
Contents
What is Fabry disease
Fabry disease is an inherited disorder of lipid (fat) metabolism that resulted in deficient activity of the enzyme alpha-galactosidase A (α-Gal A) and progressive lysosomal deposition of a particular type of fat called globotriaosylceramide (GL-3) in cells throughout the body 1. The alpha-galactosidase A (α-Gal A) enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. Alpha-galactosidase A (α-Gal A) normally breaks down a fatty substance called globotriaosylceramide (GL-3). Mutations in the GLA gene alter the structure and function of the alpha-galactosidase A (α-Gal A) enzyme, preventing it from breaking down this substance effectively. As a result, globotriaosylceramide builds up in cells throughout the body, particularly cells lining blood vessels in the skin and cells in the kidneys, heart, and nervous system. The progressive accumulation of globotriaosylceramide (GL-3) damages cells, leading to the varied signs and symptoms of Fabry disease. Fabry disease belongs to a group of diseases known as lysosomal storage disorders.
Fabry disease is caused by mutations in the GLA gene and is inherited in an X-linked manner. The GLA gene provides instructions for making an enzyme called alpha-galactosidase A (α-Gal A). In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). If only one male in a family is affected, his mother is likely heterozygous; rarely, a single affected male in a family may have a de novo pathogenic variant. A heterozygous female has a 50% chance of transmitting the GLA pathogenic variant in each pregnancy. An affected male transmits his pathogenic variant to all of his daughters. Heterozygote (carrier) testing for at-risk relatives and prenatal testing for pregnancies at increased risk are possible if the pathogenic variant in a family is known.
Fabry disease affects an estimated 1 in 40,000 to 60,000 males 2. The incidence of males with the type 1 classic Fabry disease is about 1 in 40,000 males, but varies with demography and race, ranging from about ~1 in 18,000 to 1 in 95,000 based on newborn screening studies 3. Fabry disease also occurs in females, although the prevalence is unknown. Milder, late-onset forms of Fabry disease are probably more common than the classic, severe form.
Males are typically more severely affected than females. Females have a more variable course and may be asymptomatic or as severely affected as males. There are two major disease phenotypes: the type 1 “classic” and type 2 “later-onset” subtypes. Both lead to renal failure, and/or cardiac disease, and early death 4.
Beginning in childhood, this buildup of globotriaosylceramide causes signs and symptoms that affect many parts of the body. The classic Type 1 form of Fabry disease, occurring in males with less than 1% alpha-galactosidase A (α-Gal A) enzyme activity, and marked accumulation of globotriaosylceramide (GL-3) and related glycolipids in capillaries and small blood vessels which cause the major symptoms in childhood or adolescence. The classic Type 1 Fabry disease usually has its onset in childhood or adolescence with periodic excruciating pain in the extremities hands and feet which occur with exercise, fevers, stress, etc. (acroparesthesia); the appearance of vascular cutaneous lesions as clusters of red to blue rash-like discolorations on the skin (angiokeratomas); sweating abnormalities (anhidrosis [absent sweating], hypohidrosis [markedly decreased sweating], and rarely hyperhidrosis [excessive sweating]); gastrointestinal symptoms including abdominal pain and cramping, and frequent bowel movements; characteristic corneal and lenticular opacities (star-burst pattern of the cornea seen by slit-lamp ophthalmologic examination) that does not affect vision 5; ringing in the ears (tinnitus) and hearing loss and proteinuria 1. With increasing age (usually occurs in men in the third to fifth decade), the systemic globotriaosylceramide (GL-3) deposition, especially in the heart leads to arrhythmias, left ventricular hypertrophy and then hypertrophic cardiomyopathy and in the kidneys to progressive insufficiency then to renal failure (end-stage renal disease) and/or to cerebrovascular disease including transient ischemic attacks (TIAs) and strokes. In middle age, most males successfully treated for end-stage renal disease develop heart attack and/or stroke, a major cause of morbidity and mortality 1. Prior to renal replacement therapy (i.e., dialysis and transplantation) and enzyme replacement therapy, the average age of death of affected males with the type 1 classic phenotype was ~40 years 6.
Some affected individuals have milder forms of the disorder that appear later in life and affect only the heart or kidneys. Heterozygous females typically have milder symptoms at a later age of onset than males. Rarely, they may be relatively asymptomatic throughout a normal life span or may have symptoms as severe as those observed in males with the classic phenotype.
In contrast, males with the type 2 “later-onset” Fabry disease (previously called cardiac or renal variants) have greater than 1% alpha-galactosidase A (α-Gal A) activity, lack globotriaosylceramide (GL-3) accumulation in capillaries and small blood vessels, and do not manifest the early manifestations of type 1 males (i.e., the acroparesthesias, hypohidrosis, angiokeratomas, corneal dystrophy, etc). They experience an essentially normal childhood and adolescence. They typically present with renal and/or cardiac disease in the third to seventh decades of life. Most type 2 later-onset patients have been identified by enzyme screening of patients in cardiac, hemodialysis, renal transplant, and stroke clinics 7, and recently by newborn screening. Based on these screening studies 3 the incidence of type 2 later-onset males varies by demography, ethnicity, and race, but is at least 10 times more frequent than that of the type 1 males from the same region, ethnic group, or race.
Fabry disease treatment
Treatment of manifestations: Diphenylhydantoin, carbamazepine, or gabapentin to reduce pain (acroparesthesia); ACE inhibitors or angiotensin receptor blockers to reduce proteinuria; chronic hemodialysis and/or renal transplantation for end stage renal disease.
Prevention of primary complications: The U.S. Food and Drug Administration (FDA) approved an enzyme replacement therapy (ERT) called agalsidase beta (Fabrazyme®) as a treatment for patients with Fabry disease in 2003. Fabrazyme®, which is administered intravenously, is a form of the human enzyme produced by recombinant DNA technology. This replacement of the missing enzyme reduces the accumulation of the accumulated glycolipids in cells, including the cells of the kidney and other organs. Double-blind, placebo-controlled Phase 3 and 4 clinical trials have demonstrated the safety and effectiveness of Fabrazyme® enzyme replacement therapy for Fabry disease. The role of enzyme replacement therapy in the long-term prophylaxis of renal, cardiac, and CNS manifestations is unproven; however, experts 8, 9, 10 recommend that enzyme replacement therapy be initiated as early as possible in all males with Fabry disease (including children and those with end stage renal disease undergoing dialysis and renal transplantation) and in females with significant disease because all are at high risk for cardiac, cerebrovascular, and renal complications.
Prevention of secondary complications: Prophylaxis for renovascular disease, ischemic heart disease, and cerebrovascular disease as for the general population.
Surveillance: Annual or more frequent assessment of renal function; annual cardiology and audiology evaluations; biennial brain MRI/MRA.
Agents/circumstances to avoid: Smoking.
Evaluation of relatives at risk: Early identification of affected relatives by molecular genetic testing if the pathogenic variant in the family is known in order to initiate enzyme replacement therapy as early as possible in affected individuals.
Figure 1. Fabry disease
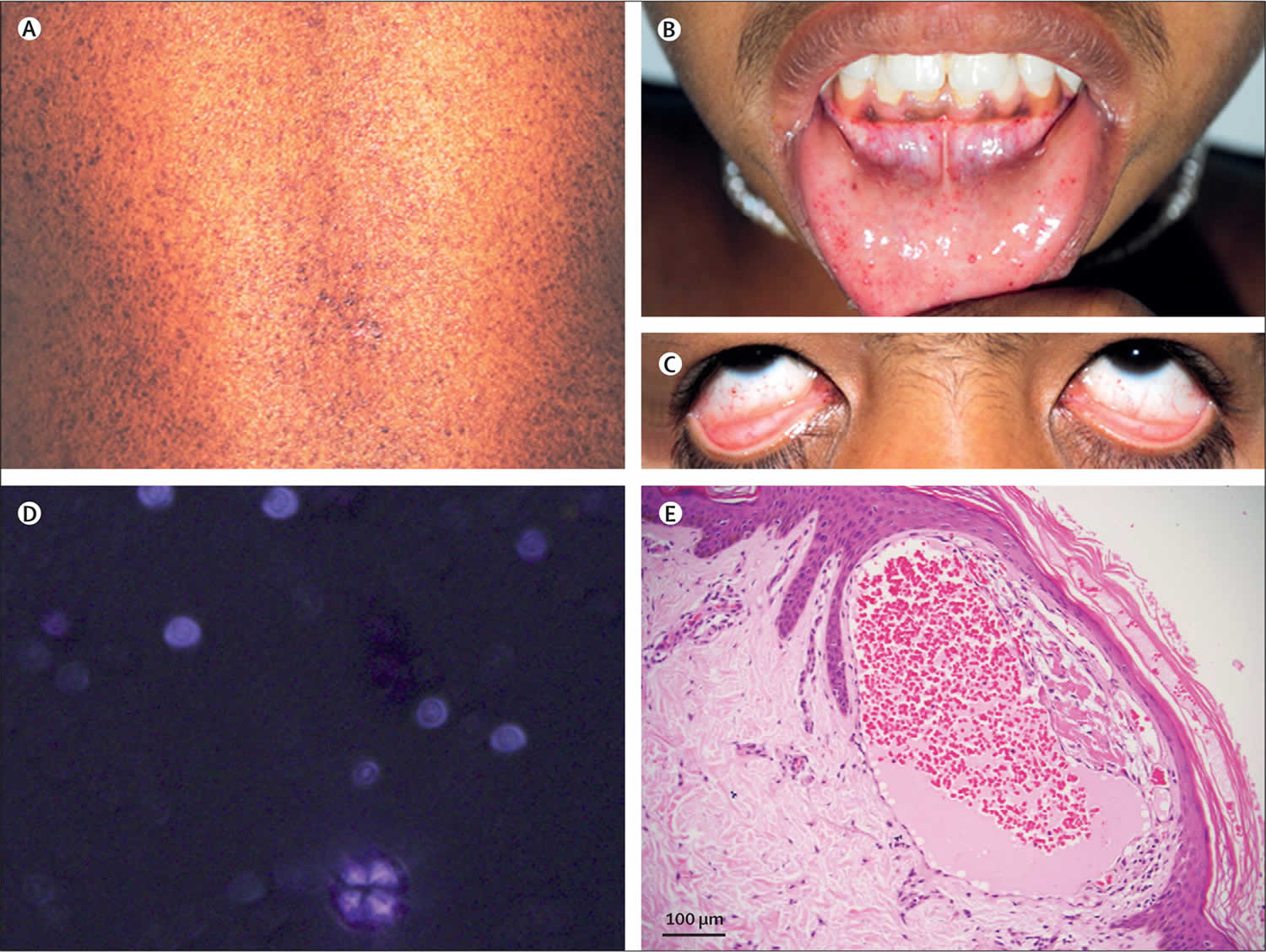
Footnote: (A–C) Telangiectasias; (D) urine analysis showing birefringent lipid molecules; and (E) skin biopsy showing angiokeratoma (haematoxylin and eosin stain).
[Source 11]Figure 2. Fabry disease eye signs
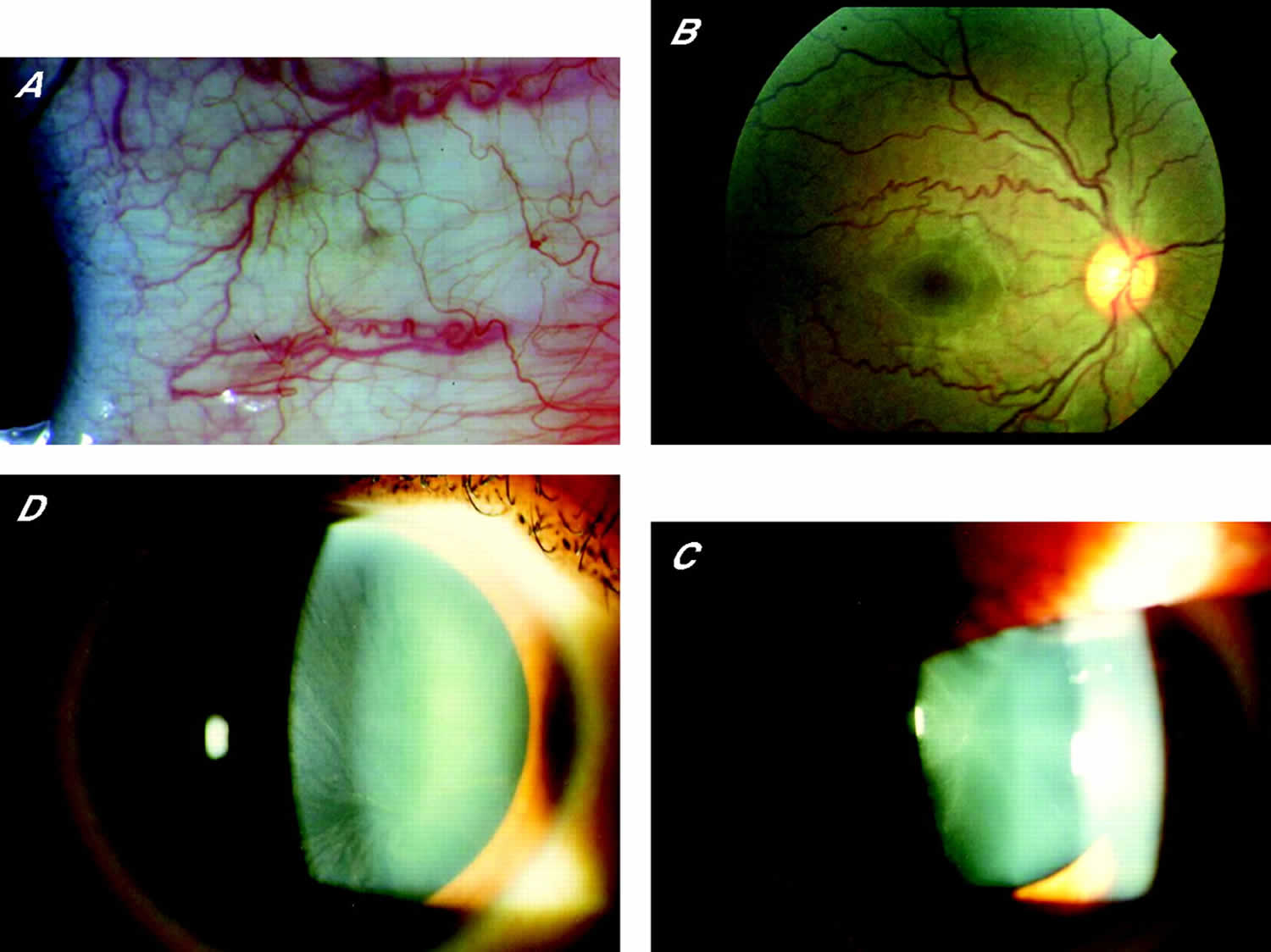
Footnote: (A) Conjunctival vessel tortuosity; (B) retinal vessel tortuosity; (C) cornea verticillata (vortex opacities located in the superficial corneal layers); and (D) Fabry cataract (posterior lens opacities with a radiating appearance).
[Source 12]Fabry disease life expectancy
Based on data from the Fabry Registry, 75 of 1422 males and 12 of 1426 females were reported to have died. The 87 deceased individuals were diagnosed at a much older age than other individuals in the Fabry Registry.
- The life expectancy of males with Fabry disease was 58.2 years, compared with 74.7 years in the general population of the United States 1. Prior to renal replacement therapy (i.e., dialysis and transplantation) and enzyme replacement therapy, the average age of death of affected males with the type 1 classic Fabry disease was ~40 years 6.
- The life expectancy of females with Fabry disease was 75.4 years, compared with 80.0 years in the United States general population 1.
The most common cause of death among both genders was cardiovascular disease 13. Most individuals (57%) who died of cardiovascular disease had previously received renal replacement therapy (e.g., dialysis or transplantation). In the Fabry Outcome Survey, the principal causes of death among 181 affected relatives (most of whom had died before 2001) were renal failure in males (42%) and cerebrovascular disease in females (25%) 14. In contrast, of the 42 individuals enrolled in the Fabry Outcome Survey whose deaths were reported between 2001 and 2007, cardiac disease was the main cause of death in both males (34%) and females (57%).
Fabry disease symptoms
Type 1 Classic Fabry disease
The signs and symptoms of males with type 1 classic Fabry disease typically begin in childhood or adolescence. Symptoms increase with age due to the progressive glycolipid accumulation in the vascular system, kidneys, and heart leading to kidney failure, heart disease, and/or strokes. Early and progressive clinical symptoms include:
- Acroparesthesias. Pain is an early symptom of the Classic subtype and may occur as early as 2-8 years old in males and during childhood or adolescence in female heterozygotes. Affected individuals may experience episodes of severe burning pain in the hands and the feet (acroparesthesia). Severe episodes of pain (Fabry’s crises) may last for hours to days and are frequently triggered by exercise, fatigue, stress, and/or fever.
- Anhidrosis or hypohidrosis. Type 1 males and some type 1 females have decreased or absent sweat production (hypohidrosis or anhidrosis) and discomfort (heat intolerance) in warm temperatures, with exercise, or fevers.
- Angiokeratomas. Early symptoms also include the appearance of a reddish to dark-blue skin rash, especially in the area between the hips and the knees. These skin lesions may be flat or raised. They often are found in the umbilical area or genitals of type 1 males. Typically, males and females with the type 2 later-onset phenotype do not have these characteristic skin lesions.
- Renal involvement. Progressive glycosphingolipid accumulation in the kidney interferes with renal function, resulting in azotemia and renal insufficiency. During childhood and adolescence, protein, casts, red cells, and birefringent lipid globules with characteristic “Maltese crosses” can be observed in the urinary sediment. Proteinuria, isosthenuria, and a gradual deterioration of tubular reabsorption, secretion, and excretion occur with advancing age. Polyuria and a syndrome similar to vasopressin-resistant diabetes insipidus occasionally develop. Gradual deterioration of renal function and the development of azotemia usually occur in the third to fifth decade of life, although end stage renal disease has been reported in the second decade. Death most often results from end stage renal disease unless chronic hemodialysis or renal transplantation is undertaken. The mean age at death of males not treated for end stage renal disease is 41 years, but occasionally an untreated male with the classic phenotype survives into the seventh decade.
- Gastrointestinal problems. Abdominal cramping, frequent bowel movements, and diarrhea may also occur, particularly after a large meal. Glycosphingolipid deposition in intestinal small vessels and in the autonomic ganglia of the bowel may cause episodic diarrhea, nausea, vomiting, bloating, cramping abdominal pain, and/or intestinal malabsorption 15. Achalasia and jejunal diverticulosis, which may lead to perforation of the small bowel, have been described. Radiographic studies may reveal thickened, edematous colonic folds, mild dilatation of the small bowel, a granular-appearing ileum, and the loss of haustral markings throughout the colon.
- Corneal dystrophy. Patients with the type 1 Classic subtype have abnormal deposits of glycolipids in their corneas resulting in a characteristic change which can be seen by an experienced ophthalmologist. These changes do not affect vision. Blood vessels in the eyes may appear twisted (cork screw-like; contorted) and/or slightly enlarged (dilated) due to the glycolipid accumulation in the vessel walls.
- Cardiac disease. Mitral insufficiency may be present in childhood or adolescence. Left ventricular enlargement and conduction abnormalities are early findings. Left ventricular hypertrophy, often associated with hypertrophy of the interventricular septum and appearing similar to hypertrophic cardiomyopathy (HCM), is progressive and occurs earlier in males than females 16. Cardiac disease is present in most males with the classic phenotype by middle age and is the major cause of morbidity and mortality in those who have undergone dialysis or transplantation for treatment of end-stage renal disease (ESRD). ECG changes including ST segment changes, T-wave inversion, and dysrhythmias such as a short PR interval and intermittent supraventricular tachycardias may be caused by infiltration of the conduction system. Echocardiography demonstrates an increased thickness of the interventricular septum and the left ventricular posterior wall 17. Magnetic resonance studies using gadolinium demonstrated late enhancement areas, corresponding to myocardial fibrosis and associated with decreased regional functioning as assessed by strain and strain-rate imaging 18. T1 mapping illustrates intramural fat deposition and posterior wall fibrosis 19. Among 714 predominantly adult individuals in the Fabry Outcome Survey 20, angina, palpitations/arrhythmia, and exertional dyspnea were found in 23%-27% of males and 22%-25% of females. Hypertension, angina pectoris, myocardial ischemia and infarction, congestive heart failure, and severe mitral regurgitation are late signs. Hypertension was found in more than 50% of males and more than 40% of females in the Fabry Outcome Survey 21.
- Cerebrovascular manifestations result primarily from multifocal small vessel involvement and may include thrombosis, transient ischemic attacks (TIA), basilar artery ischemia and aneurysm, seizures, hemiplegia, hemianesthesia, aphasia, labyrinthine disorders, or frank cerebral hemorrhage 22. Fabry Outcome Survey data indicate that stroke or TIA occur in approximately 13% of affected individuals overall (15% males, 11.5% females) 23. The Fabry Registry has reported that cerebrovascular manifestations are often a presenting feature of Fabry disease and may be more frequent than previously recognized. Rolfs et al 24 reported that in Germany a GLA pathogenic variant was identified in 21 of 432 males (4.9%) and seven of 289 females (2.4%) age 18-55 years suffering cryptogenic stroke. However, other studies have not confirmed such a high prevalence 25. Thromboembolic events are more common among individuals with Fabry disease who also have the factor V Leiden variant [Lenders et al 2015]. In addition, a distinct neurologic phenotype including decreased motor performance and nonmotor neurologic manifestations has been described. Individuals with Fabry disease showed slower gait and transfer speed, poorer fine manual dexterity, and slower hand speed than controls. Affected individuals had an increased incidence of depression, pain, and daytime sleepiness but did not exhibit extrapyramidal motor features or signs of significant cognitive impairment 26.
- Pulmonary. Several affected individuals have had pulmonary involvement, manifest clinically as chronic bronchitis, wheezing, or dyspnea. Primary pulmonary involvement has been reported in the absence of cardiac or renal disease. Pulmonary function studies may show an obstructive component 27 which has been demonstrated to stabilize with enzyme replacement therapy (ERT) 28.
- Vascular. Pitting edema of the lower extremities may be present in adulthood in the absence of hypoproteinemia, varices, or other clinically significant vascular disease. Although the pitting edema is initially reversible, progressive glycosphingolipid deposition in the lymphatic vessels and lymph nodes results in irreversible lymphedema requiring treatment with compression hosiery. Varicosities, hemorrhoids, and priapism have also been reported.
- Cranial nerve VIII involvement. High-frequency hearing loss, tinnitus, and dizziness have been reported 29.
- Psychological. Depression, anxiety, severe fatigue, and other psychosocial manifestations lead to decreased quality of life in many affected individuals 30.
- Additional Type 1 symptoms. Other symptoms that may be associated with Fabry disease include chronic fatigue, dizziness, headache, generalized weakness, nausea, and/or vomiting, delayed puberty, lack of or sparse hair growth, and rarely malformation of the joints of the fingers. Some type 1 classic males have abnormal accumulation of lymph in the feet and legs associated with swelling (lymphedema). In these cases, lymph, a body fluid containing certain white blood cells, fats, and proteins, accumulates outside blood vessels in spaces between cells and drains or flows back into the bloodstream via lymph vessels. Lymphedema results from disruption of lymph’s normal drainage due to the glycolipid accumulation in the lymphatic vessels and lymph nodes.
Type 1 and 2 Males with Fabry disease
With advancing age in type 1 males, typically in the third to fourth decades, and in type 2 males in the third to sixth decades, the progressive globotriaosylceramide (GL-3) glycolipid deposition leads to renal and heart manifestations as described below. Many of the type 2 later-onset males who lack the early manifestations in the type 1 boys, are detected in renal, heart, or stroke clinics. Patients with the type 2 later-onset subtype typically do not have the skin lesions (angiokeratoma), sweat normally, do not experience the Fabry pain or crises, and do not have heat intolerance or corneal involvement. These individuals develop heart or kidney disease later in adult life.
Signs of progressive organ involvement include:
Renal dysfunction. Progressive decrease in renal function is due to the progressive accumulation of globotriaosylceramide (GL-3) in the kidneys. There is histological evidence of this accumulation and ensuing cellular and vascular injury to renal tissue beginning in childhood and adolescence 31 in type 1 classic males and females. In the type 1 classic males the decline in kidney function progresses to kidney failure and the need for dialysis or transplantation typically by 35 to 45 years of age. In type 2 males, kidney involvement typically occurs in the fourth decade and later.
Cardiac disease. Globotriaosylceramide (GL-3) deposition can be found in all cardiac tissues, including myocytes, nerves, and coronary arteries. Heart disease includes heart enlargement, typically left ventricular hypertrophy leading to hypertrophic cardiomyopathy, rhythm abnormalities (arrhythmias), and heart failure. Left ventricular hypertrophy occurs in about 20% of males and females with an average age of diagnosis in the early 40s among males and late 40s among females. Early heart involvement in type 1 males typically includes arrhythmias and mitial insufficiency in their twenties followed by left ventricular hypertrophy leading to hypertrophic cardiomyopathy. Type 2 later-onset males develop the same heart manifestations as type 1 males and may be first diagnosed in cardiac clinics among patients with left ventricular hypertrophy or hypertrophic cardiomyopathy.
Cerebrovascular complications. As a result of the progressive globotriaosylceramide (GL-3) deposition in the small blood vessels in the brain, about 7% of males and 4% of females with Fabry disease, particularly those with the type 1 phenotype, experience ischemic or hemorrhagic strokes, occurring typically in the fourth decade of life or later 32.
Fabry disease symptoms in females
The clinical manifestations in heterozygous females range from asymptomatic throughout a normal life span to as severe as affected males. Variation in clinical manifestations is attributed to random X-chromosome inactivation 33. More severely affected females are more likely to express the X chromosome with the GLA pathogenic variant in affected organs 34.
Most heterozygous females from families in which affected males have the classic phenotype have a milder clinical course and better prognosis than affected males.
Mild manifestations include the characteristic cornea verticillata (70%-90%) and lenticular opacities that do not impair vision; acroparesthesia (50%-90%); angiokeratomas (10%-50%) that are usually isolated or sparse; and hypohidrosis. In addition, heterozygotes may have chronic abdominal pain and diarrhea 35.
With advancing age, heterozygous females with the type 1 phenotype may develop mild to moderate enlargement of the left heart (left ventricular hypertrophy) and valvular disease. More serious manifestations include significant left ventricular hypertrophy, hypertrophic cardiomyopathy, cardiomegaly, myocardial ischemia, infarction, and cardiac arrhythmias 36. Left ventricular hypertrophy occurs in about 20% of females with an average age of diagnosis in the late 40s among females. It is not clear if type 2 heterozygotes develop heart disease or if they have additionally autosomal dominant hypertrophic cardiomyopathy due to sarcomere mutations 37.
The occurrence of cerebrovascular disease including transient ischemic attacks and cerebrovascular accidents is consistent with the microvascular pathology of the disease 38. About 4% of females with Fabry disease, particularly those with the type 1 phenotype, experience ischemic or hemorrhagic strokes, occurring typically in the fourth decade of life or later 32.
Renal findings in type 1 female heterozygotes include isosthenuria, the presence of erythrocytes, leukocytes, and granular and hyaline casts in the urinary sediment, and proteinuria. According to the United States and European dialysis and transplantation registries, approximately 10% of heterozygotes develop renal failure requiring dialysis or transplantation. It is not clear what percentage of type 2 females develop renal dysfunction, if any.
Excessive guilt, fatigue, occupational difficulty, suicidal ideation, and depression have been noted in heterozygotes 39.
Fabry disease causes
Fabry disease is caused by mutations in the GLA gene. This gene provides instructions for making an enzyme called alpha-galactosidase A (α-Gal A). The alpha-galactosidase A (α-Gal A) enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. Alpha-galactosidase A normally breaks down a fatty substance called globotriaosylceramide. Mutations in the GLA gene alter the structure and function of the enzyme, preventing it from breaking down this substance effectively. As a result, globotriaosylceramide builds up in cells throughout the body, particularly cells lining blood vessels in the skin and cells in the kidneys, heart, and nervous system. The progressive accumulation of this substance damages cells, leading to the varied signs and symptoms of Fabry disease.
GLA gene mutations that result in an absence of alpha-galactosidase A activity lead to the classic, severe form of Fabry disease. Mutations that decrease but do not eliminate the enzyme’s activity usually cause the milder, late-onset forms of Fabry disease that affect only the heart or kidneys.
Fabry disease inheritance pattern
Fabry disease is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males (who have only one X chromosome), one altered copy of the GLA gene in each cell is sufficient to cause the condition. Because females have two copies of the X chromosome, one altered copy of the gene in each cell usually leads to less severe symptoms in females than in males, or rarely may cause no symptoms at all.
Unlike other X-linked disorders, Fabry disease causes significant medical problems in many females who have one altered copy of the GLA gene. These women may experience many of the classic features of the disorder, including nervous system abnormalities, kidney problems, chronic pain, and fatigue. They also have an increased risk of developing high blood pressure, heart disease, stroke, and kidney failure. The signs and symptoms of Fabry disease usually begin later in life and are milder in females than in their affected male relatives.
A small percentage of females who carry a mutation in one copy of the GLA gene never develop signs and symptoms of Fabry disease.
Figure 3. Fabry disease X-linked inheritance pattern
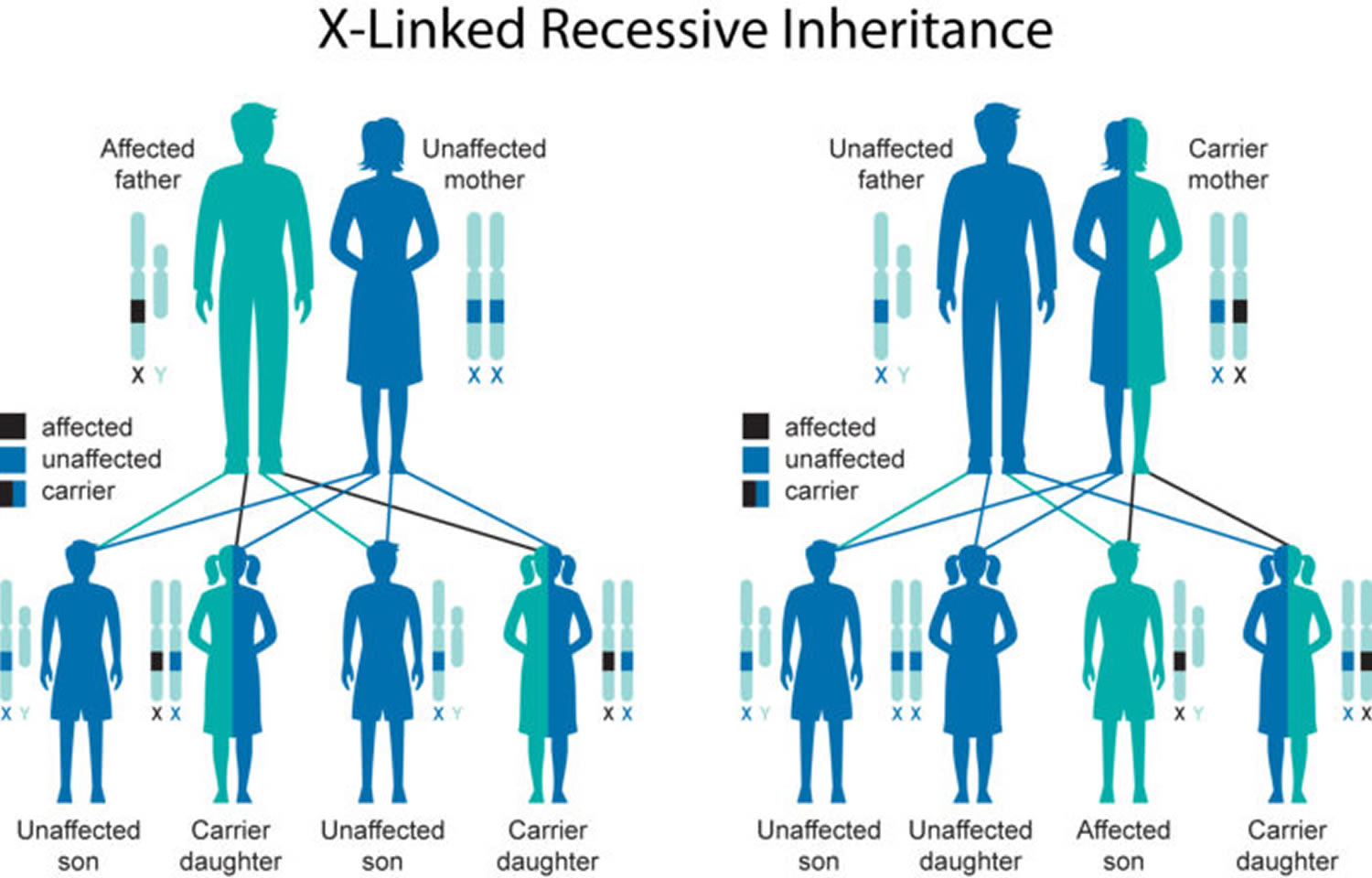
Risk to Family Members
Parents of an affected male proband
- The father of an affected male will not have the disorder nor will he be hemizygous for the GLA pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected individual, the mother of an affected male individual is an obligate heterozygote (carrier).
Note: If a woman has more than one affected child and no other affected relatives and the GLA pathogenic variant cannot be detected in her leukocyte DNA, maternal germline mosaicism should be considered as a possible explanation. - If only one individual in the family is affected, the mother of the affected male is likely heterozygous for the GLA pathogenic variant. Rarely, an affected male may have a de novo pathogenic variant, in which case the mother is not a carrier.
Siblings of an affected male proband
The risk to siblings depends on the genetic status of the mother:
- If the mother of the proband has a pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be will be heterozygotes and may be symptomatic.
- If the proband represents a simplex case (i.e., a single occurrence in a family) and if the GLA pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is low but greater than that of the general population because of the theoretic possibility of maternal germline mosaicism.
Siblings of a (symptomatic or asymptomatic) heterozygous female
- If the father of the proband has a GLA pathogenic variant, he will transmit it to all his daughters and none of his sons.
- If the mother of the proband has a GLA pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be heterozygotes (carriers) and may be symptomatic.
- Paternal germline mosaicism has been demonstrated in this condition 40. Thus, even if the GLA pathogenic variant has not been identified in paternal leukocyte DNA, female sibs of a heterozygous female may still be at increased risk of inheriting the pathogenic variant.
Offspring of an affected male. Affected males transmit the GLA pathogenic variant to:
- All of their daughters who will be heterozygotes and may be symptomatic.
- None of their sons.
Offspring of a (symptomatic or asymptomatic) heterozygous female. Women with a GLA pathogenic variant have a 50% chance of transmitting the pathogenic variant to each child:
- Males who inherit the pathogenic variant will be affected.
- Females who inherit the pathogenic variant will be heterozygotes and may be symptomatic.
Other family members. The proband’s maternal aunts may be at risk of being heterozygotes and the aunts’ offspring, depending on their gender, may be at risk of being heterozygotes and/or of being affected. At-risk females should be offered clinical examination, genetic counseling, and molecular genetic testing.
Fabry disease diagnosis
The diagnosis of Fabry disease is frequently made by physicians who recognize the pain in the extremities, absent or decreased sweating (anhidrosis or hypohidrosis), typical skin lesions (angiokeratoma), gastrointestinal abnormalities, corneal involvement, renal insufficiency, and heart symptoms present in childhood, adolescence or adulthood.
Fabry disease should be suspected in males and females with the following clinical features:
- Periodic crises of severe pain in the extremities (acroparesthesia)
- Vascular cutaneous lesions (angiokeratomas)
- Sweating abnormalities (anhidrosis, hypohidrosis, and rarely hyperhidrosis)
- Characteristic corneal and lenticular opacities
- Unexplained stroke
- Unexplained left ventricular hypertrophy
- Renal insufficiency of unknown etiology including unexplained proteinuria or microalbuminuria
The diagnosis is confirmed by demonstrating the enzyme deficiency in males and by identifying the specific GLA gene mutation in males and females.
Prenatal diagnosis of Fabry disease is made by measuring alpha-galactosidase A (α-Gal A) activity and demonstrating the family-specific GLA mutation in cells that are removed from the amniotic fluid surrounding the developing fetus at about 15 weeks of pregnancy. Early prenatal diagnosis at about 10 weeks of pregnancy can be made by a-Gal A enzyme and gene analyses of villi obtained by chronic villus sampling.
Establishing Fabry disease diagnosis
Male proband. The diagnosis of Fabry disease is established in a male proband by:
- Identification of deficient alpha-galactosidase A (α-Gal A) enzyme activity in plasma, isolated leukocytes, and/or cultured cells. The test is a fluorometric assay and uses the substrate 4-methylumbelliferyl-α-D-galactopyranoside.
- Males with classic Fabry disease have <1% α-Gal A enzyme activity.
- Males with atypical Fabry disease have residual enzyme activity >1% of normal.
Note: Both plasma and leukocyte enzyme activity should be assayed, as some pathogenic variants (e.g., p.Asn215Ser) affect intracellular trafficking or packaging/secretion of the enzyme, such that the reduction in enzyme activity in plasma is more marked than the reduction in enzyme activity in leukocytes.
- Identification of a hemizygous pathogenic variant in GLA by molecular genetic testing (see Table 1).
Female proband. The diagnosis of Fabry disease is established in a female proband by identification of a heterozygous pathogenic variant in GLA by molecular genetic testing (see Table 1).
Note: Measurement of alpha-galactosidase A (α-Gal A) enzyme activity is unreliable for identification of heterozygous females. Although demonstration of markedly decreased alpha-galactosidase A (α-Gal A) enzyme activity in a female is diagnostic of the heterozygous state, some heterozygotes have α-Gal A activity in the normal range.
Table 1. Molecular Genetic Testing Used in Fabry Disease
| Gene | Test Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by This Method |
|---|---|---|
| GLA | Sequence analysis | ~95% |
| Gene-targeted deletion/duplication analysis | ~5% |
Heterozygote (Carrier) Detection
Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the GLA pathogenic variant has been identified in the proband.
Measurement of alpha-galactosidase A (α-Gal A) enzyme activity is unreliable for carrier detection. Although demonstration of decreased α-Gal A activity in a female is diagnostic of the heterozygous state, some heterozygotes have α-Gal A activity in the normal range.
Ophthalmologic examination for the characteristic whorl-like corneal opacities by slit-lamp microscopy can be considered if enzyme analysis is uninformative and the specific GLA pathogenic variant in the family has not been identified by molecular genetic testing. However, only 80%-90% of heterozygous females have the corneal lesions.
Molecular Testing
Molecular testing approaches can include single-gene testing, use of a multigene panel, and more comprehensive genomic testing:
- Single-gene testing. Sequence analysis of GLA is performed first and followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
- Targeted analysis for the p.Ala143Pro pathogenic variant can be performed first in individuals from Nova Scotia (incidence 1:15,000).
- Targeted analysis for the IVS4+919G>A pathogenic variant can be performed first in individuals of Chinese ancestry with atypical presentation 41.
- A multigene panel that includes GLA and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- More comprehensive genomic testing may be considered if single-gene testing (and/or use of a multigene panel) has not confirmed a diagnosis in an individual with features of Fabry disease. Such testing may include exome sequencing, genome sequencing, and mitochondrial sequencing.
Prenatal Testing and Preimplantation Genetic Diagnosis
Prenatal diagnosis of Fabry disease is made by measuring a-Gal A activity and demonstrating the family-specific GLA mutation in cells that are removed from the amniotic fluid surrounding the developing fetus at about 15 weeks of pregnancy. Early prenatal diagnosis at about 10 weeks of pregnancy can be made by a-Gal A enzyme and gene analyses of villi obtained by chronic villus sampling.
Molecular genetic testing. Once the GLA pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic diagnosis are possible.
Biochemical testing. If the karyotype is 46,XY, α-Gal A enzyme activity can be measured in fetal cells. (If the GLA pathogenic variant has been identified in an affected family member, a biochemical diagnosis can be confirmed by molecular genetic testing of fetal DNA.)
Fabry disease treatment
Treatment of Manifestations
Acroparesthesia
- Diphenylhydantoin. The severe pain of such episodes in affected males and heterozygous females often responds to low-maintenance doses of diphenylhydantoin by reducing the frequency and severity of the periodic crises of excruciating pain and constant discomfort. A potential side effect of diphenylhydantoin is gingival hypertrophy.
- Carbamazepine has similar effects. The combination of the two drugs may also significantly reduce the frequency and severity of the pain. Dose-related autonomic complications with carbamazepine include urinary retention, nausea, vomiting, and ileus.
- Gabapentin has been demonstrated to improve pain 42.
Renal disease
Renal insufficiency is the most frequent and serious late complication in males with the classic phenotype. Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) should be used in those with evidence of renal involvement, especially to reduce proteinuria 43.
Chronic hemodialysis and/or renal transplantation have become lifesaving procedures. The engrafted kidney remains histologically free of glycosphingolipid deposition because the normal alpha-galactosidase A enzyme activity in the allograft catabolizes endogenous renal glycosphingolipid substrates. Therefore, successful renal transplantation corrects renal function.
Reviews of the registries of the European Renal Association-European Dialysis and Transplantation Association and the United States Renal Data System support excellent outcomes for renal transplantation in individuals with Fabry disease. For example, during the ten-year period from 1988 to 1998, 93 individuals who underwent renal transplantation were reported to the US registry. Compared to a matched control group, recipients with Fabry disease had equivalent five-year life survival (82% vs 83%) and graft survival (67% vs 75%), respectively.
Note: (1) Immune function in males with Fabry disease is similar to that in other individuals with uremia, obviating any immunologic contraindication to transplantation in this disease. Autoimmune conditions have, however, been reported to occur at an increased frequency in individuals with Fabry disease 44. (2) Transplantation of kidneys from female heterozygotes should be avoided, as the organs may already contain significant substrate deposition; all related potential donors must be evaluated to exclude affected males and heterozygous females.
Prevention of Primary Manifestations
Enzyme replacement therapy
The two enzyme replacement therapies using recombinant or gene-activated human alpha-galactosidase A enzyme that have been evaluated in clinical trials are Fabrazyme® (agalsidase beta 1 mg/kg every 2 weeks) and Replagal™ (agalsidase alfa 0.2 mg/kg every 2 weeks). Both were approved in 2001 by the European Agency for Evaluation of Medical Products; only Fabrazyme® was approved by the FDA for use in the United States.
The following is a summary of some of the clinical trials for each drug:
- Single-center double-blind placebo-controlled studies of agalsidase alfa have shown a beneficial effect of enzyme replacement therapy on neuropathic pain [Schiffmann et al 2001] and left ventricular hypertrophy [Hughes et al 2008]. Data from the Fabry Outcome Survey (FOS) suggest that enzyme replacement therapy with agalsidase alfa improves pain and quality of life, reduces the natural rate of decline of renal and cardiac function in males and females with Fabry disease [Mehta et al 2009] and may improve life expectancy [Beck et al 2015]. The enzyme is safe in children [Ramaswami et al 2006]. In persons with advanced renal disease, weekly administration of 0.2 mg/kg agalsidase alfa may be associated with a slower decline in renal function [Schiffmann et al 2007, Schiffmann et al 2015].
- A double-blind, randomized, placebo-controlled study of agalsidase beta demonstrated increased clearance of globotriaosylceramide (GL-3) from the endothelial cells of the kidney, heart, and skin among treated subjects [Eng et al 2001].
- A Phase IV extension study showed that the risk of major clinical events (a combination of death, myocardial infarction, stroke, development of ESRD, or a 33% increase in serum creatinine concentration) was reduced by 53% with agalsidase beta treatment after adjustment for baseline proteinuria (P=0.06) [Banikazemi et al 2007]. In a ten-year follow up of these individuals, with additional data from the Fabry Registry, Germain et al [2015] reported that 49 of 52 were alive and 42/52 (81%) did not experience any severe clinical events during the ten-year treatment interval. Disease progression was most likely to be observed in those individuals who initiated treatment after age 40 years and/or had advanced renal disease at baseline. A study of cardiac outcomes from the Fabry Registry of 115 males treated with agalsidase beta for at least two years reports that treated individuals fared better than 48 untreated males. Left ventricular mass fell at a slope of -3.6 g/year in 31 males aged 18-30 years but rose by 9.5 g/year in 15 males who were not treated [Germain et al 2013].
- The largest comparative study is the Canadian Fabry Disease initiative. Sirrs et al [2014] have reported five-year follow-up data on 362 subjects for a composite endpoint (death, neurologic or cardiovascular events, development of ESRD, or sustained increase in serum creatinine of 50% from baseline). Ninety-two of 178 individuals treated with ERT were randomly allocated to either agalsidase alfa or agalsidase beta. No differences were found with regard to the clinical efficacy of the two medications, and individuals who switched from agalsidase beta to agalsidase alfa during the time of Fabrazyme® shortage were stable. In comparison with the placebo group in the Banikazemi study individuals treated with ERT had a significant reduction in clinical events, which occurred at an older age. The eight-year follow-up data continued to suggest that the two medications are equivalent at their standard doses [M West, personal communication].
- Antibody formation has been reported with both agalsidase alfa and agalsidase beta in males, but not females [Linthorst et al 2005, Wilcox et al 2012]. Lenders et al [2015] reported that 40% of 68 males with Fabry disease on ERT have evidence of serum-mediated inhibition of agalsidase activity. They further reported that inhibition-positive individuals have worse clinical outcomes and higher levels of lyso-Gb3 than inhibition-negative individuals. There appeared to be no difference between agalsidase alfa and beta with regard to the development of serum inhibitors. The impact of antibody formation on the overall efficacy of treatment is currently unknown.
- During 2009-2012, a shortage of agalsidase beta resulted in the substitution of agalsidase alfa for agalsidase beta in several cohorts of affected individuals. Reports thus far have not indicated any significant difference in clinical parameters as a result of this transition [Smid et al 2011, Tsuboi & Yamamoto 2012, Pisani et al 2013, Goker-Alpan et al 2016].
- Lubanda et al 45 have shown in a small study of 21 individuals that those who have been ‘stabilized’ with agalsidase beta at 1 g/kg can thereafter be safely treated with a maintenance dose of 0.3 g/kg every other week. A study of lower-dose agalsidase beta has been conducted in children 46; it will be interesting to observe if lower doses of agalsidase beta are equally efficacious in children.
There is an emerging consensus that enzyme replacement therapy has, at best, a limited impact on the long-term outcome of Fabry disease. Studies of consecutive affected persons from individual centers suggest that cardiac, renal, and cerebrovascular outcomes are comparable among treated and untreated cohorts 47. A recent Cochrane review has also highlighted the generally poor quality of evidence in favor of enzyme replacement therapy for Fabry disease.
Despite these emerging data, a panel of physician experts have recommended that enzyme replacement therapy be initiated as early as possible in all males with Fabry disease, including children and those with end stage renal disease undergoing dialysis and renal transplantation, and in heterozygous females with significant disease 48 because all are at high risk for cardiac, cerebrovascular, and neurologic complications including transient ischemic attacks and strokes. The treatment initiation guidelines from a group of European physicians are generally more conservative 49. They emphasize the need to start enzyme replacement therapy before the advent of irreversible complications and suggest that initiation of enzyme replacement therapy after irreversible organ damage has occurred is to be avoided. Enzyme replacement therapy should be discontinued if it is making no impact on organ function in an individual; and compliance should be closely monitored.
Prevention of Secondary Complications
The prophylaxis for renovascular disease, ischemic heart disease, and cerebrovascular disease in persons with Fabry disease is the same as for the general population.
- Proteinuria/microalbuminuria should be minimized with ACE inhibitors/angiotensin receptor blockers (ARBs) 50; blood pressure control optimized; and cholesterol normalized 51.
- Aspirin and other anti-platelet agents such as clopidogrel may be recommended for the prophylaxis of stroke.
- Although evidence as to the effect on long-term outcomes is lacking, use of aspirin and lipid-lowering agents and optimal blood pressure control are recommended in persons with symptoms of cardiac ischemia 8.
The role of enzyme replacement therapy in the long-term prophylaxis of renal, cardiac, and CNS manifestations is unproven; however, on the basis of stabilization of organ function in persons with more advanced disease, some have suggested the initiation of enzyme replacement therapy in early disease stages: at first sign of disease manifestations in boys; at age 12-13 years in asymptomatic boys; and at the time of diagnosis in adult males 8. Studies indicate that late initiation of therapy when renal or cardiac manifestations are significant is associated with less effect than initiation earlier in the disease course 52.
Surveillance
The following are appropriate:
- Annual or more frequent renal function studies
- Annual cardiology evaluation
- Annual audiology evaluation
- Biennial brain MRI/MRA
Agents/Circumstances to Avoid
The obstructive lung disease, which has been documented in older hemizygous males and heterozygous females, is more severe in smokers; therefore, affected individuals should be discouraged from smoking.
Amiodarone has been reported to induce cellular and biochemical changes resulting in a phenocopy in particular of the keratopathy of Fabry disease 53. Given potential effects on cellular levels of alpha-galactosidase A enzyme activity, it has been contraindicated in persons with Fabry disease. However, little evidence of a detrimental effect in this specific group exists and the relative benefit in individuals with cardiac arrhythmia should be considered.
- Mehta A, Hughes DA. Fabry Disease. 2002 Aug 5 [Updated 2017 Jan 5]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1292[↩][↩][↩][↩][↩][↩]
- Fabry disease. https://ghr.nlm.nih.gov/condition/fabry-disease[↩]
- Liao HC, Chiang CC, Niu DM, Wang CH, Kao SM, Tsai FJ, Huang YH, Liu HC, Huang CK, Gao HJ, Yang CF, Chan MJ, Lin WD, Chen YJ. Detecting multiple lysosomal storage diseases by tandem mass spectrometry–a national newborn screening program in Taiwan. Clin Chim Acta. 2014;431:80-86.[↩][↩]
- Desnick R, Ioannou Y, Eng C. Alpha-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease.8th ed. New York, NY: McGraw-Hill; 2001:3733-3774.[↩]
- Germain DP, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007;18:1547-1557.[↩]
- Fabry Disease. https://rarediseases.org/rare-diseases/fabry-disease/[↩][↩]
- Elliott P, et al. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson-Fabry Disease survey. Heart. 2011;97:1957-1960.[↩]
- Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, Bultas J, Lee P, Sims K, Brodie SE, Pastores GM, Strotmann JM, Wilcox WR. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539–48.[↩][↩][↩]
- Germain DP, Charrow J, Desnick RJ, Guffon N, Kempf J, Lachmann RH, Lemay R, Linthorst GE, Packman S, Scott CR, Waldek S, Warnock DG, Weinreb NJ, Wilcox WR. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52:353–8[↩]
- Ortiz A, Abiose A, Bichet DG, Cabrera G, Charrow J, Germain DP, Hopkin RJ, Jovanovic A, Linhart A, Maruti SS, Mauer M, Oliveira JP, Patel MR, Politei J, Waldek S, Wanner C, Yoo HW, Warnock DG. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: data from the Fabry Registry. J Med Genet. 2016;53:495–502.[↩]
- Fabry’s disease. The Lancet Volume 378, ISSUE 9798, P1254, October 01, 2011 https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(11)60473-X/fulltext[↩]
- Sodi A, Ioannidis AS, Mehta A, et al. Ocular manifestations of Fabry’s disease: data from the Fabry Outcome Survey. British Journal of Ophthalmology 2007;91:210-214. http://dx.doi.org/10.1136/bjo.2006.100602[↩]
- Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med. 2009;11:790–6[↩]
- Mehta A, Clarke JT, Giugliani R, Elliott P, Linhart A, Beck M, Sunder-Plassmann G, Investigators FOS. Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. J Med Genet. 2009;46:548–52[↩]
- Hoffmann B, Schwarz M, Mehta A, Keshav S., Fabry Outcome Survey European Investigators. Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007;5:1447–53.[↩]
- Kampmann C, Baehner FA, Whybra C, Bajbouj M, Baron K, Knuf M, Wiethoff CM, Trübel H, Beck M. The right ventricle in Fabry disease. Acta Paediatr Suppl. 2005;94:15–8.[↩]
- Pieroni M, Chimenti C, De Cobelli F, Morgante E, Del Maschio A, Gaudio C, Russo MA, Frustaci A. Fabry’s disease cardiomyopathy: echocardiographic detection of endomyocardial glycosphingolipid compartmentalization. J Am Coll Cardiol. 2006;47:1663–71.[↩]
- Weidemann F, Breunig F, Beer M, Sandstede J, Stork S, Voelker W, Ertl G, Knoll A, Wanner C, Strotmann JM. The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. Eur Heart J. 2005;26:1221–7.[↩]
- Sado DM, White SK, Piechnik SK, Banypersad SM, Treibel T, Captur G, Fontana M, Maestrini V, Flett AS, Robson MD, Lachmann RH, Murphy E, Mehta A, Hughes D, Neubauer S, Elliott PM, Moon JC. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging. 2013 May 1;6(3):392–8.[↩]
- Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, Elliott PM. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28:1228–35[↩]
- Kleinert J, Dehout F, Schwarting A, de Lorenzo AG, Ricci R, Kampmann C, Beck M, Ramaswami U, Linhart A, Gal A, Houge G, Widmer U, Mehta A, Sunder-Plassmann G. Prevalence of uncontrolled hypertension in patients with Fabry disease. Am J Hypertens. 2006;19:782–7.[↩]
- Politei JM, Capizzano AA. Magnetic resonance image findings in 5 young patients with Fabry disease. Neurologist. 2006;12:103–5.[↩]
- Ginsberg L, Manara R, Valentine AR, Kendall B, Burlina AP. Magnetic resonance imaging changes in Fabry disease. Acta Paediatr Suppl. 2006;95:57–62.[↩]
- Rolfs A, Bottcher T, Zschiesche M, Morris P, Winchester B, Bauer P, Walter U, Mix E, Lohr M, Harzer K, Strauss U, Pahnke J, Grossmann A, Benecke R. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet. 2005;366:1794–6.[↩]
- Rolfs A, Fazekas F, Grittner U, Dichgans M, Martus P, Holzhausen M, Böttcher T, Heuschmann PU, Tatlisumak T, Tanislav C, Jungehulsing GJ, Giese AK, Putaala J, Huber R, Bodechtel U, Lichy C, Enzinger C, Schmidt R, Hennerici MG, Kaps M, Kessler C, Lackner K, Paschke E, Meyer W, Mascher H, Riess O, Kolodny E, Norrving B. Stroke in Young Fabry Patients (sifap) Investigators. Acute cerebrovascular disease in the young: the Stroke in Young Fabry Patients Study. Stroke. 2013;44:340–9.[↩]
- Löhle M, Hughes D, Milligan A, Richfield L, Reichmann H, Mehta A, Schapira AH. Clinical prodromes of neurodegeneration in Anderson-Fabry disease. Neurology. 2015;84:1454–64.[↩]
- Magage S, Lubanda JC, Susa Z, Bultas J, Karetova D, Dobrovolny R, Hrebicek M, Germain DP, Linhart A. Natural history of the respiratory involvement in Anderson-Fabry disease. J Inherit Metab Dis. 2007;30:790–9[↩]
- Odler B, Cseh Á, Constantin T, Fekete G, Losonczy G, Tamási L, Benke K, Szilveszter B, Müller V. Long time enzyme replacement therapy stabilizes obstructive lung disease and alters peripheral immune cell subsets in Fabry patients. Clin Respir J. 2017;11:942–50.[↩]
- Hegemann S, Hajioff D, Conti G, Beck M, Sunder-Plassmann G, Widmer U, Mehta A, Keilmann A. Hearing loss in Fabry disease: data from the Fabry Outcome Survey. Eur J Clin Invest. 2006;36:654–62.[↩]
- Cole AL, Lee PJ, Hughes DA, Deegan PB, Waldek S, Lachmann RH. Depression in adults with Fabry disease: a common and under-diagnosed problem. J Inherit Metab Dis. 2007;30:943–51.[↩]
- Wijburg FA, Bénichou B, Bichet DG, Clarke LA, Dostalova G, Fainboim A, Fellgiebel A, Forcelini C, An Haack K, Hopkin RJ, Mauer M, Najafian B, Scott CR, Shankar SP, Thurberg BL, Tøndel C, Tylki-Szymańska A, Ramaswami U. Characterization of early disease status in treatment-naive male paediatric patients with Fabry disease enrolled in a randomized clinical trial. PLoS One; 2015; 8;10:e0124987[↩]
- Wilcox WR, et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112-128.[↩][↩]
- Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006;43:347–52[↩]
- Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, Jabbour F, Beldjord C, De Mazancourt P, Germain DP. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89:44–54[↩]
- Gupta S, Ries M, Kotsopoulos S, Schiffmann R. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore). 2005;84:261–8[↩]
- Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112–28.[↩]
- Desnick, R.J., Macaya, D., Rehm, H., Callis, T., and Doheny, D.: Targeted Sequencing of Over 4,000 Hypertrophic Cardiomyopathy (HCM) patients for mutations causing HCM and Fabry disease: HCM mutations frequent in patients with GLA later-onset mutations, Polymorphisms, and Variants. Molec. Genet. Metabol. 111:S36-S37, 2014.[↩]
- MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–75[↩]
- Sadek J, Shellhaas R, Camfield CS, Camfield PR, Burley J. Psychiatric findings in four female carriers of Fabry disease. Psychiatr Genet. 2004;14:199–201[↩]
- Dobrovolný R, Dvoráková L, Ledvinová J, Magage S, Bultas J, Lubanda JC, Poupetová H, Elleder M, Karetová D, Hrebícek M. Recurrence of Fabry disease as a result of paternal germline mosaicism for alpha-galactosidase a gene mutation. Am J Med Genet A. 2005;134A:84–7.[↩]
- Liu HC, Perrin A, Hsu TR, Yang CF, Lin HY, Yu WC, Niu DM. Age at first cardiac symptoms in Fabry disease: association with a Chinese hotspot Fabry mutation (IVS4+919G>A), classical Fabry mutations, and sex in a Taiwanese population from the Fabry Outcome Survey (FOS). JIMD Rep. 2015;22:107–13[↩]
- Ries M, Mengel E, Kutschke G, Kim KS, Birklein F, Krummenauer F, Beck M. Use of gabapentin to reduce chronic neuropathic pain in Fabry disease. J Inherit Metab Dis. 2003;26:413–4[↩]
- Warnock DG, Thomas CP, Vujkovac B, Campbell RC, Charrow J, Laney DA, Jackson LL, Wilcox WR, Wanner C. Antiproteinuric therapy and Fabry nephropathy: factors associated with preserved kidney function during agalsidase-beta therapy. J Med Genet. 2015;52:860–6.[↩]
- Martinez P, Aggio M, Rozenfeld P. High incidence of autoantibodies in Fabry disease patients. J Inherit Metab Dis. 2007;30:365–9.[↩]
- Lubanda JC, Anijalg E, Bzdúch V, Thurberg BL, Bénichou B, Tylki-Szymanska A. Evaluation of a low dose, after a standard therapeutic dose, of agalsidase beta during enzyme replacement therapy in patients with Fabry disease. Genet Med. 2009;11:256–64.[↩]
- Wijburg FA, Bénichou B, Bichet DG, Clarke LA, Dostalova G, Fainboim A, Fellgiebel A, Forcelini C. An Haack K, Hopkin RJ, Mauer M, Najafian B, Scott CR, Shankar SP, Thurberg BL, Tøndel C, Tylki-Szymańska A, Ramaswami U. Characterization of early disease status in treatment-naive male paediatric patients with Fabry disease enrolled in a randomized clinical trial. PLoS One. 2015;10:e0124987[↩]
- Rombach SM, Hollak CE, Linthorst GE, Dijkgraaf MG. Cost-effectiveness of enzyme replacement therapy for Fabry disease. Orphanet J Rare Dis. 2013;8:29[↩]
- Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, Bultas J, Lee P, Sims K, Brodie SE, Pastores GM, Strotmann JM, Wilcox WR. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539–48[↩]
- Biegstraaten M, Arngrímsson R, Barbey F, Boks L, Cecchi F, Deegan PB, Feldt-Rasmussen U, Geberhiwot T, Germain DP, Hendriksz C, Hughes DA, Kantola I, Karabul N, Lavery C, Linthorst GE, Mehta A, van de Mheen E, Oliveira JP, Parini R, Ramaswami U, Rudnicki M, Serra A, Sommer C, Sunder-Plassmann G, Svarstad E, Sweeb A, Terryn W, Tylki-Szymanska A, Tøndel C, Vujkovac B, Weidemann F, Wijburg FA, Woolfson P, Hollak CE. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. 2015;10:36[↩]
- Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol. 2007;18:2609–17[↩]
- Waldek S, Feriozzi S. Fabry nephropathy: a review – how can we optimize the management of Fabry nephropathy? BMC Nephrol. 2014;15:72[↩]
- Ortiz A, Abiose A, Bichet DG, Cabrera G, Charrow J, Germain DP, Hopkin RJ, Jovanovic A, Linhart A, Maruti SS, Mauer M, Oliveira JP, Patel MR, Politei J, Waldek S, Wanner C, Yoo HW, Warnock DG. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: data from the Fabry Registry. J Med Genet. 2016;53:495–502[↩]
- Whitley CB, Tsai MY, Heger JJ, Prystowsky EN, Zipes DP. Amiodarone phenocopy of Fabry’s keratopathy. JAMA. 1983;249:2177–8.[↩]





{kind=link}