Contents
What is craniopharyngioma
Craniopharyngiomas are rare benign (noncancerous) tumors that are typically found near the pituitary gland and hypothalamus, which are positioned beneath the center of the brain, behind and between the eyes. Craniopharyngiomas are not cancerous, and do not spread to other parts of the brain or to other parts of the body. Craniopharyngiomas tend to become attached to the pituitary gland or to nearby tissues, including the optic nerves, the arteries that feed into the brain, and the brain itself. For this reason, craniopharyngioma is extremely difficult to control, and it is also notorious for its high rates of recurrence 1. Craniopharyngiomas grow slowly and as they grow larger, put pressure on nearby tissue. This pressure can cause health problems by interfering with the function of the brain, the pituitary gland, the optic (eye) nerves, or arteries.
Craniopharyngiomas are usually a partly solid and partly fluid-filled cyst. Craniopharyngioma is a partly cystic embryonic malformation that can occur in the sellar/parasellar region and can produce a wide array of symptomatology such as headaches, nausea and vomiting, visual disturbances, and endocrine disturbances 2.
Craniopharyngiomas are thought to form during the development of the brain and pituitary gland, possibly as a remnant of structures that are present in the early stages of the human embryo but normally disappear before birth. The two structures in the embryo thought to play a role in the formation of craniopharyngiomas are the craniopharyngeal duct and Rathke’s pouch.
Craniopharyngiomas account for ~1-5% of primary brain tumors, and can occur anywhere along the infundibulum (from the floor of the third ventricle to the pituitary gland). Craniopharyngioma has an incidence of 0.5 to 2 cases per million persons per year 2. Almost half of craniopharyngiomas cases occur during the first two decades of life. It represents 1.2% to 4% of all childhood intracranial tumors.
Although craniopharyngiomas are found in patients of all ages, there is a bimodal distribution 3. The first peak occurs between the ages of 5-15 years, consisting almost exclusively of the adamantinomatous subtype. A second, smaller peak occurs in adults aged over 40 to 74 years old, consisting of both papillary and adamantinomatous subtypes 3. The papillary subtype is found almost exclusively in adults 3.
No statistically significant differences have been described regarding demographic characteristics such as age, gender, race, and geographical location. Interesting instances of craniopharyngioma have been reported, including craniopharyngiomas of the cerebellopontine angle, malignant transformation of craniopharyngiomas and familial cases of craniopharyngiomas 4.
Craniopharyngioma has a very high recurrence rate, with reported rates as high as 50% 2. Craniopharyngioma also has high survival rates (83% to 96% five-year survival and 65% to 100% 10-year survival) but also carries similar rates of morbidity, with almost all patients developing some complications.
The goal of treatment for craniopharyngiomas is to relieve symptoms. Because the symptoms are generally caused by pressure from the growing tumor on the brain, the optic nerves or the pituitary gland, the way to relieve the symptoms is usually to remove or shrink the tumor.
- Surgery may be done to remove all or most of the tumor. Surgeons may reach the tumor through an incision in the upper lip or at the bottom of the nose (endonasal surgery), or by cutting through the skull.
- Radiation therapy may be used in some cases to kill tumor cells or shrink the tumor after surgery.
Craniopharyngioma key points
- Childhood craniopharyngiomas are benign brain tumors found near the pituitary gland.
- There are no known risk factors for childhood craniopharyngioma.
- Signs of childhood craniopharyngioma include vision changes and slow growth.
- Tests that examine the brain, vision, and hormone levels are used to detect (find) childhood craniopharyngiomas.
- Childhood craniopharyngiomas are diagnosed and may be removed in the same surgery.
- Certain factors affect prognosis (chance of recovery) and treatment options.
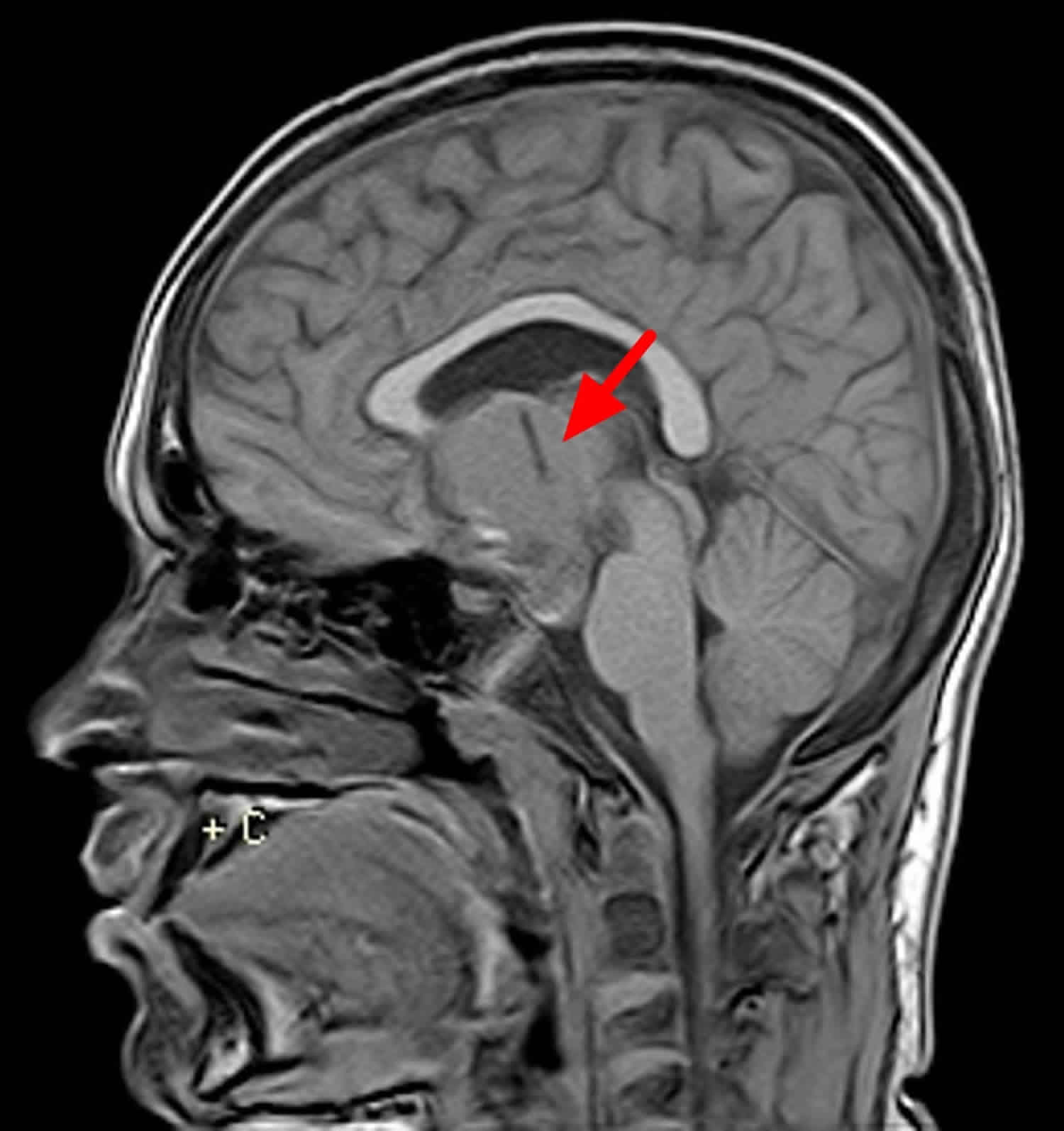
Craniopharyngioma location
In the vast majority of cases, craniopharyngiomas have a large suprasellar component (95%), with most involving both the suprasellar and intrasellar spaces (75%). A minority are purely suprasellar (20%), whereas purely intrasellar location is quite uncommon (<5%), and may be associated with the expansion of the pituitary fossa 5. Larger tumors can extend in all directions, frequently distorting the optic chiasm, or compressing the midbrain with resulting obstructive hydrocephalus.
Occasionally, craniopharyngiomas appear as intraventricular, homogeneous, soft-tissue masses without calcification (papillary subtype). The third ventricle is a particularly common location.
Rare or ectopic locations reported include: nasopharynx, posterior fossa, extension down the cervical spine.
Craniopharyngioma histological subtypes
There are two histological subtypes of craniopharyngioma: adamantinomatous and papillary, which are said to differ not only in appearance but also in prognosis and epidemiology. A mixed or transitional subtype has also been described, although imaging features and prognosis are similar to the adamantinomatous subtype 6. Whether or not they represent distinct entities or a spectrum of morphology remains a bit controversial 7.
Adamantinomatous craniopharyngioma subtype is more common than the papillary subtype by a 3-9-fold difference 3.
Adamantinomatous craniopharyngioma
Adamantinomatous craniopharyngioma is characterized by dense nodules and trabeculae of squamous epithelium bordered by a palisade of columnar epithelium sometimes referred to as a “picket fence.” These nests of squamous epithelium are surrounded by loose aggregates of squamous epithelium known as stellate reticulum. Spread between the mix, one can find cystic cavities which hold an oily proteinaceous fluid along with cholesterol, piloid gliosis, granulomatous inflammation, calcification, and nodules of “wet keratin.”
Papillary craniopharyngioma
On the other hand, papillary craniopharyngioma is characterized as well-differentiated squamous epithelium lacking surface maturation, with occasional goblet cells and ciliated epithelium. It is not nearly as organized as the adamantinomatous subtype, and calcifications are rare.
Craniopharyngioma vs Pituitary adenoma
Pituitary adenomas are benign tumor of the pituitary gland that do not spread to other parts of the body. Almost all pituitary tumors are benign (not cancer) glandular tumors called pituitary adenomas. Despite being benign, pituitary adenomas can cause major health problems because they are close to the brain, may invade nearby tissues (like the skull or the sinuses). Pituitary adenomas can make pituitary gland produce too much or too little of certain hormones, causing health problems.
Pituitary adenomas usually stay in the sella turcica (the tiny space in the skull that the pituitary gland sits in). Sometimes pituitary adenomas grow into the bony walls of the sella turcica and nearby tissues, like blood vessels, nerves, and sinuses. Pituitary adenomas don’t grow very large, but they can have a big impact on a person’s health. There is very little room for tumors to grow in this part of the skull. So, if the pituitary adenoma gets larger than about a centimeter (about half an inch) across, it may grow upward, where it can press on and damage nearby parts of the brain and the nerves that arise from it. This can lead to problems like vision changes or headaches.
Pituitary adenomas can be divided into 2 categories based on size:
- Microadenomas are smaller than 1 centimeter. Most pituitary adenomas are microadenomas. Because these tumors are small, they rarely damage the rest of the pituitary or nearby tissues. But they can cause symptoms if they make too much of a certain hormone. Many people actually have small adenomas that are never found because they don’t grow large enough or make enough hormones to cause a problem.
- Macroadenomas are 1 centimeter or larger. Macroadenomas can affect a person’s health in 2 ways. First, they can cause symptoms if they make too much of a certain hormone. Second, they can cause symptoms by pressing on normal parts of the pituitary or on nearby nerves, such as the optic nerves.
Functional versus non-functional adenoma
Pituitary adenomas are also classified by whether they make too much of a hormone and, if they do, which type they make. If a pituitary adenoma makes too much of a hormone it’s called functional or secretory tumors. If it doesn’t make enough hormones to cause problems it’s called non-functional 8.
Functional pituitary adenomas
Most of the pituitary adenomas that are found make excess hormones. The hormones can be detected by blood tests or by tests of the tumor when it is removed with surgery.
- These hormones play important roles in the healthy functioning of the body:
- Prolactin causes a woman’s breasts to make milk during and after pregnancy.
- Adrenocorticotropic hormone (ACTH) is involved in the body’s response to stress.
- Growth hormone (GH) helps control body growth and metabolism.
- Thyroid-stimulating hormone (TSH) is involved in growth, body temperature, and heart rate.
Based on these results, pituitary adenomas are classified as:
- Lactotroph adenomas make prolactin and account for about 4 out of 10 pituitary tumors.
- Somatotroph adenomas make growth hormones and make up about 2 in 10 pituitary tumors.
- Corticotroph adenomas make ACTH and account for about 1 in 10 pituitary tumors.
- Gonadotroph adenomas make LH and FSH and are very rare.
- Thyrotroph adenomas make TSH and are very rare.
- Plurihormonal adenomas make more than one hormone.
- Null cell adenomas do not make hormones. (These are non-functional adenomas.)
The kind of hormone an adenoma makes strongly affects what signs and symptoms it causes. It also affects which tests are used for diagnosis, the choice of treatment, and the patient’s outlook.
Non-functional adenomas
Pituitary adenomas that don’t make excess hormones are called non-functional adenomas or null cell adenomas. They account for about 3 in 10 of all pituitary tumors that are found. They are usually found as macroadenomas, causing symptoms because of their size as they press on nearby structures.
Pituitary adenomas signs and symptoms
The symptoms of functioning pituitary adenomas depend on the particular hormone the tumor is overproducing.
Prolactin
A pituitary tumor that produces too much prolactin may cause:
- Headache
- Some loss of vision
- Less frequent or no menstrual periods or menstrual periods with a very light flow
- Difficulty getting pregnant
- Impotence in men
- Lower sex drive
- The flow of breast milk in a woman who is not pregnant or breastfeeding
Adrenocorticotropic Hormone (ACTH)
A pituitary tumor that produces too much adrenocorticotropic hormone (ACTH) may cause:
- Headache
- Some loss of vision
- Weight gain reflected in the face, neck, and trunk of the body, but thin arms and legs
- A lump of fat on the back of the neck
- Thin skin that may include purple or pink stretch marks on the chest or abdomen
- Easy bruising
- Growth of fine hair on the face, upper back, or arms
- Bones that break easily
- Anxiety, irritability, depression
- Growth deceleration with weight gain in children
- Irregular menses
Growth Hormone
A pituitary tumor that produces too much growth hormone may cause:
- Headache
- Some loss of vision
- In adults, growth of the bones in the face, hands, and feet
- In children, excessive growth of the whole body
- Tingling or numbness in the hands and fingers
- Snoring or pauses in breathing during sleep
- Joint pain
- Sweating more than usual
- Extreme dislike of or concern about one or more parts of the body
Thyroid-Stimulating Hormone (TSH)
A pituitary tumor that produces too much TSH (through high T4) may cause:
- Irregular heartbeat
- Shakiness
- Weight loss
- Trouble sleeping
- Frequent bowel movements
- Sweating
Symptoms of Nonfunctioning Tumors
Nonfunctioning tumors press on or damage the pituitary and prevent it from secreting enough hormones. If there is too little of a particular hormone, the gland or organ it normally controls will not function correctly. Symptoms of nonfunctioning pituitary tumors are1:
- Headache
- Some loss of vision
- Loss of body hair
- In women, less frequent menstrual periods or no periods at all, or no milk from the breasts
- In men, loss of facial hair, growth of breast tissue, and impotence
- In women and men, lower sex drive
- In children, slowed growth and sexual development
Other General Symptoms of Pituitary Tumors
- Nausea and vomiting
- Confusion
- Dizziness
- Seizure
- Runny or drippy nose
Craniopharyngioma causes
Craniopharyngiomas have no clear cause. There are two major theories of the development of craniopharyngioma: the embryonic theory and the metaplastic theory. These two theories correlate with the two subtypes of craniopharyngioma which are the adamantinomatous craniopharyngiomas and the papillary craniopharyngiomas.
The embryonic theory is related to the development of adamantinomatous craniopharyngiomas, which are believed to be the most common subtype of craniopharyngioma that occurs in the pediatric population. During embryogenesis, there is an outpouching of the ectodermal roof of the stomodeum. This outpouching, known as Rathke’s pouch, extends cranially towards the floor of the diencephalon to later form the adenohypophysis or anterior pituitary gland. While migrating cranially, its extension forms the craniopharyngeal duct which later involutes. In some occasions, involution is not total, and remnants of ectodermal cells can be present. These embryonic cells can proliferate around the extension of the craniopharyngeal duct and develop into a craniopharyngioma.
The metaplastic theory is related to the development of papillary craniopharyngiomas, which are believed to be the most common subtype of craniopharyngioma in adult patients. It states that adenohypophyseal cells of the pars tuberalis can undergo metaplasia and result in the formation of squamous cell nests. These nests can then proliferate and lead to a papillary craniopharyngioma.
Craniopharyngioma symptoms
Symptoms of craniopharyngiomas typically appear in childhood, usually between the ages of 5 and 14. Symptoms vary widely, depending on the tumor’s size and location.
Craniopharyngioma most commonly manifests with signs of increased intracranial pressure including a headache and nausea and vomiting along with visual and endocrine disturbances (62% to 84% and 52% to 87%, respectively) 2. The most common visual disturbance encountered is temporal hemianopsia due to optic chiasm compression. The onset of blindness is considered a neurosurgical emergency. At the time of presentation, around 40% to 87% of patients present with at least one hormonal deficit. The hormonal deficiency is secondary to normal pituitary compression, particularly of the anterior pituitary. In some cases, posterior pituitary hormonal deficiencies can be seen, particularly diabetes insipidus. In children, failure to thrive and decreased growth rate can be the initial presentation as growth hormone is the most commonly affected hormone.
If the craniopharyngioma tumor is putting pressure on the brain, symptoms may include:
- Headache
- Nausea and vomiting
- Balance problems or trouble walking
If the craniopharyngioma tumor is putting pressure on or interfering with the function of the optic nerves, symptoms may include:
- Vision problems, such as double vision or narrowed field of vision
If the craniopharyngioma tumor is putting pressure on or interfering with the function of the pituitary gland, symptoms may include:
- Slowed growth or short stature
- Excessive thirst and urination
- Delayed puberty
- Low energy, listlessness or unusual sleepiness
- Weight gain
- Changes in personality or behavior
- Intolerance of cold temperatures
Craniopharyngioma diagnosis
Your doctor will usually begin with a physical exam and with questions about any symptoms you may have noticed.
- Height and weight will be charted to look for changes in growth patterns.
- The doctor may check for lumps.
If signs indicate the possibility of a pituitary tumor, additional tests may include:
- Neurological exam to check mental status, coordination, reflexes and muscle function
- Eye test to check for loss of vision, including narrowing field of vision
- Blood tests to check for high or low levels of hormones and blood sugar
- Magnetic resonance imaging (MRI) or computerized tomography (CT) scan to get visual images of the pituitary gland, brain and spinal cord, sometimes repeated over time to find out whether or how quickly the tumor is growing
Craniopharyngioma radiology
Adamantinomatous craniopharyngioma
Adamantinomatous craniopharyngiomas typically have a lobulated contour as a result of usually being multiple cystic lesions. Solid components are present, but often form a relatively minor part of the mass and enhance vividly on both CT and MRI. Overall, calcification is very common, but this is only true of the adamantinomatous subtype (~90% are calcified) 6.
These tumors have a predilection to being large, extending superiorly into the third ventricle, and encasing vessels, and even adhering to adjacent structures 6.
CT scan
cysts
- near-CSF density
- typically large and a dominant feature
- present in 90% of cases
solid component
- soft tissue density
- enhancement in 90%
calcification
- seen in 90%
- typically stippled and often peripheral in location
MRI scan
cysts
- T1: iso- to hyperintense to brain (due to high protein content “motor oil cysts”)
- T2: variable but ~80% are mostly or partly T2 hyperintense
solid component
- T1 C+ (Gd): vivid enhancement
- T2: variable or mixed
calcification
- difficult to appreciate on conventional imaging
- susceptible sequences may better demonstrate calcification
MR angiography: may show displacement of the A1 segment of the anterior cerebral artery (ACA)
MR spectroscopy: cyst contents may show a broad lipid spectrum, with an otherwise flat baseline 9
Papillary craniopharyngioma
Papillary craniopharyngiomas tend to be more spherical in outline and usually lack the prominent cystic component; most are either solid or contain a few smaller cysts. Calcification is uncommon or even rare in the papillary subtype, a fact often forgotten 6.
These tumors tend to displace adjacent structures.
CT scan
cysts
- small and not a significant feature
- near-CSF density
solid component
- soft tissue density
- vivid enhancement
calcification
- uncommon, rare
MRI scan
cysts
- when present, they are variable in signal
- T1: 85% are T1 hypointense
solid component
- T1: iso- to slightly hypointense to brain
- T1 C+ (Gd): vivid enhancement
- T2: variable/mixed
MR spectroscopy: cyst contents does not show a broad lipid spectrum as they are filled with aqueous fluid
Craniopharyngioma treatment
Multiple modalities can be implemented in the management of craniopharyngioma, including neurological surgery, radiotherapy, and instillation of sclerosing substances. There is no consensus on the best treatment regimen. The types of modalities chosen depend on neurosurgeon judgment and experience.
Craniopharyngioma surgery
The most common surgical approaches include pterional, subfrontal, and transsphenoidal. Transcallosal approaches for craniopharyngiomas with third ventricle extension, retrosigmoid approaches for posterior fossa craniopharyngiomas and transorbital approaches have also been described. Extension of resection is a matter of debate. Gross total resection has been associated with increased incidence of post-surgical deficits, with no clear benefits in regards to recurrence rates. Unless the tumor is clearly visualized and with no extension into neural structures including the hypothalamus, optic nerves, optic chiasm, and/or carotids, most neurosurgeons will favor a partial resection. These partial resections can be accompanied by adjuvant methods including radiotherapy and instillation of sclerosing substances. For the later, in cases of cystic tumors, the surgeon can introduce a catheter into the tumor and connect it to a reservoir. The reservoir then permits instillation of substances into the tumor as well as aspiration of cystic fluid, which can be performed as an outpatient procedure.
Radiotherapy
Radiation therapy includes various modalities: conventional external radiotherapy, proton beam therapy, stereotactic radiotherapy, radiosurgery, and brachytherapy. The goal of radiotherapy is to decrease tumor burden while protecting essential neural structures. Specific Gray doses have been designated for every radiation modality. Multiple reports have suggested decreased mortality with slightly reduced morbidity following radiation therapy. Despite this, radiation therapy has not been proven to reduce recurrence rate. Therefore, it continues to be an adjuvant modality to neurosurgical intervention.
Instillation of sclerosing substances
This method consists of instillation of different toxic substances with the endpoint of producing tumor fibrosis and sclerosis, for example, radioactive isotopes, bleomycin, interferon alpha. This method has been reported to produce significant cyst shrinkage, but prospective data are still missing, making it a promising option. A disadvantage of this option is that severe neurotoxicity can occur in some cases due to cystic leakage of the sclerosing substance.
Craniopharyngioma prognosis
For most children and adolescents with craniopharyngiomas, treatment is effective in removing or stopping the growth of the tumor.
Pituitary hormone replacements are managed by experienced pediatric endocrinologists. Treatment may include replacing thyroid hormone, growth hormone, pubertal hormones, adrenal steroids, and desmopressin (DDAVP).
Some damage caused by craniopharyngiomas, such as injury to the optic nerves, the brain or the pituitary gland, may be permanent and will not be relieved by removal or shrinkage of the tumor. In some cases, surgery to remove the tumor could damage the tissue to which the tumor is attached. If the tumor is not completely removed, the condition may return.
For children for whom surgical treatment is effective, ongoing surveillance and lifelong pituitary hormone replacement is the mainstay of therapy.
Follow-up care
Lifelong treatment or follow-up is generally needed.
- If the treatment has removed the tumor, follow-up tests and imaging will be needed periodically to make sure the tumor has not returned.
- Medication to replace hormone production may be needed if the function of the pituitary gland has been damaged by the craniopharyngioma tumor.
- Qi W, Gu F, Wu C. Growth hormone replacement therapy improves hypopituitarism-associated hypoxemia in a patient after craniopharyngioma surgery: A case report. Medicine (Baltimore). 2019 Jan;98(3):e14101[↩]
- Ortiz Torres M, Mesfin FB. Craniopharyngioma. [Updated 2019 Jan 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459371[↩][↩][↩][↩]
- Feng,Ying, Ni,Ming, Wang,Yong‑Gang, Zhong,Li‑Yong. Comparison of neuroendocrine dysfunction in patients with adamantinomatous and papillary craniopharyngiomas. (2019) Experimental and Therapeutic Medicine. 17 (1): 51. doi:10.3892/etm.2018.6953[↩][↩][↩][↩]
- Zacharia BE, Bruce SS, Goldstein H, Malone HR, Neugut AI, Bruce JN. Incidence, treatment and survival of patients with craniopharyngioma in the surveillance, epidemiology and end results program. (2012) Neuro-oncology. 14 (8): 1070-8. doi:10.1093/neuonc/nos142[↩]
- Bernstein M. Neuro-oncology, the essentials. Thieme. (2007) ISBN:1588904970.[↩]
- Sartoretti-Schefer S, Wichmann W, Aguzzi A et-al. MR differentiation of adamantinous and squamous-papillary craniopharyngiomas. AJNR Am J Neuroradiol. 1997;18 (1): 77-87.[↩][↩][↩][↩]
- Eldevik OP, Blaivas M, Gabrielsen TO et-al. Craniopharyngioma: radiologic and histologic findings and recurrence. AJNR Am J Neuroradiol. 1996;17 (8): 1427-39.[↩]
- https://www.cancer.gov/types/pituitary/patient/about-pituitary-tumors-pdq[↩]
- Keating RF, Goodrich JT, Packer RJ. Tumors of the pediatric central nervous system. George Thieme Verlag. (2001) ISBN:0865778485[↩]




{kind=link}