Contents
What is Hurler syndrome
Hurler syndrome is the most severe form of mucopolysaccharidosis type 1 (MPS type 1), a rare lysosomal storage disease, characterized by skeletal abnormalities, cognitive impairment, heart disease, respiratory problems, enlarged liver and spleen, characteristic facies and reduced life expectancy. Hurler syndrome is caused by the absence of alpha-L-iduronidase enzyme which responsible for degradation of glycosaminoglycans (GAG or mucopolysaccharides). This leads to a buildup of dermatan sulfate and heparin sulfate in multiple tissues, resulting in progressive deterioration and, eventually, death. Mucopolysaccharidosis type 1 (MPS type 1) is subdivided into three subtypes: Hurler syndrome (most severe), Hurler-Scheie syndrome (intermediate severity), and Sheie syndrome (mild).
The incidence of Hurler syndrome is approximately 1 in 100,000 births 1. Male and female children are equally affected. All races and ethnicities are at risk of inheriting the disease. The prevalence of the Hurler syndrome subtype of mucopolysaccharidosis type 1 is estimated at 1/200,000 in Europe.
Mucopolysaccharidosis type 1 (MPS type 1) is a form of mucopolysaccharidosis caused by a deficiency of the enzyme alpha-L-iduronidase. The most severe form of mucopolysaccharidosis type 1 is often called Hurler syndrome (or MPS type 1H). It is named for the physician, Gertrud Hurler, who first described the disorder in 1919. A milder form of mucopolysaccharidosis type 1 is called Scheie syndrome (or MPS type 1S), and the name Hurler-Scheie (MPS type 1H/S) is sometimes applied to an intermediate form that does not fit clearly in either the milder or more severe category.
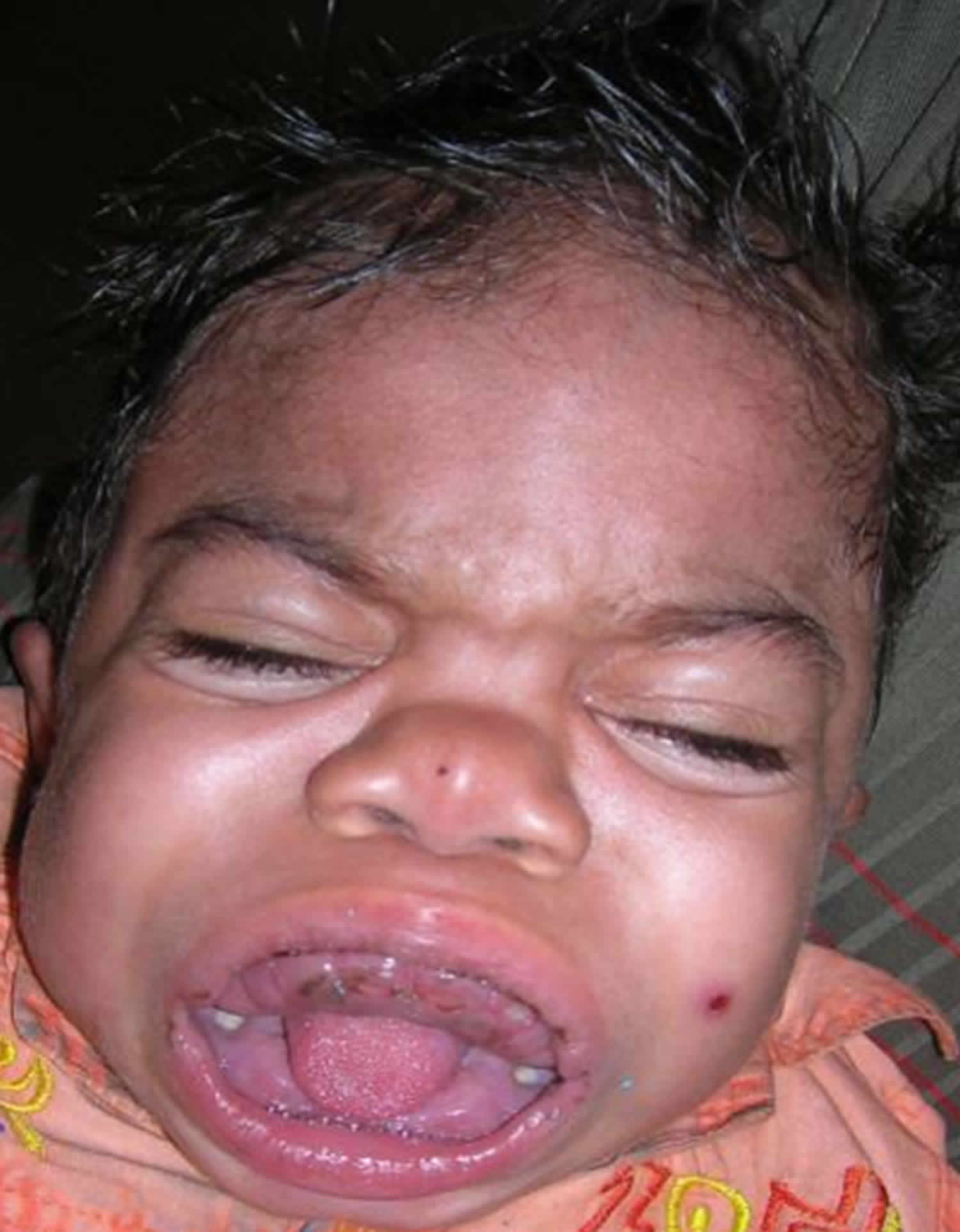
Patients present within the first year of life with musculoskeletal alterations including short stature, dysostosis multiplex, thoracic-lumbar kyphosis, progressive coarsening of the facial features (including large head with bulging frontal bones, depressed nasal bridge with broad nasal tip and anteverted nostrils, full cheeks and enlarged lips), cardiomyopathy and valvular abnormalities, neurosensorial hearing loss, enlarged tonsils and adenoids, and nasal secretion. Developmental delay is usually observed between 12 and 24 months of life and is primarily in the realm of speech with progressive cognitive and sensorial deterioration. Hydrocephaly can occur after the age of two. Diffuse corneal compromise leading to corneal opacity becomes detectable from three years of age onwards. Other manifestations include organomegaly, hernias and hirsutism.
Hurler syndrome key facts:
- Hurler syndrome is a rare autosomal recessive lysosomal storage disorder.
- Affected individuals demonstrate typical coarse facial features including a flat nasal bridge and excessive hair growth.
- Usually manifests as cognitive developmental delay, corneal clouding, cardiac disease, and characteristics musculoskeletal manifestations.
- Additional symptoms include hearing loss, recurrent respiratory tract infections, macroglossia, and claw hand deformities.
- Vertebral bodies are characteristically hypoplastic and show anteroinferior beaking on radiological examination. They usually develop typical skeletal abnormalities known as dysostosis multiplex.
- Most patients with severe cases die with heart failure before the age of 10. Cardiac evaluation is recommended every 1 or 2 years after an initial diagnosis.
- Hurler syndrome diagnosis is based on a thorough clinical examination, and measurement of urinary GAG levels is a useful screening test. Enzyme activity assays based on cultured fibroblasts, leukocytes, plasma, and serum are confirmatory and are considered a gold standard.
- A differential diagnosis for Hurler syndrome includes Hunter syndrome (mucopolysaccharidosis type 2) and Sly syndrome (mucopolysaccharidosis type 6).
- The mainstay of treatment is enzyme replacement therapy and hematopoietic stem cell transplant.
- Additionally, surgical management alleviates the symptoms, thereby reducing the morbidity and hospitalization frequency.
- Early detection of the disease and the appropriate, multidisciplinary approach to management is recommended to improve the quality of life.
Figure 1. Hurler syndrome
Hunter syndrome
Hunter syndrome is mucopolysaccharidosis type 2. Hunter syndrome patients present with similar features as in Hurler syndrome, but patients with Hunter syndrome present in later onset with a slower clinical course and absence of corneal manifestations.
Sly syndrome
Sly syndrome is mucopolysaccharidosis type 6. This a rare disorder shares similar clinical features as Hurler syndrome. Mental retardation may be mild or absent. Hydrops fetalis is a most common presentation, and usually, patients do not survive to diagnosis.
What causes Hurler syndrome
Hurler syndrome is caused by mutations in the IDUA gene that is located on chromosome 4 (4p16.3) 2, leading to a complete deficiency in the alpha-L-iduronidase (IUDA) enzyme and lysosomal accumulation of dermatan sulfate and heparan sulfate (GAG).
Hurler syndrome is caused by a deficiency of a lysosomal alpha-L-iduronidase (IUDA) enzyme, which aids in the breakdown of dermatan sulfate and heparin sulfate (GAG). This finally results in the accumulation of large amounts of heparan sulfate (GAG) in the body, eventually causing the cells to become severely dysfunctional leading to death. The deposition of heparan sulfate (GAG) causes enlargement and thickening of various organs like the heart, spleen, liver, muscles, connective tissues, joints, and the central nervous system causing severe functional impairment.
Hurler syndrome inheritance pattern
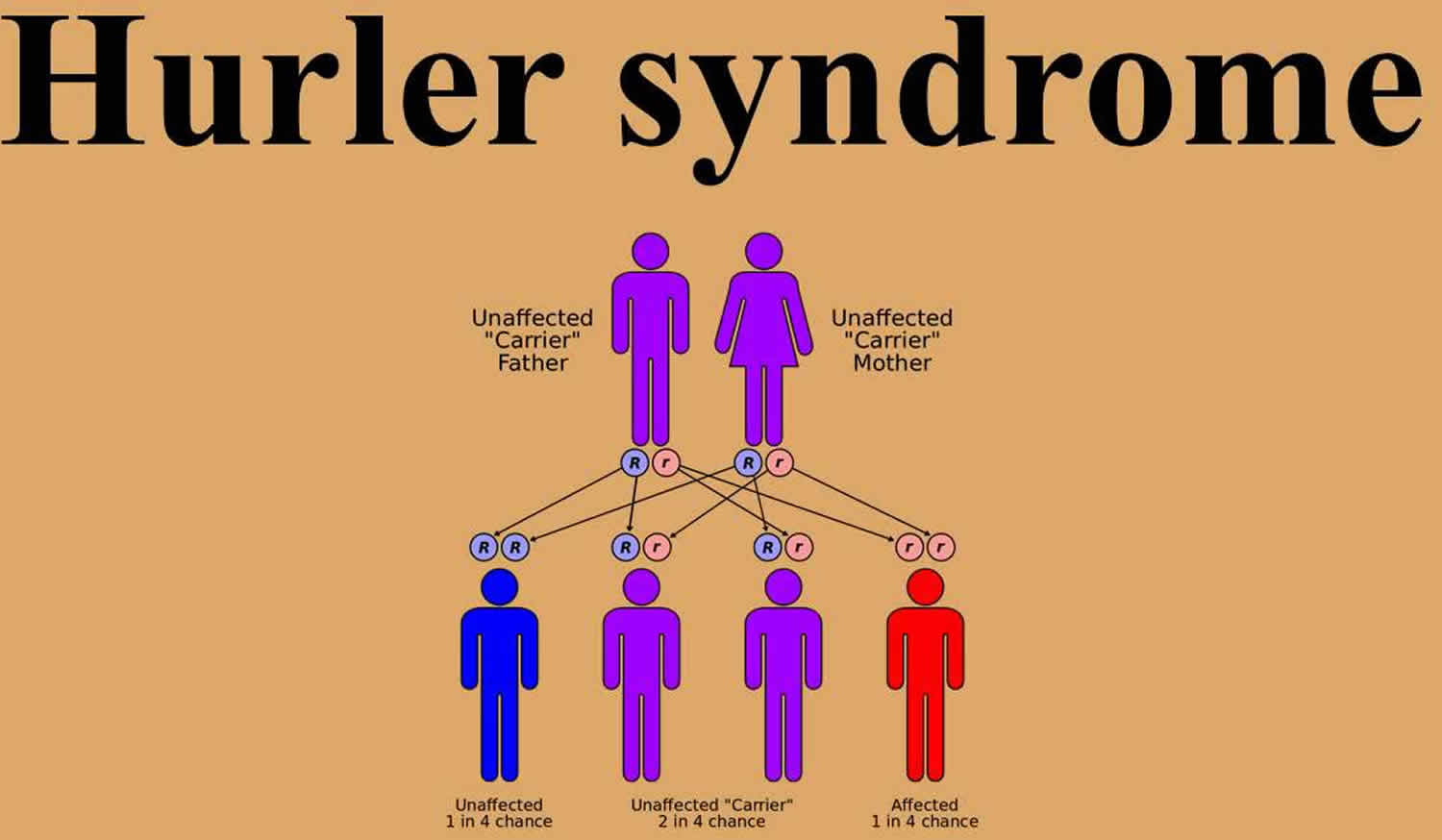
Hurler syndrome is inherited in an autosomal recessive pattern. Genetic counseling and testing should be offered to couples with a positive family history.
Autosomal recessive means two copies of the abnormal gene, one from each parent (one abnormal gene from mum and one abnormal gene from dad), is needed to cause the disorder or disease. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Autosomal recessive disorders are typically not seen in every generation of an affected family. Cystic fibrosis and sickle cell anemia are common examples of an autosomal recessive genetic disorders.
Autosomal refers to the fact that whatever gene is involved is found on one of the first 22 chromosomes (called the autosomes) and not on the X or Y chromosome (the sex chromosomes).
Recessive refers to the explanation above that you need two copies of the abnormal gene, one from mom and one from dad, in order to have a autosomal recessive condition. The copy you inherit from mom is not working AND the copy you inherit from dad is not working, resulting in zero functioning copies of that gene. With autosomal recessive conditions, if you only have one copy of the non-working gene (called a carrier), you do not have the condition and typically do not have any related symptoms.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates Hurler syndrome autosomal recessive inheritance pattern. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Hurler syndrome autosomal recessive inheritance pattern
Hurler syndrome symptoms
Children with Hurler syndrome are usually not born with signs but develop symptoms during the first year of life 3. Developmental delay may become apparent by the age of 1 to 2 years, with a maximum functional age of 2 to 4 years. The average age of mortality is 5 years, and nearly all patients die before 10 years of age 3.
- General appearance: Characteristic features include coarse facies, enlarged head with prominent frontal bones, widely placed eye sockets with protruding eyes, flat appearance of the nasal bridge, enlarged lips, and wide-open eyes. The neck is typically short and stiff. These characteristic features were formerly described traditionally in the medical literature by the term gargoylism.
- Neurological manifestations: Children have a progressive developmental delay before 2 years of age and lose previously acquired skills. GAGs are deposited in the meninges and spinal cord. This results in obstruction of CSF thereby causing high pressure communicating hydrocephalus and convulsions. Odontoid dysplasia and anterior C1-C2 subluxation occur frequently and can cause cord compression and sudden death.
- Respiratory manifestations: Patients often develop frequent ear, sinus, and pulmonary infections with thick secretions that lead to frequent emergency department visits and hospitalizations. Soft tissue thickening in the nose, pharynx, tonsils, and adenoids along with abnormalities in the tracheal cartilage causes progressive airway obstruction and sleep apnea. In some patients, sleep apnea is unrecognized and can cause significant hypoxemia at night, leading to complications like pulmonary hypertension and cor pulmonale 4.
- Cardiac manifestations: These include cardiomyopathy, endocardial fibroelastosis, valvular regurgitation, and heart failure. GAG deposition within the blood vessels causes diffuse narrowing of the coronary arteries. Some untreated patients develop irregular lesions of the aorta. Most patients with severe Hurler syndrome die from heart failure before the age of 10 5. It has been recommended that patients undergo cardiac evaluation every 1 or 2 years after an initial diagnosis of Hurler syndrome.
- Gastrointestinal manifestations: Swallowing might become difficult due to GAG deposition in the muscle tissue of the tongue, resulting in macroglossia which might impair speech. Patients also develop umbilical and inguinal hernias within several months of life, often one of the first clinical signs. Hepatosplenomegaly is also noted.
- Musculoskeletal system: Patients may be of normal height during infancy but stop growing by the age of 2 years. They may not reach a height of greater than 4 feet. Skeletal abnormalities occur by about 6 months but become more clinically obvious by 10 to 14 months. A rapidly enlarging head size due to craniosynostosis and hyperostosis of the skull is usually seen. They may experience debilitating spine and hip deformities, abnormal bone and cartilage development (particularly spine and hands), carpal tunnel syndrome, and joint stiffness. Abnormal curvature of the lower spine, giving a hunchback appearance called Gibbus (dorsal kyphosis) deformity, is common. Patients may develop progressive joint stiffness and contractures, which limit mobility and are painful. Cervical myelopathy is seen due to congenital vertebral anomalies and atlantoaxial subluxation. Vertebral bodies are characteristically hypoplastic and show anteroinferior beaking on radiological examination. Patients usually develop typical skeletal abnormalities, known as dysostosis multiplex.
- Ocular manifestations: Clouding of the cornea occurs due to structural alteration of the corneal stroma and derangement of collagen fibrils thereby leading to blindness. Retinal degeneration and optic nerve compression can also occur within the first year of life.
- Hearing manifestations: GAGs build up in tubes of the middle ear and further prevent them from draining properly, leading to recurrent ear infections. Patients usually develop combined conductive and sensorineural hearing loss 6.
- Integumentary manifestations: Hair is often coarse and more abundant than in normal children. Bluish birthmarks known as Mongolian spots are common.
The mucopolysaccharidosis type 1 registry reports the following findings in individuals with Hurler syndrome 7:
- Coarse facial features (86.4%)
- Corneal clouding (70.9%)
- Heaptomegaly (70.0%)
- Kyphosis/gibbus (70.0%)
- Hernias (58.9%)
- Airway-related symptoms, such as sleep disturbances/snoring (51.6%)
- Splenomegaly (50.9%)
- Cardiac valve abnormalities (48.9%)
- Cognitive impairment (46.4%)
- Dystosis multiplex (43.6%)
- Enlarged tongue (41.3%)
- Joint contractures (37.9%)
- Enlarged tonsils (28.6%)
Hurler syndrome diagnosis
Early diagnosis is difficult as the first clinical manifestations are not specific. Diagnosis of Hurler syndrome is based on a thorough clinical examination and measurement of urinary GAG levels which is a useful screening test. A positive test is suggestive of an mucopolysaccharidosis, but false-negative results are common 3. Positive family history is often present. Genetic testing is available.
Diagnosis is based on detection of increased urinary excretion of heparan and dermatan sulfate and confirmed by demonstration of enzymatic deficiency in leukocytes or fibroblasts.
Enzyme activity assays based on cultured fibroblasts, leukocytes, plasma, and serum are confirmatory and are considered the gold standard. By using an enzyme assay or DNA analysis, it is sometimes possible to distinguish Hurler syndrome from the other closely related mucopolysaccharidosis type 1 subtypes, along with symptom severity and age of onset should be considered, establishing a specific diagnosis.
Gene sequencing can be done to identify the mutations in families at risk so that patients can be offered genetic counseling and carrier testing to allow for more informed family planning.
Antenatal diagnosis is possible by measurement of enzymatic activity in cultivated chorionic villus or amniocytes and by genetic testing if the disease-causing mutation is known 8.
Suggestive Findings
Mucopolysaccharidosis type 1 should be suspected in individuals with the following clinical and supportive laboratory findings.
Clinical findings vary by disease severity. Clinical findings alone are not diagnostic.
Clinical findings:
- Coarse facial features
- Early frequent upper-respiratory infections including otitis media
- Inguinal or umbilical hernia
- Hepatosplenomegaly
- Characteristic skeletal and joint findings (gibbus deformity; limitation of joint range of motion)
- Characteristic ocular findings (corneal clouding)
Hurler syndrome life expectancy
The life expectancy of mucopolysaccharidosis with a median age is 8.7 years. The survival rate is varied based on bone marrow transplantation. Patients who received successful bone marrow transplantation had a 2-year survival rate of 68% and 10-year survival rate of 64% when compared to those individuals who did not receive the transplants 3. Hurler syndrome patients had a significantly decreased life expectancy, with the median age of 6.8 years. The average age of mortality is 5 years, and nearly all patients die before 10 years of age 3.
Hurler syndrome treatment
Most therapies for Hurler syndrome are directed towards treatment of complications and are not specific for an underlying abnormality.
- Enzyme replacement therapy: Recombinant human alpha L- iduronidase (Aldurazyme) is given as a weekly intravenous injection. Better outcomes can be achieved if it is given before severe complications ensue 9. It is used for patients with the Hurler and Hurler-Scheie forms of mucopolysaccharidosis type 1 and moderate-to-severe symptoms in patients with Scheie form.
- Hematopoietic stem cell transplant: Hematopoietic stem cell transplant or bone marrow transplant or blood stem cell transplant, is the progressive replacement of enzyme-deficient hematopoietic cells with donor-derived enzyme competent cells. It is the ideal treatment for patients who are under 2 years of age and in selected patients over this age limit as it can prolong the survival. Hematopoietic stem cell transplant decreases hepatosplenomegaly, airway obstruction, CSF pressures and increases joint mobility, cardiac function, and improves or stabilizes hearing (mostly in young patients). Hematopoietic stem cell transplant is more effective at preventing disease progression than reversing the established disease 9.
- Additional management of Hurler syndrome is supportive and includes surgical interventions like adenotonsillectomy; hernia repair; ventriculoperitoneal shunt; cardiac valve replacement; carpal tunnel release; spinal decompression; physical, occupational, and speech therapies; respiratory support such as continuous positive pressure ventilation with oxygen supplementation (CPAP); hearing aids; and medications for pain and gastrointestinal disturbances. Corneal transplants can be done for vision problems; however, surgery in patients with Hurler syndrome often presents with complications related to anesthetic procedures.
- Newer therapies: Research is ongoing with gene therapy in animal models, which includes the delivery of iduronidase enzyme gene by using viral vectors 10. It demonstrated correction of disease in the liver, spleen, and brain effects to a certain extent. Gene therapy may provide a future alternative human treatment for mucopolysaccharidosis type disorder.
Hematopoietic stem cell transplantation
Since 1980, hematopoietic stem cell transplantation (also known as a bone marrow transplant or blood stem cell transplant) has been used to treat Hurler syndrome. Hurler-Scheie and Scheie syndromes are not traditionally treated with hematopoietic stem cell transplant. Instead, individuals with Hurler-Scheie and Scheie syndromes receive enzyme replacement therapy with laronidase (recombinant alpha-L-iduronidase; Aldurazyme) as the primary treatment 11.
The goal of hematopoietic stem cell transplant is to cure the patient from the primary disease and restore complete physical and mental health condition; however, infertility is common after myeloablative hematopoietic stem cell transplant conditioned with total body irradiation and high doses of gonadotoxic drugs 12.
Bone marrow transplant or blood stem cell transplant, can stop the disease from causing more damage. It replaces the blood-forming cells (stem cells) that are missing the important protein with healthy ones. With healthy blood-forming cells, the body will break down the sugary substance that was building up. This keeps the organs from more damage. But, it can’t fix any damage that has already happened.
Allogeneic transplant is used for Hurler syndrome. This type of transplant uses healthy blood-forming cells donated by someone else to replace the unhealthy ones. These healthy cells can come from a family member, unrelated donor or umbilical cord blood. First, the child gets chemotherapy (chemo), with or without radiation, to kill the unhealthy cells. Then, the healthy donated cells are given to your child through an intravenous (IV) catheter. The new cells travel to the inside of the bones and begin to make healthy blood cells.
The entire transplant process, from the start of chemo or radiation, until hospital discharge, can last weeks to months. This is followed by many months of recovery near the transplant center and at home. The transplant team will closely watch your child to prevent and treat any complications.
Most children have a better chance of a cure if they have a transplant soon after diagnosis. Your child should see a transplant doctor as soon as your child is diagnosed.
Hematopoietic stem cell transplant is recommended for patients with Hurler syndrome under the age of 2 years with normal cognitive function (defined as IQ>70) 13. When successful, it is a one-time procedure that can prolong survival, preserve cognitive function, and reduce morbidity 14. Hematopoietic stem cell transplant reduces Hurler syndrome disease morbidity with substantial clinical benefit, including alleviation of hepatosplenomegaly, upper airway obstruction (including sleep apnea), cardiac symptoms, and coarse facial features. Unfortunately, early diagnosis of Hurler syndrome is limited by several factors, including: rarity of the disease, wide variability in clinical presentation and disease course, and the nonspecific nature of early disease manifestations 13.
- Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008 Sep 16;3:24[↩]
- Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008 Sep 16;3:24.[↩]
- Sakuru R, Bollu PC. Hurler Syndrome. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532261[↩][↩][↩][↩][↩]
- Harrison R, Schaefer S, Warner L, Mercer J, Jones S, Bruce I. Transnasal adenoidectomy in mucopolysaccharidosis. Int. J. Pediatr. Otorhinolaryngol. 2018 Aug;111:149-152.[↩]
- Braunlin E, Steinberger J, DeFor T, Orchard P, Kelly AS. Metabolic Syndrome and Cardiovascular Risk Factors after Hematopoietic Cell Transplantation in Severe Mucopolysaccharidosis Type I (Hurler Syndrome). Biol. Blood Marrow Transplant. 2018 Jun;24(6):1289-1293.[↩]
- Mesolella M, Cimmino M, Cantone E, Marino A, Cozzolino M, Della Casa R, Parenti G, Iengo M. Management of otolaryngological manifestations in mucopolysaccharidoses: our experience. Acta Otorhinolaryngol Ital. 2013 Aug;33(4):267-72[↩]
- Beck, M., Arn, P., Giugliani, R., Muenzer, J., Okuyama, T., Taylor, J., & Fallet, S. (2014). The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. [↩]
- Fensom AH, Benson PF. Recent advances in the prenatal diagnosis of the mucopolysaccharidoses. Prenat. Diagn. 1994 Jan;14(1):1-12.[↩]
- Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K, James DC, Lucas CF, Rogers TR, Benson PF, Tansley LR, Patrick AD, Mossman J, Young EP. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981 Oct 03;2(8249):709-12[↩][↩]
- Ponder KP, Haskins ME. Gene therapy for mucopolysaccharidosis. Expert Opin Biol Ther. 2007 Sep;7(9):1333-45.[↩]
- Beck, M., Arn, P., Giugliani, R., Muenzer, J., Okuyama, T., Taylor, J., & Fallet, S. (2014). The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med.[↩]
- Tichelli, A., & Rovo, A. (2013). Fertility issues following hematopoietic stem cell transplantation. Expert Rev Hematol., 6(4), 375-388.[↩]
- Peters, C., Balthazor, M., (1996). Outcomes of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood, 87, 4894-4902. [↩][↩]
- Hobbs, J., Hugh-Jones, K., Barrett, A., Byrom, N., Chambers, D., Henry, K., . . . Young, E. (1981). Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone marrow transplantation. Lancet, 318(8249), 709-712.[↩]




{kind=link}