 36.
Congenital erythropoietic porphyria (CEP) onset in most affected individuals occurs at birth or early infancy. The first sign of congenital erythropoietic porphyria (CEP) is often pink-to-dark red discoloration of the urine. Hemolytic anemia is common and can range from mild to severe, with some affected individuals requiring chronic blood transfusions 1. Porphyrin deposition may lead to corneal ulcers and scarring, reddish-brown discoloration of the teeth (erythrodontia), and bone loss and/or expansion of the bone marrow. However, congenital erythropoietic porphyria (CEP) signs and symptoms are broad and range from nonimmune hydrops fetalis in utero to late-onset disease with only mild skin manifestations in adulthood.
The extent of enzyme deficiency caused by the UROS gene mutation in each case is the major determinant of the age of onset and severity of symptoms seen in the disease 1.
The diagnosis of congenital erythropoietic porphyria may be suspected when the reddish-colored urine is noted at birth or later in life. This finding, or the occurrence of skin blisters on sun or light exposure, should lead to a thorough clinical evaluation and specialized laboratory tests. The diagnosis can be made by testing the urine for increased levels of specific porphyrins. Diagnostic confirmation requires the demonstration of the specific UROS enzyme activity and/or by identifying the specific mutation(s) in the UROS gene which is/are responsible for the impaired enzyme.
Prenatal and preimplantation genetic diagnoses are available for subsequent pregnancies in congenital erythropoietic porphyria families.
There is no FDA-approved treatment for congenital erythropoietic porphyria (CEP) or specific treatment for the photosensitivity 1. Currently, the only effective management is prevention of skin blistering by strict avoidance of sun and light exposure, including the long-wave ultraviolet light that passes through window glass or is emitted from artificial light sources. Therefore, the use of protective clothing, wraparound sunglasses, protective window films, reddish incandescent bulbs, filtering screens for fluorescent lights, and opaque sunscreens containing zinc oxide or titanium oxide is recommended 1. Wound care is necessary to prevent infection of opened blisters; surgical intervention may be necessary; blood transfusions with iron chelation are necessary when hemolysis is significant to maintain the hematocrit >35 to decrease reticulocytosis in transfusion-dependent patients 37, 1. Bone marrow transplantation (BMT) or hematopoietic stem cell (HSCT) transplantation is the only cure for congenital erythropoietic porphyria and should be considered in children with severe skin and bone marrow involvement 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 1.
Figure 1. Congenital erythropoietic porphyria (CEP) signs and symptoms
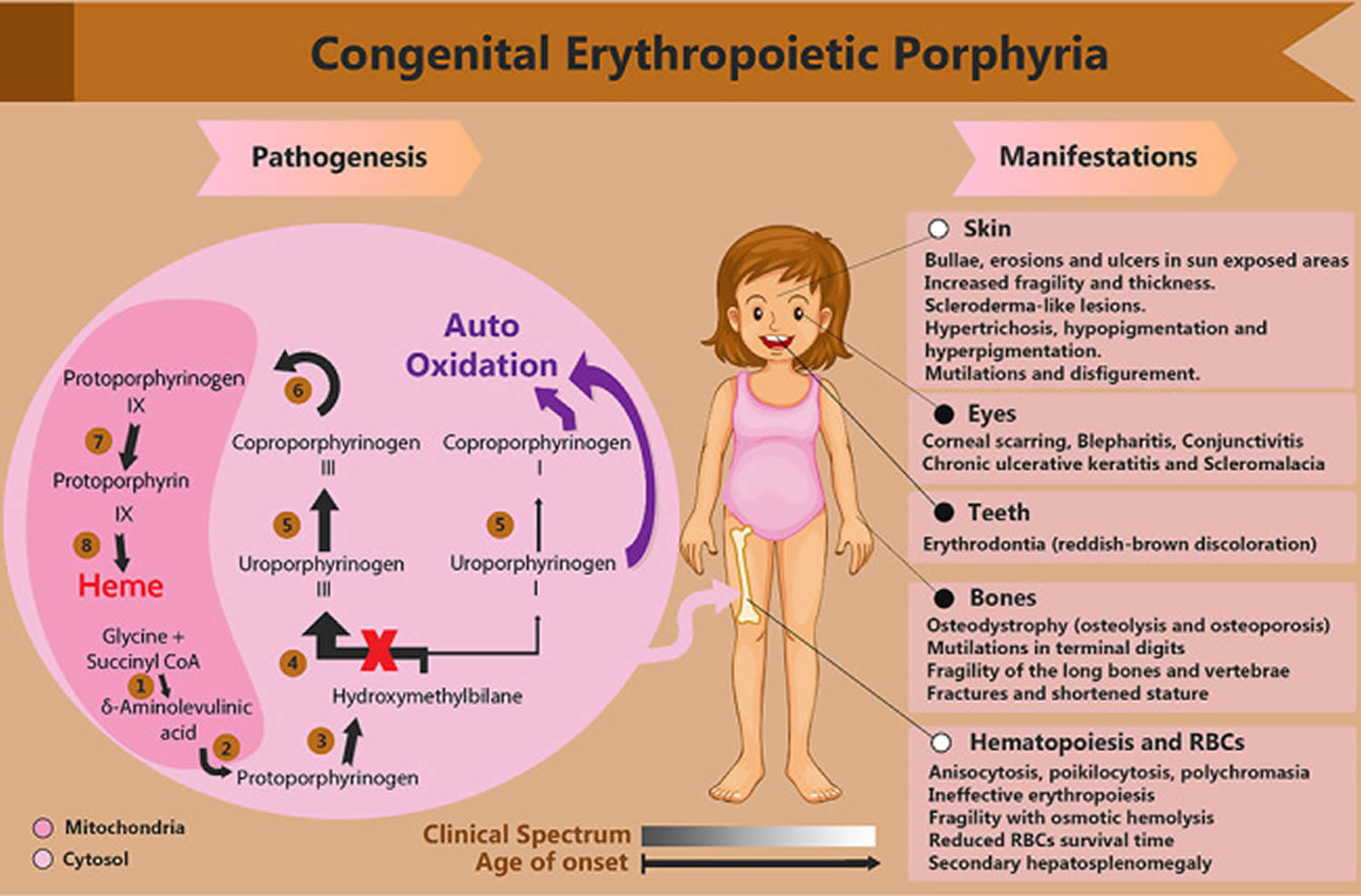
Footnotes: An illustration of the heme biosynthesis pathway and congenital erythropoietic porphyria (CEP) pathophysiology. Each number represents the enzyme of the corresponding step: (1) aminolevulinic acid synthase (ALAS), (2) aminolevulinic acid dehydratase (ALAD), (3) hydroxymethylbilane synthase (HMBS), (4) uroporphyrinogen III synthase (UROS), (5) uroporphyrinogen decarboxylase (UROD), (6) coproporphyrinogen-III oxidase (CPOX), (7) protoporphyrinogen oxidase (PPOX), (8) ferrochelatase (FECH). The gray–white rectangle represents the clinical spectrum that narrows by the increase in the age of onset, where the gray beginning expresses the concurrence of black-and-white-indexed involvements.
[Source 10 ]Figure 2. Congenital erythropoietic porphyria (CEP)
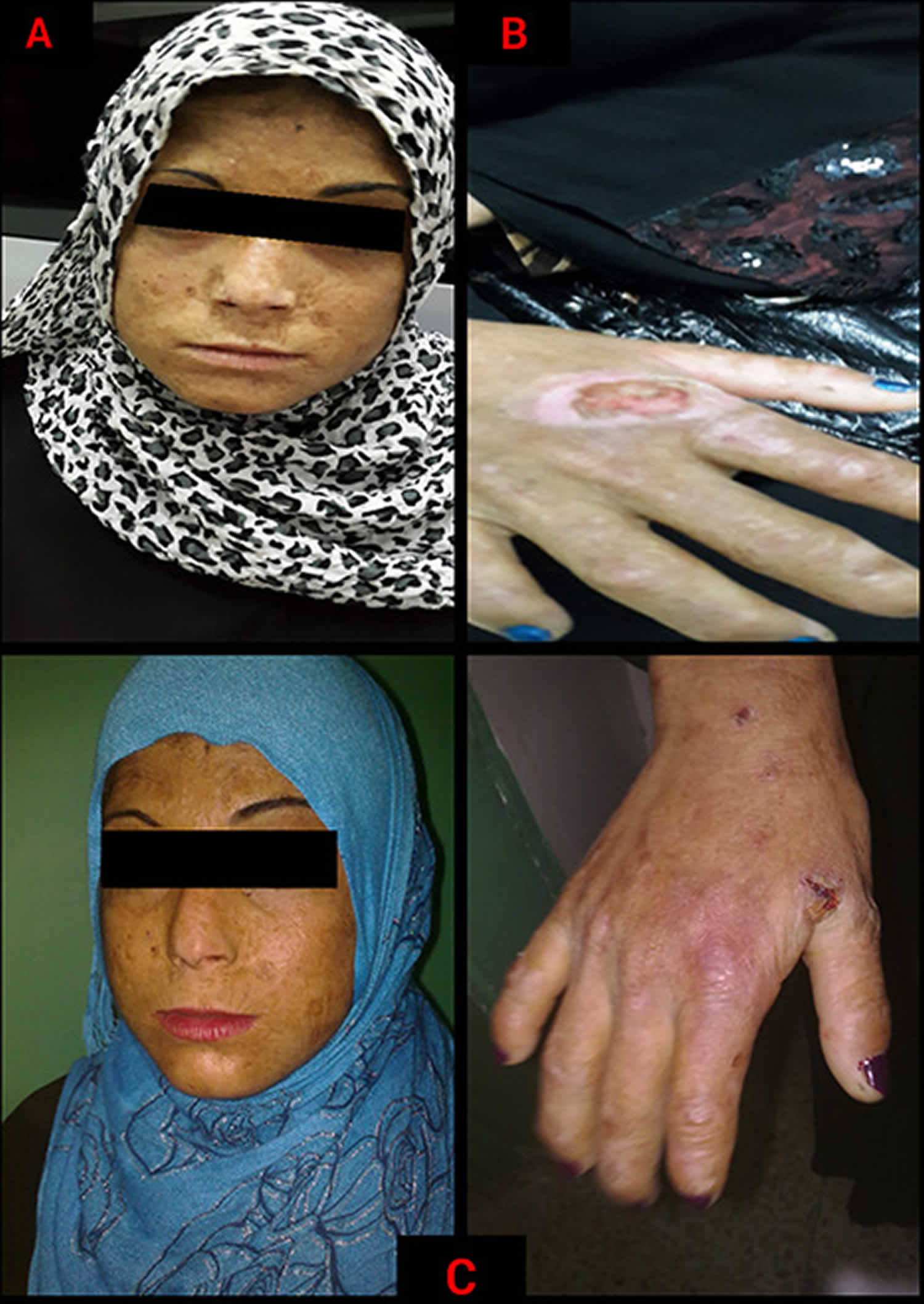
Footnotes: Congenital erythropoietic porphyria (CEP) in a 13-year-old female. (A) a dyspigmented sclerodermatous appearance of the face accompanied with linear and spotted atrophic lesions. (B) Immense areas of ulceration over the dorsum of the hand, with acrolysis of the bones and soft tissues at the ends of the fingers. (C) Healing ulcers on the face and dorsum of the hand after 1 month of treatment; the skin improved in terms of pigmentation and elasticity, with no active ulcers.
[Source 10 ]Figure 3. Congenital erythropoietic porphyria (CEP) signs
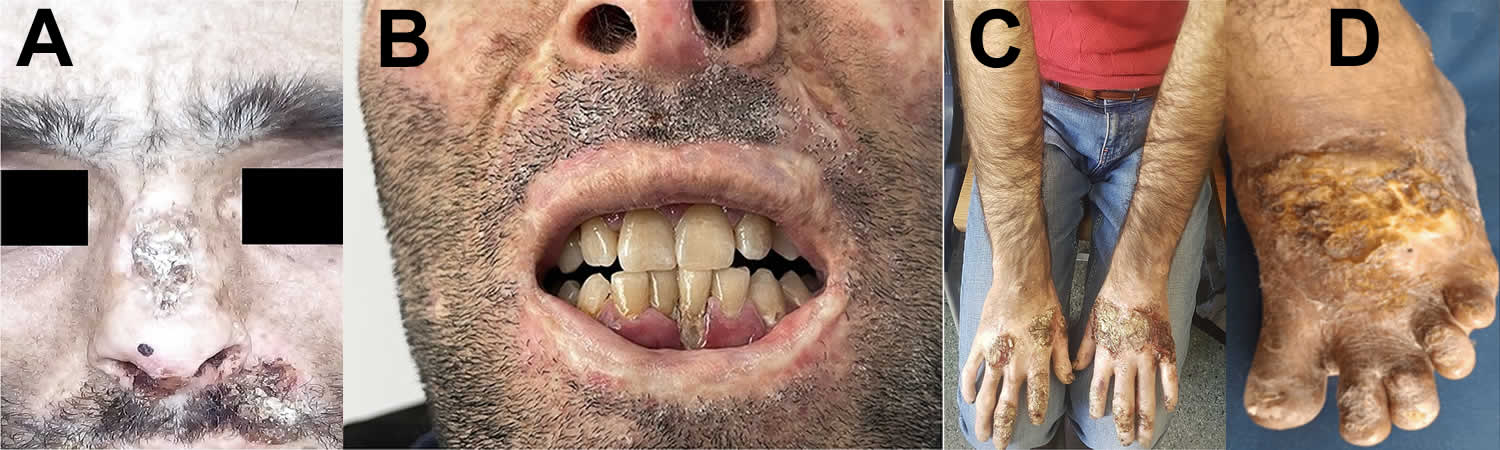
Footnotes: (A) The mutilated nose bridge; (B) Porphyrins accumulation on the gums and erythrodontia; (C) Hands with infected bullae and mutilated index fingers; (D) hypertrophied scars in sun-exposed dorsal foot areas and verrucous growth over the toes
[Sources 19, 14 ]Figure 4. Congenital erythropoietic porphyria under Wood’s lamp
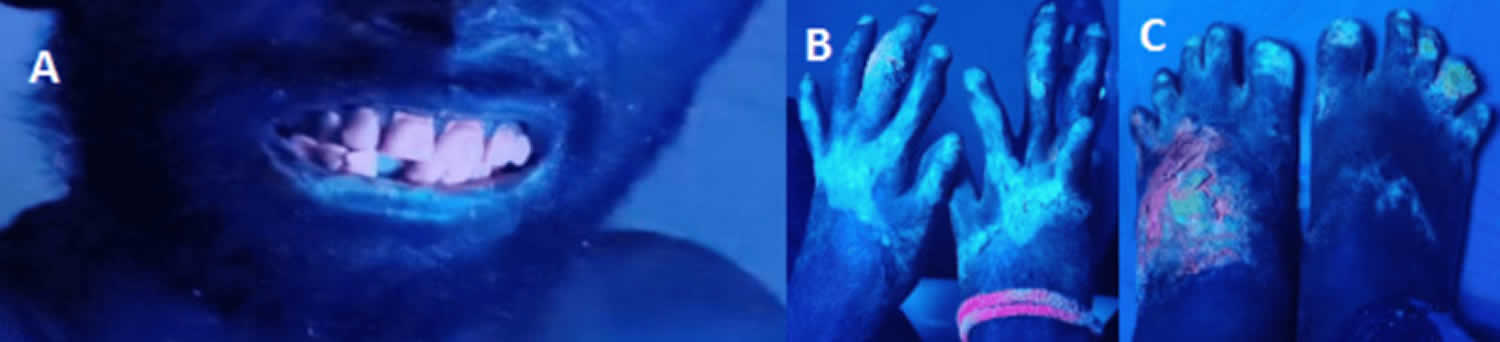
Footnotes: (A) Wood’s lamp examination of the face, (B) Wood’s lamp examination of the hands, (C) Wood’s lamp examination of the feet, showing red-pink fluorescence from teeth and blue hue from sun-exposed skin areas.
[Source 14 ]Figure 5. Congenital erythropoietic porphyria urine

Figure 6. Porphyrin molecular structure
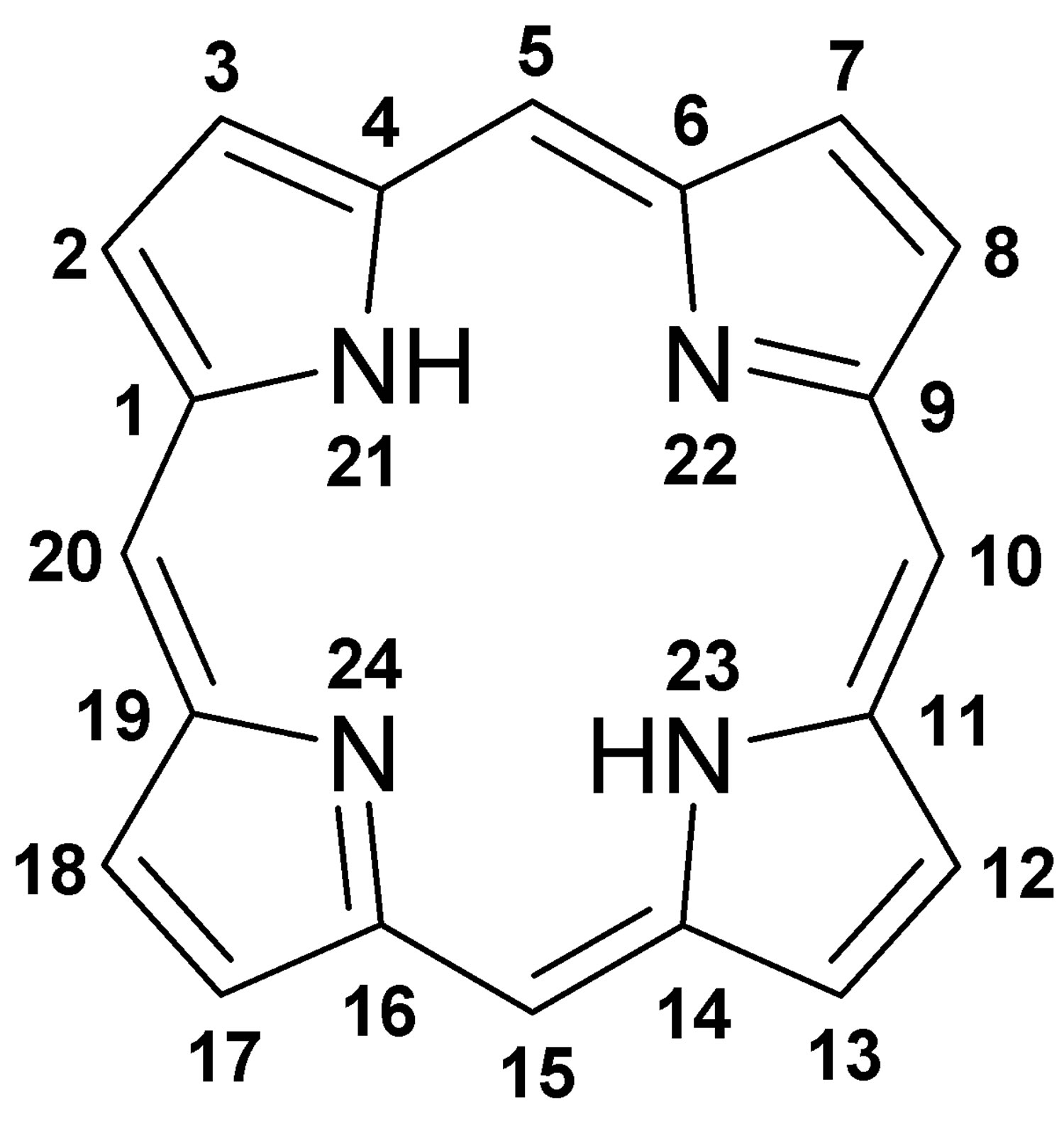
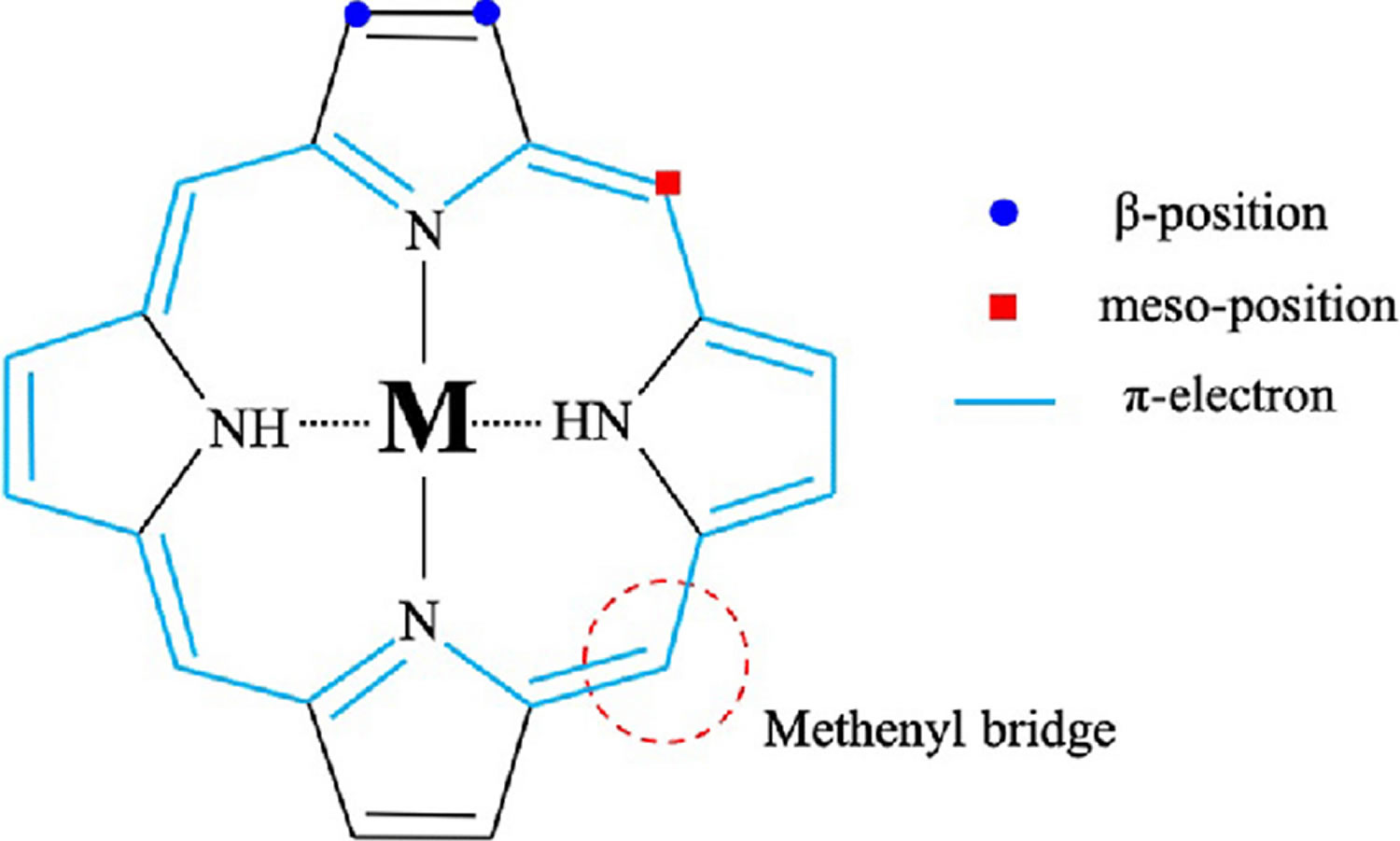
Footnote: Molecular structure of porphyrin (M represent metal ions, such as Mg, Cu, Fe, Zn, etc.).
[Source 48 ]Figure 7. Hemoglobin molecular structure
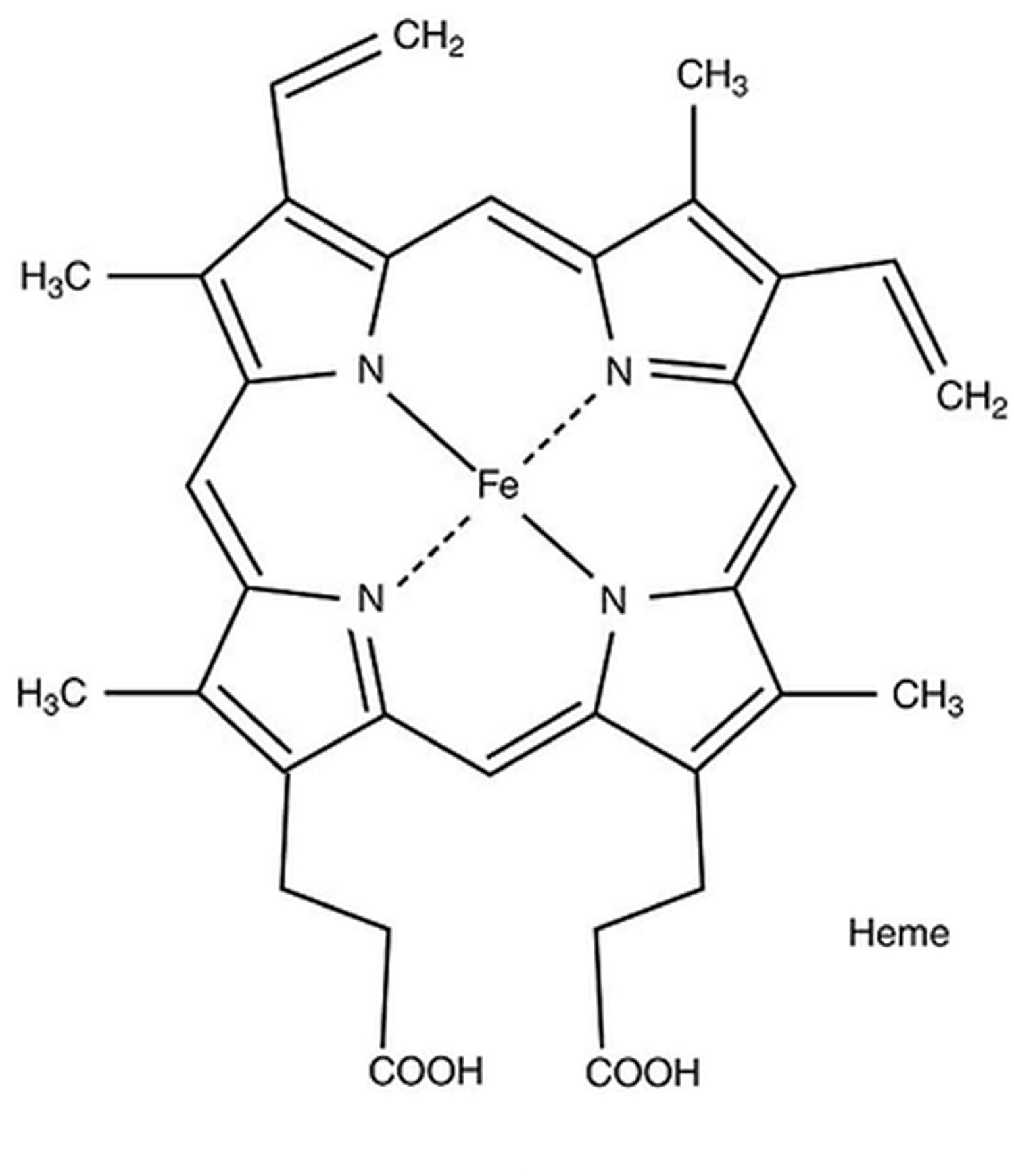
Figure 8. Heme biosynthesis pathway
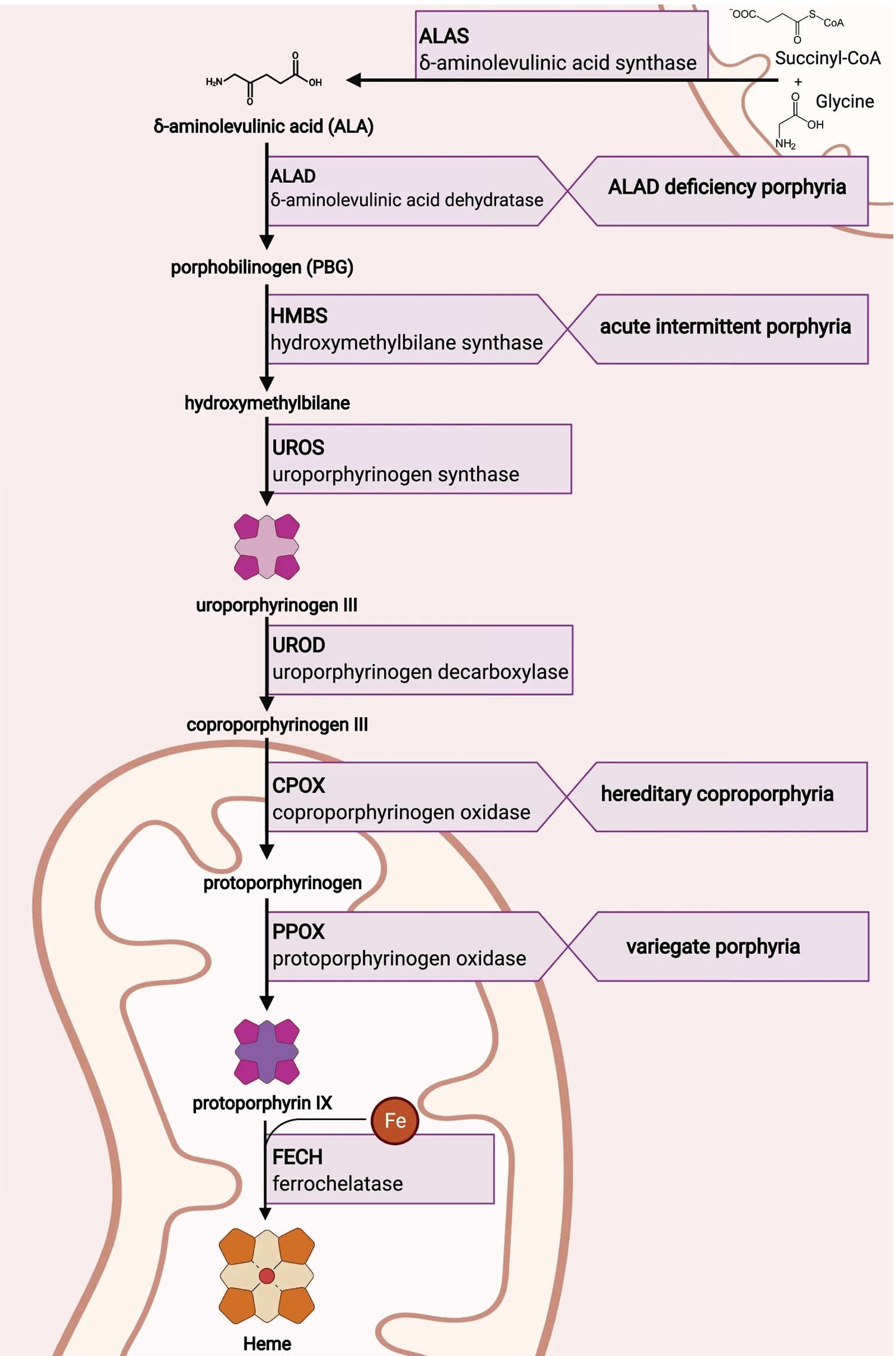
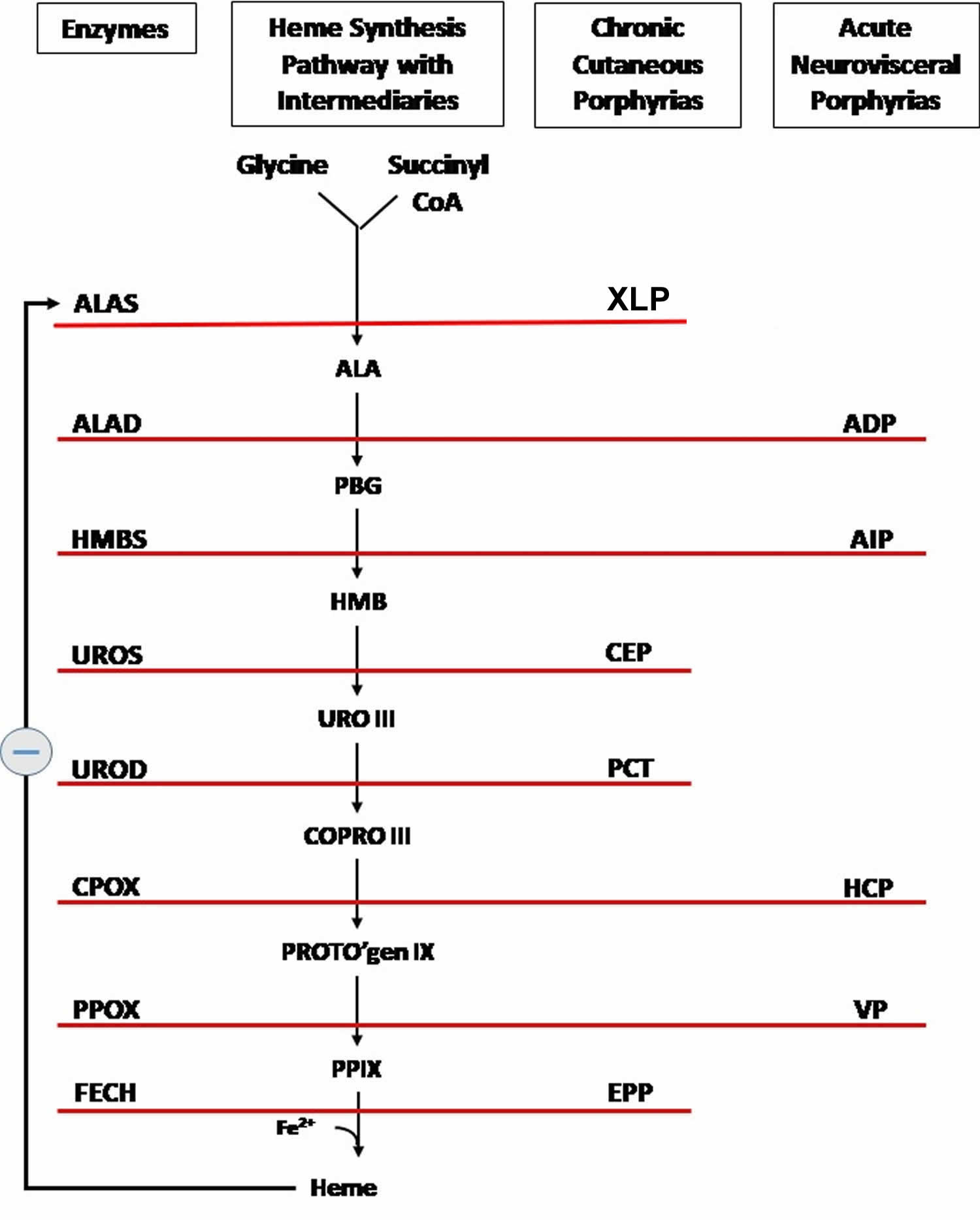
Footnotes: The heme biosynthetic pathway requires 8 enzymatic steps. Heme synthesis pathway showing the enzymes involved in the heme synthesis pathway and the associated porphyrias with the disruption of each specific enzyme. Gain-of-function variants in ALAS2 result in X-linked protoporphyria (XLP), and loss-of-functions variants in FECH result in erythropoietic protoporphyria (EPP). In both X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP), metal-free protoporphyrin IX (PPIX) accumulates in erythroblasts, erythrocytes, the plasma, and the biliary system. Metal-free protoporphyrin IX (PPIX) is photosensitive, particularly to visible light in the blue range, and the light-mediated activation of metal-free protoporphyrin IX (PPIX) produces free radicals that damage the surrounding tissues.
Enzymes, encoded by genes, catalyze each of the steps. Gene mutations cause deficient enzyme production. Disruptions are indicated by red lines connecting enzymes with the resultant porphyrias. ALAS (ALAS2) = aminolevulinate synthase (aminolevulinate synthase 2); ALAD = delta-aminolevulinic acid dehydratase; PBGD = porphobilinogen dehydratase; HMBS = hydroxymethylbilane synthase; UROS = uroporphyrinogen-III synthase; UROD = uroporphyrinogen III decarboxylase; CPOX = coproporphyrinogen-III oxidase; PPOX = protoporphyrinogen oxidase; FECH = ferrochelatase.
Porphyrias resulting from disruption of enzyme production. XLP (X-linked protoporphyria); ADP (aminolevulinic acid dehydratase porphyria); AIP (acute intermittent porphyria); CEP (congenital erythropoietic porphyria); PCT (porphyria cutanea tarda); HCP (hereditary coproporphyria); VP (variegate porphyria); EPP (erythropoietic protoporphyria).
Abbreviations: ALA = aminolevulinic acid; PBG = porphobilinogen; HMB = hydroxymethylbilane; URO III = uroporphyrinogen III; COPRO III = coproporphyrinogen III; PROTO’gen IX protoporphyrinogen IX; PPIX = protoporphyrin IX; Fe2+ = iron.
[Source 50 ]Figure 9. Uroporphyrinogen III synthase (UROS) enzyme activity
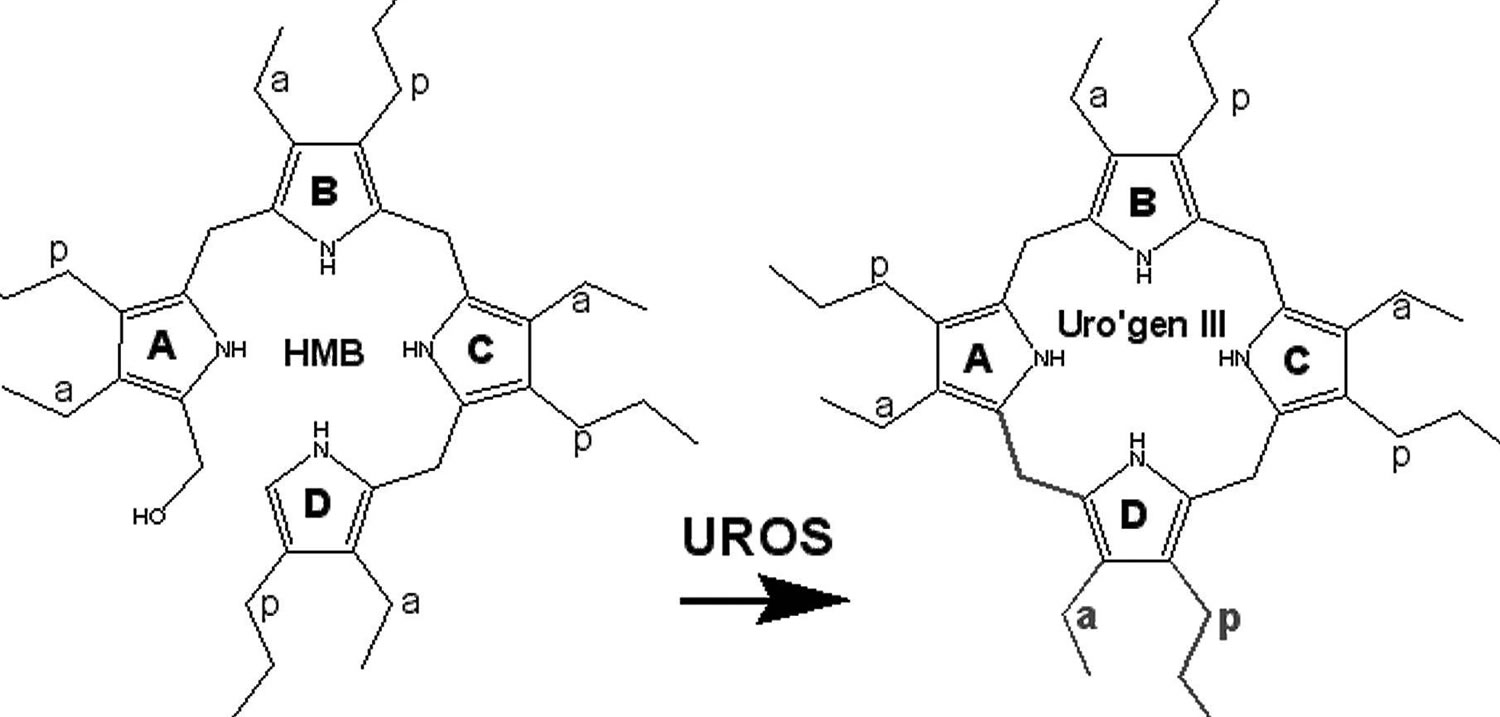
Footnotes: UROS (uroporphyrinogen III synthase) enzyme catalyzes the cyclization of hydroxymethylbilane (HMB) with concomitant inversion of ring D to yield uroporphyrinogen III (URO III) 29. Deficiency of UROS (uroporphyrinogen III synthase) enzyme results in accumulation of HMB (hydroxymethylbilane), most of which is converted nonenzymatically to non-physiological compound uroporphyrinogen I, which is then acted upon by uroporphyrinogen decarboxylase (UROD) the fifth enzyme in the heme synthetic pathway to form coproporphyrinogen I 21. Coproporphyrinogen I then oxidizes spontaneously to form the porphyrins coproporphyrin I and uroporphyrin I. In congenital erythropoietic porphyria (CEP), these toxic porphyrins mostly accumulate in red blood cell precursors in your bone marrow, as well as your teeth, bones, urine, and feces. These toxic porphyrins are not significantly elevated in your liver. This leads to disease characterized by severe skin photosensitivity and breakdown of red blood cells (hemolysis) 22.
[Source 15 ]Figure 10. Congenital erythropoietic porphyria pathophysiology
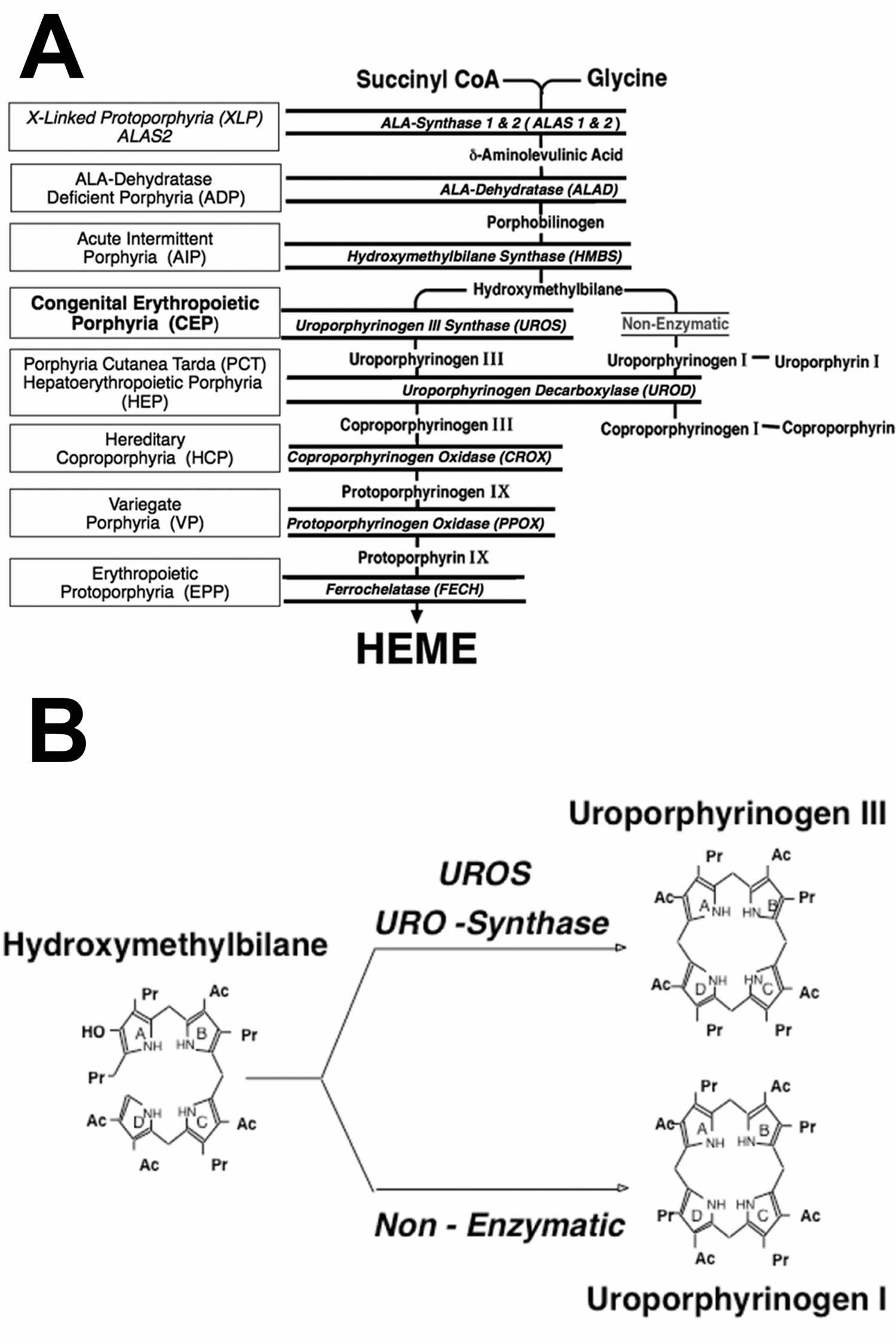
Foootnotes: (A) Heme biosynthetic pathway and the porphyrias resulting from the indicated heme biosynthetic enzyme defect. There are 8 enzymatic steps in the conversion of glycine and succinyl-CoA to heme. The heme biosynthetic enzymes are italicized, their substrates and products are indicated, and the resulting porphyrias are shown in boxes. Note that there are two aminolevulinic acid synthase (ALAS) isozymes: a housekeeping enzyme, ALAS1, encoded by a gene that is regulated by negative feedback repression by heme, and an erythroid specific enzyme, ALAS2, that is regulated by the iron response proteins and erythroid transcription binding proteins.
(B) Uroporphyrinogen synthase (UROS) normally converts hydroxymethylbilane (HMB) to uroporphyrinogen III. When the UROS activity is markedly decreased, HMB (hydroxymethylbilane) is non-enzymatically converted to uroporphyrinogen I, which is then enzymatically converted to oproporphyrinogen I by uroporphyrinogen decarboxylase (UROD). The accumulated uroporphyrinogen I and coproporphyrinogen I are oxidized to their respective porphyrins, which are photoactive and cause the sun/light-induced hemolysis and cutaneous manifestations of congenital erythropoietic porphyria (CEP).
[Source 16 ]Congenital erythropoietic porphyria cause
Congenital erythropoietic porphyria is caused by mutations in the UROS gene located on chromosome 10q26.2 25. Congenital erythropoietic porphyria (CEP) is rarely inherited in X-linked inheritance pattern due to mutation of the GATA1 gene located on the X chromosome (chromosome Xp11.23) that affected males only while the female heterozygotes were mostly asymptomatic 26, 15, 27, 22. Females who are heterozygous for GATA1 gene will be either asymptomatic or have a milder disease with predominantly hematologic abnormalities due to skewed X-chromosome inactivation 15, 27. In a small percentage of congenital erythropoietic porphyria patients (>10%), a disease-causing mutation has not been detected in the UROS or GATA1 genes, raising the possibility that additional gene(s) may play a role in congenital erythropoietic porphyria pathogenesis 28.
Disease-causing mutations in either UROS gene or GATA1 gene result in absent or markedly reduced UROS (uroporphyrinogen III synthase) enzymatic activity. UROS (uroporphyrinogen III synthase) enzyme catalyzes the cyclization of hydroxymethylbilane (HMB) with concomitant inversion of ring D to yield uroporphyrinogen III (URO III) 29. Deficiency of UROS (uroporphyrinogen III synthase) enzyme results in accumulation of HMB (hydroxymethylbilane), most of which is converted nonenzymatically to non-physiological compound uroporphyrinogen I, which is then acted upon by uroporphyrinogen decarboxylase (UROD) the fifth enzyme in the heme synthetic pathway to form coproporphyrinogen I 21. Coproporphyrinogen I then oxidizes spontaneously to form the porphyrins coproporphyrin I and uroporphyrin I. In congenital erythropoietic porphyria (CEP), these toxic porphyrins mostly accumulate in red blood cell precursors in your bone marrow, as well as your teeth, bones, urine, and feces. These toxic porphyrins are not significantly elevated in your liver. Since these toxic porphyrins are photocatalytic compounds, exposure of the skin to sunlight and other sources of long-wave ultraviolet light elicits a phototoxic reaction, resulting in blistering and vesicle formation as well as increased friability of the skin and breakdown of red blood cells (hemolysis) 22, 30, 31.
Congenital erythropoietic porphyria inheritance pattern
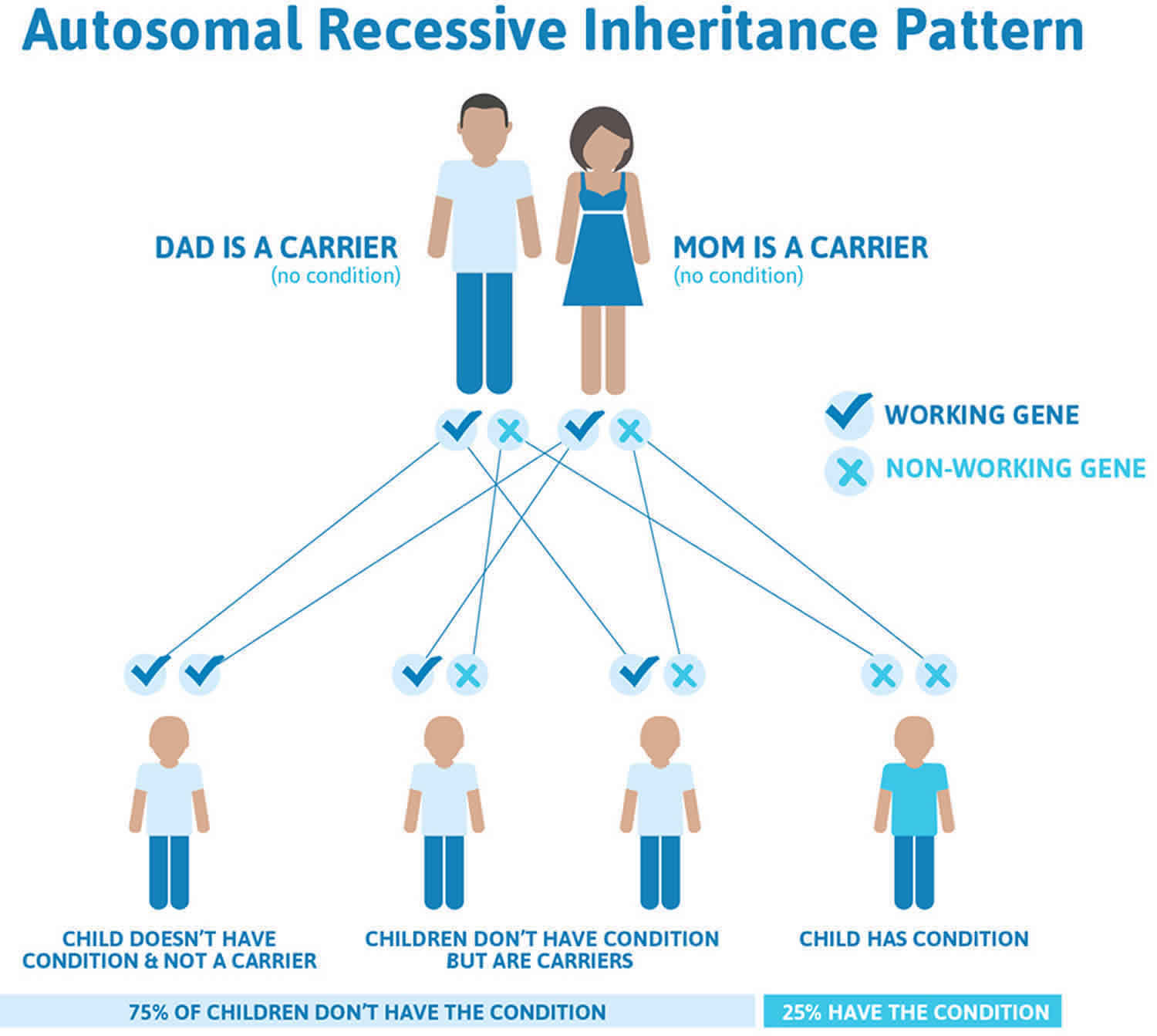
Congenital erythropoietic porphyria (CEP) is inherited as an autosomal recessive genetic condition. Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. Patients with congenital erythropoietic porphyria (CEP) are either homozygotes or compound heterozygotes for mutations of the UROS gene at 10q25.2-q26.3 that encodes uroporphyrinogen III synthase (UROS), a cytosolic enzyme that converts hydroxymethyl bilane (HMB) to uroporphyrinogen III. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, and usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Congenital erythropoietic porphyria (CEP) is rarely inherited in X-linked inheritance pattern due to mutation of the GATA1 gene located on the X chromosome that affected males, while the female heterozygotes were mostly asymptomatic 15, 27, 22. Females who are heterozygous for GATA1 gene will be either asymptomatic or have a milder disease with predominantly hematologic abnormalities due to skewed X-chromosome inactivation 15, 27.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 11. Congenital erythropoietic porphyria (CEP) autosomal recessive inheritance pattern
Congenital erythropoietic porphyria pathophysiology
The pathogenesis of congenital erythropoietic porphyria is explained by markedly deficient, but not absent uroporphyrinogen III synthase (UROS) activity. Consistent with its autosomal recessive inheritance, a very low level (≤1% of normal) appears to be sufficient for life. A uroporphyrinogen III synthase (UROS) knockout mouse model was lethal in the early embryo, indicating that the absence of UROS activity is not compatible with life 51. Deficient UROS activity leads to the accumulation of the enzyme’s substrate, hydroxymethylbilane (HMB), most of which is converted non-enzymatically to uroporphyrinogen I 16. Although uroporphyrinogen I can undergo decarboxylation by uroporphyrinogen decarboxylase (UROD) to form hepta-, hexa- and pentacarboxyl-porphyrinogen I, and, finally, coproporphyrinogen I, further metabolism cannot proceed because the next enzyme in the pathway, coproporphyrinogen oxidase (CPOX), is stereospecific for the III isomer 16. The isomer I porphyrins are non-physiologic, phototoxic, cannot be metabolized to heme, and are pathogenic when they accumulate in large amounts 22, 52, 53.
Uroporphyrin I accumulation in bone marrow, red blood cells, plasma and urine is the biochemical hallmark of congenital erythropoietic porphyria (CEP). Large amounts of isomer I porphyrinogens in bone marrow erythroid precursors (especially normoblasts and reticulocytes) and red blood cells (erythrocytes) undergo auto-oxidation to the corresponding porphyrins, which damage red blood cells and are deposited in tissues and bones. Photosensitivity occurs because these porphyrins are photocatalytic and cytotoxic compounds 52. Exposure of the skin to sunlight and other sources of long-wave ultraviolet (UV) light results in blistering and vesicle formation and increased friability of the skin 30. Damage to the red blood cells (erythrocytes) leads to hemolysis, which is almost always present, but may not be accompanied by anemia if erythroid hyperplasia is sufficient to compensate for the increased rate of erythrocyte destruction 16. The degree of compensation may vary over time but in severe cases, chronic hemolysis and ineffective erythropoiesis may result in transfusion dependence 22.
Accumulated porphyrins are excreted in large amounts in urine and feces and pink to dark-reddish urine is typically noted in the diaper shortly after birth. Urinary porphyrin concentrations are markedly increased (100–1000 times normal; up to 50–100 mg/day) and consist mostly of the uroporphyrin I and coproporphyrin I isomers, with lesser increases in hepta-, hexa- and pentacarboxylporphyrins I isomers 54. Although type I urinary porphyrin isomers predominate, type III isomers also are increased. Fecal porphyrins are also markedly increased with a predominance of coproporphyrin I. Fecal isocoproporphyrins are not increased 55. As opposed to the acute hepatic porphyrias, there is no elevation of urinary aminolevulinic acid (ALA) or porphobilinogen (PBG).
Congenital erythropoietic porphyria signs and symptoms
Congenital erythropoietic porphyria or Gunther disease severity and age at onset of symptoms are highly variable between individuals. For many, the onset of symptoms and diagnosis are often in infancy. Some do not develop symptoms until adulthood, their symptoms are usually milder and primarily skin related 56.
Congenital erythropoietic porphyria symptoms usually start in infancy or childhood and the diagnosis in most patients is suggested by the reddish color of the urine which stains the diapers and fluoresces with a Wood’s lamp (ultraviolet light). Congenital erythropoietic porphyria (CEP) or Gunther disease major symptoms are sensitivity of the skin to sunlight and some types of artificial light, and anemia which can be very severe. Skin photosensitivity may be extreme, and can lead to blistering, severe scarring and increased hair growth (hypertrichosis). The fluid-filled blisters may rupture and get infected. These infected wound can lead to scarring, bone loss, and deformities. The hands, arms, and face are the most commonly affected areas. Increased hair growth (hypertrichosis) on sun-exposed skin, brownish-colored teeth (erythrodontia), and reddish-colored urine are common. There may be bone fragility and vitamin deficiencies, especially vitamin D deficiency. Vitamin D is a fat-soluble vitamin that essential for bone development and maintenance, as it enhances calcium, magnesium, and phosphate absorption. Vitamin D deficiency due to sunlight avoidance in people with congenital erythropoietic porphyria can lead to a range of health problems, including bone disorders, muscle weakness, and an increased risk of fractures.
Red blood cells have a shortened life-span, and anemia often results. Synthesis of heme and hemoglobin are actually increased to compensate for the shortened red blood cell survival. The spleen can be enlarged due to extramedullary erythropoiesis, particularly in people with severe hemolytic anemia.
The most common symptom of congenital erythropoietic porphyria (CEP) is hypersensitivity of the skin to sunlight and some types of artificial light (photosensitivity), with blistering of the skin occurring after exposure. Affected individuals may also exhibit abnormal accumulations of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema). Affected areas of the skin may develop sac-like lesions (vesicles or bullae), scar, and/or become discolored (hyperpigmentation) if exposure to sunlight is prolonged. These affected areas of skin may become abnormally thick. In addition, in some cases, affected individuals may also exhibit malformations of the fingers and nails. The severity and degree of photosensitivity differ depending on the severity of the patient’s gene lesions which correlate with the deficient enzyme activity. In the great majority of congenital erythropoietic porphyria (CEP) cases, photosensitivity is seen from birth; however, in some cases, it may not occur until childhood, adolescence or adulthood. Patients also have brownish discolored teeth (erythrodontia), which fluoresce under ultraviolet light.
In more severe congenital erythropoietic porphyria (CEP) cases, other symptoms can include a low level of red blood cells (anemia), enlargement of the spleen (secondary hypersplenism), and increased hair growth (hypertrichosis) 16. The anemia can be severe and such patients require periodic blood transfusions to quickly increase the amount of red blood cells and iron in the blood. In severely affected patients, anemia may be present in the fetus (non-immune hydrops fetalis). Eye problems also can occur including corneal scarring, eye inflammation, and infections.
Skin involvement
Skin photosensitivity usually begins shortly after birth and is characterized by increased friability and blistering of the epidermis on the hands and face and other sun-exposed areas. Bullae and vesicles containing serous fluid which fluoresces due to their porphyrin content. Blisters are prone to rupture and become infected. Recurrent vesicle formation and secondary infection can lead to cutaneous scarring and deformities, as well as to loss of digits and facial features such as the eyelids, nose and ears. The skin may be thickened, with areas of hypo- and hyper-pigmentation and hypertrichosis of the face and extremities 57. Adult-onset congenital erythropoietic porphyria (CEP) patients have milder clinical symptoms and often exhibit only the skin manifestations of congenital erythropoietic porphyria (CEP) 58, 59, 60, 61, 62, 63. Photosensitivity symptoms are provoked mainly by visible light (400–410 nm Soret wavelength) and to a lesser degree by wavelengths in the long-wave UV region. Affected individuals are also sensitive to sunlight that passes through window glass that does not filter long-wave UVA or visible light as well as to light from artificial light sources 64.
Hemolytic anemia
Mild to severe hemolysis is accompanied by anisocytosis, poikilocytosis, polychromasia, basophilic stippling, reticulocytosis, increased nucleated red cells, absence of haptoglobin, increased unconjugated bilirubin and increased fecal urobilinogen. Plasma iron turnover also is increased 65. Hemolysis presumably results from the accumulated uroporphyrin I in erythrocytes. Secondary splenomegaly develops in response to the increased uptake of abnormal erythrocytes from the circulation, which may contribute to the anemia and also may result in leucopenia and thrombocytopenia. The latter is sometimes associated with significant bleeding and splenectomy may be beneficial in such cases. Anemia due to hemolysis can be severe. Erythroid hyperplasia and markedly ineffective erythropoiesis usually accompany hemolytic anemia in transfusion-dependent patients 54, 61, 66.
In the three male patients with congenital erythropoietic porphyria (CEP) due to GATA1 mutations, hematologic abnormalities including dyserythropoietic anemia, beta-thalassemia intermedia, thrombocytopenia, and hereditary persistence of fetal hemoglobin have been described 15, 27.
Other Clinical Features
Deposition of porphyrins can cause corneal ulcers and scarring, which can ultimately can lead to blindness 16. Other eye signs and symptoms can include scleral necrosis, necrotizing scleritis, seborrheic blepharitis, keratoconjunctivitis, sclerokeratitis, and ectropion 67, 68, 69. Porphyrins deposited in the teeth produce a reddish-brown color called erythrodontia. The teeth may fluoresce on exposure to long-wave ultraviolet light.
Deposition of porphyrins in bone causes osteopenia (lower than normal bone density) due to demineralization 37, 54, 70, 71. The risk for osteopenia and osteoporosis is further increased by vitamin D deficiency, which individuals with congenital erythropoietic porphyria (CEP) are prone to due to avoidance of sun exposure. Porphyrin accumulation in the bone can also cause expansion of the bone marrow, which can lead to hyperplastic bone marrow observed on biopsy 31, 57.
Congenital erythropoietic porphyria diagnosis
The diagnosis of congenital erythropoietic porphyria may be suspected when the reddish-colored urine is noted at birth or later in life. This finding, or the occurrence of skin blisters on sun or light exposure, should lead to a thorough clinical evaluation and specialized laboratory tests. The biochemical diagnosis of congenital erythropoietic porphyria is established by detection of markedly elevated levels of uroporphyrin I and coproporphyrin I in urine, red blood cells or amniotic fluid as well as high fecal coproporphyrin I concentrations. Diagnostic confirmation requires the demonstration of the specific UROS enzyme activity and/or by identifying the specific mutation(s) in the UROS gene which is/are responsible for the impaired enzyme 22.
Congenital erythropoietic porphyria should be considered in the differential diagnosis of non-immune hydrops fetalis, in which case the amniotic fluid surrounding the fetus will be pink, dark-red or brown, and should examined for porphyrins 72. Newborns with pink to dark-red urine-stained diapers should immediately have diagnostic studies. Congenital erythropoietic porphyria (CEP) also should be considered in children or adults who have porphyrinuria or skin blistering following exposure to sunlight or other sources of long-wave ultraviolet light. In some cases, the disease is less severe and presents in adult life with mild anemia and/or skin lesions.
Congenital erythropoietic porphyria (CEP) should be suspected in individuals with the following clinical and laboratory findings and family history 1.
- Nonimmune hydrops fetalis
- Signs of congenital erythropoietic porphyria
- Pink-to-dark red discoloration of the urine (pink or dark red urine-stained diapers are often the first sign in infants)
- Hemolytic anemia
- Severe cutaneous photosensitivity with onset usually in infancy or early childhood
- Blisters and vesicles in light-exposed areas, which are prone to rupture and infection
- Scarring and deformities (photomutilation) of digits and facial features, caused by recurrent blistering, infections, and bone resorption
- In light-exposed areas: friable skin, skin thickening, hypo- and hyperpigmentation
- Reddish-brown discoloration of teeth (fluoresce on exposure to long-wave ultraviolet light), also called erythrodontia
- Corneal ulcers and scarring
- Hypertrichosis of the face and extremities
- Laboratory findings include markedly increased levels of uroporphyrin I and coproporphyrin I isomers in red blood cells, urine, or amniotic fluid as well as coproporphyrin I in feces
Prenatal and preimplantation genetic diagnoses are available for subsequent pregnancies in congenital erythropoietic porphyria families.
Congenital erythropoietic porphyria treatment
Avoidance of sun and light exposure, including the long-wave ultraviolet light that passes through window glass or is emitted from artificial light sources is essential to prevent the skin lesions in individuals with congenital erythropoietic porphyria (CEP) 1. The use of topical sunscreens containing zinc oxide or titanium oxide, protective clothing, long sleeves, hats, gloves, and wraparound sunglasses are strongly recommended. Individuals with congenital erythropoietic porphyria will benefit from window tinting or using vinyls or films to cover the windows in their car or house and the use of reddish incandescent bulbs, filtering screens for fluorescent lights 1. Before tinting or shading car windows, affected individuals should check with their local Registry of Motor Vehicles to ensure that such measures do not violate any local laws.
Wound care is necessary to prevent infection of opened blisters; surgical intervention may be necessary; blood transfusions with iron chelation are necessary when hemolysis is significant to maintain the hematocrit >35 to decrease reticulocytosis in transfusion-dependent patients 37, 1. Chronic transfusions have been useful in decreasing the bone marrow production of the phototoxic porphyrins.
Blood transfusions and perhaps removing the spleen may reduce porphyrin production by the bone marrow. Activated charcoal given by mouth is sometimes effective.
When successful, bone marrow transplantation (BMT) or hematopoietic stem cell (HSCT) transplantation has cured patients with congenital erythropoietic porphyria, but is accompanied by specific risks of complications and should be considered in children with severe skin and bone marrow involvement 1, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47.
Evaluations Following Initial Diagnosis
To establish the extent of congenital erythropoietic porphyria disease and needs in an individual diagnosed with congenital erythropoietic porphyria (CEP), the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Hematologic indices including reticulocytes and bilirubin (to assess hemolysis) and iron profile (to assess iron storage)
- Serum calcium and vitamin D concentrations; bone densitometry
- Liver function tests, especially in transfusion-dependent individuals given the risk for liver disease due to iron storage
- Skin evaluation to assess photosensitivity, photomutilation, and secondary skin changes (thickening, hyper- or hypopigmentation, hypertrichosis)
- Eye evaluation for corneal ulcers and scarring and other ocular manifestations
- Teeth assessment for erythrodontia (reddish-brown color from porphyrin deposition)
- Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature, mode of inheritance, and implications of congenital erythropoietic porphyria (CEP) in order to facilitate medical and personal decision making
Sun Avoidance and Skin Protection
There is no FDA-approved treatment for this disease or specific treatment for the photosensitivity. In patients who have not undergone bone marrow transplantation (BMT) or hematopoietic stem cell (HSCT) transplantation, exposure to sunlight, ultraviolet light as well as light emitted by fluorescent sources, should be avoided. Sunscreen lotions containing zinc oxide or titanium oxide can be beneficial, but do not replace strict avoidance of sun and light exposure 73. Most individuals with congenital erythropoietic porphyria (CEP) try to adjust their daily life in order to reduce light exposure as much as possible, which in some cases includes choosing a profession which allows work during the night. This leads to a very restricted family and social life and quality of life is often significantly decreased.
Skin trauma should be avoided and the use of skin antiseptics should be encouraged to prevent bacterial superinfections that can complicate cutaneous blisters and result in scarring and mutilation. Further measures in an effort to minimize bacterial colonization if affected areas and decrease the risk for cutaneous complications could include bleach baths as well as topical treatment with hypochlorous acid solution 74. Severe infections such as cellulitis and bacteremia may have to be treated with intravenous antibiotics and long-term oral antibiotic therapy to suppress chronic skin infections may be required.
Pharmacologic photoprotection has been explored in erythropoietic protoporphyria (EPP), which is the third most common porphyria 75. While beta-carotene has been reported to provide some photoprotective effect in erythropoietic protoporphyria (EPP) patients, it has not proven to be beneficial in congenital erythropoietic porphyria (CEP) 76, 75. Recently, afamelanotide (Scenesse®) – a synthetic melanocortin stimulating hormone analog – was approved for erythropoietic protoporphyria (EPP) patients by the European Medicines Agency in 2014. This hormone is implanted subcutaneously and promotes melanin synthesis via the melanocortin-1 receptor. Beneficial effects have been reported in clinical trials with erythropoietic protoporphyria (EPP) patients 77. However the use of afamelanotide (Scenesse®) in congenital erythropoietic porphyria (CEP) patients has not been investigated and it is unclear if the congenital erythropoietic porphyria (CEP) cutaneous complications can be prevented. One recent case report describes an adult patient with severe congenital erythropoietic porphyria (CEP) who underwent treatment with afamelanotide over a period of one year. While improved tolerance of sun exposure was reported by the individual, no protective effect was observed in already scarred areas of hands and face 78.
Newborns with a known diagnosis of congenital erythropoietic porphyria (CEP) who develop hyperbilirubinemia should not receive photodynamic therapy to avoid its harmful photosensitizing effects and subsequent severe cutaneous blistering and scarring 55.
Hemolytic anemia treatment
In cases of severe hemolysis, frequent blood transfusions may be necessary. Chronic transfusions (every 2–4 weeks) can suppress erythropoiesis and and decrease porphyrin production, which reduces porphyrin levels and photosensitivity 37. Such therapy is likely to be successful if the hematocrit remains above 32% (target hematocrit greater than 35%) and parenteral or oral iron chelators can be administered to reduce the resulting iron overload 79. Experimental induction of iron deficiency either using treatment with iron chelators or via phlebotomies (removal of blood) improved photosensitivity and hemolysis in a few affected individuals 80, 81, 13, 82.
Treatment with hydroxyurea to reduce the bone marrow porphyrin synthesis has been used in some severe congenital erythropoietic porphyria (CEP) cases and may be considered 83. Splenectomy may be considered for patients with splenomegaly and hemolytic anemia, thrombocytopenia and leukopenia. Removing the spleen may correct blood dyscrasia, improve the red blood cell lifespan and substantially reduce transfusion requirements in some patients. It may indirectly also lead to reduced photosensitivity 37.
Efforts to reduce porphyrin levels by administration of hydroxychloroquine, by plasmapheresis, and by intravenous hematin have not shown a clear benefit. Oral charcoal may increase fecal loss of porphyrins 84 and may be considered for patients who are not transfusion-dependent and have milder disease. Oral charcoal seems less successful in more severe cases 85, 86. Iron deficiency induced by treatment with deferasirox led to better erythroid differentiation in the bone marrow and improved hemolysis as well as photosensitivity in one patient 80.
Eye treatments
- Avoidance of damage to the eyelids and cornea by wearing wraparound sunglasses
- Topical antibiotics for corneal ulcers, scleritis, and blepharitis
- Artificial tears and lubricants to help prevent dry eyes in those with ectropion
- Corrective surgery of eyelids to help protect the cornea from injury in those with ectropion 45
Bone loss treatment
Bisphosphonates can be considered in individuals with osteoporosis 45.
Monitoring
Monitor blood indices including iron profile, reticulocyte count, and bilirubin to assess hemolysis every six months. In those receiving blood transfusions, monitor for hemolysis more frequently and for iron overload. Monitor liver function and vitamin D 25-OH every six to twelve months in all affected individuals.
Agents to avoid
All affected individuals should avoid sunlight and UV light. To avoid eye complications and damage to the eyelids, wrap-around sun glasses should be worn. Corneal ulcers, scleritis, and blepharitis should be treated with topical antibiotics. In patients with ectropion, corrective surgery of the eyelid to help protect the cornea from injury may be indicated 45. In individuals undergoing surgeries, use of protective filters for artificial lights in the operating room to prevent phototoxic damage 87.
To avoid bone demineralization, vitamin D supplementation is indicated and bisphosphonates can be considered in individuals with osteoporosis 45.
In those with liver dysfunction avoid drugs that may induce cholestasis (e.g., estrogens).
Pregnancy Management
Successful pregnancies in women with congenital erythropoietic porphyria resulting in healthy and unaffected children have been described 88, 36.
Protective filters for artificial lights should be used in the delivery/operating room to prevent phototoxic damage to the mother during delivery 87.
Therapies Under Investigation
Although there are no clinical trials at the present time, therapeutic approaches under investigation include phlebotomy or iron chelation strategies to reduce the hemolysis and decrease the accumulated porphyrins and, thus, photosensitivity 80, 81, 13, 82.
A mice model of congenital erythropoietic porphyria (CEP) is being used to investigate pharmacologic chaperone therapy (i.e., administration of small-molecule drugs to enhance the residual activity of mutated enzymes that have low activities or are unstable). Specifically, use of the antimicrobial agent ciclopirox as a chaperone-stabilized UROIII-synthase and reversed congenital erythropoietic porphyria (CEP)-related findings such as abnormal URO I levels in the blood, splenomegaly, and liver porphyrins 89. Studies involving use of this medication in humans have not yet been performed.
Congenital erythropoietic porphyria prognosis
Congenital erythropoietic porphyria or Gunther disease severity and age at onset of symptoms are highly variable between individuals. For many, the onset of symptoms and diagnosis are often in infancy. Some do not develop symptoms until adulthood, their symptoms are usually milder and primarily skin related 56. Due to the hematological complications and an increased risk of infection, overall life expectancy may be markedly diminished in more severely affected patients 22. In addition, long-term damage, such as loss of fingers or toes and facial cartilage or contractures of the hands, can have a significant impact on patients’ quality of life, psychiatric well-being, and functional status with regard to daily life and ability to work.
Successful bone marrow (BMT) or hematopoietic stem cell (HSCT) transplantation is the only curative approach, but is accompanied by specific risks of complications and should be considered in children with severe skin and bone marrow involvement 1. The age of children with congenital erythropoietic porphyria (CEP) receiving bone marrow transplantation (BMT) ranges from younger than one year to 13 years 36. Some of the first individuals with congenital erythropoietic porphyria (CEP) to successfully undergo bone marrow transplantation in childhood are now in their 20s 39, 46. Although there is limited information available regarding their long-term outcome post receiving bone marrow transplantation (BMT), experts have learned that individuals successfully transplanted have essentially no photosensitivity to artificial light sources or sunlight.
Several cases of late-onset congenital erythropoietic porphyria (CEP) have been reported in patients older than 50 years of age who presented with skin congenital erythropoietic porphyria (CEP) symptoms and increased porphyrin metabolite excretion, though at a lower level than in patients with the classic infantile-onset congenital erythropoietic porphyria (CEP) presentation 16. In several cases, the occurrence of congenital erythropoietic porphyria (CEP) symptoms was associated with the presence of myelodysplastic syndrome (a group of blood cancers where the bone marrow doesn’t produce enough healthy blood cells leading to a deficiency in red blood cells, white blood cells, and platelets, causing symptoms like fatigue, increased risk of infection, and easy bruising or bleeding) and neither germline nor somatic mutations in UROS or GATA1 were detected [39–41]. However, low-level mosaicism in the bone marrow may not be picked up by sequencing methods, but may be sufficient to cause an accumulation of porphyrin metabolites resulting in an attenuated phenotype 90, 70, 91.
Recently, one 60 year-old male patient was described with late-onset skin porphyria symptoms and a urinary porphyrin metabolite pattern consistent with congenital erythropoietic porphyria (CEP). He was heterozygous for the common UROS gene mutation encoding p.C73R, but a second mutation was not detected. He did not have myelodysplastic syndrome or another underlying hematologic abnormality. While the exact pathophysiology was unclear in the absence of a second disease-causing mutation, acquired mosaicism in the bone marrow affecting the UROS gene was hypothesized to be the underlying cause 92.
- Erwin A, Balwani M, Desnick RJ; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Congenital Erythropoietic Porphyria. 2013 Sep 12 [Updated 2021 Apr 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK154652[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bhusal M, Bhattarai S, Shah M, Khadka A. Congenital erythropoietic porphyria: A case series of a rare uroporphyrinogen III synthase gene mutation in Nepalese patients. JAAD Case Rep. 2021 Feb 25;10:102-106. doi: 10.1016/j.jdcr.2021.02.017[↩]
- Enciso L. Hypertrichosis and erythrodontia in congenital erythropoietic porphyria. EJHaem. 2021 May 26;2(3):640-641. doi: 10.1002/jha2.215[↩]
- Goudet C, Ged C, Petit A, Desage C, Mahe P, Salhi A, Harzallah I, Blouin JM, Mercie P, Schmitt C, Poli A, Gouya L, Barlogis V, Richard E. Severe Perinatal Presentations of Günther’s Disease: Series of 20 Cases and Perspectives. Life (Basel). 2024 Jan 17;14(1):130. doi: 10.3390/life14010130[↩]
- Abkowitz JL. A simple Rx for congenital erythropoietic porphyria. Blood. 2020 Nov 19;136(21):2371-2372. doi: 10.1182/blood.2020007968[↩]
- Jia N, Yimin Y, Li M, Jiang L, Liu Y. Identification of a novel nonsense mutation and a recurrent missense mutation in UROS gene in a patient with congenital erythropoietic porphyria. Front Genet. 2025 Mar 31;16:1486595. doi: 10.3389/fgene.2025.1486595[↩]
- Bernardo-Seisdedos G, Charco JM, SanJuan I, García-Martínez S, Urquiza P, Eraña H, Castilla J, Millet O. Improving the Pharmacological Properties of Ciclopirox for Its Use in Congenital Erythropoietic Porphyria. J Pers Med. 2021 May 28;11(6):485. doi: 10.3390/jpm11060485[↩]
- Bragazzi Cunha J, Elenbaas JS, Maitra D, Kuo N, Azuero-Dajud R, Ferguson AC, Griffin MS, Lentz SI, Shavit JA, Omary MB. Acitretin mitigates uroporphyrin-induced bone defects in congenital erythropoietic porphyria models. Sci Rep. 2021 May 5;11(1):9601. doi: 10.1038/s41598-021-88668-9[↩]
- Khan J, Hashmi MU, Noor N, Khan AJ, Shrateh ON, Tahir MJ. Congenital erythropoietic porphyria presenting with recurrent epistaxis: a case report. J Med Case Rep. 2023 Nov 14;17(1):472. doi: 10.1186/s13256-023-04204-5[↩]
- Kahila A, Zamlout A, Mazloum A, Laila O, Badran A. Congenital erythropoietic porphyria (Gunther disease): a case report. Oxf Med Case Reports. 2020 Jul 24;2020(7):omaa051. doi: 10.1093/omcr/omaa051[↩][↩][↩]
- Sudrié-Arnaud B, Legendre M, Snanoudj S, Pelluard F, Bekri S, Tebani A. An Atypical Case of Congenital Erythropoietic Porphyria. Genes (Basel). 2021 Nov 19;12(11):1828. doi: 10.3390/genes12111828[↩]
- Gopalakrishna H, Mironova M, Malik S, Faust A, Khurram N, Koh C, Kleiner DE, Heller T. Porto-Sinusoidal Vascular Disease in Congenital Erythropoietic Porphyria Needing Liver Transplantation. ACG Case Rep J. 2024 Apr 26;11(5):e01336. doi: 10.14309/crj.0000000000001336[↩]
- Blouin JM, Ged C, Bernardo-Seisdedos G, Cabantous T, Pinson B, Poli A, Puy H, Millet O, Gouya L, Morice-Picard F, Richard E. Identification of novel UROS mutations in a patient with congenital erythropoietic porphyria and efficient treatment by phlebotomy. Mol Genet Metab Rep. 2021 Feb 11;27:100722. doi: 10.1016/j.ymgmr.2021.100722[↩][↩][↩]
- Saikrishna P, Palaniswamy G, Pillikunte Doddareddy N, Ishfaq L, Zargar MN, Wafa Eranhikkal F, Sahu S. Congenital Erythropoietic Porphyria: A Rare Inherited Disorder. Cureus. 2024 Mar 5;16(3):e55558. doi: 10.7759/cureus.55558[↩][↩][↩]
- Phillips JD, Steensma DP, Pulsipher MA, Spangrude GJ, Kushner JP. Congenital erythropoietic porphyria due to a mutation in GATA1: the first trans-acting mutation causative for a human porphyria. Blood. 2007 Mar 15;109(6):2618-21. doi: 10.1182/blood-2006-06-022848[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Erwin AL, Desnick RJ. Congenital erythropoietic porphyria: Recent advances. Mol Genet Metab. 2019 Nov;128(3):288-297. doi: 10.1016/j.ymgme.2018.12.008[↩][↩][↩][↩][↩][↩][↩][↩]
- Desjardins MP, Naccache L, Hébert A, Auger I, Teira P, Pelland-Marcotte MC. Very Early Diagnosis and Management of Congenital Erythropoietic Porphyria. Clin Pediatr (Phila). 2023 Jun;62(5):399-403. doi: 10.1177/00099228221128661[↩]
- Arora S, Harith AK, Sodhi N. Congenital Erythropoietic Porphyria with Undescended Testis. Indian J Dermatol. 2016 Jul-Aug;61(4):467. doi: 10.4103/0019-5154.185749[↩]
- Kamalyan M, Mohammadi M. Congenital erythropoietic porphyria five years observation with standard treatment: a case report. Oxf Med Case Reports. 2024 Jan 27;2024(1):omad151. doi: 10.1093/omcr/omad151[↩][↩][↩]
- Peterlin P, Bonnelye J, Garnier A, Le Bourgeois A, Guillaume T, Jullien M, Dutartre H, Le Moigne M, Schmitt C, Gouya L, Poli A, Barbarot S, Chevallier P. Successful treatment of congenital erythropoietic porphyria using matched unrelated hematopoietic stem cell transplantation in an adult: A case report. Skin Health Dis. 2024 Jan 27;4(2):e342. doi: 10.1002/ski2.342[↩]
- Chao CB, Zhang Y. Biochemistry, Uroporphyrinogen. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK546643[↩][↩][↩][↩][↩][↩][↩]
- Desnick RJ, Astrin KH. Congenital erythropoietic porphyria: advances in pathogenesis and treatment. Br J Haematol. 2002 Jun;117(4):779-95. doi: 10.1046/j.1365-2141.2002.03557.x[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Warner CA, Poh-Fitzpatrick MB, Zaider EF, Tsai SF, Desnick RJ. Congenital erythropoietic porphyria. A mild variant with low uroporphyrin I levels due to a missense mutation (A66V) encoding residual uroporphyrinogen III synthase activity. Arch Dermatol. 1992 Sep;128(9):1243-8. doi: 10.1001/archderm.128.9.1243[↩]
- R.P. Katugampola, A.V. Anstey, A.Y. Finlay, S. Whatley, J. Woolf, N. Mason, J.C. Deybach, H. Puy, C. Ged, H. de Verneuil, S. Hanneken, E. Minder, X. Schneider‐Yin, M.N. Badminton, A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases, British Journal of Dermatology, Volume 167, Issue 4, 1 October 2012, Pages 888–900, https://doi.org/10.1111/j.1365-2133.2012.11154.x[↩]
- Aizencang G, Solis C, Bishop DF, Warner C, Desnick RJ. Human uroporphyrinogen-III synthase: genomic organization, alternative promoters, and erythroid-specific expression. Genomics. 2000 Dec 1;70(2):223-31. doi: 10.1006/geno.2000.6373[↩][↩]
- Caiulo A, Nicolis S, Bianchi P, Zuffardi O, Bardoni B, Maraschio P, Ottolenghi S, Camerino G, Giglioni B. Mapping the gene encoding the human erythroid transcriptional factor NFE1-GF1 to Xp11.23. Hum Genet. 1991 Feb;86(4):388-90. doi: 10.1007/BF00201840[↩][↩]
- Di Pierro E, Russo R, Karakas Z, Brancaleoni V, Gambale A, Kurt I, Winter SS, Granata F, Czuchlewski DR, Langella C, Iolascon A, Cappellini MD. Congenital erythropoietic porphyria linked to GATA1-R216W mutation: challenges for diagnosis. Eur J Haematol. 2015 Jun;94(6):491-7. doi: 10.1111/ejh.12452[↩][↩][↩][↩][↩][↩][↩]
- Di Pierro E, Brancaleoni V, Granata F. Advances in understanding the pathogenesis of congenital erythropoietic porphyria. Br J Haematol. 2016 May;173(3):365-79. doi: 10.1111/bjh.13978[↩][↩][↩]
- Schroeder WA, Shelton JB, Shelton JR, Huynh V, Teplow DB. High performance liquid chromatographic separation of the globin chains of non-human hemoglobins. Hemoglobin. 1985;9:461–482. doi: 10.3109/03630268508997024[↩][↩][↩]
- Bickers DR, Frank J, The Porphyrias. In: Fitzpatrick’s Dermatology in General Medicine, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K (eds), 8th (ed), McGraw-Hill, New York (NY), Chapter 132, (2012). https://accessmedicine.mhmedical.com/Content.aspx?bookId=392§ionId=41138853[↩][↩][↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ, Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias In: Metabolic The and Molecular Bases of Inherited Disease, 8th (ed), Scriver CR, Beaudet AL, Sly WS and Valle D, (eds), New York (NY), McGraw-Hill, (2014) 2961–3062.[↩][↩][↩][↩]
- Shirazi N., Chauhan B.P., Jindal R., Ahmad S. Congenital erythropoietic porphyria: a rare case of photosensitivity with hemolytic anaemia and mental retardation. J Coll Physicians Surg Pak. 2019;29(6):S23–S25. doi: 10.29271/jcpsp.2019.06.S23[↩]
- Congenital Erythropoietic Porphyria (CEP). https://porphyriafoundation.org/for-patients/types-of-porphyria/cep/[↩]
- Desnick RJ, Glass IA, Xu W, Solis C, Astrin KH. Molecular genetics of congenital erythropoietic porphyria. Semin Liver Dis. 1998;18(1):77-84. doi: 10.1055/s-2007-1007143[↩]
- Schulenburg-Brand D, Katugampola R, Anstey AV, Badminton MN. The cutaneous porphyrias. Dermatol Clin. 2014 Jul;32(3):369-84, ix. doi: 10.1016/j.det.2014.03.001[↩]
- Katugampola RP, Badminton MN, Finlay AY, Whatley S, Woolf J, Mason N, Deybach JC, Puy H, Ged C, de Verneuil H, Hanneken S, Minder E, Schneider-Yin X, Anstey AV. Congenital erythropoietic porphyria: a single-observer clinical study of 29 cases. Br J Dermatol. 2012 Oct;167(4):901-13. doi: 10.1111/j.1365-2133.2012.11160.x[↩][↩][↩]
- Piomelli S, Poh-Fitzpatrick MB, Seaman C, Skolnick LM, Berdon WE. Complete suppression of the symptoms of congenital erythropoietic porphyria by long-term treatment with high-level transfusions. N Engl J Med. 1986 Apr 17;314(16):1029-31. doi: 10.1056/NEJM198604173141607[↩][↩][↩][↩][↩]
- Tezcan I, Xu W, Gurgey A, Tuncer M, Cetin M, Oner C, Yetgin S, Ersoy F, Aizencang G, Astrin KH, Desnick RJ. Congenital erythropoietic porphyria successfully treated by allogeneic bone marrow transplantation. Blood. 1998 Dec 1;92(11):4053-8. https://doi.org/10.1182/blood.V92.11.4053[↩][↩]
- Thomas C, Ged C, Nordmann Y, de Verneuil H, Pellier I, Fischer A, Blanche S. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J Pediatr. 1996 Sep;129(3):453-6. doi: 10.1016/s0022-3476(96)70082-3[↩][↩][↩]
- Harada FA, Shwayder TA, Desnick RJ, Lim HW. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J Am Acad Dermatol. 2001 Aug;45(2):279-82. doi: 10.1067/mjd.2001.114730[↩][↩]
- Shaw PH, Mancini AJ, McConnell JP, Brown D, Kletzel M. Treatment of congenital erythropoietic porphyria in children by allogeneic stem cell transplantation: a case report and review of the literature. Bone Marrow Transplant. 2001 Jan;27(1):101-5. doi: 10.1038/sj.bmt.1702738[↩][↩]
- Dupuis-Girod S, Akkari V, Ged C, Galambrun C, Kebaïli K, Deybach JC, Claudy A, Geburher L, Philippe N, de Verneuil H, Bertrand Y. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Günther disease). Eur J Pediatr. 2005 Feb;164(2):104-7. doi: 10.1007/s00431-004-1575-x[↩][↩]
- Taibjee SM, Stevenson OE, Abdullah A, Tan CY, Darbyshire P, Moss C, Goodyear H, Heagerty A, Whatley S, Badminton MN. Allogeneic bone marrow transplantation in a 7-year-old girl with congenital erythropoietic porphyria: a treatment dilemma. Br J Dermatol. 2007 Mar;156(3):567-71. doi: 10.1111/j.1365-2133.2006.07699.x[↩][↩]
- Faraci M, Morreale G, Boeri E, Lanino E, Dallorso S, Dini G, Scuderi F, Cohen A, Cappelli B. Unrelated HSCT in an adolescent affected by congenital erythropoietic porphyria. Pediatr Transplant. 2008 Feb;12(1):117-20. doi: 10.1111/j.1399-3046.2007.00842.x[↩][↩]
- Katugampola RP, Anstey AV, Finlay AY, Whatley S, Woolf J, Mason N, Deybach JC, Puy H, Ged C, de Verneuil H, Hanneken S, Minder E, Schneider-Yin X, Badminton MN. A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases. Br J Dermatol. 2012 Oct;167(4):888-900. doi: 10.1111/j.1365-2133.2012.11154.x[↩][↩][↩][↩][↩][↩]
- Zix-Kieffer I, Langer B, Eyer D, Acar G, Racadot E, Schlaeder G, Oberlin F, Lutz P. Successful cord blood stem cell transplantation for congenital erythropoietic porphyria (Gunther’s disease). Bone Marrow Transplant. 1996 Jul;18(1):217-20.[↩][↩][↩]
- Kauffman L, Evans DI, Stevens RF, Weinkove C. Bone-marrow transplantation for congenital erythropoietic porphyria. Lancet. 1991 Jun 22;337(8756):1510-1. doi: 10.1016/0140-6736(91)93198-i[↩][↩]
- Lin, Jou & Shi, Donglu. (2021). Photothermal and photovoltaic properties of transparent thin films of porphyrin compounds for energy applications. Applied Physics Reviews. 8. 011302. https://doi.org/10.1063/5.0036961[↩]
- Panawala, Lakna. (2017). What is the Function of Hemoglobin in the Human Body. https://www.researchgate.net/publication/313841668_What_is_the_Function_of_Hemoglobin_in_the_Human_Body[↩]
- Edel Y, Mamet R. Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. 2018 Apr 19;9(2):e0013. doi: 10.5041/RMMJ.10333[↩]
- Bensidhoum ML, Lemeur M, Dierich M, Costet A, Raymond P, Daniel S, De Verneuil JY, Ged CH, The Disruption of Mouse Uroporphyrinogen III Synthase (uros) Gene is Fully Lethal. Transgenics 2 (1998) 275–280.[↩]
- Poh-Fitzpatrick MB. Porphyrin-sensitized cutaneous photosensitivity: pathogenesis and treatment. Clin Dermatol. 1985 Apr-Jun;3(2):41-82. doi: 10.1016/0738-081x(85)90034-3[↩][↩]
- Shoolingin-Jordan PM. Porphobilinogen deaminase and uroporphyrinogen III synthase: structure, molecular biology, and mechanism. J Bioenerg Biomembr. 1995 Apr;27(2):181-95. doi: 10.1007/BF02110033[↩]
- Fritsch C, Bolsen K, Ruzicka T, Goerz G. Congenital erythropoietic porphyria. J Am Acad Dermatol. 1997 Apr;36(4):594-610. doi: 10.1016/s0190-9622(97)70249-4[↩][↩][↩]
- Verstraeten L, Van Regemorter N, Pardou A, de Verneuil H, Da Silva V, Rodesch F, Vermeylen D, Donner C, Noël JC, Nordmann Y, et al. Biochemical diagnosis of a fatal case of Günther’s disease in a newborn with hydrops foetalis. Eur J Clin Chem Clin Biochem. 1993 Mar;31(3):121-8. doi: 10.1515/cclm.1993.31.3.121[↩][↩]
- Warner CA, Yoo HW, Roberts AG, Desnick RJ. Congenital erythropoietic porphyria: identification and expression of exonic mutations in the uroporphyrinogen III synthase gene. J Clin Invest. 1992 Feb;89(2):693-700. https://pmc.ncbi.nlm.nih.gov/articles/instance/442904/pdf/jcinvest00046-0357.pdf[↩][↩]
- Poh-Fitzpatrick MB. The erythropoietic porphyrias. Dermatol Clin. 1986 Apr;4(2):291-6.[↩][↩]
- Fityan A, Fassihi H, Sarkany R. Congenital erythropoietic porphyria: mild presentation with late onset associated with a mutation in the UROS gene promoter sequence. Clin Exp Dermatol. 2016 Dec;41(8):953-954. doi: 10.1111/ced.12932[↩]
- Deybach JC, de Verneuil H, Phung N, Nordmann Y, Puissant A, Boffety B. Congenital erythropoietic porphyria (Günther’s disease): enzymatic studies on two cases of late onset. J Lab Clin Med. 1981 Apr;97(4):551-8.[↩]
- Murphy A, Gibson G, Elder GH, Otridge BA, Murphy GM. Adult-onset congenital erythropoietic porphyria (Günther’s disease) presenting with thrombocytopenia. J R Soc Med. 1995 Jun;88(6):357P-358P[↩]
- Pain RW, Welch FW, Woodroffe AJ, Handley DA, Lockwood WH. Erythropoietic uroporphyria of Gunther first presenting at 58 years with positive family studies. Br Med J. 1975 Sep 13;3(5984):621-3. doi: 10.1136/bmj.3.5984.621[↩][↩]
- Kramer S, Viljoen E, Meyer AM, Metz J. The anaemia of erythropoietic prophyria with the description of the disease in an elderly patient. Br J Haematol. 1965 Nov;11(6):666-75. doi: 10.1111/j.1365-2141.1965.tb00115.x[↩]
- Rao SU, Dar NR, Abbas M, Mumtaz J. Late onset erythropoietic porphyria (Gunther’s disease). J Coll Physicians Surg Pak. 2011 Sep;21(9):564-6.[↩]
- Fritsch C, Lang K, Bolsen K, Lehmann P, Ruzicka T. Congenital erythropoietic porphyria. Skin Pharmacol Appl Skin Physiol. 1998 Nov-Dec;11(6):347-57. doi: 10.1159/000029857[↩]
- Schmid R, Schwartz S, Sundberg RD, Erythropoietic (congenital) porphyria: A rare abnormality of the normoblasts. Blood 10 (1955) 416–428.[↩]
- Weston MJ, Nicholson DC, Lim CK, Clark KG, Macdonald A, Henderson MA, Williams R. Congenital erythropoietic uroporphyria (Günther’s disease) presenting in a middle aged man. Int J Biochem. 1978;9(12):921-6. doi: 10.1016/0020-711x(78)90071-x[↩]
- Venkatesh P, Garg SP, Kumaran E, Tewari HK. Congenital porphyria with necrotizing scleritis in a 9-year-old child. Clin Exp Ophthalmol. 2000 Aug;28(4):314-8. doi: 10.1046/j.1442-9071.2000.00330.x[↩]
- Siddique SS, Gonzalez-Gonzalez LA, Thakuria P, Chang PY, Foster CS. Scleral necrosis in a patient with congenital erythropoietic porphyria. Cornea. 2011 Jan;30(1):97-9. doi: 10.1097/ICO.0b013e3181e458fa[↩]
- Oguz F, Sidal M, Bayram C, Sansoy N, Hekim N. Ocular involvement in two symptomatic congenital erythropoietic porphyria. Eur J Pediatr. 1993 Aug;152(8):671-3. doi: 10.1007/BF01955245[↩]
- Kontos AP, Ozog D, Bichakjian C, Lim HW. Congenital erythropoietic porphyria associated with myelodysplasia presenting in a 72-year-old man: report of a case and review of the literature. Br J Dermatol. 2003 Jan;148(1):160-4. doi: 10.1046/j.1365-2133.2003.05040.x[↩][↩]
- Laorr A, Greenspan A. Severe osteopenia in congenital erythropoietic porphyria. Can Assoc Radiol J. 1994 Aug;45(4):307-9.[↩]
- Daïkha-Dahmane F, Dommergues M, Narcy F, Gubler MC, Dumez Y, Gauthier E, Nordmann Y, Nessmann C, Terrasse G, Muller F. Congenital erythropoietic porphyria: prenatal diagnosis and autopsy findings in two sibling fetuses. Pediatr Dev Pathol. 2001 Mar-Apr;4(2):180-4. doi: 10.1007/s100240010143[↩]
- Mathews-Roth MM. Carotenoids in erythropoietic protoporphyria and other photosensitivity diseases. Ann N Y Acad Sci. 1993 Dec 31;691:127-38. doi: 10.1111/j.1749-6632.1993.tb26164.x[↩]
- Howard M, Hall A, Ramsay D. Beneficial use of a novel topical hypochlorous acid preparation for chronic dematoses at risk of secondary infection. Australas J Dermatol. 2016 Nov;57(4):326-327. doi: 10.1111/ajd.12451[↩]
- Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic Protoporphyria, Autosomal Recessive. 2012 Sep 27 [Updated 2017 Sep 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK100826[↩][↩]
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract. 2002 May;56(4):272-8.[↩]
- Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ. Afamelanotide for Erythropoietic Protoporphyria. N Engl J Med. 2015 Jul 2;373(1):48-59. doi: 10.1056/NEJMoa1411481[↩]
- Howard M, Hall A, Ramsay D. Congenital erythropoietic porphyria (Gunther disease) – long-term follow up of a case and review. Dermatol Online J. 2017 Feb 15;23(2):13030/qt10n7k90g[↩]
- Poh-Fitzpatrick MBP, S.; Seaman C; Skolnick LM, Congenital erythropoietic porphyria: complete suppression of symptoms by long-term high-level transfusion with deferoxamine infusion iron rescue, in: Orfanos RSHGCE (Ed.), Dermatology in Five Continents, Springer-Verlag, Berlin, 1988.[↩]
- Egan DN, Yang Z, Phillips J, Abkowitz JL. Inducing iron deficiency improves erythropoiesis and photosensitivity in congenital erythropoietic porphyria. Blood. 2015 Jul 9;126(2):257-61. doi: 10.1182/blood-2014-07-584664[↩][↩][↩]
- Blouin JM, Ged C, Lalanne M, Lamrissi-Garcia I, Morice-Picard F, Costet P, Daher R, Moreau-Gaudry F, Bedel A, Puy H, Gouya L, Karim Z, Richard E. Iron chelation rescues hemolytic anemia and skin photosensitivity in congenital erythropoietic porphyria. Blood. 2020 Nov 19;136(21):2457-2468. doi: 10.1182/blood.2020006037[↩][↩]
- Mirmiran A, Poli A, Ged C, Schmitt C, Lefebvre T, Manceau H, Daher R, Moulouel B, Peoc’h K, Simonin S, Blouin JM, Deybach JC, Nicolas G, Puy H, Richard E, Gouya L. Phlebotomy as an efficient long-term treatment of congenital erythropoietic porphyria. Haematologica. 2021 Mar 1;106(3):913-917. doi: 10.3324/haematol.2019.228270[↩][↩]
- Guarini L, Piomelli S, Poh-Fitzpatrick MB. Hydroxyurea in congenital erythropoietic porphyria. N Engl J Med. 1994 Apr 14;330(15):1091-2. doi: 10.1056/NEJM199404143301519[↩]
- Tishler PV, Winston SH. Rapid improvement in the chemical pathology of congenital erythropoietic porphyria with treatment with superactivated charcoal. Methods Find Exp Clin Pharmacol. 1990 Nov;12(9):645-8.[↩]
- Minder EI, Schneider-Yin X, Möll F. Lack of effect of oral charcoal in congenital erythropoietic porphyria. N Engl J Med. 1994 Apr 14;330(15):1092-4. doi: 10.1056/NEJM199404143301520[↩]
- Gorchein A, Guo R, Lim CK, Raimundo A, Pullon HW, Bellingham AJ. Porphyrins in urine, plasma, erythrocytes, bile and faeces in a case of congenital erythropoietic porphyria (Gunther’s disease) treated with blood transfusion and iron chelation: lack of benefit from oral charcoal. Biomed Chromatogr. 1998 Nov-Dec;12(6):350-6. doi: 10.1002/(SICI)1099-0801(199811/12)12:6<350::AID-BMC761>3.0.CO;2-B[↩]
- Wahlin S, Srikanthan N, Hamre B, Harper P, Brun A. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl. 2008 Sep;14(9):1340-6. doi: 10.1002/lt.21527[↩][↩]
- Hallai N, Anstey A, Mendelsohn S, Williams J, Evans-Jones G, Malick S, Badminton MN. Pregnancy in a patient with congenital erythropoietic porphyria. N Engl J Med. 2007 Aug 9;357(6):622-3. doi: 10.1056/NEJMc070009[↩]
- Urquiza P, Laín A, Sanz-Parra A, Moreno J, Bernardo-Seisdedos G, Dubus P, González E, Gutiérrez-de-Juan V, García S, Eraña H, San Juan I, Macías I, Ben Bdira F, Pluta P, Ortega G, Oyarzábal J, González-Muñiz R, Rodríguez-Cuesta J, Anguita J, Díez E, Blouin JM, de Verneuil H, Mato JM, Richard E, Falcón-Pérez JM, Castilla J, Millet O. Repurposing ciclopirox as a pharmacological chaperone in a model of congenital erythropoietic porphyria. Sci Transl Med. 2018 Sep 19;10(459):eaat7467. doi: 10.1126/scitranslmed.aat7467[↩]
- Sarkany RP, Ibbotson SH, Whatley SD, Lawrence CM, Gover P, Mufti GJ, Murphy GM, Masters GS, Badminton MN, Elder GH. Erythropoietic uroporphyria associated with myeloid malignancy is likely distinct from autosomal recessive congenital erythropoietic porphyria. J Invest Dermatol. 2011 May;131(5):1172-5. doi: 10.1038/jid.2011.5[↩]
- Cernik C, Haller N, Mostow EN. Adult-onset erythropoietic porphyria in the setting of MDS. Arch Dermatol. 2009 Aug;145(8):948-9. doi: 10.1001/archdermatol.2009.161[↩]
- Aguilera P, Badenas C, Whatley SD, To-Figueras J. Late-onset cutaneous porphyria in a patient heterozygous for a uroporphyrinogen III synthase gene mutation. Br J Dermatol. 2016 Dec;175(6):1346-1350. doi: 10.1111/bjd.14675[↩]





{kind=link}