Contents
What is epidermolysis bullosa
Epidermolysis bullosa is a rare group of genetic connective tissue disorders that cause painful blisters to form on the skin. These blisters can cause serious problems if they become infected. Anyone can have epidermolysis bullosa. Epidermolysis bullosa affects 1 out of every 20,000 births in the United States (approximately 200 children a year are born with epidermolysis bullosa). Skin blisters are the major symptom of epidermolysis bullosa and symptoms usually first appear in babies or toddlers.
There are many genetic and symptomatic variations of epidermolysis bullosa, but all share the prominent symptom of extremely fragile skin that blisters and tears from minor friction or trauma. Internal organs and bodily systems can also be seriously affected by the disease. Some people develop blisters inside the body—in places such as the mouth, the stomach, the tube between the throat and stomach, and the bladder.
There are different forms of epidermolysis bullosa, depending upon where the blistering occurs within the different skin layers. Some people with the illness have a mild form with few blisters. Others have many blisters on the skin.
Epidermolysis bullosa is always painful, often pervasive and debilitating and in some cases lethal before the age of 30. Epidermolysis bullosa affects both genders and every racial and ethnic background equally.
Dermatologists can identify the disease by taking a small piece of skin and looking at it under a microscope. Other tests can identify defective genes in epidermolysis bullosa patients and their family members.
There is no cure for epidermolysis bullosa, although there are medicines to help prevent infection and to reduce discomfort. Treatment includes daily proper skin care to prevent blisters, pain management, treating blisters and infections, and a good diet. Surgery may be needed in more severe cases.
While many who live with milder forms of epidermolysis bullosa can lead long and productive lives, the list of manifestations and secondary complications in the more severe forms is lengthy and requires multiple interventions from a range of medical specialists. Those forms of epidermolysis bullosa result in disfigurement, disability, and in some cases early death.
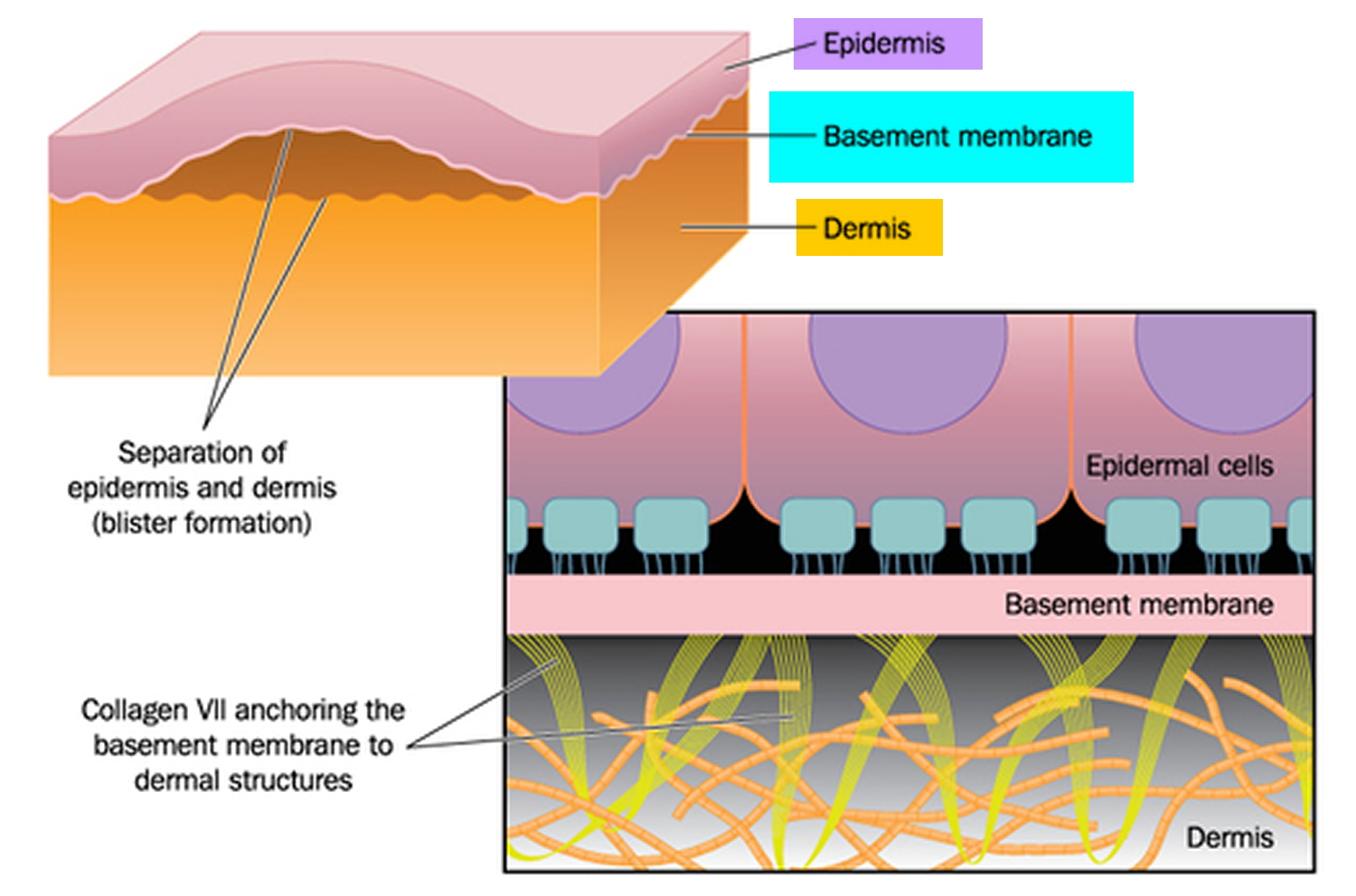
Figure 1. Epidermolysis bullosa
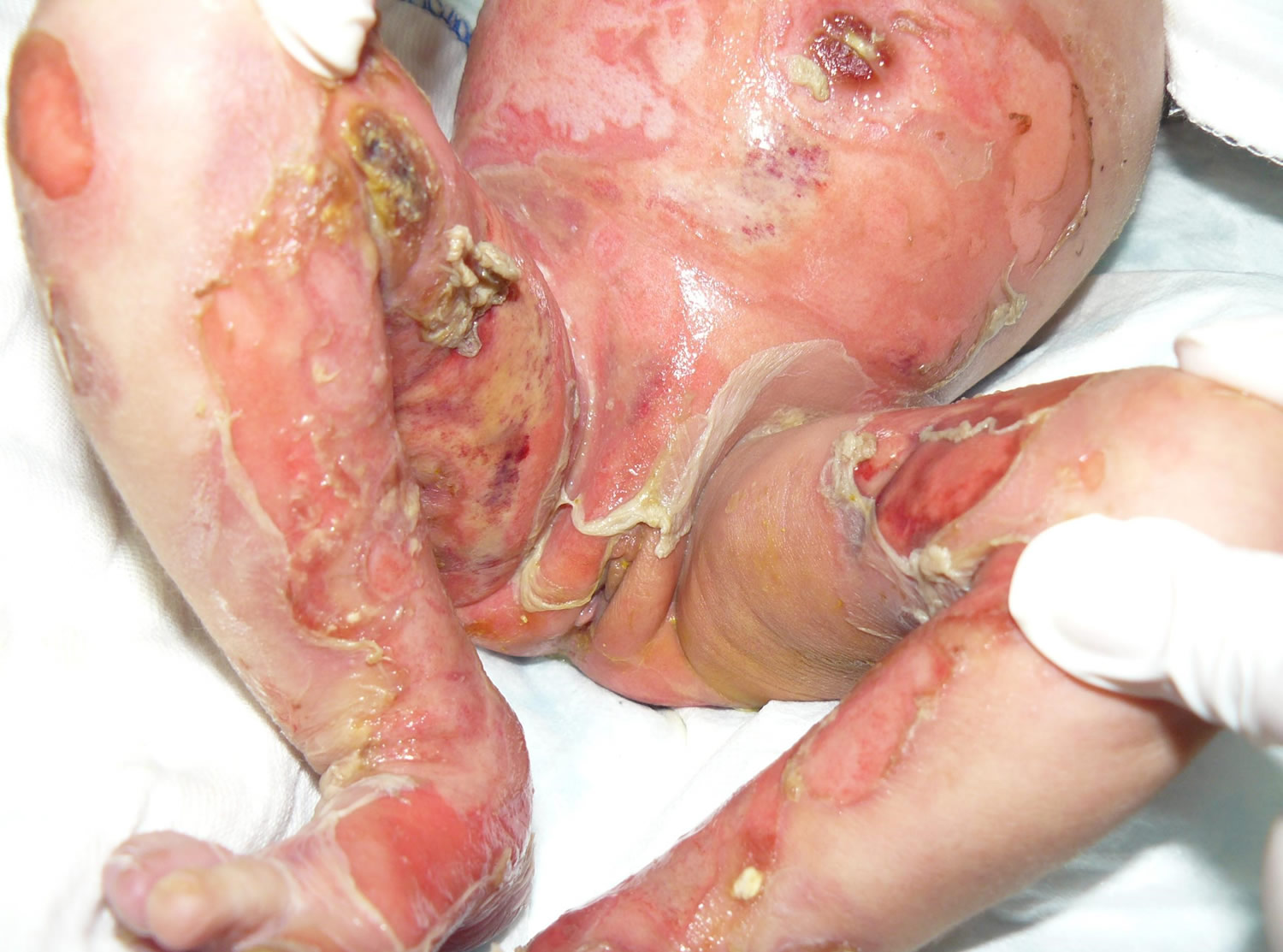
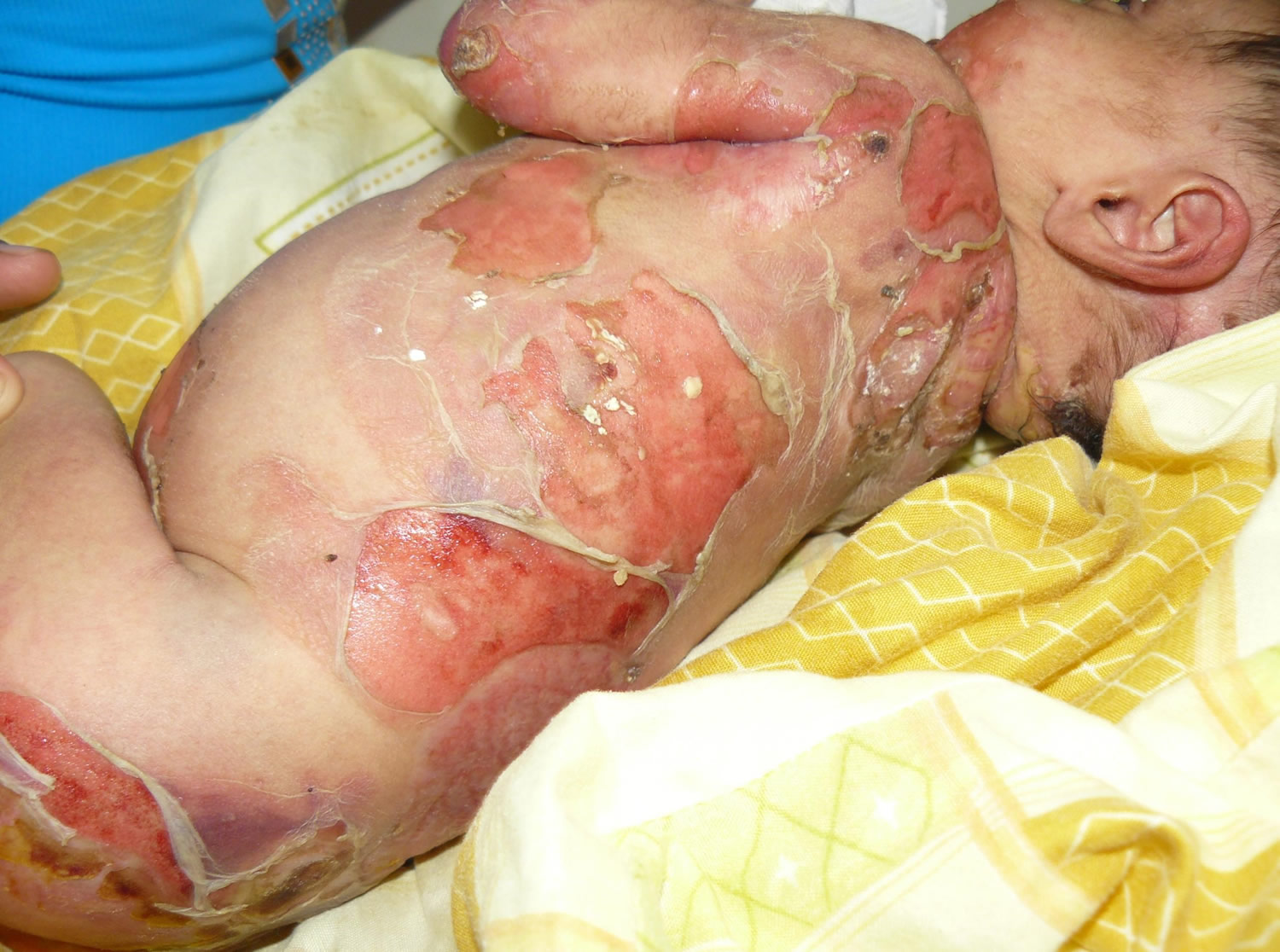
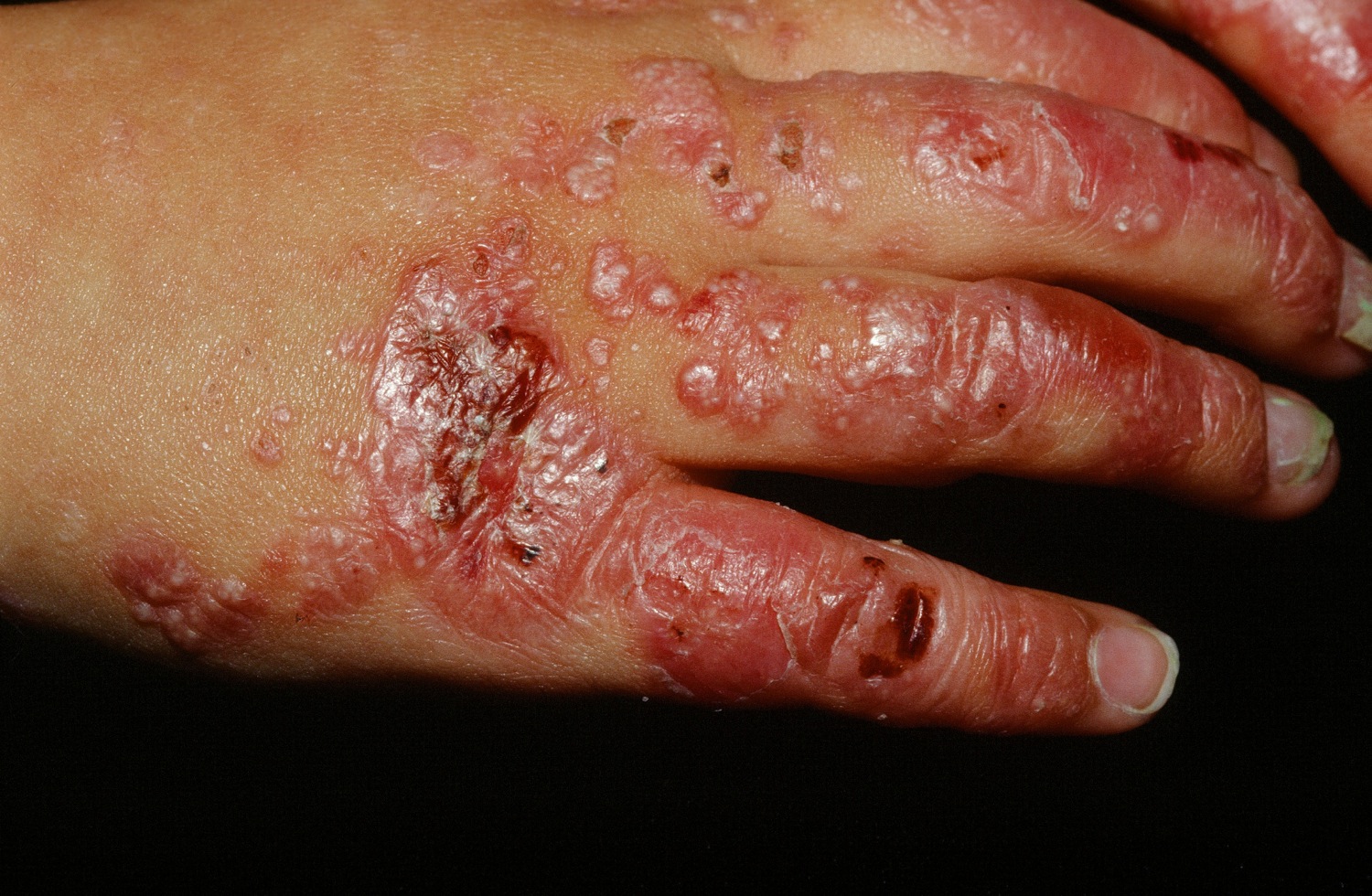
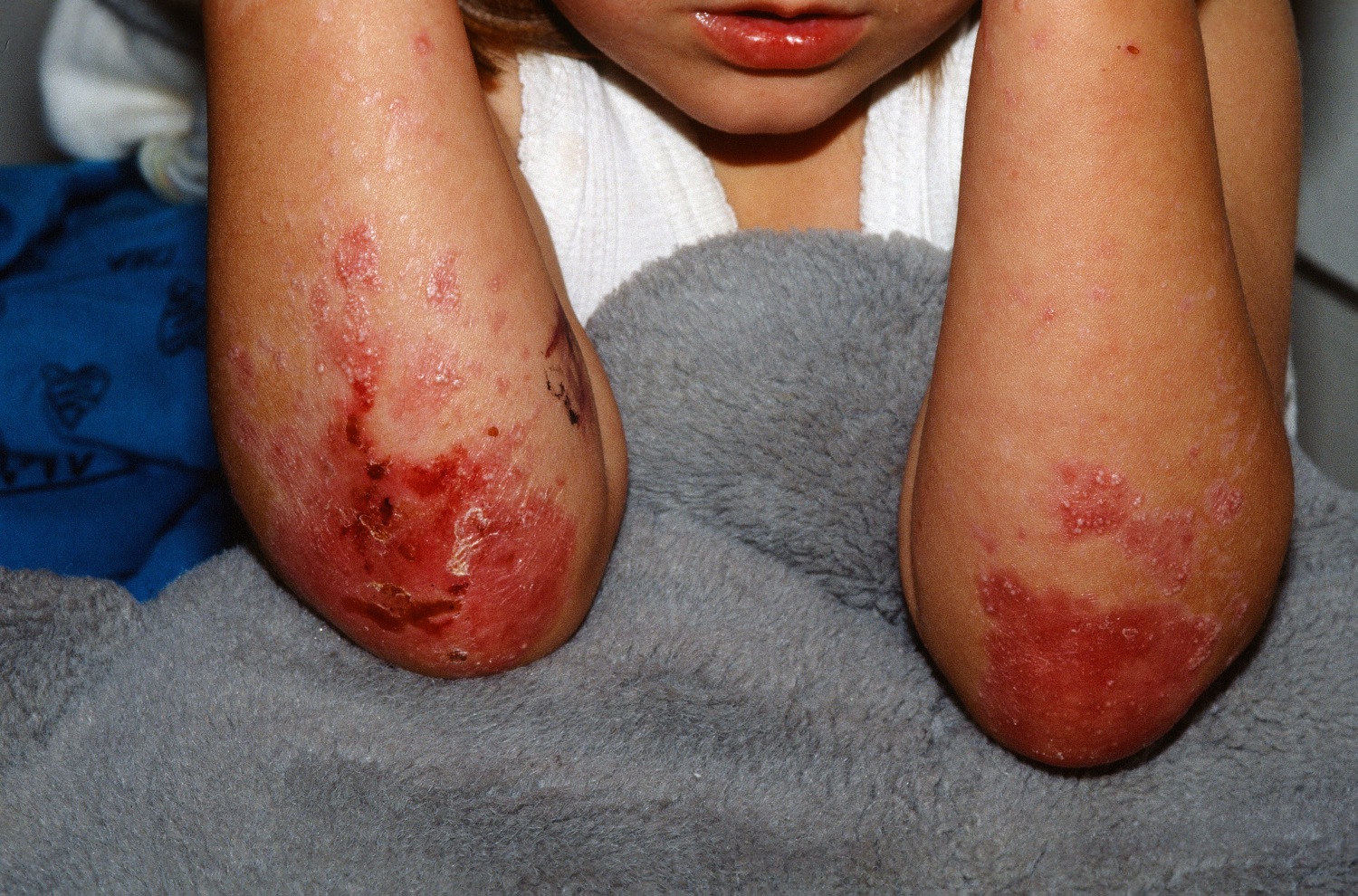
Figure 2. Skin structure
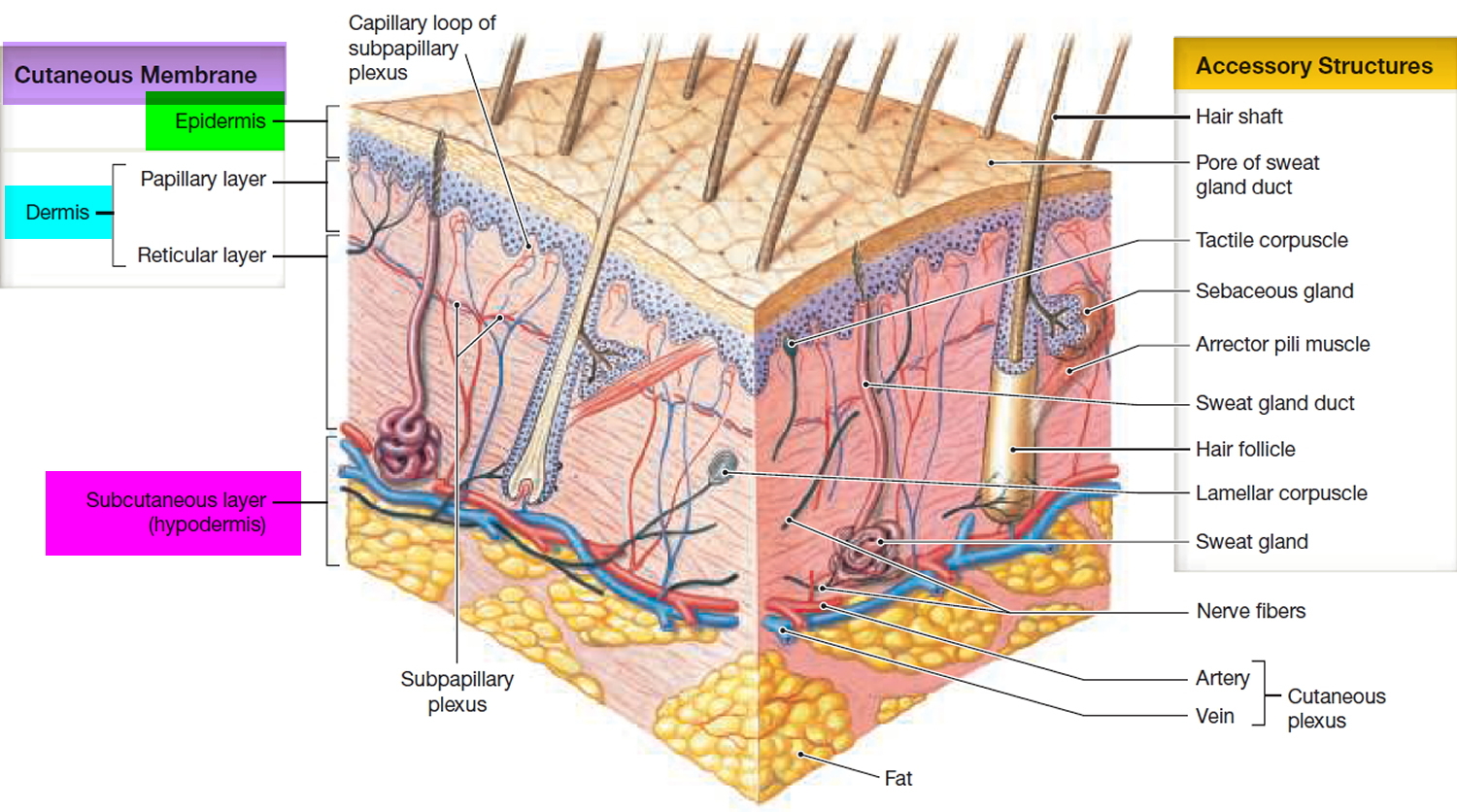
Figure 3. Structure and skin cells of the Epidermis
Figure 4. Epidermolysis bullosa autosomal dominant inheritance pattern
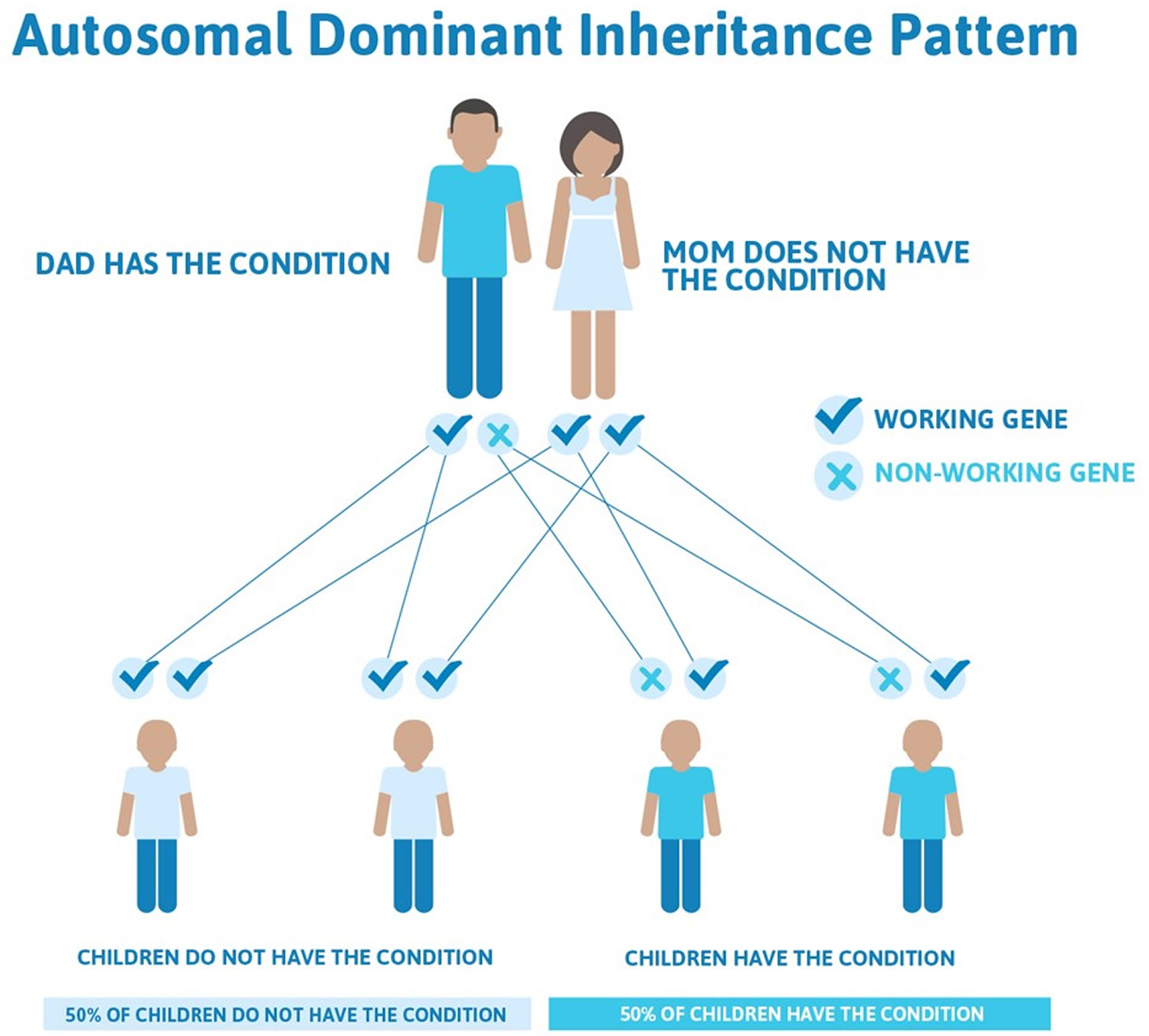
Figure 5. Epidermolysis bullosa autosomal recessive inheritance pattern
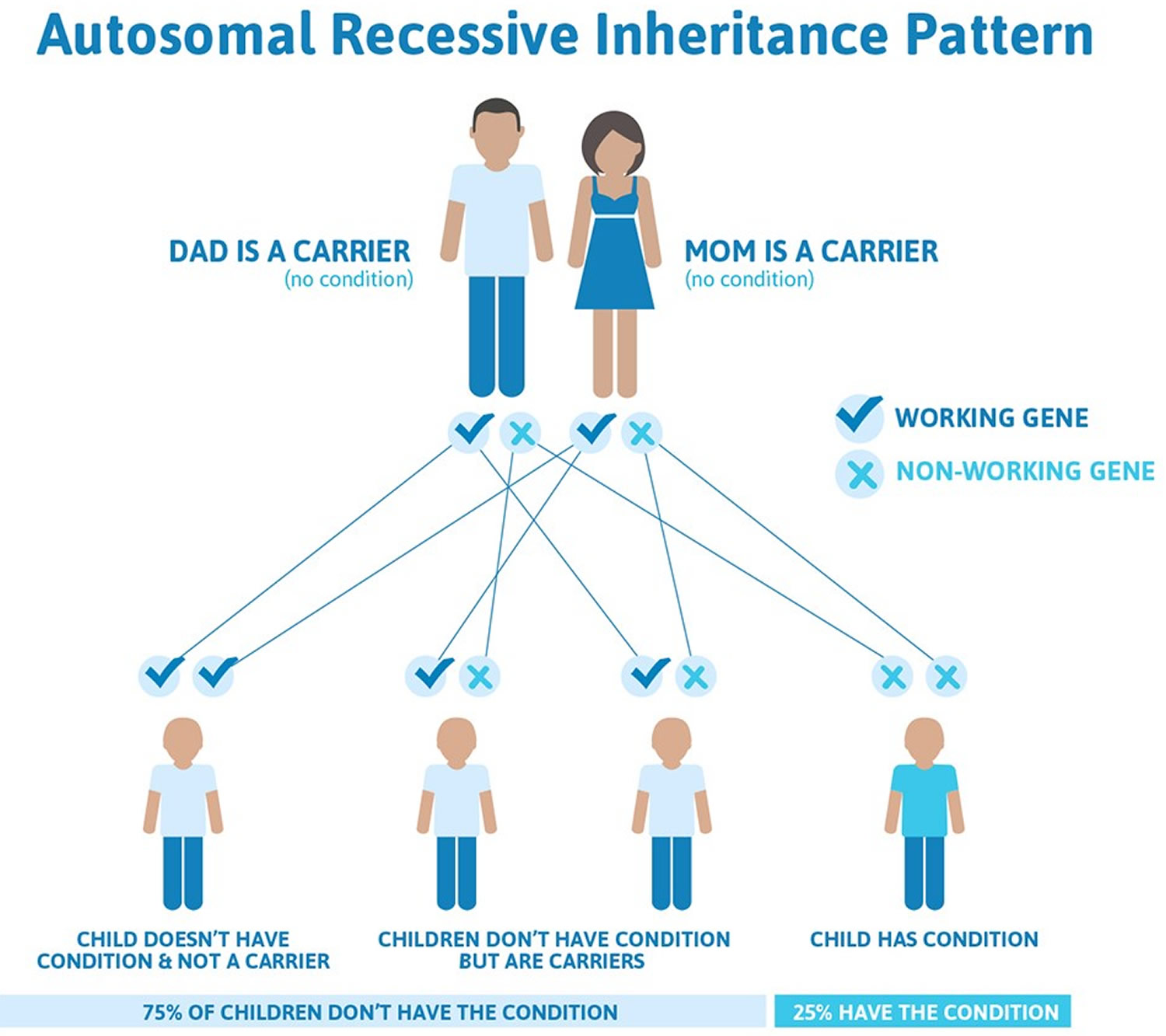
Figure 6. Epidermolysis bullosa blister formation
Epidermolysis bullosa types
Epidermolysis bullosa has been categorized as encompassing 4 major types (Simplex, Junctional, Dystrophic & Aquisita) and 31 subtypes, therefore it is commonly referred to as a group of disorders. Other manifestations of epidermolysis bullosa include: anemia, cardiomyopathy, syndactyly (fusion of the fingers and toes), renal insufficiency, dysphagia (difficulty swallowing), malnourishment, cancer, constipation, osteoporosis, muscular dystrophy, and pyloric atresia.
Although the 4 major types differ in severity, their features overlap significantly, and they are caused by mutations in the same genes. Most researchers now consider the major forms of epidermolysis bullosa to be part of a single disorder with a range of signs and symptoms.
The skin is made up of an outer layer (epidermis) and an underlying layer (dermis). The area where the layers meet is called the basement membrane. The various types of epidermolysis bullosa are largely defined by which layer the blisters form in.
Epidermolysis bullosa simplex
This is the most common form. It develops in the outer layer of skin and mainly affects the palms and the feet. The blisters usually heal without scarring. Although epidermolysis bullosa simplex is considered a non-scarring form of epidermolysis bullosa, secondary infection may cause scarring.
The mildest form of epidermolysis bullosa simplex, known as the localized type epidermolysis bullosa simplex (formerly called the Weber-Cockayne type), is characterized by skin blistering that begins anytime between childhood and adulthood and is usually limited to the hands and feet in response to friction. This type of epidermolysis bullosa simplex usually does not involve nails or mucous membranes. Most individuals seem to be more prone to blisters in warmer climates and during periods of strenuous activity such as jogging, marching or walking. With trauma or friction rarely the blistering can be (generalized) or appear on other parts of the body. Later in life, skin on the palms of the hands and soles of the feet may thicken and harden (hyperkeratosis).
The Dowling-Meara type is the most severe form of epidermolysis bullosa simplex. Extensive, severe blistering can occur anywhere on the body, including the inside of the mouth, gastrointestinal tract and rarely, the upper respiratory tree and blisters may appear in clusters. Blistering is present from birth and tends to improve with age. Since Dowling-Meara type epidermolysis bullosa simplex is the most severe form of epidermolysis bullosa simplex, the widespread blistering may lead to death in infancy. However, blistering tends to become smaller and less problematic for most patients as they grow older. Affected individuals also experience abnormal nail growth and hyperkeratosis of the palms and soles. If the thickening is severe enough it may limit the range of motion of a joint. In such cases, consultation from a surgeon may be necessary to determine the best course of treatment. Heat may exacerbate blistering. Milia (tiny cysts on skin) may be present after blisters have healed. Nail thickening and discoloration is a common feature.
Another form of epidermolysis bullosa simplex, known as the other generalized type epidermolysis bullosa simplex (formerly called the Koebner type), is associated with widespread blisters that appear at birth or in early infancy. There may be mild involvement of mucous membranes. Fingernails and toenails are sometimes involved. Localized thickening of the skin (keratoderma) on the soles of the feet and the palms of the hands may occur especially as one gets older. The blistering tends to be less severe than in the Dowling-Meara type. Though it is not a common feature of this type of epidermolysis bullosa simplex to scar on rare occasions it does happen.
Epidermolysis bullosa simplex with mottled pigmentation is characterized by patches of darker skin on the trunk, arms, and legs that fade in adulthood. This form of the disorder also involves skin blistering from early infancy, hyperkeratosis of the palms and soles, and abnormal nail growth.
In addition to the four major types described above, researchers have identified another skin condition related to epidermolysis bullosa simplex, which they call the Ogna type. It is caused by mutations in a gene that is not associated with the other types of epidermolysis bullosa simplex. It is unclear whether the Ogna type is a subtype of epidermolysis bullosa simplex or represents a separate form of epidermolysis bullosa.
Several other variants of epidermolysis bullosa simplex have been proposed, but they appear to be very rare.
The exact prevalence of epidermolysis bullosa simplex is unknown, but this condition is estimated to affect 1 in 30,000 to 50,000 people. The localized type is the most common form of the condition.
Epidermolysis bullosa simplex causes
The four major types of epidermolysis bullosa simplex can result from mutations in either the KRT5 or KRT14 gene. These genes provide instructions for making proteins called keratin 5 and keratin 14. These tough, fibrous proteins work together to provide strength and resiliency to the outer layer of the skin (the epidermis). Mutations in either the KRT5 or KRT14 gene prevent the keratin proteins from assembling into strong networks, causing cells in the epidermis to become fragile and easily damaged. As a result, the skin is less resistant to friction and minor trauma and blisters easily. In rare cases, no KRT5 or KRT14 gene mutations are identified in people with one of the four major types of epidermolysis bullosa simplex.
Mutations in another gene, PLEC, have been associated with the rare Ogna type of epidermolysis bullosa simplex. The PLEC gene provides instructions for making a protein called plectin, which helps attach the epidermis to underlying layers of skin. Researchers are working to determine how PLEC gene mutations lead to the major features of the condition.
Some precipitating factors that may cause an outbreak of blistering may include the following:
- Physical stress
- Emotional stress
- Warmer climates
- Infections
- Sexual maturation
Even though some forms of epidermolysis bullosa simplex are localized it is important to know that all skin cells are affected. Therefore, all skin surfaces are prone to develop generalized blistering.
Epidermolysis bullosa simplex inheritance pattern
Epidermolysis bullosa simplex is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder (see Figure 4 above). Some affected people inherit the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
In rare cases, epidermolysis bullosa simplex is inherited in an autosomal recessive pattern (see Figure 5 above). Autosomal recessive inheritance means the condition results when two copies of the gene in each cell are altered. The parents of an individual with an autosomal recessive disorder typically each carry one copy of the altered gene, but do not show signs and symptoms of the disorder.
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa is one of the major forms of epidermolysis bullosa. This type may be severe, with blisters beginning in infancy. A baby with junctional epidermolysis bullosa may develop a hoarse-sounding cry from continual blistering and scarring of the vocal cords.
Researchers classify junctional epidermolysis bullosa into two main types: Herlitz junctional epidermolysis bullosa and non-Herlitz junctional epidermolysis bullosa. Although the types differ in severity, their features overlap significantly, and they can be caused by mutations in the same genes. Both types of junctional epidermolysis bullosa are rare, affecting fewer than 1 per million people in the United States.
Herlitz junctional epidermolysis bullosa is the more severe form of the condition. From birth or early infancy, affected individuals have blistering over large regions of the body. Blistering also affects the mucous membranes, such as the moist lining of the mouth and digestive tract, which can make it difficult to eat and digest food. Some babies develop a hoarse cry and breathing difficulties which indicates internal involvement as well. As a result, many affected children have chronic malnutrition and slow growth. Skin blistering and ulcerations can occur spontaneously on the arms, hands, finger tips, back of the head, neck, shoulders, trunk, buttocks, legs and feet and toes (generalized distribution). Nails may be ulcerated or dystrophic. Warmer climates can exacerbate blistering. Blistering is noted on perioral (around the mouth) and mucosal surfaces as well. The extensive blistering leads to scarring and the formation of red, bumpy patches called granulation tissue. Granulation tissue bleeds easily and profusely, making affected infants susceptible to serious infections and loss of necessary proteins, minerals, and fluids. Additionally, a buildup of granulation tissue in the airway can lead to a weak, hoarse cry and difficulty breathing.
Other complications of Herlitz junctional epidermolysis bullosa can include fusion of the fingers and toes, abnormalities of the fingernails and toenails, joint deformities (contractures) that restrict movement, and hair loss (alopecia). Because the signs and symptoms of Herlitz junctional epidermolysis bullosa are so severe, these infants often die during infancy due to overwhelming infection (sepsis), malnutrition, dehydration, electrolyte imbalance or complications resulting from blistering in the respiratory, gastrointestinal or genitourinary tract.
The milder form of junctional epidermolysis bullosa is called non-Herlitz junctional epidermolysis bullosa. The blistering associated with non-Herlitz junctional epidermolysis bullosa may be limited to the hands, feet, knees, and elbows, and it often improves after the newborn period. Other characteristic features of this condition include alopecia, malformed fingernails and toenails, and irregular tooth enamel. Most affected individuals do not have extensive scarring or granulation tissue formation, so breathing difficulties and other severe complications such as infection, dehydration, electrolyte imbalances, respiratory, gastrointestinal, and/or genitourinary tract involvement are rare. These complications may lead to death. Non-Herlitz junctional epidermolysis bullosa is typically associated with a normal lifespan.
Junctional epidermolysis bullosa with Pyloric Atresia
Some infants are born with junctional epidermolysis bullosa and have been observed to have pyloric atresia, in which the opening between the stomach and the intestines fails to form. Surgery is necessary to repair the anomaly.
Generalized blistering, ulcerations of skin and mucous membranes is usually evident at birth. Blistering may be mild to severe. Erosions on finger and toenails, nail dystrophy or absence of nails may be evident. Erosions and loss of hair (alopecia ) upon the scalp and granulation tissue around mouth and nares may occur. There may be some scarring and thinning of the skin on affected areas (atrophic scarring). Warmer climates can exacerbate blistering.
The infant may suffer complications such as infection, dehydration, electrolyte imbalances, respiratory, gastrointestinal, and/or genitourinary tract involvement. These complications may lead to death.
Electron microscopic evaluation of the structure of the skin of a person affected with junctional epidermolysis bullosa-pyloric atresia reveals skin separation at the level of the lamina lucida, small hemidesmosomal plaques and reduced amount of keratin filaments with hemidesmosomes.
Mutations in junctional epidermolysis bullosa-pyloric atresia are within the genes encoding either alpha 6 or its partner beta 4 integrin. These components of the hemidesmosome are found both in skin and the stomach, explaining the failure of formation of the first part of the intestine (the pylorus).
Since epidermolysis bullosa varies in severity these manifestations may or may not be experienced by the individual affected.
Junctional epidermolysis bullosa causes
Junctional epidermolysis bullosa results from mutations in the LAMA3, LAMB3, LAMC2, and COL17A1 genes. Mutations in each of these genes can cause Herlitz junctional epidermolysis bullosa or non-Herlitz junctional epidermolysis bullosa. LAMB3 gene mutations are the most common, causing about 70 percent of all cases of junctional epidermolysis bullosa.
The LAMA3, LAMB3, and LAMC2 genes each provide instructions for making one part (subunit) of a protein called laminin 332. This protein plays an important role in strengthening and stabilizing the skin by helping to attach the top layer of skin (the epidermis) to underlying layers. Mutations in any of the three laminin 332 genes lead to the production of a defective or nonfunctional version of this protein. Without functional laminin 332, cells in the epidermis are fragile and easily damaged. Friction or other minor trauma can cause the skin layers to separate, leading to the formation of blisters.
The COL17A1 gene provides instructions for making a protein that is used to assemble type XVII collagen. Collagens are molecules that give structure and strength to connective tissues, such as skin, tendons, and ligaments, throughout the body. Type XVII collagen helps attach the epidermis to underlying layers of skin, making the skin strong and flexible. Mutations in the COL17A1 gene prevent the normal formation of collagen XVII. As a result, the skin is less resistant to friction and minor trauma and blisters easily. Most COL17A1 gene mutations cause non-Herlitz junctional epidermolysis bullosa, although a few individuals with mutations in this gene have had the more severe Herlitz junctional epidermolysis bullosa.
Junctional epidermolysis bullosa inheritance pattern
Both types of junctional epidermolysis bullosa are inherited in an autosomal recessive pattern (see Figure 5 above), which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Dystrophic epidermolysis bullosa
Dystrophic epidermolysis bullosa is one of the major forms of epidermolysis bullosa. Dystrophic epidermolysis bullosa is related to a flaw in the gene that helps produce a type of collagen that provides strength to the pig-skin like dermis layer of the skin. If this substance is missing or doesn’t function, the layers of the skin won’t join properly. The signs and symptoms of this condition vary widely among affected individuals. In mild cases, blistering may primarily affect the hands, feet, knees, and elbows. Severe cases of this condition involve widespread blistering that can lead to vision loss, disfigurement, and other serious medical problems.
Researchers classify dystrophic epidermolysis bullosa into three major types. Although the types differ in severity, their features overlap significantly and they are caused by mutations in the same gene.
- Recessive Dystrophic Epidermolysis Bullosa – Hallopeau-Siemens type
- Recessive Dystrophic Epidermolysis Bullosa – Non-Hallopeau-Siemens type
- Autosomal Dominant Dystrophic Epidermolysis Bullosa (Dominant Dystrophic Epidermolysis Bullosa)
Autosomal recessive dystrophic epidermolysis bullosa, Hallopeau-Siemens type is the most severe, classic form of the condition. Affected infants are typically born with widespread blistering and areas of missing skin, often caused by trauma during birth. Most often, blisters are present over the whole body and affect mucous membranes such as the moist lining of the mouth and digestive tract. As the blisters heal, they result in severe scarring. Scarring in the mouth and esophagus can make it difficult to chew and swallow food, leading to chronic malnutrition and slow growth. Additional complications of progressive scarring can include fusion of the fingers and toes, loss of fingernails and toenails, joint deformities (contractures) that restrict movement, and eye inflammation leading to vision loss. Additionally, young adults with the classic form of dystrophic epidermolysis bullosa have a very high risk of developing a form of skin cancer called squamous cell carcinoma, which tends to be unusually aggressive and is often life-threatening.
A second type of autosomal recessive dystrophic epidermolysis bullosa is known as the non-Hallopeau-Siemens type. This form of the condition is somewhat less severe than the classic type and includes a range of subtypes. Blistering is limited to the hands, feet, knees, and elbows in mild cases, but may be widespread in more severe cases. Affected people often have malformed fingernails and toenails. Non-Hallopeau-Siemens dystrophic epidermolysis bullosa type involves scarring in the areas where blisters occur, but this form of the condition does not cause the severe scarring characteristic of the classic type.
The third major type of dystrophic epidermolysis bullosa is known as the autosomal dominant dystrophic epidermolysis bullosa type (Dominant Dystrophic Epidermolysis Bullosa). The signs and symptoms of this condition tend to be milder than those of the autosomal recessive forms, with blistering often limited to the hands, feet, knees, and elbows. The blisters heal with scarring, but it is less severe. Most affected people have malformed fingernails and toenails, and the nails may be lost over time. In the mildest cases, abnormal nails are the only sign of the condition.
Considered together, the incidence of all types of dystrophic epidermolysis bullosa is estimated to be 6.5 per million newborns in the United States. The severe autosomal recessive forms of this disorder affect fewer than 1 per million newborns.
Dystrophic epidermolysis bullosa causes
Mutations in the COL7A1 gene cause all three major forms of dystrophic epidermolysis bullosa. This gene provides instructions for making a protein that is used to assemble type VII collagen. Collagens are molecules that give structure and strength to connective tissues, such as skin, tendons, and ligaments, throughout the body. Type VII collagen plays an important role in strengthening and stabilizing the skin. It is the main component of structures called anchoring fibrils, which anchor the top layer of skin, called the epidermis, to an underlying layer called the dermis.
COL7A1 mutations alter the structure or disrupt the production of type VII collagen, which impairs its ability to help connect the epidermis to the dermis. When type VII collagen is abnormal or missing, friction or other minor trauma can cause the two skin layers to separate. This separation leads to the formation of blisters, which can cause extensive scarring as they heal. Researchers are working to determine how abnormalities of type VII collagen also underlie the increased risk of skin cancer seen in the severe form of dystrophic epidermolysis bullosa.
Dystrophic epidermolysis bullosa inheritence pattern
The most severe types of dystrophic epidermolysis bullosa are inherited in an autosomal recessive pattern. Autosomal recessive inheritance means that both copies of the COL7A1 gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
A milder form of dystrophic epidermolysis bullosa has an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. About 70 percent of all people with autosomal dominant dystrophic epidermolysis bullosa have inherited an altered COL7A1 gene from an affected parent. The remaining 30 percent of affected people have the condition as a result of a new mutation in the COL7A1 gene. These cases occur in people with no history of the disorder in their family.
Epidermolysis bullosa acquisita
Epidermolysis bullosa acquisita is a chronic mucocutaneous autoimmune skin blistering disease 1. The pathogenic relevance of autoantibodies targeting type VII collagen (COL7) has been well-documented. Therefore, epidermolysis bullosa acquisita is a prototypical autoimmune disease with a well-characterized pathogenic relevance of autoantibody binding to the target antigen. Epidermolysis bullosa acquisita is a rare disease with an incidence of 0.2 new cases per million and per year 1.
Epidermolysis bullosa acquisita is distinguished from other epidermolysis bullosa diseases on the basis of distinctive clinical and histological features diagnostic criteria for the disease. Specifically, these included (i) clinical lesions resembling dystrophic epidermolysis bullosa, (ii) adult onset of disease, (iii) a negative family history of dystrophic epidermolysis bullosa and (iv) exclusion of other bullous diseases 2. The diagnosis of epidermolysis bullosa acquisita is based on the clinical presentation, the detection of tissue-bound antibodies by direct immunofluorescence microscopy, and the detection of circulating antibodies directed against COL7 and/or a u-serrated pattern in direct immunofluorescence microscopy. Additional tests such as transmission electron microscopy or antigen mapping may be performed in unclear cases. The detection of a linear IgG and/or IgA deposition along the basement membrane in a perilesional skin lesion from the patient is observed in almost all epidermolysis bullosa acquisita cases. In detail, these deposits can be detected in at least 93% of all epidermolysis bullosa acquisita patients. If the direct immunofluorescence sections are observed at high magnification, n- and u-serrated patterns can be differentiated. The n-serrated pattern is seen in several subepidermal autoimmune bullous dermatosis, but the u-serrated pattern is unique to epidermolysis bullosa acquisita (Figure 7). If this criterion is included for epidermolysis bullosa acquisita diagnosis, the diagnostic sensitivity can be greatly increased 3.
Figure 7. Epidermolysis bullosa acquisita u-serrated pattern in direct immunofluorescence microscopy
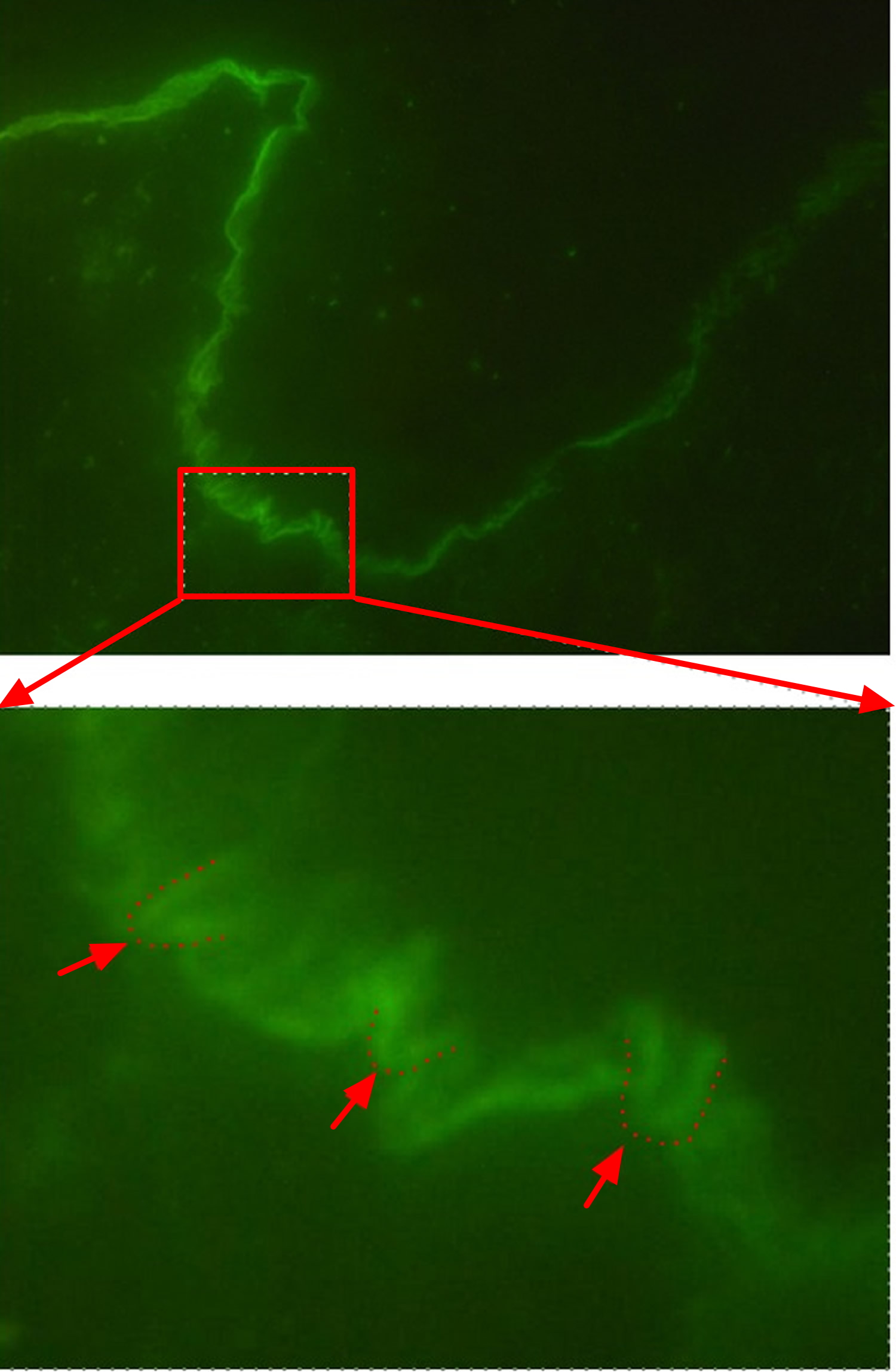
The cutaneous manifestations in epidermolysis bullosa acquisita patients are heterogeneous. However, epidermolysis bullosa acquisita patients can be classified into two major clinical subtypes: noninflammatory (classical or mechanobullous) and inflammatory epidermolysis bullosa acquisita, which is characterized by cutaneous inflammation resembling bullous pemphigoid, linear IgA disease, mucous membrane pemphigoid, or Brunsting-Perry pemphigoid 4. The clinical presentation of an individual epidermolysis bullosa acquisita patient may change during the course of the disease, or the same patients may present with two different forms simultaneously.
The current treatment of epidermolysis bullosa acquisita relies on general immunosuppressive therapy, which does not lead to remission in all cases.
Due to the low prevalence, no controlled clinical trials on the treatment of epidermolysis bullosa acquisita have been performed. Current recommendations for epidermolysis bullosa acquisita treatment are therefore solely based on the clinical expertise by clinicians specialized in autoimmune bullous dermatoses 5. In addition, the clinical phenotype and the disease severity have to be taken into account when selecting treatment for epidermolysis bullosa acquisita patients. However, the increased understanding of the diseases’ pathogenesis has identified several potential therapeutic targets. However, and of note, the treatment of epidermolysis bullosa acquisita is challenging. In a cohort of 30 epidermolysis bullosa acquisita patients, who were initially treated with a combination of methylprednisolone, dapsone, and colchicine, remission was achieved after a median of 9 months on therapy. Long-term followup of these patients showed complete remission in 46% and incomplete remission in another 46% of the patients 6 years after initiation of treatment 6. Overall, most experts recommend colchicine as first line treatment, as it has fewer adverse events than most of the other medications used for epidermolysis bullosa acquisita treatment.
Epidermolysis bullosa acquisita treatment
Colchicine
Colchicine has been used in well over 40 epidermolysis bullosa acquisita patients 6. As most reports state beneficial effects of colchicine, it should be used as first line treatment for patients with (inflammatory-type) epidermolysis bullosa acquisita.
Corticosteroids
Most epidermolysis bullosa acquisita patients are treated with systemic corticosteroids. Initial doses range from 0.5 to 1.5 mg/kg per day 7. In most cases, corticosteroid treatment is combined with other immunosuppressive/modulatory agents to lower the corticosteroid dose. These include almost all of the treatment modalities described below. As steroid treatment often leads to improvement of epidermolysis bullosa acquisita, it has to be considered as effective. Furthermore, corticosteroid treatment is also effective in antibody transfer-induced epidermolysis bullosa acquisita in mice 8. However, due to its known high number of adverse events, other treatment modalities should be taken into consideration before systemic corticosteroid treatment whenever possible. Furthermore, it is tempting to speculate that patients with inflammatory-type epidermolysis bullosa acquisita respond better to corticosteroids compared to patients with noninflammatory epidermolysis bullosa acquisita.
Methotrexate and Azathioprine
Both compounds are used as steroid-sparing agents. There are no reports on single use of these compounds in epidermolysis bullosa acquisita patients. Furthermore, no data is available documenting the effectiveness of either methotrexate or azathioprine as steroid-sparing agents in epidermolysis bullosa acquisita. Therefore, both compounds should be used only in the treatment of refractory cases.
Cyclosporine
Since its first use in 1987 for epidermolysis bullosa acquisita 9, cyclosporine has been used in a total of 11 epidermolysis bullosa acquisita patients 10. In all patients, the use of cyclosporine was reported to have improved epidermolysis bullosa acquisita. Of note, in at least 3 patients, cyclosporine was used as monotherapy, leading to remission of epidermolysis bullosa acquisita. Hence, cyclosporine is most likely an effective treatment of epidermolysis bullosa acquisita.
High Dose Intravenous Immunoglobulin (IVIG)
Immunoglobulin IgG preparations, isolated from human serum, have been used as substitution treatment for patients with antibody deficiencies and severe infections for many decades 11. Later, intravenous application of high doses of IgG (IVIG) has been established as an effective therapy for many autoimmune diseases, including immune thrombocytopenia, Guillain-Barre syndrome, multiple sclerosis, myasthenia gravis, pemphigus disease, and Kawasaki disease 12. Until 2010, 12 patients with extensive treatment-resistant epidermolysis bullosa acquisita had been treated with IVIG, and the reported response was usually favorable 13. In 2012, results on additional 10 IVIG-treated epidermolysis bullosa acquisita patients were reported. These patients received IVIG in addition to the previous medication that had failed to control the disease. These earlier drugs were withdrawn over a 5–9-month period, and IVIG was continued as monotherapy for a total duration of 30–52 months. In all 10 patients, a satisfactory clinical response was observed, and during followup (29–123 months), no relapse was observed 14. Collectively, this data strongly supports the assumption that IVIG is an effective treatment option for epidermolysis bullosa acquisita.
Dapsone
Several case reports have stated the use of dapsone in epidermolysis bullosa acquisita 6. Of note, dapsone monotherapy has been shown to be effective in one epidermolysis bullosa acquisita patient 15. Further data, like for all applied treatments in epidermolysis bullosa acquisita, is needed before final conclusions of the efficacy of dapsone can be drawn.
Cyclophosphamide
Cyclophosphamide has been rarely used in epidermolysis bullosa acquisita. Therefore, no interpretation on its efficacy for controlling epidermolysis bullosa acquisita can be drawn 16.
Rituximab
Anti-CD20 treatment with rituximab has been increasingly used for the treatment of autoimmune bullous dermatoses 17. Regarding epidermolysis bullosa acquisita, 12 cases have been reported with rituximab treatment. In most cases, a favorable outcome was reported. Most patients treated with rituximab, several previous treatments had failed. In most cases, anti-CD20 was administered in an adjuvant setting. One patient, unresponsive to previous treatments, was treated with immunoadsorption to lower antibody titers rapidly, followed by rituximab monotherapy. This led to rapid and lasting clinical remission. Another patient was treated with additional rituximab as azathioprine monotherapy had not improved epidermolysis bullosa acquisita. Approximately 2 weeks later, the patient died of bacterial pneumonia 18. Hence, anti-CD20 treatment should be taken into account when selecting treatments for patients with either severe and/or relapsing epidermolysis bullosa acquisita.
Plasmapheresis and Immunoadsorption
Although several reports and case report series have documented a beneficial effect of plasmapheresis and immunoadsorption in autoimmune bullous dermatoses—especially in pemphigus 19, only very limited experience with this treatment modality has been described in epidermolysis bullosa acquisita. Taken together, 3 cases of either plasmapheresis or immunoadsorption in epidermolysis bullosa acquisita patients have been published so far, and in all cases, plasmapheresis and immunoadsorption have been used in addition to other treatments 20. Despite the improvement that was reported in all 3 cases, the currently available data does not allow drawing a final conclusion on the effectiveness of these methods for epidermolysis bullosa acquisita patients.
Extracorporeal Photochemotherapy
In principle, similar conclusions like for plasmapheresis and immunoadsorbtion can be drawn for extracorporeal photochemotherapy. Currently, data on 8 extracorporeal photochemotherapy patients successfully treated with extracorporeal photochemotherapy has been reported 21. Hence, this method seems promising for the management of treatment-refractory patients.
Kindler syndrome
Kindler syndrome is a rare subtype of inherited epidermolysis bullosa, is characterized by a mixed pattern of blistering on multiple levels within and/or beneath the basement membrane zone. Skin fragility and acral blister formation beginning at birth, diffuse cutaneous atrophy, photosensitivity (which is most prominent during childhood and usually decreases after adolescence), poikiloderma, diffuse palmoplantar hyperkeratosis, and pseudosyndactyly. Mucosal manifestations are also common and include hemorrhagic mucositis and gingivitis, periodontal disease, premature loss of teeth, and labial leukokeratosis. Other mucosal findings can include ectropion, esophageal strictures/stenosis, anal stenosis, colitis, urethral stenosis/strictures, and severe phimosis. Severe long-term complications of Kindler syndrome include periodontitis, mucosal strictures, and aggressive squamous cell carcinomas. Manifestations can range from mild to severe.
Kindler syndrome is inherited as an autosomal recessive disorder (see Figure 5 above). This means that an abnormal gene must be inherited from each parent. On average, one-in-four children in a family are affected, and the familial nature of the disorder may be unnoticed.
Clinical features of Kindler syndrome
- Blistering and photosensitivity beginning in infancy or early childhood
- Gradual poikiloderma (altered pigmentation) and cutaneous atrophy (wasting)
- Trauma related blistering on hands and feet
- Can also develop mucosal involvement, ophthalmic and dental abnormalities
- Early development of actinic keratoses
The diagnosis of Kindler syndrome is established in the index case with characteristic clinical findings and identification of either biallelic FERMT1 gene mutation variants on molecular genetic testing or suggestive histologic findings and/or immunolabeling on skin biopsy. Mutational analysis (blood testing of genes), although not currently considered the first-line diagnostic test, is also available in some countries.
Kindler syndrome treatment: See treatment of epidermolysis bullosa below.
Epidermolysis bullosa causes
Epidermolysis bullosa is usually inherited. Epidermolysis bullosa can result from a genetic mutation in one of 18 genes. These mutations, or errors in the genetic code, do not allow the body to either produce an essential protein or produce a working form of the protein thus resulting in extremely fragile skin. The disease gene may be passed on from one parent who has the disease (autosomal dominant inheritance). Or it may be passed on from both parents (autosomal recessive inheritance) or arise as a new mutation in the affected person that can be passed on.
Epidermolysis bullosa can also be an autoimmune disease in which the body produces antibodies to the structural components of the skin.
The severity of epidermolysis bullosa is generally dependent upon many factors including type, subtype, and inheritance pattern.
Epidermolysis bullosa prevention
It’s not possible to prevent epidermolysis bullosa. But you can take steps to help prevent blisters and infection.
- Handle your child gently. Your infant or child needs cuddling, but be very gentle. To pick up your child, place him or her on soft material, such as cotton, and support under the buttocks and behind the neck. Don’t lift your child from under his or her arms.
- Take special care with the diaper area. If your child wears diapers, remove the elastic bands and avoid cleansing wipes. Line the diaper with a nonstick dressing or spread it with a thick layer of zinc oxide paste.
- Keep the home environment cool. Set your thermostat so that your home remains cool and the temperature remains steady.
- Keep the skin moist. Gently apply lubricants, such as petroleum jelly.
- Dress your child in soft clothes. Use soft clothing that’s simple to get on and off. It may help to remove labels and put clothing on seam-side out to minimize scratching. Try sewing foam pads into the lining of clothing by elbows, knees and other pressure points. Use soft special shoes, if possible.
- Prevent scratching. Trim your child’s fingernails regularly. Consider putting mittens on him or her at bedtime to help prevent scratching and infection.
- Encourage your child to be active. As your child grows, encourage him or her to be involved in activities that don’t cause skin injury. Swimming is a good option. For children with mild forms of epidermolysis bullosa, they can protect their skin by wearing long pants and sleeves for outdoor activities.
- Cover hard surfaces. For example, place sheepskin on car seats and line the bathing tub with a thick towel.
Epidermolysis bullosa complications
Complications of epidermolysis bullosa may include:
- Infection. Blistering skin is vulnerable to bacterial infection.
- Sepsis. Sepsis occurs when bacteria from a massive infection enter the bloodstream and spread throughout the body. Sepsis is a rapidly progressing, life-threatening condition that can cause shock and organ failure.
- Fusion of fingers and changes in the joints. Severe forms of epidermolysis bullosa can cause fusion of fingers or toes and abnormal bending of joints (contractures). This can affect the function of the fingers, knees and elbows.
- Problems with nutrition. Blisters in the mouth can make eating difficult and lead to malnutrition and anemia (such as low iron levels in the blood). Problems with nutrition can also cause delayed wound healing and, in children, slowed growth. It is important to work with a nutritionist experienced in the care of special needs patients. Treatment for iron deficiency anemia is often necessary. Other patients have selenium and carnitine or vitamin D deficiencies which may predispose them to cardiomyopathy and osteoporosis. Many patients develop failure to thrive and require feeding gastrostomies.
- Constipation. Difficulty passing stool may be due to painful blisters in the anal area. It can also be caused by not ingesting enough liquids or high-fiber foods, such as fruits and vegetables.
- Dental problems. Tooth decay and problems with tissues inside the mouth are common with some types of epidermolysis bullosa.
- Skin cancer. Adolescents and adults with certain types of epidermolysis bullosa are at high risk of developing a type of skin cancer known as squamous cell carcinoma. Squamous cell carcinoma is the leading cause of death in epidermolysis bullosa usually occurring after the 2nd decade of life. Patients with recessive dystrophic epidermolysis bullosa and junctional epidermolysis bullosa are at increased risk of developing skin cancers during their lifetimes. It is very important that all epidermolysis bullosa patients have at least yearly examination of all skin areas.
- Death. Infants with a severe form of junctional epidermolysis bullosa are at high risk of infections and loss of body fluids from widespread blistering. Their survival also may be threatened because of blistering, which may hamper their ability to eat and breathe. Many of these infants die in childhood.
Epidermolysis bullosa symptoms
Epidermolysis bullosa signs and symptoms vary depending on type. They include:
- Fragile skin that blisters easily, especially on the hands and feet
- Nails that are thick or don’t form
- Blisters inside the mouth and throat
- Thickened skin on the palms and soles of the feet
- Scalp blistering, scarring and hair loss (scarring alopecia)
- Thin-appearing skin (atrophic scarring)
- Tiny white skin bumps or pimples (milia)
- Dental problems, such as tooth decay from poorly formed enamel
- Difficulty swallowing (dysphagia)
- Itchy, painful skin
Epidermolysis bullosa blisters may not appear until a toddler first begins to walk or until an older child begins new physical activities that trigger more intense friction on the feet.
Epidermolysis bullosa diagnosis
A doctor may suspect epidermolysis bullosa from the appearance of the affected skin. He or she will likely have your child undergo laboratory tests to confirm the diagnosis. They may include:
- Skin biopsy for immunofluorescent mapping. With this technique, a small sample of affected skin is removed and examined with a microscope and reflected light to identify the layer or layers of skin involved. This test also identifies whether the proteins needed for skin growth are functioning.
- Genetic testing. Genetic testing is sometimes used to confirm the diagnosis because most forms of epidermolysis bullosa are inherited. A small sample of blood is taken and sent to a lab for analysis.
- Prenatal testing. Families with a history of epidermolysis bullosa may want to consider prenatal testing and genetic counseling.
Epidermolysis bullosa treatment
There is no cure for epidermolysis bullosa, although there are medicines to help prevent infection and to reduce discomfort.
Goals of treatment include preventing blisters, caring for blistered skin, treating infection, and treating nutritional problems.
Dietitians can find recipes for food that is nutritious and easy to eat, and recommend diets to prevent stomach problems, constipation, or diarrhea.
Medications
Medications can help control pain and itching and treat complications such as infection in the bloodstream (sepsis). The doctor may prescribe oral antibiotics if the wounds show signs of widespread infection (fever, weakness, swollen lymph glands).
Surgery
Surgery may be necessary in some cases:
- If the tube from your mouth to stomach has narrowed because of scarring, your doctor may suggest surgery to make it larger.
- If you are unable to eat, your doctor may suggest a feeding tube so that food can go right into your stomach.
- If blisters have caused your fingers or toes to join together, your doctor may suggest surgery to separate them.
- If scarring has affected the function of the hand, the doctor may suggest a skin graft.
Rehabilitation therapy
Working with a rehabilitation specialist (physical therapist, occupational therapist) can help ease the limitations on motion caused by scarring and contractures.
Home remedies
Coping with the symptoms of epidermolysis bullosa can be difficult. You do not have to handle the disease alone. There are doctors, nurses, social workers, clergy members, psychologists, dietitians (people who study food and nutrition), and support groups that can help.
Preventing blisters. There are a few things you can do to protect your skin from blistering. These steps will also help protect your baby’s skin during cuddling.
- Avoid getting too hot by keeping rooms at an even temperature.
- Apply lotion to the skin to reduce rubbing and keep the skin moist.
- Wear soft clothing.
- Use sheepskin on car seats and other hard surfaces.
- Wear mittens at bedtime to help prevent scratching.
Treating blisters. Talk to your doctor about how to treat blisters when they appear. Steps your doctor might take include:
- Explaining how to safely break a new blister.
- Prescribing a mild painkiller so that changing bandages won’t hurt as much.
- Recommending special bandages that will help the blisters to heal, lessen the pain, and prevent infection.
Caring for blisters
Your doctor can show you how to care for blisters properly and advise you on ways to prevent them. Talk to your doctor about safe ways to break and drain blisters before they get too large. Your doctor can also recommend products to help keep the affected areas moist, which helps promote heling and prevent infection.
In general, take these steps:
- Wash your hands. Wash your hands before touching your child’s blisters or changing dressings.
- Control pain. About 30 minutes before a dressing change or other painful procedure, older children and adults may take a prescription-strength pain medication. For people who don’t respond to pain relievers, other options include anti-seizure drugs such as gabapentin and pregabalin.
- Cleanse skin daily. To cleanse a wound, soak it for five to 10 minutes in a mild solution of salt and water. Other options are mild solutions of diluted vinegar or bleach. Soaking loosens stuck bandages and helps reduce the pain of changing bandages. Rinse with lukewarm water.
- Puncture new blisters. This prevents them from spreading. Use a sterile needle to puncture each new blister in two spots. But leave the roof of the blister intact to allow for drainage while protecting the underlying skin.
- Apply treated dressings. Spread petroleum jelly or other moisturizing substance on a nonstick bandage (Mepilex, Telfa, Vaseline gauze). Then gently place the bandage on the wound. Secure the pad with rolled gauze if needed.
- Wrap blistered hands and feet daily. With some severe forms of this condition, daily wraps help prevent contractures and fusion of the fingers and toes. Special wraps and gauze dressings are useful for this treatment.
- Watch for signs of infection. If you notice redness, heat, pus or a red line leading from the blister, talk with your doctor about prescription antibiotics.
- Keep cool. Blistering is often worsened by heat and warm conditions.
Dressing Basics
After lancing and gently draining epidermolysis bullosa blisters, dressings are applied. In most cases, dressings consist of three layers:
- The first or primary layer is non-adherent and the products used are variable and primarily based on individual tolerance and family preference.
- The secondary layer stabilizes the primary layer and provides padding and protection.
- The third layer is used to maintain the dressing in place. It is usually somewhat elastic in nature.
First or Contact Layer Dressings
The contact layer sits on top of a wound protect against trauma during dressing changes and allow for the passage of drainage through the holes onto an outer secondary dressing.
One of the benefits of using contact layers is the fact that they can stay on the wound bed for several days without disturbing the newly healed skin. Depending on the condition of the wound, the surrounding skin and the presence of infection, the physician may modify the frequency of dressing changes.
These dressings are made up of a single layer, non-adherent woven mesh like material. Examples include:
- Mepitel is made of a silicone safetac material
- N-Terface is made of a high density polyethylene
- Conformant 2 is made of a high density polyethylene
- Restore Contact Layer is a non-occlusive, fine polyester mesh with a petrolatum-based formula
Hydrogels
Hydrogels may be in the form of either a gel, impregnated into gauze or in the form of a hydrated sheet dressing. Basically made up of glycerin or water, they hydrate wounds providing a moist environment enhancing re-epithelialization. These dressings may be cooled in the refrigerator to provide a soothing effect on painful wounds.
Hydrogels may be helpful as primary or secondary dressings to a varied number of wounds. They are not usually recommended for wounds with heavy exudate. Please note in some instances hydrogels may dehydrate if not covered appropriately. If this happens, the dressing will need to be moistened for removal. Please review product application carefully.
- Vigilon
- DermaGuaze
- Normlgel (Note: Normlgel contains normal saline, has been known to sting.)
- Restore Hydrogel
Secondary Dressings
If a contact layer product is not being used a secondary dressing will go directly on the wound area
Impregnated Gauze:
The gauze dressings listed below are impregnated with Vaseline, Aquaphor or Zinc Oxide. They can be used as primary dressings. Adding additional ointment will help prevent the gauze from drying out and hardening.
- Vaseline Gauze
- Aquaphor Gauze
- Viscopaste Zinc Oxide Paste Gauze
Basic Non-Adherent Gauze:
Provides a non-stick covering for the wounds. Can be used with or without a contact layer.
- Telfa
- Release
- Non Adhesive Pads
Foams:
These dressings are non-adherent and absorbent.
Most foams provide insulation and a moist environment. Foam dressings are used on wounds with light to heavy amounts of drainage. Please keep in mind if the dressing becomes saturated with drainage, it could irritate the surrounding skin.
Foams can also be used for heavier draining, non-infected wounds and can be used over a contact layer.
Depending on the condition of the wound, the surrounding skin and the presence of infection, the physician may modify the frequency of dressing changes.
- Allevyn
- Cutinova Foam
- Gentleheal
- Mepilex
- Mepilex Transfer
- Mepilex Lite
- Mepilex Border
- Mepilex Border Lite
- PolyMem
- Restore Foam
Specialty Absorptive Dressings:
These dressings provide a non shearing, non-adherent, absorbent environment that allows for the wicking away of drainage. They can be utilized on wounds with mild or heavy drainage and can be used to provide a cushion/padding for areas of the skin that can come into contact with pressure such as crib, playpen, high chair and play areas. Special use: some hospitals will use a large sheet of Exu-dry under a newborn.
Exu-dry: Two non-adherent wound contact layers which helps to prevent friction and shear. The drainage is wicked away into an absorbent layer.
Hydrofiber Wound Dressing:
When applied to a wound it creates a gel after coming into contact with wound exudate. This provides a moist environment and helps reduce damage of newly forming tissue upon removal and should only be used on moderately to heavily draining wounds. These dressings are not indicated for dry (non-draining) wounds. The gel can be rinsed or soaked away with saline, allowing removal of the dressing without re-traumatizing the wound bed and minimizing pain. Depending on the condition of the wound, the surrounding skin and the presence of infection, the physician may modify the frequency of dressing changes.
- Aquacel: A sterile, absorbent pad made from sodium carboxymethylcellulose fibers
- Restore Calcium Alginat: Alginates are made of soft non-woven fibers which are derived from seaweed
Gauze:
Cotton mesh interwoven material
Rolled gauze: can be used to hold dressings in place. Available in both sterile and non-sterile packaging, in various length and sizes by many venders
Kerlix
Gauze pads can be used for cleansing and covering. It is available from many vendors and comes in various widths.
Retention Gauze:
The outermost layer, these help hold the dressings in place. These mesh dressings are packaged as a roll, and are cut to length based on the bandaged area. They are washable and can be used more than once. There are many options:
- Promed Elastic Net: This is packaged as a roll, and is cut to length based on the bandaged area
- Surilast
- Tubifast: This is also available in garments, though in most cases these are unlikely to be covered by insurance
Similar in look to a roller bandage, these options come in many colors and the layers will adhere to itself, which helps it stay in place.
- Coban: Similar in look to a roller bandage, this comes in many colors and will adhere to itself, which helps it stay in place
- Coflex: Similar in look to a roller bandage, this comes in many colors and will adhere to itself, which helps it stay in place
Adhesive removers:
While it is better to avoid having adhesives and tape applied to epidermolysis bullosa skin, accidents happen. These are some suggestions of products that can help remove things that stick to skin and wounds.
- AllKare Adhesive Remover Wipes
- Uni-Solve Adhesive Remover
- Detachol
- Trio Niltac Medical Adhesive Remover (currently not available in the USA or Canada)
Advanced Wound Care
For non-healing wounds, your medical provider may decide to use a specialized product to help close the wounds. With the advancement of science there are many useful wound care products that are either collagen based or even live cells. All of these products are expensive and should only be used in those individuals with chronic wound that will not heal with simple good wound care. Your healthcare provider, or a specialist in wound care, can help you determine if one of these advance wound care products are right for a non-healing wound.
Treating infection. Infections may develop even when blisters are treated. Signs of infection are:
- Redness and heat around open parts of the skin.
- Pus.
- Crusting on top of the sore.
- A red line or streak under the skin that spreads away from blister.
- A sore that does not heal.
- Fever or chills.
If you get an infection, your doctor may treat it with:
- A soaking liquid.
- An antibiotic cream or pill.
- A special covering (for sores that don’t heal).
Wound Care Products
Wound care is part of the supportive care given to those with epidermolysis bullosa. It is important to note that no one product works for every person. Wound healing is primary goal.
Cleansers and Topical Products:
Cleansers (mild):
Some cleansers used successfully in epidermolysis bullosa include:
- Cetaphil mild non soap cleanser
- Dove mild soap
- Restore Wound Cleanser
It is important to use gentle cleaners and wash the wounds and skin routinely. We have found that if bathing is painful, the addition of one pound of pool salt in a tub of water really decrease the pain with bathing.
Topical:
1)Moisturizers:
- Emu oil : purified from the refined fat of an Emu
- Restore Dimethicreme: dimethicone-based protective layer
- A&D ( Schering-Plough HealthCare Products, Inc)
- Aquaphor (Beiersdorf, Inc.)
- White petroleum
These are some of the non-medicated topicals that when used alone, or on a dressing, help maintain a moist environment on wound bed. When used on dressings, it helps to prevent sticking to the wound.
2) Antibiotic Ointments:
A) Over the counter (OTC) antibiotic ointments:
- Bacitracin
- Polysporin
Apply topical antibiotics to lesions: Mild, over-the-counter antibiotics (Polysporin, Bacitracin) are effective in preventing infection and may be rotated every month or two to discourage bacterial resistance.
It is important to note that some data suggests that white petrolatum is a better antibacterial agent than those listed above and it does not cause allergic contact dermatitis. There are epidermolysis bullosa providers who advocate its use for that reason.
B) Prescription antibiotic ointments:
- Bactroban (Mupricin) (GlaxoSmithKline)
- Centany (Mupricin) (OrthoNeutrogena)
Mupricin should be used only when infection is present. (Signs of infection include increased redness, swelling, pain and warmth).
Prolonged use of antibiotics has been associated with the development of resistant Staph infections.
3) Antimicrobial Topicals – Honey:
MediHoney (DermaScience)
Recently approved for use in the USA, these products can be helpful in cleaning up draining wounds. The ointment can be applied to the dressings already used by the family.
*It is very important to remember that you must use medical grade honey and that honey form your local grocery store is not to be used under any circumstances.*.
4) Antimicrobial Topicals – Silver (AG) Products:
- Silvadine cream 1% ( silver sulfadiazine ) – a soft, white, water-miscible cream
- Restore Silver contact layer and foam
- Mepilex AG
- Acticoat
- Aquacel AG
- SilvaSorb
- Silverlon
- Contreet
Different modes of silver delivery can lead to varying rates of absorption.
There are many unanswered questions about the safety of long-term use of silver products and there is not consensus among epidermolysis bullosa providers regarding its use in epidermolysis bullosa. Until clinical trials are conducted that prove these products are safe to use on a regular long-term basis, individuals should use the products cautiously and for limited periods of time.
With improper use there have been a few documented cases in the laboratory of bacteria becoming resistant to silver.
5) Additional Antimicrobial Dressings:
Xeroform contains a medication called Bismuthtribromophenate. Please consult physician prior to using. It may not be appropriate for infants and children.
Providing good nutrition
A varied, nutritious diet promotes growth and development in children and helps wounds heal. If blisters in the mouth or throat make it difficult for your child to eat, here are some suggestions:
- For babies, try bottle nipples designed for premature infants, a syringe or a rubber-tipped medicine dropper.
- For older children, serve nutritious, soft foods that are easy to swallow, such as vegetable soup and fruit smoothies. Puree solid foods with broth or milk.
- Serve food and beverages lukewarm, at room temperature or cold.
- Talk with a dietitian or doctor about using supplements to minimize nutrient and vitamin deficiencies.
Preventing nutritional problems. In some people with epidermolysis bullosa, blisters may appear in the mouth and in the tube leading from the mouth to the stomach. This makes it hard to chew and swallow and can lead to nutritional problems. Because nutrition is so important for proper growth and development, it is important that children with the disease eat well.
Steps you can take to help prevent nutritional problems in children with the disease include:
- Feed infants using a bottle with a special nipple, an eyedropper, or a syringe.
- Add extra liquid to finely mashed food to make it easier to swallow.
- Give your children soups, milk drinks, mashed potatoes, custards, and puddings.
- Never serve food that is too hot.
Dietitians can help by:
- Finding recipes for food that is nutritious and easy to eat.
- Suggesting you take certain vitamins.
- Recommending diet changes to prevent stomach problems, constipation, or diarrhea.
- Ludwig RJ. Clinical Presentation, Pathogenesis, Diagnosis, and Treatment of Epidermolysis Bullosa Acquisita. ISRN Dermatology. 2013;2013:812029. doi:10.1155/2013/812029. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3727188/[↩][↩]
- Roenigk HH, Jr., Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Archives of Dermatology. 1971;103(1):1–10. https://www.ncbi.nlm.nih.gov/pubmed/4321800[↩]
- Buijsrogge JJA, Diercks GFH, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. British Journal of Dermatology. 2011;165(1):92–98. https://www.ncbi.nlm.nih.gov/pubmed/21457208[↩]
- Schmidt E, Zillikens D. Pemphigoid diseases. The Lancet. 2013;381:320–332. https://www.ncbi.nlm.nih.gov/pubmed/23237497[↩]
- Kim JH, Kim SC. Epidermolysis bullosa acquisita. Journal of the European Academy of Dermatology and Venereology. 2013 https://www.ncbi.nlm.nih.gov/pubmed/23368767[↩]
- Kim JH, Kim YH, Kim S-C. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Dermato-Venereologica. 2011;91(3):307–312. https://www.ncbi.nlm.nih.gov/pubmed/21394418[↩][↩][↩]
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? Journal of Dermatology. 2010;37(3):220–230 https://www.ncbi.nlm.nih.gov/pubmed/20507385[↩]
- Hellberg L, Holdorf K, Hänsel M, et al. Methylprednisolone blocks autoantibody-induced tissue damage through inhibition of neutrophil activation. Journal of Investigative Dermatology. 2013 https://www.ncbi.nlm.nih.gov/pubmed/23448878[↩]
- Connolly SM, Sander HM. Treatment of epidermolysis bullosa acquisita with cyclosporine. Journal of the American Academy of Dermatology. 1987;16(4):p. 890. https://www.ncbi.nlm.nih.gov/pubmed/3571559[↩]
- Maize JC, Jr., Cohen JB. Cyclosporine controls epidermolysis bullosa acquisita co-occuring with acquired factor VIII deficiency. International Journal of Dermatology. 2005;44(8):692–694. https://www.ncbi.nlm.nih.gov/pubmed/16101876[↩]
- Driessen G, van der Burg M. Educational paper: primary antibody deficiencies. European Journal of Pediatrics. 2011;170(6):693–702 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3098982/[↩]
- Cines DB, Blanchette VS. Medical progress: immune thrombocytopenic purpura. New England Journal of Medicine. 2002;346(13):995–1008 https://www.ncbi.nlm.nih.gov/pubmed/11919310[↩]
- Ishii N, Hashimoto T, Zillikens D, Ludwig RJ. High-dose intravenous immunoglobulin (IVIG) therapy in autoimmune skin blistering diseases. Clinical Reviews in Allergy and Immunology. 2010;38(2-3):186–195 https://www.ncbi.nlm.nih.gov/pubmed/19557317[↩]
- Ahmed AR, Gürcan HM. Treatment of epidermolysis bullosa acquisita with intravenous immunoglobulin in patients non-responsive to conventional therapy: clinical outcome and post-treatment long-term follow-up. Journal of the European Academy of Dermatology and Venereology. 2012;26:1074–1083 https://www.ncbi.nlm.nih.gov/pubmed/21819451[↩]
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. Journal of Cutaneous Medicine and Surgery. 2001;5(5):397–399 https://www.ncbi.nlm.nih.gov/pubmed/11907850[↩]
- Kirtschig G, Murrell D, Wojnarowska F, Khumalo N. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database of Systematic Reviews. 2003;(1)CD004056 https://www.ncbi.nlm.nih.gov/pubmed/12535507[↩]
- Joly P, Mouquet H, Roujeau J-C, et al. A single cycle of rituximab for the treatment of severe pemphigus. New England Journal of Medicine. 2007;357(6):545–552 https://www.ncbi.nlm.nih.gov/pubmed/17687130[↩]
- McKinley SK, Huang JT, Tan J, Kroshinsky D, Gellis S. A case of recalcitrant epidermolysis bullosa acquisita responsive to rituximab therapy. Pediatric Dermatology. 2012 https://www.ncbi.nlm.nih.gov/pubmed/23106762[↩]
- Schmidt E, Zillikens D. Immunoadsorption in dermatology. Archives of Dermatological Research. 2010;302(4):241–253. https://www.ncbi.nlm.nih.gov/pubmed/20049466[↩]
- Kubisch I, Diessenbacher P, Schmidt E, Gollnick H, Leverkus M. Premonitory epidermolysis bullosa acquisita mimicking eyelid dermatitis: successful treatment with rituximab and protein a immunoapheresis. American Journal of Clinical Dermatology. 2010;11(4):289–293. https://www.ncbi.nlm.nih.gov/pubmed/20373827[↩]
- Miller JL, Stricklin GP, Fine JD, King LE, del Carmen Arzubiaga M, Ellis DL. Remission of severe epidermolysis bullosa acquisita induced by extracorporeal photochemotherapy. British Journal of Dermatology. 1995;133(3):467–471. https://www.ncbi.nlm.nih.gov/pubmed/8547007[↩]




{kind=link}