Contents
Duchenne muscular dystrophy
Duchenne muscular dystrophy is one of the most severe forms of inherited muscular dystrophies that is characterized by progressive muscle degeneration and weakness due to the alterations of a protein called dystrophin that helps keep muscle cells intact. Duchenne muscular dystrophy primarily affects the skeletal muscles, which are used for movement, and heart (cardiac) muscle. Duchenne muscular dystrophy occurs almost exclusively in males, but in rare cases it can affect girls. Duchenne muscular dystrophy does not exhibit a predilection for any race or ethnic group.
Duchenne muscular dystrophy is one of four conditions known as muscular dystrophies or dystrophinopathies. The other three diseases that belong to muscular dystrophies group are Becker muscular dystrophy, a mild form of Duchenne muscular dystrophy; an intermediate clinical presentation between Duchenne muscular dystrophy and Becker muscular dystrophy; and Duchenne muscular dystrophy-associated dilated cardiomyopathy (heart-disease) with little or no clinical skeletal, or voluntary, muscle disease.
Duchenne muscular dystrophy and Becker muscular dystrophy have similar signs and symptoms and are caused by different mutations in the same DMD gene or dystrophin gene. The two conditions differ in their severity, age of onset, and rate of progression. In boys with Duchenne muscular dystrophy, muscle weakness tends to appear in early childhood, usually between ages 2 and 3 and worsen rapidly. Affected children may have delayed motor skills, such as sitting, standing, and walking. They are usually wheelchair-dependent by adolescence. The signs and symptoms of Becker muscular dystrophy are usually milder and more varied. In most cases, muscle weakness becomes apparent later in childhood or in adolescence and worsens at a much slower rate.
Duchenne muscular dystrophy is usually first diagnosed when a child is three to four years old, although symptoms are common earlier than this. Early signs of Duchenne muscular dystrophy include:
- toe-walking – children start walking on their tip toes
- larger than normal calf muscles, which is called pseudohypertrophy
- a waddling type of walk
- inability to run or climb stairs
- an unusual way of getting off the floor, called a Gowers sign.
Some children with Duchenne muscular dystrophy also have delay in their speech development, and many will not walk until after 18 months of age.
Both the Duchenne and Becker forms of muscular dystrophy are associated with a heart condition called cardiomyopathy. This form of heart disease weakens the cardiac muscle, preventing the heart from pumping blood efficiently. In both Duchenne and Becker muscular dystrophy, cardiomyopathy typically begins in adolescence. Later, the heart muscle becomes enlarged, and the heart problems develop into a condition known as dilated cardiomyopathy. Signs and symptoms of dilated cardiomyopathy can include an irregular heartbeat (arrhythmia), shortness of breath, extreme tiredness (fatigue), and swelling of the legs and feet. These heart problems worsen rapidly and become life-threatening in most cases. Males with Duchenne muscular dystrophy typically live into their twenties, while males with Becker muscular dystrophy can survive into their forties or beyond.
A related condition called X-linked dilated cardiomyopathy is a form of heart disease caused by mutations in the same gene as Duchenne and Becker muscular dystrophy, and it is sometimes classified as subclinical Becker muscular dystrophy. People with X-linked dilated cardiomyopathy typically do not have any skeletal muscle weakness or wasting, although they may have subtle changes in their skeletal muscle cells that are detectable through laboratory testing.
The estimated incidence is 1 in 3600 male live-born infants. In Europe and North America, the prevalence of Duchenne muscular dystrophy is approximately 6 per 100,000 individuals 1. Some studies have estimated the prevalence of DMD as 2 per 10,000 in the United States. Between 400 and 600 boys in the United States are born with Duchenne muscular dystrophy and Becker muscular dystrophy each year.
Current therapy is centered on treatment with glucocorticoids and physiotherapy to prevent orthopedic complications 2.
Duchenne muscular dystrophy key points to remember
- Duchenne muscular dystrophy is a progressive disease causing increasing weakness of the muscles of the arms and legs, the breathing muscles and the heart.
- Duchenne muscular dystrophy can be inherited or may occur in only one family member. Genetic testing is recommended, especially if you have a family history of neuromuscular disease.
- It is essential to keep regular appointments with a neurologist, physiotherapist and other health care professionals.
- Duchenne muscular dystrophy is a progressive disorder and your child’s needs will increase as they get older.
Can my son attend a normal kindergarten and school?
Most children with Duchenne muscular dystrophy are able to attend mainstream kindergartens and schools, though appropriate adjustments need to be made for their physical and learning needs. Your child will have an Individualised Learning Plan and may qualify for additional support, in the form of an aide or school modifications.
Are any learning problems associated with Duchenne muscular dystrophy?
Up to one third of boys with Duchenne muscular dystrophy have a learning problem, but these are unlikely to be significant. There are also increased risks of ADHD, dyslexia and cognitive skills.
What is Gower’s sign?
Gower’s sign is an unusual way of standing up from the floor, where a child uses their upper limbs to compensate for weak lower limbs. A child will push themselves up on their arms and knees, then use their hands to ‘walk up’ their legs before standing upright.
Duchenne muscular dystrophy causes
Mutations in the DMD gene (dystrophin gene) located on chromosome Xp21, cause the Duchenne muscular dystrophy and Becker muscular dystrophy 3. Most mutations are deletions and duplications, and this accounts for 70% to 80% of the mutations. Point mutations are seen in 20% to 30% of patients. DMD gene is the largest known human gene (containing 79 exons of a coding sequence and 2.5 Mb of DNA), provides instructions for making a protein called dystrophin. This protein is located primarily in muscles used for movement (skeletal muscles) and in heart (cardiac) muscle, where it helps stabilize and protect muscle fibers. Small amounts of dystrophin are present in nerve cells in the brain 4. Dystrophin may also play a role in chemical signaling within cells.
In skeletal and cardiac muscles, dystrophin is part of a group of proteins (a protein complex) that work together to strengthen muscle fibers and protect them from injury as muscles contract and relax. The dystrophin complex acts as an anchor, connecting each muscle cell’s structural framework (cytoskeleton) with the lattice of proteins and other molecules outside the cell (extracellular matrix). The dystrophin complex may also play a role in cell signaling by interacting with proteins that send and receive chemical signals.
Little is known about the function of dystrophin in nerve cells. Research suggests that the protein is important for the normal structure and function of synapses, which are specialized connections between nerve cells where cell-to-cell communication occurs.
Mutations in the DMD gene alter the structure or function of dystrophin or prevent any functional dystrophin from being produced. Muscle cells without enough of this protein become damaged as muscles repeatedly contract and relax with use. The damaged fibers weaken and die over time, leading to the muscle weakness and heart problems characteristic of Duchenne and Becker muscular dystrophies. Mutations that lead to an abnormal version of dystrophin that retains some function usually cause Becker muscular dystrophy, while mutations that prevent the production of any functional dystrophin tend to cause Duchenne muscular dystrophy.
Because Duchenne and Becker muscular dystrophies result from faulty or missing dystrophin, these conditions are classified as dystrophinopathies.
Duchenne muscular dystrophy inheritance pattern
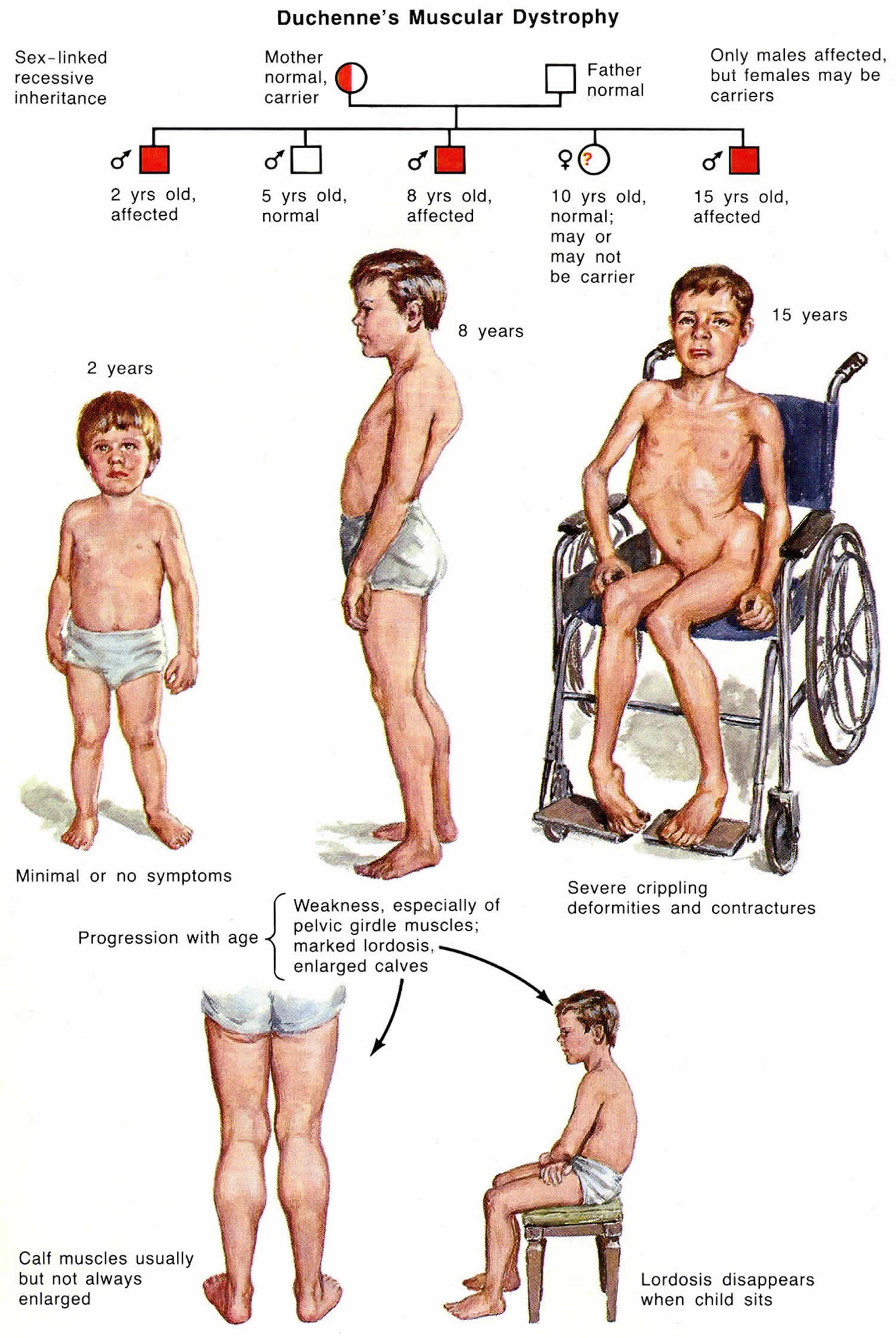
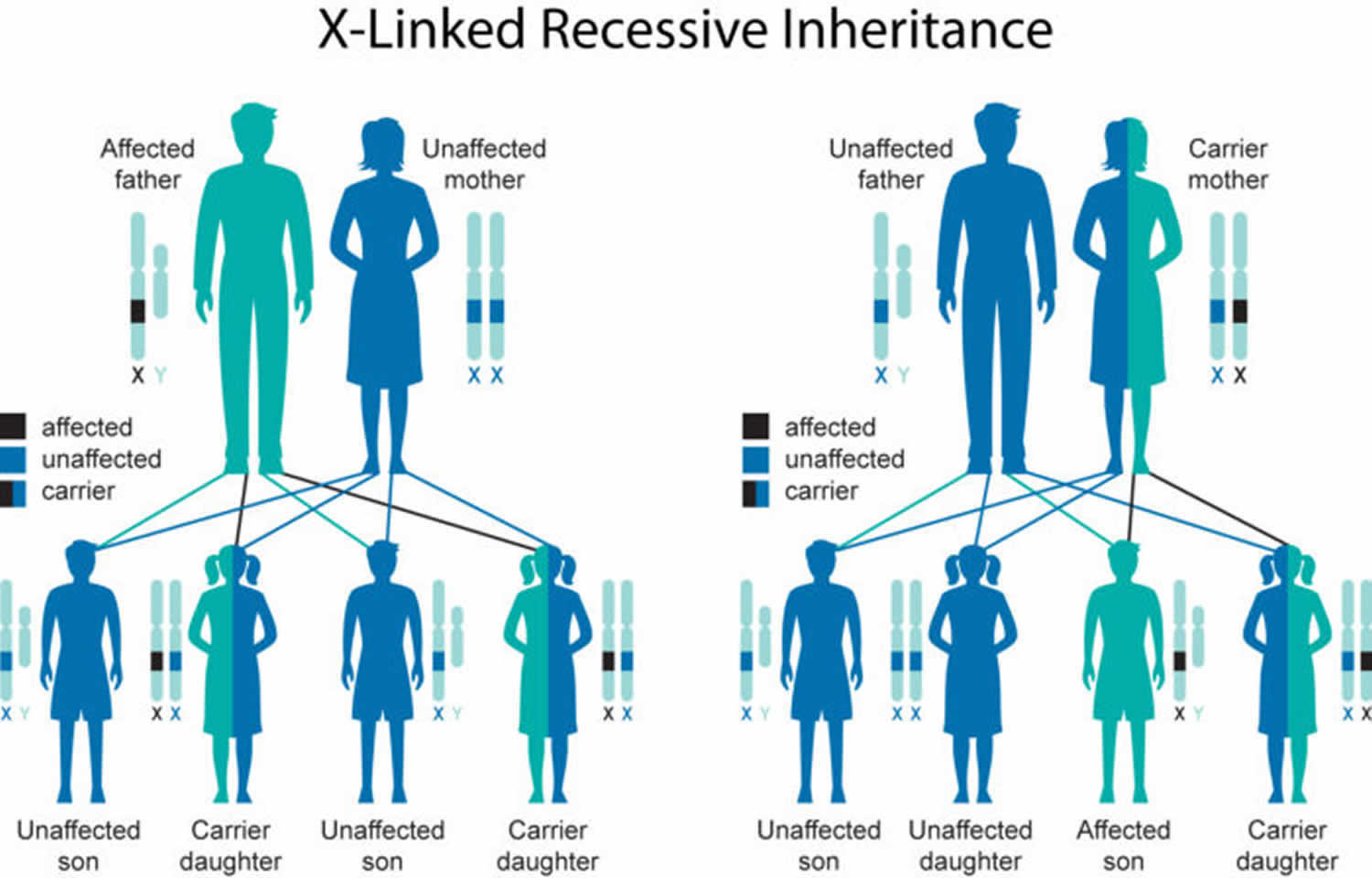
Duchenne muscular dystrophy is inherited in an X-linked recessive pattern; however, approximately 30% of cases are due to new mutations 5. The DMD gene associated with Duchenne muscular dystrophy is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause Duchenne muscular dystrophy. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause Duchenne muscular dystrophy. Because it is unlikely that females will have two altered copies of the DMD gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In many cases, an affected male inherits the mutation from his mother, who carries one altered copy of the DMD gene. The remainder of cases probably result from new mutations in the gene in affected males and are not inherited.
In X-linked recessive inheritance, a female with one mutated copy of the gene in each cell is called a carrier. She can pass on the altered gene but usually does not experience signs and symptoms of the disorder. Carrier females show no evidence of muscular weakness; however, symptomatic female carriers have been described. About 2.5% to 20% of female carriers (females who carry a DMD gene mutation) may have muscle weakness and cramping. This can be explained by the Lyon hypothesis in which the normal X chromosome becomes inactivated, and the X chromosome with the mutation is expressed 6. These symptoms are typically milder than the severe muscle weakness and atrophy seen in affected males. Females who carry a DMD gene mutation also have an increased risk of developing heart abnormalities including cardiomyopathy.
Female carriers can become symptomatic if they are associated with Turners syndrome (45X) or mosaic Turner karyotype, balanced X autosome translocations with breakpoints within the dystrophin gene and preferential inactivation of the normal X, and females with a normal karyotype but with nonrandom X chromosome inactivation with diminished expression of the normal dystrophin allele 6.
Figure 1. Duchenne muscular dystrophy X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Duchenne muscular dystrophy symptoms
Duchenne muscular dystrophy usually becomes apparent early during childhood. Weakness related to Duchenne muscular dystrophy selectively affects the limb muscles close to the trunk before the ones far from it; the legs are affected before the arms. Development in the first few years of life is typically normal, with milestones achieved at a slightly delayed if not normal rate. Growth velocity with Duchenne muscular dystrophy is typically slower than normal in the first years of life, leading to short stature. Mild hypotonia in an infant may be present, and poor head control in an infant may be an initial sign. Patients do not have atypical facies, but with the onset of facial muscle weakness, a transverse or horizontal sign may be seen in later childhood. Weakness and difficulty in ambulation in typically first noted between 2 and three years of life. This manifests as toe walking, difficulty running, climbing up stairs, and frequently falling. Weakness is more pronounced in proximal than distal muscles and the lower limb more than the upper limb.
Boys with Duchenne muscular dystrophy are often late walkers. Patients must use a wheelchair by the age of 12. In ambulatory children, an increased incidence of fractures is noted as a consequence of the frequent falls.
Affected children develop weakness and wasting (atrophy) of the muscles closest to the trunk (proximal muscles) such as those of the upper legs and pelvic area and upper arms and shoulder area. However, a few other muscles appear disproportionally bulky. As the disease progresses, muscle weakness and atrophy spread to affect the lower legs, forearms, neck and trunk. The rate of progression is quite similar from person to person but individual variation may happen.
In children with Duchenne muscular dystrophy, initial findings may include delays in reaching developmental milestones such as sitting or standing without assistance; toe walking; an unusual, waddling manner of walking (Trendelenburg gait); difficulty climbing stairs or rising from a sitting position (Gower’s sign); and repeated falling. Toddlers and young children may seem awkward and clumsy and may exhibit abnormal enlargement of the calves due to scarring of muscles (pseudohypertrophy). Aside from the calves, hypertrophy of the tongue and muscles of the forearm may be seen but are less classical. Parents may be falsely encouraged by an apparent improvement between the ages of 3 and 5, but this may be due to natural growth and development. As the disease progresses, additional abnormalities may develop such as progressive curvature of the spine (scoliosis or lumbar lordosis), wasting of thigh and pectoral muscles, and abnormal fixation of certain joints (contractures). A contracture occurs when thickening and shortening of tissue such as muscle fibers causes deformity and restricts movement of affected areas, especially the joints. Contractures of the ankles, knees, hips, and elbows may be seen. Without physical therapy treatment, leg braces may be needed by age 8-9 to assist affected individuals to walk. By approximately ages 10 to 12, most affected individuals require a wheelchair. As a result of scoliosis, pulmonary function may be impaired, which can lead to pulmonary compromise.
Children with Duchenne muscular dystrophy have reduced bone density and an increased risk of developing fractures of certain bones, such as hips and spine. Many affected individuals will display mild to moderate degrees of non-progressive intellectual impairment and learning disabilities.
By the late teens, Duchenne muscular dystrophy may also be characterized by additional potentially life-threatening complications including weakness and deterioration of the heart muscle (cardiomyopathy). Cardiomyopathy can result in impairment in the ability of the heart to pump blood, irregular heartbeats (arrhythmias), and heart failure. Another serious complication associated with Duchenne muscular dystrophy is weakness and deterioration of muscles in the rib cage. This can result in an increased susceptibility to respiratory infections (e.g., pneumonia), difficulty coughing, and, ultimately, respiratory failure.
Pharyngeal weakness can result in episodes of aspiration, nasal regurgitation of liquids, and a nasal quality of voice.
Involvement of muscles within the gastrointestinal tract may result in dysmotility, a condition in which the passage of food through the digestive tract usually because of slow and uncoordinated movements of the muscles of the digestive tract. Gastrointestinal dysmotility may result in constipation and diarrhea.
Incontinence of urine and stools due to urethral and anal sphincter weakness is uncommon and, if present, is a late manifestation.
One third of patients with Duchenne muscular dystrophy may have various degree of cognitive impairment including learning disability, attention deficit and autistic spectrum disorder.
Rarely, malignant hyperthermia after anesthesia may be a presenting sign.
Symptomatic female carriers may have an early onset and progressive muscular dystrophy.
Intellectual disability
Intellectual impairment is seen in all patients; however, only 20% to 30% of patients have an intelligence quotient (IQ) less than 70. The degree of impairment does not correlate with disease severity. Most patients have only a mild form of learning impairment and can function in a regular classroom. Epilepsy is more common than in the general population, and uncommonly, autism-like behavior has been described.
Duchenne muscular dystrophy associated cardiomyopathy
Symptoms of cardiomyopathy can develop in the early teens and are present in almost all patients in their twenties. Persistent tachycardia and heart failure, maybe presenting signs. In affected patients, dilated cardiomyopathy is characterized by extensive fibrosis of the posterobasal left the ventricular wall. As the disease progresses, fibrosis can spread to the lateral free wall of the left ventricle. With the involvement of the posterior papillary muscle, significant mitral regurgitation can occur. Inter and intraatrial conduction abnormalities, possibly involving the AV node, can be seen. Arrhythmias, particularly supraventricular arrhythmias, are also associated with the developing cardiomyopathy.
Physical exam shows pseudohypertrophy of the calf muscle and occasionally the quadriceps muscle. Shortening of the Achilles tendon may be noted, and the patient may have hyporeflexia or areflexia. Ankle reflexes are preserved till late in the disease unless contractures develop. Knee deep tendon reflexes are less brisk than the ankle and can be lost by age 6. The brachioradialis reflex is brisker than the biceps or triceps deep tendon reflexes. Typically, children are noted to use their arms to lift themselves from a seated position on the ground. This is known as Gowers sign.
Duchenne muscular dystrophy diagnosis
A diagnosis of Duchenne muscular dystrophy is made based upon a thorough clinical evaluation, a detailed patient history, and a variety of specialized tests including molecular genetic tests. If the genetic tests are not informative, surgical removal and microscopic examination (biopsy) of affected muscle tissue that may reveal characteristic changes to muscle fibers. Specialized blood tests (e.g. creatine kinase) that evaluate the presence and levels of certain proteins in muscle (immunohistochemistry) are also used 7.
Molecular genetic tests involve the examination of deoxyribonucleic acid (DNA) to identify specific a genetic mutation including deletions, duplications or single point mutations. Samples of blood or muscles cells may be tested. These techniques can also be used to diagnosis Duchenne muscular dystrophy before birth (prenatally).
Blood tests may reveal elevated levels of the creatine kinase (CK), an enzyme that is found in abnormally high levels when muscle is damaged. The detection of elevated CK levels (usually in the thousands or ten thousands range) can confirm that muscle is damaged or inflamed, but cannot confirm a diagnosis of Duchenne muscular dystrophy.
In some cases, a specialized test can be performed on muscle biopsy samples that can determine the presence and levels of specific proteins within cells. Various techniques such as immunostaining, immunofluorescence or Western blot (immunoblot) can be used. These tests involve the use of certain antibodies that react to certain proteins such as dystrophin. Tissue samples from muscle biopsies are exposed to these antibodies and the results can determine whether a specific muscle protein is present in the cells and in what quantity or what size.
Serum creatine kinase
Serum creatine kinase (CK) measurements are elevated before the development of clinical symptoms and signs and may also be elevated in newborns. Levels peak by age two and can be more than 10 to 20 times above the upper limit of normal. As age and disease progress, serum creatine kinase levels decrease as fibrosis and fat progressively replace muscle. Other muscle enzymes, such as aldolase levels and AST levels, may also elevate.
Asymptomatic carriers may also have elevated creatine kinase levels. This is seen in about 80% of cases, and the highest levels are noted between ages 8 and 12.
Muscle biopsy
A muscle biopsy will demonstrate endomysial connective tissue proliferation, scattered degeneration, and regeneration of myofibers, muscle fiber necrosis with a mononuclear cell infiltrate, and replacement of muscle with adipose tissue and fat.
The muscled biopsied are the quadriceps femoris and the gastrocnemius.
Electromyography
Characteristic myopathic features can be seen; however, this is nonspecific. Motor and sensory nerve conduction velocities are normal, and denervation is not present.
Gene analysis
Patients with Duchenne muscular dystrophy demonstrate the complete or near-complete absence of dystrophin gene. Dystrophin immunoblotting can be used to predict the severity of the disease. In Duchenne muscular dystrophy, patients are found to have less than 5% of the normal quantity of dystrophin.
Polymerase chain reactions (PCR) can also be used and detect up to 98% of mutations. Multiplex ligation-dependent probe amplification (MPLA) is also used to identify duplications and deletions. Duplications can lead to in-frame or out of frame transcription products. Fluorescence in situ hybridization (FISH) is used less frequently but is useful to identify small point mutations.
Dystrophin immunocytochemistry can also be sued to detect cases not identifies by PCR.
Electrocardiogram (ECG)
Characteristic ECG changes are tall R waves in V1-V6 with an increased R/S ratio and deep Q waves in leads I,aVL, and V5-6. Conduction abnormalities with arrhythmias may be identified with telemetry. As mentioned previously, supraventricular arrhythmias are more common. Intra-atrial conduction abnormalities are more common than AV or infra-nodal defects in Duchenne muscular dystrophy.
Echocardiogram
Evidence of dilated cardiomyopathy is present in almost all patients by the end of their teens or in their 20s.
Duchenne muscular dystrophy treatment
No medical cure exists for Duchenne muscular dystrophy and the disease has a poor prognosis. Treatments are aimed at the specific symptoms present in each individual. Treatment options should include physical therapy and active and passive exercise to build muscle strength and prevent contractures. Surgery may be recommended in some patients to treat contractures or scoliosis. Braces may be used to prevent the development of contractures. The use of mechanical aids (e.g., canes, braces, and wheelchairs) may become necessary to aid walking (ambulation).
Corticosteroids are used as standard of care to treat individuals with Duchenne muscular dystrophy 8. These drugs slow the progression of muscle weakness in affected individuals and delay the loss of ambulation by 2-3 years. Two common corticosteroid drugs used to treat individuals with Duchenne muscular dystrophy are prednisone and deflazacort (which is not available in the United States).
In 2016, Exondys 51 (eteplirsen) injection was FDA approved to treat Duchenne muscular dystrophy and is the first drug approved for this condition. Exondys 51 is specifically indicated for patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which affects about 13 percent of the population with Duchenne muscular dystrophy. Exondys 51 is made by Sarepta Therapeutics.
In 2017, Emflaza (deflazacort) was FDA approved to treat patients age 5 years and older with Duchenne muscular dystrophy. Emflaza is marketed by PTC Therapeutics.
Glucocorticoid therapy
Glucocorticoid therapy decreases the rate of apoptosis of myotubes and can decelerate myofiber necrosis. Prednisone is used in patients four years and older in whom muscle function is declining or plateauing.
Prednisone is recommended at a dosage of (0.75 mg/kg per day or 10 mg/kg per week is given over two weekend days).
Deflazacort, an oxazoline derivative of prednisone, is sometimes preferred over prednisone as it has a better side effect profile and has an estimated dosage equivalency of 1:1.3 compared with prednisone. The recommended dosage is 0.9 mg/kg/day.
Studies have shown that glucocorticoid treatment is associated with improved pulmonary function, delayed development of scoliosis reduces incidence and progression of cardiomyopathy and overall improved mortality.
Cardiomyopathy
Treatment with angiotensin-converting enzyme (ACE) inhibitors and/or beta-blockers is recommended. Early studies suggest that early treatment with ACE inhibitors may slow progression of the disease and prevent the onset of heart failure.
Overt heart failure is treated with digoxins and diuretics as in other patients with cardiomyopathy.
Surveillance consists of a cardiology assessment with ECG and echocardiogram. This should be performed at the time of diagnosis or by the age of 6 years. Routine surveillance should be performed once every two years until the age of 10 and then yearly after that. If evidence of cardiomyopathy is present, surveillance every six months is indicated.
Pulmonary interventions
Pulmonary function must be tested prior to the exclusive use of a wheelchair. This should be repeated twice a year once the patient reaches 12 years of age, must use a wheelchair or vital capacity is found to be less than 80% of predicted.
Orthopedic interventions
Physiotherapy to prevent contractures is the mainstay of the orthopedic interventions. Based upon patient requirements, passive stretching exercises, plastic ankle-foot orthosis during sleep, long leg braces to assist in ambulation may be used. Surgery to release contractures may be required for advanced disease. Surgery to correct scoliosis may improve pulmonary function.
Nutrition
Patients are at risk for malnutrition, including obesity. Calcium and vitamin D should be supplemented to prevent osteoporosis secondary to chronic steroid use. DEXA scanning should be obtained at age three and then repeated yearly.
Exercise
Guidelines recommend all patients participate in a gentle exercise to avoid disuse atrophy. A combination of swimming pool and recreation-based exercises is recommended. Activity should be reduced if myoglobinuria is noted or significant muscle pain develops.
Novel therapies
Gene therapies include medications that bind RNA and skip over the defective codon. This produces a shorter but potentially functional protein. Eteplirsen us an exon 51 skipping antisense oligonucleotides medications used for this purpose. Eteplirsen has been approved by the FDA for this purpose.
Care at home
Modifications to your house may be needed over time to accommodate a wheelchair to help your child remain mobile and independent. Items that can improve your child’s comfort and independence include soft pressure care mattresses, ramps and modified taps. Your child’s physiotherapist or occupational therapist can advise you.
Exercise
Gentle exercise and participation in physical activity have both psychological and physical benefits for children with Duchenne muscular dystrophy. Exercise can slow muscle degeneration and help to strengthen your child.
In addition to exercises recommended by your child’s physiotherapist, activities like playing at the park, riding a bike, swimming or hydrotherapy help flexibility, muscle strength and confidence. An increase in strength can improve performance of daily activities such as stair climbing and walking, and also help postural muscles needed to keep the spine straight. Swimming can still be enjoyed after walking and riding are no longer possible.
Regular play and incidental exercise will help keep your child participating in activities with their friends at school. As they become less comfortable walking, a wheelchair or electric scooter may help them to continue to participate in social and sporting activities and maintain independence.
Other benefits of exercise for children with Duchenne muscular dystrophy include maintaining range of motion in joints, and preventing spinal curvature (scoliosis) and obesity.
It is important to avoid your child becoming over-exerted or exhausted, however, as this can worsen their muscle damage.
Duchenne muscular dystrophy life expectancy
Duchenne muscular dystrophy prognosis is typically poor. Muscle weakness may present initially with difficulty in ambulation but progressively advances to such an extent that affected patients are unable to carry out activities of daily living and must use wheelchairs. Patients are often wheelchair dependent by the age of 12 years. Death occurs as a result of respiratory muscle weakness or cardiac complications in the teens or 20s 6. Other causes of death are pneumonia, aspiration, or airway obstruction.
- Ryder, S. et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet Journal of Rare Diseases (2017). doi:10.1186/s13023-017-0631-3[↩]
- Bello L, Pegoraro E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J Clin Med. 2019 May 10;8(5).[↩]
- Duchenne and Becker muscular dystrophy. https://ghr.nlm.nih.gov/condition/duchenne-and-becker-muscular-dystrophy[↩]
- Jones D. Duchenne muscular dystrophy awaits gene therapy. Nat. Biotechnol. 2019 Apr;37(4):335-337.[↩]
- Cai A, Kong X. Development of CRISPR-Mediated Systems in the Study of Duchenne Muscular Dystrophy. Hum Gene Ther Methods. 2019 Jun;30(3):71-80.[↩]
- Venugopal V, Pavlakis S. Duchenne Muscular Dystrophy. [Updated 2019 Nov 15]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482346[↩][↩][↩]
- Ke Q, Zhao ZY, Mendell JR, Baker M, Wiley V, Kwon JM, Alfano LN, Connolly AM, Jay C, Polari H, Ciafaloni E, Qi M, Griggs RC, Gatheridge MA. Progress in treatment and newborn screening for Duchenne muscular dystrophy and spinal muscular atrophy. World J Pediatr. 2019 Jun;15(3):219-225.[↩]
- Shimizu-Motohashi Y, Komaki H, Motohashi N, Takeda S, Yokota T, Aoki Y. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy: Current Status of Therapeutic Approaches. J Pers Med. 2019 Jan 07;9(1).[↩]




{kind=link}