Contents
- Hereditary spastic paraplegia
- Hereditary spastic paraplegia types
- Uncomplicated (pure) hereditary spastic paraplegia
- Complex hereditary spastic paraplegia
- Spastic paraplegia 1
- Spastic paraplegia 2
- Spastic Paraplegia 3A
- Spastic Paraplegia 4
- Spastic paraplegia type 5A
- Spastic paraplegia 6
- Spastic Paraplegia 7
- Spastic Paraplegia 8
- Spastic paraplegia 9A
- Spastic paraplegia 10
- Spastic Paraplegia 11
- Spastic paraplegia 12
- Spastic paraplegia 13
- Spastic paraplegia 14
- Spastic Paraplegia 15
- Spastic paraplegia 16
- Spastic paraplegia 17
- Spastic paraplegia 18
- Spastic paraplegia 19
- Spastic paraplegia 23
- Spastic paraplegia 24
- Spastic paraplegia 25
- Spastic paraplegia 26
- Spastic paraplegia 29
- Spastic paraplegia 31
- Spastic paraplegia 32
- Spastic paraplegia 39
- Spastic Paraplegia 47
- Spastic Paraplegia 49
- Spastic Paraplegia 50
- Spastic paraplegia 51
- Spastic Paraplegia 52
- Hereditary spastic paraplegia causes
- Table 3. Chromosomes, loci, and genes involved in hereditary spastic paraplegias and their subtypes
- Autosomal Dominant hereditary spastic paraplegia
- Autosomal Recessive hereditary spastic paraplegia
- X-Linked Recessive hereditary spastic paraplegia
- Maternal (Mitochondrial) Inheritance hereditary spastic paraplegia
- Hereditary spastic paraplegia pathophysiology
- Hereditary spastic paraplegia symptoms
- Hereditary spastic paraplegia complications
- Hereditary spastic paraplegia diagnosis
- Hereditary spastic paraplegia differential diagnosis
- Hereditary spastic paraplegia treatment
- Hereditary spastic paraplegia prognosis
- Hereditary spastic paraplegia types
Hereditary spastic paraplegia
Hereditary spastic paraplegia (HSP) also known as hereditary spastic paraparesis, familial spastic paraplegia, familial spastic paraparesis or Strümpell-Lorrain syndrome is a group of rare inherited neurodegenerative disorders that cause weakness and stiffness in the leg muscles 1, 2, 3, 4, 5, 6, 7, 8, 9. There are more than 80 different genetic types of hereditary spastic paraplegia. Hereditary spastic paraplegia primary symptoms are difficulty walking due to muscle weakness and muscle tightness (spasticity) in the legs. The clinical manifestation of hereditary spastic paraplegia starts in infancy and continues into adulthood with slow progression 10, 11. The symptoms gradually get worse over time.
Hereditary spastic paraplegia is associated with significant disability and a negative impact on quality of life 12.
There may be significant variation in the severity of leg weakness (varying from none to marked), the degree of spasticity (varying from minimal to severe), and the occurrence of other neurologic symptoms between different genetic types of hereditary spastic paraplegia; as well differences in the nature and severity of symptoms between individuals who have exactly the same genetic type of hereditary spastic paraplegia 13.
Various types of hereditary spastic paraplegia are classified according to 14, 15, 13:
- a) the mode of inheritance (autosomal dominant, autosomal recessive, X-linked recessive, mitochondrial, or maternal);
- b) the gene in which the mutation occurs; and
- c) the clinical syndrome (pattern of symptoms and neurological findings).
There are more than 80 genetic types of hereditary spastic paraplegia. Genetically, hereditary spastic paraplegia categorization relies on the positions of causative genes (“loci”) with the designation “spastic paraplegia genes (SPGs)”, and numbered in order of their discovery (for example, SPG1 through SPG80).
Hereditary spastic paraplegia syndromes are classified as “uncomplicated” or “pure” hereditary spastic paraplegia when symptoms are confined to leg weakness and tightness (spasticity) and urinary urgency. Hereditary spastic paraplegia syndromes are classified as “complicated” or “complex” hereditary spastic paraplegia when leg weakness and tightness (spasticity) are accompanied by other neurological disturbance such as peripheral nerve impairment, muscle atrophy, or intellectual impairment 16.
Hereditary spastic paraplegia prevalence ranges from 0.1 to 9.6 per 100,000 individuals reported around the world 17, 18, 19, 20, 21, 22. Hereditary spastic paraplegia affects males and females of all ethnic groups from around the world. It’s difficult to know exactly how many people have hereditary spastic paraplegia because hereditary spastic paraplegia has high clinical and genetic heterogeneity and is prone to misdiagnosis.
Hereditary spastic paraplegia is mainly observed in autosomal dominant pure form in about 80% of the North American and north European hereditary spastic paraplegia populations, with SPG4/SPAST mutations in 40%, SPG3A/ATL1 mutations in 10% at hereditary spastic paraplegias’ early beginning, about 10% SPG31/REEP1 mutations, and almost 3% SPG10/KIF5A mutations 17, 23. The mutation mentioned above is seen in complex hereditary spastic paraplegias and other neuropathies involving motor and sensory neurons 24.
On the other hand, autosomal recessive hereditary spastic paraplegias are more complex and seen in a high degree of the consanguineous marriages (marriage between individuals who are closely related) population, with nearly 30% registered hereditary spastic paraplegias from the Middle East and northern Africa. SPG11 and SPG15 constitute a significant chunk of autosomal recessive forms of hereditary spastic paraplegias 25. The common phenotypic characters include thinning corpus callosum, periventricular white matter change in the ear lynx, early development of parkinsonism, cognitive ability slacking, moderate ataxia, retina abnormalities, and prominent paraplegia 17. In addition, SPG35/FA2H and SPG45/C19orf12 are prevalent forms of autosomal recessive hereditary spastic paraplegias 26, 27. SPG5A shows CYP7B1 pathological variant in 7.3% autosomal recessive hereditary spastic paraplegia and 3% sporadic pure hereditary spastic paraplegia 28. 5–12% of autosomal recessive hereditary spastic paraplegia cases account for SPG7 29 and about 3–5% of hereditary spastic paraplegia individuals show SPG11 variants in autosomal recessive mode.
X-linked hereditary hereditary spastic paraplegias shows complex phenotypes with few cases, and five hereditary spastic paraplegias are known to date with three genes identified as SPG22/SLC16A2, SPG1/L1CAM, and SPG2/PLP1.
Late spastic paraplegia-like symptoms are due to alterations (m.9176T>C) in the ATP6 of mitochondrial DNA 30. Similarly, with their colleagues, Sánchez-Ferrero et al. 31 reported alterations in mitochondrial MT-CO3 and MT-T1 causing hereditary spastic paraplegia. About 1–2% of cases show X and mitochondrial chromosomes mutations.
Testing for hereditary spastic paraplegia genes is available and performed for individual hereditary spastic paraplegia genes, for panels containing dozens of hereditary spastic paraplegia genes, and by analysis of all genes (whole exome and whole genome analysis). Genetic testing is often helpful to confirm the clinical diagnosis of hereditary spastic paraplegia. Genetic testing is most often able to find causative gene mutations for subjects with hereditary spastic paraplegia who have a family history of a similarly affected first-degree relative.
Despite discovery of more than 80 genes in which mutations cause various types of hereditary spastic paraplegia, many individuals with hereditary spastic paraplegia do not have an identified gene mutation. This is because: a) genes for all types of hereditary spastic paraplegia have not been discovered and furthermore, some discovered genes are not yet included in clinical testing panels; b) methods of gene sequencing typically used to analyze large panels of genes do not analyze all regions of genes. Furthermore, while sensitively detecting gene sequence changes, these “next generation sequencing” methods are less sensitive in detecting gene insertions and deletions that do not change the sequence of the remaining portion of the gene.
Genetic testing is expensive and not all insurance companies provide reimbursement for this analysis. Identifying a causative gene mutation can bring closure to a diagnostic odyssey, contribute insight into the prognosis and can be applied to genetic counseling and prenatal diagnosis. Nonetheless at present, genetic testing results very rarely influence treatment which is largely directed toward reducing symptoms.
Interpreting hereditary spastic paraplegia genetic test results may be straightforward. Genetic testing may identify a gene mutation that is known to be associated with hereditary spastic paraplegia in other subjects, absent in unaffected subjects, and known or predicted to change the protein function. These mutations are termed “likely pathogenic” (likely to be disease causing). On the other hand, genetic testing may also identify gene variations that are considered normal variations (for example, they may be present in subjects who do not have hereditary spastic paraplegia and may be predicted to not change the protein function). Such mutations are considered “benign” variations and are not likely to cause hereditary spastic paraplegia.
In addition to “likely pathogenic” and “likely benign” mutations”, it is not uncommon for genetic testing to identify gene variations that are “of uncertain significance”. Such mutations may not have been reported to be associated with the disorder or may not be predicted to disturb the function of the protein. By definition, it is not known if gene variations of uncertain significance cause hereditary spastic paraplegia (i.e. are pathogenic) or are actually normal variations that are of no medical consequence. Individuals seeking more information regarding results of genetic testing are recommended to consult a medical geneticist or genetic counselor.
At present, there is no specific treatment to prevent or reverse nerve degeneration in hereditary spastic paraplegia. Treatments are directed at reducing symptoms and improving balance, strength, and agility. Individuals should be evaluated periodically (annually or as needed) by a neurologist and physiatrist (medical doctor who specializes in pain management and rehabilitation) to assess progression and develop treatment strategies to maximize walking ability and reduce symptoms.
Current treatment recommendations for hereditary spastic paraplegia include:
- Daily regimen of physical therapy directed toward improving cardiovascular fitness, maintaining and improving muscle strength and gait, and reducing spasticity
- Occupational therapy, assistive walking devices, and ankle-foot orthotics as needed
- Drugs to reduce muscle spasticity (e.g., Lioresal [oral or intrathecal], tizanidine, dantrolene, botulinum A and B toxin injections [Botox, Dysport, Xeomin, or Myoblock]) and urinary urgency (e.g., oxybutynin, solifenacin, mirabegron, or intrabladder injections with Botox)
Individuals with hereditary spastic paraplegia are recommended to pursue active lifestyles including physical rehabilitation in order to maintain and improve functional abilities and cardiovascular fitness. In addition, genetic counselling in patients and their families to understand the transmission risk of hereditary spastic paraplegia in successive generations and the proband state (Table 2) are recommended.
Figure 1. Hereditary spastic paraplegia
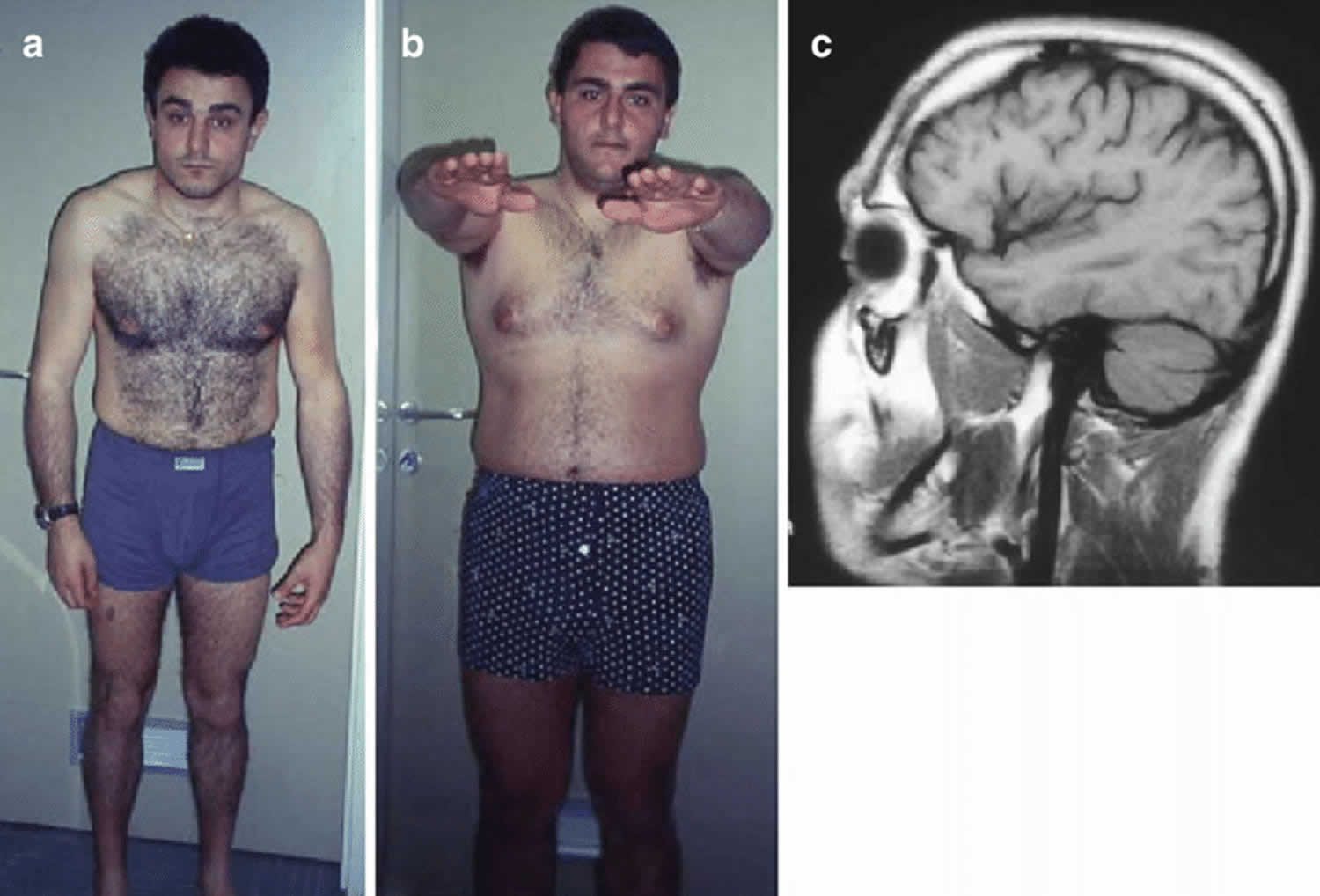
Footnotes: (a, b) Two siblings with hereditary spastic paraplegia. Note bended position of the shoulders with abnormal position of the left arm (a) and tendency to uneven position in Mingazzini I (b). (c) On brain MRI there are both cortical atrophy and cerebellar atrophy.
[Source 32 ]Figure 2. Hereditary spastic paraplegia genes
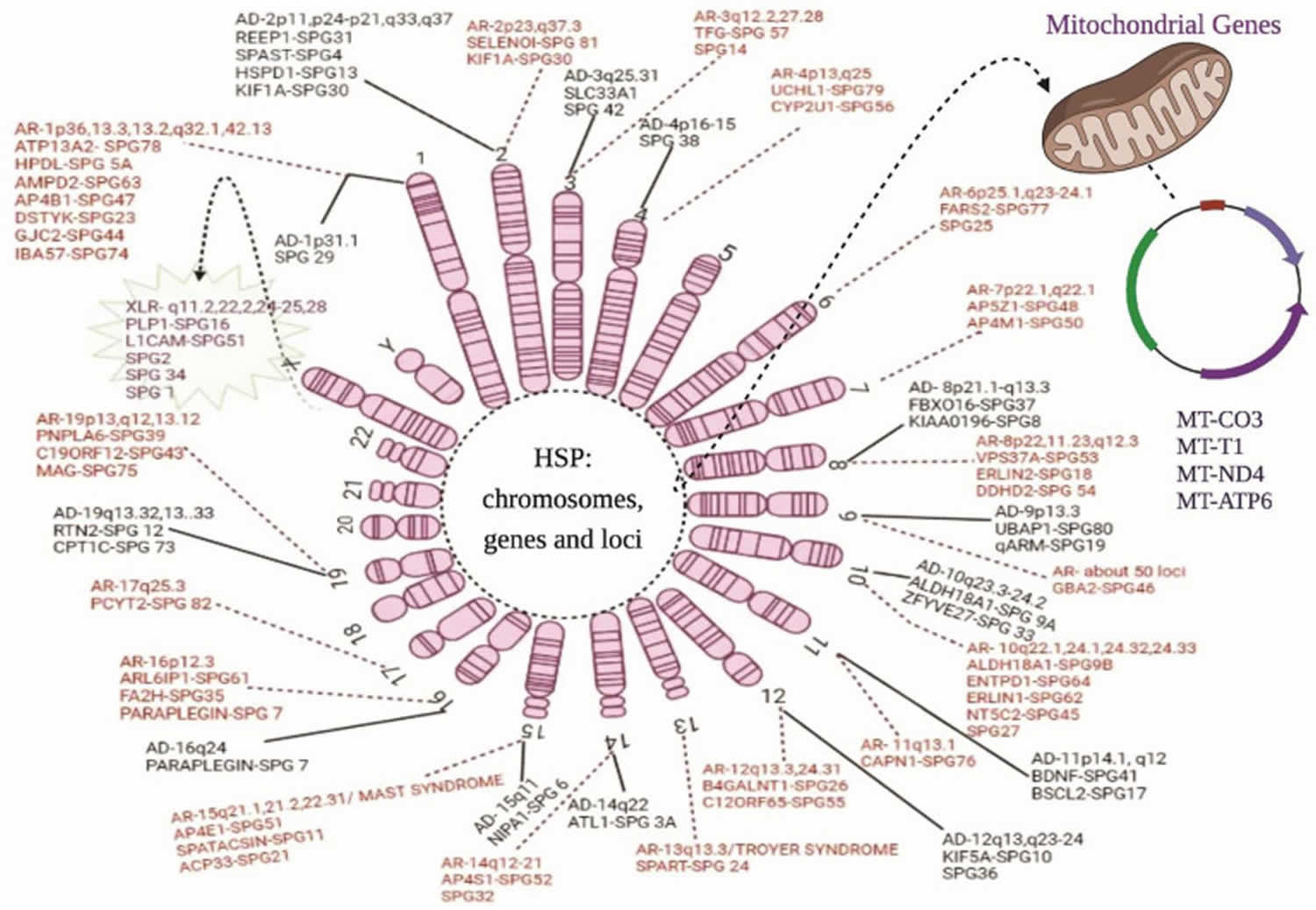
Footnotes: Chromosomal and genetic markers in hereditary spastic paraplegia. The above diagram illustrates all the genes and their location on the chromosomes related to the hereditary spastic paraplegia phenotypes: AD = autosomal dominant forms; AR = autosomal recessive forms; XLR = X-linked recessive forms; and maternal or mitochondrial inheritance. The solid line represents the autosomal dominant forms of hereditary spastic paraplegia, whereas the dotted lines show the autosomal recessive forms. The spiky green cloud with a dotted line represents the X-linked recessive hereditary spastic paraplegia form.
[Source 7 ]Figure 3. Hereditary spastic paraplegia pathophysiology
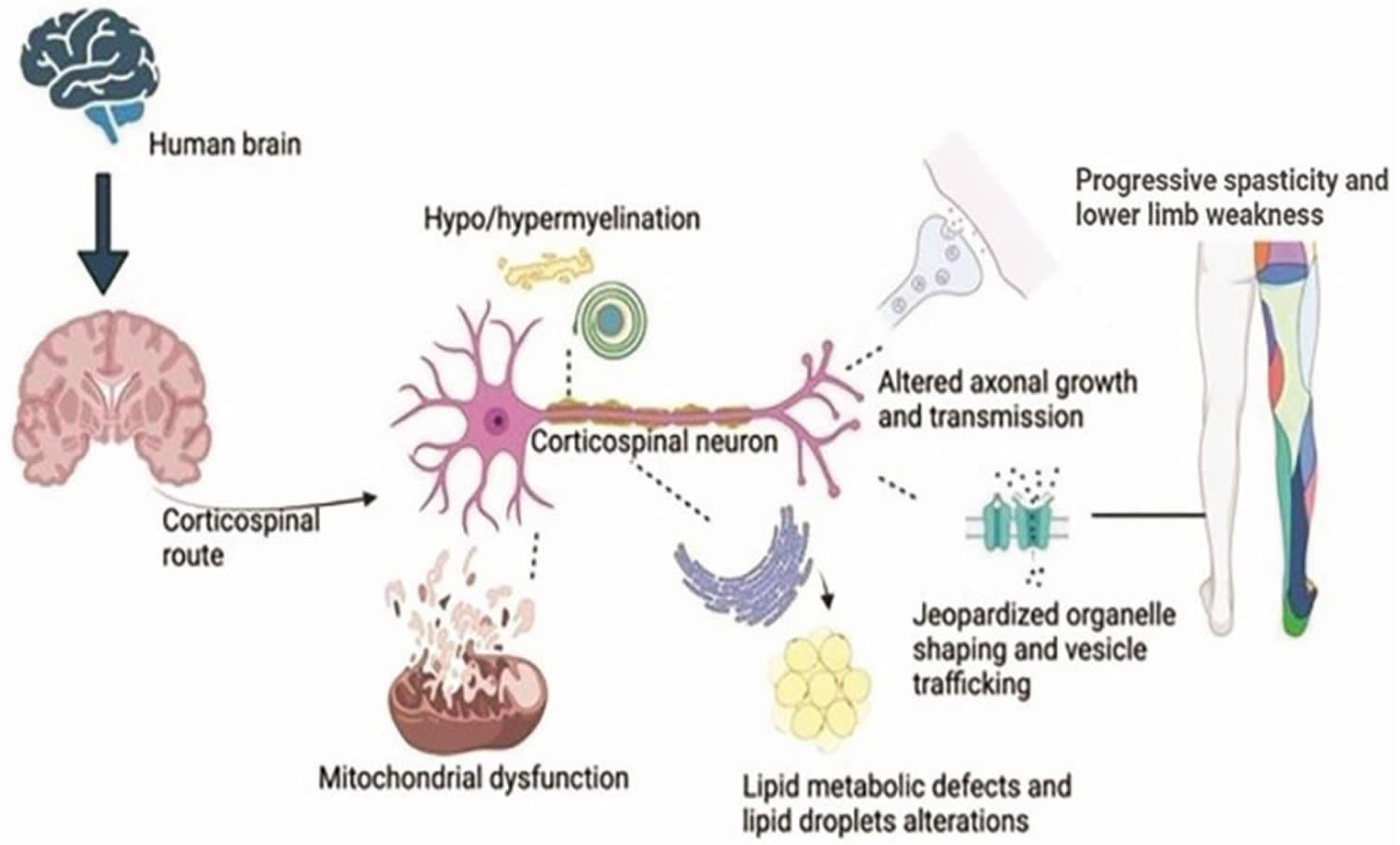
Footnotes: Hereditary spastic paraplegia pathophysiology. The neurons in the corticospinal zone of the brain undergo mutations in the genes, causing a breakdown of organelle shaping and trafficking and dysfunction in the mitochondrial cells at the neuron’s nuclear region. Few gene mutations lead to faulty transmission in the axons, and some mutations cause degeneration of the myelin sheath of the corticospinal neuron. Likewise, an endoplasmic reticulum shaping genes’ mutation causes defective metabolism, especially lipid droplet formations. All these characteristics lead to lower limb spasticity and weakness, causing hereditary spastic paraplegia phenotypes.
[Source 7 ]Figure 4. Hereditary spastic paraplegia differential diagnosis
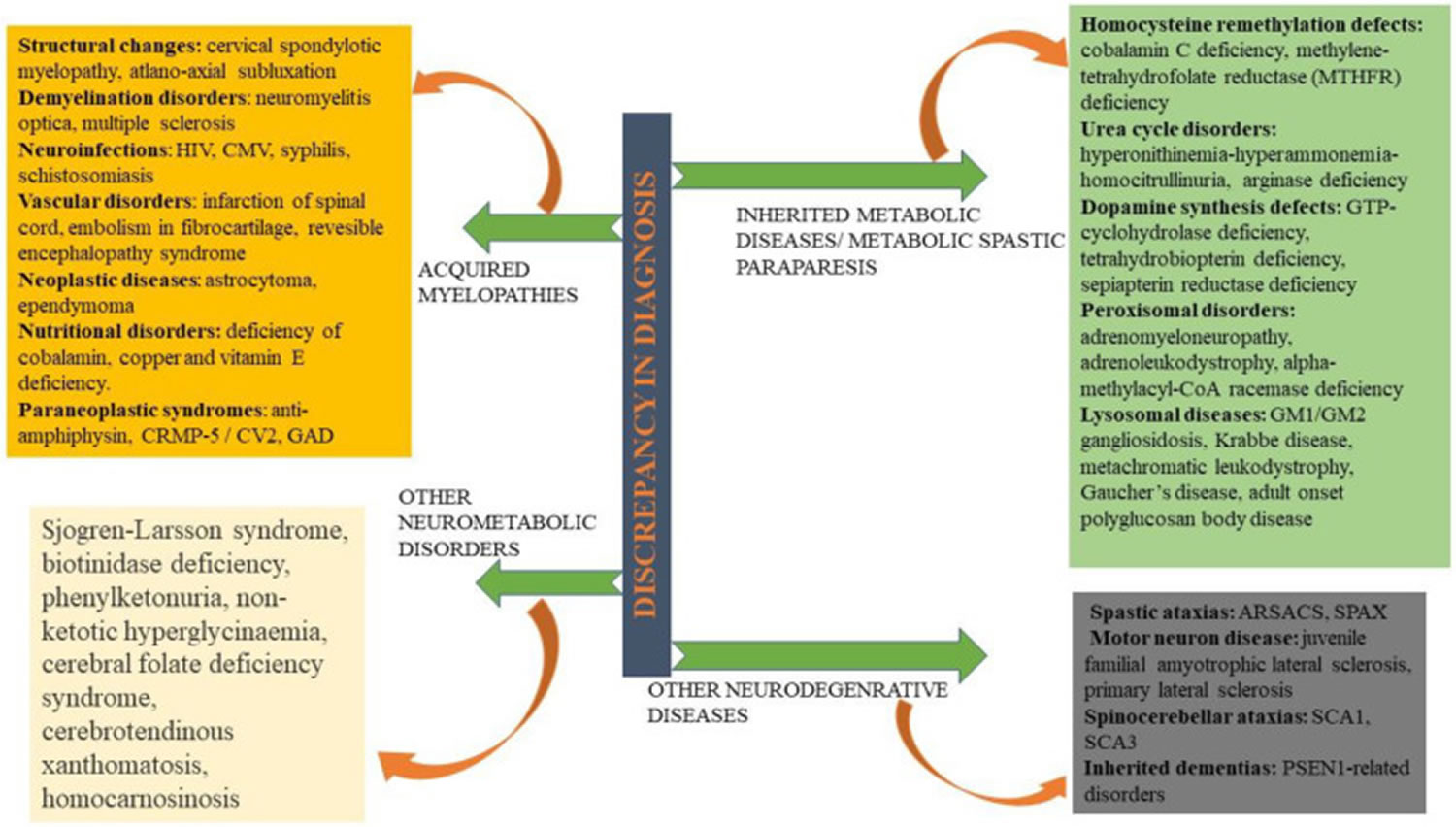
Footnotes: Hereditary spastic paraplegia differential diagnosis. Being an extremely heterogeneous disorder in clinical and genetic aspects, hereditary spastic paraplegia is often misinterpreted with acquired myelopathies, neurometabolic disorders, inherited metabolic paraparesis, and some neurodegenerative disorders.
Abbreviations: ARSACS = Autosomal recessive spastic ataxia of Charlevoix–Saguenay; SCAs = Spinocerebellar ataxias; CMV = Cytomegalovirus; CRMP-5 = Collapsin response mediator protein-5; GAD = Glutamate decarboxylase; HTLV = Human T lymphotropic virus; HIV = Human immunodeficiency virus; PSEN = Presenilin; SPAX: Spastic ataxia.
[Source 7 ]Figure 5. Hereditary spastic paraplegia treatment
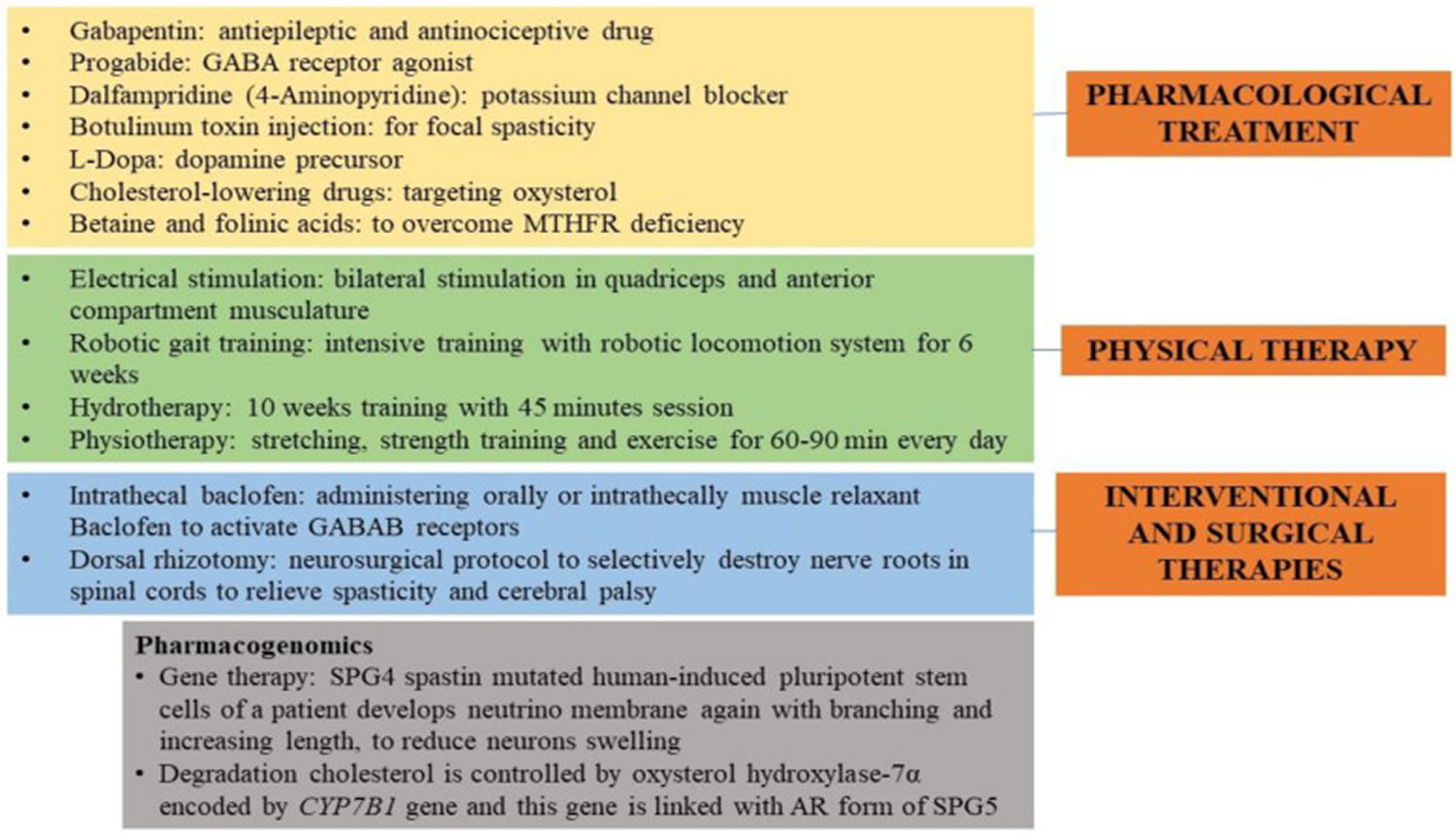
Footnote: Hereditary spastic paraplegia therapies and treatments. The image illustrates the pharmacological, physical, interventional, and surgical treatments given to hereditary spastic paraplegia (HSP) people to soothe the spasticity and improve their gait. A pharmacogenomics approach is also reported.
[Source 7 ]Hereditary spastic paraplegia types
Various types of hereditary spastic paraplegia are classified according to 14, 15, 13:
- A) Mode of inheritance (autosomal dominant, autosomal recessive, X-linked recessive, mitochondrial, or maternal inheritance);
- B) The gene in which the mutation occurs; and
- C) The clinical syndrome (pattern of symptoms and neurological findings).
Clinically, hereditary spastic paraplegia can be categorized into “uncomplicated” or “pure” hereditary spastic paraplegia when symptoms are confined to leg weakness, tightness (spasticity), impaired vibration sense, and urinary urgency. Hereditary spastic paraplegia syndromes are classified as “complicated” or “complex” hereditary spastic paraplegia when leg weakness and tightness (spasticity) are accompanied by other neurological disturbance such as peripheral nerve impairment, muscle atrophy, or intellectual impairment 33.
Table 1. Pure and complex hereditary spastic paraplegia types
| Pure hereditary spastic paraplegia | Complex hereditary spastic paraplegia |
|---|---|
| Impairments present in uncomplicated hereditary spastic paraplegia plus other neurologic findings such as:
|
Table 2. Hereditary spastic paraplegia classification
| Criteria for Classification | Types |
|---|---|
| Symptoms and signs (Harding’s classification) |
|
| Age and onset of spasticity (Harding’s classification) |
|
| Inheritance pattern |
|
| Intracellular involvement |
|
Uncomplicated (pure) hereditary spastic paraplegia
Uncomplicated (or “pure”) hereditary spastic paraplegia is characterized by neurologic impairment limited to progressive lower-extremity spastic weakness, hypertonic urinary bladder disturbance, and mild diminution of lower-extremity vibration sensation. Individuals with uncomplicated hereditary spastic paraplegia experience the following:
- Difficulty walking (may either be non-progressive or worsen insidiously)
- Often, the need for canes, walkers, or wheelchairs
- Possible urinary urgency and lower-extremity paresthesias (the sensation of pins and needles)
- Typically, normal strength and dexterity of the upper extremities
- No involvement of speech, chewing, or swallowing
Though symptoms may be disabling, life span is not shortened.
Complex hereditary spastic paraplegia
Complex hereditary spastic paraplegia is characterized by the impairments present in uncomplicated “pure” hereditary spastic paraplegia plus other system involvement or other neurological findings including any of the following:*
- Ataxia
- Seizures
- Intellectual disability
- Dementia
- Muscle atrophy
- Extrapyramidal disturbance
- Peripheral neuropathy
Note:* In the absence of other causes for these additional features.
Spastic paraplegia 1
Spastic paraplegia 1 (SPG 1) also called Mental Retardation Aphasia Shuffling Gait Adducted Thumbs (MASA) syndrome, Gareis-Mason syndrome, Crash syndrome, clasped thumb and mental retardation or thumb congenital clasped with mental retardation is characterized by mild to moderate intellectual disability, delayed development of speech, hypotonia progressing to spasticity or spastic paraplegia, adducted thumbs, and mild to moderate distension of the cerebral ventricles 34, 35, 36.
Males with spastic paraplegia 1 (SPG 1) are born with severe hydrocephalus, adducted thumbs, and spasticity; intellectual disability is severe. In less severely affected males, hydrocephalus may be subclinically present and documented only because of developmental delay; intellectual disability ranges from mild (IQ: 50-70) to moderate (IQ: 30-50) 36. It is important to note that all phenotypes can be observed in affected individuals within the same family.
Spastic paraplegia 2
Spastic paraplegia 2 (SPG 2) also called spastic paraparesis type 2 or SPPX2 is a rare, X-linked leukodystrophy characterized primarily by spastic gait and autonomic dysfunction 37, 38. When additional central nervous system (CNS) signs, such as intellectual deficit, ataxia, or extrapyramidal signs, are present, the syndrome is referred to as complicated spastic paraplegia.
Spastic Paraplegia 3A
Spastic paraplegia 3A (SPG3A) also known as ATL1-hereditary spastic paraplegia is characterized by progressive bilateral and mostly symmetric spasticity and weakness of the legs 39. Compared to other forms of autosomal dominant hereditary spastic paraplegia, in which diminished vibration sense caused by degeneration of the corticospinal tracts and dorsal columns and urinary bladder hyperactivity are present in all affected individuals, these findings occur in a minority of individuals with spastic paraplegia 3A (SPG3A). The average age of onset is four years. More than 80% of reported individuals manifest spastic gait before the end of the first decade of life. Most persons with early-onset ATL1-hereditary spastic paraplegia have a “pure” (“uncomplicated”) hereditary spastic paraplegia; however, complicated hereditary spastic paraplegia with axonal motor neuropathy and/or distal amyotrophy with lower motor neuron involvement (Silver syndrome phenotype) has been observed. The rate of progression in ATL1-hereditary spastic paraplegia is slow, and wheelchair dependency or need for a walking aid (cane, walker, or wheelchair) is relatively rare.
Spastic paraplegia 3A (SPG3A) should be suspected in individuals with the following clinical findings and family history 39:
Clinical findings
- Early age of onset, from infancy to ten years (average age: 4 years)
- Progressive bilateral and mostly symmetric lower-extremity weakness and spasticity resulting from axonal degeneration of the corticospinal tracts
- Diminished vibration sense caused by impairment of dorsal columns
- Urinary bladder hyperactivity
Family history consistent with autosomal dominant inheritance, including affected males and females in multiple generations and simplex cases (i.e., a single occurrence in a family). Absence of a known family history does not rule out the diagnosis.
Spastic Paraplegia 4
Spastic paraplegia 4 (SPG4) also known as SPAST-hereditary spastic paraplegia is characterized by insidiously progressive bilateral lower-limb gait spasticity 40. More than 50% of affected individuals have some weakness in the legs and impaired vibration sense at the ankles 40. Sphincter disturbances are very common. Onset is insidious, mostly in young adulthood, although symptoms may start as early as age one year and as late as age 76 years. Intrafamilial variation is considerable.
Spastic paraplegia 4 (SPG4) should be suspected in individuals with the following 40:
- Characteristic clinical symptoms of insidiously progressive bilateral leg stiffness affecting gait with or without spasticity at rest and mild proximal weakness, often accompanied by urinary urgency
- Neurologic examination demonstrating corticospinal tract deficits affecting both legs (spastic weakness, hyperreflexia, and extensor plantar responses). Mildly impaired vibration sensation in the ankles is present in the majority of individuals.
- Family history consistent with autosomal dominant inheritance, or exclusion of other causes of spastic paraplegia in simplex cases (i.e., a single occurrence in a family)
Note: The presence of other signs/symptoms suggestive of complicated hereditary spastic paraplegia does not exclude spastic paraplegia 4 (SPG4), although it reduces its probability.
Brain and spinal cord MRI
- Often normal in individuals with spastic paraplegia 4 (SPG4)
- Spinal cord atrophy can occur in spastic paraplegia 4 (SPG4), but is less pronounced than in other genetic causes of hereditary spastic paraplegia.
- Mild vermis atrophy, a thin corpus callosum, subtle white matter changes, and/or cerebellar atrophy have been reported 41, 42.
Note: The MRI is useful in identifying anomalies of the brain, cerebro-medullary junction, and medulla that are characteristic of disorders discussed in Differential Diagnosis.
Electromyography (EMG) with nerve conduction velocities (NCV) is used to exclude peripheral nervous system involvement, which could raise the possibility of an alternative diagnosis as severe polyneuropathy is not a frequent symptom of SPAST-HSP. Karle et al performed neurophysiologic examinations of 128 individuals with hereditary spastic paraplegia, including 35 individuals with spastic paraplegia 4 (SPG4), and showed that massively elongated central motor conduction time argued against spastic paraplegia 4 (SPG4); however, reduced amplitudes and prolonged latencies were reported, in particular in individuals with a spastic paraplegia 4 (SPG4) pathogenic missense variant 43.
Spastic paraplegia type 5A
Spastic paraplegia type 5A (SPG5A) is a form of hereditary spastic paraplegia that is characterized by either a pure form of hereditary spastic paraplegia of slowly progressive spastic paraplegia of the lower extremities with bladder dysfunction and pes cavus or a complex form of hereditary spastic paraplegia with additional manifestations including cerebellar signs, nystagmus, distal or generalized muscle atrophy and cognitive impairment 44, 45. Age of onset is highly variable, ranging from early childhood to adulthood. White matter hyperintensity and cerebellar and spinal cord atrophy may be noted, on brain magnetic resonance imaging, in some patients.
Spastic paraplegia 6
Spastic paraplegia 6 (SPG 6) is a rare, pure or complex form of hereditary spastic paraplegia typically characterized by presentation in late adolescence or early adulthood as a pure phenotype of lower limb spasticity with hyperreflexia and extensor plantar responses, as well as mild bladder disturbances and pes cavus 46, 47. Rarely, it can present as a complex phenotype with additional manifestations including epilepsy, variable peripheral neuropathy and/or memory impairment 46, 47.
Spastic Paraplegia 7
Spastic paraplegia 7 (SPG7) is characterized by insidiously progressive bilateral leg weakness and spasticity 48. Most affected individuals have decreased vibration sense and cerebellar signs 48. Onset is mostly in adulthood, although symptoms may start as early as age 11 years and as late as age 72 years 48. Additional features including ataxia (gait and limbs), spastic dysarthria, dysphagia, pale optic disks, ataxia, nystagmus, strabismus, ptosis, hearing loss, motor and sensory neuropathy, amyotrophy, scoliosis, pes cavus, and urinary sphincter disturbances may be observed 48.
Spastic paraplegia 7 (SPG7) should be suspected in individuals with the following 48:
- Insidiously progressive bilateral leg weakness
- Spasticity
- Decreased vibratory sense
- Cerebellar signs
- Neurologic examination demonstrating EITHER of the following:
- A pure clinical presentation of spastic paraplegia with hyperreflexia, extensor plantar responses, and mildly impaired vibration sensation in the distal legs
- A complicated phenotype of spastic paraplegia including optic neuropathy, progressive external ophthalmoplegia/ptosis slowed speech, swallowing difficulties, palatal tremor, subtle cognitive impairment, urinary urgency, ataxia, nystagmus, strabismus, decreased hearing, scoliosis, pes cavus, motor and sensory neuropathy, and amyotrophy 49, 50, 51, 52
- Neuroimaging findings of cerebellar atrophy (MRI) or white matter changes as detected by diffusion tensor imaging in the frontal lobes, the corticospinal tracts, and the brain stem
- Family history consistent with autosomal recessive inheritance
Spastic Paraplegia 8
Spastic paraplegia 8 (SPG8) is a slowly progressive pure spastic paraplegia of the lower limbs (i.e., pyramidal signs including hyperreflexia, spasticity, and occasionally clonus without other neurologic findings) 53. Some affected individuals have urinary urgency that usually becomes apparent at the same time as the spasticity 53. Onset is between ages 10 and 59 years 53. Affected individuals often become wheelchair dependent. While intra- and interfamilial phenotypic variability is high, SPG8 is typically more severe than other types of hereditary spastic paraplegia.
Spastic paraplegia 8 (SPG8) should be suspected or considered in individuals with the following clinical and neuroimaging findings and family history 54, 55, 56, 57:
Clinical findings
- Onset in the 20s and 30s (range: age 20-60 years)
- Slowly progressive “pure” spastic paraplegia of the lower limbs (i.e., pyramidal signs including hyperreflexia, spasticity, and occasionally clonus without other neurologic findings)
- Mild distal decreased vibration sense
- Urinary urgency
Neuroimaging findings
Brain MRI is generally normal. In one moderately affected individual spine MRI showed significant atrophy of the thoracic spinal cord as determined by cross-sectional area measurements 58.
Other studies. Normal:
- Cerebrospinal fluid
- Electrophysiologic studies:
- Nerve conduction velocity
- Electromyography
- Biochemical testing
- Vitamin B12
- Very long chain fatty acids
- Lactate 59
Family history consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations). Absence of a known family history does not rule our the diagnosis.
Spastic paraplegia 9A
Spastic paraplegia-9A (SPG9A) is a rare complex hereditary spastic paraplegia that is characterized by progressive spasticity and weakness of the lower limbs frequently associated mild cerebellar signs, congenital bilateral cataract, gastroesophageal reflux, pes cavus, urinary urgency and occasionally short stature 60, 61, 62. Spastic paraplegia-9A (SPG9A) age at onset usually ranges from adolescence to adulthood, and patients have gait difficulties, motor neuropathy, and spastic dysarthria 63.
Spastic paraplegia 10
Spastic paraplegia 10 (SPG 10) is a rare, hereditary spastic paraplegia that can present as either a pure or complex hereditary spastic paraplegia 64, 65. The pure form hereditary spastic paraplegia is characterized by lower limb spasticity, hyperreflexia and extensor plantar responses, presenting in childhood or adolescence. The complex form hereditary spastic paraplegia is characterized by the association with additional manifestations including peripheral neuropathy with upper limb muscle atrophy, moderate intellectual disability and parkinsonism. Deafness and retinitis pigmentosa have also been reported 64, 65.
Spastic Paraplegia 11
Spastic paraplegia 11 (SPG11) is characterized by progressive spasticity and weakness of the lower limbs frequently associated with the following: mild intellectual disability with learning difficulties in childhood and/or progressive cognitive decline; peripheral neuropathy; pseudobulbar involvement; and increased reflexes in the upper limbs 66. Less frequent findings include: cerebellar signs (ataxia, nystagmus, saccadic pursuit); retinal degeneration; pes cavus; scoliosis; and parkinsonism with characteristic brain MRI features that include thinning of the corpus callosum 66. Onset occurs mainly during infancy or adolescence (range: age 1-31 years) and in rare cases as late as age 60 years. Most affected individuals become wheelchair bound one or two decades after disease onset.
Spastic paraplegia 11 (SPG11) should be suspected in individuals with the following clinical and imaging findings 66:
Frequent clinical findings
- Progressive spasticity and weakness of the lower limbs
- Mild intellectual disability with learning difficulties in childhood and/or progressive cognitive decline with onset in the first to third decade
- Axonal, motor, or sensorimotor peripheral neuropathy (>80% of individuals) 67, 68, 69, 70, 71, 72
- Pseudobulbar involvement with dysarthria and/or dysphagia
- Increased reflexes in the upper limbs
Less frequent clinical findings
- Cerebellar signs (ataxia or ocular signs including nystagmus and/or saccadic pursuit)
- Retinal degeneration (Kjellin syndrome). Kjellin syndrome is characterized by retinal degeneration, autosomal recessive hereditary spastic paraplegia, and thin corpus callosum initially associated with spastic paraplegia 15 (SPG15) but more often occurring in individuals with SPG11 73
- Pes cavus
- Scoliosis
- Extrapyramidal signs such as parkinsonism 74, 75
Imaging findings on brain and spinal cord MRI
- Thinning of the corpus callosum (TCC) (>90% of individuals) 67 particularly with long T1 and T2 values in the forceps minor of the corpus callosum, the so-called “ear of the lynx” sign which appears hyperintense on FLAIR and hypointense on T1-weighted images 76
- Cortical atrophy is frequently observed.
- White matter hyperintensities 77
- Atrophy of both the brain stem and the cerebellum can be observed 79.
- The basal ganglia may also be affected 80.
Note: 60% of individuals with thinning of the corpus callosum, cognitive impairment, and spastic paraparesis were found to have biallelic SPG11 pathogenic variants 67.
Spastic paraplegia 12
Spastic paraplegia 12 (SPG 12) is a pure form of hereditary spastic paraplegia characterized by a childhood- to adulthood-onset of slowly progressive lower limb spasticity and hyperreflexia of lower extremities, extensor plantar reflexes, distal sensory impairment, variable urinary dysfunction and pes cavus 81, 82.
Spastic paraplegia 13
Spastic paraplegia 13 (SPG 13) is a rare, pure or complex form of hereditary spastic paraplegia characterized by progressive spastic paraplegia with pyramidal signs in the upper and lower limbs, and decreased vibration sense 83, 84.
Spastic paraplegia 14
Spastic paraplegia 14 (SPG 14) is a rare, complex hereditary spastic paraplegia characterized by adulthood-onset of slowly progressive spastic paraplegia of lower limbs presenting with spastic gait, hyperreflexia, and mild lower limb hypertonicity associated with mild intellectual disability, visual agnosia, short and long-term memory deficiency and mild distal motor neuropathy 85, 86. Bilateral pes cavus and extensor plantar responses are also associated 85, 86.
Spastic Paraplegia 15
Spastic paraplegia 15 (SPG15) is typically an early-onset complex hereditary spastic paraplegia that is characterized by progressive spasticity that begins in the lower extremities and is associated with several manifestations resulting from central and peripheral nervous system dysfunction 87. While onset of spasticity is typically in mid- to late childhood or adolescence (i.e., between ages 5 and 18 years), other signs and symptoms, such as developmental delay or learning disability, may be present earlier, often preceding motor involvement 87. Individuals with adult onset have also been reported 87.
Spastic paraplegia 15 (SPG15) should be suspected in individuals with the following clinical and brain imaging findings and family history 87:
Clinical findings
- Spasticity and weakness with progression from a spastic diplegia to a spastic tetraplegia with associated pyramidal signs (Babinski sign, hyperreflexia, ankle clonus)
- Learning disability or intellectual disability, progressive cognitive impairment
- Dysarthria and cerebellar signs
- Peripheral neuropathy (axonal sensorimotor neuropathy)
- Distal amyotrophy / loss of muscle bulk
- Less common:
- Extrapyramidal movement disorders including focal dystonia and parkinsonism
- Retinopathy
- Sensorineural hearing impairment
- Epilepsy
- Cataracts
Laboratory findings
- Nerve conduction studies show an axonal sensorimotor neuropathy in a subset of affected individuals.
- Electroretinography may show changes consistent with retinopathy in a subset of affected individuals.
Brain imaging
- Thinning of the corpus callosum (most commonly the anterior parts) (70%)
- Periventricular white matter signal changes (>50%). The periventricular white matter signal changes in SPG15 can have a characteristic appearance involving the forceps minor. This is known as the “ears of the lynx” sign, as hypointense signal on T1-weighted and hyperintense signal on FLAIR images on axial views resemble the shape of the ears of a lynx with its characteristic apical hair tuft 76.
- Cerebral and cerebellar atrophy (~25%)
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not rule out the diagnosis.
Spastic paraplegia 16
Spastic paraplegia 16 (SPG 16) also called X-linked spastic paraplegia type 16, is a complex, hereditary, spastic paraplegia characterized by delayed motor development, spasticity, and inability to walk, later progressing to quadriplegia, motor aphasia, bowel and bladder dysfunction 88, 89. Patients also present with vision problems and mild intellectual disability. Spastic paraplegia 16 (SPG 16) affects only males 88, 89.
Spastic paraplegia 17
Spastic paraplegia 17 (SPG 17) also called Silver spastic paraplegia syndrome or spastic paraplegia with amyotrophy (progressive wasting of muscle tissues) of hands and feet is a complex hereditary spastic paraplegia characterized by progressive spastic paraplegia, upper and lower limb muscle atrophy, hyperreflexia, extensor plantar responses, pes cavus and occasionally impaired vibration sense 90, 91. Association with hand muscles amyotrophy typical 90, 91.
Spastic paraplegia 18
Spastic paraplegia 18 (SPG 18) is a rare, complex type of hereditary spastic paraplegia characterized by progressive spastic paraplegia (presenting in early childhood) associated with delayed motor development, severe intellectual disability and joint contractures 92, 93. A thin corpus callosum is equally noted on brain magnetic resonance imaging. SPG18 is caused by a mutation in the ERLIN2 gene (8p11.2) encoding the protein, Erlin-2 92, 93.
Spastic paraplegia 19
Spastic paraplegia 19 (SPG 19) is a pure form of hereditary spastic paraplegia characterized by a slowly progressive and relatively benign spastic paraplegia presenting in adulthood with spastic gait, lower limb hyperreflexia, extensor plantar responses, bladder dysfunction (urinary urgency and/or incontinence), and mild sensory and motor peripheral neuropathy 94, 95.
Spastic paraplegia 23
Spastic paraplegia 23 (SPG 23) also called spastic paraplegia and pigmentary abnormalities or Lison syndrome is a rare, complex type of hereditary spastic paraplegia that presents in childhood with progressive spastic paraplegia, associated with peripheral neuropathy, skin pigment abnormalities (i.e. vitiligo, hyperpigmentation, diffuse lentigines), premature graying of hair, and characteristic facies (i.e. thin with ”sharp” features) 96, 97. The spastic paraplegia 23 (SPG 23) phenotype has been mapped to a locus on chromosome 1q24-q32 96, 97.
Spastic paraplegia 24
Spastic paraplegia 24 (SPG 24) is a very rare, pure form of spastic paraplegia characterized by an onset in infancy of lower limb spasticity associated with gait disturbances, scissor gait, tiptoe walking, clonus and increased deep tendon reflexes 98, 99. Mild upper limb involvement may occasionally also be associated 98, 99.
Spastic paraplegia 25
Spastic paraplegia 25 (SPG 25) also called spinal disc herniation with autosomal recessive spastic paraplegia is a rare, complex type of hereditary spastic paraplegia characterized by adult-onset spastic paraplegia associated with spinal pain that radiates to the upper or lower limbs and is related to disk herniation (with minor spondylosis), as well as mild sensorimotor neuropathy 100, 101. The SPG25 phenotype has been mapped to a locus on chromosome 6q23-q24.1 100, 101.
Spastic paraplegia 26
Spastic paraplegia 26 (SPG 26) is a rare, complex type of hereditary spastic paraplegia characterized by the onset in childhood/adolescence (ages 2-19) of progressive spastic paraplegia associated mainly with mild to moderate cognitive impairment and developmental delay, cerebellar ataxia, dysarthria, and peripheral neuropathy 102, 103. Less commonly reported manifestations include skeletal abnormalities (i.e. pes cavus, scoliosis), dyskinesia, dystonia, cataracts, cerebellar signs (i.e. saccadic dysfunction, nystagmus, dysmetria), bladder disturbances, and behavioral problems 102, 103. Spastic paraplegia 26 (SPG 26) is caused by mutations in the B4GALNT1 gene (12q13.3), encoding Beta-1, 4 N-acetylgalactosaminyltransferase 1.
Spastic paraplegia 29
Spastic paraplegia 29 (SPG 29) is a complex form of hereditary spastic paraplegia characterized by a spastic paraplegia presenting in adolescence, associated with the additional manifestations of sensorial hearing impairment due to auditory neuropathy and persistent vomiting due to a hiatal or paraesophageal hernia 104, 105.
Spastic paraplegia 31
Spastic paraplegia 31 (SPG 31) ia a rare type of hereditary spastic paraplegia usually characterized by a pure phenotype of proximal weakness of the lower extremities with spastic gait and brisk reflexes, with a bimodal age of onset of either childhood or adulthood (>30 years) 106, 107. In some cases, it can present as a complex phenotype with additional associated manifestations including peripheral neuropathy, bulbar palsy (with dysarthria and dysphagia), distal amyotrophy, and impaired distal vibration sense 106, 107.
Spastic paraplegia 32
Spastic paraplegia 32 (SPG 32) is a rare, complex type of hereditary spastic paraplegia characterized by a slowly progressive spastic paraplegia (with walking difficulties appearing at onset at 6-7 years of age) associated with mild intellectual disability 108, 109. Brain imaging reveals thin corpus callosum, cortical and cerebellar atrophy, and pontine dysraphia. The SPG32 phenotype has been mapped to a locus on chromosome 14q12-q21 108, 109.
Spastic paraplegia 39
Spastic paraplegia 39 (SPG 39) also called NTE related motor neuron disorder is a rare autosomal recessive spastic paraplegia characterized by upper motor neuron involvement, progressive spastic paraplegia, peripheral neuropathy, distal muscle wasting and sometimes reduced cognitive functioning and/or cerebellar ataxia 110. Spastic paraplegia 39 (SPG 39) onset between childhood and early adulthood 111. Patients present with progressive spasticity, hyperreflexia, and distal upper and lower muscle wasting. Reduced cognitive functioning and cerebellar ataxia have also been reported 111. MR imaging may reveal cerebellar and/or spinal cord atrophy.
Spastic Paraplegia 47
Spastic paraplegia 47 (SPG47) also called AP4B1-associated hereditary spastic paraplegia or AP-4 deficiency syndrome is both a neurodevelopmental and a slowly progressive neurological disorder that generally presents with global developmental delay, moderate to severe intellectual disability, impaired/absent speech, small head size (microcephaly), seizures and progressive motor symptoms 112. Hypotonia (low-muscle tone) develops into hypertonia (high-muscle tone), resulting in spasticity of the legs that leads to the inability to walk (non-ambulation) and wheelchair reliance. Spasticity may progress to the upper extremities, leading to the partial or total loss of use of all four limbs and torso (tetraplegia) 112.
Most children with spastic paraplegia 47 (SPG47) have:
- a “floppy” appearance in infancy due to low muscle tone
- delayed motor development
- increasing spasticity and paralysis in the lower limbs starting in early childhood
- microcephaly
- intellectual disability
- poor or absent speech development
Other known features of spastic paraplegia 47 (SPG47) can include the following (not every child will have these features):
- short stature
- late walking and later loss of the ability to walk independently.
- dystonia (involuntary muscle contractions)
- ataxia (impaired balance and coordination)
- seizures including frequent seizures in the setting of fever.
Since many of the initial clinical manifestations of spastic paraplegia 47 (SPG47) are nonspecific and may resemble other disorders characterized by spasticity, developmental delay / intellectual disability and seizures, the diagnosis is often only made after further diagnostic testing. This may include a brain MRI showing characteristic features such as a thin corpus callosum, wide lateral ventricles and changes in the white matter. A definitive diagnosis is reached by genetic testing.
Spastic Paraplegia 49
Spastic Paraplegia 49 (SPG49) also called autosomal recessive spastic paraplegia type 49 or hereditary sensory and autonomic neuropathy due to TECPR2 mutation is a rare genetic condition that affects the nerves outside the brain and spinal cord 113, 114, 115. Spastic Paraplegia 49 (SPG49) is characterized by several symptoms, including: Early hypotonia (low muscle tone) that evolves to spastic paraparesis (weakness and stiffness in the legs), Areflexia (absence of reflexes), Decreased pain and temperature sensitivity, Autonomic neuropathy (damage to the nerves that control involuntary body functions), Gastroesophageal reflux disease (GERD). Recurrent pneumonia and respiratory problems, Intellectual disability, Dysmorphic features, including mild brachycephalic microcephaly (a small head with a short, broad shape), short broad neck, low anterior hairline, and coarse facial features. Spastic Paraplegia 49 (SPG49) is caused by mutations in the TECPR2 gene. When functioning normally, this gene helps the body break down and recycle cells that are damaged or unnecessary. Mutations prevent the TECPR2 gene from working correctly, leading to disturbances in the secretory pathway and nerve damage.
Spastic Paraplegia 49 (SPG49) should be suspected in individuals with the following clinical findings and family history 115:
Clinical findings
- Developmental delay / intellectual disability (mainly in the moderate to severe range)
- Neurologic findings
- Muscular hypotonia
- Gait ataxia
- Hyporeflexia / areflexia of the lower limbs
- Impaired pain sensitivity
- Dysarthria
- Autonomic dysfunction
- Impaired temperature regulation
- Impaired blood pressure regulation
- Central nocturnal and/or daytime hypoventilation
- Dysphagia
- Abnormal gastrointestinal mobility resulting in gastroesophageal reflux and/or constipation
- Recurrent respiratory infections resulting from aspiration as a result of gastroesophageal reflux and dysphagia
- Behavioral abnormalities (hyperactivity, aggressiveness, autism spectrum disorder)
- Distinctive facial features that may include brachycephaly, synophrys, thick eyebrows, hypotelorism, epicanthus, round or triangular-shaped face, and dental crowding 116, 117
Brain imaging
- Thin or dysplastic corpus callosum
- Mild ventriculomegaly (often asymmetric)
- Delayed myelination (i.e., delayed appearance of signal changes related to final myelination of white matter pathways in affected children)
- Mild cerebral atrophy or mild atrophy of the cerebellar hemispheres or vermis
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity) and/or a Bukharian or Ashkenazi Jewish background. Absence of a known family history and/or of Bukharian or Ashkenazi Jewish background does not preclude the diagnosis.
Spastic Paraplegia 50
Spastic Paraplegia 50 (SPG 50) also called AP4M1-associated hereditary spastic paraplegia or AP-4 deficiency syndrome is both a neurodevelopmental and a slowly progressive neurological disorder that generally presents with global developmental delay, moderate to severe intellectual disability, impaired/absent speech, small head size (microcephaly), seizures and progressive motor symptoms 118. Hypotonia (low-muscle tone) develops into hypertonia (high-muscle tone), resulting in spasticity of the legs that leads to non-ambulation and wheelchair reliance. Spasticity may progress to the upper extremities, leading to the partial or total loss of use of all four limbs and torso (tetraplegia).
Most children with spastic paraplegia 50 (SPG 50) have:
- a “floppy” appearance in infancy due to low muscle tone
- delayed motor development
- increasing spasticity and paralysis in the lower limbs starting in early childhood
- microcephaly
- intellectual disability
- poor or absent speech development
Other known features of spastic paraplegia 50 (SPG 50) can include the following (not every child will have these features):
- short stature
- late walking and later loss of the ability to walk independently
- dystonia (involuntary muscle contractions)
- ataxia (impaired balance and coordination)
- seizures including frequent seizures in the setting of fever
Since many of the initial clinical signs and symptoms of spastic paraplegia 50 (SPG 50) are nonspecific and may resemble other disorders characterized by spasticity, developmental delay / intellectual disability and seizures, the diagnosis is often only made after further diagnostic testing. This may include a brain MRI showing characteristic features such as a thin corpus callosum, wide lateral ventricles and changes in the white matter. A definitive diagnosis is reached by genetic testing.
Spastic paraplegia 51
Spastic paraplegia 51 (SPG 51) also known as AP-4 deficiency syndrome or AP-4-associated hereditary spastic paraplegia is a group of neurodegenerative disorders characterized by a progressive, complex spastic paraplegia with severe intellectual disability with onset typically in infancy or early childhood 119. Early-onset hypotonia evolves into progressive lower-extremity spasticity. The majority of children become non-ambulatory and usually wheelchair bound 119. Over time spasticity progresses to involve the upper extremities, resulting in a spastic quadriplegia (tetraplegia) 119. Associated complications include dysphagia, contractures, foot deformities, dysregulation of bladder and bowel function, and a pseudobulbar affect. About 50% of affected individuals develop seizures. Postnatal microcephaly (usually in the -2SD to -3SD range) is common 119. All have developmental delay. Speech development is significantly impaired and many affected individuals remain nonverbal. Intellectual disability in older children is usually moderate to severe 119.
Formal diagnostic criteria for spastic paraplegia 51 (SPG 51) have not been established 119.
AP-4-associated hereditary spastic paraplegia should be suspected in individuals with the following clinical findings and characteristic brain imaging findings 120, 121, 122, 123:
Clinical characteristic findings:
- Progressive spastic paraplegia with progression to tetraplegia in the later stages (94%, 58/62) *
- Early-onset developmental delay (100%, 68/68) *
- Delayed motor milestones (100%, 54/54) *
- Failure to achieve or loss of independent ambulation (93%, 41/44) *
- Impaired or absent speech development (98%, 51/52) *
- Neonatal/infantile hypotonia (usually mild) (100%, 41/41) *
- Postnatal microcephaly (77%, 47/61) (usually in -2SD to -3SD range) *
- Early-onset seizures including frequent febrile seizures (42%, 25/59) *
Less frequent findings:
- Short statue (65%, 17/26) *
- Nonspecific dysmorphic facial features (82%, 41/50) *
- Episodes of stereotypic laughter [Ebrahimi-Fakhari et al 2018]
- Foot deformities (i.e., clubfoot)
Brain imaging characteristic findings:
- Thinning of the corpus callosum (with prominent thinning of the posterior parts) (88%, 37/42) *
- Delayed myelination and nonspecific loss of the periventricular white matter (69%, 29/42) *
- Ex-vacuo ventriculomegaly, often with prominent enlargement of the posterior horns of the lateral ventricles (60%, 24/40) *
Less frequent findings:
Note: * Data from the International Registry and Natural History Study of Adaptor-Protein 4-Related Hereditary Spastic Paraplegia.
Spastic Paraplegia 52
Spastic Paraplegia 52 (SPG 52) also called AP-4 deficiency syndrome or AP4S1-associated hereditary spastic paraplegia is a slowly-progressing neurodegenerative disorder that generally presents with global developmental delay, moderate to severe intellectual disability, impaired/absent speech, small head size (microcephaly), seizures, and progressive motor symptoms 126. Hypotonia (low-muscle tone) develops into hypertonia (high-muscle tone), resulting in spasticity of the legs that leads to non-ambulation and wheelchair reliance 126. Spasticity may progress to the upper extremities, leading to the partial or total loss of all four limbs and torso (tetraplegia) 126.
Most children with spastic paraplegia 52 (SPG 52) have 126:
- a “floppy” appearance in infancy due to low muscle tone
- increasing spasticity and paralysis in the lower limbs starting in early childhood
- intellectual disability
- microcephaly
- delayed motor development
- poor or absent speech development
Other known features of spastic paraplegia 52 (SPG 52) can include the following (not every child will have these features):
- short stature
- late walking and later loss of the ability to walk independently
- dystonia (involuntary muscle contractions)
- ataxia (impaired balance and coordination)
- seizures including frequent seizures in the setting of fever
Some children may also have facial differences that can include:
- high palate
- wide nasal bridge
- bulbous nose
- wide mouth
- protruding tongue
- short philtrum (the groove between the bottom of the nose and top of the lips)
- narrow forehead
- flat feet or club feet
Since many of the initial clinical manifestations of SPG52 are nonspecific and may resemble other disorders characterized by spasticity, developmental delay / intellectual disability, and seizure, the diagnosis is often only made after further diagnostic testing. This may include a brain MRI showing characteristic features such as a thin corpus callosum, wide lateral ventricles and changes in the white matter. A definitive diagnosis is reached by genetic testing.
Hereditary spastic paraplegia causes
Hereditary spastic paraplegias are caused by gene mutations. To date, more than 80 genetic types of hereditary spastic paraplegia have been defined by genetic linkage analysis and identification of hereditary spastic paraplegia-related gene variants (Table 3). Underlying causes of hereditary spastic paraplegia: Each of the more than 80 genetic types of hereditary spastic paraplegia is due to mutations in a different gene. Each genetic type of hereditary spastic paraplegia is due to a mutation in a specific “hereditary spastic paraplegia gene”. For example, mutations in SPG3A/atlastin, SPG4/spastin, and SPG7/paraplegin genes cause SPG3A, SPG4, and SPG7 hereditary spastic paraplegia, respectively. These genes encode proteins that have diverse molecular functions including movement of chemicals from one part of the cell to another (“axon transport”), energy production (“mitochondrial disturbance”), and disorders of specific lipid metabolism, among others 127, 128.
Disturbance in some of these functions appears to lead to altered nerve cell (neuron) development. For these types of hereditary spastic paraplegia, the disorder is not a degenerative process, but rather a developmental disturbance in which the formation of selected nerve pathways during intra-uterine development was abnormal.
For other genetic types, hereditary spastic paraplegia gene mutations cause the ends of very long nerve processes (axons) to slowly degenerate within the spinal cord. This impairs nerve transmission from the brain through the spinal cord. To be clear, the entire spinal cord is not degenerating. Rather, the abnormalities in hereditary spastic paraplegia appear to selectively affect only specific nerve pathways, particularly the very long nerve processes (axons) that carry signals from the brain motor cortex to the lower part of the thoracic spinal cord. In some types, this disturbance is not limited to the spinal cord but also affects nerves in the legs (and arms, to a lesser extent). This latter process is termed “peripheral neuropathy”.
Depending on the genetic type of hereditary spastic paraplegia, hereditary spastic paraplegia may be transmitted to offspring (and inherited from parents) as Autosomal dominant, Autosomal recessive, X-Linked Inheritance, and “maternal” or mitochondrial inheritance. The various genetic types of hereditary spastic paraplegia and their inheritance patterns are summarized in the Table 3 below.
The following discussion of inheritance patterns is intended as an overview. Individuals seeking genetic counseling for hereditary spastic paraplegia are recommended to consult a genetic counselor or medical geneticist for specific information. In general, autosomal dominantly inherited forms of hereditary spastic paraplegia can be transmitted by (or inherited from) an individual who has the disorder. In general, each child of an individual who has a autosomal dominantly inherited form of hereditary spastic paraplegia has a 50% chance of inheriting the gene mutation and a similar (approximately 50% chance) of developing the condition. Occasionally, autosomal dominantly inherited hereditary spastic paraplegia “skips” a generation. (i.e. genetic penetrance is very high, exceeding 90%, but is occasionally incomplete). Although the chance of inheriting the condition can be estimated, it is difficult to predict with certainty the age at which symptoms would begin or their severity. There may be significant differences in the severity of the disorder between family members.
For autosomal recessive inherited forms of hereditary spastic paraplegia, both parents are usually carriers of the gene mutation and usually do not have symptoms (there are exceptions to this generalization: occasionally, parents who are carriers of some forms of autosomal recessive hereditary spastic paraplegia have had symptoms of hereditary spastic paraplegia). In general, if one individual in a family has a autosomal recessively inherited disorder, each of this individual’s full siblings (for example, another child in this family) has approximately a 25% chance of having the same disorder. In general, individuals who have autosomal recessive inherited disorders do not transmit the disorder to their children. There have been some reported exceptions to this however.
X-linked inheritance disorders are transmitted from women to their sons. Daughters may carry X-linked gene mutations, but like their mothers, usually do not have symptoms although they may have mild symptoms and rarely, may have more significant symptoms of the disorder.
Maternally or mitochondrial transmitted disorders are those in which the gene mutation involves a mitochondrial gene, are transmitted from mothers to sons or daughters (not transmitted from males).
Table 3. Chromosomes, loci, and genes involved in hereditary spastic paraplegias and their subtypes
| Inheritance Mode | Chromosome Number | Locus | Genes | Hereditary Spastic Paraplegia Subtype |
|---|---|---|---|---|
| Autosomal dominance (AD) | 1 | 1p31.1 | NERG1 * | SPG29 |
| 2 | 2p11.2 2p22.3 2q33.1 2q37.3 | REEP1 SPAST HSPD1 KIF1A | SPG31 SPG4 SPG13 SPG30 | |
| 3 | 3q25.31 | SLC33A1 | SPG42 | |
| 4 | 4p16-15 | JAKM1P1 * | SPG38 | |
| 8 | 8p21.1-q13.3 8q24.13 | FBXO16 KIAA0196 | SPG37 SPG8 | |
| 9 | 9p13.3 9q | UBAP1 – | SPG80 SPG19 | |
| 10 | 10q24.1 10q24.2 | ALDH18A1 ZFYVE27 | SPG9A SPG33 | |
| 11 | 11p14.1-p11.2 11q12.3 | BDNF * BSCL2 | SPG41 SPG17 | |
| 12 | 12q13.3 12q23-24 | KIF5A CKAP4 * | SPG10 SPG36 | |
| 14 | 14q22.1 | ATL1 | SPG3A | |
| 15 | 15q11.2 | NIPA1 | SPG6 | |
| 16 | 16q24.3 | PARAPLEGIN | SPG7 | |
| 19 | 19q13.32 19q13.33 | RTN2 CPT1C | SPG12 SPG73 | |
| Autosomal recessive (AR) | 1 | 1p36.13 1p34.1 1p13.3 1p13.2 1q32.1 1q42.13 1q42.13 | ATP13A2 HPDL AMPD2 AP4B1 DSTYK GJC2 IBA57 | SPG78 SPG83 SPG63 SPG47 SPG21 SPG44 SPG74 |
| 2 | 2p23.3 2q37.3 | SELENOI KIF1A | SPG81 SPG30 | |
| 3 | 3q12.2 3q27-q28 | TFG – | SPG57 SPG14 | |
| 4 | 4p13 4q25 | UCHL1 CYP2U1 | SPG79 SPG56 | |
| 6 | 6p25.1 6q23-24.1 | FARS2 – | SPG77 SPG25 | |
| 7 | 7p22.1 7q22.1 | AP5Z1 AP4M1 | SPG48 SPG50 | |
| 8 | 8p11.23 8p11.23 8p22 8q12.3 | ERLIN2 DDHD2 VPS37A CYP7B1 | SPG18 SPG54 SPG53 SPG5A | |
| 9 | 9p13.3 | GBA2 | SPG46 | |
| 10 | 10q22.1-q24.1 10q24.1 10q24.1 10q24.31 10q24.31-10q24.33 | – ALDH18A1 ENTPD1 ERLIN1 NT5C2 | SPG27 SPG9B SPG64 SPG62 SPG45 | |
| 11 | 11q13.1 | CAPN1 | SPG76 | |
| 12 | 12q13.3 12q24.31 | B4GALNT1 C12ORF65 | SPG26 SPG55 | |
| 13 | 13q13.3 (TROYER SYNDROME) 13q14 | SPART – | SPG20 SPG24 | |
| 14 | 14q22.1 14q24.1 14q32.31 | DDHD1 ZFYVE26 TECPR2 | SPG28 SPG15 SPG49 | |
| 15 | 15q21.1 15q21.2 15q22.31 (MAST SYNDROME) | KIAA1840 AP4E1 ACP33 | SPG11 SPG51 SPG21 | |
| 16 | 16p12.3 16q23.1 16q24.3 | ARL6IP1 FA2H PARAPLEGIN | SPG61 SPG35 SPG7 | |
| 17 | 17q25.3 | PCYT2 | SPG 82 | |
| 19 | 19p13.2 19q12 19q13.12 | PNPLA6 C19ORF12 MAG | SPG39 SPG43 SPG75 | |
| X-Linked Inheritance (XLR) | X | Xq11.2 Xq11.2 Xq22.2 Xq24-25 Xq28 | MTMR8 * ZC4H2 * PLP1 GLUD2 L1CAM | SPG16 SPG16 SPG2 SPG34 SPG1 |
| Mitochondrial or Maternal Inheritance | Mitochondria | – – – – | MT-CO3 MT-T1 MT-ND4 MT-ATP6 | – – – – |
Footnotes: * The predicted genes that might induce hereditary spastic paraplegia phenotypes are in bold letters.
[Source 7 ]Autosomal Dominant hereditary spastic paraplegia
Autosomal dominant hereditary spastic paraplegia is the most common type of hereditary spastic paraplegia affecting 75 to 80% of registered cases with SPG4 (SPAST mutation) as the most common type, accounting for approximately 40% of all autosomal dominant hereditary spastic paraplegia 129. The following chromosomal markers and genes (Table 3) are involved in autosomal dominant hereditary spastic paraplegia (Figure 2).
- SPG3A caused by ATL1 mutation, is the second most common type of autosomal dominant hereditary spastic paraplegia, accounting for approximately 10%-15% of all autosomal dominant hereditary spastic paraplegia. SPG3A is the main cause of autosomal dominant hereditary spastic paraplegia with early onset before the age of 10 (occurs in >75% of individuals in this category) 130, 9.
- SPG30 caused by KIF1A mutation and SPG31 caused REEP1 mutation are both relatively common, each accounting for about 5% of all autosomal dominant hereditary spastic paraplegia 9.
- Other types of autosomal dominant hereditary spastic paraplegia SPG6/NIPA1 and SPG8/KIAA0196 with a predominantly adult onset are relatively rare and most of them account for 1% or less of all autosomal dominant hereditary spastic paraplegia 131, 132, 9.
- Mutation in the heat shock protein HSP60 (SPG13), SPG17/BSCL2 are known to induce hereditary motor neuropathies 133. Other prevalent autosomal dominant forms of hereditary spastic paraplegia have been observed due to mutations in the SPG31/REEP1 134 and SPG33/ZFYVE27 genes 135.
The mean age of patients upon manifestation is 31.7 years, with an exception of 70 years of age in a few cases with spasticity of the lower limbs along with/without bladder and sensory dysfunction 136, 137, 40.
Autosomal Recessive hereditary spastic paraplegia
Autosomal Recessive hereditary spastic paraplegias are rare and confined to single families or single persons with heterogeneity and an ever-growing list of recently observed genes. The chromosomal markers linked to autosomal recessive pattern are discussed with their loci and the gene (Table 3) involved in the hereditary spastic paraplegias progression (Figure 2). Consanguineous marriage (marriage between individuals who are closely related) increases the frequency of autosomal recessive hereditary spastic paraplegias in a given community, and about less than 30% of the registered hereditary spastic paraplegias cases show recessive inheritance patterns. Usually, the phenotypic characters are different between each family member but are complex invariably 18.
SPG11 is the most prevalent form of Autosomal Recessive hereditary spastic paraplegias seen in about 8% of the registered cases ranging from 4 to 36 years of age. The instances with autosomal recessive pattern usually show cases with learning disabilities and spasticity in the lower extremes in the second decade of the patient’s life. Table 3 lists out the chromosomes, their locus, and genes involved in the autosomal recessive pattern of hereditary spastic paraplegia. About 50% show neuropathy of motor axons, ataxia, dysarthria, progressive spasticity in the upper body, and visual failure 138.
X-Linked Recessive hereditary spastic paraplegia
X-Linked Recessive hereditary spastic paraplegia represents about 1–2% of hereditary spastic paraplegia 7. X-Linked Recessive hereditary spastic paraplegia shows a distinct phenotype of pure hereditary spastic paraplegia and complicated hereditary spastic paraplegia with spasticity and weakness in a slow progressive way in the lower legs in pure form, whereas dementia, ataxia, retinopathy, epilepsy, ichthyosis are observed in complicated cases 7. About four loci have been registered to date to induce hereditary spastic paraplegia characteristics on the X chromosome (Figure 2). The loci q11.2, q22.2, q24-25, and q28 are SPG16, SPG2, SPG34, and SPG1, respectively (Table 3).
Mutation in proteolipid protein (PLP1) at q22.2 induces lower extremity spasticity severely. Animal and cell transfection models showed oligodendrocyte apoptosis and PLP accumulation in endoplasmic reticulum, leading to unfolded protein response causing hereditary spastic paraplegia characteristics 139.
The usual brain function is controlled by the temporal and structural association between the cells with L1 cell adhesion molecules members (SPG1/L1CAM) through hetero- and homophilic interactions. Any alterations of q28 in L1CAM (usually loss of function) lead to development abnormalities, with cognitive impairment, limbs spasticity, corpus callosum aplasia, axons outgrowth, hydrocephalus, and myelination anomalies collectively known as CRASH or L1 syndrome 140.
Maternal (Mitochondrial) Inheritance hereditary spastic paraplegia
Being sporadic, maternal (mitochondrial) inheritance is mainly seen in complex hereditary spastic paraplegias, primarily directing the focus towards mitochondrial disabilities 7. Maternal (mitochondrial) inheritance are the rarest hereditary spastic paraplegia kinds and affect nearly 1–2% of hereditary spastic paraplegia cases 7. Usually, alterations linked to ATP6 gene’s “m.9176 T>C” are associated with mitochondrial function impairment for the hereditary spastic paraplegia phenotype 7. Similar alteration is observed in Leigh syndrome, implicating the role of modifying environmental and lifestyle factors to express various phenotypes for the same alteration 141. Mutations in MT-CO3 gene encoding for Cytochrome c oxidase III/complex IV—respiratory chain complex IV subunit; MT-T1 gene associated with Isoleucine transfer RNA; and MT-ND4 and MT-ATP6 genes encoding for Complex V, ATP synthase, and subunit ATPase 6—respiratory chain complex V subunit are shown to impart mental retardation, cerebellar ataxias, loss of hearing, chronic progressive external ophthalmoplegia, and neuropathy in few rare hereditary spastic paraplegias 142.
Hereditary spastic paraplegia pathophysiology
Hereditary spastic paraplegias are mainly due to neuronal degeneration 1. The core phenotypic features of hereditary spastic paraplegia result from axonal degeneration of neurons of the pyramidal motor system which is responsible for the voluntary movements in humans. The neurons of the pyramidal tracts extend from the layer V of the cerebral motor cortex and, after synaptic connection in the spinal cord with the secondary motor neuron, finally innervate the skeletal muscles at the neuromuscular junctions. Neurons are the principal category of cells that degenerate in hereditary spastic paraplegia, even though the contribution of oligodendrocytes and other glial cells to the pathology of hereditary spastic paraplegia cannot be ignored since several hereditary spastic paraplegia genes are expressed in non-neuronal cells 143.
Neurons are polarized cells 144 and the pyramidal tract neurons can be injured in a length dependent manner through a dying-back mechanism of their axons. Since the longest axons are more susceptible, this leads to the primary clinical involvement of the lower limbs 145, 146. In the complex hereditary spastic paraplegias, further regions of the central and peripheral nervous systems alongside other extra-neurological organs and systems can also be involved. In these other tissues, the degeneration of cell bodies occurs in interneurons with shorter axons as well and then the dying-back hypothesis is less evident.
Lateral corticospinal axonal degradation is predominantly observed in hereditary spastic paraplegia postmortem studies with a higher depletion rate in the spinal cord’s thoracic zone distal end and cervical zone mainly due to axons degeneration in fasciculus gracilis fibres to demyelinating 147. Sometimes, degeneration can extend to the rostrum, internal capsule, peduncles of the cerebellum, pons, and medullary zone, with declined Betz cells concentration (pyramidal neurons) 133.
The central nervous system (CNS) long axons are hotspots and the first site of hereditary spastic paraplegia axonopathy. Peripheral neuropathy, a common hereditary spastic paraplegia subgroup symptom, is caused by the depletion of other neurons. Due to neuropathy in specific locations, shorter neurons in the basal ganglia, cerebellum, anterior horn cells, and Clarke’s column cause hereditary spastic paraplegia features 148. Neuronal region genetic mutations cause deformities predominantly in the myelin layer, and other studies revealed cerebellar atrophy and central nervous system (CNS) myelination, along with corticospinal axonal degeneration and developmental disorders, smaller spinal cord diameter, and thin corpus callosum as classical developmental abnormality signs in hereditary spastic paraplegias (see Figure 2) 142.
Table 4. Hereditary spastic paraplegia pathogenesis
| Cellular Models Involved in hereditary spastic paraplegia Pathogenesis | Proteins | SPG Forms |
|---|---|---|
| 1. Organelle’s morphogenesis/membrane structure Microtubule/membrane structure-associated ATPaseER tubules linking GTPaseER morphology organizerModulating lipid metabolism and droplet formationRegenerating lysosomeAdaptor proteinsTubule formation in endosomesDefects in organelles morphology inducing axonal pathology (lysosome and ER endosomes) | SPASTAtlastin SPAST AIP4/Spartin Spatacsin AP-4 for trafficking precursors of amyloids Strumpellin REEP1 | SPG4SPG3A SPG4 SPG20 SPG11 SPG47 SPG8 SPG31 |
| 2. Bone morphogenic proteins | NIPA1 Atlastin-1 Spastin Acetyl-CoA transporter | SPG6 SPG42 SPG3A SPG4 |
| 3. Motor proteins transportation | KIF5A KIF1C KIF1A | SPG10 SPG58 SPG30 |
| 4. Mitochondrial failure | Paraplegin HSP60 IBA57 Spartin | SPG7 SPG13 SPG74/SPG77 SPG20 |
| 5. Axon elongation path | L1CAM | SPG1 |
| 6. Myelination errors | PLP1 GJC2 MAG | SPG2 SPG44 SPG75 |
| 7. Nucleotide’s metabolism | AMPD2 NT5C2 ENTPD1 | SPG63 SPG65 SPG64 |
Hereditary spastic paraplegia symptoms
The primary symptom of hereditary spastic paraplegia is difficulty walking due to weakness and tightness (spasticity) in the legs. Both legs are affected, usually to a relatively similar degree.
The term “paraplegia” means severe weakness in both legs including paralysis. “Paraparesis” indicates weakness in both legs of lesser severity than paraplegia. Although HSP is typically referred to as hereditary spastic paraplegia the degree of weakness is variable and ranges from no weakness (full strength) to marked weakness (paraplegia).
When present, weakness does not affect all leg muscles, but rather is most obvious in muscles of hip flexion (iliopsoas), hip abduction (gluteus medius), knee flexion (hamstrings), and foot dorsiflexsion (bending the foot back toward the shin via tibialis anterior muscle). In contrast, muscles of leg extension (quadriceps) and foot extension (gastrocnemius-soleus) usually are not affected in uncomplicated hereditary spastic paraplegia.
Spasticity primarily affects muscles of leg extension (quadriceps), knee flexion (hamstrings), hip adduction (bringing the knees together, thigh adductor muscles), and muscles that extend the feet (gastrocnemius-soleus [Achilles tendon]).
Walking pattern described as “spastic gait” occurs in which the following elements are present, each to variable degree in different individuals:
- a) heel strike is shifted forward (landing on the mid-foot or even further forward on the balls of the feet);
- b) there is reduced foot dorsiflexion (not bending the toes up, but instead tending to drag the toes, often catching them on carpet or when stepping over curbs, and causing the toes of the shoes to be worn out);
- c) stride length may become shorter;
- d) there may be “circumduction” or “scissoring”, with one leg crossing into the path of the other;
- e) there is a tendency for the knees to be maintained flexed (not fully extended in mid-stride),
- f) for thighs to be close together (adductor tightness), and
- g) hip flexion (knee lifting) to be reduced.
Balance difficulty, often worse when walking in the dark or on uneven surfaces is not uncommon in individuals with hereditary spastic paraplegia.
Tightness in the legs and leg muscle spasm (often at night) are common.
The consequences of abnormal walking pattern cause strain on the ankles, knees, hips, and back and often cause pain in these areas.
Urinary urgency, the symptom of experiencing a very short interval between the sensation of need to urinate and difficulty remaining continent, is very common in hereditary spastic paraplegia and occasionally may be an early symptom. Bowel urgency is less common but may occur. Medications such as oxybutynin may reduce urinary urgency. If urinary urgency is severe or accompanied by difficulty initiating urination, consultation with a urologist is recommended.
Additional symptoms
Some genetic types of hereditary spastic paraplegia tend to cause only spastic weakness in the legs and urinary urgency. These syndromes are referred to as “uncomplicated hereditary spastic paraplegia”. Other genetic types of hereditary spastic paraplegia tend to be associated with additional symptoms (“complicated hereditary spastic paraplegia”) including difficulty with coordination (“ataxia”), impaired vision, seizures (epilepsy), muscle atrophy, disturbance of the nerves in the arms and legs (neuropathy), and disturbance cognitive ability (intellectual impairment and dementia). Previously, it was considered that hereditary spastic paraplegia caused symptoms only in the legs, and therefore, did not affect the strength or coordination of the arms and hands, or speech or swallowing. As the number of hereditary spastic paraplegia types has grown, it is now recognized that the arms, hands, and speech and swallowing may be affected in some genetic types of complicated hereditary spastic paraplegia
Pattern of symptom progression
When hereditary spastic paraplegia begins in very early childhood (before age two years, for example), symptoms may not worsen even over many years or decades. Individuals with this “non-progressive” (non-worsening) pattern may resemble subjects with spastic cerebral palsy, a life-long disorder that also remains relatively stable. One caveat however: although early childhood-onset forms of hereditary spastic paraplegia may be “non-progressive”, the degree of spasticity may increase slowly if adequate range-of-motion is not maintained through stretching exercises and muscle spasticity reduction.
In contrast, when hereditary spastic paraplegia symptoms begin after early childhood (in adolescence or adulthood), symptoms usually worsen very slowly over a number of years. Sudden onset or rapid worsening over weeks or months is not typical of hereditary spastic paraplegia and suggests an alternate disorder or co-existing condition. After a number of years of very gradual worsening, the rate of worsening appears to slow down for many (not all) subjects. These subjects seem to reach a “functional plateau” beyond which the degree of worsening seems to be similar to that expected for age and similar degrees of physical exercise. Nevertheless, not all patients reach an apparent “leveling off” or functional plateau but instead experience continuous worsening of walking ability due to very slowly progressive muscle weakness and tightness .
Variability in the type of symptoms and their severity
There may be significant variability in the type of symptoms and their severity. For example, symptoms may remain mild in some patients or become quite severe in others patients. This variability may occur between different genetic types of hereditary spastic paraplegia as well as in between individuals with the same genetic type of hereditary spastic paraplegia including family members who share not only the same genetic type of hereditary spastic paraplegia but also precisely the same genetic mutation.
There is not a perfect correlation between the genetic type of hereditary spastic paraplegia and the pattern of symptoms. For example, while some genetic types of hereditary spastic paraplegia (e.g. dominantly inherited hereditary spastic paraplegia due to SPG4 or spastin mutation) usually are associated with “uncomplicated” hereditary spastic paraplegia syndromes, some patients with these types of hereditary spastic paraplegia develop additional neurologic symptoms. As another example, although SPG7 and SPG11 typically are associated with additional neurologic symptoms (ataxia, neuropathy, cognitive impairment, for example), some subjects with mutations in these genes have uncomplicated hereditary spastic paraplegia (only spastic weakness in the legs). There also may be variation in severity and the nature of symptoms between affected family members. Therefore, it is generally not possible to predict with certainty the severity or exact nature of symptoms associated with given genetic type of hereditary spastic paraplegia. A cautious, “wait and see” approach, combined with pro-active, individualized physical therapy is recommended.
Hereditary spastic paraplegia complications
Possible complications of hereditary spastic paraplegia include:
- shortening and hardening of the calf muscles – having regular physiotherapy may help prevent this
- cold feet – this is fairly common and occurs as a result of the deterioration of the nerves in the spine
- extreme tiredness (fatigue) – this may be because of the extra effort needed for walking and the symptoms interrupting sleep
- back and knee pain – caused by the muscle weakness and walking problems
- stress and depression
Hereditary spastic paraplegia diagnosis
Hereditary spastic paraplegia is diagnosed by the following 16:
- Typical symptoms lower extremity spastic weakness that may be non-worsening (early childhood onset) or slowly progressive over many years. The clinical manifestations are spastic paraplegia of both lower limbs, but cases with increased tendon reflexes, slight impairment of rapid alternating movements or mild distal amyotrophy in the upper limbs are not excluded; the cranial nerves and language are not affected.
- Findings on neurologic examination lower extremity hyperreflexia usually accompanied by some degree of spasticity and sometimes a specific pattern of muscle weakness; and
- The exclusion of alternate disorders by history, examination, neuroimaging, and laboratory studies as needed.
- Laboratory testing: Common biomarkers are normal.
- Imaging: Brain and spinal cord magnetic resonance imaging (MRI) are usually normal, but some patients may present with dysgenesis of the corpus callosum or tapering of the spinal cord.
The occurrence of similarly affected family members is helpful in recognizing hereditary spastic paraplegia but is not required for the diagnosis of hereditary spastic paraplegia. Many individuals with hereditary spastic paraplegia do not have similarly affected family members. Such individuals could represent the first occurrence of a genetic mutation (“de novo mutation”). Depending on the genetic type of hereditary spastic paraplegia (dominant, recessive, X-linked, or maternal transmission), there may be a possibility that the disorder could be transmitted to the offspring of these individuals. Genetic testing is often helpful in confirming the clinical diagnosis of hereditary spastic paraplegia and in determining the genetic type of hereditary spastic paraplegia. Results of genetic testing can be used, together with clinical information, to provide genetic counseling.
Hereditary spastic paraplegia diagnostic tests
Neurologic examination is important for patients with symptoms of hereditary spastic paraplegia. First, this establishes the diagnosis and excludes alternative and co-existing disorders, some of which may have specific treatments. Second, neurologic examination helps identify the specific features of an individual’s walking disturbance. Knowing which specific muscles need strengthening, which specific muscles need spasticity-reduction (through medication, Botox injection, and stretching), and the degree of impairment of balance, speed, and precision of movement helps neurologists and physiatrists develop a proactive therapy approach to improve and maintain the ability to walk; and limit the cumulative impact of abnormal walking patterns on ankles, knees, hips, and spine.
Laboratory tests, neurophysiologic testing, and neuroimaging: Routine laboratory studies (such as blood counts, serum electrolytes, and tests of kidney, liver, and endocrine functions) including analysis of cerebrospinal fluid (obtained by “spinal tap”) are normal in most types of hereditary spastic paraplegia. The primary role of such testing is to help exclude alternate and co-existing diagnoses.
Magnetic resonance imaging (MRI) scans of the brain and spinal cord are important in diagnosing hereditary spastic paraplegia because they help exclude other disorders such as multiple sclerosis and structural abnormalities of the brain and spinal cord. Routine magnetic resonance imaging (MRI) of the brain is usually normal in uncomplicated hereditary spastic paraplegia, and, depending on the genetic type and its neurologic features, in many forms of complicated hereditary spastic paraplegia. In contrast to routine brain MRI, which is usually normal in uncomplicated hereditary spastic paraplegia, special MRI techniques such as diffusion tensor imaging, reserved primarily for research purposes often show more widespread nerve pathway abnormalities in uncomplicated and complicated hereditary spastic paraplegia.
In contrast to the typically normal brain MRI in subjects with uncomplicated hereditary spastic paraplegia, there are many types of complicated hereditary spastic paraplegia in which brain MRI demonstrates specific abnormalities including reduced size of the corpus callosum (a structure containing nerve fibers that transit from one brain hemisphere to the other).Thin corpus callosum is a frequent (but not constant) feature of SPG11 and SPG15 and has also been present in many other types of hereditary spastic paraplegia (including SPG3A, SPG4, SPG7, SPG15, SPG21, SPG32, SPG47, PG49, SPG54, and SPG56) 149, 150. In addition to thin corpus callosum, many genetic types of hereditary spastic paraplegia have abnormal appearing brain white matter (e.g. due SPG5/CYPB7, SPG7/paraplegin, SPG21/maspardin, and SPG35/FA2H gene mutations) 149, 150.
Spinal cord MRI scan in hereditary spastic paraplegia is usually normal although may show somewhat smaller diameter of the thoracic spinal cord 151.
Genetic testing
There are different approaches available to test hereditary spastic paraplegia genetically, and the most cost-effective and widely available one is the next generation sequencing (NGS), as it comprises of screening the whole exons to find the number of genes linked to hereditary spastic paraplegia phenotypes but still possess limitations for differentiating the variants of large deletions, duplications, alterations in the promotor or intronic regions, and cases of triplet repeat disorders 152. To obtain normal results, the multiplex ligation-dependent probe amplification is utilized for genes such as SPAST with exon deletions 153. In a few places, first-generation sequencing is initially carried out with a set of targeted genes, and next generation sequencing is considered later 18. The algorithm varies from clinician to clinician; some centres prefer next generation sequencing (NGS) panel sequencing as a first-line investigatory protocol, even in the absence of finding a pathogenic variant.
The clinician must conclude to find the gene for multiple ligation-dependent probe amplification or relate the uncertain variant with the phenotype. In addition, clinicians should have knowledge on the other monogenic diseases with the same phenotype of slowly progressing spasticity on lower limbs with no spinal cord imaging abnormalities not to be categorized in spastic paraplegia gene (SPG). The next generation sequencing (NGS) panel does not comprehensively cover hereditary spastic paraplegia alone, and they include the panels for ataxias (Spinocerebellar, AR, spastic), myelination defects, and other neurometabolic disorders 18.
Hereditary spastic paraplegia differential diagnosis
Hereditary spastic paraplegia is often misdiagnosed and confused with several disorders, predominantly with leukodystrophies 154, primary lateral sclerosis, X-linked adrenomyeloneuropathy 155 and rarely with peripheral neuropathy, Parkinson’s disease, hereditary ataxia, or amyotrophic lateral sclerosis (ALS) (see Figure 4). Specific disorder’s gene panels are used to provide an accurate clinic classification to improve diagnostic yield.
Hereditary spastic paraplegia treatment
Despite encouraging progress in many research laboratories, treatment for hereditary spastic paraplegia is presently limited to reducing symptoms of muscle weakness, improving balance, strength, agility, spasticity, and urinary urgency 156. Individuals should be evaluated periodically (annually or as needed) by a neurologist and physiatrist (medical doctor who specializes in pain management and rehabilitation) to assess progression and develop treatment strategies to maximize walking ability and reduce symptoms.
Current treatment recommendations for hereditary spastic paraplegia include:
- Daily regimen of physical therapy directed toward improving cardiovascular fitness, maintaining and improving muscle strength and gait, and reducing spasticity
- Occupational therapy, assistive walking devices, and ankle-foot orthotics as needed
- Drugs to reduce muscle spasticity (e.g., Lioresal [oral or intrathecal], tizanidine, dantrolene, botulinum A and B toxin injections [Botox, Dysport, Xeomin, or Myoblock]) and urinary urgency (e.g., oxybutynin, solifenacin, mirabegron, or intrabladder injections with Botox)
Individuals with hereditary spastic paraplegia are recommended to pursue active lifestyles including physical rehabilitation in order to maintain and improve functional abilities and cardiovascular fitness.
Hereditary spastic paraplegia physical therapy
Physiotherapy with daily physical exercise program especially foot and ankle orthotics and peroneal nerve stimulation guided by physical therapist or personal trainer and developed for each patient’s unique type of symptoms is recommended for lower limb building and strength, reducing toe dragging, and enhancing the cardiovascular system’s functioning 147. This recommendation is based not on peer-reviewed scientific publications but rather on the reports of large numbers of hereditary spastic paraplegia subjects who state that exercise helps and that periods of reduced exercise are associated with increased symptoms. Hereditary spastic paraplegia symptoms are variable. One type of exercise may not benefit all individuals. Exercise programs should be developed by a neurologist, physiatrist, physical therapist or personal trainer who is experienced with hereditary spastic paraplegia or similar disorders and should focus on the specific factors that make walking difficult for the specific individual. Individuals are advised to consult their primary care physician before beginning exercise programs, to begin with low intensity, increase slowly, set small goals, keep records of their progress, add variety, and be creative.
Exercise goals are to:
- Improve and maintain cardiovascular fitness;
- Reverse the reduced functional capacity, stiffness, and weakness due to relatively sedentary lifestyle that often accompanies chronic gait disorders and which are superimposed on walking disturbance due to hereditary spastic paraplegia;
- Improve the mechanics of walking, facilitate neurologic circuits underlying walking reflexes, reduce falling, and maintain bone and joint fitness; and
- Maximize an individual’s independence and sense of control.
For some patients, weakness in certain muscles is the most significant factor making walking difficult. For these patients, daily exercise programs should focus on resistance exercises designed to maintain and very gradually increase the strength of these weak muscles. For others, spasticity is a more significant factor than weakness in limiting the ability to walk. For these patients, stretching exercises and muscle relaxant medication may be more beneficial than muscle strengthening exercises alone. A variety of exercises is recommended including walking, for example in shallow swimming pool, water aerobics, swimming, bicycling (including pedaling in reverse to exercise the strength and speed of hip flexion), yoga, dance, core exercises, balance exercises, and therapeutic horseback riding.
Muscle relaxing medication
Patients with significant degrees of spasticity may benefit from antispasmodic (skeletal muscle relaxants) medications such as tizanidine and baclofen. In general, reducing spasticity through medication improves walking primarily when spasticity, not weakness is the primary factor limiting the ability to walk. Early intrathecal baclofen improves gait in severe spastic patients wheelchair-ridden by declining pain, disability, and muscle tone 157. When weakness is the major factor, markedly reducing spasticity (e.g. through intrathecal baclofen pump) may make the legs very relaxed (even hypotonic or “floppy”) but actually make walking and standing more difficult. Patients considering intrathecal baclofen pumps should undergo at least one trial in which the important criteria is not simply if spasticity reduction occurred, but rather if spasticity reduction resulted in improved walking ability. Botulinum toxin (“Botox” or botulinum toxin type-A) injection may be helpful when muscle tightness particular affects a limited number of muscles (e.g. adductors or ankles), with stretching exercises of ankle, hips, and knees, increases gait velocity by reducing calf muscle tone to maintain strength and balance 158. In addition, the Botulinum toxin efficacy has not been accessed in the common nonmotor manifestations observed in hereditary spastic paraplegia cases such as pain, fatigue, depression, and excessive daytime sleepiness, but studies by Servelhere et al. 159 found significant improvement for fatigue after Botox treatment in hereditary spastic paraplegia patients by relieving spasticity and improving gait biomechanics to reduce fatigue. Yet studies are required to identify the analgesic response of the Botox in more hereditary spastic paraplegia cases to obtain conclusive knowledge.
Oxybutynin and related medications may reduce urinary urgency and infection 18. Individuals with more advanced bladder or bowel symptoms are recommended to consult urology or gastroenterology specialists, respectively.
A positive response is observed in a few to 4-aminpyridine (Dalfampridine), which is awaiting conclusive results from larger group studies 160.
Orthotics
Ankle-foot orthotics and heel raise orthotics may be useful to reduce the tendency for the feet to be extended (toes down) causing toe dragging and tripping 18. Ankle-foot orthotics are often used in combination with medications (e.g. Lioresal or Botox) that reduce muscle spasticity.
Potential future treatments
Treatment based on the genotype has paved its way in neurological diseases in general for Huntington’s disease or spinal muscular atrophy. However, hereditary spastic paraplegia is lagging due to its complex genetics, mechanism diversity, various subtypes with rare forms, and slower disease progression 18. Through gene therapy, microtubule loss can be corrected in SPG4 phenotypes 161. Havlicek et al. 162 showed that spastin-mutated human-induced pluripotent stem cells of the patient develop neutrino membrane again with branching and increasing the length to reduce neuron swelling. Cholesterol breakdown is controlled by oxysterol hydroxylase-7α encoding the CYP7B1, and this gene is linked with AR SPG5. Primarily lowering cholesterol levels is recommended for managing hereditary spastic paraplegia phenotypes using atorvastatin. Deeper studies are required to obtain reliable results for utilising combined therapies 163.
The paraplegin gene mediates the fast opening of the transition pore, and it can be modulated using medications 18. One study showed that intramuscular paraplegin administration in hereditary spastic paraplegia mice model prevents pathology progression in neurons and protects mitochondrial structure in the peripheral nerves 164. Gene therapy for the recessive hereditary spastic paraplegia forms such as SPG11, SPG15, and SPG7 can allow editing genes at their target location or for replacement. Currently, the collaboration between world researchers and randomized control studies on large groups is required to obtain efficacious treatment for this neuromuscular pathology 18.
Hereditary spastic paraplegia prognosis
There is significant variation in hereditary spastic paraplegia symptoms and their severity. This limits the certainty of making predictions. In general however, some genetic types of hereditary spastic paraplegia are usually associated with only leg weakness, spasticity, and urinary urgency (“uncomplicated hereditary spastic paraplegia”). Other types of hereditary spastic paraplegia are usually associated with other neurological disturbances in addition to these symptoms (“complicated hereditary spastic paraplegia”). Although there are exceptions, an individual with pure hereditary spastic paraplegia (uncomplicated hereditary spastic paraplegia) patient survival are unaffected. Complicated hereditary spastic paraplegia or complex hereditary spastic paraplegia can also be associated with extrapyramidal symptoms, cerebellar or cognitive dysfunction and peripheral neuropathy. The survival of complex hereditary spastic paraplegia patients is often related to the severity of other non-limb symptoms 165, 10, 166.
Symptoms of hereditary spastic paraplegia vary from mild to severe. Individuals with severe symptoms may be unable to walk independently. In general, hereditary spastic paraplegia does not shorten lifespan.
- Elsayed LEO, Eltazi IZ, Ahmed AE, Stevanin G. Insights into Clinical, Genetic, and Pathological Aspects of Hereditary Spastic Paraplegias: A Comprehensive Overview. Front Mol Biosci. 2021 Nov 26;8:690899. doi: 10.3389/fmolb.2021.690899[↩][↩]
- Yu W, He J, Liu X, Wu J, Cai X, Zhang Y, Liu X, Fan D. Clinical features and genetic spectrum of Chinese patients with hereditary spastic paraplegia: A 14-year study. Front Genet. 2023 Feb 27;14:1085442. doi: 10.3389/fgene.2023.1085442[↩]
- van Gelder LMA, Bonci T, Buckley EE, Price K, Salis F, Hadjivassiliou M, Mazzà C, Hewamadduma C. A Single-Sensor Approach to Quantify Gait in Patients with Hereditary Spastic Paraplegia. Sensors (Basel). 2023 Jul 20;23(14):6563. doi: 10.3390/s23146563[↩]
- Regensburger M, Spatz IT, Ollenschläger M, Martindale CF, Lindeburg P, Kohl Z, Eskofier B, Klucken J, Schüle R, Klebe S, Winkler J, Gaßner H. Inertial Gait Sensors to Measure Mobility and Functioning in Hereditary Spastic Paraplegia: A Cross-Sectional Multicenter Clinical Study. Neurology. 2022 Jun 6;99(10):e1079–89. doi: 10.1212/WNL.0000000000200819[↩]
- Siow SF, Yeow D, Rudaks LI, Jia F, Wali G, Sue CM, Kumar KR. Outcome Measures and Biomarkers for Clinical Trials in Hereditary Spastic Paraplegia: A Scoping Review. Genes (Basel). 2023 Sep 3;14(9):1756. doi: 10.3390/genes14091756[↩]
- Toupenet Marchesi L, Leblanc M, Stevanin G. Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells. 2021 Jul 2;10(7):1678. doi: 10.3390/cells10071678[↩]
- Meyyazhagan A, Orlacchio A. Hereditary Spastic Paraplegia: An Update. Int J Mol Sci. 2022 Feb 1;23(3):1697. doi: 10.3390/ijms23031697[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- De Beukelaer N., Bar-On L., Hanssen B., Peeters N., Prinsen S., Ortibus E., Desloovere K., Van Campenhout A. Muscle characteristics in pediatric hereditary spastic paraplegia vs. bilateral spastic cerebral palsy: An exploratory study. Front. Neurol. 2021;12:635032. doi: 10.3389/fneur.2021.635032[↩]
- Hedera P. Hereditary Spastic Paraplegia Overview. 2000 Aug 15 [Updated 2021 Feb 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1509[↩][↩][↩][↩]
- Fink J. K. (2013). Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. ACTA NEUROPATHOL. 126 (3), 307–328. 10.1007/s00401-013-1115-8[↩][↩]
- Shi Y., Wang A., Chen B., Wang X., Niu S., Li W., et al. (2022). Clinical features and genetic spectrum of patients with clinically suspected hereditary progressive spastic paraplegia. Front. Neurol. 13, 872927. 10.3389/fneur.2022.872927[↩]
- Kerstens H.C.J.W., Lith B.J.H.v., Nijkrake M.J., Swart B.J.M.d., Bemd L.A.C.v.d., Smeets R., Klemens F., Warrenburg B.P.C.v.d., Wees P.J.v.d., Geurts A.C.H. Healthcare needs, expectations, utilization, and experienced treatment effects in patients with hereditary spastic paraplegia: A web-based survey in the Netherlands. Orphanet J. Rare Dis. 2021;16:1–283. doi: 10.1186/s13023-021-01915-0[↩]
- Hereditary Spastic Paraplegia. https://rarediseases.org/rare-diseases/hereditary-spastic-paraplegia[↩][↩][↩]
- Koh K., Ishiura H., Tsuji S., Takiyama Y. JASPAC: Japan spastic paraplegia research consortium. Brain Sci. 2018;8:153. doi: 10.3390/brainsci8080153[↩][↩]
- Schüle R., Wiethoff S., Martus P., Karle K.N., Otto S., Klebe S., Klimpe S., Gallenmüller C., Kurzwelly D., Henkel D., et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016;79:646–658. doi: 10.1002/ana.24611[↩][↩]
- Harding A. E. (1983). Classification of the hereditary ataxias and paraplegias. LANCET 1 (8334), 1151–1155. 10.1016/s0140-6736(83)92879-9[↩][↩]
- Blackstone C. Hereditary spastic paraplegia. Handb Clin Neurol. 2018;148:633-652. doi: 10.1016/B978-0-444-64076-5.00041-7[↩][↩][↩]
- Shribman S., Reid E., Crosby A.H., Houlden H., Warner T.T. Hereditary spastic paraplegia: From diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18:1136–1146. doi: 10.1016/S1474-4422(19)30235-2[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Ruano L., Melo C., Silva M.C., Coutinho P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology. 2014;42:174–183. doi: 10.1159/000358801[↩]
- Erichsen A.K., Koht J., Stray-Pedersen A., Abdelnoor M., Tallaksen C.M.E. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: A population-based study. Brain. 2009;132:1577–1588. doi: 10.1093/brain/awp056[↩]
- Coutinho P., Ruano L., Loureiro J.L., Cruz V.T., Barros J., Tuna A., Barbot C., Guimarães J., Alonso I., Silveira I., et al. Hereditary ataxia and spastic paraplegia in Portugal: A population-based prevalence study. JAMA Neurol. 2013;70:746–755. doi: 10.1001/jamaneurol.2013.1707[↩]
- Vander Stichele G., Durr A., Yoon G., Schüle R., Blackstone C., Esposito G., Buffel C., Oliveira I., Freitag C., van Rooijen S., et al. An integrated modelling methodology for estimating global incidence and prevalence of hereditary spastic paraplegia subtypes SPG4, SPG7, SPG11, and SPG15. BMC Neurol. 2022;22:115. doi: 10.1186/s12883-022-02595-4[↩]
- Tesson C., Koht J., Stevanin G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015;134:511–538. doi: 10.1007/s00439-015-1536-7[↩]
- Beetz C., Pieber T.R., Hertel N., Schabhüttl M., Fischer C., Trajanoski S., Graf E., Keiner S., Kurth I., Wieland T., et al. Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type v. Am. J. Hum. Genet. 2012;91:139–145. doi: 10.1016/j.ajhg.2012.05.007[↩]
- Ruano L., Melo C., Silva M.C., Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology. 2014;42:174–183. doi: 10.1159/000358801[↩]
- Novarino G., Fenstermaker A.G., Zaki M.S., Hofree M., Silhavy J.L., Heiberg A.D., Abdellateef M., Rosti B., Scott E., Mansour L., et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363[↩]
- Klebe S., Stevanin G., Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev. Neurol. 2015;171:505–530. doi: 10.1016/j.neurol.2015.02.017[↩]
- Schüle R., Siddique S., Deng X., Yang Y., Donkervoort S., Hansson M., Madrid R.E., Siddique N., Schöls L., Björkhem I. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J. Lipid Res. 2010;51:819–823. doi: 10.1194/jlr.M002543[↩]
- Casari G., Marconi R. Spastic Paraplegia. Br. Med. J. 2018;1:642. doi: 10.1136/bmj.1.3456.642[↩]
- Verny C., Guegen N., Desquiret V., Chevrollier A., Prundean A., Dubas F., Cassereau J., Ferre M., Amati-Bonneau P., Bonneau D., et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion. 2011;11:70–75. doi: 10.1016/j.mito.2010.07.006[↩]
- Sánchez-Ferrero E., Coto E., Corao A.I., Díaz M., Gámez J., Esteban J., Gonzalo J.F., Pascual-Pascual S.I., De Munaín A.L., Morís G., et al. Mitochondrial DNA polymorphisms/haplogroups in hereditary spastic paraplegia. J. Neurol. 2012;259:246–250. doi: 10.1007/s00415-011-6155-1[↩]
- Angelini, Corrado. (2014). Spastic Paraparesis Type 7. 10.1007/978-3-319-07500-6_82[↩]
- Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/S0140-6736(83)92879-9[↩]
- MASA syndrome. https://rarediseases.info.nih.gov/diseases/6986/spastic-paraplegia-1[↩]
- MASA syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=2466[↩]
- Stumpel C, Vos YJ. L1 Syndrome. 2004 Apr 28 [Updated 2021 Jan 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1484[↩][↩]
- Spastic paraplegia type 2. https://rarediseases.info.nih.gov/diseases/4923/spastic-paraplegia-2[↩]
- Spastic paraplegia type 2. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=99015[↩]
- Hedera P. Spastic Paraplegia 3A. 2010 Sep 21 [Updated 2020 Jun 18]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK45978[↩][↩]
- Parodi L, Rydning SL, Tallaksen C, et al. Spastic Paraplegia 4. 2003 Apr 17 [Updated 2019 Jun 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1160[↩][↩][↩][↩]
- Duning T, Warnecke T, Schirmacher A, Schiffbauer H, Lohmann H, Mohammadi S, Young P, Deppe M. Specific pattern of early white-matter changes in pure hereditary spastic paraplegia. Mov Disord. 2010 Sep 15;25(12):1986-92. doi: 10.1002/mds.23211[↩]
- da Graça FF, de Rezende TJR, Vasconcellos LFR, Pedroso JL, Barsottini OGP, França MC Jr. Neuroimaging in Hereditary Spastic Paraplegias: Current Use and Future Perspectives. Front Neurol. 2019 Jan 16;9:1117. doi: 10.3389/fneur.2018.01117[↩]
- Karle KN, Schüle R, Klebe S, Otto S, Frischholz C, Liepelt-Scarfone I, Schöls L. Electrophysiological characterisation of motor and sensory tracts in patients with hereditary spastic paraplegia (HSP). Orphanet J Rare Dis. 2013 Oct 9;8:158. doi: 10.1186/1750-1172-8-158[↩]
- Autosomal recessive spastic paraplegia type 5A. https://rarediseases.info.nih.gov/diseases/4926/spastic-paraplegia-5a[↩]
- Autosomal recessive spastic paraplegia type 5A. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100986[↩]
- Autosomal dominant spastic paraplegia type 6. https://rarediseases.info.nih.gov/diseases/4928/spastic-paraplegia-6[↩][↩]
- Autosomal dominant spastic paraplegia type 6. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100988[↩][↩]
- Casari G, Marconi R. Spastic Paraplegia 7. 2006 Aug 24 [Updated 2018 Oct 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1107[↩][↩][↩][↩][↩]
- Brugman F, Scheffer H, Wokke JH, Nillesen WM, de Visser M, Aronica E, Veldink JH, van den Berg LH. Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology. 2008 Nov 4;71(19):1500-5. doi: 10.1212/01.wnl.0000319700.11606.21[↩]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008 Dec;7(12):1127-38. doi: 10.1016/S1474-4422(08)70258-8[↩]
- Warnecke T, Duning T, Schirmacher A, Mohammadi S, Schwindt W, Lohmann H, Dziewas R, Deppe M, Ringelstein EB, Young P. A novel splice site mutation in the SPG7 gene causing widespread fiber damage in homozygous and heterozygous subjects. Mov Disord. 2010 Mar 15;25(4):413-20. doi: 10.1002/mds.22949[↩]
- Pfeffer G, Gorman GS, Griffin H, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain. 2014 May;137(Pt 5):1323-36. doi: 10.1093/brain/awu060[↩]
- Meijer IA, Valdmanis PN, Rouleau GA. Spastic Paraplegia 8. 2008 Aug 13 [Updated 2020 May 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1827[↩][↩][↩]
- Reid E, Dearlove AM, Whiteford ML, Rhodes M, Rubinsztein DC. Autosomal dominant spastic paraplegia: refined SPG8 locus and additional genetic heterogeneity. Neurology. 1999 Nov 10;53(8):1844-9. doi: 10.1212/wnl.53.8.1844[↩]
- Rocco P, Vainzof M, Froehner SC, Peters MF, Marie SK, Passos-Bueno MR, Zatz M. Brazilian family with pure autosomal dominant spastic paraplegia maps to 8q: analysis of muscle beta 1 syntrophin. Am J Med Genet. 2000 May 15;92(2):122-7. doi: 10.1002/(sici)1096-8628(20000515)92:2<122::aid-ajmg8>3.0.co;2-b[↩]
- Valdmanis PN, Meijer IA, Reynolds A, Lei A, MacLeod P, Schlesinger D, Zatz M, Reid E, Dion PA, Drapeau P, Rouleau GA. Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am J Hum Genet. 2007 Jan;80(1):152-61. doi: 10.1086/510782[↩]
- de Bot ST, Vermeer S, Buijsman W, Heister A, Voorendt M, Verrips A, Scheffer H, Kremer HP, van de Warrenburg BP, Kamsteeg EJ. Pure adult-onset spastic paraplegia caused by a novel mutation in the KIAA0196 (SPG8) gene. J Neurol. 2013 Jul;260(7):1765-9. doi: 10.1007/s00415-013-6870-x[↩]
- Hedera P, Rainier S, Alvarado D, Zhao X, Williamson J, Otterud B, Leppert M, Fink JK. Novel locus for autosomal dominant hereditary spastic paraplegia, on chromosome 8q. Am J Hum Genet. 1999 Feb;64(2):563-9. doi: 10.1086/302258[↩]
- Gasser T, Finsterer J, Baets J, Van Broeckhoven C, Di Donato S, Fontaine B, De Jonghe P, Lossos A, Lynch T, Mariotti C, Schöls L, Spinazzola A, Szolnoki Z, Tabrizi SJ, Tallaksen CM, Zeviani M, Burgunder JM, Harbo HF; EFNS. EFNS guidelines on the molecular diagnosis of ataxias and spastic paraplegias. Eur J Neurol. 2010 Feb;17(2):179-88. doi: 10.1111/j.1468-1331.2009.02873.x[↩]
- Koh K, Ishiura H, Beppu M, Shimazaki H, Ichinose Y, Mitsui J, Kuwabara S, Tsuji S, Takiyama Y; Japan Spastic Paraplegia Research Consortium. Novel mutations in the ALDH18A1 gene in complicated hereditary spastic paraplegia with cerebellar ataxia and cognitive impairment. J Hum Genet. 2018 Sep;63(9):1009-1013. doi: 10.1038/s10038-018-0477-0[↩]
- Coutelier M, Blesneac I, Monteil A, Monin ML, Ando K, Mundwiller E, Brusco A, Le Ber I, Anheim M, Castrioto A, Duyckaerts C, Brice A, Durr A, Lory P, Stevanin G. A Recurrent Mutation in CACNA1G Alters Cav3.1 T-Type Calcium-Channel Conduction and Causes Autosomal-Dominant Cerebellar Ataxia. Am J Hum Genet. 2015 Nov 5;97(5):726-37. doi: 10.1016/j.ajhg.2015.09.007[↩]
- Autosomal dominant spastic paraplegia type 9A. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=447753[↩]
- Autosomal dominant spastic paraplegia type 9A. https://rarediseases.info.nih.gov/diseases/9583/spastic-paraplegia-9[↩]
- Autosomal dominant spastic paraplegia type 10. https://rarediseases.info.nih.gov/diseases/9590/spastic-paraplegia-10[↩][↩]
- Autosomal dominant spastic paraplegia type 10. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100991[↩][↩]
- Stevanin G. Spastic Paraplegia 11. 2008 Mar 27 [Updated 2019 Dec 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1210[↩][↩][↩]
- Stevanin G, Azzedine H, Denora P, et al. SPATAX consortium. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. 2008 Mar;131(Pt 3):772-84. doi: 10.1093/brain/awm293[↩][↩][↩][↩]
- Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S, Munhoz RP, Rogaeva EA, St George-Hyslop PH, Bernardi G, Kawarai T. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain. 2010 Feb;133(Pt 2):591-8. doi: 10.1093/brain/awp325[↩]
- Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A, Tranchant C, Dion P, Rouleau GA. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging. 2012 Apr;33(4):839.e5-9. doi: 10.1016/j.neurobiolaging.2011.11.012[↩]
- Özoğuz A, Uyan Ö, Birdal G, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging. 2015 Apr;36(4):1764.e9-1764.e18. doi: 10.1016/j.neurobiolaging.2014.12.032[↩]
- Manole A, Chelban V, Haridy NA, Hamed SA, Berardo A, Reilly MM, Houlden H. Severe axonal neuropathy is a late manifestation of SPG11. J Neurol. 2016 Nov;263(11):2278-2286. doi: 10.1007/s00415-016-8254-5[↩]
- Montecchiani C, Pedace L, Lo Giudice T, Casella A, Mearini M, Gaudiello F, Pedroso JL, Terracciano C, Caltagirone C, Massa R, St George-Hyslop PH, Barsottini OG, Kawarai T, Orlacchio A. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain. 2016 Jan;139(Pt 1):73-85. doi: 10.1093/brain/awv320[↩]
- Puech B, Lacour A, Stevanin G, Sautiere BG, Devos D, Depienne C, Denis E, Mundwiller E, Ferriby D, Vermersch P, Defoort-Dhellemmes S. Kjellin syndrome: long-term neuro-ophthalmologic follow-up and novel mutations in the SPG11 gene. Ophthalmology. 2011 Mar;118(3):564-73. doi: 10.1016/j.ophtha.2010.07.024[↩]
- Anheim M, Lagier-Tourenne C, Stevanin G, Fleury M, Durr A, Namer IJ, Denora P, Brice A, Mandel JL, Koenig M, Tranchant C. SPG11 spastic paraplegia. A new cause of juvenile parkinsonism. J Neurol. 2009 Jan;256(1):104-8. doi: 10.1007/s00415-009-0083-3[↩]
- Faber I, Martinez ARM, Martins CR Jr, Maia ML, Souza JP, Lourenço CM, Marques W Jr, Montecchiani C, Orlacchio A, Pedroso JL, Barsottini OGP, Ramos CD, Lopes-Cendes Í, Friedman JH, Amorim BJ, França MC Jr. SPG11-related parkinsonism: Clinical profile, molecular imaging and l-dopa response. Mov Disord. 2018 Oct;33(10):1650-1656. doi: 10.1002/mds.27491[↩]
- Pascual B, de Bot ST, Daniels MR, França MC Jr, Toro C, Riverol M, Hedera P, Bassi MT, Bresolin N, van de Warrenburg BP, Kremer B, Nicolai J, Charles P, Xu J, Singh S, Patronas NJ, Fung SH, Gregory MD, Masdeu JC. “Ears of the Lynx” MRI Sign Is Associated with SPG11 and SPG15 Hereditary Spastic Paraplegia. AJNR Am J Neuroradiol. 2019 Jan;40(1):199-203. doi: 10.3174/ajnr.A5935[↩][↩]
- França MC Jr, Yasuda CL, Pereira FR, D’Abreu A, Lopes-Ramos CM, Rosa MV, Cendes F, Lopes-Cendes I. White and grey matter abnormalities in patients with SPG11 mutations. J Neurol Neurosurg Psychiatry. 2012 Aug;83(8):828-33. doi: 10.1136/jnnp-2011-300129[↩]
- Hehr U, Bauer P, Winner B, Schule R, Olmez A, Koehler W, Uyanik G, Engel A, Lenz D, Seibel A, Hehr A, Ploetz S, Gamez J, Rolfs A, Weis J, Ringer TM, Bonin M, Schuierer G, Marienhagen J, Bogdahn U, Weber BH, Topaloglu H, Schols L, Riess O, Winkler J. Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol. 2007 Dec;62(6):656-65. doi: 10.1002/ana.21310[↩]
- Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, Martin E, Ouvrard-Hernandez AM, Tessa A, Bouslam N, Lossos A, Charles P, Loureiro JL, Elleuch N, Confavreux C, Cruz VT, Ruberg M, Leguern E, Grid D, Tazir M, Fontaine B, Filla A, Bertini E, Durr A, Brice A. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007 Mar;39(3):366-72. doi: 10.1038/ng1980[↩]
- Faber I, Martinez ARM, de Rezende TJR, Martins CR Jr, Martins MP, Lourenço CM, Marques W Jr, Montecchiani C, Orlacchio A, Pedroso JL, Barsottini OGP, Lopes-Cendes Í, França MC Jr. SPG11 mutations cause widespread white matter and basal ganglia abnormalities, but restricted cortical damage. Neuroimage Clin. 2018 Jun 9;19:848-857. doi: 10.1016/j.nicl.2018.05.031[↩]
- Autosomal dominant spastic paraplegia type 12. https://rarediseases.info.nih.gov/diseases/9586/spastic-paraplegia-12[↩]
- Autosomal dominant spastic paraplegia type 12. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100993[↩]
- Autosomal dominant spastic paraplegia type 13. https://rarediseases.info.nih.gov/diseases/9616/spastic-paraplegia-13[↩]
- Autosomal dominant spastic paraplegia type 13. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100994[↩]
- Autosomal recessive spastic paraplegia type 14. https://rarediseases.info.nih.gov/diseases/9589/spastic-paraplegia-14[↩][↩]
- Autosomal recessive spastic paraplegia type 14. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100995[↩][↩]
- Ebrahimi-Fakhari D, Alecu JE, Blackstone C. Spastic Paraplegia 15. 2021 May 27. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK570772[↩][↩][↩][↩]
- X-linked spastic paraplegia type 16. https://rarediseases.info.nih.gov/diseases/9585/spastic-paraplegia-16[↩][↩]
- X-linked spastic paraplegia type 16. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100997[↩][↩]
- Autosomal dominant spastic paraplegia type 17. https://rarediseases.info.nih.gov/diseases/4219/spastic-paraplegia-17[↩][↩]
- Autosomal dominant spastic paraplegia type 17. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100998[↩][↩]
- Autosomal recessive spastic paraplegia type 18. https://rarediseases.info.nih.gov/diseases/4922/spastic-paraplegia-18[↩][↩]
- Autosomal spastic paraplegia type 18. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=209951[↩][↩]
- Autosomal dominant spastic paraplegia type 19. https://rarediseases.info.nih.gov/diseases/9588/spastic-paraplegia-19[↩]
- Autosomal dominant spastic paraplegia type 19. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=100999[↩]
- Autosomal recessive spastic paraplegia type 23. https://rarediseases.info.nih.gov/diseases/336/spastic-paraplegia-23[↩][↩]
- Autosomal recessive spastic paraplegia type 23. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=101003[↩][↩]
- Autosomal recessive spastic paraplegia type 24. https://rarediseases.info.nih.gov/diseases/9296/spastic-paraplegia-24[↩][↩]
- Autosomal recessive spastic paraplegia type 24. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=101004[↩][↩]
- Autosomal recessive spastic paraplegia type 25. https://rarediseases.info.nih.gov/diseases/9582/spastic-paraplegia-25[↩][↩]
- Autosomal recessive spastic paraplegia type 25. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=101005[↩][↩]
- Autosomal recessive spastic paraplegia type 26. https://rarediseases.info.nih.gov/diseases/9587/spastic-paraplegia-26[↩][↩]
- Autosomal recessive spastic paraplegia type 26. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=101006[↩][↩]
- Autosomal dominant spastic paraplegia type 29. https://rarediseases.info.nih.gov/diseases/9729/spastic-paraplegia-29[↩]
- Autosomal dominant spastic paraplegia type 29. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=101009[↩]
- Autosomal dominant spastic paraplegia type 31. https://rarediseases.info.nih.gov/diseases/10817/spastic-paraplegia-31[↩][↩]
- Autosomal dominant spastic paraplegia type 31. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=101011[↩][↩]
- Autosomal recessive spastic paraplegia type 32. https://rarediseases.info.nih.gov/diseases/12749/spastic-paraplegia-32[↩][↩]
- Autosomal recessive spastic paraplegia type 32. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=171622[↩][↩]
- Synofzik M, Hufnagel RB, Züchner S. PNPLA6 Disorders. 2014 Oct 9 [Updated 2021 Jun 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK247161[↩]
- Autosomal recessive spastic paraplegia type 39. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=139480[↩][↩]
- Spastic Paraplegia 47. https://rarediseases.org/rare-diseases/spastic-paraplegia-47[↩][↩]
- Hereditary sensory and autonomic neuropathy due to TECPR2 mutation. https://rarediseases.info.nih.gov/diseases/13568/autosomal-recessive-spastic-paraplegia-type-49[↩]
- Hereditary sensory and autonomic neuropathy due to TECPR2 mutation. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=320385[↩]
- Heimer G, Neuser S, Ben-Zeev B, et al. TECPR2-Related Hereditary Sensory and Autonomic Neuropathy with Intellectual Disability. 2022 Sep 22. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK584409[↩][↩]
- Heimer G, Oz-Levi D, Eyal E, Edvardson S, Nissenkorn A, Ruzzo EK, Szeinberg A, Maayan C, Mai-Zahav M, Efrati O, Pras E, Reznik-Wolf H, Lancet D, Goldstein DB, Anikster Y, Shalev SA, Elpeleg O, Ben Zeev B. TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur J Paediatr Neurol. 2016 Jan;20(1):69-79. doi: 10.1016/j.ejpn.2015.10.003[↩]
- Neuser S, Brechmann B, Heimer G, et al. Clinical, neuroimaging, and molecular spectrum of TECPR2-associated hereditary sensory and autonomic neuropathy with intellectual disability. Hum Mutat. 2021 Jun;42(6):762-776. doi: 10.1002/humu.24206[↩]
- Spastic Paraplegia 50. https://rarediseases.org/rare-diseases/spastic-paraplegia-50[↩]
- Ebrahimi-Fakhari D, Behne R, Davies AK, et al. AP-4-Associated Hereditary Spastic Paraplegia. 2018 Dec 13. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK535153[↩][↩][↩][↩][↩][↩]
- Verkerk AJ, Schot R, Dumee B, Schellekens K, Swagemakers S, Bertoli-Avella AM, Lequin MH, Dudink J, Govaert P, van Zwol AL, Hirst J, Wessels MW, Catsman-Berrevoets C, Verheijen FW, de Graaff E, de Coo IF, Kros JM, Willemsen R, Willems PJ, van der Spek PJ, Mancini GM. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. 2009 Jul;85(1):40-52. doi: 10.1016/j.ajhg.2009.06.004[↩]
- Abou Jamra R, Philippe O, Raas-Rothschild A, Eck SH, Graf E, Buchert R, Borck G, Ekici A, Brockschmidt FF, Nöthen MM, Munnich A, Strom TM, Reis A, Colleaux L. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet. 2011 Jun 10;88(6):788-795. doi: 10.1016/j.ajhg.2011.04.019[↩]
- Moreno-De-Luca A, Helmers SL, Mao H, Burns TG, Melton AM, Schmidt KR, Fernhoff PM, Ledbetter DH, Martin CL. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J Med Genet. 2011 Feb;48(2):141-4. doi: 10.1136/jmg.2010.082263[↩]
- Ebrahimi-Fakhari D, Cheng C, Dies K, Diplock A, Pier DB, Ryan CS, Lanpher BC, Hirst J, Chung WK, Sahin M, Rosser E, Darras B, Bennett JT; CureSPG47. Clinical and genetic characterization of AP4B1-associated SPG47. Am J Med Genet A. 2018 Feb;176(2):311-318. doi: 10.1002/ajmg.a.38561[↩]
- Vill K, Müller-Felber W, Alhaddad B, Strom TM, Teusch V, Weigand H, Blaschek A, Meitinger T, Haack TB. A homozygous splice variant in AP4S1 mimicking neurodegeneration with brain iron accumulation. Mov Disord. 2017 May;32(5):797-799. doi: 10.1002/mds.26922[↩]
- Roubertie A, Hieu N, Roux CJ, Leboucq N, Manes G, Charif M, Echenne B, Goizet C, Guissart C, Meyer P, Marelli C, Rivier F, Burglen L, Horvath R, Hamel CP, Lenaers G. AP4 deficiency: A novel form of neurodegeneration with brain iron accumulation? Neurol Genet. 2018 Jan 24;4(1):e217. doi: 10.1212/NXG.0000000000000217[↩]
- Spastic Paraplegia 52. https://rarediseases.org/rare-diseases/spastic-paraplegia-52[↩][↩][↩][↩]
- Fink JK. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013 Sep;126(3):307-28. doi: 10.1007/s00401-013-1115-8[↩]
- Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014 Jan 31;343(6170):506-511. doi: 10.1126/science.1247363[↩]
- Balicza P., Grosz Z., Gonzalez M.A., Bencsik R., Pentelenyi K., Gal A., Varga E., Klivenyi P., Koller J., Züchner S., et al. Genetic background of the hereditary spastic paraplegia phenotypes in Hungary—An analysis of 58 probands. J. Neurol. Sci. 2016;364:116–121. doi: 10.1016/j.jns.2016.03.018[↩]
- Namekawa M., Ribai P., Nelson I., Forlani S., Fellmann F., Goizet C., Depienne C., Stevanin G., Ruberg M., Dürr A., et al. SPG3A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology. 2006;66:112–114. doi: 10.1212/01.wnl.0000191390.20564.8e[↩]
- Reed J.A., Wilkinson P.A., Patel H., Simpson M.A., Chatonnet A., Robay D., Patton M.A., Crosby A.H., Warner T.T. A novel NIPA1 mutation associated with a pure form of autosomal dominant hereditary spastic paraplegia. Neurogenetics. 2005;6:79–84. doi: 10.1007/s10048-004-0209-9[↩]
- Valdmanis P.N., Meijer I.A., Reynolds A., Lei A., MacLeod P., Schlesinger D., Zatz M., Reid E., Dion P.A., Drapeau P., et al. Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am. J. Hum. Genet. 2007;80:152–161. doi: 10.1086/510782[↩]
- Salinas S., Proukakis C., Crosby A., Warner T.T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8[↩][↩]
- Beetz C., Schüle R., Deconinck T., Tran-Viet K.N., Zhu H., Kremer B.P.H., Frints S.G.M., Van Zelst-Stams W.A.G., Byrne P., Otto S., et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain. 2008;131:1078–1086. doi: 10.1093/brain/awn026[↩]
- Martignoni M., Riano E., Rugarli E.I. The Role of ZFYVE27/Protrudin in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2008;83:127–128. doi: 10.1016/j.ajhg.2008.05.014[↩]
- Chrestian N., Dupré N., Gan-Or Z., Szuto A., Chen S., Venkitachalam A., Brisson J.D., Warman-Chardon J., Ahmed S., Ashtiani S., et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol. Genet. 2016;3:e122. doi: 10.1212/NXG.0000000000000122[↩]
- Parodi L., Coarelli G., Stevanin G., Brice A., Durr A. (2018a). Hereditary Ataxias and Paraparesias: Clinical and Genetic Update. Curr. Opin. Neurol. 31, 462–471. 10.1097/WCO.0000000000000585[↩]
- Kara E., Tucci A., Manzoni C., Lynch D.S., Elpidorou M., Bettencourt C., Chelban V., Manole A., Hamed S.A., Haridy N.A., et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. 2016;139:1904–1918. doi: 10.1093/brain/aww111[↩]
- Cloake N., Yan J., Aminian A., Pender M., Greer J. PLP1 mutations in patients with multiple sclerosis: Identification of a new mutation and potential pathogenicity of the mutations. J. Clin. Med. 2018;7:342. doi: 10.3390/jcm7100342[↩]
- Linneberg C., Toft C.L.F., Kjaer-Sorensen K., Laursen L.S. L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing. Sci. Rep. 2019;9:1–14. doi: 10.1038/s41598-019-39884-x[↩]
- Finsterer J., Löscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025[↩]
- Murala S., Nagarajan E., Bollu P.C. Hereditary spastic paraplegia. Neurol. Sci. 2021;42:883–894. doi: 10.1007/s10072-020-04981-7[↩][↩]
- Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012;35:25-47. doi: 10.1146/annurev-neuro-062111-150400[↩]
- Kevenaar JT, Hoogenraad CC. The axonal cytoskeleton: from organization to function. Front Mol Neurosci. 2015 Aug 14;8:44. doi: 10.3389/fnmol.2015.00044[↩]
- Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004 Dec;30(6):576-84. doi: 10.1111/j.1365-2990.2004.00587.x[↩]
- Blackstone C. Converging cellular themes for the hereditary spastic paraplegias. Curr Opin Neurobiol. 2018 Aug;51:139-146. doi: 10.1016/j.conb.2018.04.025[↩]
- Fink J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–328. doi: 10.1007/s00401-013-1115-8[↩][↩]
- White K.D., Ince P.G., Lusher M., Lindsey J., Cookson M., Bashir R., Shaw P.J., Bushby K.M.D. Clinical and pathologic findings in hereditary spastic paraparesis with spastin mutation. Neurology. 2000;55:89–94. doi: 10.1212/WNL.55.1.89[↩]
- Fink JK. Upper Motor Neuron Disorders: Hereditary Spastic Paraplegia and Primary Lateral Sclerosis. In: Johnson MV, Adams HP, Fatemi A, eds. Neurobiology of Disease.Oxford University Press, 2016.[↩][↩]
- Fink JK. The Hereditary Spastic Paraplegias. In: Rosenberg R, ed. Molecular and Genetic Basis of Neurologic and Psychiatric Disease, 5th edition ed Lippincott Williams & Wilkins, 2014.[↩][↩]
- Hedera P, Eldevik OP, Maly P, Rainier S, Fink JK. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology. 2005 Oct;47(10):730-4. doi: 10.1007/s00234-005-1415-3[↩]
- Hensiek A., Kirker S., Reid E. Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J. Neurol. 2015;272:1601–1612. doi: 10.1007/s00415-014-7598-y[↩]
- Boone P.M., Liu P., Zhang F., Carvalho C.M., Towne C.F., Batish S.D., Lupski J.R. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet. Med. 2011;13:582–592. doi: 10.1097/GIM.0b013e3182106775[↩]
- Zhang W., Gong J., Ding L., Zhang Z., Pan X., Chen X., Guo W., Zhang X., Yang X., Peng G., et al. Functional validation of a human GLUD2 variant in a murine model of Parkinson’s disease. Cell Death Dis. 2020;11:1–17. doi: 10.1038/s41419-019-2182-0[↩]
- Da Graça F.F., De Rezende T.J.R., Vasconcellos L.F.R., Pedroso J.L., Barsottini O.G.P., França M.C. Neuroimaging in Hereditary Spastic Paraplegias: Current Use and Future Perspectives. Front. Neurol. 2019;9:1117. doi: 10.3389/fneur.2018.01117[↩]
- Julien C, Lissouba A, Madabattula S, Fardghassemi Y, Rosenfelt C, Androschuk A, Strautman J, Wong C, Bysice A, O’sullivan J, Rouleau GA, Drapeau P, Parker JA, Bolduc FV. Conserved pharmacological rescue of hereditary spastic paraplegia-related phenotypes across model organisms. Hum Mol Genet. 2016 Mar 15;25(6):1088-99. doi: 10.1093/hmg/ddv632[↩]
- Margetis K., Korfias S., Boutos N., Gatzonis S., Themistocleous M., Siatouni A., Dalivigka Z., Flaskas T., Stranjalis G., Boviatsis E., et al. Intrathecal baclofen therapy for the symptomatic treatment of hereditary spastic paraplegia. Clin. Neurol. Neurosurg. 2014;123:142–145. doi: 10.1016/j.clineuro.2014.05.024[↩]
- De Niet M., De Bot S.T., Van De Warrenburg B.P.C., Weerdesteyn V., Geurts A.C. Functional efects of botulinum toxin type-A treatment and subsequent stretching of spastic calf muscles: A study in patients with hereditary spastic paraplegia. J. Rehabil. Med. 2015;47:147–153. doi: 10.2340/16501977-1909[↩]
- Servelhere K.R., Faber I., Martinez A., Nickel R., Moro A., Germiniani F.M.B., Moscovich M., Blume T.R., Munhoz R.P., Teive H.A.G., et al. Botulinum toxin for hereditary spastic paraplegia: Effects on motor and non-motor manifestations. Arq. Neuropsiquiatr. 2018;76:183–188. doi: 10.1590/0004-282×20180013[↩]
- Béreau M., Anheim M., Chanson J.B., Tio G., Echaniz-Laguna A., Depienne C., Collongues N., de Sèze J. Dalfampridine in hereditary spastic paraplegia: A prospective, open study. J. Neurol. 2015;262:1285–1288. doi: 10.1007/s00415-015-7707-6[↩]
- Mohan N., Qiang L., Morfini G., Baas P.W. Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia. Brain Sci. 2021;11:1081. doi: 10.3390/brainsci11081081[↩]
- Havlicek S., Kohl Z., Mishra H.K., Prots I., Eberhardt E., Denguir N., Wend H., Plötz S., Boyer L., Marchetto M.C.N., et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 2014;23:2527–2541. doi: 10.1093/hmg/ddt644[↩]
- Xue H., Yu A., Chen X., Lin N., Lin M., Huang H., Xu L. Prenatal diagnosis of PLP1 duplication by single nucleotide polymorphism array in a family with Pelizaeus-Merzbacher disease. Aging. 2021;13:1488–1497. doi: 10.18632/aging.202477[↩]
- Pirozzi M., Quattrini A., Andolfi G., Dina G., Malaguti M.C., Auricchio A., Rugarli E.I. Intramuscular viral delivery of paraplegin rescues peripheral axonopathy in a model of hereditary spastic paraplegia. J. Clin. Investig. 2006;116:202–208. doi: 10.1172/JCI26210[↩]
- Coutinho P., Ruano L., Loureiro J. L., Cruz V. T., Barros J., Tuna A., et al. (2013). Hereditary ataxia and spastic paraplegia in Portugal: A population-based prevalence study. JAMA NEUROL. 70 (6), 746–755. 10.1001/jamaneurol.2013.1707[↩]
- Dong E., Wang C., Wu S., Lu Y. Q., Lin X. H., Su H. Z., et al. (2018). Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 13 (1), 36. 10.1186/s13024-018-0269-1[↩]




{kind=link}