Contents
- Erythropoietic protoporphyria
Erythropoietic protoporphyria
Erythropoietic protoporphyria also called EPP is a rare inherited disorder of the heme biosynthetic pathway resulting in the accumulation of protoporphyrin IX (PPIX) in red blood cells that causes painful, non-blistering photosensitivity to sunlight and artificial light (phototoxicity) that is usually noticed in early childhood and occurs throughout life and potential liver disease 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13. On sun exposure, patients with erythropoietic protoporphyria (EPP) may first experience tingling, itching, burning of their skin. After continued exposure to light, the skin may become red and swollen. The hands, arms, and face are the most commonly affected areas. Protoporphyrin IX (PPIX) accumulates first in the bone marrow in erythropoietic protoporphyria (EPP) and then in red blood cells, plasma and sometimes the liver.
Protoporphyrin IX (PPIX) is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. Protoporphyrin IX (PPIX) is not soluble in water so is not excreted in the urine. Excess protoporphyrin IX is excreted by the liver into bile, where it becomes insoluble and can crystallize, forming gallstones and obstructing bile flow. Biliary obstruction impairs protoporphyrin IX excretion, resulting in further protoporphyrin IX accumulation and escalating protoporphyrin IX-mediated liver damage. Protoporphyrin IX-mediated liver dysfunction has been reported in over 50% of patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), which may progress to liver failure in up to 5% of patients and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation 14, 15.
Erythropoietic protoporphyria (EPP) is the third most common type of porphyria, and the most common in childhood. Erythropoietic protoporphyria (EPP) causes very painful photosensitivity and can greatly impair quality of life. Delay in diagnosis is greater than with any other type of porphyria.
Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Usually, these symptoms subside in 12 to 24 hours and heal without significant scarring. Blistering and scarring are characteristic of other types of cutaneous porphyria but are unusual in Erythropoietic protoporphyria. Skin manifestations generally begin early childhood and are more severe in the summer.
Erythropoietic protoporphyria (EPP) is caused by mutations in the FECH gene. The FECH gene provides instructions for making an enzyme known as ferrochelatase 16. The ferrochelatase (FECH) enzyme is involved in the production of a molecule called heme. The production of heme is a multi-step process that requires eight different enzymes. Ferrochelatase is responsible for the eighth and final step in this process, in which an iron (Fe2+) atom is inserted into the center of protoporphyrin IX (the product of the seventh step) to form heme. Heme is vital for all of your body’s organs, although it is most abundant in your blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Due to abnormally low levels of ferrochelatase (FECH) enzyme, excessive amounts of protoporphyrin IX (PPIX) build up in the bone marrow, blood plasma, and red blood cells 17. Protoporphyrin IX (PPIX) compounds are formed during the normal process of heme production, but reduced activity of ferrochelatase allows them to accumulate to toxic levels. The excess porphyrins can leak out of developing red blood cells and be transported through the bloodstream to your skin and other tissues. High levels of porphyrins compounds in the skin cause the oversensitivity to sunlight that is characteristic of erythropoietic protoporphyria (EPP). Large amounts of porphyrins in the gallbladder can also cause gallstones. Less commonly, a buildup of porphyrins in the liver can result in liver damage that leads to cirrhosis and liver failure.
Some patients with symptoms of protoporphyria have a genetic change (gain-of-function variants in exon 11 of ALAS2) in a different gene called ALAS2 gene (delta-aminolevulinic acid synthase-2 gene) and follows an X-linked inheritance pattern 18, 19. When a patient has a genetic change in the ALAS2 gene located on the X chromosome, the condition is referred to as X-linked protoporphyria (XLP) 20. XLP accounts for 2–10% of protoporphyria cases 1. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 19. This version of the enzyme is found only in developing red blood cells called erythroblasts. ALA-synthase enzyme also plays an important role in the production of heme. The production of heme is a multi-step process that requires eight different enzymes. ALA-synthase is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA). The excess delta-aminolevulinic acid (δ-ALA) is converted by other enzymes to compounds called porphyrins. If porphyrins compounds build up in erythroblasts, they can leak out and be transported through the bloodstream to your skin and other tissues. High levels of porphyrins in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP).
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are characterized by a buildup of protoporphyrin in the skin, blood, and liver. It typically presents in early childhood with immediate pain and crying upon exposure to bright sunlight (e.g., babies may cry in the sun or in the car). Erythropoietic protoporphyria signs and symptoms is seasonal in nature with symptoms principally occurring in the spring and summer season.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) signs and symptoms include:
- Painful, non-blistering skin reactions to sunlight or artificial light, most often on the tops of the hands and feet, face and ears. Most individuals with EPP develop acute cutaneous photosensitivity within 30 minutes after exposure to sun or ultraviolet light. Pain can be severe and and can last up to a week after sun exposure 6.
- Itching, burning, or redness of the skin.
- Swelling or edema in the affected areas.
- Persistent redness or inflammation of the skin.
- Pregnancy has been associated with decreased protoporphyrin levels and increased tolerance to sun exposure, but these are inconsistent 21, 22, 23, 24, 25, 26, 27, 28.
- Over the years, the skin on the backs of the hands and cheeks can have some thickening with subtle pitted scarring.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) causes skin pain on exposure to sunlight. There may not be anything to see at the time. Prolonged exposure can result in some redness and swelling, and uncommonly in blistering and crusting.
EPP is a very rare inherited disorder that affects males and females in equal numbers. It is estimated that erythropoietic protoporphyria (EPP) occurs in about 1 in about 75,000 to 1 in 200,000 individuals in Europe, with prevalence figures ranging between 1 in 75,000 (The Netherlands) and 1 in 200,000 (Wales) 29, 30, 9. However, recent genetic evidence has revealed erythropoietic protoporphyria true prevalence is ∼1 in 17,000 31, 32, 33. The number of patients affected by these disorders in the US is unknown. Erythropoietic protoporphyria (EPP) seems that males and females are equally affected 34.
X-linked protoporphyria (XLP) accounts for about 10% of erythropoietic protoporphyria cases in the United States. X-linked protoporphyria (XLP) is more likely to present in males. Females with XLP may or may not have symptoms 9.
The onset of symptoms affecting the skin usually occurs in infancy, with an average of diagnosis at age 4; however, in some cases, onset may not occur until adolescence or rarely even adulthood. A clinical diagnosis of EPP is often made during childhood. The blood needs to be sent for porphyrin analysis in a tube protected from light with aluminium foil.
- The patient’s red blood cells may be noted to fluoresce by ultraviolet microscopy.
- The characteristic change is the elevation of the red cell protoporphyrin.
- Genetic testing for mutations in the ferrochelatase (FECH) gene can be performed.
- A skin biopsy is rarely performed as the skin often appears normal; however, EPP has some characteristic features on histopathology.
Once the diagnosis has been made, regular checks of liver function are required with intermittent liver imaging. Genetic counseling is recommended for affected individuals and their families.
Erythropoietic protoporphyria is a lifelong disease, and repeated phototoxic reactions eventually lead to thickening of the skin and wax-like scarring on the face. In a small number of patients with erythropoietic protoporphyria, the accumulation of protoporphyrins in the liver leads to cirrhosis and liver failure. Onset in adulthood is rare, but an acquired form has been identified, in which clones of cells with mutated ferrochelatase expand in the setting of the myelodysplastic or myeloproliferative syndrome 35.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) diagnosis is made by finding increased levels of the protoporphyrin in the plasma or red blood cells. High performance liquid chromatography or extraction methods that measure total, metal-free, and zinc protoporphyrin are recommended for the diagnosis of protoporphyria 36. Current lists of laboratories that perform such testing can be found on websites for the United Porphyrias Association, the European Porphyria Network, and the American Porphyria Foundation. These laboratories can also give advice regarding the optimal laboratory testing or differential diagnosis of biochemical abnormalities. As sample materials and pre‐analytical specifications depend on the specific diagnostic methods, it is advisable to contact the respective laboratory prior to sample collection. Skin biopsies are not required for the diagnosis of protoporphyrias or any other type of cutaneous porphyria. Genetic testing is useful to confirm the diagnosis.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
For the initial screening for porphyrias with skin symptoms, a plasma fluorescence scan detecting increased plasma porphyrin concentrations is recommended. This approach offers the added benefit of recognizing other forms of porphyrias associated with cutaneous symptoms, that is, porphyria cutanea tarda (PCT), porphyria variegata (VP), hereditary coproporphyria (HC) and congenital erythropoietic porphyria (CEP). Moreover, a negative test result excludes any form of porphyria as the underlying cause of concurrent skin symptoms. In the case of the protoporphyrias, protoporphyrin IX (PPIX) present in the plasma typically causes a positive plasma‐fluorescence scan with a peak at around 633–635 nm 37. The diagnosis of the protoporphyrias is confirmed by the quantification of protoporphyrin IX (PPIX) in red blood cells (erythrocytes), whereby metal‐free PPIX and zinc protoporphyrin IX (ZnPP) are determined separately. A metal‐free PPIX ≥3 times the upper limit of normal (ULN) establishes the diagnosis of protoporphyrias.
To reliably perform a plasma fluorescence scan and the protoporphyrin IX (PPIX) measurement in red blood cells (erythrocytes), the vial of blood (5 mL) needs to be protected from light with an aluminium foil. Exposure of the blood tube to light can lead to photobleaching of PPIX, resulting in a decrease in measured PPIX levels in the sample as compared to the actual levels in the subject. This effect is related to the ability of conjugated double bonds in the porphyrin rings to absorb the energy from visible light 38. To avoid diagnostic delay, plasma porphyrins should be measured at the same time with erythrocyte protoporphyrin 39.
Other test materials used for the diagnosis of other forms of porphyrias are urine and faeces. Urinary porphyrins are not diagnostic in protoporphyrias as, due to its hydrophobicity, excess PPIX is excreted by the biliary route and faeces. Therefore, in the faeces, an increase in PPIX can be present, but the diagnosis requires confirmation by a significant increase of PPIX and zinc protoporphyrin IX (ZnPP)P in the erythrocytes.
Erythropoietic protoporphyria treatment includes the following:
- Avoid sunlight and wear protective clothing.
- Use topical anesthetic creams for the treatment of phototoxic symptoms. Anecdotally, many patients choose to self-treat with ice, cold water, or cold compresses, resulting in minor relief of phototoxic symptoms. However, there is no evidence of benefit with narcotic analgesics, oral or topical corticosteroids, antihistamines, acetaminophen and non-steroidal anti-inflammatory drugs 1.
- Take vitamin D supplements. There is an increased recognition of prevalence of vitamin D deficiency in protoporphyria patients due to lifelong sunlight avoidance. Routine screening for vitamin D deficiency and supplementation as per population guidelines are recommended 40, 41
- Avoid alcohol to prevent additional cause of liver damage 1. Additionally, immunization against hepatitis A and B is recommended.
- Narrowband UVB phototherapy. Narrowband UVB phototherapy does not cause the EPP pain. It is given 3 days a week over 6 weeks in Spring. It increases melanin content causing a tan and induces skin thickening so to provide some level of protection from the sun.
- Scenesse (afamelanotide). Scenesse (afamelanotide) is an alpha-melanocyte stimulating hormone that is given by subcutaneous implantation and works by increasing skin pigmentation which provides protection and improves sun tolerance 10. There is no data on the safety of Scenesse (afamelanotide) during pregnancy so this cannot be recommended for pregnant women.
- In patients undergoing surgery of prolonged duration, such as liver transplantation, light filters that limit transmission of the wavelengths 340–470 nm (i.e., acrylate yellow filter) are recommended 42, 43. No specific anesthetic agents or other medications are contraindicated in protoporphyria.
- Due to insufficient data related to efficacy, the following therapies are not recommended for the prevention of phototoxic symptoms: beta-carotene, cysteine, cimetidine, isoniazid, warfarin, quinacrine, oral zinc, N-acetylcysteine, vitamin C, omega-3 fatty acids, oral adenosine monophosphate, canthaxanthine, terfenadine, inosine, dithiothreitol (DTT) and glycerol, pyridoxine, and hydroxyethylrutosides 44, 45, 46, 1. Although several previous studies have investigated beta carotene in EPP, the evidence shows unclear or no benefit 1. Dersimelagon is a synthetic, orally administered, small molecule agonist of melanocortin-1 receptor (MC1R) being tested for the prevention of protoporphyria phototoxicity. A positive Phase 2 clinical trial has been completed showing promising results, and a Phase 3 study is ongoing 47. There are no approved therapies for children patients.
Figure 1. Erythropoietic protoporphyria
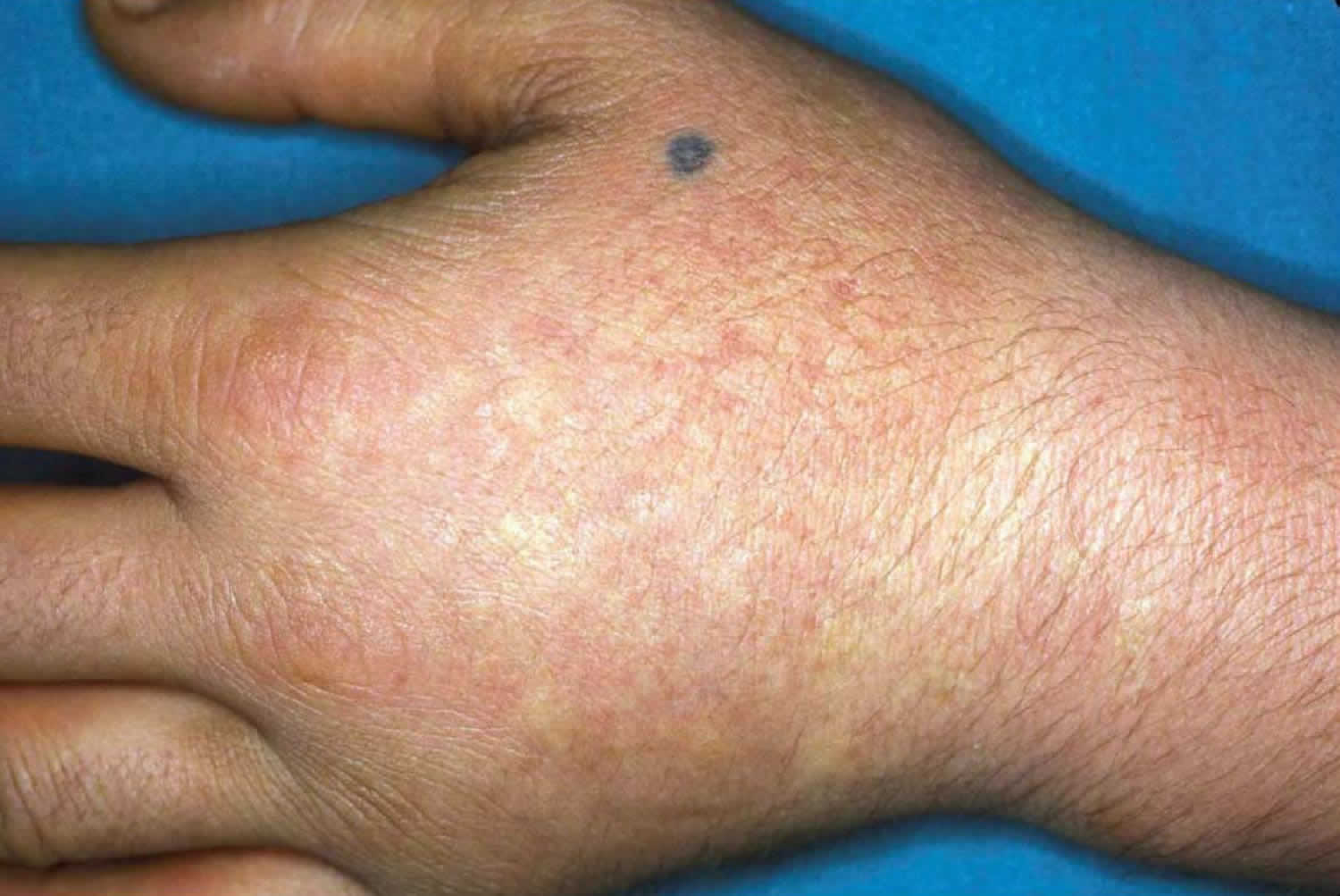
Footnote: Acute photosensitivity reaction in erythropoietic protoporphyria (EPP).
[Source 6 ]Figure 2. Erythropoietic protoporphyria
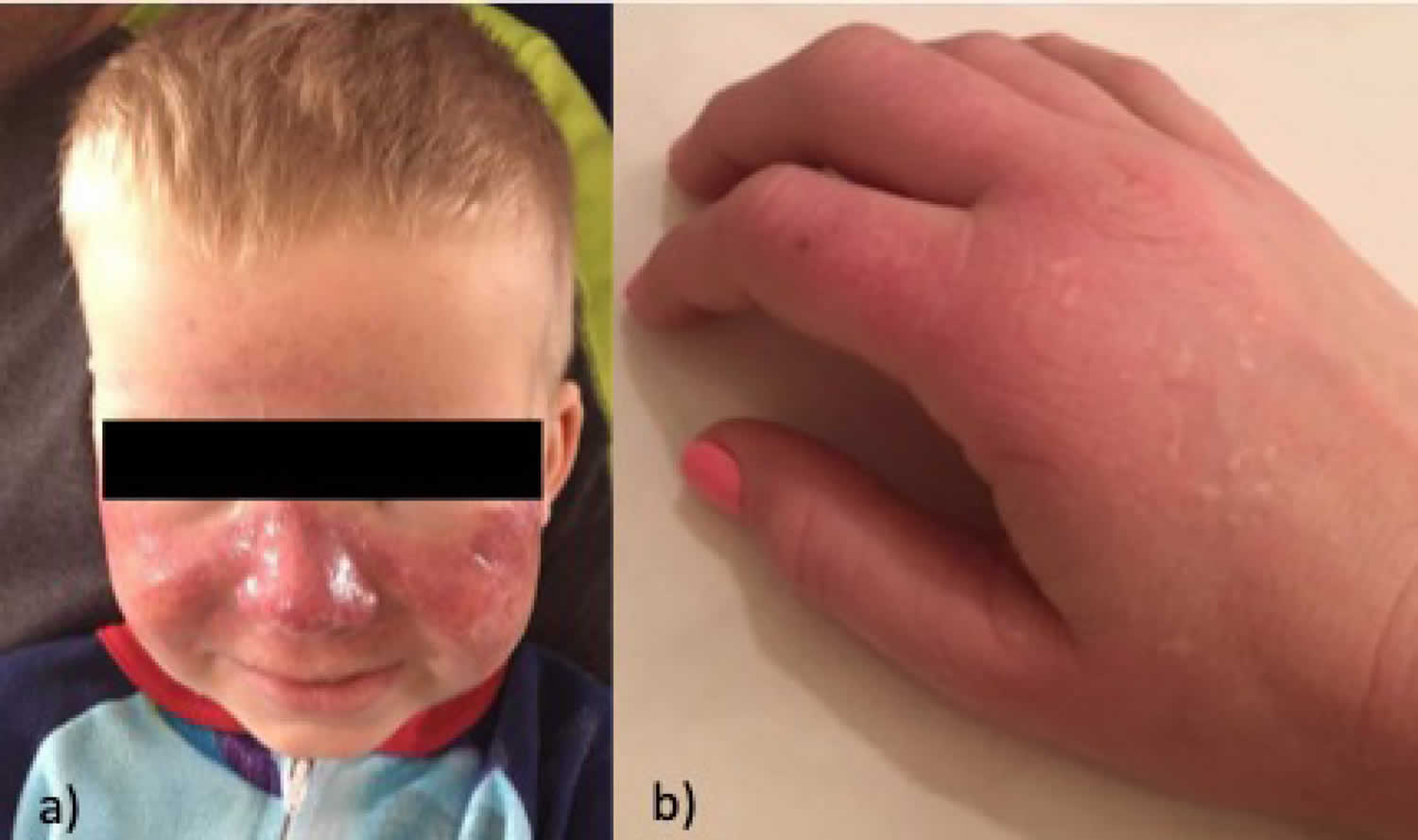
Footnotes: a) Child with extensive edema of the face with erythema and petechiae; b) Adult patient with skin redness (erythema) and skin swelling (edema) during phototoxic episode, with hypopigmented scars and skin thickening present
[Source 1 ]Figure 3. Chronic erythropoietic protoporphyria skin lesions
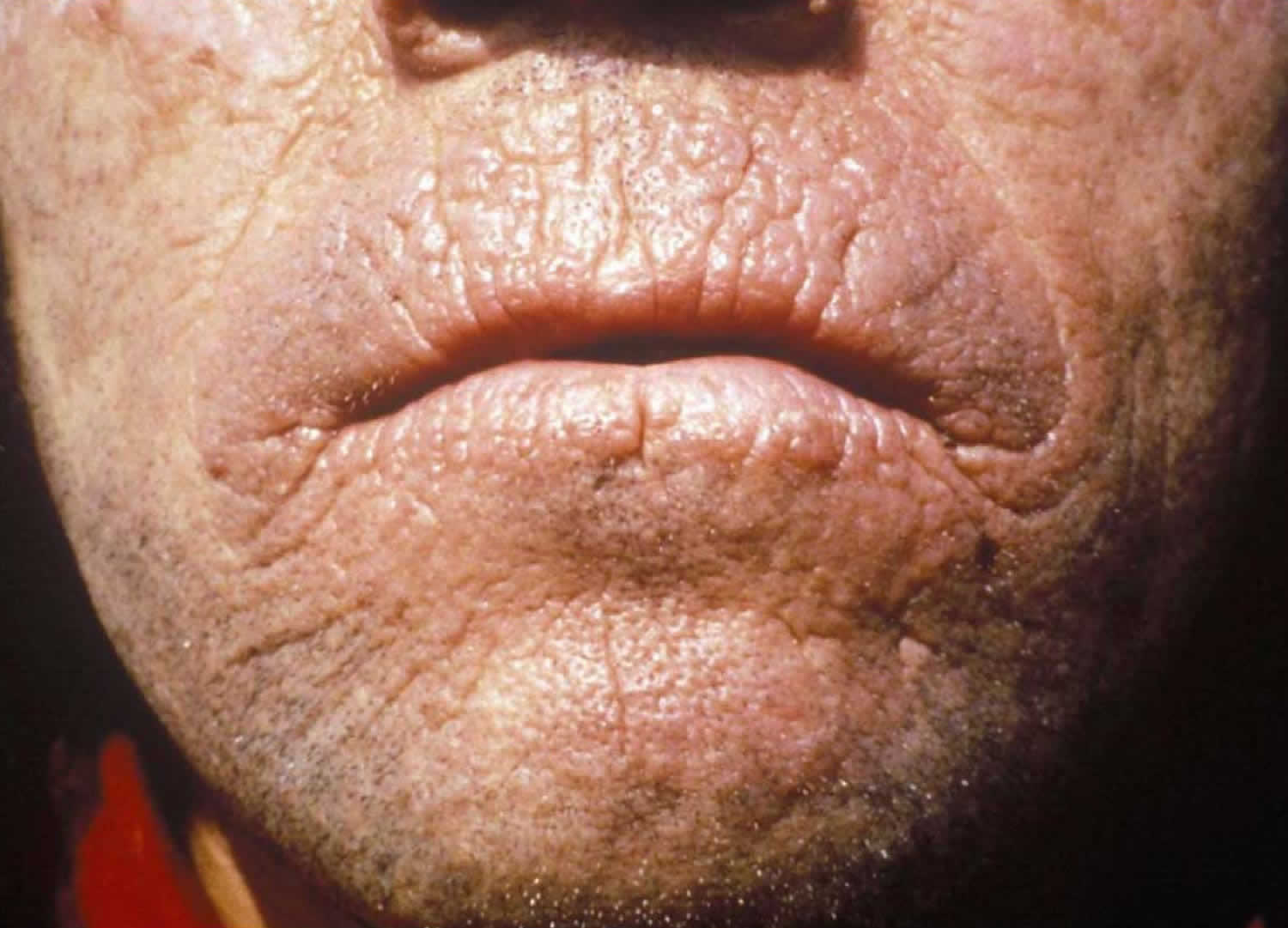
Figure 4. Heme chemical structure
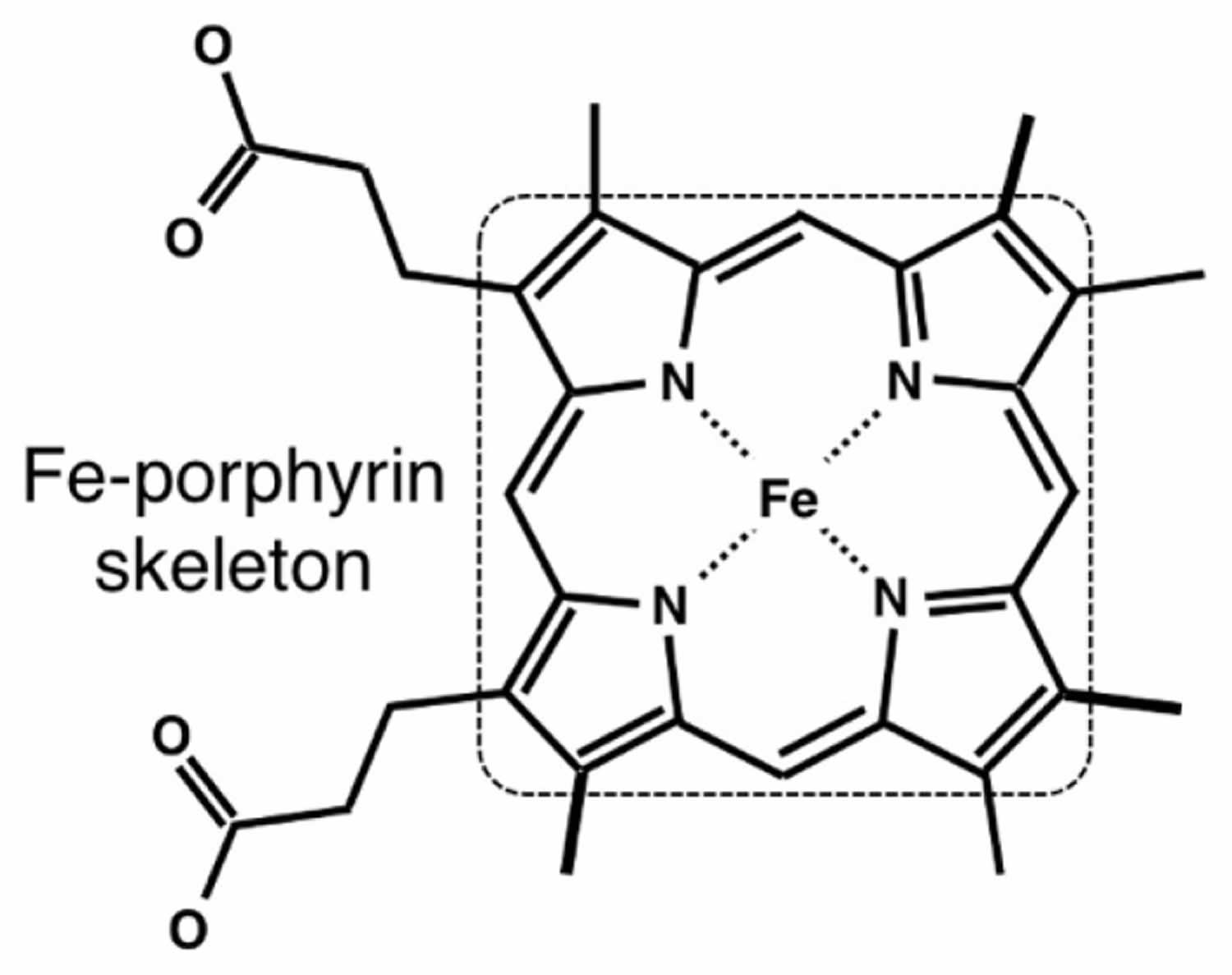
Figure 5. Heme synthesis pathway
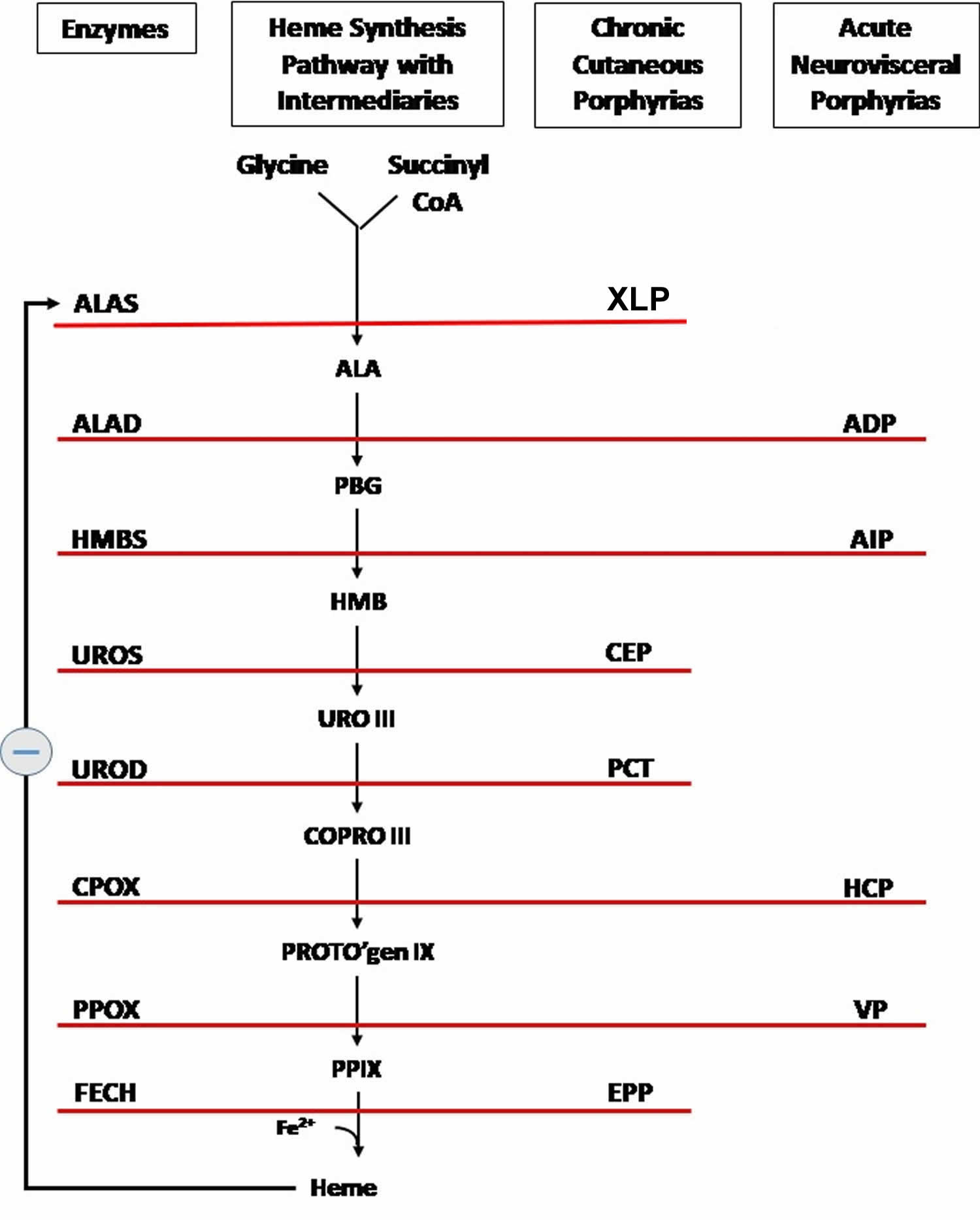
Footnotes: The heme biosynthetic pathway requires 8 enzymatic steps. Heme synthesis pathway showing the enzymes involved in the heme synthesis pathway and the associated porphyrias with the disruption of each specific enzyme. Gain-of-function variants in ALAS2 result in X-linked protoporphyria (XLP), and loss-of-functions variants in FECH result in erythropoietic protoporphyria (EPP). In both X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP), metal-free protoporphyrin IX (PPIX) accumulates in erythroblasts, erythrocytes, the plasma, and the biliary system. Metal-free protoporphyrin IX (PPIX) is photosensitive, particularly to visible light in the blue range, and the light-mediated activation of metal-free protoporphyrin IX (PPIX) produces free radicals that damage the surrounding tissues.
Enzymes, encoded by genes, catalyze each of the steps. Gene mutations cause deficient enzyme production. Disruptions are indicated by red lines connecting enzymes with the resultant porphyrias. ALAS (ALAS2) = aminolevulinate synthase (aminolevulinate synthase 2); ALAD = delta-aminolevulinic acid dehydratase; PBGD = porphobilinogen dehydratase; HMBS = hydroxymethylbilane synthase; UROS = uroporphyrinogen-III synthase; UROD = uroporphyrinogen III decarboxylase; CPOX = coproporphyrinogen-III oxidase; PPOX = protoporphyrinogen oxidase; FECH = ferrochelatase.
Porphyrias resulting from disruption of enzyme production. XLP (X-linked protoporphyria); ADP (aminolevulinic acid dehydratase porphyria); AIP (acute intermittent porphyria); CEP (congenital erythropoietic porphyria); PCT (porphyria cutanea tarda); HCP (hereditary coproporphyria); VP (variegate porphyria); EPP (erythropoietic protoporphyria).
Abbreviations: ALA = aminolevulinic acid; PBG = porphobilinogen; HMB = hydroxymethylbilane; URO III = uroporphyrinogen III; COPRO III = coproporphyrinogen III; PROTO’gen IX protoporphyrinogen IX; PPIX = protoporphyrin IX; Fe2+ = iron.
[Source 39 ]Erythropoietic protoporphyria causes
Erythropoietic protoporphyria (EPP) is caused by mutations in the FECH gene. The vast majority of patients with EPP are compound heterozygotes for both a rare pathogenic FECH variant and a common low expression FECH allele that is present in ~10% of the Caucasian population (c.315-48T>C), a combination that results in ~30% of normal enzyme activity 49, 50. Homozygosity for FECH c.315-48T>C does not cause EPP 51. The FECH gene provides instructions for making an enzyme known as ferrochelatase 16. The ferrochelatase (FECH) enzyme is involved in the production of a molecule called heme. The production of heme is a multi-step process that requires eight different enzymes. Ferrochelatase is responsible for the eighth and final step in this process, in which an iron (Fe2+) atom is inserted into the center of protoporphyrin IX (the product of the seventh step) to form heme. Heme is vital for all of your body’s organs, although it is most abundant in your blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Due to abnormally low levels of ferrochelatase (FECH) enzyme, excessive amounts of protoporphyrin build up in the bone marrow, blood plasma, and red blood cells. Porphyrins compounds are formed during the normal process of heme production, but reduced activity of ferrochelatase allows them to accumulate to toxic levels. The excess porphyrins can leak out of developing red blood cells and be transported through the bloodstream to your skin and other tissues such as your liver. High levels of porphyrins compounds in the skin cause the oversensitivity to sunlight that causes severe pain and inflammation resulting in symptoms of EPP. Large amounts of porphyrins in the gallbladder can also cause gallstones. Less commonly, a buildup of porphyrins in the liver can result in liver damage that leads to cirrhosis and liver failure.
Some patients with symptoms of protoporphyria have a genetic change (gain-of-function variants in exon 11 of ALAS2) in a different gene called ALAS2, a gene located on the X chromosome and follows an X-linked inheritance pattern 18. When a patient has a genetic change in the ALAS2 gene, the condition is referred to as X-linked protoporphyria (XLP). XLP accounts for 2–10% of protoporphyria cases 1. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 19. This version of the enzyme is found only in developing red blood cells called erythroblasts. ALA-synthase enzyme also plays an important role in the production of heme. The production of heme is a multi-step process that requires eight different enzymes. ALA-synthase is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA). The excess delta-aminolevulinic acid (δ-ALA) is converted by other enzymes to compounds called porphyrins. If porphyrins compounds build up in erythroblasts, they can leak out and be transported through the bloodstream to your skin and other tissues. High levels of porphyrins in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP).
About 4 to 6% of patients with the symptoms of erythropoietic protoporphyria (EPP) and elevated red blood cell protoporphyrin IX (PPIX) levels will not have mutations in ferrochelatase (FECH) or 5-aminolevulinate synthase 2 (ALAS2) 52, 53.
Recently, an autosomal dominant mutation in human CLPX, a modulator of heme biosynthesis, was found to result in the accumulation of delta-aminolevulinate synthase (ALAS) and protoporphyrin IX (PPIX) and symptoms of protoporphyria in an affected family 54. Acquired somatic ferrochelatase (FECH) mutations have been identified in a small number of patients in whom erythropoietic protoporphyria (EPP) has developed after the age of 40 years in association with myelodysplasia or myeloproliferative disorder 55, 56. A case of late-onset erythropoietic protoporphyria (EPP) with myelodysplastic syndrome has also been reported in a patient who had the homozygous IVS3–48T>C polymorphism in the ferrochelatase (FECH) gene 57. Late onset X-linked protoporphyria (XLP) has also been reported in a case of early myelodysplastic syndrome with somatic mosaicism in the bone marrow 58.
Erythropoietic protoporphyria inheritance pattern
Erythropoietic protoporphyria (EPP) is inherited in an autosomal recessive manner 59, 9. Everyone has two copies of the FECH gene, one inherited from the mother and one from the father. Most individuals with EPP have a different gene mutation on each copy of the FECH genes. On one copy, the change, called a mutation, has stopped this copy of the FECH gene from working properly. On the other copy, there is a small change called a “low-expression allele” or a polymorphism. This alteration still affects the way the FECH gene works; it produces less ferrochelatase enzyme than normal. This small change is common in the general population, with up to 10% of Caucasians with one copy of this change. This alteration will not cause EPP by itself, and people who have the alteration on each copy of the FECH gene will NOT develop EPP. But when someone inherits the small alteration from one parent and a mutation from the other, they will develop EPP, because there will not be enough ferrochelatase (FECH) enzyme made. Most patients with EPP have the low-expression alteration on one copy of the FECH gene and a mutation on the other copy. The risk for patients with EPP to have a child who also has the condition depends on the genetic changes in their partner.
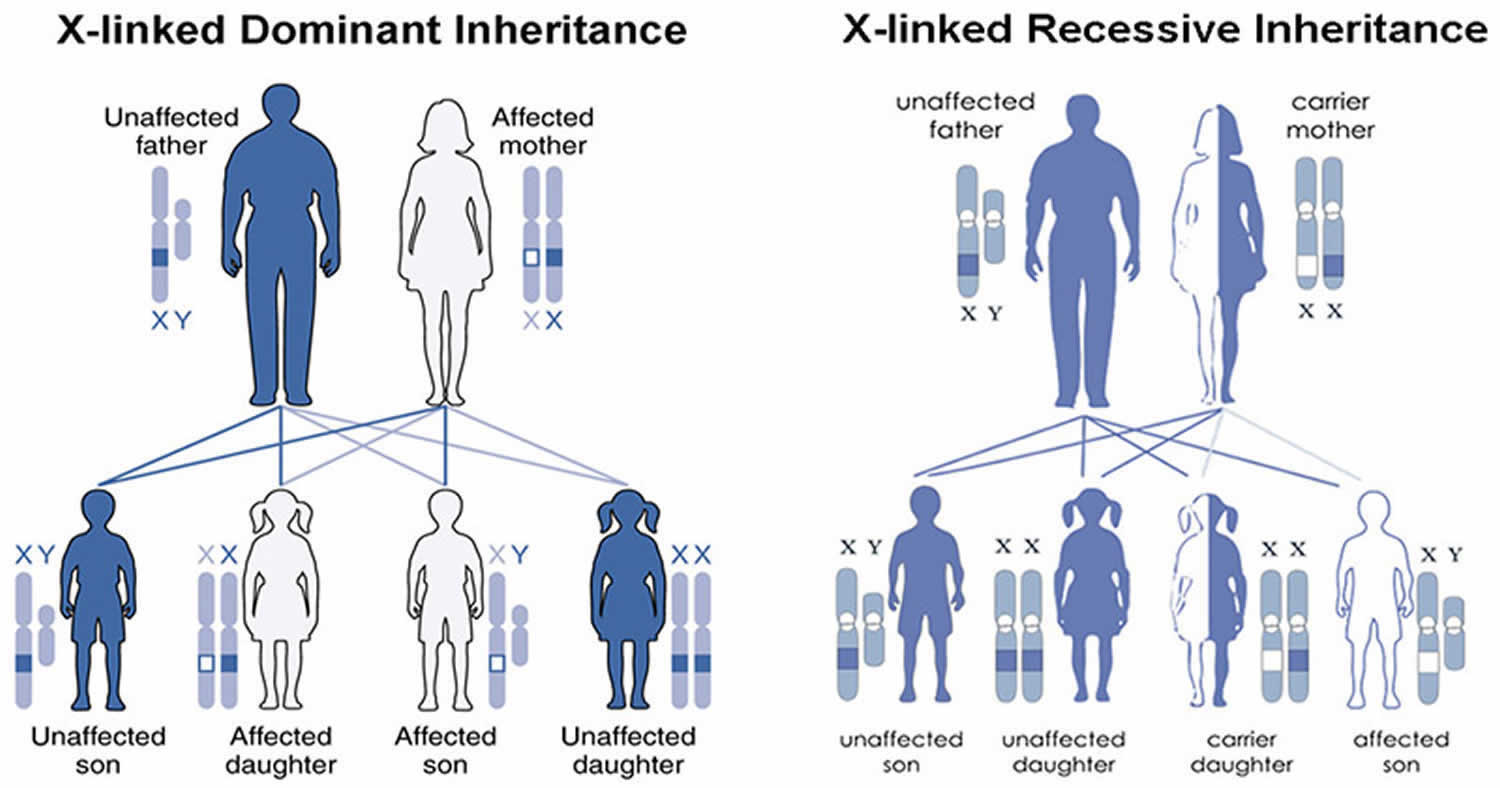
X-linked protoporphyria (XLP) is passed down through families in an X-linked manner 9. Males have one X chromosome and one Y chromosome, while females have two X chromosomes.This means that males have only one copy of the ALAS2 gene and females have two copies of the ALAS2 gene. When a male has a mutation is his single copy of ALAS2 gene, he is expected to have symptoms of X-linked protoporphyria (XLP). In a woman with a mutation in one of her ALAS2 genes, the second functioning copy of the ALAS2 gene can help compensate and may lead to less severe symptoms or no symptoms at all. It is not possible to predict or control the severity of X-linked protoporphyria (XLP) in females. Men with XLP pass on their X chromosome to their daughters and their Y chromosome to their sons. Therefore, a man with X-linked protoporphyria (XLP) with pass on his genetic change to all his daughters, and none of his sons. For a female with XLP, she will pass on the X chromosome with the genetic change 50% of the time. Thus, in each pregnancy, there is a 50% chance of having a child with a mutation in ALAS2 gene.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 6. Erythropoietic protoporphyria (EPP) autosomal recessive inheritance pattern
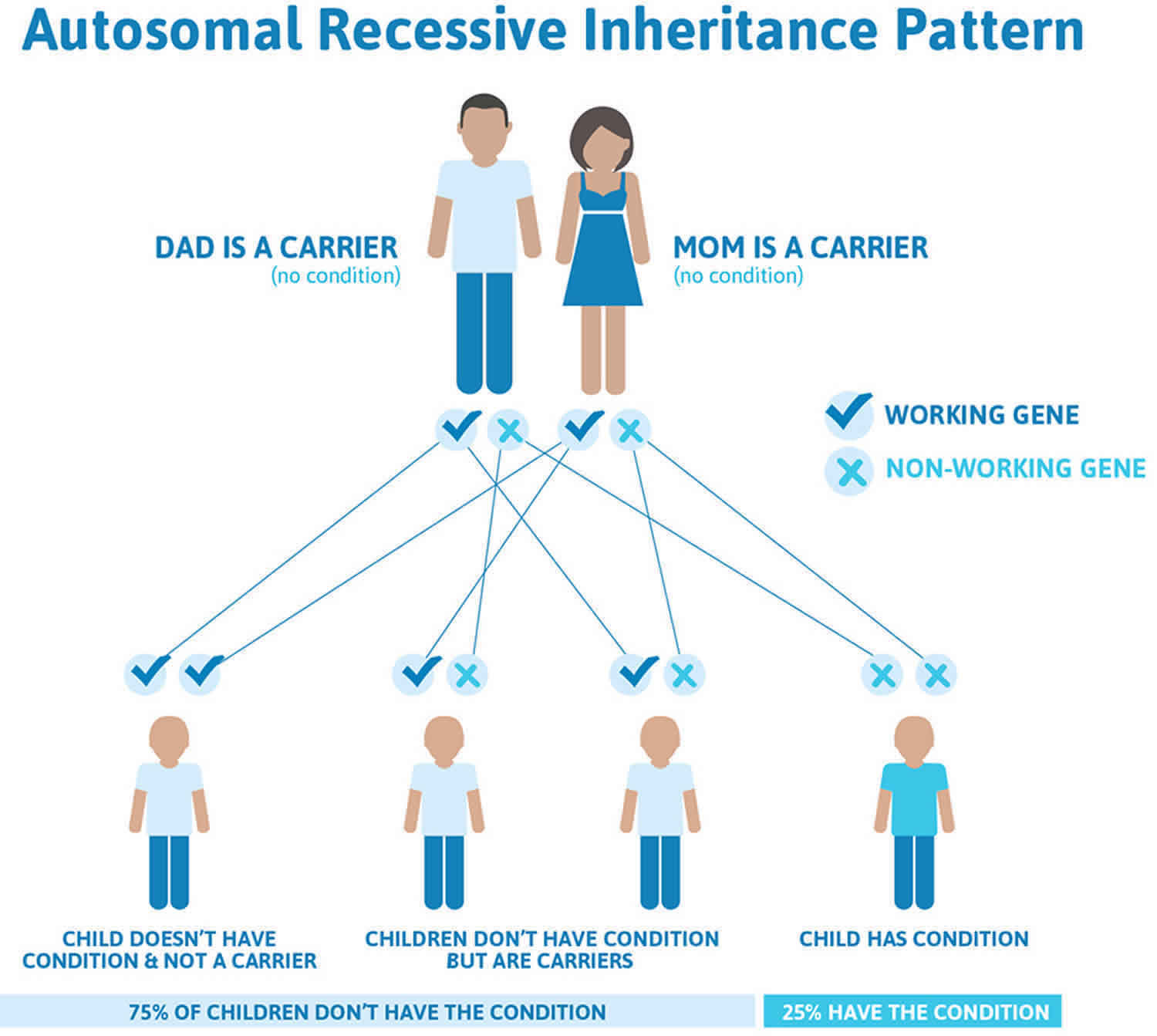
Figure 7. X-linked protoporphyria (XLP) inheritance pattern
Erythropoietic protoporphyria signs and symptoms
The most common symptom of erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) is hypersensitivity of the skin to sunlight and some types of artificial light (photosensitivity), with pain, itching, and/or burning of the skin occurring after exposure to sunlight and occasionally to fluorescent light. Some patients may also be sensitive to some types of artificial light such as fluorescent lights. When the skin is exposed to sun, people with erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) first develop tingling, itching, and/or burning of the skin 9. These symptoms serve as warning signs as longer exposure can result in severe pain. Affected individuals may also have an abnormal accumulation of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema) 9. In rare cases, affected areas of the skin may develop sac-like lesions (vesicles or bullae) and scar if exposure to sunlight is prolonged 9. However, scarring and/or discoloring of the skin (hyperpigmentation) is uncommon and rarely severe 9. The affected areas of skin may become abnormally thick. In addition, in some cases, affected individuals may also exhibit malformations of the nails. The severity and degree of photosensitivity is different from case to case. Photosensitivity is often seen during infancy; however, in some cases, it may not occur until adolescence or adulthood.
The severity and degree of symptoms is different from case to case. Some patients may only be able to tolerate a few minutes of sun exposure while others may be able to tolerate longer sun exposure without symptoms. The amount of sun tolerated may also be different based on weather conditions. Symptoms are often seen during infancy; however, in some cases, it may not occur until adolescence or rarely in adulthood.
Symptoms usually start in childhood but diagnosis is often delayed since skin blistering is not common and because the porphyrins are insoluble, they cannot be detected on urine analysis. The diagnosis is made upon finding increased levels of the protoporphyrin IX (PPIX) in the plasma or red blood cells.
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, gallstones can cause obstruction and/or inflammation of the gallbladder (cholecystitis).
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, such stones can cause obstruction and/or inflammation of the gallbladder (cholecystitis).
Rarely, affected individuals may also develop liver damage that, in very severe cases, may lead to liver failure requiring liver transplantation. As liver transplantation does not cure erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP), a bone marrow transplant following liver transplant may be necessary in some cases 9.
Patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) may also have mild anemia (low blood counts). In many cases, this may be due to low iron stores (iron deficiency). They may also have high levels of liver enzymes on blood tests.
Liver involvement
Patients with erythropoietic protoporphyria (EPP) may also have liver disease that require specialist medical treatment and possibly liver transplantation. Liver disease develops in association with erythropoietic protoporphyria (EPP) in 1 to 4% of cases 30, 34, with usual features of visceral enlargement and portal hypertension. Liver disease in protoporphyria is related to the excess protoporphyrin cleared by the entero-hepatic circulation leading to paracrystalin protoporphyrin deposition in liver cells (hepatocytes) and precipitation in the biliary canaliculiy. The percentage of patients who will develop liver disease is not established, nor specific factors that may influence its development. In EPP, the degree of severity of the liver disease is variable. Liver disease in EPP may include: gallstones (cholelithiasis) with possible obstructive episodes and chronic liver disease evolving to rapid acute liver failure 60, 61.
The incidence of gallstones (cholelithiasis) is frequent in about 20% of EPP patients EPP 6. Gallstones with high protoporphyrin content are generated due to the accumulation of insoluble protoporphyrin and increased biliary protoporphyrin concentration 62.
It is not possible to predict whether or not acute liver failure will occur. Studies have revealed that an increase in coproporphyrin urinary excretion, together with a change in isomer predominance from isomer III to isomer I, and increasing levels of protoporphyrinaemia may precede this complication 63.
Progression of protoporphyric liver deterioration leads to splenomegaly, splenic sequestration of erythrocytes with haemolysis (which increases erythropoiesis) and protoporphyrin generation ending in fulminant liver failure 60, 63, 64.
Erythropoietic protoporphyria complications
The main serious complication associated with EPP is protoporphyrin-related liver disease, which may be fatal 8. Severe liver disease affects 2%–5% of protoporphyia patients 65. Liver injury occurs from the crystallization of protoporphyrin in the bile ducts 66. This obstruction of the biliary system further increases plasma protoporphyrin, which is typically excreted in the bile, escalating the protoporphyrin-mediated liver damage that may progress rapidly 66. Previous studies indicate that higher protoporphyrin levels may be associated with an increased risk of liver disease or progression 66. Genetic sequencing to identify EPP patients with two pathogenic FECH variants other than c.315-48T>C may be considered, as these patients may be more likely to progress to liver failure 67. Therefore, liver function tests should be performed at the time of diagnosis and at least yearly thereafter 68, 69, 70. This may allow for early identification, evaluation for additional factors contributing to liver disease, and medical management.
The extent to which alcohol use worsens protoporphyria-related liver disease or whether a safe limit alcohol use exists remains unclear 1. Avoidance of excessive alcohol intake is recommended for all people with EPP or XLP 1. Furthermore, immunization against hepatitis A and B is recommended. Although protoporphyria patients are at an increased risk for gallstones (cholelithiasis), in asymptomatic patients with normal liver chemistries, a screening ultrasound is not recommended 1. Surgery to remove your gallbladder (cholecystectomy) is not recommended for asymptomatic gallstones (cholelithiasis).
In the later stages of liver disease, the patient may develop peripheral nerve damage (neuropathy), which mimics the peripheral neuropathy of acute porphyria and may lead to breathing failure 8. On repeated exposure to sunlight, the skin over the face, dorsum of hands (knuckles), can become thickened or lichenified along with loss of lunulae of fingernails. Due to regular sunlight avoidance, patients with EPP are more prone to develop a vitamin-D deficiency, which can lead to osteoporosis 20.
Erythropoietic protoporphyria diagnosis
The diagnosis of erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) may be made by a thorough clinical evaluation, and specialized laboratory tests. Total red blood cell (erythrocyte) protoporphyrin concentration, including proportions of metal-free and zinc-bound protoporphyrin, is the recommended test for the diagnosis of protoporphyria 71, 72, 73. High performance liquid chromatography or extraction methods that measure total, metal-free, and zinc protoporphyrin are recommended for the diagnosis of protoporphyria 36. Current lists of laboratories that perform such testing can be found on websites for the United Porphyrias Association, the European Porphyria Network, and the American Porphyria Foundation. These laboratories can also give advice regarding the optimal laboratory testing or differential diagnosis of biochemical abnormalities. As sample materials and pre‐analytical specifications depend on the specific diagnostic methods, it is advisable to contact the respective laboratory prior to sample collection. Genetic testing is useful to confirm the diagnosis.
The diagnosis of erythropoietic protoporphyria is established by finding an abnormally high level of total red blood cell (erythrocyte) protoporphyrin, and showing that this increase is mostly free protoporphyrin rather than zinc protoporphyrin. There is considerable confusion about which test to order. Sometimes laboratories have measured only zinc protoporphyrin and reported results incorrectly as “protoporphyrin” or “free erythrocyte protoporphyrin (FEP)”.
Porphyrins are almost always elevated in plasma in erythropoietic protoporphyria, but may be normal in mild cases. Fecal porphyrins may be normal or increased.
An experienced biochemical laboratory can usually distinguish between patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), because erythropoietic protoporphyria (EPP) have much less zinc protoporphyrin in their erythrocytes. This can be explained because in the bone marrow the enzyme ferrochelatase not only normally makes heme (iron protoporphyrin) from protoporphyrin and iron, but can also make zinc protoporphyrin, especially when excess protoporphyrin is present or iron is deficient. However, this does not replace DNA studies or genetic testing.
Genetic testing of the FECH and ALAS2 genes is useful to confirm the diagnosis and identify if it is erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) 49. This information is useful for genetic counseling and testing family members as both are inherited in a different manner. Genetic counseling is recommended for couples with a personal or family history of protoporphyria who are planning to have children.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are usually diagnosed during infancy or early childhood, due to characteristic symptoms, and by testing the red blood cells (erythrocytes) for increased levels of protoporphyrin IX (PPIX). The blood needs to be sent for porphyrin analysis in a tube protected from light with aluminium foil. Exposure of the blood tube to light can lead to photobleaching of protoporphyrin IX (PPIX), resulting in a decrease in measured protoporphyrin IX (PPIX) levels in the sample as compared to the actual levels in the subject. This effect is related to the ability of conjugated double bonds in the porphyrin rings to absorb the energy from visible light 38. To avoid diagnostic delay, plasma porphyrins should be measured at the same time with erythrocyte protoporphyrin 39.
The normal total protoporphyrin level in red blood cells (erythrocytes) is 80 mcg/dL, but in a patient with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), protoporphyrin level in red blood cells (erythrocytes) is elevated up to 300 to 8000 mcg/dL 8. There is an increased percentage of red blood cell (erythrocyte) metal-free protoporphyrin rather than zinc protoporphyrin 8. In patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), the urinary porphyrin levels are normal.
Rarely, erythropoietic protoporphyria (EPP) develops in adults in the presence of a bone marrow disorder such as polycythemia vera, and is due to expansion of a clone of red blood cell precursors in the marrow that is deficient in ferrochelase.
DNA studies or genetic testings are important for confirming the diagnosis of erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) and for genetic counseling. This should be completed first in a person known to have the disease, and the information about the mutations in that individual used to guide testing of family members.
When erythropoietic protoporphyria (EPP) is due to a FECH mutation the inheritance is described as autosomal recessive. It is most common to find that one severe mutation is inherited from one parent and another weak mutation inherited from the other parent. The weak mutation is quite common in normal Caucasians, rare in Blacks and even more common in Japanese and Chinese populations. This mutation is sometime referred to as “hypomorphic” because it results in formation of a less than normal amount of ferrochelatase. But is does not cause Erythropoietic protoporphyria unless it is paired with a severe mutation. The severe mutation is characteristic for an Erythropoietic protoporphyria family and is present in all affected individuals. “Carriers” of the severe mutation are not affected because they do not have the weak mutation. Affected individuals and unaffected carriers can transmit the severe mutation to the next generation. Some of their children will have Erythropoietic protoporphyria if the other parent has a copy of the weak mutation. Rarely, the weak mutation is absent in an Erythropoietic protoporphyria family and two severe mutations are found, with at least one producing some ferrochelatase.
In X-linked protoporphyria (XLP), mutations of the ALAS2 gene, which is found on the X chromosome, causes an increase in the production of the enzyme ALAS2 in the bone marrow. Several of these “gain of function” mutations have been described in different X-linked protoporphyria families. In X-linked protoporphyria protoporphyrin production exceeds that needed for heme and hemoglobin formation. Like hemophilia and other X linked genetic diseases, X-linked protoporphyria is more common in men. Women have two X chromosomes and are usually not affected because they have a normal as well as a mutated ALAS2 gene. Men have only one X chromosome and will be affected if they inherit an ALAS2 mutation. Women with an ALAS2 mutation will, on average, pass that mutation to half of their daughters (who will usually be unaffected carriers) and to half of their sons (who will be affected).
Figure 8. Erythropoietic protoporphyria diagnostic algorithm
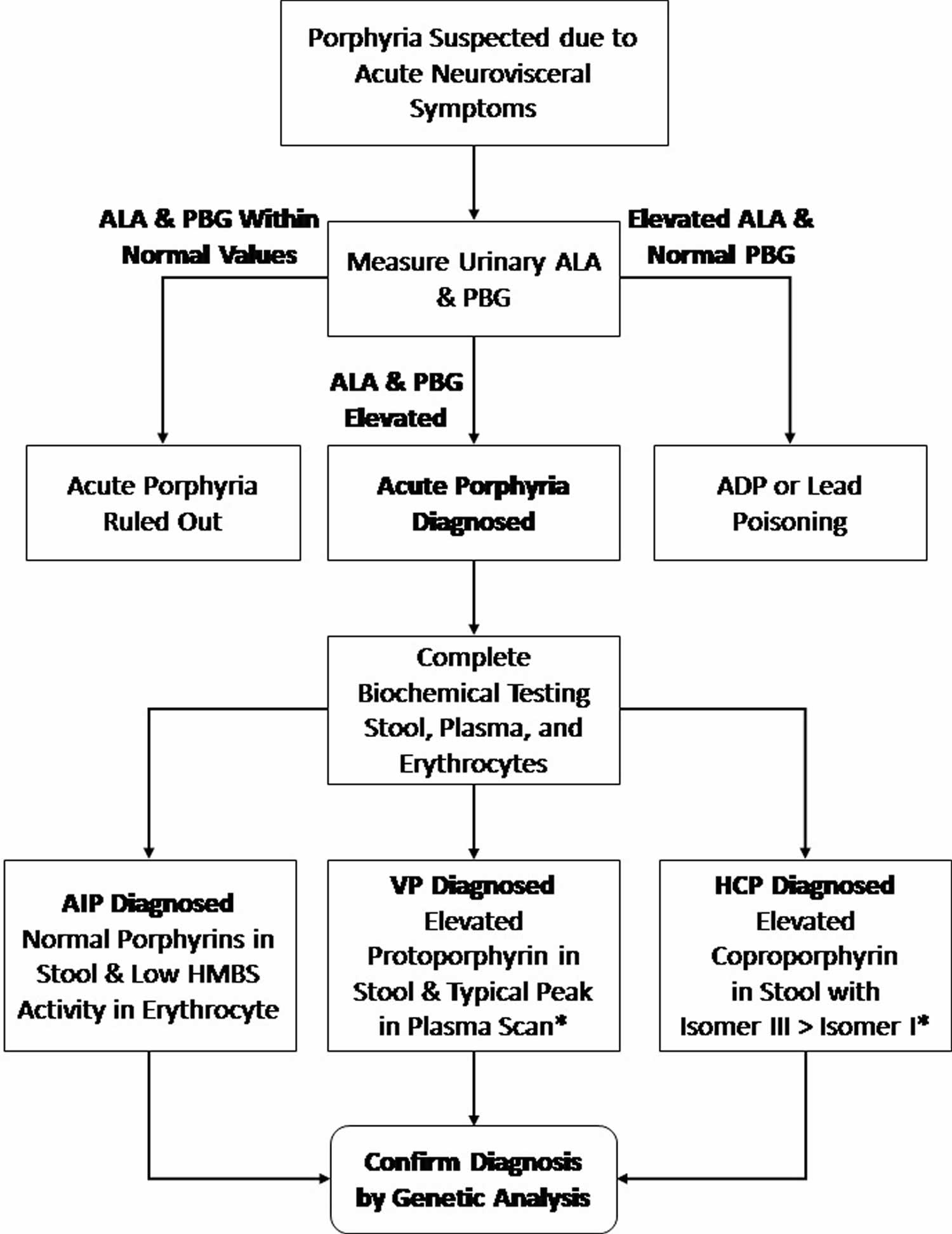
Abbreviations: ADP = aminolevulinic acid dehydratase porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; HCP = hereditary coproporphyria; HMBS = hydroxymethylbilane synthase; PBG = porphobilinogen; VP = variegate porphyria.
[Source 39 ]Pitfalls of testing
The biochemical diagnosis of protoporphyrias is based on the measurement of red blood cell (erythrocyte) total and metal-free protoporphyrin 3. The latter comprises 85% to 100% of total red blood cell (erythrocyte) protoporphyrin in EPP, and 50% to 85% in XLP 74, 36. Zinc protoporphyrin predominates in other conditions that increase red blood cell (erythrocyte) protoporphyrin. Plasma and fecal porphyrin levels can be variably increased, and urine porphyrin levels are normal. Genetic testing is recommended to distinguish between EPP and XLP and to confirm the diagnosis 74.
Measurement of red blood cell (erythrocyte) protoporphyrin levels is fraught with a lack of standardized nomenclature and methodology 75. Some large commercial laboratories measure protoporphyrin by hematofluorometry, a method that only measures zinc protoporphyrin and that was developed to screen for lead poisoning and iron deficiency. Hematofluorometers report the molar ratio of zinc protoporphyrin to heme by fluorescence but do not measure total or metal-free protoporphyrin 36. Nevertheless, these major laboratories report “free erythrocyte protoporphyrin” (FEP) levels that, in actuality, reflect only zinc protoporphyrin, which may not be increased in EPP. This inappropriate method and inaccurate terminology propagates confusion that can lead to missed diagnoses of protoporphyria 75. Accurate testing reports should only refer to erythrocyte total protoporphyrin, zinc protoporphyrin, and metal-free protoporphyrin, and comment whether the results are consistent with protoporphyria. The term FEP became unclear after the discovery of zinc protoporphyrin in erythrocytes in the 1970s and should no longer be used 3.
High performance liquid chromatography or extraction methods that measure total, metal-free, and zinc protoporphyrin are recommended for the diagnosis of protoporphyria 36. Current lists of laboratories that perform such testing can be found on websites for the United Porphyrias Association, the European Porphyria Network, and the American Porphyria Foundation. These laboratories can also give advice regarding the optimal laboratory testing or differential diagnosis of biochemical abnormalities. As sample materials and pre‐analytical specifications depend on the specific diagnostic methods, it is advisable to contact the respective laboratory prior to sample collection.
Erythropoietic protoporphyria differential diagnosis
Erythropoietic protoporphyria differential diagnosis include 8:
- Phototoxic drug reaction: Phototoxic drug reaction is a non-immunologic skin reaction that appears acutely within minutes to hours on sun-exposed skin after taking photosensitising medications. There must be a history of the introduction of any new drug. Phototoxic skin damage begins when the drug or its metabolite within the skin absorbs ultraviolet radiation (UVR) or visible light. Patients experience painful reddish skin immediately after sun exposure 76, 77.
- Hydroa vacciniforme: Hydroa vacciniforme is one of the rarest forms of photosensitivity dermatoses. Hydroa vacciniforme affects sun-exposed skin and is characterized by recurrent fluid-filled blisters (‘hydroa’) over sun-exposed sites that heal with pox-like (‘vacciniform’) scars 78.
- Solar urticaria: Solar urticaria is a condition in which exposure to sunlight or an artificial light source emitting ultraviolet radiation causes urticaria 79. Like EPP, symptoms often develop within minutes. Symptoms of solar urticaria are often itchy rather than painful. The reaction may subside within a few minutes or it may persist for up to an hour or more where it can become very disabling. The cause of solar urticaria is not clearly defined but may be due to an antigen-antibody reaction 79. It seems that a chemical created in the body (a photoallergen) reacts with UV radiation to cause an allergic reaction that manifests as urticaria.
- Polymorphic light eruption (PMLE): Polymorphic light eruption also called a sun allergy or sun poisoning. Polymorphic light eruption is a common seasonal, acquired, idiopathic photodermatosis occurring in spring and early summer that typically occurs during the first three decades of life 80. Symptoms occur in sun-exposed areas. Patients present with discrete lesions such as pruritic papules, vesicles, or plaques on sun-exposed areas.
- Discoid lupus erythematosus: It presents as scaly erythematous plaques on sun-exposed areas.
- Sunburn: It is a transient inflammatory skin response to ultraviolet radiation from sunlight or artificial sources. Sunburn can occur in individuals without an underlying dermatologic condition, with sensitivity depending on the degree of skin pigmentation and hair and eye color 81.
Erythropoietic protoporphyria treatment
There is no cure for erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP). Lifelong photosensitivity is the main problem. To reduce pain, avoid unnecessary exposure to sunlight will be of benefit to individuals with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP). The use of sun protective clothing such as long sleeves, wide-brimmed hats, and sunglasses will also benefit individuals with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP). Tanning creams which increase skin pigmentation or sunscreens which contain physical reflecting agents such as zinc oxide or titanium dioxide that reflect visible light may be beneficial to some patients. Individuals with EPP and XLP may also benefit from window tinting or using films to cover the windows in their car or house. Other sources of light may also cause symptoms, including fluorescent and halogen lights. Protect the skin from exposure to operating visible light and ultraviolet A (UVA) light during a surgical procedure. Light filters that limit transmission of the wavelengths 340–470 nm (i.e., acrylate yellow filter) are recommended and should be used in operating rooms to protect the patient, especially in case of long surgical procedures like liver transplantation in a patient with erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) 42, 43, 82. No specific anesthetic agents or other medications are contraindicated in protoporphyria.
Vitamin D supplementation to prevent vitamin D deficiency is appropriate in erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) patients that strictly avoid exposure to sunlight. There is an increased recognition of prevalence of vitamin D deficiency in protoporphyria patients due to lifelong sunlight avoidance. Routine screening for vitamin D deficiency and supplementation as per population guidelines are recommended 40, 41.
Avoidance of alcohol is important to reduce the risk of liver damage and liver failure. A drug called Prevalite (cholestyramine) or activated charcoal maybe prescribed to interrupt the circulation of protoporphyrin through the liver and intestines in patients with liver disease.
Once the pain has started, pain relief can be difficult to achieve. Most patients immerse the affected areas in cold water or use ice packs. Topical anesthetic creams can be helpful.
In 2019, the Food and Drug Administration (FDA) approved Scenesse (afamelanotide) for the treatment of adult patients with EPP. Scenesse (afamelanotide) is an alpha-melanocyte stimulating hormone that is given by subcutaneous implantation and works by increasing skin pigmentation which provides protection and improves sun tolerance 10. Afamelanotide binds to the melanocortin-1 receptor (MC1R) and increases eumelanin production from dermal cells and melanocytes, thus increasing pigmentation and providing photoprotection. Melanin is additionally a strong antioxidant thought to inactivate the reactive oxygen species (ROS) produced during phototoxic reactions 83. In the setting of erythropoietic protoporphyria (EPP), melanin production may provide defense against oxidative stress by neutralizing free radicals and the reactive oxygen species produced, thus helping decrease symptom severity 84 Scenesse (afamelanotide) was available in Europe for a period of time before its approval in the United States. There is no data on the safety of Scenesse (afamelanotide) during pregnancy so this cannot be recommended for pregnant women.
Iron supplementation should be avoided unless severe iron deficiency is present — as iron can increase photosensitivity in EPP. Both clinical improvement and increased photosensitivity have been reported during iron replacement therapy 85, 86, 87, 88, 89, 90, 91, 92, 93, 94.
- Iron deficiency in ferrochelatase-deficient EPP: If symptomatic from iron deficiency and/or have hemoglobin levels <10g/dL and ferritin <10ug/L. Iron supplementation to a goal ferritin of 50–100ug/L
- Iron deficiency in XLP: If any iron deficiency: Oral iron to a goal ferritin of 50–100 ug/L
Trials of treatment for erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) have been difficult to assess. Effective treatment should reduce pain and increase time outdoors without pain.
- Narrowband UVB phototherapy does not cause the EPP pain. It is given 3 days a week over 6 weeks in Spring. It increases melanin content (causing a tan) and induces skin thickening so to provide some level of protection from the sun.
- Oral antioxidants such as beta-carotene, Polypodium leucotomas extract, warfarin and N-acetyl cysteine have been used but studies showing effectiveness are lacking. In EPP, a high potency form of oral Lumitene (oral beta-carotene) 120-180 mg/dL has been used to improve an affected individual’s tolerance of sunlight. The dose of Lumitene depends on age, ranging from two to ten 30-mg capsules per day and usually started six to eight weeks before summer. While some patients report improvement, recent studies show that there is no data to support the benefit of this treatment 95.
In addition, individuals with high levels of protoporphyrin in the plasma and red blood cells should be observed closely by a physician for possible liver malfunction that could eventually lead to liver failure.
Liver transplantation has been performed as a life-saving measure in patients with EPP and XLP related liver failure. Bone marrow transplant can also be performed after liver transplant to prevent further damage to the liver.
EPP and XLP patients should also receive vaccination against hepatitis A and B to prevent other causes of liver damage.
Patients should be seen at least yearly to monitor protoporphyrin levels, anemia, liver enzymes, iron and vitamin D levels.
Other treatment is symptomatic and supportive.
1. Sunlight protection
Protection from sunlight is the mainstay of management of erythropoietic protoporphyria, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in Erythropoietic protoporphyria. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons Erythropoietic protoporphyria can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
2. Other considerations
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. Erythropoietic protoporphyria patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow), and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with Erythropoietic protoporphyria.
Because erythropoietic protoporphyria is a rare condition, most physicians are not knowledgeable about it. A Medic Alert bracelet with instructions to contact a specialist if needed is a worthwhile precaution.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyry levels are expected to be stable and liver tests to remain normal. Erythropoietic protoporphyria patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, Erythropoietic protoporphyria patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from erythropoietic protoporphyria. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in erythropoietic protoporphyria patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other porphyrias are not known to make erythropoietic protoporphyria worse, but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity, but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. According to Dr. Roth, laser treatments for hair removal or eye surgery have not been a problem in erythropoietic protoporphyria people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
Children with erythropoietic protoporphyria. Avoiding sunlight can be difficult for children with erythropoietic protoporphyria who have less sunlight tolerance than their friends.
Liver disease treatment
2%–5% of affected individuals develop severe liver complications that are difficult to treat, often requiring liver transplantation 65, 27. Liver complications may be accompanied by motor neuropathy.
Rapidly progressive and severe liver disease can be treated with a regimen that includes intravenous hemin (to reduce plasma porphyrin level), plasmapheresis, ursodeoxycholic acid, cholestyramine, vitamin-E (400IU), and correction of anemia 8, 96. Cholestyramine and other porphyrin absorbents, such as activated charcoal, may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement 97.
Liver transplantation is the treatment choice for severe protoporphyric liver damage or for patients who develop liver cirrhosis as a life-saving measure in individuals with severe protoporphyric liver disease 98, 99, 100. However, liver transplant recipients may experience a recurrence of protoporphyric liver disease in the transplanted liver. Combined bone marrow and liver transplantation is indicated in patients with liver failure to prevent future damage to the allografts 101.
Bone marrow transplant is a newer treatment that may be done in conjunction with liver transplantation to prevent the recurrence of EPP, and reports show that it may be able to reverse or treat both photosensitivity and protoporphyric hepatopathy 102, 103. The presumed mechanism is related to a majority of heme synthesis in the bone marrow, thus allowing for a possibility of curative treatment in some case reports, particularly as some case reports show recurrence of erythropoietic protoporphyria activity leading to fatal outcomes rapidly 104, 103, 105.
Erythropoietic protoporphyria prognosis
Erythropoietic protoporphyria (EPP) is a lifelong disease. Liver damage is the major risk, so regular liver follow-up is important. Up to 5% of affected individuals may develop more advanced liver disease, most notably cholestatic liver failure 66, 59. In most of these individuals, underlying liver cirrhosis is already present; however, some may present with rapidly progressive cholestatic liver failure 59.
Life expectancy is usually normal in patients with EPP unless liver damage develops due to hepatotoxic effects of protoporphyrins that lead to liver dysfunction. EPP generally does not decrease life expectancy but does have a great influence on the quality of life. Since the pain after photosensitivity is intense and acute, it is necessary for the patient to modify lifestyle and employment 106.
Annual assessment of red blood cell (erythrocyte) protoporphyrin levels (free and zinc-chelated), hematologic indices, and iron profile including iron, total iron binding capacity (TIBC), and ferritin is appropriate.
Liver function test including aspartate aminotransferase (AST), alanine aminotransferase (AST), total bilirubin, alkaline phosphatase (ALP) should be monitored every 6 to 12 months. Liver imaging studies including abdominal ultrasound are indicated if gallstones (cholelithiasis) is suspected.
Vitamin D 25-OH levels should be monitored in all patients whether or not they are receiving supplements.
- Dickey AK, Naik H, Keel SB, et al. Porphyrias Consortium of the Rare Diseases Clinical Research Network. Evidence-based consensus guidelines for the diagnosis and management of erythropoietic protoporphyria and X-linked protoporphyria. J Am Acad Dermatol. 2023 Dec;89(6):1227-1237. doi: 10.1016/j.jaad.2022.08.036[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Minder AE, Kluijver LG, Barman-Aksözen J, Minder EI, Langendonk JG. Erythropoietic protoporphyrias: Pathogenesis, diagnosis and management. Liver Int. 2025 Jan;45(1):e16027. doi: 10.1111/liv.16027. Epub 2024 Jul 16. Erratum in: Liver Int. 2024 Dec;44(12):3292. doi: 10.1111/liv.16104[↩]
- Leaf RK, Dickey AK. How I treat erythropoietic protoporphyria and X-linked protoporphyria. Blood. 2023 Jun 15;141(24):2921-2931. doi: 10.1182/blood.2022018688[↩][↩][↩]
- Balwani M, Naik H, Anderson KE, et al. Clinical, Biochemical, and Genetic Characterization of North American Patients With Erythropoietic Protoporphyria and X-linked Protoporphyria. JAMA Dermatol 2017;153(8):789–796. doi: 10.1001/jamadermatol.2017.1557[↩]
- Kluijver LG, Wensink D, Wagenmakers MAEM, Huidekoper HH, Witters P, Rymen D, Langendonk JG. Quality of life in children with erythropoietic protoporphyria: a case-control study. J Dermatol. 2024 Aug;51(8):1068-1078. doi: 10.1111/1346-8138.17348[↩]
- Lecha M, Puy H, Deybach JC. Erythropoietic protoporphyria. Orphanet J Rare Dis. 2009 Sep 10;4:19. doi: 10.1186/1750-1172-4-19[↩][↩][↩][↩][↩]
- Ardalan ZS, Chandran S, Vasudevan A, Angus PW, Grigg A, He S, Macdonald GA, Strasser SI, Tate CJ, Kennedy GA, Testro AG, Gow PJ. Management of Patients With Erythropoietic Protoporphyria-Related Progressive Liver Disease. Liver Transpl. 2019 Nov;25(11):1620-1633. doi: 10.1002/lt.25632[↩]
- Ahmed jan N, Masood S. Erythropoietic Protoporphyria. [Updated 2023 Feb 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK563141[↩][↩][↩][↩][↩][↩][↩]
- Erythropoietic Protoporphyria and X-Linked Protoporphyria. https://rarediseases.org/rare-diseases/erythropoietic-protoporphyria[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- McNeil MM, Nahhas AF, Braunberger TL, Hamzavi IH. Afamelanotide in the Treatment of Dermatologic Disease. Skin Therapy Lett. 2018 Nov;23(6):6-10. https://www.skintherapyletter.com/dermatology/afamelanotide[↩][↩][↩]
- Magnus IA, Jarret A, Prankerd TAJ, Rimington C. Erythropoietic protoporphyria: a new porphyria syndrome with solar urticaria due to protoporphyrinæmia. Lancet. 1961;278:574–581. doi: 10.1016/s0140-6736(61)92427-8[↩]
- Cox TM. Protoporpnhyria. In: Kadish KM, Smith KM, Guilard R, editor. The Porphyrin Handbook Medical Aspects of Poprhyrias, Chap 90. Vol. 14. Academic Press. San Diego; 2003. pp. 121–149.[↩]
- Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408–14. doi: 10.1016/j.ajhg.2008.08.003[↩]
- Levy C, Naik H, Overbey J, et al. Porphyrias Consortium of the Rare Diseases Clinical Research Network. Liver involvement in a large cohort of patients with erythropoietic protoporphyria or X-linked protoporphyria. Hepatol Commun. 2025 Feb 19;9(3):e0657. doi: 10.1097/HC9.0000000000000657[↩]
- Levy C, Dickey AK, Wang B, Thapar M, et al. Porphyrias Consortium of the Rare Diseases Clinical Network. Evidence-based consensus guidelines for the diagnosis and management of protoporphyria-related liver dysfunction in erythropoietic protoporphyria and X-linked protoporphyria. Hepatology. 2024 Mar 1;79(3):731-743. doi: 10.1097/HEP.0000000000000546[↩]
- FECH gene. https://medlineplus.gov/genetics/gene/fech[↩][↩]
- Snast I, Kaftory R, Sherman S, Edel Y, Hodak E, Levi A, Lapidoth M. Acquired erythropoietic protoporphyria: A systematic review of the literature. Photodermatol Photoimmunol Photomed. 2020 Jan;36(1):29-33. doi: 10.1111/phpp.12501[↩]
- Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet 2008;83(3):408–414. doi: 10.1016/j.ajhg.2008.08.003[↩][↩]
- ALAS2 gene. https://medlineplus.gov/genetics/gene/alas2[↩][↩][↩]
- Balwani M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: pathophysiology, genetics, clinical manifestations, and management. Mol Genet Metab. 2019 Nov;128(3):298-303. doi: 10.1016/j.ymgme.2019.01.020[↩][↩]
- Madu AE, Whittaker SJ. Erythropoietic protoporphyria in pregnancy. J Obstet Gynaecol 2006;26(7):687–688. doi: 10.1080/01443610600930670[↩]
- Jacquemyn Y Erythropoietic protoporphyria in pregnancy. J Obstet Gynaecol 2003;23(2):196. doi: 10.1080/0144361031000074817[↩]
- Nevins EG, Wijesiriwardana A. Erythropoietic protoporphyria in pregnancy. J Obstet Gynaecol Published online 2020:1–2. doi: 10.1080/01443615.2020.1777954[↩]
- Bewley AP, Keefe M, White JE. Erythropoietic protoporphyria improving during pregnancy. Br J Dermatol 1998;139(1):145–147. doi: 10.1046/j.1365-2133.1998.02333.x[↩]
- Poh-Fitzpatrick MB. Human protoporphyria: reduced cutaneous photosensitivity and lower erythrocyte porphyrin levels during pregnancy. J Am Acad Dermatol 1997;36(1):40–43. doi: 10.1016/s0190-9622(97)70323-2[↩]
- Heerfordt IM, Wulf HC. Patients with erythropoietic protoporphyria have reduced erythrocyte protoporphyrin IX from early in pregnancy. Br J Dermatol 2017;177(3):e38–e40. doi: 10.1111/bjd.15228[↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ: Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, eds. The Metabolic and Molecular Basis of Inherited Disease. 8 ed. New York, NY: McGraw-Hill; 2001:2991-3062.[↩][↩]
- Wahlin S, Floderus Y, Stål P, Harper P. Erythropoietic protoporphyria in Sweden: demographic, clinical, biochemical and genetic characteristics. J Intern Med. 2011 Mar;269(3):278-88. doi: 10.1111/j.1365-2796.2010.02236.x[↩]
- Went L, Klasen EC. Genetic aspects of erythropoietic protoporphyria. Ann Hum Genet. 1984;48:105–117. doi: 10.1111/j.1469-1809.1984.tb01006.x[↩]
- Elder GH, Smith SG, Smyth SJ. Laboratory investigation of the porphyrias. Ann Clin Biochem. 1990;27:395–412. doi: 10.1177/000456329002700501[↩][↩]
- Balwani M, Bloomer R, Desnick R, Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network . In: GeneReviews. Adam MP, Mirzaa GM, Pagon RA, editors. University of Seattle; 2017. Erythropoietic protoporphyria, autosomal recessive; pp. 1993–2023.[↩]
- Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013;36(5):849–857. doi: 10.1007/s10545-012-9544-4[↩]
- Dickey AK, Quick C, Ducamp S, et al. Evidence in the UK Biobank for the underdiagnosis of erythropoietic protoporphyria. Genet Med. 2021;23(1):140–148. doi: 10.1038/s41436-020-00951-8[↩]
- Holme SA, Anstey AV, Finlay AY, Elder GH, Badminton MN. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155:574–581. doi: 10.1111/j.1365-2133.2006.07472.x[↩][↩]
- Wulf HC, Nissen CV, Philipsen PA. Inactivation of protoporphyrin IX in erythrocytes in patients with erythropoietic protoporphyria: A new treatment modality. Photodiagnosis Photodyn Ther. 2020 Mar;29:101582. doi: 10.1016/j.pdpdt.2019.101582[↩]
- Gou EW, Balwani M, Bissell DM, et al. Pitfalls in erythrocyte protoporphyrin measurement for diagnosis and monitoring of protoporphyrias. Clin Chem. 2015;61(12):1453–1456. doi: 10.1373/clinchem.2015.245456[↩][↩][↩][↩][↩]
- Minder EI, Schneider‐Yin X. Laboratory Guide to the Methods in Biochemical Genetics. Springer Berlin Heidelberg; 2008.[↩]
- Ericson MB, Grapengiesser S, Gudmundson F, Wennberg AM, Larkö O, Moan J, Rosén A. A spectroscopic study of the photobleaching of protoporphyrin IX in solution. Lasers Med Sci. 2003;18(1):56-62. doi: 10.1007/s10103-002-0254-2[↩][↩]
- Edel Y, Mamet R. Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. 2018 Apr 19;9(2):e0013. doi: 10.5041/RMMJ.10333[↩][↩][↩][↩]
- Holme SA, Anstey A v, Badminton MN, Elder GH. Serum 25-hydroxyvitamin D in erythropoietic protoporphyria. Br J Dermatol 2008;159(1):211–213. doi: 10.1111/j.1365-2133.2008.08616.x[↩][↩]
- Spelt JM, de Rooij FW, Wilson JH, Zandbergen AA. Vitamin D deficiency in patients with erythropoietic protoporphyria. J Inherit Metab Dis 2010;33:S1–4. doi: 10.1007/s10545-008-1037-0[↩][↩]
- Wahlin S, Srikanthan N, Hamre B, Harper P, Brun A. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl 2008;14(9):1340–1346. doi: 10.1002/lt.21527[↩][↩]
- Levoska MA, Griffith JL, Nagai S, Collins K, Lim HW. A multi-disciplinary approach utilizing filters for surgical procedures in erythropoietic protoporphyria. J Am Acad Dermatol 2020;83:e329–330. doi: 10.1016/j.jaad.2020.02.024[↩][↩]
- Minder EI, Schneider-Yin X, Steurer J, Bachmann LM. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cellular and Molecular Biology 2009;55(1):84–97. doi: 10.1170/T841[↩]
- Mathews-Roth MM, Kass EH, Fitzpatrick TB, Pathak MA, Harber LC. Phototesting as an objective measure of improvement in erythropoietic protoporphyria. Arch Dermatol 1979;115(12):1391–1392. doi: 10.1001/archderm.1979.04010120001002[↩]
- Mathews-Roth MM, Pathak MA, Fitzpatrick TB, Harber LH, Kass EH. Beta Carotene Therapy for Erythropoietic Protoporphyria and Other Photosensitivity Diseases. Arch Dermatol. 1977;113(9):1229–1232. doi:10.1001/archderm.1977.01640090077011[↩]
- Balwani M, Bonkovsky HL, Belongie KJ, et al. Erythropoietic Protoporphyria: Phase 2 Clinical Trial Results Evaluating the Safety and Effectiveness of Dersimelagon (MT-7117), an Oral MC1R Agonist. Blood 2020;136:51–51. doi: 10.1182/BLOOD-2020-142467[↩]
- Kondo HX, Iizuka H, Masumoto G, Kabaya Y, Kanematsu Y, Takano Y. Prediction of Protein Function from Tertiary Structure of the Active Site in Heme Proteins by Convolutional Neural Network. Biomolecules. 2023 Jan 9;13(1):137. doi: 10.3390/biom13010137[↩]
- Balwani M, Doheny D, Bishop DF, et al. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med 2013;19(1):26–35. doi: 10.2119/molmed.2012.00340[↩][↩]
- Dickey AK, Quick C, Ducamp S, et al. Evidence in the UK Biobank for the underdiagnosis of erythropoietic protoporphyria. Genetics in Medicine 2021;23(1):140–148. doi: 10.1038/s41436-020-00951-8[↩]
- Gouya L, Martin-Schmitt C, Robreau AM, et al. Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. American Journal of Human Genetics 2006;78(1):2–14. doi: 10.1086/498620[↩]
- Whatley SD, Mason NG, Holme SA, Anstey AV, Elder GH, Badminton MN. Molecular epidemiology of erythropoietic protoporphyria in the U.K. Br J Dermatol. 2010 Mar;162(3):642-6. doi: 10.1111/j.1365-2133.2010.09631.x[↩]
- Balwani M, Naik H, Anderson KE, Bissell DM, Bloomer J, Bonkovsky HL, Phillips JD, Overbey JR, Wang B, Singal AK, Liu LU, Desnick RJ. Clinical, Biochemical, and Genetic Characterization of North American Patients With Erythropoietic Protoporphyria and X-linked Protoporphyria. JAMA Dermatol. 2017 Aug 1;153(8):789-796. doi: 10.1001/jamadermatol.2017.1557[↩]
- Yien YY, Ducamp S, van der Vorm LN, Kardon JR, Manceau H, Kannengiesser C, Bergonia HA, Kafina MD, Karim Z, Gouya L, Baker TA, Puy H, Phillips JD, Nicolas G, Paw BH. Mutation in human CLPX elevates levels of δ-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc Natl Acad Sci U S A. 2017 Sep 19;114(38):E8045-E8052. doi: 10.1073/pnas.1700632114[↩]
- Blagojevic D, Schenk T, Haas O, Zierhofer B, Konnaris C, Trautinger F. Acquired erythropoietic protoporphyria. Ann Hematol. 2010 Jul;89(7):743-4. doi: 10.1007/s00277-009-0859-7[↩]
- Aplin C, Whatley SD, Thompson P, Hoy T, Fisher P, Singer C, Lovell CR, Elder GH. Late-onset erythropoietic porphyria caused by a chromosome 18q deletion in erythroid cells. J Invest Dermatol. 2001 Dec;117(6):1647-9. doi: 10.1046/j.0022-202x.2001.01560.x[↩]
- Suzuki H, Kikuchi K, Fukuhara N, Nakano H, Aiba S. Case of late-onset erythropoietic protoporphyria with myelodysplastic syndrome who has homozygous IVS3-48C polymorphism in the ferrochelatase gene. J Dermatol. 2017 Jun;44(6):651-655. doi: 10.1111/1346-8138.13709[↩]
- Livideanu CB, Ducamp S, Lamant L, Gouya L, Rauzy OB, Deybach JC, Paul C, Puy H, Marguery MC. Late-onset X-linked dominant protoporphyria: an etiology of photosensitivity in the elderly. J Invest Dermatol. 2013 Jun;133(6):1688-90. doi: 10.1038/jid.2012.467[↩]
- Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic Protoporphyria, Autosomal Recessive. 2012 Sep 27 [Updated 2017 Sep 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK100826[↩][↩][↩]
- Bruguera M, Herrero C. [Liver disease in erythropoietic protoporphyria] Gastroenterol Hepatol. 2005;28:632–636. doi: 10.1016/s0210-5705(05)71529-6[↩][↩]
- Meerman L. Erythropoietic protoporphyria. An overview with emphasis on the liver. Scand J Gastroenterol Suppl. 2000;(232):79-85.[↩]
- Thunell S, Harper P, Brun A. Porphyrins, porphyrin metabolism and porphyrias. IV. Pathophysiology of erythyropoietic protoporphyria–diagnosis, care and monitoring of the patient. Scand J Clin Lab Invest. 2000;60:581–604. doi: 10.1080/003655100448310[↩]
- Todd DJ. Erythropoietic protoporphyria. Br J Dermtol. 1994;131:751–756. doi: 10.1111/j.1365-2133.1994.tb08577.x[↩][↩]
- Bonkovsky HL, Schned AR. Fatal liver failure in protoporphyria. Synergism between ethanol excess and the genetic defect. Gastroenterology. 1986 Jan;90(1):191-201. https://www.gastrojournal.org/article/0016-5085(86)90093-4/pdf?referrer=https%3A%2F%2Fpubmed.ncbi.nlm.nih.gov%2F[↩]
- Anstey AV, Hift RJ. Liver disease in erythropoietic protoporphyria: insights and implications for management. Postgrad Med J. 2007 Dec;83(986):739-48. doi: 10.1136/gut.2006.097576[↩][↩]
- Anstey A v., Hift RJ. Liver disease in erythropoietic protoporphyria: Insights and implications for management. Gut 2007;56(7):1009–1018. doi: 10.1136/gut.2006.097576[↩][↩][↩][↩]
- Whatley SD, Mason NG, Khan M, et al. Autosomal recessive erythropoietic protoporphyria in the United Kingdom: prevalence and relationship to liver disease. J Med Genet 2004;41(8):e105. doi: 10.1136/jmg.2003.016121[↩]
- Coffey A, Leung DH, Quintanilla NM. Erythropoietic protoporphyria: Initial diagnosis with cholestatic liver disease. Pediatrics 2018;141:S445–S450. doi: 10.1542/peds.2016-1625[↩]
- Cripps DJ, Goldfarb SS. Erythropoietic protoporphyria: hepatic cirrhosis. Br J Dermatol 1978;98(3):349–354. doi: 10.1111/j.1365-2133.1978.tb06163.x[↩]
- Khalili MJ, Farahmand F, Hirbod-Mobarakeh A, Yousefi A, Sotoudeh S, Monajemzadeh M, Razaghian A, Rezaei N. Erythropoietic protoporphyria and early onset of cholestasis. Turk J Pediatr. 2012 Nov-Dec;54(6):645-50.[↩]
- Yao YJ, Li SFY. Determination of erythrocyte porphyrins by epi-illumination fluorescence microscope with capillary electrophoresis. J Liq Chromatogr Relat Technol 1996;19(1):1–15. doi: 10.1080/10826079608006285[↩]
- Hennig G, Gruber C, Vogeser M, et al. Dual-wavelength excitation for fluorescence-based quantification of zinc protoporphyrin IX and protoporphyrin IX in whole blood. J Biophotonics 2014;7(7):514–524. doi: 10.1002/jbio.201200228[↩]
- Gou EW, Balwani M, Bissell DM, et al. Pitfalls in Erythrocyte Protoporphyrin Measurement for Diagnosis and Monitoring of Protoporphyrias. Clin Chem 2015;61(12):1453–1456. doi: 10.1373/clinchem.2015.245456[↩]
- Balwani M, Naik H, Anderson KE, et al. Clinical, biochemical, and genetic characterization of North American patients with erythropoietic protoporphyria and X-linked protoporphyria. JAMA Dermatol. 2017;153(8):789–796. doi: 10.1001/jamadermatol.2017.1557[↩][↩]
- Dickey AK, Naik H, Keel SB, et al. Evidence-based consensus guidelines for the diagnosis and management of erythropoietic protoporphyria and X-linked protoporphyria. J Am Acad Dermatol. Published online 27 August 2022 doi: 10.1016/j.jaad.2022.08.036[↩][↩]
- Coffey A, Leung DH, Quintanilla NM. Erythropoietic Protoporphyria: Initial Diagnosis With Cholestatic Liver Disease. Pediatrics. 2018 Apr;141(Suppl 5):S445-S450. doi: 10.1542/peds.2016-1625[↩]
- Drug-induced photosensitivity. https://dermnetnz.org/topics/drug-induced-photosensitivity[↩]
- Hydroa vacciniforme. https://dermnetnz.org/topics/hydroa-vacciniforme[↩]
- Solar urticaria. https://dermnetnz.org/topics/solar-urticaria[↩][↩]
- Polymorphic light eruption. https://dermnetnz.org/topics/polymorphic-light-eruption[↩]
- de Castro Maqueda G, Gutiérrez-Manzanedo JV, González-Montesinos JL, Vaz Pardal C, Rivas Ruiz F, de Troya Martín M. Sun Exposure and Photoprotection: Habits, Knowledge and Attitudes Among Elite Kitesurfers. J Cancer Educ. 2022 Jun;37(3):517-523. doi: 10.1007/s13187-020-01838-7[↩]
- Edel Y, Mamet R, Snast I, Kaftory R, Mazor S, Hodak E, Lapidoth M, Elis A, Molad Y, Levi A. Epidemiology of cutaneous porphyria in Israel: a nationwide cohort study. J Eur Acad Dermatol Venereol. 2020 Jan;34(1):184-187. doi: 10.1111/jdv.15769[↩]
- Solano F. Photoprotection and Skin Pigmentation: Melanin-Related Molecules and Some Other New Agents Obtained from Natural Sources. Molecules 2020;25(7). doi: 10.3390/MOLECULES25071537[↩]
- Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for Erythropoietic Protoporphyria. N Engl J Med. 2015 Jul 2;373(1):48-59. doi: 10.1056/NEJMoa1411481[↩]
- Holme SA, Thomas CL, Whatley SD, Bentley DP, Anstey AV, Badminton MN. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J Am Acad Dermatol. 2007 Jun;56(6):1070-2. doi: 10.1016/j.jaad.2006.11.030[↩]
- Lyoumi S, Abitbol M, Andrieu V, Henin D, Robert E, Schmitt C, Gouya L, de Verneuil H, Deybach JC, Montagutelli X, Beaumont C, Puy H. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood. 2007 Jan 15;109(2):811-8. doi: 10.1182/blood-2006-04-014142[↩]
- Schmidt H, Snitker G, Thomsen K, Lintrup J. Erythropoietic protoporphyria. A clinical study based on 29 cases in 14 families. Arch Dermatol 1974;110(1):58–64. doi: 10.1001/archderm.110.1.58[↩]
- Holme SA, Worwood M, Anstey A v, Elder GH, Badminton MN. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood 2007;110(12):4108–4110. doi: 10.1182/blood-2007-04-088120[↩]
- Barman-Aksözen J, Minder EI, Schubiger C, Biolcati G, Schneider-Yin X. In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol Dis 2015;54(1):71–77. doi: 10.1016/j.bcmd.2014.07.017[↩]
- Lyoumi S, Abitbol M, Andrieu V, et al. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood 2007;109(2):811–818. doi: 10.1182/blood-2006-04-014142[↩]
- Barman-Aksoezen J, Girelli D, Aurizi C, et al. Disturbed iron metabolism in erythropoietic protoporphyria and association of GDF15 and gender with disease severity. J Inherit Metab Dis 2017;40(3):433–441. doi: 10.1007/s10545-017-0017-7[↩]
- Bossi K, Lee J, Schmeltzer P, et al. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur J Clin Invest 2015;45(10):1032–1041. doi: 10.1111/eci.12503[↩]
- Bentley DP, Meek EM. Clinical and biochemical improvement following low-dose intravenous iron therapy in a patient with erythropoietic protoporphyria. British Journal of Haematology 2013;163(2):289–291. doi: 10.1111/bjh.12485[↩]
- Delaby C, Lyoumi S, Ducamp S, Martin-Schmitt C, Gouya L, Deybach JC, Beaumont C, Puy H. Excessive erythrocyte PPIX influences the hematologic status and iron metabolism in patients with dominant erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):45-52.[↩]
- Minder EI, Schneider-Yin X, Steurer J, Bachmann LM. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):84-97.[↩]
- Do KD, Banner BF, Katz E, Szymanski IO, Bonkovsky HL. Benefits of chronic plasmapheresis and intravenous heme-albumin in erythropoietic protoporphyria after orthotopic liver transplantation. Transplantation. 2002 Feb 15;73(3):469-72. doi: 10.1097/00007890-200202150-00024[↩]
- McCullough AJ, Barron D, Mullen KD, Petrelli M, Park MC, Mukhtar H, Bickers DR. Fecal protoporphyrin excretion in erythropoietic protoporphyria: effect of cholestyramine and bile acid feeding. Gastroenterology. 1988 Jan;94(1):177-81. doi: 10.1016/0016-5085(88)90627-0[↩]
- McGuire BM, Bonkovsky HL, Carithers RL Jr, Chung RT, Goldstein LI, Lake JR, Lok AS, Potter CJ, Rand E, Voigt MD, Davis PR, Bloomer JR. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005 Dec;11(12):1590-6. doi: 10.1002/lt.20620[↩]
- Wahlin S, Stal P, Adam R, Karam V, Porte R, Seehofer D, Gunson BK, Hillingsø J, Klempnauer JL, Schmidt J, Alexander G, O’Grady J, Clavien PA, Salizzoni M, Paul A, Rolles K, Ericzon BG, Harper P; European Liver and Intestine Transplant Association. Liver transplantation for erythropoietic protoporphyria in Europe. Liver Transpl. 2011 Sep;17(9):1021-6. doi: 10.1002/lt.22341[↩]
- Weiss Y, Balwani M, Chen B, Yasuda M, Nazarenko I, Desnick RJ. Congenital erythropoietic porphyria and erythropoietic protoporphyria: Identification of 7 uroporphyrinogen III synthase and 20 ferrochelatase novel mutations. Mol Genet Metab. 2019 Nov;128(3):358-362. doi: 10.1016/j.ymgme.2018.08.015[↩]
- Rand EB, Bunin N, Cochran W, Ruchelli E, Olthoff KM, Bloomer JR. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006 Dec;118(6):e1896-9. doi: 10.1542/peds.2006-0833[↩]
- Wang YM, Gloude NJ, Davies SM, Lucky AW, Nelson AS. Hematopoietic stem cell transplant for erythropoietic porphyrias in pediatric patients. Pediatr Blood Cancer. 2021 Sep;68(9):e29231. doi: 10.1002/pbc.29231[↩]
- Hashmi SK, Harstead E, Sachdev M, Black DD, Clark I, Ortanca I, Triplett BM, Talleur AC. Hematopoietic cell transplant for reversal of liver fibrosis in a pediatric patient with erythropoietic protoporphyria. Pediatr Transplant. 2021 Sep;25(6):e13966. doi: 10.1111/petr.13966[↩][↩]
- Endo Y, Hibi T, Shinoda M, Obara H, Kitago M, Yagi H, Abe Y, Hasegawa Y, Matsubara K, Hori S, Tanaka M, Makiuchi S, Nakano Y, Itano O, Kuroda T, Kitagawa Y. Reappraisal of liver transplantation for erythropoietic protoporphyria: A deadly combination of disease recurrence and biliary complication. Pediatr Transplant. 2022 Jun;26(4):e14261. doi: 10.1111/petr.14261[↩]
- Ricci A, Di Betto G, Bergamini E, Buzzetti E, Corradini E, Ventura P. Iron Metabolism in the Disorders of Heme Biosynthesis. Metabolites. 2022 Aug 31;12(9):819. doi: 10.3390/metabo12090819[↩]
- Rutter KJ, Ashraf I, Cordingley L, Rhodes LE. Quality of life and psychological impact in the photodermatoses: a systematic review. Br J Dermatol. 2020 May;182(5):1092-1102. doi: 10.1111/bjd.18326[↩]





 & X-linked protoporphyria (XLP). Causes & symptoms of erythropoietic protoporphyria. How is erythropoietic protoporphyria diagnosed & best treated){kind=link}