
Contents
- Familial hemophagocytic lymphohistiocytosis
- Familial hemophagocytic lymphohistiocytosis types
- Familial hemophagocytic lymphohistiocytosis type 2
- Familial hemophagocytic lymphohistiocytosis type 3
- Familial hemophagocytic lymphohistiocytosis type 4
- Familial hemophagocytic lymphohistiocytosis type 5
- X-linked lymphoproliferative disease 1 (Duncan disease)
- X-linked lymphoproliferative syndrome type 2
- Immunodeficiencies associated with HLH and partial albinism
- Familial hemophagocytic lymphohistiocytosis causes
- Familial hemophagocytic lymphohistiocytosis pathophysiology
- Familial hemophagocytic lymphohistiocytosis signs and symptoms
- Familial hemophagocytic lymphohistiocytosis diagnosis
- Familial hemophagocytic lymphohistiocytosis treatment
- Familial hemophagocytic lymphohistiocytosis prognosis
- Familial hemophagocytic lymphohistiocytosis types
Familial hemophagocytic lymphohistiocytosis
Familial hemophagocytic lymphohistiocytosis (FHL) also called primary hemophagocytic lymphohistiocytosis is a rare, genetic inborn errors of immunity causing aggressive life-threatening intense immune activation syndrome in which your body makes too many activated immune cells called macrophages (histiocytes) and lymphocytes (T cells, natural killer cells and B cells) 1, 2, 3, 4, 5, 6. Hemophagocytic lymphohistiocytosis (HLH) is caused by an overactive and abnormal immune response resulting in an uncontrolled excessive inflammatory and ineffective immune response resulting in progressive multi-organ damage and potentially death if untreated 7, 8. In hemophagocytic lymphohistiocytosis (HLH), the immune system responds to a stimulus or ‘trigger’, often an infection, cancer or rheumatologic disease, but the response is ineffective and abnormal. This ineffective, abnormal response, causes a variety of signs and symptoms, which, if not treated, can potentially become life-threatening. Consequently, infants with HLH are born seemingly healthy, but upon encountering an immunological insult such as a viral infection (e.g., Epstein-Barr virus [EBV]), their lymphocytes cannot execute cytotoxic degranulation, leading to a positive feedback loop of massive inflammation without clearing the infectious trigger 9, 10, 11. Left untreated, familial hemophagocytic lymphohistiocytosis is fatal 12, 13.
Most children with familial hemophagocytic lymphohistiocytosis (FHL) are born healthy and develop the illness in the first 2 to 6 months of life 14, 15. Familial hemophagocytic lymphohistiocytosis (FHL) with onset in the neonatal period is rare 16. Up to 20% of patients present at more than 2 years of age, and in rare cases, patients with FHL remain asymptomatic until adulthood 17, 18, 19, 20.
Familial hemophagocytic lymphohistiocytosis (FHL) was first described in 1952 by Farquhar and Claireaux, who identified the disease in two fatal cases involving siblings, with the major findings being a marked increase of histiocytes in the bone marrow at the expense of blood-forming cells 21. Familial hemophagocytic lymphohistiocytosis (FHL) was shown to be more prevalent in consanguineous marriages (marriage between two blood-related individuals who are second cousins or closer), suggesting an autosomal recessive pattern of inheritance 22, 23, 24, 25, 26, 5, 27, 28.
Familial hemophagocytic lymphohistiocytosis (FHL) is caused by mutations to the PRF1, UNC13D, STX11, STXBP2, and other genes that are associated with the exocytosis of lytic granules from cytotoxic cells 28, 29. Familial hemophagocytic lymphohistiocytosis is classified into six different types based on genetic linkage analysis and chromosomal localization; five specific genetic defects have been identified, which account for approximately 90% of all patients 28. Familial hemophagocytic lymphohistiocytosis type 1 is due to as yet unidentified gene defect located on chromosome 9. Familial hemophagocytic lymphohistiocytosis type 2 is caused by mutations in the perforin (PRF1) gene, familial hemophagocytic lymphohistiocytosis type 3 by mutations in the Munc-13–4 (UNC13D) gene, familial hemophagocytic lymphohistiocytosis type 4 by mutations in the syntaxin 11 (STX11) gene and familial hemophagocytic lymphohistiocytosis type 5 due to mutations in the gene encoding syntaxin binding protein 2 (STXBP-2) 28. Twenty to fifty per cent of FHL cases worldwide are caused by mutations in perforin (PRF1) gene. Perforin is a critical protein for cytotoxicity mediated by granules present in NK cells and cytotoxic T lymphocytes, and it is required to fight viral infections as well as to keep the immune system, especially histiocytes, under control. Furtjermore, at least three genetic diseases other than FHL have been associated with the hemophagocytic syndrome, including mutations in SH2D1A (X-linked lymphoproliferative disease) 30, 31, mutations in CHS1 (Chediak-Higashi disease) 32 and mutations in RAB27A gene (Griscelli syndrome type-2) 32.
The reported incidence of FHL varies among different studies, probably reflecting different prevalence within different ethnic groups and the higher index of suspicion and improved diagnosis in more recent studies. Familial hemophagocytic lymphohistiocytosis (FHL) has been reported to be 0·12 to 1 cases per 100,000 children per year in Sweden 33, whereas it was found to be 0·342 per 100,000 in Japan 34. The frequency of familial hemophagocytic lymphohistiocytosis (FHL) among hospitalized patients in Turkey is 7·5 per 10,000, which is due most probably to the high rate of consanguineous marriage in Turkish pedigrees 35.
HLH should be suspected in all patients with prolonged high-grade fever associated with abnormally large spleen (splenomegaly) and multiple organ involvement 36. The clinical spectrum of hemophagocytic lymphohistiocytosis disease is wide, ranging from mild organ dysfunction to multiorgan failure requiring intensive care. Central nervous system (CNS) involvement is frequent and often severe, even though it is not included in the official diagnostic criteria 36.
The typical patient is an infant under 1 year of age, ill-appearing (toxic aspect), occasionally with a critical sepsis-like aspect 4. A younger age at onset suggests an underlying genetic basis, as seen in familial hemophagocytic lymphohistiocytosis (F-HLH) or in HLH forms arising from genetic primary immunodeficiencies; however, familial HLH can present at any age, including adulthood 37.
Progressive splenomegaly (abnormally large spleen) is typically observed in patients, and can be associated with liver involvement, neurological signs, respiratory and kidney failure 38. Skin rashes, reddening of the skin because of inflammation (erythroderma), swelling (edema) or tiny spots on the skin due to bleeding under the skin (petechiae) have been reported 38. Atypical forms, usually seen in children older than 1 year, include isolated fever of unknown origin (FUOs), isolated central nervous system (CNS) involvement 39 or isolated acute liver failure 40, 41. Enlarged lymph node (lymphadenopathy) is uncommon in patients and indicates a potential underlying lymphoma 42.
Typical laboratory findings include cytopenias (low levels of red blood cells [anemia], white blood cells [leukopenia] or platelets [thrombocytopenia]), high level of triglycerides in blood (hypertriglyceridemia), low fibrinogen in blood (hypofibrinogenemia) are suggestive of HLH in the context of general inflammation and hyperferritinemia. Liver function tests are frequently altered 36. Leukocytosis (elevated white blood cell (WBC) count) is not typical of HLH except in HLH-associated with defined rheumatological conditions or macrophage activation syndrome-HLH (MAS-HLH) 36. Hemophagocytosis describes the pathognomonic findings where highly activated macrophages taking up different cells including lymphocytes, erythrocytes, leukocytes, and platelets in different tissues, producing excessive cytokines and an uncontrolled inflammatory reaction 7, 36, 43.
To date, the only curative treatment available for familial hemophagocytic lymphohistiocytosis is an allogenic hematopoietic stem cell transplantation (HSCT) 36, 44. However, inflammation must be adequately controlled before undergoing hematopoietic stem cell transplantation (HSCT), and despite recommendations for intensification of therapy in the HLH-2004 international treatment study, the reported pre-HSCT mortality was still high at 19% 13. Survival rates vary depending on the conditioning protocol 3, 45, 46, 47, 48, 49, 50, 51, 52, 53. Thus far, results from clinical trial NCT01998633 suggest that reduced intensity conditioning can improve overall 1-year survival of HLH patients to 82.4%, although larger and longer term studies will be needed to follow-up on these results 54. For patients who do receive hematopoietic stem cell transplantation (HSCT), the 5-year survival rate has improved from 54% to 61% 3, 55, 52, 53.
Figure 1. Familial hemophagocytic lymphohistiocytosis pathogenesis
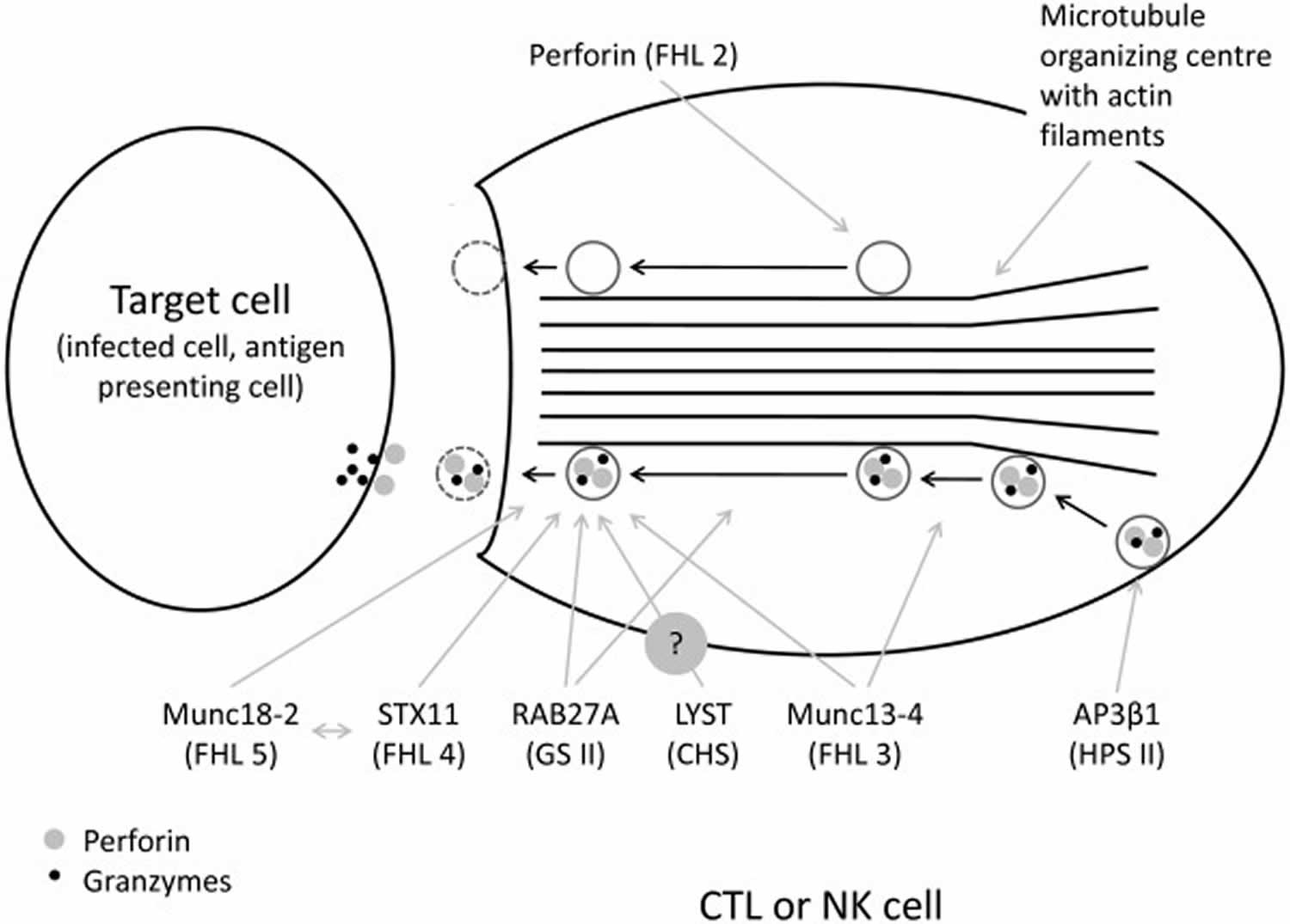
Footnote: The function of LYST gene, probably important for correct size and function of lytic granules, is not entirely understood. Note the empty granula in perforin deficiency. Hemophagocytic lymphohistiocytosis (HLH) is thought to develop due to abnormal reciprocal activation of mononuclear phagocytes (MNPs; monocytes, macrophages, and dendritic cells) and type 1 lymphocytes (NK cells and Th1, CD8+ cytotoxic T lymphocyte, and NKT cells). T cells with normal cytotoxic granules release them to induce mononuclear phagocyte apoptosis and terminate the synapse, whereas impaired or perforin-deficient granules (gray) cannot terminate mononuclear phagocyte activation. Immune synapse prolongation and excess IL-18 (from mononuclear phagocyte and/or epithelial sources) both amplify production of lymphocyte cytokines like IFN-γ, which in turn further activates mononuclear phagocytes and promotes hemophagocytosis and release of HLH biomarkers like ferritin, CXCL9, and interleukin-18-binding protein (IL-18BP). Activated lymphocytes upregulate the IL-2 receptor, which is cleaved by proteases released by activated mononuclear phagocytes. The absence of X-linked inhibitor of apoptosis protein (XIAP) may permit pathogenic inflammasome activation and lymphocyte apoptosis, while SLAM-associated protein (SAP) deficiency impairs restimulation-induced cell death (RICD) and, like CD27 and CD70 deficiency, prevents normal killing of EBV-infected B cells
Abbreviations: CHS = Chédiak Higashi syndrome; CTL = CD8+ cytotoxic T lymphocyte; FHL = familial hemophagocytic lymphohistiocytosis; GSII = Griscelli syndrome type 2; HPSII = Hermansky-Pudlak syndrome type 2; NK = natural killer.
[Source 15 ]Familial hemophagocytic lymphohistiocytosis types
Familial hemophagocytic lymphohistiocytosis (F-HLH) is subclassified based on the underlying genetic mutation (Table 1). Familial hemophagocytic lymphohistiocytosis is a genetically heterogeneous disorder caused by mutations in genes involved in the secretory lysosome-dependent exocytosis pathway 56. More than 60 years after the first FHL case was reported, five independent, FHL-causing loci have been identified and the underlying genetic defect has been described for four of them accounting for over 90% of familial hemophagocytic lymphohistiocytosis (Table 1) 57, 58, 59, 60, 61, 62, 63. They encode the proteins perforin, MUNC13-4, syntaxin-11 and MUNC18-2, all of which play a key role in lymphocyte cytotoxicity (Figure 1) 64.
Around 70% of familial hemophagocytic lymphohistiocytosis cases are explained by mutations in only two genes: PRF1 and UNC13D, which cause familial hemophagocytic lymphohistiocytosis type 2 and familial hemophagocytic lymphohistiocytosis type 3 subgroups, respectively 65. In addition to the described four subgroups of familial hemophagocytic lymphohistiocytosis, in which HLH is usually the primary manifestation, a few additional genetic conditions and other immune deficiencies may cause a clinical syndrome largely overlapping that of FHL but in which additional, distinctive clinical features occur.
Table 1. Familial hemophagocytic lymphohistiocytosis genetic causes and types
| HLH type/association | Gene or locus (protein) | HLH-related dysfunction | Unique clinical features |
|---|---|---|---|
| Familial HLH | |||
| Type 1 (F-HLH 1) | 9q21.3-q22 (unknown) | Unknown | N/A |
| Type 2 (F-HLH 2) | PRF1 (perforin) | Cytolytic pore formation | None |
| Type 3 (F-HLH 3) | UNC13D (Unc-13 homolog D) | Cytolytic vesicle priming | Increased incidence of central nervous system (CNS) disease |
| Type 4 (F-HLH 4) | STX11 (Syntaxin 11) | Cytolytic vesicle fusion | Mild, recurrent HLH, colitis |
| Type 5 (F-HLH 5) | STXBP2 (Syntaxin binding protein 2) | Cytolytic vesicle fusion | Colitis; hypogammaglobulinemia, and sensorineural hearing loss |
| Primary Immunodeficiency Syndromes | |||
| Chédiak-Higashi syndrome | LYST (Lysosomal trafficking regulator) | Cytolytic vesicle trafficking | Partial oculocutaneous albinism, recurrent pyogenic infections, and bleeding tendency |
| Hermansky-Pudlak syndrome type 2 | AP3B1 (Adaptor Related Protein Complex 3 Subunit Beta 1) | Cytolytic vesicle trafficking | Partial albinism; immunodeficiency, bleeding tendency, and platelet storage pool deficiency |
| Griscelli syndrome type 2 | RAB27A (Rab-27A protein) | Cytolytic vesicle docking | Partial albinism and silvery grey hair |
| Epstein-Barr virus (EBV) triggered | |||
| X-linked lymphoproliferative disorder type 1 (XLP1) | SH2D1A (SH2 Domain Containing 1A) | Defective killing of EBV-infected B cells | Severe HLH; Hypogammaglobulinemia, and lymphoma |
| X-linked lymphoproliferative disorder type 2 (XLP2) | XIAP (X-linked inhibitor of apoptosis protein) | Increased inhibition of apoptosis in EBV-infected cells (gain of function) | Mild, recurrent HLH (may be triggered by non-EBV viruses) |
| IL-2-inducible T cell kinase deficiency | ITK (IL2 inducible T cell kinase) | Impaired proliferation of EBV-specific T cells | Hodgkin lymphoma |
| CD27 deficiency | CD27 | Impaired proliferation of EBV-specific T cells | Combined immunodeficiency |
| X-linked immunodeficiency with magnesium defect (XMEM) | MAGT1 (Magnesium transporter 1) | Impaired proliferation of EBV-specific T cells | Combined immunodeficiency, chronic viral infections, and lymphoma |
| Inflammasome activation | |||
| Autoinflammatory disease | NLRC4 (NLR Family CARD Domain Containing 4) | Increased inflammasome activation (gain of function) | Mild and recurrent HLH |
Abbreviations: CNS = central nervous system; EBV = Epstein-Barr virus.
[Source 66, 67 ]| Primary HLH | Gene | Protein | Pathophysiology | Functional testing |
|---|---|---|---|---|
| Familial HLH (FHL) | ||||
| FHL-1 | Unknown | Unknown | ||
| FHL-2 | PFR1 | Perforin | Lack of perforin expression in lytic granules | Perforin expression |
| FHL-3 | UNC13D | Munc13-4 | Deficiency in fusion of lytic granule with plasma membrane | Degranulation |
| FHL-4 | STX11 | Syntaxin11 | Deficiency in fusion of lytic granule with plasma membrane | Degranulation |
| FHL-5 | STXBP2 | Munc18-2 | Deficiency in fusion of lytic granule with plasma membrane | Degranulation |
| Other immunodeficiency syndromes with defect in degranulation | ||||
| GS-II | RAB27A | Rab27a | Deficiency in docking of lytic granule to the plasma membrane | Degranulation hair microscopy |
| CHS | LYST | Lyst | Defect in maturation of vesicles into secretory cytotoxic granules | Degranulation hair microscopy |
| — | ||||
| Other inborn errors of immunity | Gene | Protein | Pathophysiology | Functional testing |
| Immunodeficiency syndromes with HLH as a frequent manifestation | ||||
| XLP-1 | SH2D1A | SAP | Defective killing of EBV infected B-cells by CD8 and NK cells | SAP expression |
| XLP-2 | BIRC4 | XIAP | Impaired inhibition of inflammasome activity | XIAP expression L18MDP assay |
| TIM3 deficiency | HAVCR2 | TIM3 | Persistent T cell activation and increased production of inflammatory cytokines | TIM3 expression |
| Immunodeficiency syndromes with HLH as an occasional manifestation | ||||
| Chronic granulomatous disease (CGD) | CYBB, CYBA, NCF1, NCF2, NCF4 | Components of NADPH oxidase | Excessive inflammatory responses due to altered inflammasome regulation by NADPH oxidase? | Oxidative Burst |
| (S)CID | >50 genes | various | Lack of pathogen control | Lymphocyte phenotyping |
| Wiskott-Aldrich syndrome | WAS | WASP | Lack of pathogen control Impaired cytoskeleton-inflammasome interaction? | WASP expression (FACS) |
| CD27 deficiency | CD27 | CD27 | Impaired co-stimulation of T cells Lack of EBV control | CD27 expression |
| ITK deficiency | ITK | ITK | Impaired TCR mediated signaling Lack of EBV control | ITK expression |
| IFNγ receptor deficiency | IFNGR1 IFNGR2 | IFN-gamma receptor | Lack of pathogen control (mycobacteria, salmonella) | STAT1 phosphorylation |
| ALPS | FAS (het) FASLG | FAS FASLG | Defects in Fas ligand-mediated elimination of activated lymphocytes | TCR DNT Vitamin B12, soluble FasL |
| Autoinflammatory diseases with HLH as a frequent manifestation | ||||
| NLRC4 gain of function | NLRC4 (het) | NLRC4 | Constitutive inflammasome activation IL-1β/IL-18 production | Genetic testing |
| CDC42 mutations | CDC42 (het) | CDC42 | Impaired cytoskeleton-inflammasome interaction? | Genetic testing |
Familial hemophagocytic lymphohistiocytosis type 2
Familial hemophagocytic lymphohistiocytosis type 2 accounts for 20 to 50% of all FHL cases, depending on the cohort studied 68, 69, 70. Mutations in the perforin gene (PRF1) are responsible for familial hemophagocytic lymphohistiocytosis type 2 69, 70, 71. Perforin (PRF1) gene has three exons: exon 2 and 3 code a 555 amino-acid polypeptide. Over 120 different mutations have been identified to date: 101 missense/non-sense mutations and 21 deletion/insertion mutations 69, 71, 72, 73, 74. Since the pathogenic role of some mutations, especially some single amino-acid substitutions, is difficult to assess, their functional characterization is still required 56. In a recent comprehensive in silico analysis of 76 missense mutations in PRF1, An et al. 75 used a structural approach to explain the effect of a mutation on the ability of perforin to oligomerize, thereby offering an explanation for the observed defect in cytotoxicity in familial hemophagocytic lymphohistiocytosis type 2 patients.
Accumulation of a sufficient number of cases helped to establish connections between specific mutations and particular ethnic groups: c.1122G > A (p.W374X) was found to have a high incidence in Turkish patients 70; mutation c.50delT (p.L17FsX) is very frequent in patients of African-American origin 76; while the c.1090-1091delCT (p.L364fsX) mutation has only been identified in Japanese patients 71.
Mutation c.272C > T (p.A91V) has a particularly high frequency in Southern European population, ranging from 2.5 up to 10% 77, 78. It seems to be present at a very low frequency in African-American subjects and Sub-Saharan Africans, with no reported cases of the polymorphism in Japan, supporting the concept of a Mediterranean origin of the mutation. Although its impact on the protein structure and function has been documented 79, 80, its pathogenic role in FHL has been considered controversial. It is now clear that patients with Familial hemophagocytic lymphohistiocytosis type 2 frequently show A91V mutation in combination with another pathogenic mutation; association of A91V with another missense/hypomorphic mutation frequently results in a late-onset of familial hemophagocytic lymphohistiocytosis type 2 81. Interestingly, in addition to the 106 mutations described in Familial hemophagocytic lymphohistiocytosis type 2 patients, other mutations have been associated with different phenotypes.
Familial hemophagocytic lymphohistiocytosis type 3
Familial hemophagocytic lymphohistiocytosis type 3 is caused by mutations in the UNC13D gene, which encodes the 1,090 amino-acids-long protein Munc13-4. Munc13-4 is involved in the priming of secretory granules and their fusion with the plasma membrane, and its loss-of-function consequently impairs the release of perforin and granzyme into the synaptic cleft 82.
Familial hemophagocytic lymphohistiocytosis type 3 covers between 30 and 40% of FHL patients, based on different geographic areas and ethnic groups 56. The clinical picture of familial hemophagocytic lymphohistiocytosis type 3 patients is undistinguishable from that of familial hemophagocytic lymphohistiocytosis type 2 patients. However, evaluation of patient cytotoxic T lymphocyte (CTL) and NK cells by flow-cytometry showed a clear difference: while familial hemophagocytic lymphohistiocytosis type 2 patients have no perforin but show no degranulation defect, cells from familial hemophagocytic lymphohistiocytosis type 3 patients have normal perforin expression but reduced to absent degranulation 83.
UNC13D consists of 32 coding exons. To date at least 112 different mutations in UNC13D have been reported as a cause of Familial hemophagocytic lymphohistiocytosis type 3: 60 missense/non-sense, 25 splicing/regulatory, 25 deletion/insertion mutations, as well as 2 complex gene rearrangements 82, 83, 84, 85. After some initial difficulties in recognizing biallelic mutations in patients with familial hemophagocytic lymphohistiocytosis type 3, it became evident that not only exonic mutations, but also variations outside exons and splice sites are a common cause of familial hemophagocytic lymphohistiocytosis type 3 86. Since then, additional contribution confirmed this issue. In 2011, Meeths et al. 87 described two novel mutations frequently occurring in Northern European populations: the deep intronic mutation c.118-308G > A selectively impairs UNC13D transcription in lymphocytes, thus abolishing Munc13-4 expression while the 253-kb inversion affects the 3′-end of the transcript, also abolishing Munc13-4 expression. Another deep intronic mutation, c.118-307G > A, was recently reported in a Chinese patient and documented to impair UNC13D transcription, possibly by disrupting a transcription factor binding-site or enhancer element 88. Two mutations have been described in specific populations: the c.1596 + 1G > C mutation is described as the most common UNC13D mutation in Japan 86 while mutation c.754-1G > C is predominantly found in Korean patients 89. Altogether, the mutation analysis of patients with familial hemophagocytic lymphohistiocytosis type 3 turned out to be far more engaging than that of patients with familial hemophagocytic lymphohistiocytosis type 2.
Recently in a genotype–phenotype study of 84 patients with familial hemophagocytic lymphohistiocytosis type 3 from Italy, Germany, and Sweden, Sieni et al. 85 described that central nervous system (CNS) involvement is more common in patients with familial hemophagocytic lymphohistiocytosis type 3 than with familial hemophagocytic lymphohistiocytosis type 2. Moreover, the combination of fever, splenomegaly, thrombocytopenia, and hyperferritinemia appears to be the most easily and frequently recognized clinical pattern in familial hemophagocytic lymphohistiocytosis type 3, and in association with a defective granule release assay may lead to clinical suspicion of familial hemophagocytic lymphohistiocytosis type 3 85.
While familial hemophagocytic lymphohistiocytosis type 2 and familial hemophagocytic lymphohistiocytosis type 3 subgroups account for the majority of patients with FHL, additional genetic subgroups have been progressively identified.
Familial hemophagocytic lymphohistiocytosis type 4
Familial hemophagocytic lymphohistiocytosis type 4 is caused by mutations in the STX11 gene 90. The STX11 gene consists of two exons and encodes the 287 amino-acid-long SNARE protein Syntaxin 11 (Stx11).
Although original reports of familial hemophagocytic lymphohistiocytosis type 4 were restricted to families of Turkish or Kurdish origin, more recently patients of different origins have been identified with a defect in STX11. Despite the initial clustering of cases, at least 12 different STX11 mutations have been described to date: 5 missense/non-sense mutations, 5 small deletions, 1 small deletion, and 1 gross deletion 91, 92, 93. Patients with familial hemophagocytic lymphohistiocytosis type 4 seem to have a later onset and a less severe clinical presentation of the disease compared to familial hemophagocytic lymphohistiocytosis type 2 and familial hemophagocytic lymphohistiocytosis type 3 94. Mutations in STX11 have never been associated with variant phenotypes, different from familial hemophagocytic lymphohistiocytosis type 4.
Recently, Sepulveda et al. 95 tried to elucidate the role of STX11 mutations in the pathogenesis of FHL. They generated a Stx11-deficient (Stx11−/−) murine model that faithfully reproduced the manifestations of HLH and represented a suitable model for studying familial hemophagocytic lymphohistiocytosis type 4 in vivo and the role of Stx11 in vitro. By comparing the severity of HLH in Stx11−/− mice with that observed in Rab27a−/− and Prf1−/− mice, they established a correlation between the murine mutants and the age at HLH onset in their human counterparts 95. Furthermore, in a recent report the STX11 L58P mutation revealed that both the N-terminus and Habc domain of Stx11 are required for binding to Munc18-2, implying similarity to the dynamic binary binding of neuronal syntaxin 1 to Munc18-1 96.
Familial hemophagocytic lymphohistiocytosis type 5
Familial hemophagocytic lymphohistiocytosis type 5 is due to mutations in STXBP2 (also named MUNC18-2) 97, 98 and has been reported to account for up to 20% of cases with FHL in the German series 99. Since 2010, 40 different mutations of STXBP2 have been described: 15 missense/non-sense, 10 splicing/regulatory, and 15 deletion/insertion mutations 98, 100, 101, 102, 103. Familial hemophagocytic lymphohistiocytosis type 5 does not appear to be restricted to a specific geographic region.
In contrast to what was observed for the comparison between familial hemophagocytic lymphohistiocytosis type 2 and familial hemophagocytic lymphohistiocytosis type 3, some clinical presentations of familial hemophagocytic lymphohistiocytosis type 5 seem to be different from other classical manifestations of FHL. Gastrointestinal symptoms, such as chronic diarrhea, gastro-esophageal reflux, and abdominal pain are present in a significant number of patients. Renal tubular dysfunction was also observed in one patient. This could be explained by an impaired expression and function of Munc18-2/STXBP2 protein in cells other than cytotoxic lymphocytes, including intestinal and renal epithelium 104. The defect caused by insufficient function of STXBP2 protein in the neutrophils is associated with defective mobilization of the granules. As a result, the cell is unable to kill bacteria; insufficient clearance of E. coli might be one or the main reason for the frequency of gastrointestinal symptoms in patients with familial hemophagocytic lymphohistiocytosis type 5 99, 103, 104, 105.
Since platelets contain syntaxin-binding proteins with non-redundant functional roles, platelets from familial hemophagocytic lymphohistiocytosis type 5 patients have defective secretion, with decreased Munc18-2 and Stx11 levels. These data demonstrated a key role for Munc18-2, perhaps as a limiting factor, in platelet exocytosis, suggesting that it regulates Stx11 106. These data together with those of Pagel et al. 99 suggest that, although bleeding histories may be too variable to be a sufficient diagnostic criteria, platelet function assays may be worth investigating in patients with FHL.
X-linked lymphoproliferative disease 1 (Duncan disease)
X-linked lymphoproliferative disease 1 (XLP-1) is a rare congenital immunodeficiency caused by mutations in SH2D1A (Xq25), the gene encoding the signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) 107, 108.
In immune-competent individuals Epstein–Barr virus (EBV) causes infectious mononucleosis, a common, usually self-limited disease. In X-linked lymphoproliferative disease 1 (XLP-1), the lack (or dysfunction) of SLAM-associated protein (SAP) causes the selective inability to control infection by EBV, a γ-herpes virus that infects B-cells 109, 110, 111, 112. Several immunological defects have been identified, including defective NK and CD8+ T-cell-mediated cytolytic responses against EBV-infected cells, which lead to B-cell accumulation and persistence of reactive inflammatory responses 113. In the absence of SAP, 2B4 receptor (member of SLAM family), when engaged by its ligand CD48, delivers inhibitory instead of activating signals 110. It has been recently demonstrated that in X-linked lymphoproliferative disease 1 (XLP-1) NK cells the co-engagement of 2B4 with different activating receptors inhibits NCR, CD16, and activating KIRs, characterized by ITAM-dependent signaling pathways. In contrast, the 2B4 dysfunction does not affect the activity of DNAM-1 and NKG2D triggering receptors. Thus, while CD48+ B-EBV and lymphoma B-cells devoid of NKG2D and DNAM-1 ligands were resistant to lysis, the preferential usage of these receptors allowed XLP-1 NK cells to kill lymphomas that expressed sufficient amounts of the specific ligands 114. Better knowledge of the underlying dysfunction could be turned into a diagnostic tool. Patients with XLP-1 may present with different phenotypes: fulminant mononucleosis, B-cell lymphoma, lymphoproliferation, and dysgammaglobulinemia 113, 115, 116, but also with hemophagocytic lymphohistiocytosis 117. Thus, differential diagnosis is relevant. To this issue, immunological screening for intra-cytoplasmic SAP expression and rapid assays to examine 2B4 receptor function, which is inhibitory instead of activating in SH2D1A mutated patients 114, 110, may be applied.
In the literature, 100 SH2D1A mutations were found: 46 missense/non-sense, 14 splicing, 2 regulatory mutations, 9 small deletions, 6 small insertions, and 23 gross deletions 115, 118. Interestingly, this gene is characterized by a high number of deletions including the entire gene. Intronic mutations have also been described affecting SH2D1A transcription but not mRNA splicing, and leading to markedly reduced level of SAP protein 118. Therefore, the strategy of mutation analysis of SH2D1A gene must be designed to include these possible variants.
X-linked lymphoproliferative syndrome type 2
A subset of patients with an X-linked lymphoproliferative-like phenotype was recently found to have mutations in BIRC4, the gene encoding the X-linked inhibitor of apoptosis protein (XIAP) and has been linked to another subgroup, named X-linked lymphoproliferative syndrome type 2 (XLP-2) 119.
X-linked inhibitor of apoptosis protein is an essential ubiquitin ligase for pro-inflammatory signaling downstream of the nucleotide-binding oligomerization domain containing (NOD)-1 and -2 pattern recognition receptors. Recently, the X-linked inhibitor of apoptosis protein (XIAP) baculovirus IAP repeat (BIR2) domain was recognized as a hotspot for missense mutations in X-linked lymphoproliferative syndrome type 2 (XLP-2). XLP-2-BIR2 mutations severely impair NOD-1/2-dependent immune signaling in primary cells from X-linked lymphoproliferative syndrome type 2 (XLP-2) patients and in reconstituted XIAP deficient cell lines. XLP-2-BIR2 mutations abolish the XIAP–RIPK2 interaction resulting in impaired ubiquitylation of RIPK2 and recruitment of linear ubiquitin chain assembly complex (LUBAC) to the NOD-2-complex. These new findings document that impaired immune signaling in response to NOD-1/2 stimulation is a general defect in X-linked lymphoproliferative syndrome type 2 (XLP-2) and demonstrate that the XIAP BIR2–RIPK2 interaction might be even targeted pharmacologically to modulate inflammatory signaling 120.
In a comparison of the clinical phenotypes associated with X-linked lymphoproliferative disease 1 (XLP-1) and X-linked lymphoproliferative syndrome type 2 (XLP-2), EBV infection was the common trigger of HLH in 92% of XLP-1 and 83% of XLP-2. HLH (XLP-1, 55%; XLP-2, 76%) and hypogammaglobulinemia (XLP-1, 67%; XLP-2, 33%) occurred in both groups, although with different proportions. Survival rates and mean ages at the first HLH episode did not differ for both groups, but HLH was more severe with lethal outcome in X-linked lymphoproliferative disease 1 (XLP-1) 121. Only X-linked lymphoproliferative disease 1 (XLP-1) patients developed lymphomas while X-linked lymphoproliferative syndrome type 2 (XLP-2) patients preferentially displayed chronic hemorrhagic colitis, recurrent splenomegaly often associated with cytopenia and fever 121.
In a similar study, Marsh et al. 122 reported an early disease onset during infancy for X-linked lymphoproliferative syndrome type 2 (XLP-2)-linked HLH and a high relapse rate; however, this seemed to occur even in the absence of an EBV infection. Some X-linked lymphoproliferative syndrome type 2 (XLP-2) patients develop hypo/dysgammaglobulinemia resulting from humoral immune system derangement. Intriguingly, and in contrast to X-linked lymphoproliferative disease 1 (XLP-1), XLP-2 was never associated with common variable immunodeficiency 122.
In a third consortium review of 25 patients 123, the majority initially presented with manifestations other than HLH, such as Crohn-like bowel disease (n = 6), severe infectious mononucleosis (n = 4), isolated splenomegaly (n = 3), uveitis (n = 1), periodic fever (n = 1), fistulating skin abscesses (n = 1), and severe Giardia enteritis (n = 1). Subsequent manifestations included celiac-like disease, antibody deficiency, splenomegaly, and partial HLH. Screening by flow-cytometry identified 14 of 17 patients in this cohort 123. Given these clinical differences, XIAP deficiency must be considered in a wide range of clinical presentations. It has recently been suggested that XIAP deficiency would be better classified if defined as an X-linked subtype of FHL, rather than as a second type of X-linked lymphoproliferative disease 121.
To date, 41 mutations are known in BIRC4: 20 missense/non-sense, 2 splicing mutations, 2 regulatory mutations, 16 deletions/insertions, and 1 complex rearrangement 119, 121, 123, 124. The phenotypic differences may be the result of differences in the molecular basis of each disease. However, neither genotype, nor protein expression, nor results from cell death studies were clearly associated with the clinical phenotype.
Although HSCT remains the milestone for cure of FHL, some discrepancy recently emerged in the outcome of patients with X-linked lymphoproliferative syndrome type 2 (XLP-2). In an international survey of 19 patients, 7 received myeloablative (MAC) regimens, 1 received an intermediate-intensity regimen, and 11 received reduced intensity conditioning (RIC) regimens predominantly consisting of alemtuzumab, fludarabine, and melphalan. The probability of survival was very low in the MAC group, with all but one patient dying from transplantation-related toxicities (especially veno-occlusive disease and pulmonary hemorrhage); otherwise, 55% of those who received RIC survived at a median of 570 days after HSCT. The probability of surviving in the RIC was enhanced by disease inactivity at the time of HSCT. Based on these findings, MAC regimens should not be used for patients with XIAP deficiency. The reason may be connected with the loss of XIAP anti-apoptotic functions in X-linked lymphoproliferative syndrome type 2 (XLP-2) patients 125.
Immunodeficiencies associated with HLH and partial albinism
FHL associated with albinism is found as having abnormal lymphocyte cytotoxicity. Similarities were noted between molecular vesicle trafficking, especially pigment transport in the skin and hair. Moreover, the degranulation of platelets, mast cells, and perforin was shown to be impaired 15. At present three syndromes are known to cause HLH and manifest with partial albinism.
Chédiak–Higashi syndrome
Around 85% of Chédiak–Higashi syndrome patients develop HLH in the first decade of life. They show markedly defective cytotoxicity of both NK cells and CTL. The genetic defect is caused by mutations in the LYST gene (69), which encodes a 3,801 amino-acid protein. Each clinical manifestation of Chédiak–Higashi syndrome (albinism, bleeding tendency, recurrent bacterial infections, neurologic dysfunction, and HLH) (70, 71) is associated with a defect of a specific cell type and the formation of enlarged lysosomes in these cells.
The presence of giant inclusion bodies of lysosomal origin in a variety of granule-containing cells, including hematopoietic cells and melanocytes, has thus become the hallmark of the disease (72). This feature together with HLH and oculo-cutaneous albinism can address clinical suspicion toward Chédiak–Higashi syndrome and direct molecular analysis to LYST sequencing. In our revision of the available literature, a total of 56 mutations were found to be reported at the time of writing: 23 missense/non-sense, 4 splicing, 20 small deletions, 8 small insertions, and 1 gross deletion. As expected, disruptive mutations correlated with the severe form of the disease (73). The wider diffusion of mutation analysis, despite the big size of the gene, provided an increased number of reports from different geographic areas during the last few years, confirming that Chédiak–Higashi syndrome has no ethnic or geographic boundaries.
A rare neurologic disorder, named hereditary spastic paraplegia (HSP) and characterized by leg spasticity, weakness, hyperreflexia, and additional neurological symptoms, was recently reported in two adult siblings with hereditary spastic paraplegia and homozygous LYST pathogenic mutation. Large peroxidase-positive granules were observed in both patients’ granulocytes, while pigment deficiency, immune deficiency, and bleeding tendency were not observed. This example illustrates nicely how the clinical spectrum of Chédiak–Higashi syndrome may be much broader than recognized at present (74).
Griscelli syndrome type 2
Griscelli syndrome is a rare autosomal-recessive disorder that results in pigmentary dilution of the skin and hair, the presence of large clumps of pigment in hair shafts, and an accumulation of melanosomes in melanocytes and is characterized by partial oculo-cutaneous albinism and HLH 126. Among Griscelli syndrome subtypes, only patients with Griscelli syndrome type 2 (GS2) develop hemophagocytic lymphohistiocytosis (HLH) 56. Mutations causing Griscelli syndrome type 2 (GS2) were mapped to the RAB27A gene located on chromosome 15q15-21.1, which is composed of 5 coding exons and encodes for a 221 amino-acid protein that belongs to the superfamily of small Rab GTPase proteins 56. Cytotoxic T lymphocytes (CTLs) and NK cell activity defect results from the inability of cytotoxic granules to dock to the plasma membrane whereas hypopigmentation is accounted for by a defective release of melanosomes from melanocyte dendrites 56. Change in phagosomal function and antigen cross-presentation of Rab27a-deficient dendritic cells has been reported in vitro. In the mouse model (ashen mice), the dendritic cells are unable to perform a sufficient antigen cross-presentation 127.
To date 34 RAB27A mutations are known: 15 missense/non-sense, 4 splicing, and 15 deletions/insertion. In 2010, Meeths et al. 128 sequenced RAB27A in patients diagnosed as HLH and found one mutated family. Since the clinical picture of the two syndromes is indistinguishable, they concluded that the diagnosis of Griscelli syndrome type 2 (GS2) may be overlooked, particularly in fair-haired patients with hemophagocytic syndromes 128.
Hermansky–Pudlak type 2
The term Hermansky-Pudlak syndrome encompasses nine different human autosomal-recessive genetic disorders, sharing partial oculo-cutaneous albinism and bleeding disorders (78, 79). Furthermore, patients with Hermansky–Pudlak type 2 (Hermansky-Pudlak syndrome type 2) also show an increased susceptibility to infections, resulting from both congenital neutropenia and impaired cytotoxic activity. Mutations of the gene encoding the β-3A subunit of adaptor protein-3 (AP-3) complex are the cause of Hermansky-Pudlak syndrome type 2 (80). To date, 20 mutations are known in this gene and are associated with Hermansky-Pudlak syndrome type 2: 7 missense/non-sense, 1 splicing, 11 deletions/insertion, 1 complex rearrangement, and a chromosome 5 inversion that disrupts the gene sequence (81). To date, only one Hermansky-Pudlak syndrome type 2 patient has been reported who developed HLH. However, since this patient also carried a potentially contributing heterozygous RAB27A mutation, the risk to develop HLH in Hermansky-Pudlak syndrome type 2 remains unclear (82). The pearl mouse model of Hermansky-Pudlak syndrome type 2, upon infection with lymphocytic choriomeningitis virus, developed all the key features of the disease, which yet was only transient (82). In a cohort of 22 Hermansky-Pudlak syndrome type 2 patients, only one additional patient with HLH was identified; two developed incomplete, transient HLH-like episodes, although the cytotoxicity or degranulation capacity was impaired in all 16 patients tested (83). Although future reports might clarify the genotype–phenotype correlations, the risk for HLH in Hermansky-Pudlak syndrome type 2 appears lower than in Griscelli or Chédiak–Higashi syndrome (79, 82).
Familial hemophagocytic lymphohistiocytosis causes
Familial hemophagocytic lymphohistiocytosis (F-HLH) or primary HLH is caused by changes or mutations of specific areas within a person’s genes. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, absent, or overproduced. Depending upon the functions of the particular protein, this can affect many organ systems of the body.
Several genes have been shown to be abnormal in different people with familial hemophagocytic lymphohistiocytosis (FHL) and researchers are actively working to discover these genes. At least four different genes (i.e., PRF1, UNC13D, STX11 and STXBP2) have been identified that result in a genetic predisposition to developing hemophagocytic lymphohistiocytosis. A genetic predisposition means a person has a gene or genes for a particular disorder, but the disorder will not develop unless other factors help to trigger the disorder. Because familial hemophagocytic lymphohistiocytosis manifests shortly after birth (usually in the first few weeks to months), and varies between siblings with identical mutations, it is thought that an infectious trigger is required for disease expression, even though an infectious trigger is usually not identified.
Interestingly, familial hemophagocytic lymphohistiocytosis may develop in utero 129, suggesting HLH-related mutations may be important in maintaining immune homeostasis in the absence of infection 67.
Primary hemophagocytic lymphohistiocytosis associated with lymphocyte cytotoxic defects:
- The genes that are associated with familial hemophagocytic lymphohistiocytosis (FHL) are 28, 130:
- Unidentified gene defect located on chromosome 9 in Familial hemophagocytic lymphocytosis type 1,
- Perforin (PRF1) gene in Familial hemophagocytic lymphocytosis type 2,
- Munc-13–4 (UNC13D) in Familial hemophagocytic lymphocytosis type 3,
- Syntaxin-11 (STX11) in Familial hemophagocytic lymphocytosis type 4,
- Syntaxin binding protein 2 (STXBP-2) in Familial hemophagocytic lymphocytosis type 5.
- SH2D1A gene in X-Linked Lymphoproliferative Disease 1 (XLP1)
- RAB27A gene in Griscelli Syndrome type 2
- LYST gene (also known as CHS1) in Chediak-Higashi syndrome
- Ethnic variations in causative mutations have also been reported, with 47% of white familial hemophagocytic lymphohistiocytosis (FHL) patients having mutations in UNC13D, 22% in STXBP2, and 20% in PRF1. In contrast, Hispanics and blacks were more likely to have PRF1 mutations (71% and 98%, respectively), versus Arabs, who had a 36% PRF1 mutation rate 131.
Primary hemophagocytic lymphohistiocytosis associated with abnormalities of inflammasome activation 130:
- BIRC4 gene in XIAP deficiency
- NLR family CARD domain containing 4 protein (NLRC4) gene in familial cold autoinflammatory syndrome (FCAS)
Primary hemophagocytic lymphohistiocytosis associated with defined Mendelian disorders affecting inflammation
- SLC7A7 gene in Lysinuric protein intolerance
- Familial (apparently Mendelian) HLH of unknown origin
Identified causative mutations in familial hemophagocytic lymphohistiocytosis (F-HLH) result in impaired CD8+ T cell and NK cell cytotoxicity or altered lymphocyte activation and survival 132. Around 70% of familial hemophagocytic lymphohistiocytosis (F-HLH) is caused by the loss of function mutations in PRF1, the gene encoding perforin, a key cargo of cytolytic vesicles, or in UNC13D, encoding Munc13-4, which is critical for the release of cytolytic vesicles from T and NK cells 133. Mutations in other genes involved in cytolysis, such as STX11 and STXP2, are associated with familial hemophagocytic lymphohistiocytosis as well 134. Cytolytic dysfunction caused by abnormal granule exocytosis causes certain primary immunodeficiency (PID) syndromes associated with HLH, such as Chédiak-Higashi syndrome (CHS), Griscelli syndrome type 2 (GS2), and Hermansky-Pudlak syndrome type 2 (HPS2). Familial hemophagocytic lymphohistiocytosis type 1 to 5 develops within the first year of life 135.
Familial hemophagocytic lymphohistiocytosis inheritance pattern
Apart from the egg and sperm cells, each cell of your body normally has two working copies of each gene. One copy of each gene comes from your biological mother, and one comes from your biological father. Most of the time, familial hemophagocytic lymphohistiocytosis (F-HLH) or primary HLH is an autosomal recessive disorder. This means that for HLH to occur, both copies of the same HLH-associated genes must be mutated. In this situation, the parents are considered HLH “carriers” because they carry one normal and one altered copy of an HLH-associated gene in the cells of their bodies. Although they carry an altered gene copy, HLH carriers usually remain healthy and do not develop the signs and symptoms of HLH. However, they have a 50% chance of passing on the defective gene to their children.
Familial hemophagocytic lymphohistiocytosis autosomal recessive inheritance pattern means that a person must inherit two changed copies of the same gene (one abnormal gene from each parent) in order to have the condition. If a person inherits one abnormal gene and one normal gene, then in most cases that person will be a healthy carrier because the normal gene compensates for the abnormal gene. Being a carrier means that you do not have the condition, but carry a changed copy of the gene on one of a pair of genes.
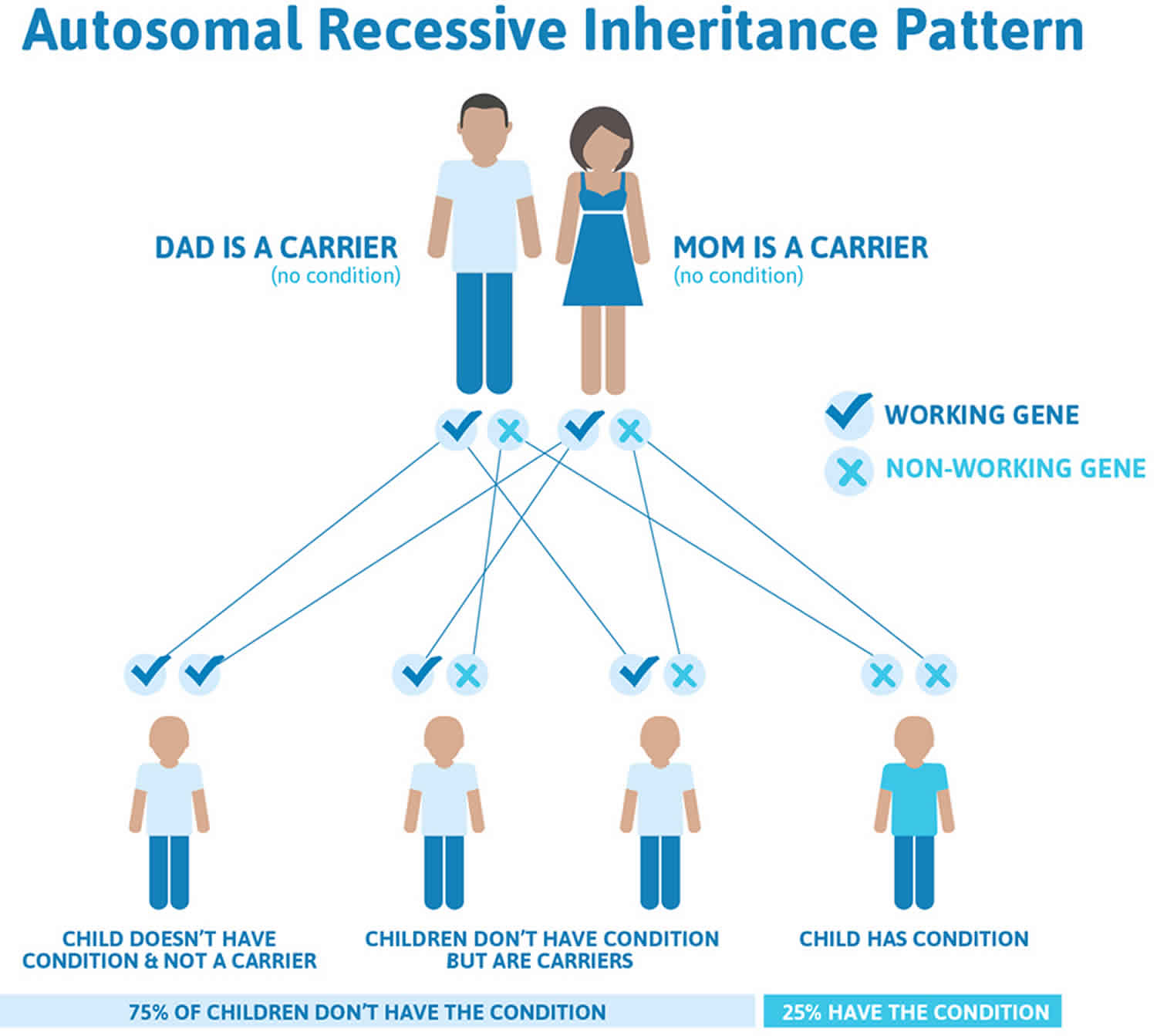
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Key points to remember
- A person must inherit two copies of a abnormal gene, one from each parent, in order to be affected by the condition (25% chance). If a person inherits only one abnormal gene then they will be a carrier (50% chance). These outcomes occur randomly. They remain the same in every pregnancy and are the same for boys and girls.
- A abnormal gene cannot be corrected – it is present for life.
- A abnormal gene is not something that can be caught from other people. They can still be a blood donor, for example.
- People often feel guilty about a genetic condition which runs in the family. It is important to remember that it is no-one’s fault and no-one has done anything to cause it to happen.
Figure 2. Familial hemophagocytic lymphohistiocytosis autosomal recessive inheritance pattern
Familial hemophagocytic lymphohistiocytosis pathophysiology
The pathogenesis of HLH has mostly been studied in familial hemophagocytic lymphohistiocytosis (FHL). An inherited defect in the perforin/granzyme pathway or in the fusion of cytotoxic lytic granules with the surface of natural killer (NK) cells causes, in the presence of an external trigger, an over-response by cytotoxic CD8+ T lymphocytes 1. Viral infections are the most common triggers 136.
Cytotoxic CD8+ T cells produce large amounts of interferon gamma (IFN-γ), which in turn activates macrophages 1. Overstimulated macrophages release large amounts of inflammatory cytokines, such as interleukin-1 beta (IL-1β), interleukin-6 (IL-6), interleukin-12 (IL-12), interleukin-18 (IL-18) and tumor necrosis factor alpha (TNFα). There is also an increased production of interleukin-10 (IL-10), with inhibitory activity, but not sufficient to limit the phenomenon 1. IL-12 and IL-18 produced by macrophages in turn stimulate CD8+ T cells, amplifying the inflammatory response 6. The resulting tissue damage causes a release of interleukin-33 (IL-33) and IL-1β, which further activates the macrophages. Activated macrophages engulf blood cells and produce large amounts of ferritin. The ‘cytokine storm’ causes all the clinical manifestations of HLH, from endothelial damage to coagulopathy and multi-organ failure 6, 37, 137, 138, 139. The central role of interferon gamma (IFN-γ) in the pathogenesis of familial hemophagocytic lymphohistiocytosis has been demonstrated in a perforin-deficient mouse model 140.
A similar pathogenesis of HLH can also be observed in patients with primary immunodeficiencies involving granule trafficking or exocytosis, such as Hermansky-Pudlak syndrome type 2, Griscelli syndrome type 2 and Chediak-Higashi syndrome 2, all with reduced cytotoxic T lymphocytes (CTL) cytotoxicity 141, 142. Advances in genetic diagnosis suggest that cell killing by cytotoxic T lymphocytes (CTLs) and NK cells can be affected from mildly to severely, thus explaining the different HLH phenotypes as a continuum 143. The known mutations in genes related to granule-mediated killing account for familial hemophagocytic lymphohistiocytosis (FHL) and primary forms in general 36. Minor alterations of the cytotoxic T lymphocytes (CTL) and NK cells activity, together with an external trigger, account for secondary HLH, severe sepsis, multi-organ failure and HLH in the context of rheumatologic diseases 143.
In all cases, a cytokine storm causing a devastating inflammation is the primal agent of the multiorgan failure, regardless of the underlying defect 144, 143, 145, 146, 147, 148 and HLH should be considered a clinical syndrome of hyperinflammation with different phenotypes 149. Moreover, this finding likely explains the numerous similarities and overlaps between HLH and other systemic inflammatory syndromes, such as septic shock, cytokine release syndrome following viral infections 148 and acute liver failure 150.
The case of malignancy-associated HLH includes two completely heterogeneous mechanisms. Firstly, when HLH is a manifestation of the underlying malignancy, particularly in the case of lymphomas, the overproduction of inflammatory cytokines such as INF-γ and TNF-α is likely caused by neoplastic cells 6, 151. Secondly, when HLH develops as the consequence of chemotherapy, the process is more likely to be caused by the association of drug-induced cytotoxic T lymphocytes (CTL) suppression and infection 152.
Figure 3. Familial hemophagocytic lymphohistiocytosis pathophysiology
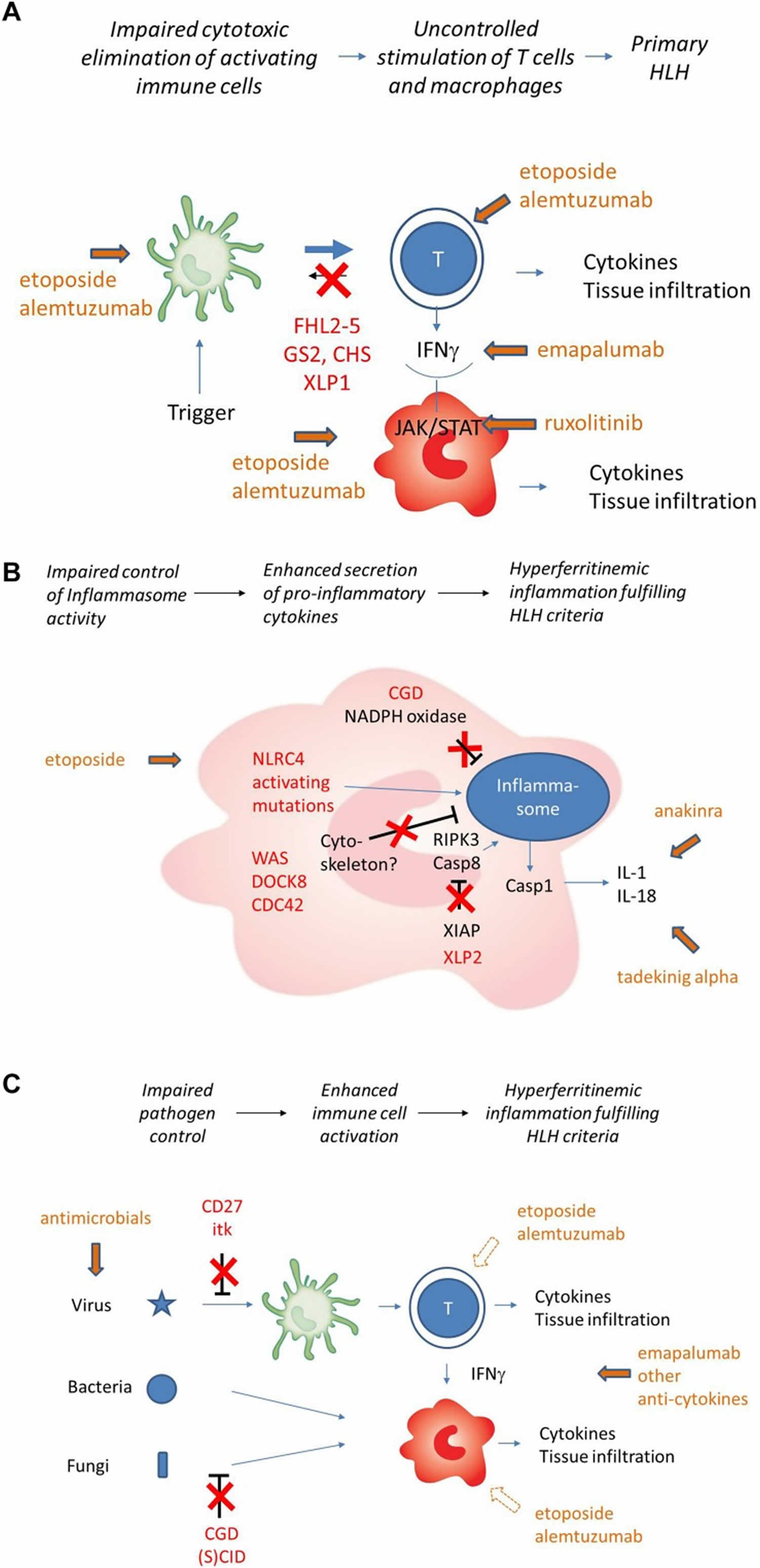
Footnotes: (A) Impaired cytotoxicity (red) leads to uncontrolled T cell activation by antigen-presenting cell (APC). T cell secreted IFN-gamma (IFN-γ) is the key driver of macrophage activation. Cellular and cytokine targets of therapy are indicated in green. (B) Pathogenesis of HLH in the context of impaired inflammasome homeostasis (simplified). Inborn errors of immunity shown (NLRC4, XIAP) or assumed (Cytoskeletal disorders, CGD) to be involved in inflammasome homeostasis are indicated in red. Cytokine targets of therapy are indicated in green. Macrophages are the key cells involved, T cells play a less prominent role. Cell-directed therapies are rarely used. (C) HLH pathogenesis in the context of inborn errors of immunity with impaired pathogen control (simplified). Inborn errors of immunity impairing virus control (mainly EBV) and/or bacterial/fungal control are indicated in red. Subsequent immune stimulation leads to hypersecretion of variable cytokines. Macrophage activation can occur in the absence of T cells, but T cells can be involved depending on the genetic defect and the trigger. Cell-directed therapies further impair pathogen control and should only be used in exceptional cases.
[Source 66 ]Familial hemophagocytic lymphohistiocytosis signs and symptoms
The onset and severity of hemophagocytic lymphohistiocytosis can vary greatly from one person to another 153. The specific symptoms that develop can also vary greatly, although HLH often causes multiorgan involvement. Generally, affected individuals develop fevers, a rash, an abnormally large liver (hepatomegaly), and an abnormally large spleen (splenomegaly). Fevers of unknown origin may be prolonged and persistent, often failing to respond to antibiotics 153. Sometimes, the lymph nodes are also abnormally large (lymphadenopathy). Lymph nodes are part of your lymphatic system, a circulatory network of vessels, ducts, and nodes that filter and distribute certain protein-rich (lymph) and blood cells throughout the body. Lymph nodes are small structures, found in groups throughout the body, that help to filter or drain out harmful substances from the body.
These initial sign and symptoms are described as nonspecific. This means that these signs and symptoms are common to many other different disorders or conditions, which can make getting a correct diagnosis difficult.
Affected individuals may also have low levels circulating red blood cells (anemia) and low levels of circulating platelets (thrombocytopenia). Red blood cells deliver oxygen to the body and platelets allow the body to form clots to stop bleeding. Individuals with anemia may experience tiredness, increased need for sleep, weakness, lightheadedness, dizziness, irritability, headaches, pale skin color, difficulty breathing (dyspnea), and cardiac symptoms. Individuals with thrombocytopenia (low levels of circulating platelets) are more susceptible to excessive bruising following minimal injury and to spontaneous bleeding from the mucous membranes, especially those of the gums and nose.
Some affected individuals may develop neurological symptoms including seizures, changes in mental status and irritability, paralysis (palsy) of certain cranial nerves, and problems coordinating voluntary movements (ataxia). Affected individuals are at risk of developing posterior reversible encephalopathy syndrome, which causes a rapid onset of headaches, altered consciousness, seizures, and disturbances in vision. Neurological problems are most common with familial hemophagocytic lymphohistiocytosis.
Additional symptoms can occur depending upon the specific organ system involved in an individual. These symptoms can include significant problems breathing (lung dysfunction), severe low blood pressure (hypotension), liver inflammation (hepatitis), kidney dysfunction, yellowing of the skin and whites of the eyes (jaundice), swelling due to fluid accumulation (edema), abdominal swelling due to fluid accumulation (ascites), and a variety of skin problems including widespread, reddening of the skin because of inflammation (erythroderma), rashes, blood spots (purpura), and tiny spots on the skin due to bleeding under the skin (petechiae).
Infant with fever
While “classic” familial hemophagocytic lymphohistiocytosis may present at almost any age, it most commonly presents in infants under 1 year of age, and neonatal or in utero presentations are well described 154. This “typical” patient with HLH usually presents with fever and is then noted to have cytopenias and splenomegaly. Further investigation reveals an illappearing patient, sometimes with a critical sepsis-like appearance. Marrow biopsy is often obtained to evaluate for leukemia and may (variably) reveal hemophagocytosis, while not displaying malignancy. Though infection and malignancy must be carefully considered, a triggering infection may not be identified 129. While these patients are well described by current diagnostic criteria, their most striking features are typically the degree of cytopenias, hypofibrinogenemia (in the face of inflammation), and hepatosplenomegaly. This patient population is capable of sudden, severe, and unpredictable worsening, necessitating rapid diagnosis and aggressive intervention.
Older children with infection
Children over the age of 1 year with hemophagocytic lymphohistiocytosis typically have definable triggers, most commonly viral infection. Epstein-Barr virus (EBV) is the most frequently identified infection triggering HLH, followed by cytomegalovirus (CMV), though numerous other viral, bacterial, parasitic, and fungal infections have been reported 155, 156, 157. Though underlying genetic mutations may not be found in patients with EBV-HLH, disease is often responsive to standard treatments and rituximab may be a helpful adjunct, as it may decrease viral burden 158. EBV-associated HLH is usually seen with primary infection, though some patients may present with HLH (or HLH recurrence) associated with chronic active EBV infection 159. These patients typically have predominant EBV infection of T cells (less often NK cells), instead of B cells. Overall, clinical presentations in older patients are similar to those in younger patients, though they may vary widely in severity. While patients presenting with relatively milder acuity may only require corticosteroids or intravenous immune globulin, aggressive treatment may be appropriate, as early initiation of etoposide is associated with improved survival in severe EBV-HLH 160, 161, 162. Ferritin values are consistently high in older patients, and sCD25 is always elevated, though often not to the degree seen in infants.
Central nervous system involvement
Patients with familial hemophagocytic lymphohistiocytosis present with central nervous system (CNS) involvement about one-third of the time with symptoms including seizures, meningismus, signs of cranial nerve involvement, ataxia, dysarthria, and encephalopathy 55, 163. However, patients may also rarely present with isolated central nervous system (CNS) inflammation, without complete or even any systemic signs of HLH. Though rare, this isolated disease is an important forme fruste of HLH, which often presents insidiously with patients experiencing substantial delays in diagnosis 164. Cerebrospinal fluid analysis shows pleocytosis and hyperproteinemia, and brain biopsy reveals T-cell and histiocytic infiltrates. MRI findings in isolated or non-isolated CNS HLH typically include increased signal in T2-weighted images of widespread areas, with symmetric periventricular white matter hyperintensity, meningeal enhancement, diffusion restriction, and may progress to include necrosis 165, 166, 167. Age of onset of isolated CNS HLH varies greatly, but is most often seen in children over a year of age, as younger children will usually have accompanying systemic HLH 4.
Familial hemophagocytic lymphohistiocytosis diagnosis
Diagnosing familial hemophagocytic lymphohistiocytosis can be challenging because many of its initial symptoms mimic other common conditions. Symptoms such as persistent fevers, respiratory issues, rash, anemia, seizures and enlarged liver, spleen or lymph nodes may be cause other conditions.
Familial hemophagocytic lymphohistiocytosis diagnosis is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. Guidelines have been published that detail the criteria necessary for a diagnosis of hemophagocytic lymphohistiocytosis (see Table 1).
Familial hemophagocytic lymphohistiocytosis is diagnosed when an individual meets established HLH-2004 diagnostic criteria 26. HLH-2004 criteria for diagnosis of HLH requires either 1 or 2.
- The presence of a known HLH-causing genetic mutation, OR
- Has five or more of the clinical or laboratory findings listed below:
- High and often prolonged periods of fever (more than 7 days);
- Rash, irritability and/or seizures;
- An abnormally large spleen (splenomegaly);
- Low blood cells (cytopenias): red blood cells (hemoglobin less than 9 g/dL, hemoglobin less than 10 g/dL in infants <4 week), white blood cells (neutrophils less than 1 × 109/L), or platelets (platelets count less than 100 × 109/L);
- Abnormally high levels of a type of fat called a triglyceride in the blood (hypertriglyceridemia) triglyceride ≥ 3.0 mmol/L (≥ 265 mg/dl);
- Abnormally low levels of fibrinogen (a specific blood clotting protein) (hypofibrinogenemia) serum fibrinogen < 150 mg/dL;
- Destruction of blood cells by macrophages (hemophagocytosis) in the bone marrow, spleen, or lymph nodes and/or cerebrospinal fluid with no evidence of malignancy;
- Low or absent natural killer (NK) cell activity;
- Abnormal high levels of ferritin (a protein that binds to iron) in the blood (ferritin ≥ 500mcg/L); and
- Elevated soluble interleukin-2 receptor alpha chain (sCD25 ≥ 2400 U/mL), a specialized protein that builds up in the blood when the immune system is stimulated.
- Reduced expression of proteins such as perforin, signaling lymphocytic activation molecule-associated protein (SAP), X-linked inhibitor of apoptosis protein (XIAP), and/or depressed cell functions
On occasion, HLH may be strongly considered, and HLH-directed therapy may be initiated, even though the 5 HLH-2004 diagnostic criteria are not fulfilled 168.
If hemophagocytic activity is not proven at the time of presentation, further search for hemophagocytic activity is encouraged. If the bone marrow specimen is not conclusive, material may be obtained from other organs. Serial marrow aspirates over time may also be helpful. The following findings may provide strong supportive evidence for the diagnosis: spinal fluid pleocytosis (mononuclear cells) and/or elevated spinal fluid protein and histological picture in the liver resembling chronic persistent hepatitis (biopsy). Other abnormal clinical and laboratory findings consistent with the diagnosis are cerebromeningeal symptoms, lymph node enlargement, jaundice, edema, skin rash, hepatic enzyme abnormalities, hypoproteinemia, hyponatremia, and elevated very low-density lipoprotein (VLDL↑)/low high-density lipoprotein (HDL↓).
Figure 3. Hemophagocytic lymphohistiocytosis diagnostic algorithm
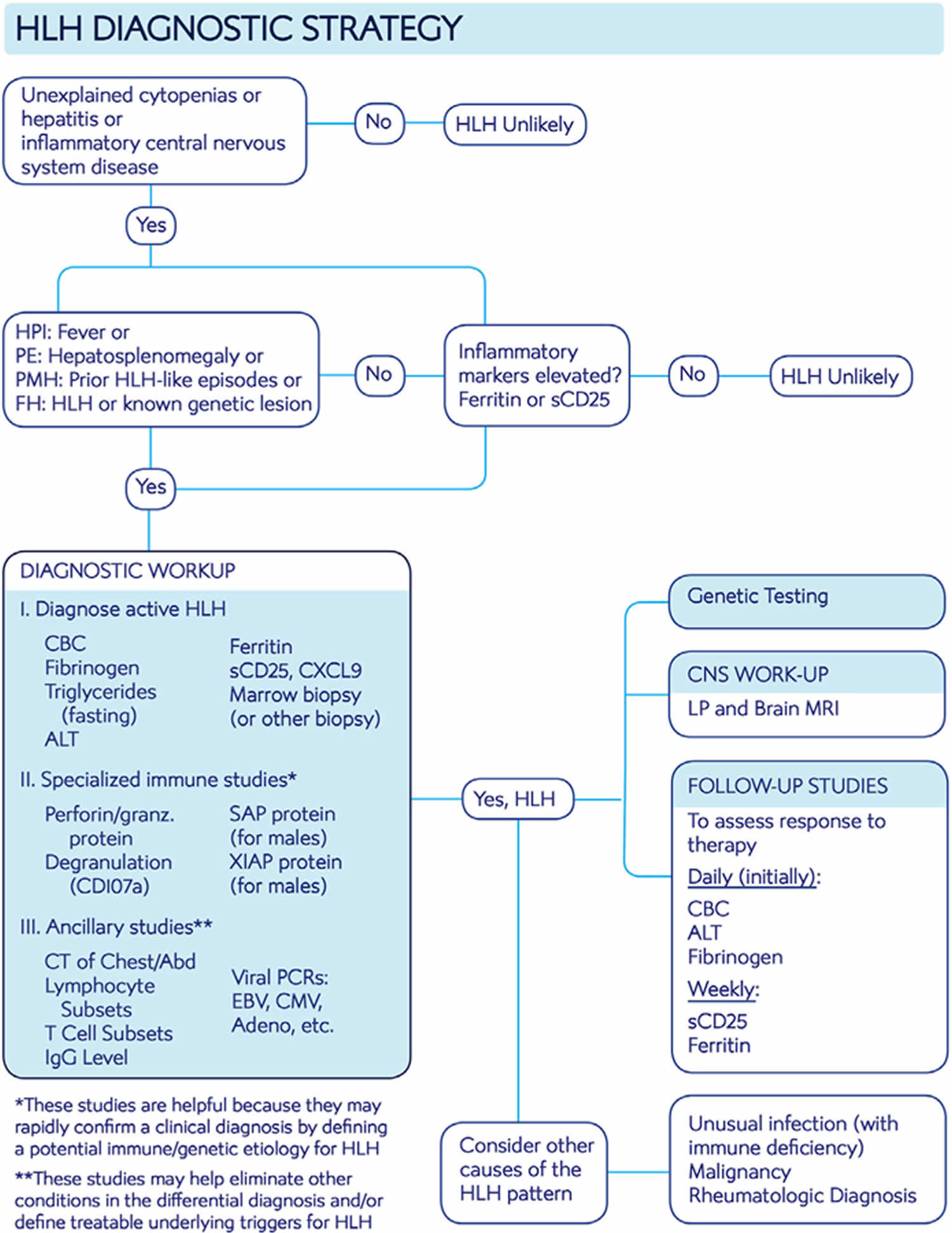
Table 2. Hemophagocytic lymphohistiocytosis diagnostic criteria
| HLH2004 entry criteria | Comment |
|---|---|
| A. Molecular diagnosis consistent with HLH: Pathologic mutations of PRF1, UNC13D, STXBP2, Rab27a, STX11, SH2D1A, or XIAP or | In a patient with known genetic defects, treatment before full development of HLH may be appropriate, but genetic studies usually just help to define HLH recurrence risk, not the presence of an active disease state. |
| B. Five of the eight criteria listed below are fulfilled | |
| 1. Fever ≥38.3°C | Nearly universal in untreated HLH. |
| 2. Splenomegaly | While splenomegaly and hepatomegaly are very common in HLH, adenopathy is not. |
| 3. Cytopenias (affecting at least two of three lineages in the peripheral blood): Hemoglobin <9 g/dL (in infants <4 weeks: hemoglobin <10 g/dL) Platelets <100 × 10³/mL Neutrophils < 1 × 10³/mL | Cytopenias are ubiquitous in HLH. Lack of cytopenias should make one doubt a diagnosis of HLH, except in the special case of isolated, CNS-only disease. |
| 4. Hypertriglyceridemia (>265 mg/dL) and/or hypofibrinogenemia (<150 mg/dL) | Low fibrinogen in the context of inflammation is paradoxical and one of the more distinctive features of HLH. |
| 5. Hemophagocytosis in bone marrow or spleen or lymph nodes or liver | Not specific to HLH, or essential for the diagnosis, but helpful as a disease marker. Of note, it is often not evident early after disease onset. |
| 6. Low or absent NK-cell activity | More modern and robust assays measuring perforin levels and its degranulation should replace this assay for reliable diagnosis of HLH. This assay is not specific for primary HLH. |
| 7. Ferritin >500 ng/mL | Most patients have much higher levels than this threshold suggests. |
| 8. Elevated Soluble CD25 (soluble IL-2 receptor alpha) | As HLH is a T-cell driven disease, this assay is extremely informative for diagnosis and response to therapy. See Clinical Testing and Workup for patients with suspected HLH disease for more information. |
Clinical testing and workup for patients with suspected HLH disease
The initial workup is two-pronged and aims (a) to establish the diagnosis of HLH promptly and (b) to identify mimickers of HLH disease (if present), or potentially treatable underlying HLH disease triggers.
Common laboratory studies
- Complete blood count (CBC), hepatic panel, fibrinogen, triglycerides. Essential to define typical features of HLH. Though not part of diagnostic criteria, hepatitis is extremely common in HLH.
- Ferritin. Usually (but not always) very elevated
- Search for triggering infection with cultures, viral PCRs: EBV, CMV, adenovirus
Specialized laboratory studies
- Markers of immune activation: soluble interleukin-2 receptor alpha chain (sCD25), granzyme B expression and CXCL9. Essentially always elevated in untreated HLH; very sensitive, but not specific. sCD25, and perhaps CXCL9, are also useful for monitoring response to therapy
- Interlukin-18 (IL-18) level. Elevated in HLH associated with inflammasome-opathies (XIAP, NLRC4, systemic onset juvenile arthritis)
- Measurement of proteins affected in familial HLH: perforin, SAP, XIAP. Helpful to quickly confirm a suspected familial HLH diagnosis
- Functional studies: lymphocyte degranulation (CD107a mobilization), NK cell function. May help to fulfill diagnostic criteria. Degranulation is the preferred functional assay over NK cell function 170.
- Lymphocyte subsets, T-cell subsets, immune globulin levels. Screening studies for primary immune deficiency
- Genetic testing (multigene panel or whole exome). Essential for defining HLH recurrence risk
Imaging studies (should be routinely performed to help rule out malignancy, or unusual infections if trigger is unknown)
- Body cavity CT’s (chest/abdomen/pelvis)
- Consider 18F-fluorodeoxyglucose (FDG) positron emission tomography-computed tomography (PET-CT) if suspicion of lymphoma
- MRI of brain. Brain MRI complements lumbar puncture for central nervous system (CNS) assessment
Tissue sampling (essential to rule out malignancy, identify hemophagocytosis, and identify central nervous system (CNS) involvement)
- Bone marrow biopsy
- Lumbar puncture
- Other tissue biopsies as appropriate: liver, lymph node, masses
Physicians may order blood tests to take a complete blood cell count, which will measure the levels of red cells, white cells and platelets. Blood tests can also reveal abnormally high ferritin levels, or abnormal high levels of triglycerides. Physicians may also use blood tests to look for signs of infection in the blood and conduct tests to determine how well the blood clots (coagulation studies). Physicians may also order tests that can assess the health and function of the liver.
Sometimes, a bone marrow biopsy (the surgical removal and microscopic examination of a tissue sample) may be taken and studied for signs of hemophagocytosis, signs of infection or infectious organisms, and the accumulation of macrophages.
Molecular genetic testing can confirm a diagnosis of hemophagocytic lymphohistiocytosis in certain people. Molecular genetic testing can detect mutations in one of the four specific genes known to cause familial forms of this disorder, but is available only as a diagnostic service at specialized laboratories.
It should be emphasized that since T-cell activation is central to HLH pathogenesis, elevated soluble IL2 receptor alpha chain (sCD25) should always be observed in untreated HLH 171, 172, 173. If sCD25 is not elevated, then one should doubt a diagnosis of HLH 4. Similarly, though not as well established because HLH appears to be largely driven by interferon gamma (IFN-γ), elevations of CXCL9 (a sensitive indicator of IFN-γ bioactivity) should be seen in untreated cases of HLH disease 174, 175, 176. Additionally, elevated expression of granzyme B in NK cells has been shown to be similarly ubiquitous in HLH; normal expression levels would suggest that HLH is not the correct diagnosis 177. Ferritin levels above 10,000 ng/mL appear to be relatively specific for HLH but are not very sensitive 178. While specialized immunologic testing may facilitate diagnosis, if a diagnosis can be made without them then treatment should not be delayed pending these results. Likewise, treatment should not be delayed for assessment of central nervous system (CNS) involvement, though this should always be conducted (once a lumbar puncture may be safely performed).
Practical examples of when to not diagnose HLH disease
Certain clinical features may suggest mimics of HLH disease 4:
- Despite the common presence of splenomegaly, prominent or abnormal lymphadenopathy is not typically seen in HLH. This finding strongly suggests lymphoma, Castleman disease, or unusual infection such as HIV, histoplasmosis or mycobacterial disease 179. Similarly, leukocytosis is not typical of HLH (except in R-HLH/MAS) and should prompt a search for an alternative diagnosis.
- An extremely elevated sCD25 (>10- to 20-fold above normal) in a noninfantile patient suggests undiagnosed lymphoma, especially when ferritin is not similarly elevated 179, 180.
- The early occurrence of isolated and aggressive “CNS relapse” during treatment of systemic HLH may suggest an undiagnosed malignant disorder relapsing in the CNS.
- A normal or only modestly elevated sCD25, despite extremely elevated ferritin, suggests disseminated infection in the context of primary immune deficiency, especially in an infant 181.
Other diagnostic tools
Other features supporting an HLH diagnosis that are not part of the HLH-2004 diagnostic criteria include hyperbilirubinemia, hepatomegaly, transaminitis (present in the vast majority of patients with HLH), and elevated lactate dehydrogenase and d-dimer levels, with the latter usually elevated even when international normalized ratio, partial thromboplastin time, and fibrinogen are normal 182. These findings may help to discriminate HLH from septic shock and conditions such as autoimmune hemolytic anemia, and they are also useful in assessing response to therapy.
H Score
The HLH-probability calculator (HScore), with graded clinical and laboratory parameters, is a Web-based online calculator developed retrospectively in adult patients that may be a helpful diagnostic tool (Table 3) 183. The pattern of inflammatory cytokines (elevated levels of interferon-γ [IFN-γ] and IL-10, with only modestly elevated IL-6 levels) has high diagnostic accuracy for secondary hemophagocytic lymphohistiocytosis and may be a useful approach to differentiate HLH from infection and to monitor patients; however, the utility of this pattern of changes needs to be verified in children and adults outside of China 184, 185, 186.
Table 3. Parameters and points in the HScore
| Parameter | No. of points (criteria for scoring) |
|---|---|
| Known underlying immunosuppression* | 0 (no) or 18 (yes) |
| Temperature (°C) | 0 (<38.4), 33 (38.4–39.4), or 49 (>39.4) |
| Organomegaly | 0 (no), 23 (hepatomegaly or splenomegaly), or 38 (hepatomegaly and splenomegaly) |
| No. of cytopenias† | 0 (1 lineage), 24 (2 lineages), or 34 (3 lineages) |
| Ferritin (μg/L) | 0 (<2000), 35 (2000-6000), or 50 (>6000) |
| Triglyceride (mmol/L) | 0 (<1.5), 44 (1.5-4), or 64 (>4) |
| Fibrinogen (g/L) | 0 (>2.5) or 30 (≤2.5) |
| Aspartate aminotransferase (U/L) | 0 (<30) or 19 (≥30) |
| Hemophagocytosis on bone marrow aspirate | 0 (no) or 35 (yes) |
Footnote:
* HIV positive or receiving long-term immunosuppressive therapy (ie, glucocorticoids, cyclosporine A, azathioprine).
† Defined as a hemoglobin level of 9.2 g/L and/or a leukocyte count ≤5 × 109/L and/or a platelet count ≤110 × 109/L.
Familial hemophagocytic lymphohistiocytosis treatment
The treatment of hemophagocytic lymphohistiocytosis is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, specialists in diagnosing and treating blood disorders (hematologists), specialists in diagnosing and treating cancer (oncologists), specialists in diagnosing and treating immune system diseases (immunologists), geneticists (for familial forms), social workers, and other healthcare professionals may need to systematically and comprehensively plan treatment. Psychosocial support for the entire family is essential as well. Genetic counseling may be of benefit for affected individuals and their families. At most institutions, the treatment of children with familial hemophagocytic lymphohistiocytosis is managed by hematologists.
The aim of hemophagocytic lymphohistiocytosis treatment is to suppress the life-threatening inflammation that leads to organ damage 139. Specific therapeutic procedures and interventions may vary, depending upon numerous factors, such as the underlying cause; the presence or absence of certain symptoms; the overall severity of the symptoms and the disorder; an individual’s age and general health; and/or other elements. Decisions concerning the use of particular drug regimens and/or other treatments should be made by physicians and other members of the health care team in careful consultation with the patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
For children with familial hemophagocytic lymphohistiocytosis, the first step in treatment is generally to suppress the overactive immune system. This is often done using a combination of steroids and chemotherapy, with the goal of putting the disease into remission. The Histiocyte Society first published international treatment protocol for HLH in 1994 (HLH-94), which combined chemotherapy and immunotherapy (etoposide and cyclosporine A [CsA]) for 8 weeks with or without intrathecal methotrexate in select patients to control the inflammatory response as a bridge to hematopoietic stem cell transplantation (HSCT) 44, 36. The HLH-94 protocol was reserved for patients with familial, relapsing, or severe/persistent HLH 55. HLH-94 dramatically improved survival in familial hemophagocytic lymphohistiocytosis from an estimated 5-year rate of 22% to an estimated 3-year rate of 51% ±20 55. The HLH-94 protocol (minus HSCT) was also found to be beneficial in secondary forms of HLH 13. For this reason, HLH-94 is often recommended by oncologists for the treatment of nonfamilial forms of HLH that are severe or relapsing 67.
Based on experience with HLH-94, the Histiocyte Society in 2004 modified the protocol (HLH-2004) in several ways, most notably by adding early and continuous cyclosporine A (CsA) therapy 26. During therapy with either protocol (HLH-94 or HLH-2004), patients should be monitored closely for clinical and laboratory response, and relapses should be expected as steroids are weaned (around week 6 in HLH-94) 4. HSCT remains the only long-term curative treatment, and achieving disease remission of HLH prior to transplant is essential for good prognosis, because HSCT complications (infection, excessive bleeding, organ failure, graft rejection, and graft versus host disease) and mortality are associated with HLH disease activity 187.
The HLH-94 induction chemotherapy is based on steroids and etoposide administration for 8 weeks with or without intrathecal methotrexate, followed by maintenance with cyclosporine A (CSA), etoposide and dexamethasone pulses while waiting for allogeneic hematopoietic stem cell transplantation (allo-HSCT) 44, 36. Chemotherapy might be suspended after 8 weeks of treatment once clinical remission is obtained and a clear genetic base is excluded 44. Otherwise, if a clear genetic base is proved, or in case of reactivation, hematopoietic stem cell transplantation (HSCT) is recommended 44.
After initial treatment, children with familial hemophagocytic lymphohistiocytosis usually undergo an allogeneic stem cell transplant (allo-HSCT), which replaces their defective immune system with a healthy one from a donor. Donor cells may come from a sibling, a parent or an unrelated donor. A stem cell transplant offers familial hemophagocytic lymphohistiocytosis patients the best chance for a cure 36, 44.
For disease refractory to HLH-94/2004, an anti-CD52 monoclonal antibody called alemtuzumab (which depletes T and B cells) has shown effectiveness as a salvage therapy 4, 188. Salvage therapy should be directed by oncologists and hematologists 67.
Figure 4. HLH-94 protocol
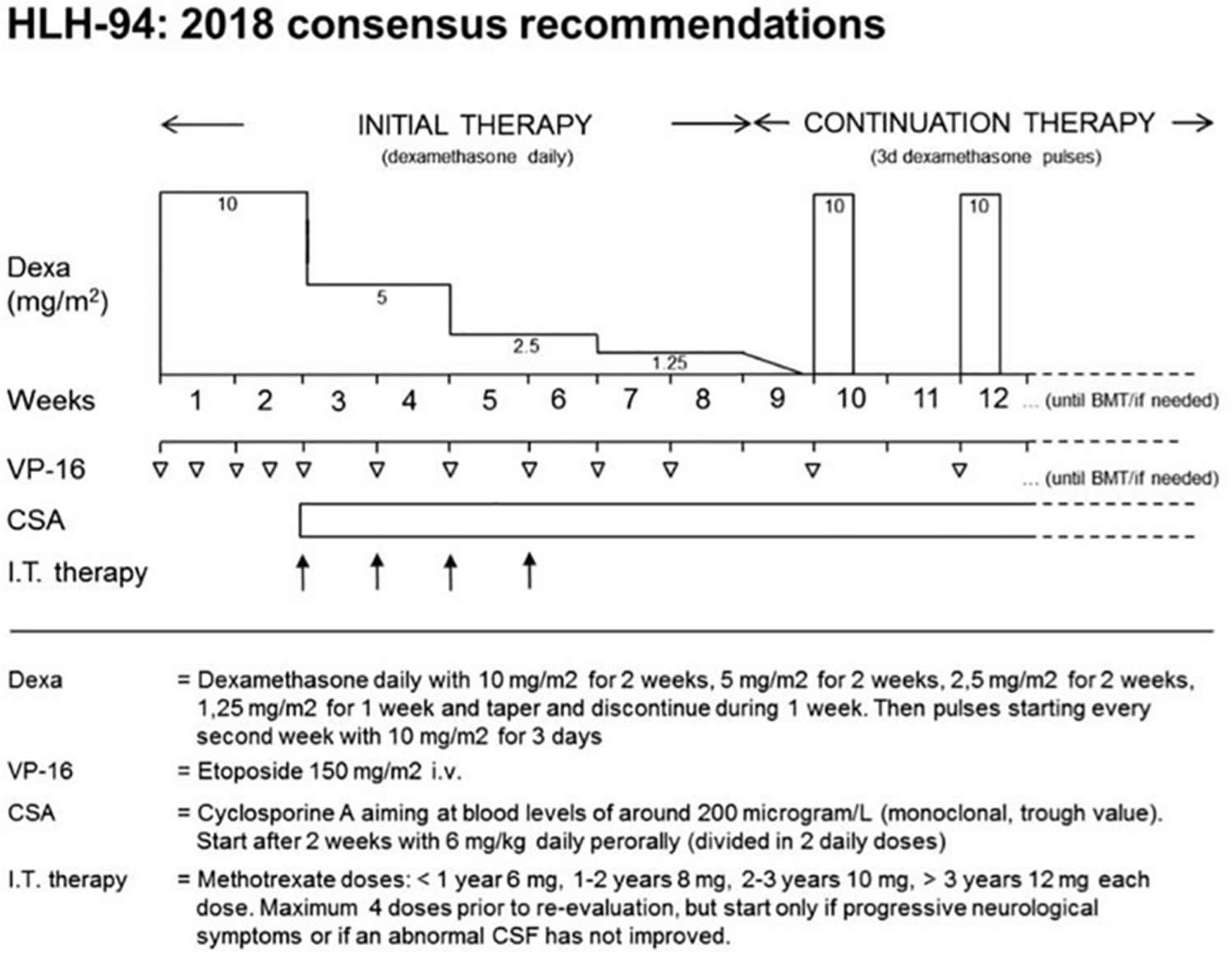
Footnotes: The HLH-94 protocol is based on immunochemotherapy including dexamethasone, etoposide, and cyclosporine A (CSA). After an intensive phase of 2 weeks with high doses of dexamethasone and twice weekly administration of etoposide, dexamethasone is tapered until week nine. Cyclosporine A (CSA) is used from week three onward to prevent reactivation. Intrathecal therapy with methotrexate is recommended in patients with central nervous system (CNS) involvement. Immunological testing and genetic confirmation of the underlying genetic disease is required in all patients and should provide the basis for hematopoietic stem cell transplantation (HSCT) within 8 weeks.
[Source 66 ]HLH-94 protocol
2018 consensus statements by the HLH Steering Committee of the Histiocyte Society recommending the use of HLH-94. The HLH-94 protocol is based on immuno-chemotherapy including dexamethasone, etoposide and cyclosporine A to achieve remission of the hyperinflammatory state and to maintain remission until hematopoietic stem cell transplantation (HSCT) can be performed 189. Functionally, all agents target lymphocytes, macrophages and antigen presenting cells. The cytostatic agent etoposide induces cell death mainly in activated T cells 190, but also in macrophages and dendritic cells. The use of the calcineurin inhibitor cyclosporine A leads to an inhibition of the transcription factor NFAT (nuclear factor of activated T-cells) and thus to a reduced activation and proliferation of T cells. Steroids slow down inflammation by reducing cytokine secretion, but in addition, they have a moderate cytotoxic effect on activated T cells. It is recommended to treat patients with central nervous system (CNS) involvement also with intrathecal methotrexate 191, although there is no clear evidence of benefit. The protocol has a 2-week intensive phase with dexamethasone and twice weekly administration of etoposide, followed by 6 weeks of weekly etoposide and steroid tapering. In this second phase, cyclosporine A is used to prevent reactivation 192. Rapid immunological testing followed by genetic confirmation of the underlying genetic disease is required in all patients and should provide the basis for hematopoietic stem cell transplantation (HSCT) within these 8 weeks. In the international multicenter registry-based HLH-2004 study, 5-year probability of survival for children with genetically verified familial HLH treated with this protocol was 59% 193. Dexamethasone/etoposide-based protocols have been successfully used in X-linked lymphoproliferative disorder or patients with TIM3 deficiency. It remains an ultimate choice also in other inborn errors of immunity, but the toxicity and immunosuppression associated with etoposide asks for more targeted therapies in these conditions.
Hematopoietic Stem Cell Transplantation (HSCT)
To prevent recurrences, allogenic stem cell transplantation (HSCT) should be carried out as soon as possible after achieving initial remission in the “primary” HLH 192, 194. Hematopoietic stem cell transplantation (HSCT) remains the only curative option for familial hemophagocytic lymphohistiocytosis. The timing of HSCT has to balance the risks between achieving full remission versus reactivation. Although active disease at conditioning remains a risk factor, full remission of all clinical symptoms is not required for successful HSCT. In particular, active neurological disease should prompt aggressive management including early HSCT 191.
Allogeneic stem cell transplantation (HSCT) is also the definitive treatment of choice for HLH in the context of several other inborn errors of immunity, including X-linked lymphoproliferative disorder type 1 (XLP1) or X-linked lymphoproliferative disorder type 2 (XLP2) and patients with TIM3 deficiency 195, 196. Furthermore, allogenic stem cell transplantation (HSCT) has been successfully performed in a patient with a CDC42 mutation 197. However, there has been no report of HSCT in NLRC4 deficiency, where it is unlikely to impact on IL-18 hypersecretion by intestinal epithelial cells 198. Careful and broad genetic and functional evaluation is therefore mandatory before proceeding to HSCT based on clinical grounds in rare cases of familial or recurrent HLH without detection of a genetic cause.
In cases with suspected “primary” HLH, donor search and pretransplantation diagnostics should be carried out promptly during the initial presentation 66. Bi-allelic mutations should be ruled out in potential related donors 66. In autosomal-recessive disease, heterozygous carriers are in most cases appropriate donors 66. In the X-linked conditions, skewed X-inactivation should be excluded in potential female carrier donors 199.
Antithymocyte Globulin (ATG)
Antithymocyte globulin directly targets T cells and other lymphocytes, to a minor extent also granulocytes and monocytes 200. In a retrospective single-center analysis of 38 patients with familial hemophagocytosis, a protocol consisting of steroids, cyclosporine A and first-line ATG resulted in a higher initial remission rate compared to the HLH-94 protocol (active disease in 26% of patients versus 53%, after 2 months of therapy) but was associated with a higher percentage of relapses before HSCT (32% versus 13%) 201. In an attempt to combine advantages of both protocols, a trial combining ATG and etoposide has been performed (NCT01104025), but the results have not yet been reported. There is no clear role for ATG beyond “primary” HLH 66.
Alemtuzumab (Anti-CD52)
More recently, the humanized monoclonal anti-CD52 antibody alemtuzumab has been used in patients with “primary” HLH 202. It is directed against the CD52 antigen (CAMPATH 1) which is a surface protein on mature lymphocytes and antigen-presenting cells (APCs). First promising results have been achieved when used in a bridging to transplant setting 203. Excellent initial results have been orally reported from a trial evaluating Alemtuzumab as first-line treatment for “primary” HLH (in combination with methyl-prednisolone and cyclosporine A) (NCT02472054). The profound and long-lasting immune suppression and limits this drug to “primary” HLH, where induction of remission is rapidly followed by HSCT. The problem of viral (re-)activation is an important caveat when using alemtuzumab in X-linked lymphoproliferative disorder.
Emapalumab
Emapalumab is a recombinant human monoclonal antibody against interferon gamma (IFN-γ) 204. It has received FDA approval in November 2018 for the treatment of pediatric and adult patients with primary HLH with refractory, recurrent, or progressive disease or intolerance to HLH therapy 205. An international multicenter follow-up study to further assess the efficacy and safety of emapalumab is still ongoing (NCT03312751). This trial will also provide data on its use as first-line therapy in “primary” HLH. Serum levels of CXCL9 emerge as an interesting biomarker for increased interferon gamma (IFN-γ) activity 206 and may be particularly helpful when considering the use of emapalumab in first-line treatment of HLH in the context of other inborn errors of immunity. Notably, a patient with CDC42 mutation who did not respond to steroids, cyclosporine A, and anakinra was successfully treated with emapalumab 197.
Ruxolitinib
Janus kinase inhibitors represent interesting therapeutic compounds in the context of HLH, since they not only inhibit signaling downstream of interferon gamma (IFN-γ), but also of several other pro-inflammatory cytokines. In the mouse-model of LCMV-induced “primary” HLH, the disease manifestations, including CNS involvement, were reduced upon JAK1/2 blockade by ruxolitinib 207, 208. The successful individual use of ruxolitinib reported in single cases of secondary HLH 209, 210, 211 has resulted in its prospective evaluation for this indication (NCT03795909). Preliminary results of a single-center phase 2 pilot study on the efficacy of ruxolitinib in secondary HLH demonstrate good tolerance to ruxolitinib in a small cohort of five patients 212. However, “primary” HLH is excluded in these studies. A trial investigating the benefit of JAK inhibition in first-line treatment of human “primary” HLH is in preparation.
IL-18 Binding Protein
IL-18 is released by activated macrophages and can induce interferon gamma (IFN-γ) and other pro- inflammatory cytokines 213. Several reports have found elevated free IL-18 concentrations (i.e., IL-18 not bound to its binding protein IL-18BP) in the serum of patients with both “primary” and secondary HLH as well as in animal models and IL- 18 levels correlated with the presence of HLH-criteria and disease progression 214. In a murine model of “primary” HLH it was shown that treatment with IL-18BP can reduce severe organ damage, but does not improve survival 215. An ongoing multicenter, double-blind, placebo-controlled, randomized withdrawal trial evaluates efficacy and safety of IL-18 Binding Protein (tadekinig alfa) in pediatric patients with NLRC4 associated hyperinflammation including HLH or X-linked inhibitor of apoptosis (XIAP) deficiency, diseases, in which IL-18 levels are particularly elevated (NCT03113760). While this treatment seems promising to attenuate the autoinflammatory manifestations of X-linked inhibitor of apoptosis (XIAP) deficiency, it remains to be seen whether it also has a role in acute EBV-induced HLH in this disease.
IL-1, IL-6, and TNF alpha blockade
The pro-inflammatory cytokines elevated in “primary” HLH also include IL-1, IL-6, and TNF alpha, which can be targeted by monoclonal antibodies and other blocking agents. They have been used successfully in the context of secondary HLH 216, 217 and related conditions associated with a “cytokine storm” such as hyperinflammation associated with CAR-T cell therapy 218. Moreover, case reports have illustrated a partial effect of IL-1 blockade in patients with NLRC4 or CDC42 mutations 219, 197 and in patients with chronic granulomatous disease 220. Anecdotal reports have reported efficacy of IL-6 blockade in manifestations of X-linked inhibitor of apoptosis (XIAP) deficiency different from acute EBV-induced HLH 221. However, efficacy of IL-1, IL-6, or TNF alpha blockade in “primary” HLH has not been clearly documented, neither in first-line nor in rescue therapy 66.
Control of Triggers
Infections should be diagnosed and treated aggressively in all forms of HLH. When active EBV infection is present, rituximab (anti-CD20 antibody) can help controlling the immune stimulation by eliminating EBV infected B cells 222, 131. However, in some states of persistent EBV replication, EBV has been demonstrated in T or NK cells leading to resistance against rituximab treatment 223. Cell-targeted therapy results in significant immunosuppression, such that reverse isolation, aspergillus-effective antifungal and Pneumocystis jirovecii prophylaxis should be administered 224. Weekly monitoring for infection or reactivation of latent pathogens (EBV, CMV, adenovirus, aspergillus antigen) is recommended 192.
Monitoring treatment response
Monitoring response to therapy and detecting early signs of reactivation is crucial in patients with “primary” HLH 131, 26. Fever, aminotransferase levels, ferritin, and coagulation parameters are practical, sensitive, and dynamic measures useful in the day-to-day monitoring of HLH patients 67, 225. Blood counts must be interpreted with the consideration of effects of myelosuppressive therapies (i.e., etoposide). Soluble IL-2 receptor (sCD25) and sCD163 appear to be useful indicators of active disease, but the restricted availability of these assays limits their utility in day-to-day management 67. Monitoring coagulation parameters is essential, but they may not show marked abnormalities until late in the disease course 67. Other biomarkers for disease activity such as free IL-18 or CXCL9 are being explored. If more rapid turnaround times can be achieved, they might be valuable for guiding therapy in the future 66.
In general, persistent or progressive disease in the face of standard initial therapy, should prompt continued investigation for an underlying infectious or malignant trigger (especially in adults). At the same time, HLH-targeted therapies should be intensified under the guidance of an experienced oncologist and/or hematologist 67.
Salvage therapy in Refractory HLH
Early mortality of acute HLH remains a major concern. 25–50% of patients with acute “primary” HLH fail to achieve rapid and sustained initial remission after etoposide-based therapy. If cytopenia [in particular thrombocytopenia with platelets less 50 x 109/L (less than 50,000 platelets per microliter)] and ferritin and/or sCD25 fail to respond after 2 weeks, the risk for an adverse outcome increases, justifying consideration of alternative (salvage) therapy 192. There are no standard recommendations for the treatment of relapsing or refractory HLH. The salvage therapies published so far include alemtuzumab, anakinra, antithymocyte globulin (ATG), and regimens consisting of liposomal doxorubicin, etoposide, and dexamethasone 226. In an observational study reporting on treatment of 22 patients with refractory HLH with alemtuzumab, 86 percent of patients showed partial response, and 77 percent were able to receive HSCT 227. Notably, CMV and adenovirus viremia occurred in 23–32% of patients.
Emapalumab (GAMIFANT), a neutralizing antibody against interferon gamma (INF-γ), has recently been licensed as the first drug for the treatment patients with “primary” hemophagocytic lymphohistiocytosis (HLH) with refractory, recurrent or progressive disease or intolerance with conventional HLH therapy 228. The recommended starting dose is 1 mg/kg twice per week with dexamethasone as a background treatment, but doses can be increased up to 10 mg/kg based on clinical response 205. Due to the risk of serious infections (frequent during therapy of primary HLH patients and observed in 32% of patients in the trial) patients should receive prophylaxis for Herpes Zoster, Pneumocystis jirovecii, and fungal infections and should be monitored for tuberculosis, adenovirus, EBV and CMV.
The study included 27 patients with a mean age of 1 year (range: 0.1 to 13 years), with a “primary” HLH in 82% of patients. Patients had received various combinations of dexamethasone, etoposide, cyclosporine A, and anti-thymocyte globulin (ATG) prior to emapalumab. Full response was defined as normalization of all, while partial response was defined as normalization of ≥3 HLH parameters and HLH improvement was defined as ≥3 HLH abnormalities improved by at least 50% from baseline. Twenty patients completed the 8-week study, while seven were prematurely withdrawn. Seventy percent (19/27) of patients proceeded to HSCT. The overall response rate was 63%, the median time to response was 8 days. A complete response was achieved in 7 patients, partial response in 8 patients and HLH improvement in 2 patients 229, 205. Since refractory primary HLH has poor prognosis, these data are encouraging. However, the exact place of this drug in the context of existing and emerging therapies remains to be defined 66.
Familial hemophagocytic lymphohistiocytosis prognosis
The advent of HLH-94 and HLH-2004 treatment protocols markedly improved the survival of children with familial hemophagocytic lymphohistiocytosis. Nevertheless, familial hemophagocytic lymphohistiocytosis survivors may suffer long-term side effects such as cognitive and growth delay, hearing impairment, and obstructive lung disease 55. Prognosis for adults with HLH remains poor. Overall mortality rates ranged between 42 and 88% 230. In a series of 68 adult patients with HLH from multiple tertiary care centers, half of whom had an underlying malignancy, only 31% of patients were alive at a median follow up of 32 months 231. Predictors of mortality in adult HLH include older age and underlying lymphoma 161. These data illustrate a pressing need for better diagnostics and therapies for HLH, particularly in the adult population 67.
- Benevenuta C, Mussinatto I, Orsi C, Timeus FS. Secondary hemophagocytic lymphohistiocytosis in children (Review). Exp Ther Med. 2023 Jul 17;26(3):423. doi: 10.3892/etm.2023.12122[↩][↩][↩][↩]
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233-46. doi: 10.1146/annurev-med-041610-134208[↩]
- Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, Ishii E, Janka G, Ladisch S, Lehmberg K, McClain KL, Minkov M, Montgomery S, Nanduri V, Rosso D, Henter JI. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017 Dec 21;130(25):2728-2738. doi: 10.1182/blood-2017-06-788349[↩][↩][↩]
- Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, Hines M, Eckstein O, Ladisch S, Nichols KE, Rodriguez-Galindo C, Wistinghausen B, McClain KL. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019 Nov;66(11):e27929. doi: 10.1002/pbc.27929[↩][↩][↩][↩][↩][↩][↩][↩]
- Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. 2020 Apr 16;135(16):1332-1343. doi: 10.1182/blood.2019000936[↩][↩]
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34(101515) doi: 10.1016/j.berh.2020.101515[↩][↩][↩][↩]
- Janka GE, Lehmberg K. Hemophagocytic syndromes – An update. Blood Rev. 2014;28(4):135–42. doi: 10.1016/j.blre.2014.03.002[↩][↩]
- Madkaikar M, Shabrish S, Desai M. Current Updates on Classification, Diagnosis and Treatment of Hemophagocytic Lymphohistiocytosis (HLH) Indian J Pediatr. 2016;83(5):434–43. doi: 10.1007/s12098-016-2037-y[↩]
- Vick EJ, Patel K, Prouet P, Martin MG. Proliferation through activation: hemophagocytic lymphohistiocytosis in hematologic malignancy. Blood Adv. 2017 May 9;1(12):779-791. doi: 10.1182/bloodadvances.2017005561[↩]
- Eife R, Janka GE, Belohradsky BH, Holtmann H. Natural killer cell function and interferon production in familial hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol. 1989;6(3):265-72. doi: 10.3109/08880018909034296[↩]
- Kim T, Kulick CG, Kortepeter CM, Brinker A, Waldron P. Hemophagocytic lymphohistiocytosis associated with the use of lamotrigine. Neurology. 2019 May 21;92(21):e2401-e2405. doi: 10.1212/WNL.0000000000007517[↩]
- Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachée-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Deist F, Fischer A, de Saint Basile G. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003 Nov 14;115(4):461-73. doi: 10.1016/s0092-8674(03)00855-9[↩]
- Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, Webb D, Janka G; Histocyte Society. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002 Oct 1;100(7):2367-73. doi: 10.1182/blood-2002-01-0172[↩][↩][↩]
- Bechara E, Dijoud F, de Saint Basile G, Bertrand Y, Pondarré C. Hemophagocytic lymphohistiocytosis with Munc13-4 mutation: a cause of recurrent fatal hydrops fetalis. Pediatrics. 2011 Jul;128(1):e251-4. doi: 10.1542/peds.2010-0764[↩]
- Bode SF, Lehmberg K, Maul-Pavicic A, Vraetz T, Janka G, Stadt UZ, Ehl S. Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis. Arthritis Res Ther. 2012 Jun 8;14(3):213. doi: 10.1186/ar3843[↩][↩][↩]
- Yang Y, Luo Z, Yuan T. Familial hemophagocytic lymphohistiocytosis in a neonate: Case report and literature review. Medicine (Baltimore). 2021 Nov 24;100(47):e27786. doi: 10.1097/MD.0000000000027786[↩]
- Janka GE. Hemophagocytic syndromes. Blood Rev. 2007;21:245–253. doi: 10.1016/j.blre.2007.05.001[↩]
- Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, Villanueva J, Risma KA, Wei Q, Klein PS, Filipovich AH. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial hemophagocytic lymphohistiocytosis. Blood. 2011;118:5794–5798. doi: 10.1182/blood-2011-07-370148[↩]
- Clementi R, Emmi L, Maccario R, Liotta F, Moretta L, Danesino C, Arico M. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. 2002;100:2266–2267. doi: 10.1182/blood-2002-04-1030[↩]
- Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, Bondzio I, Gross-Wieltsch U, Schündeln M, Schütz B, Woessmann W, Groll AH, Strahm B, Pagel J, Speckmann C, Janka G, Griffiths G, Schwarz K, zur Stadt U, Ehl S. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. 2010;95:2080–2087. doi: 10.3324/haematol.2010.029389[↩]
- Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27:519–25. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1988563/pdf/archdisch01417-0015.pdf[↩]
- Aricò M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, Martinetti M, Rusca MP. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996 Feb;10(2):197-203.[↩]
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007 Feb;166(2):95-109. doi: 10.1007/s00431-006-0258-1[↩]
- Janka GE. Hemophagocytic syndromes. Blood Rev. 2007 Sep;21(5):245-53. doi: 10.1016/j.blre.2007.05.001[↩]
- Janka GE, Lehmberg K. Hemophagocytic syndromes–an update. Blood Rev. 2014 Jul;28(4):135-42. doi: 10.1016/j.blre.2014.03.002[↩]
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb;48(2):124-31. doi: 10.1002/pbc.21039[↩][↩][↩][↩]
- Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am. 2008 May;28(2):293-313, viii. doi: 10.1016/j.iac.2008.01.010[↩]
- Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011 Mar;163(3):271-83. doi: 10.1111/j.1365-2249.2010.04302.x[↩][↩][↩][↩][↩]
- Sun S, Guo X, Zhu Y, Yang X, Li Q, Gao J. [Analysis of clinical phenotype and genetic mutations of a pedigree of familial hemophagocytic lymphohistiocytosis]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014 Oct;31(5):570-3. Chinese. doi: 10.3760/cma.j.issn.1003-9406.2014.01.006[↩]
- Nakajima H, Cella M, Bouchon A, Grierson HL, Lewis J, Duckett CS, Cohen JI, Colonna M. Patients with X-linked lymphoproliferative disease have a defect in 2B4 receptor-mediated NK cell cytotoxicity. Eur J Immunol. 2000 Nov;30(11):3309-18. doi: 10.1002/1521-4141(200011)30:11<3309::AID-IMMU3309>3.0.CO;2-3[↩]
- Latour S, Gish G, Helgason CD, Humphries RK, Pawson T, Veillette A. Regulation of SLAM-mediated signal transduction by SAP, the X-linked lymphoproliferative gene product. Nat Immunol. 2001 Aug;2(8):681-90. doi: 10.1038/90615[↩]
- Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, Brandt SJ, Chotai D, Hodgman C, Solari RC, Lovett M, Kingsmore SF. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996 Jul 18;382(6588):262-5. doi: 10.1038/382262a0. Erratum in: Nature 1997 Jan 2;385(6611):97.[↩][↩]
- Henter JI, Elinder G, Söder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991 Apr;80(4):428-35. doi: 10.1111/j.1651-2227.1991.tb11878.x[↩]
- Ishii E, Ohga S, Tanimura M, Imashuku S, Sako M, Mizutani S, Miyazaki S. Clinical and epidemiologic studies of familial hemophagocytic lymphohistiocytosis in Japan. Japan LCH Study Group. Med Pediatr Oncol. 1998 May;30(5):276-83. doi: 10.1002/(sici)1096-911x(199805)30:5<276::aid-mpo3>3.0.co;2-c[↩]
- Gürgey A, Göğüş S, Ozyürek E, Aslan D, Gümrük F, Cetin M, Yüce A, Ceyhan M, Seçmeer G, Yetgin S, Hiçsönmez G. Primary hemophagocytic lymphohistiocytosis in Turkish children. Pediatr Hematol Oncol. 2003 Jul-Aug;20(5):367-71.[↩]
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Sieni E, Cetica V, Piccin A, Gherlinzoni F, Sasso FC, Rabusin M, Attard L, Bosi A, Pende D, Moretta L, Aricò M. Familial hemophagocytic lymphohistiocytosis may present during adulthood: Clinical and genetic features of a small series. PLoS One. 2012;7(e44649) doi: 10.1371/journal.pone.0044649[↩][↩]
- Malinowska I, Machaczka M, Popko K, Siwicka A, Salamonowicz M, Nasiłowska-Adamska B. Hemophagocytic syndrome in children and adults. Arch Immunol Ther Exp (Warsz) 2014;62:385–394. doi: 10.1007/s00005-014-0274-1[↩][↩]
- Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, Bielekova B, Kennedy AL, Rivkin MJ, Davies KJ, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. 2019;6(e560) doi: 10.1212/NXI.0000000000000560[↩]
- de Kerguenec C, Hillaire S, Molinié V, Gardin C, Degott C, Erlinger S, Valla D. Hepatic manifestations of hemophagocytic syndrome: A study of 30 cases. Am J Gastroenterol. 2001;96:852–857. doi: 10.1111/j.1572-0241.2001.03632.x[↩]
- Amir AZ, Ling SC, Naqvi A, Weitzman S, Fecteau A, Grant D, Ghanekar A, Cattral M, Nalli N, Cutz E, et al. Liver transplantation for children with acute liver failure associated with secondary hemophagocytic lymphohistiocytosis. Liver Transpl. 2016;22:1245–1253. doi: 10.1002/lt.24485[↩]
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383:1503–1516. doi: 10.1016/S0140-6736(13)61048-X[↩]
- Macheta M, Will AM, Houghton JB, Wynn RF. Prominent dyserythropoiesis in four cases of haemophagocytic lymphohistiocytosis. J Clin Pathol. 2001;54:961–963. doi: 10.1136/jcp.54.12.961[↩]
- Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, Gadner H, Imashuku S, Komp D, Ladisch S, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. doi: 10.1182/blood-2002-01-0172[↩][↩][↩][↩][↩][↩]
- Malinowska I, Machaczka M, Popko K, Siwicka A, Salamonowicz M, Nasiłowska-Adamska B. Hemophagocytic syndrome in children and adults. Arch Immunol Ther Exp (Warsz). 2014 Oct;62(5):385-94. doi: 10.1007/s00005-014-0274-1[↩]
- Ouachée-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, Neven B, Casanova JL, Tardieu M, Cavazzana-Calvo M, Blanche S, Fischer A. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006 Apr;117(4):e743-50. doi: 10.1542/peds.2005-1789[↩]
- Baker KS, DeLaat CA, Steinbuch M, Gross TG, Shapiro RS, Loechelt B, Harris R, Filipovich AH. Successful correction of hemophagocytic lymphohistiocytosis with related or unrelated bone marrow transplantation. Blood. 1997 May 15;89(10):3857-63.[↩]
- Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, Locatelli F, Montgomery SM, Webb D, Winiarski J, Filipovich AH, Henter JI; Histiocyte Society. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005 Jun;129(5):622-30. doi: 10.1111/j.1365-2141.2005.05501.x[↩]
- Imashuku S, Hibi S, Todo S, Sako M, Inoue M, Kawa K, Koike K, Iwai A, Tsuchiya S, Akiyama Y, Kotani T, Kawamura Y, Hirosawa M, Hasegawa D, Kosaka Y, Yamaguchi H, Ishii E, Kato K, Ishii M, Kigasawa H. Allogeneic hematopoietic stem cell transplantation for patients with hemophagocytic syndrome (HPS) in Japan. Bone Marrow Transplant. 1999 Mar;23(6):569-72. doi: 10.1038/sj.bmt.1701620[↩]
- Cesaro S, Locatelli F, Lanino E, Porta F, Di Maio L, Messina C, Prete A, Ripaldi M, Maximova N, Giorgiani G, Rondelli R, Aricò M, Fagioli F. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: a retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP). Haematologica. 2008 Nov;93(11):1694-701. doi: 10.3324/haematol.13142[↩]
- Dürken M, Horstmann M, Bieling P, Erttmann R, Kabisch H, Löliger C, Schneider EM, Hellwege HH, Krüger W, Kröger N, Zander AR, Janka GE. Improved outcome in haemophagocytic lymphohistiocytosis after bone marrow transplantation from related and unrelated donors: a single-centre experience of 12 patients. Br J Haematol. 1999 Sep;106(4):1052-8. doi: 10.1046/j.1365-2141.1999.01625.x[↩]
- Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, Davies G, Webb D, Veys P, Amrolia P. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006 Feb 1;107(3):1233-6. doi: 10.1182/blood-2005-05-1819[↩][↩]
- Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. 2008 Oct;42 Suppl 2:S47-50. doi: 10.1038/bmt.2008.283[↩][↩]
- Allen CE, Marsh R, Dawson P, Bollard CM, Shenoy S, Roehrs P, Hanna R, Burroughs L, Kean L, Talano JA, Schultz KR, Pai SY, Baker KS, Andolina JR, Stenger EO, Connelly J, Ramirez A, Bryant C, Eapen M, Pulsipher MA. Reduced-intensity conditioning for hematopoietic cell transplant for HLH and primary immune deficiencies. Blood. 2018 Sep 27;132(13):1438-1451. doi: 10.1182/blood-2018-01-828277[↩]
- Trottestam H, Horne A, Aricò M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI; Histiocyte Society. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011 Oct 27;118(17):4577-84. doi: 10.1182/blood-2011-06-356261[↩][↩][↩][↩][↩]
- Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B, Pende D, Griffiths G, Aricò M. Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. 2014 Apr 16;5:167. doi: 10.3389/fimmu.2014.00167[↩][↩][↩][↩][↩][↩]
- zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, Kabisch H, Schneppenheim R, Nürnberg P, Janka G, Hennies HC. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14:827–834. doi: 10.1093/hmg/ddi076[↩]
- zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, Strauss J, Kasper B, Nürnberg G, Becker C, Maul-Pavicic A, Beutel K, Janka G, Griffiths G, Ehl S, Hennies HC. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85:482–492. doi: 10.1016/j.ajhg.2009.09.005[↩]
- Ohadi M, Lalloz MR, Sham P, Zhao J, Dearlove AM, Shiach C, Kinsey S, Rhodes M, Layton DM. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999;64:165–171. doi: 10.1086/302187[↩]
- Cetica V, Pende D, Griffiths GM, Arico M. Molecular basis of familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95:538–541. doi: 10.3324/haematol.2009.019562[↩]
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G, Kumar V. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957[↩]
- Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, Al-Manjomi F, Al-Harbi M, Alangari A, Le Deist F, Gennery AR, Prince N, Cariou A, Nitschke P, Blank U, El-Ghazali G, Ménasché G, Latour S, Fischer A, de Saint Basile G. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119:3765–3773. doi: 10.1172/JCI40732[↩]
- Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachée-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Deist F, Fischer A, de Saint Basile G. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115:461–473. doi: 10.1016/S0092-8674(03)00855-9[↩]
- de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10:568–579. doi: 10.1038/nri2803[↩]
- Sieni E, Cetica V, Mastrodicasa E, Pende D, Moretta L, Griffiths G, et al. Familial hemophagocytic lymphohistiocytosis: a model for understanding the human machinery of cellular cytotoxicity. Cell Mol Life Sci (2012) 69(1):29–40 10.1007/s00018-011-0835-y[↩]
- Wegehaupt O, Wustrau K, Lehmberg K, Ehl S. Cell Versus Cytokine – Directed Therapies for Hemophagocytic Lymphohistiocytosis (HLH) in Inborn Errors of Immunity. Front Immunol. 2020 May 8;11:808. doi: 10.3389/fimmu.2020.00808[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020 Aug;34(4):101515. https://doi.org/10.1016/j.berh.2020.101515[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Cetica V, Pende D, Griffiths GM, Aricò M. Molecular basis of familial hemophagocytic lymphohistiocytosis. Haematologica (2010) 95:538–41 10.3324/haematol.2009.019562[↩]
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286(5446):1957–9 10.1126/science.286.5446.1957[↩][↩][↩]
- Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat (2006) 27(1):62–8 10.1002/humu.20274[↩][↩][↩]
- Trizzino A, zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, et al. Genoptype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet (2008) 45:15–21 10.1136/jmg.2007.052670[↩][↩][↩]
- Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood (2002) 99(1):61–6 10.1182/blood.V99.1.61[↩]
- Suga N, Takada H, Nomura A, Ohga S, Ishii E, Ihara K, et al. Perforin defects of primary haemophagocytic lymphohistiocytosis in Japan. Br J Haematol (2002) 116:346–9 10.1046/j.1365-2141.2002.03266.x[↩]
- Molleran Lee S, Villanueva J, Sumegi J, Zhang K, Kogawa K, Davis J, et al. Characterisation of diverse PRF1 mutations leading to decreased killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet (2004) 41:137–44 10.1136/jmg.2003.011528[↩]
- An O, Gursoy A, Gurgey A, Keskin O. Structural and functional analysis of perforin mutations in association with clinical data of familial hemophagocytic lymphohistiocytosis type 2 (FHL2) patients. Protein Sci (2013) 22(6):823–39 10.1002/pro.2265[↩]
- Lee SM, Sumegi J, Villanueva J, Tabata Y, Zhang K, Chakraborty R, et al. Patients of African ancestry with hemophagocytic lymphohistiocytosis share a common haplotype of PRF1 with a 50delT mutation. J Pediatr (2006) 149(1):134–7 10.1016/j.jpeds.2006.03.003[↩]
- Trapani JA, Thia KY, Andrews M, Davis ID, Gedye C, Parente P, et al. Human perforin mutations and susceptibility to multiple primary cancers. Oncoimmunology (2013) 2(4):e24185. 10.4161/onci.24185[↩]
- Santoro A, Cannella S, Trizzino A, Lo Nigro L, Corsello G, Aricò M. A single amino acid change A91V in perforin: a novel, frequent predisposing factor to childhood acute lymphoblastic leukemia? Haematologica. 2005 May;90(5):697-8.[↩]
- Trambas C, Gallo F, Pende D, Marcenaro S, Moretta L, De Fusco C, et al. A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin. Blood (2005) 106(3):932–7 10.1182/blood-2004-09-3713[↩]
- Martínez-Pomar N, Lanio N, Romo N, Lopez-Botet M, Matamoros N. Functional impact of A91V mutation of the PRF1 perforin gene. Hum Immunol (2013) 74(1):14–7 10.1016/j.humimm.2012.10.011[↩]
- Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood (2011) 118(22):5794–8 10.1182/blood-2011-07-370148[↩]
- Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell (2003) 115(4):461–73 10.1016/S0092-8674(03)00855-9[↩][↩]
- Rudd E, Bryceson YT, Zheng C, Edner J, Wood SM, Ramme K, et al. Spectrum, and clinical and functional implications of UNC13D mutations in familial haemophagocytic lymphohistiocytosis. J Med Genet (2008) 45(3):134–41 10.1136/jmg.2007.054288[↩][↩]
- Santoro A, Cannella S, Bossi G, Gallo F, Trizzino A, Pende D, et al. Novel Munc13-4 mutations in children and young adult patients with haemophagocytic lymphohistiocytosis. J Med Genet (2006) 43(12):953–60 10.1136/jmg.2006.041863[↩]
- Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, et al. Genoptype-phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet (2011) 48(5):343–52 10.1136/jmg.2010.085456[↩][↩][↩]
- Santoro A, Cannella S, Trizzino A, Bruno G, De Fusco C, Notarangelo LD, et al. Mutations affecting mRNA splicing are the most common molecular defect in patients with familial hemophagocytic lymphohistiocytosis type 3. Haematologica (2008) 93(7):1086–90 10.3324/haematol.12622[↩][↩]
- Meeths M, Chiang SC, Wood SM, Entesarian M, Schlums H, Bang B, et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood (2011) 118(22):5783–93 10.1182/blood-2011-07-369090[↩]
- Entesarian M, Chiang SC, Schlums H, Meeths M, Chan MY, Mya SN, et al. Novel deep intronic and missense UNC13D mutations in familial haemophagocytic lymphohistiocytosis type 3. Br J Haematol (2013) 162(3):415–8 10.1111/bjh.12371[↩]
- Yoon HS, Kim HJ, Yoo KH, Sung KW, Koo HH, Kang HJ, et al. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica (2010) 95:622–6 10.3324/haematol.2009.016949[↩]
- zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type 4 to chromosome 6q24 and identification of mutation in syntaxin 11. Hum Mol Genet (2005) 14:827–34 10.1093/hmg/ddi076[↩]
- Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, et al. Defective cytotoxic lymphocyte egranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood (2007) 110:1906–15 10.1182/blood-2007-02-074468[↩]
- Rudd E, Göransdotter Ericson K, Zheng C, Uysal Z, Ozkan A, Gürgey A, et al. Spectrum and clinical implications of syntaxin 11 gene mutations in familial haemophagocytic lymphohistiocytosis: association with disease-free remissions and haematopoietic malignancies. J Med Genet (2006) 43(4):e14. 10.1136/jmg.2005.035253[↩]
- Marsh RA, Satake N, Biroschak J, Jacobs T, Johnson J, Jordan MB, et al. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr Blood Cancer (2010) 55(1):134–40 10.1002/pbc.22499[↩]
- Horne A, Ramme KG, Rudd E, Zheng C, Wali Y, al-Lamki Z, et al. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol (2008) 143(1):75–83 10.1111/j.1365-2141.2008.07315.x[↩]
- Sepulveda FE, Debeurme F, Ménasché G, Kurowska M, Côte M, Pachlopnik Schmid J, et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood (2013) 121:595–603 10.1182/blood-2012-07-440339[↩][↩]
- Müller ML, Chiang SC, Meeths M, Tesi B, Entesarian M, Nilsson D, et al. An N-terminal missense mutation in STX11 causative of FHL4 abrogates syntaxin-11 binding to Munc18-2. Front Immunol (2014) 4:515. 10.3389/fimmu.2013.00515[↩]
- Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest (2009) 119(12):3765–73 10.1172/JCI40732[↩]
- zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet (2009) 85(4):482–92 10.1016/j.ajhg.2009.09.005[↩][↩]
- Pagel J, Beutel K, Lehmberg K. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood (2012) 119(25):6016–24 10.1182/blood-2011-12-398958[↩][↩][↩]
- Nagai K, Yamamoto K, Fujiwara H, An J, Ochi T, Suemori K, et al. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic and functional analyses of cytotoxic T lymphocytes. PLoS One (2010) 5(11):e14173. 10.1371/journal.pone.0014173[↩]
- Cetica V, Santoro A, Gilmour KC, Sieni E, Beutel K, Pende D, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet (2010) 47:595–600 10.1136/jmg.2009.075341[↩]
- Stepensky P, Bartram J, Barth TF, Lehmberg K, Walther P, Amann K, et al. Persistent defective membrane trafficking in epithelial cells of patients with familial hemophagocytic lymphohistiocytosis type 5 due to STXBP2/MUNC18-2 mutations. Pediatr Blood Cancer (2013) 60(7):1215–22 10.1002/pbc.24475[↩]
- Zhao XW, Gazendam RP, Drewniak A, van Houdt M, Tool AT, van Hamme JL, et al. Defects in neutrophil granule mobilization and bactericidal activity in familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) syndrome caused by STXBP2/Munc18-2 mutations. Blood (2013) 122:109–11 10.1182/blood-2013-03-494039[↩][↩]
- Sandrock K, Nakamura L, Vraetz T, Beutel K, Ehl S, Zieger B. Platelet secretion defect in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL-5). Blood (2010) 116:6148–50 10.1182/blood-2010-08-302943[↩][↩]
- Meeths M, Entesarian M, Al-Herz W, Chiang SC, Wood SM, Al-Ateeqi W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood (2010) 116:2635–43 10.1182/blood-2010-05-282541[↩]
- Al Hawas R, Ren Q, Ye S, Karim ZA, Filipovich AH, Whiteheart SW. Munc18b/STXBP2 is required for platelet secretion. Blood (2012) 120:2493–500 10.1182/blood-2012-05-430629[↩]
- Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet (1998) 20:129–35 10.1038/2424[↩]
- Filipovich AH, Zhang K, Snow AL, Marsh RA. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood (2010) 116(18):3398–408 10.1182/blood-2010-03-275909[↩]
- Palendira U, Low C, Chan A, Hislop AD, Ho E, Phan TG, et al. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol (2011) 9(11):e1001187. 10.1371/journal.pbio.1001187[↩]
- Parolini S, Bottino C, Falco M, Augugliaro R, Giliani S, Franceschini R, et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med (2000) 192:337–46 10.1084/jem.192.3.337[↩][↩][↩]
- Hislop AD, Palendira U, Leese AM, Arkwright PD, Rohrlich PS, Tangye SG, et al. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood (2010) 116(17):3249–57 10.1182/blood-2009-09-238832[↩]
- Dupré L, Andolfi G, Tangye SG, Clementi R, Locatelli F, Aricò M, et al. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood (2005) 105(11):4383–9 10.1182/blood-2004-08-3269[↩]
- Rezaei N, Mahmoudi E, Aghamohammadi A, Das R, Nichols KE. X-linked lymphoproliferative syndrome: a genetic condition typified by the triad of infection, immunodeficiency and lymphoma. Br J Haematol (2011) 152(1):13–30 10.1111/j.1365-2141.2010.08442.x[↩][↩]
- Meazza R, Tuberosa C, Cetica V, Falco M, Loiacono F, Parolini S, et al. XLP1 inhibitory effect by 2B4 does not affect DNAM-1 and NKG2D activating pathways in NK cells. Eur J Immunol (2014). 10.1002/eji.201344312[↩][↩]
- Nichols KE, Ma CS, Cannons JL, Schwartzberg PL, Tangye SG. Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol Rev (2005) 203:180–99 10.1111/j.0105-2896.2005.00230.x[↩][↩]
- Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood (2011) 117:53–62 10.1182/blood-2010-06-284935[↩]
- Arico M, Imashuku S, Clementi R, Hibi S, Teramura T, Danesino C, et al. Hemophagocytic lymphohistiocytosis due to germline mutations in SH2D1A, the X-linked lymphoproliferative disease gene. Blood (2001) 97(4):1131–3 10.1182/blood.V97.4.1131[↩]
- Recher M, Fried AJ, Massaad MJ, Kim HY, Rizzini M, Frugoni F, et al. Intronic SH2D1A mutation with impaired SAP expression and agammaglobulinemia. Clin Immunol (2013) 146(2):84–9 10.1016/j.clim.2012.11.007[↩][↩]
- Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature (2006) 444:110–4 10.1038/nature05257[↩][↩]
- Damgaard RB, Fiil BK, Speckmann C, Yabal M, zur Stadt U, Bekker-Jensen S, et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol Med (2013) 5(8):1278–95 10.1002/emmm.201303090[↩]
- Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood (2011) 117:1522–9 10.1182/blood-2010-07-298372[↩][↩][↩][↩]
- Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood (2010) 116:1079–82 10.1182/blood-2010-01-256099[↩][↩]
- Speckmann C, Lehmberg K, Albert MH, Damgaard RB, Fritsch M, Gyrd-Hansen M, et al. X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol (2013) 149(1):133–41 10.1016/j.clim.2013.07.004[↩][↩][↩]
- Marsh RA, Villanueva J, Kim MO, Zhang K, Marmer D, Risma KA, et al. Patients with X-linked lymphoproliferative disease due to BIRC4 mutation have normal invariant natural killer T-cell populations. Clin Immunol (2009) 132(1):116–23 10.1016/j.clim.2009.03.517[↩]
- Marsh RA, Kim MO, Liu C, Bellman D, Hart L, Grimley M, et al. An intermediate alemtuzumab schedule reduces the incidence of mixed chimerism following reduced-intensity conditioning hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Biol Blood Marrow Transplant (2013) 19:1625–31 10.1016/j.bbmt.2013.09.001[↩]
- Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, Prunieras M. A syndrome associating partial albinism and immunodeficiency. Am J Med (1978) 65:691–702 10.1016/0002-9343(78)90858-6[↩]
- Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PM, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol (2007) 9(4):367–78 10.1038/ncb1552[↩]
- Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, Ramme K, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer (2010) 54(4):563–72 10.1002/pbc.22357[↩][↩]
- Heeg M, Ammann S, Klemann C, Panning M, Falcone V, Hengel H, Lehmberg K, Zur Stadt U, Wustrau K, Janka G, Ehl S. Is an infectious trigger always required for primary hemophagocytic lymphohistiocytosis? Lessons from in utero and neonatal disease. Pediatr Blood Cancer. 2018 Nov;65(11):e27344. doi: 10.1002/pbc.27344[↩][↩]
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, Charlotte F, Diamond EL, Egeler RM, Fischer A, Herrera JG, Henter JI, Janku F, Merad M, Picarsic J, Rodriguez-Galindo C, Rollins BJ, Tazi A, Vassallo R, Weiss LM; Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016 Jun 2;127(22):2672-81. doi: 10.1182/blood-2016-01-690636[↩][↩]
- Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011 Oct 13;118(15):4041-52. doi: 10.1182/blood-2011-03-278127[↩][↩][↩]
- Ishii E. Hemophagocytic Lymphohistiocytosis in Children: Pathogenesis and Treatment. Front Pediatr. 2016 May 13;4:47. doi: 10.3389/fped.2016.00047[↩]
- Sieni E, Cetica V, Mastrodicasa E, Pende D, Moretta L, Griffiths G, Aricò M. Familial hemophagocytic lymphohistiocytosis: a model for understanding the human machinery of cellular cytotoxicity. Cell Mol Life Sci. 2012 Jan;69(1):29-40. doi: 10.1007/s00018-011-0835-y[↩]
- Filipovich AH, Chandrakasan S. Pathogenesis of Hemophagocytic Lymphohistiocytosis. Hematol Oncol Clin North Am. 2015 Oct;29(5):895-902. doi: 10.1016/j.hoc.2015.06.007[↩]
- Al-Samkari H, Berliner N. Hemophagocytic Lymphohistiocytosis. Annu Rev Pathol. 2018 Jan 24;13:27-49. doi: 10.1146/annurev-pathol-020117-043625[↩]
- Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. 2018;13:27–49. doi: 10.1146/annurev-pathol-020117-043625[↩]
- Ishii E. Hemophagocytic lymphohistiocytosis in children: Pathogenesis and treatment. Front Pediatr. 2016;4(47) doi: 10.3389/fped.2016.00047[↩]
- Skinner J, Yankey B, Shelton BK. Hemophagocytic lymphohistiocytosis. AACN Adv Crit Care. 2019;30:151–164. doi: 10.4037/aacnacc2019463[↩]
- Morimoto A, Nakazawa Y, Ishii E. Hemophagocytic lymphohistiocytosis: Pathogenesis, diagnosis, and management. Pediatr Int. 2016;58:817–825. doi: 10.1111/ped.13064[↩][↩]
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413[↩]
- Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: Differences and similarities. Br J Haematol. 2016;174:203–217. doi: 10.1111/bjh.14147[↩]
- Brisse E, Matthys P, Wouters CH. Understanding the spectrum of haemophagocytic lymphohistiocytosis: Update on diagnostic challenges and therapeutic options. Br J Haematol. 2016;174:175–187. doi: 10.1111/bjh.14144[↩]
- Castillo L, Carcillo J. Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med. 2009;10:387–392. doi: 10.1097/PCC.0b013e3181a1ae08[↩][↩][↩]
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004 Aug 1;104(3):735-43. doi: 10.1182/blood-2003-10-3413[↩]
- Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12:259–268. doi: 10.1038/nrrheum.2015.179[↩]
- Canna SW, Behrens EM. Making sense of the cytokine storm: A conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin North Am. 2012;59:329–344. doi: 10.1016/j.pcl.2012.03.002[↩]
- Jordan MB. Emergence of targeted therapy for hemophagocytic lymphohistiocytosis. Hematologist. 2018;15[↩]
- Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255–2273. doi: 10.1056/NEJMra2026131[↩][↩]
- Freeman HR, Ramanan AV. Review of haemophagocytic lymphohistiocytosis. Arch Dis Child. 2011;96:688–693. doi: 10.1136/adc.2009.176610[↩]
- Alonso EM, Horslen SP, Behrens EM, Doo E. Pediatric acute liver failure of undetermined cause: A research workshop. Hepatology. 2017;65:1026–1037. doi: 10.1002/hep.28944[↩]
- Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, Blechacz B, Wang S, Minkov M, Jordan MB, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123:3229–3240. doi: 10.1002/cncr.30826[↩]
- Lehmberg K, Nichols KE, Henter JI, Girschikofsky M, Greenwood T, Jordan M, Kumar A, Minkov M, La Rosée P, Weitzman S; Study Group on Hemophagocytic Lymphohistiocytosis Subtypes of the Histiocyte Society. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica. 2015 Aug;100(8):997-1004. doi: 10.3324/haematol.2015.123562[↩]
- Hemophagocytic Lymphohistiocytosis. https://rarediseases.org/rare-diseases/hemophagocytic-lymphohistiocytosis[↩][↩]
- Shah AJ, Kapoor N, Cooper RM, Crooks GM, Lenarsky C, Abdel-Azim H, Wu SQ, Wilson K, Weinberg KI, Parkman R, Kohn DB. Pre- and post-natal treatment of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2009 Jan;52(1):139-42. doi: 10.1002/pbc.21778[↩]
- Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH Jr, Simmons RL, Brunning RD. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979 Sep;44(3):993-1002. doi: 10.1002/1097-0142(197909)44:3<993::aid-cncr2820440329>3.0.co;2-5[↩]
- Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, Mehta PA, Davies SM, Jordan MB, Bleesing JJ, Filipovich AH. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010 Dec 23;116(26):5824-31. doi: 10.1182/blood-2010-04-282392[↩]
- Xu XJ, Wang HS, Ju XL, Xiao PF, Xiao Y, Xue HM, Shi HY, Gao YJ, Jia GC, Li XR, Zhao WH, Wang NL, Tang YM; Histiocytosis Study Group of the Chinese Pediatric Society. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: A retrospective multicenter study. Pediatr Blood Cancer. 2017 Apr;64(4). doi: 10.1002/pbc.26264[↩]
- Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007 Aug;29(8):569-73. doi: 10.1097/MPH.0b013e3180f61be3[↩]
- Imashuku S. Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002 Dec;44(3):259-72. doi: 10.1016/s1040-8428(02)00117-8[↩]
- Shiraishi A, Ohga S, Doi T, Ishimura M, Takimoto T, Takada H, Miyamoto T, Abe Y, Hara T. Treatment choice of immunotherapy or further chemotherapy for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2012 Aug;59(2):265-70. doi: 10.1002/pbc.24039[↩]
- Arca M, Fardet L, Galicier L, Rivière S, Marzac C, Aumont C, Lambotte O, Coppo P. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol. 2015 Jan;168(1):63-8. doi: 10.1111/bjh.13102[↩][↩]
- Imashuku S, Kuriyama K, Teramura T, Ishii E, Kinugawa N, Kato M, Sako M, Hibi S. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol. 2001 May 15;19(10):2665-73. doi: 10.1200/JCO.2001.19.10.2665[↩]
- Horne A, Wickström R, Jordan MB, Yeh EA, Naqvi A, Henter JI, Janka G. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis? Curr Treat Options Neurol. 2017 Jan;19(1):3. doi: 10.1007/s11940-017-0439-4[↩]
- Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, Bielekova B, Kennedy AL, Rivkin MJ, Davies KJ, Hsu AP, Holland SM, Gahl WA, Sundel RP, Lehmann LE, Lee MA, Alexandrescu S, Degar BA, Duncan CN, Gorman MP. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. 2019 Apr 8;6(3):e560. doi: 10.1212/NXI.0000000000000560[↩]
- Haddad E, Sulis ML, Jabado N, Blanche S, Fischer A, Tardieu M. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis. Blood. 1997 Feb 1;89(3):794-800.[↩]
- Lounder DT, Khandelwal P, Chandra S, Jordan MB, Kumar AR, Grimley MS, Davies SM, Bleesing JJ, Marsh RA. Incidence and Outcomes of Central Nervous System Hemophagocytic Lymphohistiocytosis Relapse after Reduced-Intensity Conditioning Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2017 May;23(5):857-860. doi: 10.1016/j.bbmt.2017.02.011[↩]
- Rego I, Severino M, Micalizzi C, Faraci M, Pende D, Dufour C, Aricò M, Rossi A. Neuroradiologic findings and follow-up with magnetic resonance imaging of the genetic forms of haemophagocytic lymphohistiocytosis with CNS involvement. Pediatr Blood Cancer. 2012 May;58(5):810-4. doi: 10.1002/pbc.23405[↩]
- Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, Janka G, Jordan MB, La Rosée P, Lehmberg K, Machowicz R, Nichols KE, Sieni E, Wang Z, Henter JI. Recommendations for the Use of Etoposide-Based Therapy and Bone Marrow Transplantation for the Treatment of HLH: Consensus Statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018 Sep-Oct;6(5):1508-1517. doi: 10.1016/j.jaip.2018.05.031[↩]
- Henter, J.-I., Horne, A., Aricó, M., Egeler, R.M., Filipovich, A.H., Imashuku, S., Ladisch, S., McClain, K., Webb, D., Winiarski, J. and Janka, G. (2007), HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer, 48: 124-131. https://doi.org/10.1002/pbc.21039[↩]
- Rubin TS, Zhang K, Gifford C, Lane A, Choo S, Bleesing JJ, Marsh RA. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood. 2017 Jun 1;129(22):2993-2999. doi: 10.1182/blood-2016-12-753830[↩]
- Hayden A, Lin M, Park S, Pudek M, Schneider M, Jordan MB, Mattman A, Chen LYC. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017 Dec 6;1(26):2529-2534. doi: 10.1182/bloodadvances.2017012310[↩]
- Lin M, Park S, Hayden A, Giustini D, Trinkaus M, Pudek M, Mattman A, Schneider M, Chen LYC. Clinical utility of soluble interleukin-2 receptor in hemophagocytic syndromes: a systematic scoping review. Ann Hematol. 2017 Aug;96(8):1241-1251. doi: 10.1007/s00277-017-2993-y[↩]
- Meeths M, Horne A, Sabel M, Bryceson YT, Henter JI. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015 Feb;62(2):346-352. doi: 10.1002/pbc.25308[↩]
- Bracaglia C, de Graaf K, Pires Marafon D, Guilhot F, Ferlin W, Prencipe G, Caiello I, Davì S, Schulert G, Ravelli A, Grom AA, de Min C, De Benedetti F. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017 Jan;76(1):166-172. doi: 10.1136/annrheumdis-2015-209020[↩]
- Buatois V, Chatel L, Cons L, Lory S, Richard F, Guilhot F, Johnson Z, Bracaglia C, De Benedetti F, de Min C, Kosco-Vilbois MH, Ferlin WG. Use of a mouse model to identify a blood biomarker for IFNγ activity in pediatric secondary hemophagocytic lymphohistiocytosis. Transl Res. 2017 Feb;180:37-52.e2. doi: 10.1016/j.trsl.2016.07.023[↩]
- Prencipe G, Caiello I, Pascarella A, Grom AA, Bracaglia C, Chatel L, Ferlin WG, Marasco E, Strippoli R, de Min C, De Benedetti F. Neutralization of IFN-γ reverts clinical and laboratory features in a mouse model of macrophage activation syndrome. J Allergy Clin Immunol. 2018 Apr;141(4):1439-1449. doi: 10.1016/j.jaci.2017.07.021[↩]
- Mellor-Heineke S, Villanueva J, Jordan MB, Marsh R, Zhang K, Bleesing JJ, Filipovich AH, Risma KA. Elevated Granzyme B in Cytotoxic Lymphocytes is a Signature of Immune Activation in Hemophagocytic Lymphohistiocytosis. Front Immunol. 2013 Mar 22;4:72. doi: 10.3389/fimmu.2013.00072[↩]
- Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008 Jun;50(6):1227-35. doi: 10.1002/pbc.21423[↩]
- Gurunathan A, Boucher AA, Mark M, Prus KM, O’Brien MM, Breese EH, Mizukawa BE, Absalon MJ, Nelson AS, Jordan MB, Grimley MS, Lorsbach RB, Rotz SJ, Mathanda R, Kumar AR. Limitations of HLH-2004 criteria in distinguishing malignancy-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2018 Dec;65(12):e27400. doi: 10.1002/pbc.27400[↩][↩]
- Tsuji T, Hirano T, Yamasaki H, Tsuji M, Tsuda H. A high sIL-2R/ferritin ratio is a useful marker for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann Hematol. 2014 May;93(5):821-6. doi: 10.1007/s00277-013-1925-8[↩]
- Bode SF, Ammann S, Al-Herz W, et al. Inborn Errors Working Party of the EBMT. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica. 2015 Jul;100(7):978-88. doi: 10.3324/haematol.2014.121608[↩]
- La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J, Girschikofsky M, Jordan MB, Kumar A, van Laar JAM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019 Jun 6;133(23):2465-2477. https://doi.org/10.1182/blood.2018894618[↩][↩]
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, Coppo P, Hejblum G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014 Sep;66(9):2613-20. doi: 10.1002/art.38690[↩]
- Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, Shi SW, Shen HP, Mao JQ, Zhang LY, Pan BH. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012 Jun;160(6):984-90.e1. doi: 10.1016/j.jpeds.2011.11.046[↩]
- Han XC, Ye Q, Zhang WY, Tang YM, Xu XJ, Zhang T. Cytokine profiles as novel diagnostic markers of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children. J Crit Care. 2017 Jun;39:72-77. doi: 10.1016/j.jcrc.2017.02.018[↩]
- Zondag TC, Rokx C, van Lom K, van den Berg AR, Sonneveld P, Dik WA, van Doornum GJ, Lam KH, van Laar JA. Cytokine and viral load kinetics in human herpesvirus 8-associated multicentric Castleman’s disease complicated by hemophagocytic lymphohistiocytosis. Int J Hematol. 2016 Apr;103(4):469-72. doi: 10.1007/s12185-015-1928-4[↩]
- Jordan MB, Filipovich AH. Hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis: a journey of a thousand miles begins with a single (big) step. Bone Marrow Transplant. 2008 Oct;42(7):433-7. doi: 10.1038/bmt.2008.232[↩]
- Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, Lee ND, Khan SP, Lawrence J, Mo JQ, Bleesing JJ, Filipovich AH, Jordan MB. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013 Jan;60(1):101-9. doi: 10.1002/pbc.24188[↩]
- Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. (2002) 100:2367–73. 10.1182/blood-2002-01-0172[↩]
- Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. (2014) 192:84–91. 10.4049/jimmunol.1302282[↩]
- Horne A, Wickström R, Jordan MB, Ann Yeh E, Naqvi A, Henter JI, et al. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis? Curr Treat Options Neurol. (2017) 19:3 10.1007/s11940-017-0439-4[↩][↩]
- Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract. (2018) 6:1508–17. 10.1016/j.jaip.2018.05.031[↩][↩][↩][↩]
- Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. (2017) 130:2728–38. 10.1182/blood-2017-06-788349[↩]
- Jabado N, de Graeff-Meeder ER, Cavazzana-Calvo M, Haddad E, Le Deist F, Benkerrou M, Dufourcq R, Caillat S, Blanche S, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with bone marrow transplantation from HLA genetically nonidentical donors. Blood. 1997 Dec 15;90(12):4743-8.[↩]
- Sonigo G, Battistella M, Beylot-Barry M, Ingen-Housz-Oro S, Franck N, Barete S, et al. HAVCR2 mutations are associated with severe hemophagocytic syndrome in subcutaneous panniculitis-like T-Cell lymphoma. Blood. (2020) 135:1058–61. 10.1182/blood.2019003811[↩]
- Wegehaupt O, Groß M, Wehr C, Marks R, Schmitt-Graeff A, Uhl M, Lorenz M, Schwarz K, Kratz C, Niemeyer C, Ehl S. TIM-3 deficiency presenting with two clonally unrelated episodes of mesenteric and subcutaneous panniculitis-like T-cell lymphoma and hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2020 Jun;67(6):e28302. doi: 10.1002/pbc.28302[↩]
- Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp Med. (2019) 216:2778–99. 10.1084/jem.20190147[↩][↩][↩]
- Rauch I, Deets RKA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and -8. Immunity. (2017) 46:649–59. 10.1016/j.immuni.2017.03.016[↩]
- Holle JR, Marsh RA, Holdcroft AM, Davies SM, Wang L, Zhang K, et al. Hemophagocytic lymphohistiocytosis in a female patient due to a heterozygous XIAP mutation and skewed X chromosome inactivation. Pediatr Blood Cancer. (2015) 62:1288–90. 10.1002/pbc.25483[↩]
- Popow I, Leitner PJ, Grabmeier-Pfistershammer K, Majdic O, Zlabinger G-J, Kundi M, et al. A comprehensive and quantitative analysis of the major specificities in rabbit antithymocyte globulin preparations. Am J Transplant. (2013) 13:3103–13. 10.1111/ajt.12514[↩]
- Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, Neven B, Picard C, Blanche S, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. (2007) 120:e622–8. 10.1542/peds.2006-3164[↩]
- Moshous D, Briand C, Castelle M, Dupic L, Morelle G, Chahla WA, et al. Alemtuzumab as first line treatment in children with familial lymphohistiocytosis. Blood. (2019) 134(Suppl. 1:80). 10.1182/blood-2019-124477[↩]
- Strout MP, Seropian S, Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol. (2010) 7:415–20. 10.1038/nrclinonc.2010.40[↩]
- Al-Salama ZT. Emapalumab: first global approval. Drugs. (2019) 79:99–103. 10.1007/s40265-018-1046-8[↩]
- FDA approves emapalumab for hemophagocytic lymphohistiocytosis. https://www.fda.gov/drugs/fda-approves-emapalumab-hemophagocytic-lymphohistiocytosis[↩][↩][↩]
- Takada H, Takahata Y, Nomura A, Ohga S, Mizuno Y, Hara T. Increased serum levels of interferon-gamma-inducible protein 10 and monokine induced by gamma interferon in patients with haemophagocytic lymphohistiocytosis. Clin Exp Immunol. (2003) 133:448–53. 10.1046/j.1365-2249.2003.02237.x[↩]
- Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood. (2016) 128:60–71. 10.1182/blood-2016-02-700013[↩]
- Das R, Guan P, Sprague L, Verbist K, Tedrick P, An QA, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. (2016) 127:1666–75. 10.1182/blood-2015-12-684399[↩]
- Slostad J, Hoversten P, Haddox CL, Cisak K, Paludo J, Tefferi A. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: a single patient experience. Am J Hematol. (2018) 93:E47–9. 10.1002/ajh.24971[↩]
- Sin JH, Zangardi ML. Ruxolitinib for secondary hemophagocytic lymphohistiocytosis: first case report. Hematol Oncol Stem Cell Ther. (2019) 12:166–70. 10.1016/j.hemonc.2017.07.002[↩]
- Broglie L, Pommert L, Rao S, Thakar M, Phelan R, Margolis D, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv. (2017) 1:1533–6. 10.1182/bloodadvances.2017007526[↩]
- Ahmed A, Merrill SA, Alsawah F, Bockenstedt P, Campagnaro E, Devata S, et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol. (2019) 6:e630–7. 10.1016/S2352-3026(19)30156-5[↩]
- Kaplanski G. Interleukin-18: biological properties and role in disease pathogenesis. Immunol Rev. (2018) 281:138–53. 10.1111/imr.12616[↩]
- Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. (2018) 131:1442–55. 10.1182/blood-2017-12-820852[↩]
- Chiossone L, Audonnet S, Chetaille B, Chasson L, Farnarier C, Berda-Haddad Y, et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front Immunol. (2012) 3:239 10.3389/fimmu.2012.00239[↩]
- Grom A, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. (2016) 12:259–68. 10.1038/nrrheum.2015.179[↩]
- Chamseddin B, Marks E, Dominguez A, Wysocki C, Vandergriff T. Refractory macrophage activation syndrome in the setting of adult−onset still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutaneous Pathol. (2019) 46:528–31. 10.1111/cup.13466[↩]
- Kotch C, Barrett D, Teachey DT. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol. (2019) 15:813–22. 10.1080/1744666X.2019.1629904[↩]
- Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. (2014) 46:1140–6. 10.1038/ng.3089[↩]
- de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. (2014) 111:3526–31. 10.1073/pnas.1322831111[↩]
- Wustrau K, Gross M, Lenhartz H, Oyen F, Ehl S, Müller I, et al. Tocilizumab Strikingly Abates Inflammation in a Girl with Symptomatic Deficiency of the X-Linked Inhibitor of Apoptosis (XIAP) with Extremely Skewed X-Inactivation. Hamburg: Katharina Wustrau’s research works; (2019).[↩]
- Milone MC, Tsai DE, Hodinka RL, Silverman LB, Malbran A, Wasik MA, et al. Treatment of primary Epstein-Barr virus infection in patients with X-linked lymphoproliferative disease using B-cell-directed therapy. Blood. (2005) 105:994–6. 10.1182/blood-2004-07-2965[↩]
- Kasahara Y, Yachie A, Takei K, Kanegane C, Okada K, Ohta K, et al. Differential cellular targets of Epstein-Barr virus. (EBV) infection between acute EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Blood. (2001) 98:1882–8. 10.1182/blood.v98.6.1882[↩]
- Sung L, King SM, Carcao M, Trebo M, Weitzman SS. Adverse outcomes in primary hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. (2002) 24:550–4. 10.1097/00043426-200210000-00011[↩]
- Trottestam H, Berglöf E, Horne A, Onelöv E, Beutel K, Lehmberg K, et al. Risk factors for early death in children with haemophagocytic lymphohistiocytosis: outcome predictors in HLH. Acta Paediatr. (2012) 101:313–8. 10.1111/j.1651-2227.2011.02501.x[↩]
- Marsh RA, Jordan MB, Talano JA, Nichols KE, Kumar A, Naqvi A, et al. Salvage therapy for refractory hemophagocytic lymphohistiocytosis: a review of the published experience. Pediatr Blood Cancer. (2017) 64:e26308 10.1002/pbc.26308[↩]
- Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. (2013) 60:101–9. 10.1002/pbc.24188[↩]
- Vallurupalli M, Berliner N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood. (2019) 134:1783–6. 10.1182/blood.2019002289[↩]
- Lounder DT, Bin Q, de Min C, Jordan MB. Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv. (2019) 3:47–50. 10.1182/bloodadvances.2018025858[↩]
- Carter SJ, Tattersall RS, Ramanan AV. Macrophage activation syndrome in adults: recent advances in pathophysiology, diagnosis and treatment. Rheumatology (Oxford). 2019 Jan 1;58(1):5-17. doi: 10.1093/rheumatology/key006[↩]
- Schram AM, Comstock P, Campo M, Gorovets D, Mullally A, Bodio K, Arnason J, Berliner N. Haemophagocytic lymphohistiocytosis in adults: a multicentre case series over 7 years. Br J Haematol. 2016 Feb;172(3):412-9. doi: 10.1111/bjh.13837[↩]





{kind=link}