Contents
What is galactosemia
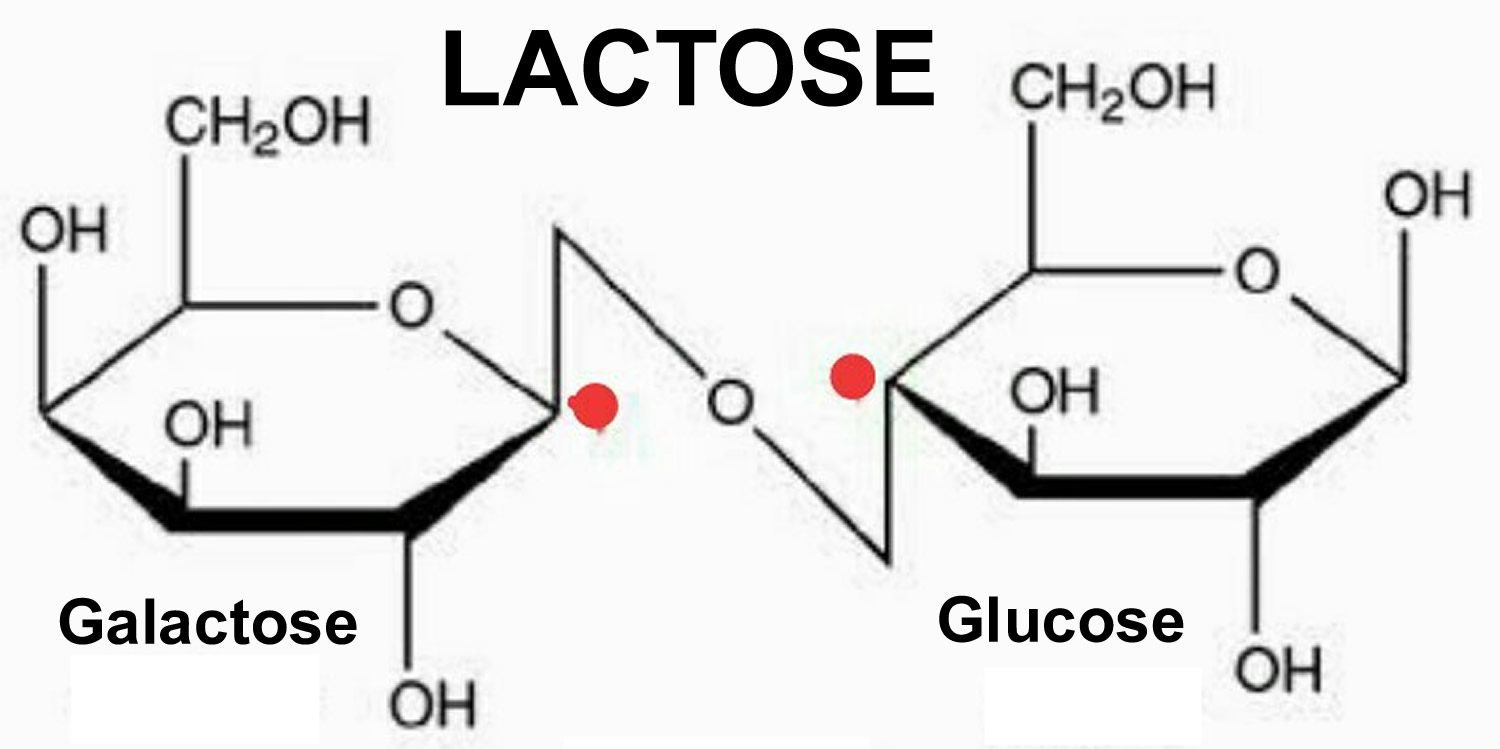
Galactosemia is a rare, inherited disorder of carbohydrate metabolism that affects the body’s ability to convert galactose (a sugar contained in milk, including human mother’s milk) to glucose (a different type of sugar). A small amount of galactose is present in many foods. Galactose is primarily part of a larger sugar called lactose, which is found in all dairy products and many baby formulas. Lactose is a disaccharide, a sugar composed of galactose and glucose. Galactosemia is caused by a deficiency of an enzyme galactose-1-phosphate uridyl transferase (GALT) which is vital to this process 1. The signs and symptoms of galactosemia result from an inability to use galactose to produce energy. If an infant with galactosemia is given milk, substances made from galactose build up in the infant’s system. These substances damage the liver, brain, kidneys, and eyes. People with galactosemia cannot tolerate any form of milk (human or animal). They must be careful about eating other foods containing galactose. Early diagnosis and treatment with a lactose-free diet is absolutely essential to avoid profound intellectual disability, liver failure and death in the newborn period. Galactosemia is inherited as an autosomal recessive genetic condition.
Researchers have identified 4 types of galactosemia. These conditions are each caused by mutations in a particular gene and affect different enzymes involved in breaking down galactose.
The 4 forms of galactosemia are:
- Galactose-1 phosphate uridyl transferase deficiency: Classic galactosemia also known as type I, the most common and most severe form
- Deficiency of galactose kinase, also known as galactosemia type II
- Deficiency of galactose-6-phosphate epimerase, also known as galactosemia type III
- Duarte variant galactosemia or Duarte galactosemia
Classic galactosemia, also known as type I, is the most common and most severe form of the condition. If infants with classic galactosemia are not treated promptly with a low-galactose diet, life-threatening complications appear within a few days after birth. Affected infants typically develop feeding difficulties, a lack of energy (lethargy), a failure to gain weight and grow as expected (failure to thrive), yellowing of the skin and whites of the eyes (jaundice), liver damage, and abnormal bleeding. Other serious complications of this condition can include overwhelming bacterial infections (sepsis) and shock. Affected children are also at increased risk of delayed development, clouding of the lens of the eye (cataract), speech difficulties, and intellectual disability. Females with classic galactosemia may develop reproductive problems caused by an early loss of function of the ovaries (premature ovarian insufficiency).
Galactosemia type II (also called galactokinase deficiency) and type III (also called galactose epimerase deficiency) cause different patterns of signs and symptoms. Galactosemia type II causes fewer medical problems than the classic type I galactosemia. Affected infants develop cataracts but otherwise experience few long-term complications. The signs and symptoms of galactosemia type III (galactose epimerase deficiency) vary from mild to severe and can include cataracts, delayed growth and development, intellectual disability, liver disease, and kidney problems.
Duarte variant galactosemia, sometimes called just Duarte galactosemia, is much more common than classic or clinical variant galactosemia in many populations and also results from mutations in the GALT gene 2. However, instead of carrying severe mutations in both copies of their GALT gene, patients with Duarte variant galactosemia carry one GALT copy with a severe (G) mutation and a second GALT copy that is only very mildly impaired and that shows a collection of characteristic sequence changes that classify it as “Duarte” (also called D or D2). A child with Duarte variant galactosemia therefore generally has one parent who is a carrier for a severe (G) GALT mutation, and one parent who is a carrier for the Duarte variant. Patients with Duarte variant galactosemia usually show about 25% the normal level of GALT activity in red blood cells. Newborns with Duarte variant galactosemia may not show any symptoms, such as jaundice, while drinking milk. Experts therefore disagree about whether infants with Duarte variant galactosemia should be put on a lactose/galactose restricted diet, and no one knows whether older children or adults with Duarte variant galactosemia are at increased risk for long-term complications. Reported studies give mixed or inconclusive results, so that more research is needed to answer the question. If long-term developmental complications do occur in patients with Duarte variant galactosemia they are generally believed to be milder than those experienced by patients with classic galactosemia. Also, girls and women with Duarte variant galactosemia are not believed to be at risk for premature ovarian insufficiency.
Newborns with Duarte variant galactosemia may or may not be detected by the same newborn screening test that detects classic or clinical variant galactosemia. Specifically, some newborn screening protocols are designed to detect Duarte variant galactosemia, while others do not. Receiving a “normal” newborn screening result for galactosemia therefore may not rule out a diagnosis of Duarte variant galactosemia 2.
Classic galactosemia occurs in 1 in 30,000 to 60,000 newborns 3 through newborn screening programs around the world, depending on the diagnostic criteria used by the program. Galactosemia has been reported in all ethnic groups. An increased frequency of galactosemia occurs in individuals of Irish ancestry. Clinical variant galactosemia occurs most often in African Americans and native Africans in South Africa who have a specific GALT gene mutation.
Galactosemia type II and type III are less common; type II probably affects fewer than 1 in 100,000 newborns and type III appears to be very rare 3.
Classic galactosemia (type I galactosemia) and clinical variant galactosemia can both result in life-threatening health problems unless treatment is started shortly after birth. A biochemical variant form of galactosemia termed Duarte galactosemia is not thought to cause clinical disease 1.
Other names for galactosemia:
- classic galactosemia
- epimerase deficiency galactosemia
- galactokinase deficiency disease
- galactose-1-phosphate uridyl-transferase deficiency disease
- galactose epimerase deficiency
- GALE deficiency
- GALK deficiency
- GALT deficiency
- UDP-galactose-4-epimerase deficiency disease
- UTP hexose-1-phosphate uridylyltransferase deficiency
Figure 1. Galactose
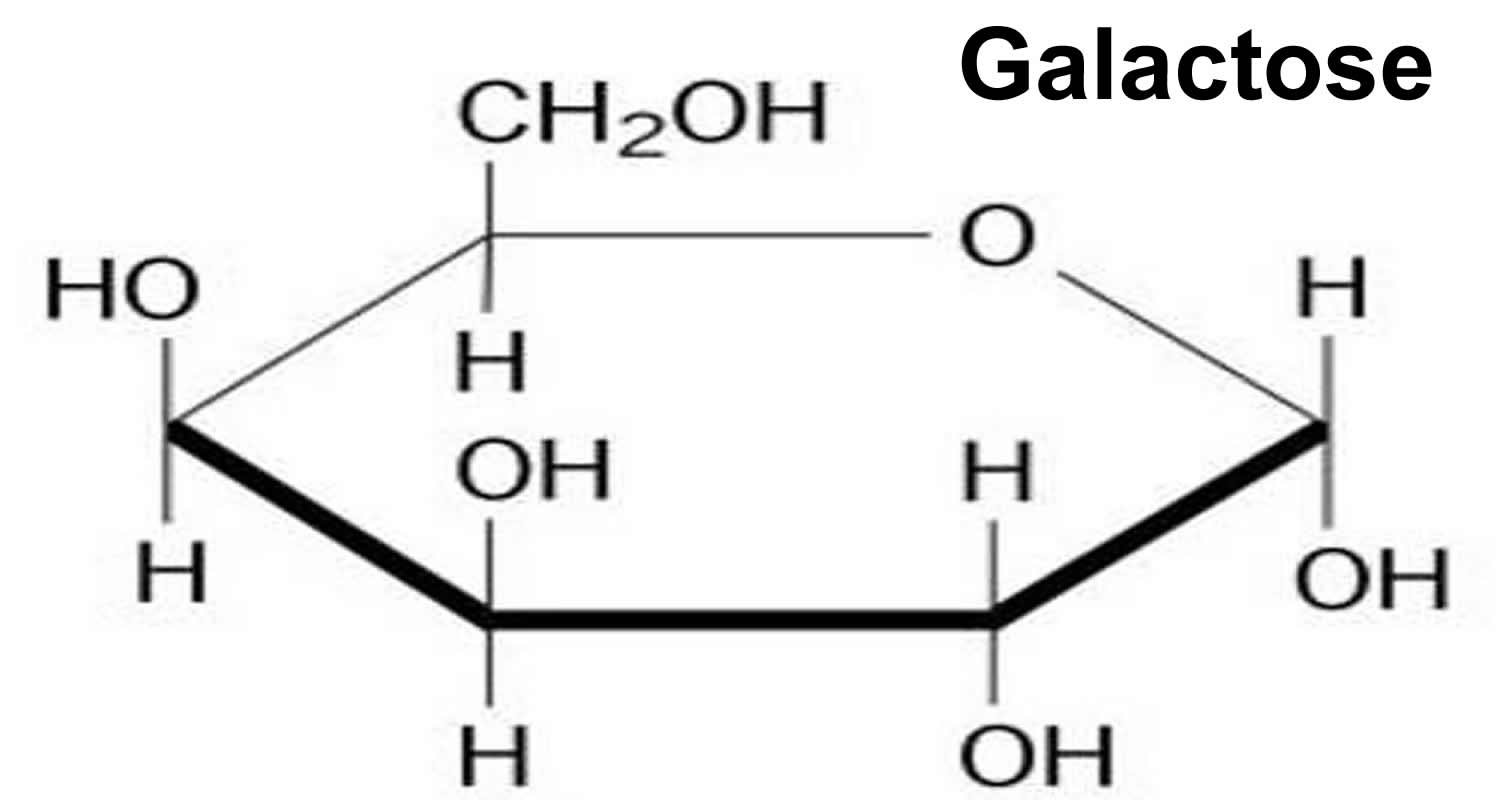
Figure 2. Lactose
Galactosemia life expectancy
People who are diagnosed early and strictly avoid milk products can live a relatively normal life. However, mild mental impairment may develop, even in people who avoid galactose 4.
Galactosemia possible complications
These complications can develop:
- Cataracts
- Cirrhosis of the liver
- Delayed speech development
- Irregular menstrual periods, reduced function of ovaries leading to ovarian failure
- Mental disability
- Severe infection with bacteria (E coli sepsis)
- Tremors (shaking) and uncontrollable motor functions
- Death (if there is galactose in the diet)
Learning disabilities
A learning disability is a disorder that interferes with a person’s ability to master a skill (such as reading, writing, arithmetic concepts, etc.). Learning disabilities can show up in many ways such as, specific difficulties with spoken and written language, coordination, self-control, or attention.
Learning disabilities can be lifelong conditions that, in some cases, affect many parts of a person’s life: school or work, daily routines, family and social life. Some people, may have overlapping learning disabilities. Other people may have a single, isolated learning problem that has little or no impact on other areas of their lives. Also, some individuals can manage, or even overcome, learning diabilities with focused therapy or specialized instruction, and/or medication.
Although no one really knows exactly why, there have been some specific learning disabilities associated with classic galactosemia. (Note – there have been no learning disabilities positively associated with Duarte Galactosemia) 5. Even some children who were diagnosed relatively quickly after birth and who are following the “restricted diet” have developed learning disabilities. Some of the learning disabilities associated with galactosemia include: speech and language difficulties, fine and/or gross motor difficulties, and difficulty with math or reading in school. Unfortunately, there are no firm numbers to quantify the percentage of galactosemics who experience learning disabilites.
It is important to know that not all children with galactosemia have learning disabilities. Because many galactosemic children do have problems, it is something very important to be aware of in observing a child’s development.
One aspect of learning disabilities and galactosemia that is important for parents to keep in mind is that neurological impairments (e.g. fine motor difficulties) can sometimes present themselves “disguised” as a learning disability. For example, a child with trouble writing numerals or pointing may appear to have a learning disability with regard to arithmetic concepts, when the case may well be that the child understands the math concept just fine, but simply cannot control his/her writing sufficiently well enough to demonstrate mastery of the concept. It is important to note that a child may in fact have both problems.
How do I know if my child has a learning disability? (Ages 0 – 3)
Because every child develops differently, this can be a difficult question to answer. Your child’s pediatrician should be your first source of information. He or she should be able to, with your input, notice any signs of problems in your child’s development. Be sure to inform your doctor of the types of problems that have been associated with galactosemia. (Because galactosemia is very rare, your doctor may know very little, if anything, about galactosemia). If he/she knows what to “look for”, it can be very helpful to him/her. Also, sometimes parents know best, so if you suspect a problem, and your doctor does not agree with you, get a second opinion. Research shows that the earlier a problem is treated, the better it is for the child. Remember to keep in mind that children develop at different rates – learning disability is defined as a significant gap between a person’s intelligence and the skills the person has achieved at each age.
What kind of services should I get for my child if I suspect a developmental problem or learning disability?
Many local school systems offer free programs for young children (infants and toddlers) as well as for older children. You can call your local school system to ask for an evaluation for your child and/or to ask that your child be put into a program that would be appropriate for him. If you are unable to get services form you local school system, try a private therapist/teacher or institution. Some medical insurance companies may pay for all or part of these services. Check with your own policy.
Neurological impairments
Along with well documented speech and language disorders, neurodevelopmental delays are also sometimes observed in galactosemic patients. By some estimates, galactosemics experience trouble with gait, balance, and fine motor tremors in anywhere from 13 to 20 %.
In one study, 45 individuals with galactosemia were examined. In that study, 12 individuals were observed to have neurological symptoms that included ataxia, tremors, and dysmetria.
Ataxia is a total or partial inability to coordinate voluntary bodily movements (as in walking, etc.).
Tremors are rhythmic, involuntary muscular contractions characterized by oscillations (to-and-fro movements) of a part of the body. The most common of all involuntary movements, tremor can affect various body parts such as the hands, head, facial structures, vocal cords, trunk, and legs; most tremors, however, occur in the hands. Although the disorder is not life-threatening, it can be responsible for functional disability and social embarrassment. There are different types of tremors. One type is Kinetic or Intention tremor which occurs during purposeful goal-oriented tasks, for example finger-to-nose testing.
Dysmetria is improper estimation of distance during muscular activity. Dysmetria includes both hypo- and hypermetria. With hypermetria, voluntary muscular movement overreaches the intended goal; with hypometria, voluntary movement falls short of the intended goal. Hypermetria is more commonly recognized than hypometria.
If you suspect that your child may be exhibiting symptoms of any of these conditions, consult your child’s medical professional and alert them to the possibility that such a condition may be related to galactosemia. Treatment for these conditions include various types of physical therapy and/or medications.
Primary Ovarian Insufficiency
A majority of girls/women who have classic galactosemia experience Primary Ovarian Insufficiency. However, there are women with classic galactosemia who have successfully conceived and given birth. To date, the reason for the high rate of ovarian failure is not known. Talk with your child’s geneticist to get the latest information about this issue. There are some tests (hormone level testing) which may be performed to check the condition of the ovaries.
(Note: There is no evidence that galactosemia has any negative effect on the reproductive health of boys.)
Speech disorders
It is believed that approximately, 60% of classic galactosemic children have speech problems. Problems range from mild to moderate or severe. One type of speech disorder that has been associated with classic galactosemia is apraxia of speech, often referred to as dyspraxia. Dyspraxia is not a developmental delay of speech. It is considered a “motor speech disorder”.
Verbal dyspraxia is defined below:
Verbal Dyspraxia: A sensory motor disorder of articulation characterized by impaired capacity to plan the positioning of speech musculature and muscle movements for the production of speech sounds.
While it is primarily an articulation disorder, there are a number of other related communication problems associated with dyspraxia, such as: problems of syntax (word order), language organization, and pragmatics (set of rules governing conversation). (Note: reading, writing, spelling, and spatial awareness can also be affected.)
If you suspect that your child has this disorder, or some other speech problem, have your child evaluated as soon as possible. The sooner speech therapy is started for a child who needs it, the better. Hearing tests are usually performed first to rule out any kind of hearing impairment. If your child’s hearing has been checked, and a hearing problem has been ruled out as a cause for speech problems, have your child evaluated by a qualified speech pathologist. You may be able to find a qualified speech pathologist through your local school system (early intervention program.) If you suspect dyspraxia, be sure to find a speech language pathologist who is qualified, and has experience with diagnosing and treating a motor speech disorder (or oral motor function disorder).
Cataracts
A cataract is a clouding of the lens of the eye. The lens is a crystal-clear, flexible structure near the front of the eyeball. It helps to keep vision in focus, and screens and refracts light rays. The lens has no blood supply. It is nourished by the vitreous (the watery substance that surrounds it). Cataracts may form in one or both eyes. If they form in both eyes, their growth rate may be very different. Cataracts are not cancerous.
Appropriate health care includes treatment by an ophthalmologist or surgery to remove the lens.
Cataracts are one of the possible complications of classic galactosemia. Cataracts are mostly observed in newborns but can also occur in adults. It is thought that 10-30% of newborns with classic galactosemia develop cataracts in the first few days or weeks of life. Once a newborn is put on a galactose-restricted diet, cataracts usually clear up on their own. Surgery is sometimes necessary in rarer cases.
It is believed that if the galactose restricted diet is followed, cataracts do not develop in galactosemic children.
Many patients have eye examinations to check for the presence of cataracts on a regular basis. More frequent during the first year of life (e.g every 3-4 months), such exams can be reduced in frequency (e.g. 1 or 2 times a year) in older children. It is a good idea to have an eye exam if for some reason Gal-1-P levels are observed to rise above a ‘target’ range.
Galactosemia symptoms
Galactosemia occurs due to disruptions or changes (mutations) of the GALT gene. The abnormal accumulation of galactose-related chemicals in various organs of the body causes the signs and symptoms and physical findings of galactosemia.
An infant with galactosemia appears normal at birth, but within a few days or weeks loses his or her appetite (anorexia) and starts vomiting excessively if they eat infant formula or breast milk that contains lactose. Yellowing of the skin, mucous membranes, and whites of the eyes (jaundice), enlargement of the liver (hepatomegaly), appearance of amino acids and protein in the urine, growth failure, and, ultimately, accumulation of fluid in the abdominal cavity (ascites) with abdominal swelling (edema) may also occur. Diarrhea, irritability, lethargy and a bacterial infection may also be early signs of galactosemia. In time, wasting of body tissues, marked weakness, and extreme weight loss occur unless lactose is removed from the diet.
Symptoms of galactosemia are:
- Convulsions
- Irritability
- Lethargy
- Poor feeding — baby refuses to eat formula containing milk
- Poor weight gain
- Yellow skin and whites of the eyes (jaundice)
- Vomiting
Children with galactosemia who have not received early treatment may show arrested physical and mental development and are particularly susceptible to cataracts in infancy or childhood. In severe cases, overwhelming infection can cause life-threatening complications, but mild cases present few symptoms and no serious impairment(s).
In order to avoid the consequences of galactosemia, which may include liver failure and kidney dysfunction, brain damage and/or cataracts, infants must be treated promptly by removing lactose from the diet. Children treated with this special diet usually show satisfactory general health and growth. They can make reasonable, though often not optimal, intellectual progress. Speech and learning difficulties and some behavioral problems are still likely to occur. Ovarian impairment is almost always seen in girls with classic galactosemia and is associated with an increase in the blood level of the gonadotropin hormone follicle-stimulating hormone (FSH); males with galactosemia do not usually exhibit abnormalities in gonadal function.
The above-mentioned complications associated with classic galactosemia and clinical variant galactosemia have not occurred in individuals with Duarte variant galactosemia, which is the best example of biochemical variant galactosemia subdivision. Significant debate exists in the medical literature as to whether individuals with Duarte variant galactosemia need to maintain a special diet.
Galactosemia causes
Galactosemia is an autosomal recessive hereditary disorder caused by a deficiency of the enzyme galactose-1- phosphate uridyl transferase (GALT) that is needed for the breakdown of the milk sugar, galactose. Deficiency of this enzyme results in the accumulation of toxic products: galactose-1-phosphate (a derivative of galactose), and galactitol (an alcohol derivative of galactose). Galactitol accumulates in the lens of the eye where it causes lens swelling and protein precipitation and, subsequently, cataracts. Accumulation of galactose-1-phosphate is thought to cause the other signs and symptoms of disease.
Mutations in the GALT, GALK1, and GALE genes cause galactosemia. These genes provide instructions for making enzymes that are essential for processing galactose obtained from the diet. These enzymes break down galactose into another simple sugar, glucose, and other molecules that the body can store or use for energy.
Mutations in the GALT gene cause classic galactosemia (type I). Most of these genetic changes almost completely eliminate the activity of the enzyme produced from the GALT gene, preventing the normal processing of galactose and resulting in the life-threatening signs and symptoms of this disorder. Another GALT gene mutation, known as the Duarte variant, reduces but does not eliminate the activity of the enzyme. People with the Duarte variant tend to have much milder features of galactosemia.
Galactosemia type II results from mutations in the GALK1 gene, while mutations in the GALE gene underlie galactosemia type III. Like the enzyme produced from the GALT gene, the enzymes made from the GALK1 and GALE genes play important roles in processing galactose. A shortage of any of these critical enzymes allows galactose and related compounds to build up to toxic levels in the body. The accumulation of these substances damages tissues and organs, leading to the characteristic features of galactosemia.
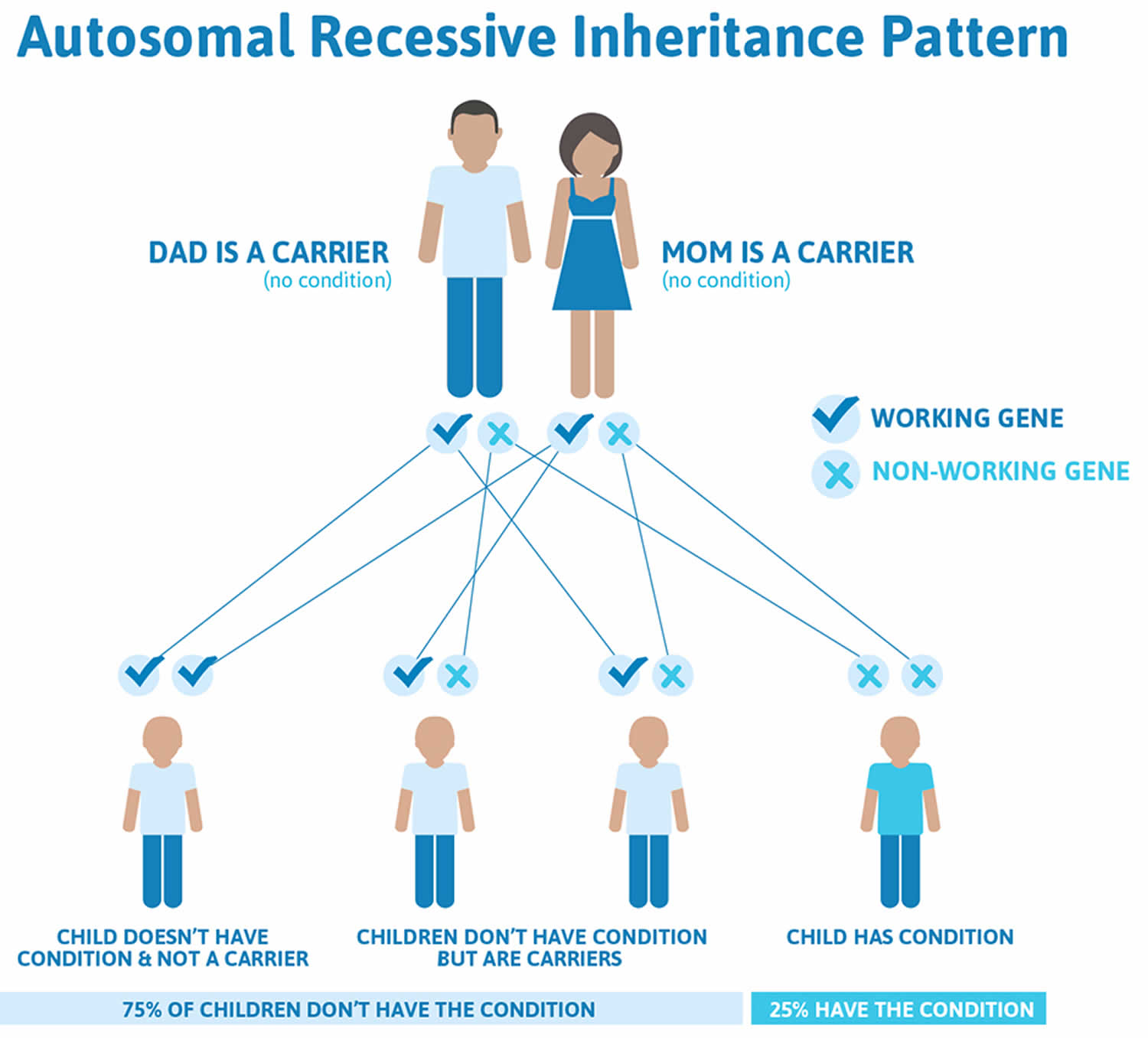
Recessive genetic diseases are determined by two genes, one received from the father and one from the mother. They occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%.
Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
If you’re a carrier of the affected genes and have a baby with a partner who’s also a carrier, your baby has:
- a 1 in 4 chance of developing the condition
- a 1 in 2 chance of being a carrier of maple syrup urine disease
- a 1 in 4 chance of receiving a pair of normal genes
Figure 3. Galactosemia autosomal recessive inheritance pattern
Galactosemia prevention
It is helpful to know your family history. If you have a family history of galactosemia and want to have children, genetic counseling will help you make decisions about pregnancy and prenatal testing. Once the diagnosis of galactosemia is made, genetic counseling is recommended for other members of the family.
Many states screen all newborns for galactosemia. If the newborn test shows possible galactosemia, they should stop giving their infant milk products right away and ask their provider about having blood tests that can be done to confirm a diagnosis of galactosemia.
Galactosemia diagnosis
Classic galactosemia and clinical variant galactosemia are diagnosed when galactose-1-phosphate is elevated in red blood cells and GALT enzyme activity (erythrocyte galactose-1-phosphate uridylyltransferase (GALT) enzyme activity) is reduced. Molecular genetic testing is also available to identify mutations in the GALT gene.
Nearly 100% of infants with galactosemia can be diagnosed in newborn screening programs using a blood sample from the heel stick. Infants with clinical variant galactosemia can be missed at newborn screening if GALT enzyme activity is not measured.
Galactosemia test
Tests to check for galactosemia include:
- Blood culture for bacterial infection (E coli sepsis)
- Enzyme activity in the red blood cells
- Ketones in the urine
- Prenatal diagnosis by directly measuring the enzyme galactose-1-phosphate uridyl transferase
- “Reducing substances” in the infant’s urine, and normal or low blood sugar while the infant is being fed breast milk or a formula containing lactose
Newborn screening tests in many states check for galactosemia.
Test results may show:
- Amino acids in the urine or blood plasma
- Enlarged liver
- Fluid in the abdomen
- Low blood sugar
Galactosemia treatment
Currently there is no therapy that permits a person with Galactosemia to eat an unrestricted diet, without concern of complications. People with galactosemia must avoid all milk, products that contain milk (including dry milk), and other foods that contain galactose, for life. Read product labels to make sure you or your child with the condition are not eating foods that contain galactose.
Infants can be fed:
- Soy formula
- Another lactose-free formula
- Meat-based formula or Nutramigen (a protein hydrolysate formula)
Calcium supplements are recommended.
Infants and children with galactosemia should reduce galactose intake and have a diet that contains lactose-free milk substitutes and other foods such as soy bean products.
A lactose tolerance test should NOT be administered to galactosemic children. Fortunately the body of an infant with galactosemia can synthesize galactolipids and other essential galactose-containing compounds without the presence of galactose in food. Therefore, satisfactory physical development is possible if a strict diet is followed.
Speech therapy may be necessary for children with speech apraxia or dysarthria. For school age children, individual education plans and/or professional help with learning skills may be necessary for some individuals, depending on psychological developmental assessments. Hormone replacement therapies may also be used in cases of delayed puberty.
Appropriate treatment (i.e., antibiotic drugs) may be used to control infection. The emotional effects of the strict diet may require attention and supportive measures throughout childhood. Genetic counseling is recommended for families with children who have galactosemia.
Galactosemia diet
Below is a list of diet resources that some parents follow, however, please always check with your healthcare provider for diet advice.
UNACCEPTABLE INGREDIENTS
Since actual products change on a regular basis it is imperative that you re-read every label every time you buy. Various parents and dietitians have put together this list of unacceptable ingredients in an attempt to simplify the ingredient dilemma.
Please, remember to always check with your clinic and dietitian. Gather all information available and then make your own decision.
Food Ingredients which are UNACCEPTABLE in the diet for Galactosemia:
- Butter
- Buttermilk
- Buttermilk Solids
- Cheese (EXCEPTIONS: Jarlsberg, Gruyere, Emmentaler, Swiss, Tilster, grated 100% Parmesan, Parmesan aged >10 months, and sharp Cheddar cheese)
- Cream
- Dough Conditioners*
- Dry Milk
- Dry Milk Protein
- Dry Milk Solids
- Ghee
- Hydrolyzed Whey**
- Ice Cream
- Lactalbumin
- Lactose
- Lactoglobulin
- Lactostearin
- Margarine***
- Milk
- Milk Chocolate
- Milk Solids
- Milk Derivatives
- MSG (Monosodium Glutamate)****
- Nonfat Milk
- Nonfat Dry Milk
- Nonfat Dry Milk Solids
- Organ Meats (liver, heart, kidney, brains, pancreas)
- Sherbert
- Sour Cream
- Fermented Soy products and Soy Sauce*****
- Whey and Whey Solids
- Yogurt
- Tragacanth Gum
NOTE: Lactate, Lactic acid and Lactylate do not contain lactose and are acceptable ingredients.
* Dough Conditioners may include caseinates which are UNACCEPTABLE. Most labels specify the name of the conditioner which is added to the product. If not, contact the company to make sure that all are acceptable.
** Hydrolyzed protein is UNACCEPTABLE and is commonly found in canned meats, like tuna. Hydrolyzed vegetable protein, however, is acceptable.
*** A few diet margarine’s do not contain milk. Check labels before using any brand. If “margarine” is listed as an ingredient in any processed food, consider the product UNACCEPTABLE.
**** MSG or Monosodium Glutamate itself is acceptable; however, some MSG’s contain lactose extenders. It is best to avoid MSG whenever possible.
***** Soy sauce is UNACCEPTABLE if it is fermented. Brands must be checked before including this in the Galactosemic diet.
Calcium supplementation recommendations
Given the necessary restriction on dairy items in the galactosemic diet, parents sometimes wonder whether their child with Galactosemia is getting sufficient calcium.
Ask your doctor or clinic what the recommended calcium intake is for your child. If your child sees a nutritionist, you may ask him/her to perform a three-day diet analysis to determine if your child is getting enough calcium (as well as other nutrients).
There are a number of natural food sources of calcium among the foods acceptable for a galactosemic diet. If you are advised to increase your child’s daily intake of calcium, it may be best to try to increase these natural sources in your child’s diet before turning to supplements.
Advice on calcium supplements varies from clinic to clinic. Below is a list of calcium supplements that some parents are using. Keep in mind that this list just represents some of the types of calcium supplements given to children with galactosemia and is NOT an endorsement for any of the products. As with anything, check labels carefully (for restricted ingredients) and always check with your own doctor/clinic before giving any supplement to your child. Keep in mind companies frequently change ingredients in their products.
- Tums – some are still using, although there has been talk about Tums being bad for tooth enamel among other things
- Centrum Vitamins – “Bone Health” – suppose to be lactose and dairy-free (It is like a regular vitamin)
- CalQuick (Twin Labs) – liquid calcium supplement (600 mg/tablespoon)
- Liquid Cal Mag+ (KAL) (600 mg/tablespoon)
- Multi-vitamins with extra calcium
Keep in mind that there are different forms of calcium used in supplements (e.g. calcium carbonate, calcium phosphate, calcium citrate, calcium gluconate, etc.). Some forms of calcium are thought to be more easily absorbed into the body than others. Another factor to consider is that levels of calcium in the blood may not always indicate the actual calcium used by the body to increase bone density. Ask your nutritionist for advice on this matter.
Calcium recommendations vary depending on age and special needs. In addition, levels of estrogen can affect calcium needs in women. Note: Calcium requires adequate Vitamin D to be absorbed into the body
Duarte Galactosemia treatment
Currently no agreement exists as to whether infants with Duarte variant galactosemia benefit from dietary galactose restriction during infancy and early childhood 2. Thus, healthcare providers or parents may choose either to restrict dietary galactose in the first year of life or not. When dietary galactose is restricted in infancy, it is recommended that the child undergo a galactose challenge around age one year followed by measurement of the erythrocyte galactose-1-phosphate level 2. If the level is within the normal range (<1.0 mg/dL), dietary restriction of galactose is generally discontinued 2. When dietary galactose is not restricted in infancy, some healthcare providers choose to check the erythrocyte galactose-1-phosphate level at age one year to assure that the level is approaching the normal range.
Surveillance: For infants on dietary restriction of galactose, if the erythrocyte galactose-1-phosphate level is >1.0 mg/dL following a galactose challenge at age one year, galactose restriction may be resumed 2. In this case, the galactose challenge and measurement of erythrocyte galactose-1-phosphate level may be repeated every 4-6 months until the erythrocyte galactose-1-phosphate level stabilizes at <1.0 mg/dL 2.
Agents/circumstances to avoid: Opinion varies as to whether avoidance of all sources of milk and dairy products until age one year is warranted 2.
Evaluation of relatives at risk: If families with one child with Duarte variant galactosemia wish to evaluate their other children for Duarte variant galactosemia, molecular genetic testing for the GALT variants identified in the family can be performed.
Genetic counseling
Duarte variant galactosemia is inherited in an autosomal recessive manner. When one parent is heterozygous for the GALT D2 allele and the other parent is heterozygous for a GALT pathogenic variant, each child has, at conception, a 25% chance of having Duarte variant galactosemia, a 25% chance of being an asymptomatic carrier of the D2 allele, a 25% chance of being an asymptomatic carrier of a GALT pathogenic variant, and a 25% chance of being unaffected and not a carrier of either GALT variant. Carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk requires prior identification of the GALT variants in the family and determination of the parental origin of each allele. Requests for prenatal testing for conditions such as Duarte variant galactosemia are not common.
- Galactosemia. https://rarediseases.org/rare-diseases/galactosemia/[↩][↩]
- Fridovich-Keil JL, Gambello MJ, Singh RH, et al. Duarte Variant Galactosemia. 2014 Dec 4. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK258640[↩][↩][↩][↩][↩][↩][↩][↩]
- Galactosemia. https://ghr.nlm.nih.gov/condition/galactosemia[↩][↩]
- Galactosemia. https://medlineplus.gov/ency/article/000366.htm[↩]
- Galactosemia Potential Long-Term Complications. http://www.galactosemia.org/potential-complications/[↩]




{kind=link}