Contents
- Hypokalemic periodic paralysis
- What is Potassium?
- Hypokalemic periodic paralysis causes
- Hypokalemic periodic paralysis pathophysiology
- Hypokalemic periodic paralysis prevention
- Hypokalemic periodic paralysis symptoms
- Hypokalemic periodic paralysis complications
- Hypokalemic periodic paralysis differential diagnosis
- Hypokalemic periodic paralysis diagnosis
- Hypokalemic periodic paralysis treatment
- Hypokalemic periodic paralysis prognosis
Hypokalemic periodic paralysis
Hypokalemic periodic paralysis also called HypoPP, HypoKPP or Westphall disease, is a rare inherited neuromuscular disorder that causes temporary episodes of muscle weakness or paralysis that is associated with a fall in blood potassium levels or hypokalemia (serum potassium less than 3.5 mmol/L) 1, 2, 3, 4, 5, 6, 7. People with hypokalemic periodic paralysis (HypoPP) typically have reduced levels of potassium in their blood (hypokalemia) during episodes of muscle weakness. Researchers are investigating how low potassium levels or hypokalemia may be related to the muscle abnormalities in hypokalemic periodic paralysis (HypoPP).
The episodes of muscle weakness or paralysis typically involve a temporary inability to move muscles in the arms and legs. The first attack usually occurs in childhood or adolescence and are triggered by strenuous exercise, high carbohydrate meals, injection of insulin, glucose, or epinephrine 1. Attacks can last for hours or days, and the frequency of attacks varies among people with hypokalemic periodic paralysis (HypoPP). The frequency is usually highest between the ages of 15 and 35, and then decreases with age. Some people with hypokalemic periodic paralysis (HypoPP) also develop chronic muscle weakness later in life or late-onset proximal myopathy affecting muscles of the trunk, shoulders, and thighs. Patients with late-onset proximal myopathy will have difficulty combing hair, difficulty climbing up the stairs, difficulty standing from a sitting position, and/or difficulty in getting up from bed 1, 8.
Hypokalemic periodic paralysis (HypoPP) is the most common type of periodic paralysis, and may be primary hypokalemic periodic paralysis (familial or idiopathic HypoPP) or secondary hypokalemic periodic paralysis (acquired HypoKPP) 9, 10, 11.
Primary hypokalemic periodic paralysis (familial or idiopathic HypoPP) is mainly caused by genetic changes in the CACNA1S or SCN4A gene where the genetic mutations occur in muscle ion channels producing potassium intracellular translocation 12, 13, 14. Both are voltage-dependent channels of the skeletal muscle fiber membrane. A mutation-induced aberrant current leads to a paradoxical membrane depolarization that renders muscle fibers unexcitable 15. Primary hypokalemic periodic paralysis (familial HypoPP) inheritance pattern is autosomal dominant.
Secondary hypokalemic periodic paralysis (acquired HypoKPP) is caused by the loss of potassium from kidneys, gastrointestinal tract or skin 5, 16, 17, 18, 19, 20. Hypokalemic periodic paralysis (HypoPP) cases related to thyroid disorders, more frequently thyrotoxicosis, and several autoimmune diseases have been previously reported 9, 16, 10. Thyrotoxicosis happens when you have too much thyroid hormone in your body in general. You could have too much thyroid hormone from taking too much thyroid medication, for example.
Although its exact prevalence is unknown, hypokalemic periodic paralysis (HypoPP) is estimated to affect 1 in 100,000 people 21, 22. However, a demographic survey in England, relying on the data of the national specialist channelopathy service, reported a minimum point prevalence of 0.13 per 100,000 people 23. Furthermore, men tend to experience symptoms of hypokalemic periodic paralysis (HypoPP) more often than women 24.
Hypokalemic periodic paralysis (HypoPP) treatment varies depending on the intensity and duration of the paralytic attacks. The goals of treatment are to relieve symptoms and prevent further attacks. Minor attacks may go away on their own 6. Moderate attacks may be self-treated in a non-medical setting by ingestion of oral potassium salts 6. But if weakness is severe, potassium may need to be given through a vein (IV). Muscle weakness that involves the breathing or swallowing muscles is an emergency situation. Dangerous irregular heartbeats (heart arrhythmias) may also occur during attacks. Any of these must be treated right away. Severe attacks typically require more intensive medical management with intravenous (IV) potassium infusion, serial measurement of serum potassium concentration, clinical evaluation of possible respiratory involvement, and continuous electrocardiogram (ECG) monitoring 6. There is no known curative treatment for hypokalemic periodic paralysis-related myopathy; physiotherapy may help to maintain strength and motor skills 6.
What is Potassium?
Potassium (K+) is a mineral that is vital to cell metabolism. Potassium is a major intracellular cation (positively charged ion) and a type of electrolyte that plays a significant role in the regulation of fluid volume and maintenance of the water-electrolyte balance 25, 26, 27. Potassium is present in all body tissues and is required for normal cell function because of its role in maintaining intracellular fluid volume and transmembrane electrochemical gradients 28, 29. Potassium helps transport nutrients into cells and removes waste products out of cells. Potassium is essential for the proper functioning of the heart, kidneys, muscles, nerves, and digestive system. Potassium is also important in muscle function, helping to transmit messages between nerves and muscles 28.
Potassium (K+) is a positively charged ion (cation), which is present throughout your body in both intracellular and extracellular fluids. The majority of body potassium, > 90%, are intracellular. It moves freely from intracellular fluid (ICF) to extracellular fluid (ECF) and vice versa when adenosine triphosphate (ATP) increases the permeability of the cell membrane. Potassium (K+) is mainly replaced inside or outside the cells by another cation, sodium (Na+). The movement of potassium into or out of the cells is linked to certain body hormones and also to certain physiological states. Standard laboratory tests measure extracellular fluid (ECF) potassium. Potassium enters the body rapidly during food ingestion. Insulin is produced when a meal is eaten; this causes the temporary movement of potassium from extracellular fluid (ECF) to intracellular fluid (ICF). Over the ensuing hours, the kidneys excrete the ingested potassium and homeostasis is returned. In the critically ill patient, suffering from high potassium level or hyperkalemia, this mechanism can be manipulated beneficially by administering high concentration (50%) intravenous glucose. Insulin can be added to the glucose, but glucose alone will stimulate insulin production and cause movement of potassium from extracellular fluid (ECF) to intracellular fluid (ICF). The stimulation of alpha receptors causes increased movement of potassium from intracellular fluid (ICF) to extracellular fluid (ECF). A noradrenaline infusion can elevate serum potassium levels. An adrenaline infusion, or elevated adrenaline levels, can lower serum potassium levels. Metabolic acidosis causes a rise in extracellular potassium levels (hyperkalemia). In this situation, excess of hydrogen ions (H+) are exchanged for intracellular potassium ions, probably as a result of the cellular response to a falling blood pH. Metabolic alkalosis causes the opposite effect, with potassium moving into the cells 30.
Potassium (K+), along with other electrolytes such as sodium (Na+), chloride (Cl–), and bicarbonate (HCO3–), helps regulate the amount of fluid in your body and maintains a stable acid-base balance. Potassium is present in all body fluids, but most potassium is found within the cells (intracellularly). Only a small amount is present in fluids outside the cells and in the liquid part of the blood (called serum or plasma).
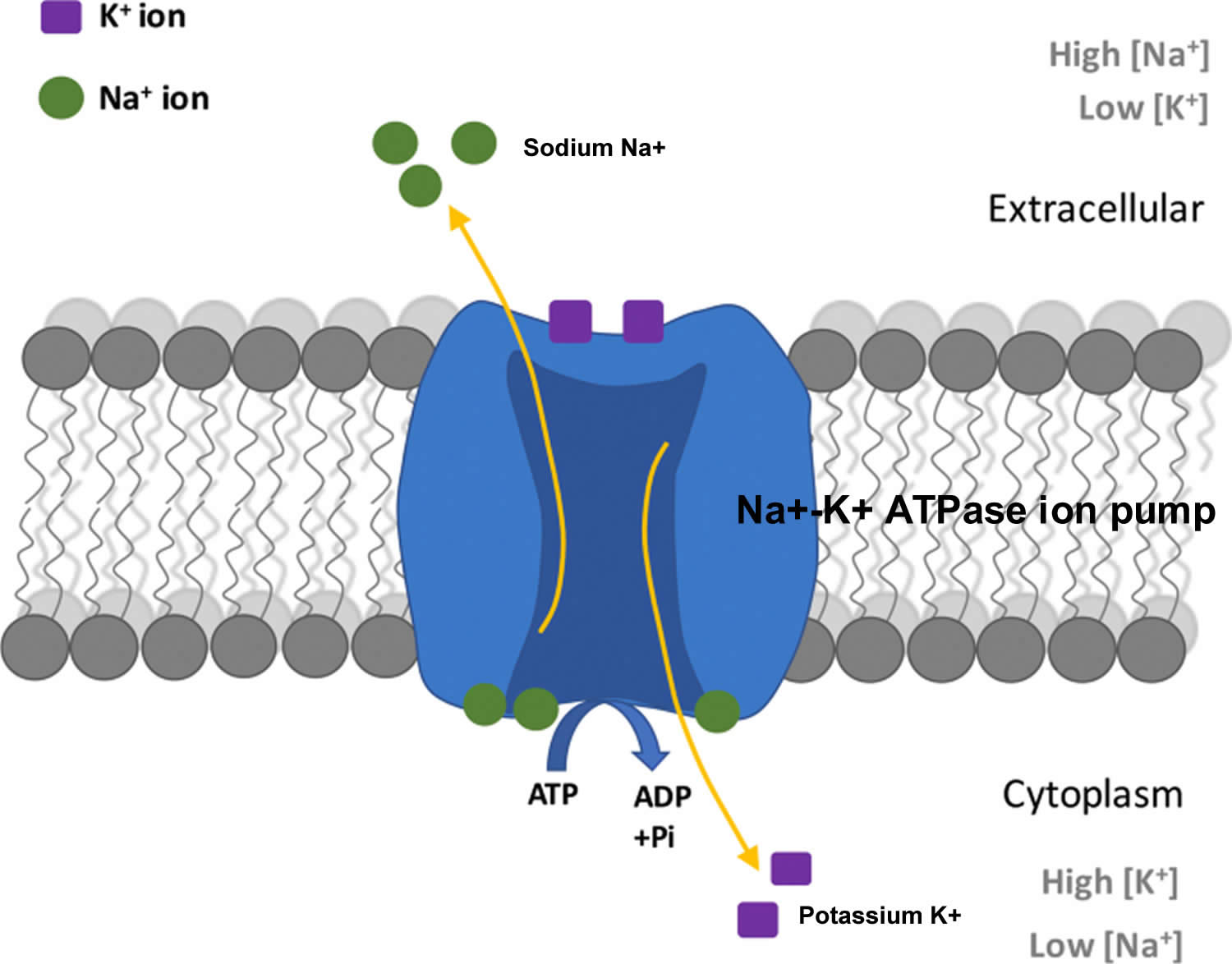
The total amount of potassium (K+) in the adult body is about 45 millimole (mmol)/kg body weight (about 140 g for a 175 pound adult; 1 millimole [mmol] = 1 milliequivalent [mEq] = 39.1 mg of potassium) 31. Most potassium are found within the cells (intracellularly) and a small amount is in extracellular fluid. The intracellular concentration of potassium is about 30 times higher than the extracellular concentration, and this difference forms a transmembrane electrochemical gradient that is maintained via the Sodium-Potassium ATPase pumps (Na+-K+ ATPase ion pumps) 32. When activated, the sodium-potassium ATPase pump (Na+-K+ ATPase ion pumps) exchanges 2 extracellular potassium (K+) ions for 3 intracellular sodium (Na+) ions, influencing membrane potential based on physiological excitation or inhibition. These sodium-potassium ATPase pumps (Na+-K+ ATPase ion pumps) are partially responsible, along with the sodium-potassium-chloride (Na+-K+-2Cl) co-transporter and sodium-calcium (Ca) exchanger, for maintaining the potential difference across the resting cell membrane as well. In addition to maintaining cellular tonicity, this gradient is required for proper nerve transmission, muscle contraction, and kidney function 28, 33, 34.
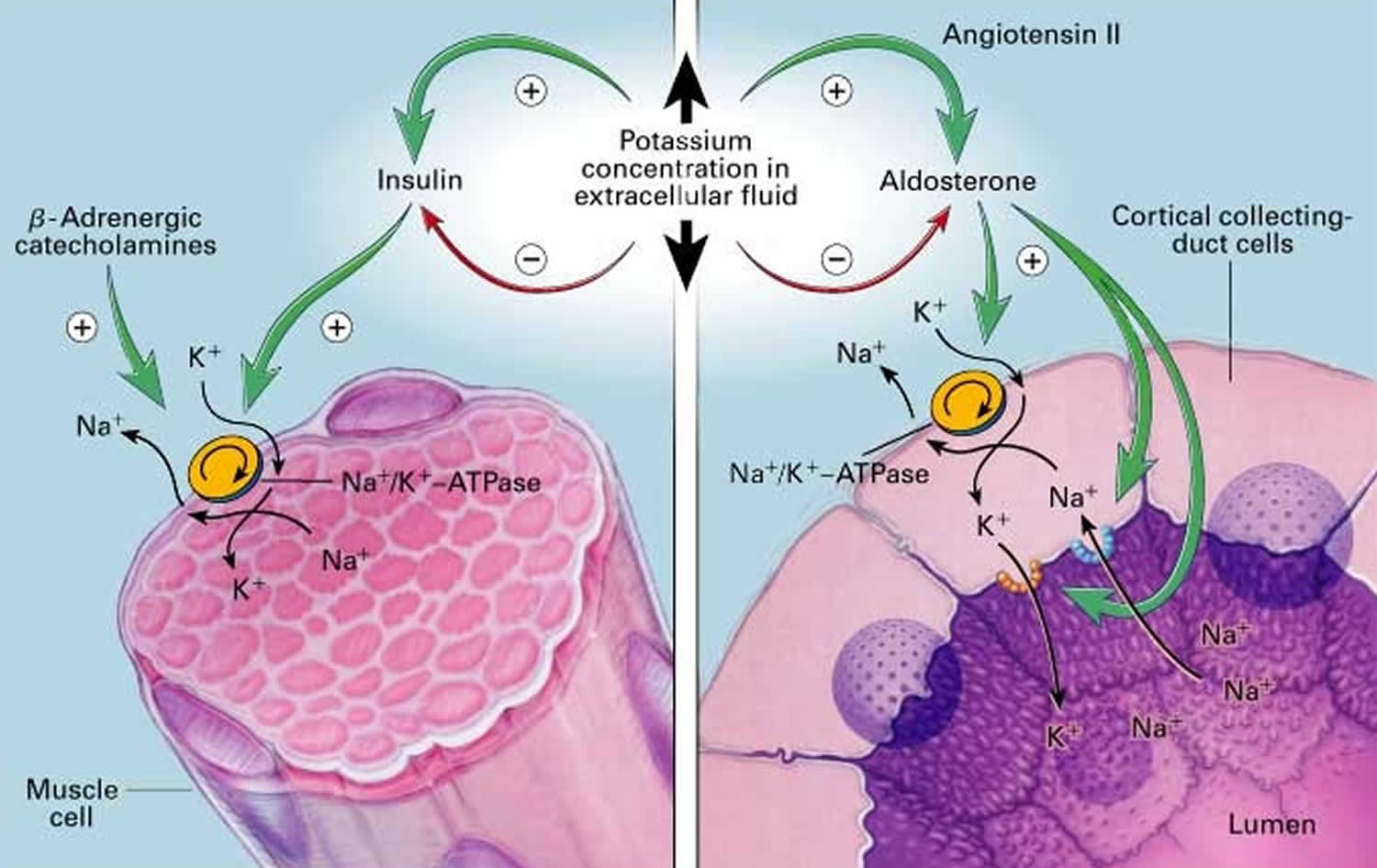
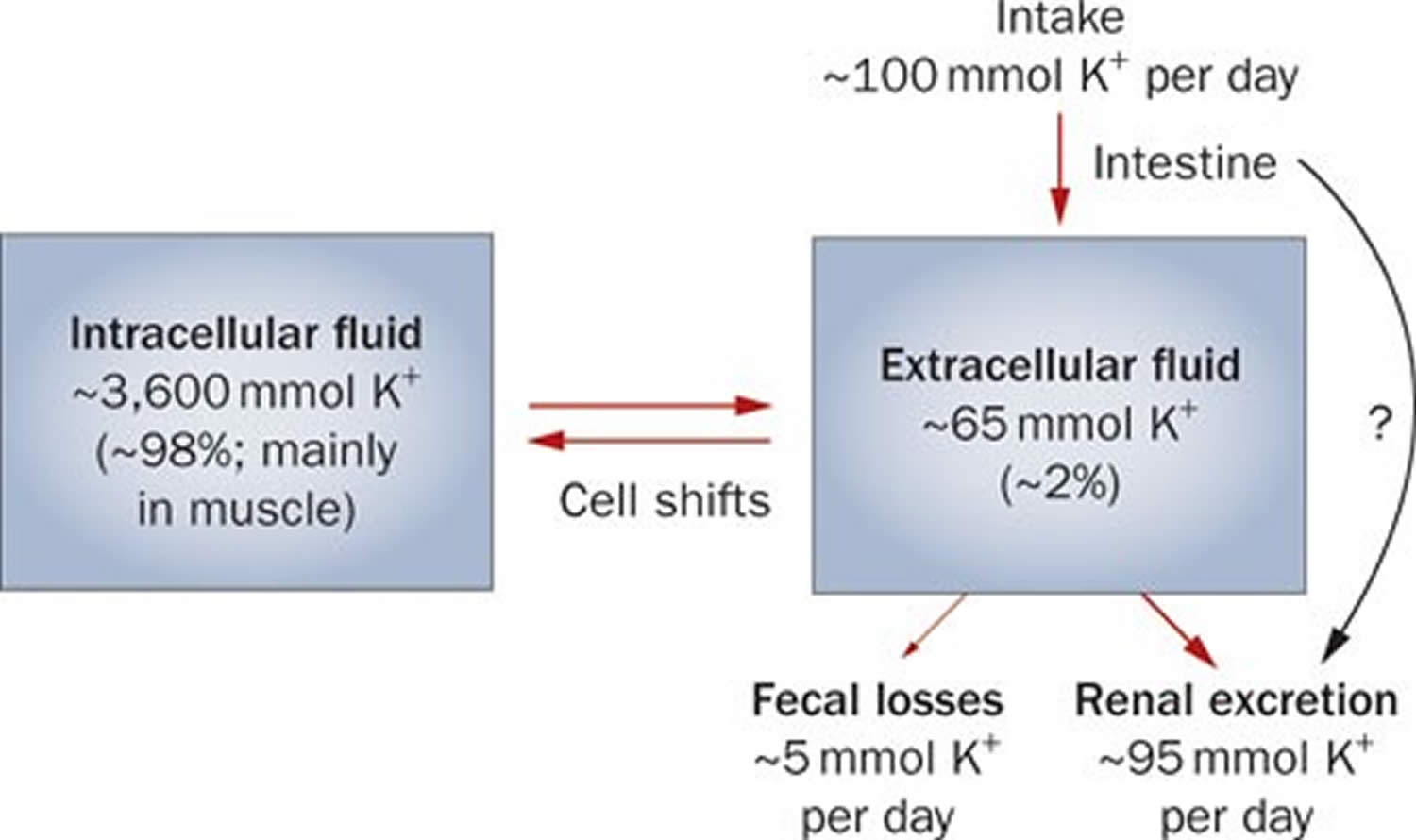
Potassium (K+) homeostasis depends on external balance (dietary intake [typically 100 mmol per day] versus excretion [95% via the kidney; 5% via the colon]) and internal balance (the distribution of potassium (K+) between intracellular and extracellular fluid compartments). The uneven distribution of potassium (K+) across cell membranes means that a mere 1% shift in its distribution can cause a 50% change in plasma potassium (K+) concentration 27. Hormonal mechanisms involving insulin, beta-adrenergic agonists and aldosterone modulate potassium (K+) distribution by promoting rapid transfer of potassium (K+) across the plasma membrane 35. Your body uses what potassium (K+) it requires and your kidneys eliminate the rest in the urine. Your body tries to keep the blood potassium level within a very narrow range. Levels are mainly controlled by aldosterone, a hormone produced by the adrenal glands in the kidneys. Extrarenal potassium (K+) losses from the body are usually small, but can be marked in individuals with chronic diarrhea, severe burns or prolonged sweating 27, 35. Under normal circumstances, the kidney’s distal nephron secretes potassium (K+) and determines final urinary excretion. In patients with low potassium levels or hypokalemia (plasma K+ concentration <3.5 mmol/l), after the exclusion of extrarenal causes, alterations in sodium ion (Na+) delivery to the distal nephron, aldosterone status, or a specific inherited or acquired defect in distal nephron function (each of which affects distal nephron K+ secretion), should be considered 27.
Figure 1. Potassium physiology
Because most potassium (K+) ions are found within the cells (a major intracellular cation), it is widely distributed in foods once derived from living tissues. Potassium concentration is higher in fruits and vegetables than in cereals and meat. You get most of the potassium you need from the foods that you eat and most people have an adequate intake of potassium. Recommended adequate intakes for potassium were set by the Food and Nutrition Board of the Institute of Medicine at 4700 mg/day 29. However it should be noted that the Food and Nutrition Board of the Institute of Medicine Recommended adequate intakes (AIs) for potassium at 4700 mg/day for adults is substantially higher than the World Health Organization’s (WHO) guidelines, which recommend 3150 mg/day for adults 36. The National Health and Nutrition Examination Survey (NHANES) data indicates that 99.2% of potassium in the US diet is naturally occurring, with the remaining 0.8% coming from fortified foods 37. These naturally occurring potassium sources include milk and other non-alcoholic beverages, as well as potatoes and fruit, which rank highest as sources of potassium intake among American adults 38. In addition, Western dietary practices with higher consumption of cereal, low nutrient density processed foods and lower consumption of fruits and vegetables has led to a diet lower in potassium and higher in sodium in recent decades 29. Salting foods and discarding the liquid induces sodium (Na+) for potassium (K+) exchange and reduces the potassium content of foods. Few Americans meet the recommended intakes; the average intake is 2591 ± 9 mg/day 37. This large gap between potassium intakes and recommended intakes led to potassium being called a shortfall nutrient in the Dietary Guidelines for Americans 39.
Actual potassium requirements would vary with an individual’s genetics, blood pressure (BP) status, and sodium intake 28. Blood pressure is currently the primary criterion for determining potassium requirements, with African Americans being more vulnerable to high blood pressure (hypertension) and more responsive to potassium supplementation than whites; individuals with high blood pressure (hypertension) are more responsive to increasing potassium intakes than individuals with normal blood pressure, and potassium having a greater benefit for those consuming a high salt diet 40. Other benefits of increasing potassium consumption may include improved blood sugar (glucose) control, glucose intolerance and insulin resistance becoming a concern for individuals with high blood pressure (hypertension) prescribed potassium wasting diuretics (water pills) 41. These differences support personalized nutrition approaches. Understanding movement of potassium within the body may help to improve these health outcomes.

Potassium is absorbed via passive diffusion, primarily in the small intestine 32. About 90% of ingested potassium is absorbed and used to maintain its normal intracellular and extracellular concentrations 42. There is around 50 mEq/kg of potassium (K+) in the body such that total body potassium (K+) in a 70-kg person is 3,500 mEq. Around 98% of potassium (K+) is found mainly within cells, and about 2% of the bodies’ potassium (K+) is in the extracellular fluid. The normal concentration of potassium (K+) in the extracellular fluid is 3.5–5.3 mEq/L. Large deviations from these values are not compatible with life.
Approximately 90% of the daily potassium (K+) intake is excreted in the urine, whereas a smaller percentage (10%) is excreted by the gastrointestinal tract in the stool and a very small amount is lost in sweat 43, 35, 44. Therefore, within the body, the kidney is the major organ responsible for potassium (K+) homeostasis. The kidneys control potassium excretion in response to changes in dietary intakes, and potassium excretion increases rapidly in healthy people after potassium consumption, unless body stores are depleted 28. The kidney facilitates potassium (K+) homeostasis by adjusting renal potassium (K+) excretion over several hours in response to a potassium load. Initial changes in extracellular potassium (K+) concentration are buffered by movement of potassium (K+) into or out of skeletal muscle cells. Internal potassium (K+) balance is a term used to refer to regulation of potassium (K+) distribution between the intracellular and extracellular space. Insulin, catecholamines, and, to a lesser extent, aldosterone are critical factors responsible for maintaining the normal internal distribution of potassium (K+) 35, 44.
The kidneys can adapt to variable potassium intakes in healthy individuals even in the setting of high dietary intake, but a minimum of 5 mmol (about 195 mg) potassium is excreted daily in urine 31. To demonstrate this, studies have shown potassium (K+) levels are kept within the normal range even when there are increases to ~15 g daily of dietary potassium (K+) intake sustained for 20 days 45, 46. Recent findings have identified the presence of an enteric potassium (K+) sensing mechanism that initiates the renal secretory process upon K+ entry into the gastrointestinal tract 44. The distal convoluted tubule has been identified as a site critical for potassium (K+) homeostasis, where it acts as a potassium (K+) sensor capable of initiating potassium (K+) excretion independent of mineralocorticoid activity 44. Combined with other obligatory losses, potassium balance cannot be achieved with intakes less than about 400–800 mg/day 35, 44.
Assessing potassium status is not routinely done in clinical practice, and it is difficult to do because most potassium in the body is inside cells 47. Although blood potassium levels can provide some indication of potassium status, they often correlate poorly with tissue potassium stores 31, 48, 49. Other methods to measure potassium status include collecting balance data (measuring net potassium retention and loss); measuring the total amount of potassium or the total amount of exchangeable potassium in the body; and conducting tissue analyses (e.g., muscle biopsies), but all have limitations 48.
Normal serum concentrations of potassium range from about 3.6 to 5.0 mmol/L and are regulated by a variety of mechanisms 31, 50. Diarrhea, vomiting, kidney disease, use of certain medications, and other conditions that alter potassium excretion or cause transcellular potassium shifts can cause low potassium level also called hypokalemia (serum potassium levels below 3.6 mmol/L) or high potassium level also called hyperkalemia (serum potassium levels above 5.0 mmol/L) 50. Otherwise, in healthy individuals with normal kidney function, abnormally low or high blood levels of potassium are rare.
Because the blood concentration of potassium is so small, minor changes can have significant consequences. If potassium levels are too low (serum potassium levels below 3.6 mmol/L) or too high (serum potassium levels above 5.0 mmol/L), there can be serious health consequences; a person may be at risk for developing shock, respiratory failure, or heart rhythm disturbances. An abnormal potassium level can alter the function of the nerves and muscles; for example, the heart muscle may lose its ability to contract.
Your body needs potassium to:
- Build proteins
- Break down and use carbohydrates
- Build muscle
- Maintain normal body growth
- Control the electrical activity of the heart
- Control the acid-base balance
Reduced potassium consumption has been associated with hypertension and cardiovascular diseases, and appropriate consumption levels could be protective against these conditions 51. A recent meta-analysis including 11 cohort studies reported an inverse association between potassium intake and risk of stroke 52. Additionally, two meta-analyses of trials comparing increased potassium to lower potassium intake found that increased potassium intake lowers blood pressure 53, 54. These results were further supported by a systematic review without a meta-analysis, which concluded that increased potassium intake results in decreased blood pressure in adults 55. Thus, a public health intervention aimed at increasing potassium intake from food could be a cost-effective strategy to reduce the burden of cardiovascular morbidity and mortality. Moreover, increasing potassium consumption from food in the population is safe; in individuals without renal impairment caused by medical conditions or drug therapy, the body is able to efficiently adapt and excrete excess potassium via the urine when consumption 56.
The American Heart Association recommended potassium intake for an average adult is 4,700 milligrams (mg) per day. Most of us aren’t getting nearly that much. On average, adult males eat almost 3,200 mg/day, and adult females eat about 2,400 mg/day 57. Remember that potassium is only part of an overall heart-healthy eating pattern. Other dietary factors that may affect blood pressure include amount and type of dietary fat; cholesterol; protein, sugar and fiber; calcium and magnesium, and of course, sodium.
For example, the DASH (Dietary Approaches to Stop Hypertension) diet study found that a diet rich in fruits, vegetables, fat-free or low-fat (1 percent) milk and milk products, whole-grain foods, fish, poultry, beans, seeds and unsalted nuts reduced blood pressure compared to a typical American diet. The DASH eating plan also had less sodium; sweets, added sugars and sugar-containing beverages; saturated and trans fats; and red meats than the typical American diet.
People with kidney problems, especially those on dialysis, should not eat too many potassium-rich foods. Your health care provider will recommend a special diet.
What does potassium do?
Potassium (K+) is the principal positively charged ion (cation) in the fluid inside of cells, while sodium (Na+) is the principal cation in the extracellular fluid. Potassium (K+) concentrations are about 30 times higher inside than outside cells, while sodium (Na+) concentrations are more than 10 times lower inside than outside cells 58. The concentration differences of these charged particles causes a difference in electric potential between the inside and outside of cells, known as the membrane potential. A cell’s membrane potential is maintained by ion pumps in the cell membrane, especially the Sodium-Potassium ATPase pumps (Na+-K+ ATPase ion pumps). These sodium-potassium ATPase pumps (Na+-K+ ATPase ion pumps) use ATP (energy) to pump sodium (Na+) of the cell and potassium (K+) into the cell, leading to a potassium (K+) gradient across the cell membrane [potassium (K+) in > potassium (K+) out], which is partially responsible for maintaining the cell membrane potential (Figure 2). The sodium-potassium ATPase pumps (Na+-K+ ATPase ion pumps) activity has been estimated to account for 20%-40% of the resting energy consumption in a typical adult 58. The large proportion of energy dedicated to maintaining sodium/potassium concentration gradients emphasizes the importance of this function in sustaining life 58. The cell membrane potential created by potassium and sodium ions allows the cell generate an action potential–a “spike” of electrical discharge. The ability of cells to produce electrical discharge is critical for body functions such as nerve impulse transmission, muscle contraction, and heart function 59, 60, 61.
Potassium is also an essential mineral needed to regulate water balance, blood pressure and levels of acidity 62. The more potassium you eat, the more sodium you pass out of the body through urine. Increased potassium intake has no adverse effect on blood lipid concentration, catecholamine concentrations, or renal function in apparently healthy adults without impaired renal handling of potassium 57. The largest benefit was detected when sodium intake was more than 4 g/day, which is the intake of most populations globally 63, so increased potassium intake should benefit most people in most countries. However, the authors also found a statistically significant decrease in blood pressure with increased potassium when sodium intake was 2-4 g/day. Therefore, increased potassium can continue to be beneficial in terms of blood pressure even as individuals and populations decrease their sodium intake. Studies examining both nutrients simultaneously support this concept, showing an increased benefit with simultaneous reduction in sodium and increase in potassium compared with changes in one nutrient individually 64, 65.
Potassium also helps relax blood vessel walls, which helps lower blood pressure 57.
World Health Organization recommends an increase in potassium intake from food to reduce blood pressure and risk of cardiovascular disease, stroke and coronary heart disease in adults. World Health Organization suggests a potassium intake of at least 90 mmol/day (3510 mg/day) for adults (conditional recommendation) 56.
Potassium also acts as a cofactor for some enzymes activity. For example, the activation of Na+/K+-ATPase requires the presence of sodium and potassium. The presence of potassium is also required for the activity of pyruvate kinase, an important enzyme in carbohydrate metabolism 66.
Figure 2. Sodium-Potassium ATPase pump
What are normal potassium levels?
Normal serum potassium values are between 3.5 to 5.0 millimoles/L (mmol/L) or 3.5 to 5.0 milliequivalent/L (mEq/L) 25, 67, 68, 69. However, there can be slight variation between laboratories and for this reason, it is important to look for the specific reference interval listed on your test report. Potassium levels outside this range, 3.5 to 5.0 millimoles/L (mmol/L) or 3.5 to 5.0 milliequivalent/L (mEq/L), are not compatible with life with increased rates of death from several causes 70, 71.
Your health care provider may order a potassium blood test as part of your regular checkup or to monitor an existing condition, such as diabetes, kidney disease, or adrenal gland disorders. You may also need this test if you take medicines that could affect your potassium levels or if you have symptoms of having too much or too little potassium.
Interpretation of a potassium test requires carefully considering the result, the laboratory reference range, and your health situation. Because potassium is frequently measured with other electrolytes, levels may be evaluated together. For a blood test, the report should list the amount of potassium measured in either milliequivalents per liter (mEq/L) or millimoles per liter (mmol/L). The test report will also show a reference range, which the laboratory considers an expected range for potassium levels.
What are low potassium levels?
Low potassium levels also known as hypokalemia is defined as serum potassium level less than 3.6 mEq/L or less than 3.6 mmol/L (Kim MJ, Valerio C, Knobloch GK. Potassium Disorders: Hypokalemia and Hyperkalemia. Am Fam Physician. 2023 Jan;107(1):59-70.https://www.aafp.org/pubs/afp/issues/2023/0100/potassium-disorders-hypokalemia-hyperkalemia.html)).
If your potassium levels are too low or hypokalemia, your symptoms may include 72:
- Irregular heartbeat
- Muscle cramps
- Weak or twitching muscles
- Fatigue
- Nausea
- Constipation
- Severe hypokalemia may result in muscular paralysis or abnormal heart rhythms (cardiac arrhythmias) that can be fatal 73, 74
- Chronic low potassium levels (chronic hypokalemia) is associated with high blood pressure (hypertension) and kidney stone formation.
Too little potassium in the blood or hypokalemia may be a sign of 72:
- Use of prescription diuretics (water pills)
- Fluid loss from diarrhea, vomiting, or heavy sweating
- Using too many laxatives
- Adrenal gland disorders, including Cushing’s syndrome and aldosteronism
- Kidney disease
- Alcohol use disorder
- Eating a lot of real licorice, which comes from licorice plants. Most licorice products sold in the U.S. don’t contain any real licorice. Check the package ingredient label to be sure.
- A diet too low in potassium (not common). Bananas, apricots, green leafy vegetables, avocados and many other foods are good sources of potassium that are part of a healthy diet.
Hypokalemic periodic paralysis causes
Both hereditary or familial and acquired causes of hypokalemic periodic paralysis (HypoPP) have been identified 6, 7. Acquired hypokalemic periodic paralysis has been associated with thyrotoxicosis. Periodic muscle weakness can also result from hypokalemia secondary to renal and gastrointestinal potassium loss as in renal tubular acidosis, gastroenteritis, or secondary to endocrine causes.
Familial hypokalemic periodic paralysis is caused by a mutation in either of two genes, calcium or sodium ion channel gene mutation. Over the last few decades, several mutations in CACNA1S, SCN4A, and KCNJ2 genes have been identified, which underlie almost 70% to 80% of cases of hypokalemic periodic paralysis, while rest remain genetically undetermined 75, 76. The most common familial hypokalemic periodic paralysis, Hypokalemic periodic paralysis 1 or type 1 HypoPP, has a mutation in the dihydropyridine-sensitive, skeletal muscle calcium voltage-gated channel subunit alpha1 S (CACNA1S) gene 13, 14. While the other familial hypokalemic periodic paralysis, Hypokalemic periodic paralysis 2 or type 2 HypoPP, has mutations in the skeletal muscle sodium voltage-gated channel alpha subunit 4 (SCN4A) gene 13, 14. Disease-causing mutations in the gene KCNJ2 and KCNJ18, code for inward rectifier potassium (Kir) channel, have also been identified 76, 77, 78, 79, 75. The familial hypokalemic periodic paralysis and thyrotoxic hypokalemic periodic paralysis constitute the primary hypokalemic periodic paralysis (HypoPP) 7.
- The CACNA1S and SCN4A genes provide instructions for making proteins that play essential roles in muscles used for movement (skeletal muscles). For the body to move normally, skeletal muscles must tense (contract) and relax in a coordinated way. Muscle contractions are triggered by the flow of certain positively charged atoms (ions) into muscle cells. The CACNA1S and SCN4A proteins form channels that control the flow of these ions. The channel formed by the CACNA1S protein transports calcium ions (Ca2+) into cells (hypokalemic periodic paralysis 1), while the channel formed by the SCN4A protein transports sodium ions (Na+) (hypokalemic periodic paralysis 2). Mutations in the CACNA1S or SCN4A gene alter the usual structure and function of calcium or sodium channels. The altered channels are “leaky,” allowing ions to flow slowly but continually into muscle cells, which reduces the ability of skeletal muscles to contract. Because muscle contraction is needed for movement, a disruption in normal ion transport leads to episodes of severe muscle weakness or paralysis.
- CACNA1S gene provides instructions for making the main piece (subunit) of a structure called voltage-gated calcium channel Cav1.1 (Hypokalemic periodic paralysis 1) 80, 81, 82. Channels containing the CACNA1S protein are found in muscles used for movement (skeletal muscles). These skeletal muscle calcium channels play a key role in a process called excitation-contraction coupling, by which electrical signals (excitation) trigger muscle tensing (contraction). Calcium channels made with the CACNA1S subunit are located in the outer membrane of muscle cells, so they can transmit electrical signals from the cell surface to inside the cell. The channels interact with another type of calcium channel called ryanodine receptor 1 (RYR1) channels (produced from the RYR1 gene). RYR1 channels are located in the membrane of a structure inside the cell that stores calcium ions. Signals transmitted by CACNA1S-containing channels turn on (activate) RYR1 channels, which then release calcium ions inside the cells. The resulting increase in calcium ion concentration within muscle cells stimulates muscles to contract, allowing the body to move.
- SCN4A gene belongs to a family of genes that provide instructions for making voltage-gated sodium channel Nav1.4 (Hypokalemic periodic paralysis 2) 81, 82. These sodium channels, which transport positively charged sodium ions (Na+) into cells, play a key role in a cell’s ability to generate and transmit electrical signals 83. The SCN4A gene provides instructions for making a critical part (the alpha subunit) of sodium channels that are abundant in muscles used for movement (skeletal muscles). For the body to move, these muscles must tense (contract) and relax in a coordinated way. Muscle contractions are triggered by the flow of ions, including sodium, into skeletal muscle cells. Channels made with the SCN4A protein control the flow of sodium ions into these cells.
- Almost all mutations neutralize a positively charged amino acid in one of the outermost arginines or lysines of a voltage sensor. The voltage-gated sodium channel Nav1.4 mutations (hypokalemic periodic paralysis 2) are situated in the voltage sensors of repeats I, II and III 84, 81. In vivo, the muscles from these patients exhibited an intracellular sodium accumulation and edema 77.
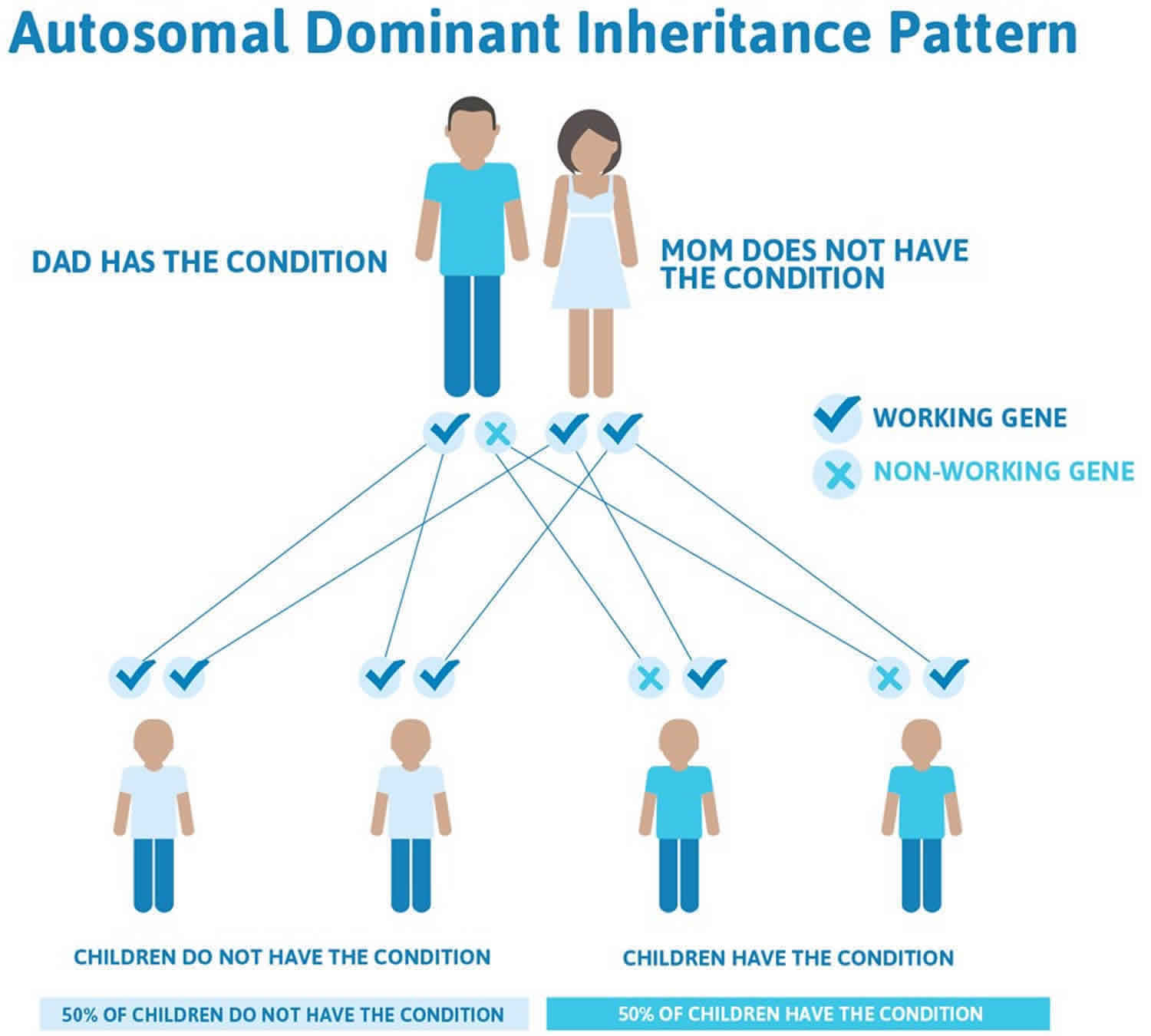
- In most cases, hypokalemic periodic paralysis is passed down through families (inherited) as an autosomal dominant disorder. In other words, only one parent needs to pass the defective gene related to hypokalemic periodic paralysis on to their child in order for the child to be affected.
A small percentage of people with the characteristic features of hypokalemic periodic paralysis do not have identified mutations in the CACNA1S or SCN4A gene. In these cases, the cause of the condition is unknown. These minor cases of hypokalemic periodic paralysis may be the result of a genetic problem that is not inherited.
Unlike other forms of periodic paralysis, people with hypokalemic periodic paralysis have normal thyroid function. But they have a very low blood level of potassium during episodes of weakness. This results from potassium moving from the blood into muscle cells in an abnormal way.
Risk factors include having other family members with periodic paralysis. The risk is slightly higher in Asian men who also have thyroid disorders.
Hypokalemic periodic paralysis inheritance
Most familial hypokalemic periodic paralysis is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Familial hypokalemic periodic paralysis has lower clinical expression in females because of the lower penetrance and attack rate compared to males 85. And also women tend to have fewer attacks of muscle weakness than men.
A small percentage of people with the characteristic features of hypokalemic periodic paralysis do not have identified mutations in the CACNA1S or SCN4A gene (sporadic cases). In these sporadic cases, the cause of the condition is unknown. These minor cases of hypokalemic periodic paralysis may be the result of a genetic problem that is not inherited or represents new mutations 75, 86.
Most cases of thyrotoxic hypokalemic periodic paralysis have been identified as sporadic and is more prevalent among Asian descents with a male predominance of 9 to 1 22.
Figure 3. Hypokalemic periodic paralysis autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Hypokalemic periodic paralysis pathophysiology
The most common genetic abnormality in hypokalemic periodic paralysis is the missense mutation in the positively charged residues, i.e., arginine, in the S4 domain of the alpha subunit (voltage sensor domain) of the skeletal muscle ion channel, most commonly L-type calcium channel (Cav1.1) and less commonly voltage-gated sodium channel (Nav1.4) 13, 14. Disease-causing mutations in the gene KCNJ2 and KCNJ18, code for inward rectifier potassium (Kir) channel, have also been identified 76, 77, 78, 79, 75. The final common mechanism for all mutations is the formation of anomalous gating pore current itself through the voltage sensor domain of ion channel that makes sarcolemmal muscle inexcitable, resulting in failure of muscle action potential and occurrence of subsequent attacks of flaccid paralysis 79, 76, 87, 88. In 90% of identified cases, arginine mutation in the S4 segment remains the primary cause 78. The other possible hypokalemic periodic paralysis mutations are yet to be determined.
The presence of gating pore current is mostly studied and understood in sodium channels. Many experiments demonstrated the presence of anomalous gating pore current in the setting of SCN4A mutation in sodium channels during the resting phase. The anomalous gating pore current results inward nonselective cation leak causing aberrant depolarization, which is sufficient to make the resting potential of the muscle fibers unstable 79, 76. And when serum potassium level drops below 3.0 mM, the affected muscle fibers paradoxically undergo sustained depolarization making muscle electrically inexcitable, whereas normal muscle fibers undergo hyperpolarization at this level of drop in serum potassium. Normally inward rectifying potassium (Kir) channel and membrane Na-K-ATPase maintains the normal negative resting membrane potential. In the presence of CACNA1S and SCN4A mutations, the depolarization induced by the gating pore current, at the modest drop of serum potassium level to around 3.0 mM, counterbalance the Kir current leading to sustained depolarization 76, 77, 21.
There are fewer experimental studies to demonstrate the evidence of gating pore current in calcium channels. But as the phenotypic expression of hypokalemic periodic paralysis in sodium and calcium channel mutations are similar, it is believed that the gating pore current does exist in calcium channel. While it is still unclear, there are numerous observations from different experimental studies to explain the possible underlying mechanisms behind muscle weakness with underlying calcium channel defects:
- The calcium channel mutations manifest as loss of function. Electrophysiologic studies have demonstrated slower activation of calcium channels and diminished calcium current density 75, 89. However, this observation does not correlate with episodes of depolarization, hypokalemia, and attacks of muscle weakness.
- In an experimental study, muscle biopsies taken from three hypokalemic periodic paralysis patients having R528H mutation of calcium channel (Cav1.1) showed the abnormal function of sarcolemmal ATP sensitive K+ (KATP) channel, supported by the fact that magnesium adenosine diphosphate (MgADP) did not stimulate the channel. The KATP channel showed reduced opening and reduced conductance state, i.e., reduced K current 75. The reduced K current is more likely related to depolarization with hypokalemia. Altered Ca2+ homeostasis resulting from the calcium channel mutation is likely the reason behind the altered function of the KATP channel. This observation hints toward the presence of a possible secondary channelopathy in patients with hypokalemic periodic paralysis.
Hypokalemic periodic paralysis prevention
Hypokalemic periodic paralysis (HypoPP) cannot be prevented. Because hypokalemic periodic paralysis (HypoPP) can be inherited, genetic counseling may be advised for couples at risk of the disorder. Treatment prevents attacks of weakness. Before an attack, there may be leg stiffness or heaviness in the legs. Doing mild exercise when these symptoms start may help prevent a full-blown attack. Taking potassium supplements may help prevent muscle weakness.
A diet rather low in sodium and carbohydrate and rich in potassium is recommended and may help decrease symptoms.
- Oral intake of potassium salts (10-20 mmol/dose, 3 doses/day) can prevent attacks, especially if the dose of potassium is taken some hours before the usual time of the attack (i.e., a nocturnal dose if crises occur at awakening).
- For individuals receiving chronic potassium supplementation for hypokalemic periodic paralysis, magnesium might be added, which can be helpful to promote renal retention of potassium and, therefore, reduce the potassium dose 21.
A carbonic anhydrase inhibitors medicine called acetazolamide and diclorphenamide may be prescribed to prevent attacks. Your doctor may tell you to also take potassium supplements because acetazolamide may cause your body to lose potassium. There is no standardized treatment regimen and no consensus as to when to start treatment with acetazolamide.
- Typical dosage for acetazolamide in adults is between 125 mg/day and 1000 mg/day (usually 250-500 mg/day), divided into three doses and taken with meals; in children a dose of 5-10 mg/kg/day, divided into three doses and taken with meals, is used.
- Acetazolamide treatment:
- Is beneficial in approximately 50% of individuals with hypokalemic periodic paralysis
- Has no effect in 30% of affected individuals
- May worsens hypokalemic periodic paralysis in individuals with who have a pathogenic variant in SCN4A
- In some affected persons, long-lasting interictal weakness may be partly reversed and muscle strength may be improved by acetazolamide treatment 90
- Whether acetazolamide treatment prevents or treats myopathy and the resulting fixed weakness that occurs with age is unknown.
- Further studies are needed to evaluate the effect of preventive acetazolamide treatment on attack rate, severity-weighted attack rate, long-lasting interictal weakness, and myopathy.
- Common side effects of carbonic anhydrase inhibitors include paresthesia, fatigue, mild, reversible cognitive disturbances and an increased risk of kidney stone (nephrolithiasis).
Dichlorphenamide was recently approved by the FDA for the treatment of periodic paralysis. Dichlorphenamide has been evaluated in four randomized, placebo- controlled studies, two each in patients with hypokalemic periodic paralysis (HypoPP) and hyperkalemic periodic paralysis (HyperPP) 91
- While randomized controlled trials of dichlorphenamide were performed in adults, the same approach is taken for children; dose adjustments may be required based on age.
- These studies demonstrated a significant reduction in the frequency and severity of the attacks. During a 52-week extension, in which all remaining participants received open-label dichlorphenamide, continued improvement in outcomes was observed in both placebo and dichlorphenamide groups.
- The dose of dichlorphenamide was 50 mg twice daily for treatment-naıve patients.
- Individuals already on dichlorphenamide before the study continued on the same dose during the study.
- In those taking acetazolamide before the study, the dose of dichlorphenamide was set at 20% of the acetazolamide dose.
- Dose reduction for tolerability was permitted.
- The mean dose of dichlorphenamide at week 9 was 82 mg/day.
- The most common side effects with dichlorphenamide were paresthesias, cognitive disorder, dysgeusia, headache, fatigue, hypoesthesia, and muscle spasms, generally not requiring discontinuation of dichlorphenamide, and reversible with drug discontinuation.
In the recent study of diclorphenamide 91, quality of life was assessed at 9 weeks and significant improvement was reported for the physical component and physical functioning, working time, bodily pain, vitality, and social functioning in those with hypokalemic periodic paralysis (HypoPP).
If carbonic anhydrase inhibitors are not tolerated or not effective after prolonged use, alternatives include potassium sparing diuretics like triamterene 50–150 mg/day, spironolactone 25–100 mg/day or eplerenone 50– 100mg daily.
- Because spironolactone is associated with a long half-life for substrate degradation, the individual can become hyperkalemic and weaker, develop cardiac arrhythmias, and suffer from hair loss. Additionally, spironolactone has androgenic side effects.
- The modern spironolactone derivate Eplerenone may be preferred because it causes fewer androgenic side effects. In addition, it has a very high repolarizing power, the parameter considered as most relevant for a beneficial effect.
- For individuals with hypokalemic periodic paralysis, potassium supplementation and a potassium-sparing diuretic may be used concomitantly, but potassium levels should be routinely monitored.
Avoid anything that can trigger paralytic attacks in the individual case, including the following:
- Unusually strenuous effort
- Excess of carbohydrate-rich meals or sweets
- Cold
- Stress/excitement/fear
- High salt intake
- Prolonged immobility
- Oral or intravenous glucosteroids
- Use of cooling, glucose and/or mannitol infusion, excessive sodium- containing fluids and certain anesthetics such as succinylcholine during anesthesia
- Use of alcohol
Hypokalemic periodic paralysis symptoms
Hypokalemic periodic paralysis can have its onset anywhere from early childhood to adulthood, the mean age of presentation of attacks is the first or second decade of life, usually the late childhood or teenage years 92. The age of onset of the first attack ranges from two to 30 years 7. Attacks usually begin in the teen years, but they can occur before age 10. However, in the case of thyrotoxic hypokalemic periodic paralysis, the onset is usually after age 20. Hypokalemic periodic paralysis symptoms include attacks of muscle weakness or loss of muscle movement (paralysis) that come and go (periodic paralysis). The paralytic attacks are characterized by decreased muscle tone (flaccidity) more marked proximally than distally with normal to decreased deep tendon reflexes 6. The paralytic episodes develop over minutes to hours and last several minutes to several days (paralytic episodes ranges from one to 72 hours with an average of nearly 24 hours) with spontaneous recovery. There is normal muscle strength between attacks. Attacks occur suddenly and are episodic. Some individuals may experience a milder form of muscle weakness between attacks that fluctuates and improves with mild exercise 22.
How often the attacks occur varies. Some people have attacks every day. Others have them once a year. During attacks the person remains alert. The frequency of attacks tends to decrease with age 75, 21. Long-lasting interval between episodes of muscle weakness may occur in some affected individuals and in some stages of the disease and in myopathic muscle changes. A myopathy may occur independent of paralytic symptoms and may be the sole manifestation of hypokalemic periodic paralysis.
The muscle weakness or paralysis:
- Most commonly occurs at the shoulders and hips
- May also affect the arms, legs, muscles of the eyes, and muscles that help with breathing and swallowing
- Occurs off and on
- Most commonly occurs on awakening or after sleep or rest
- Is rare during exercise, but may be triggered by resting after exercise
- May be triggered by high-carbohydrate, high-salt meals, stress, pregnancy, heavy exercise, and cold
- An attack usually lasts for several hours up to a day
Another symptom may include eyelid myotonia (a condition in which after opening and closing the eyes, they cannot be opened for a short time).
Patients usually present with attacks of generalized severe muscle weakness, with proximal muscle involvement more marked than distal and a profound decrease in serum potassium level (serum potassium less than 2.5 mmol/L) 21. Usually, patients go to bed in the normal state of health and wake up in the middle of the night or the morning, experiencing an attack of muscle weakness 75. Many patients also experience prodromal symptoms like fatigue, paresthesias, behavioral changes a day before an attack of muscle weakness 75. However, when incomplete, it predominantly involves lower limbs than the upper limbs. Bulbar, ocular, and respiratory muscles are usually spared, but respiratory muscle involvement can prove fatal when involved in severe cases 92, 22. The pattern of muscle weakness is similar in both familial and thyrotoxic hypokalemic periodic paralysis, and signs of overactive thyroid (hyperthyroidism) are clinically obvious in most cases of thyrotoxic hypokalemic periodic paralysis but are not always present. And attacks of muscle weakness occur during the state of hyperthyroidism and never when the thyroid function is normal.
The frequency of attacks of muscle weakness is very variable and infrequent. Some individuals have only one episode in a lifetime; more commonly, attacks occur repeatedly: daily, weekly, monthly, or less often 75, 6. And the duration of each attack also varies, ranging from minutes to days and can last up to several hours before they resolve spontaneously. Women tend to have fewer attacks than men.
The major triggering factors are rest following strenuous exercise and consumption of carbohydrate-rich meals 6, 75, 21. It is hypothesized that these triggering factors cause a rise in plasma epinephrine level or insulin level, causing an intracellular shift of potassium, resulting in lower serum potassium level, thus triggering the episode of weakness 22.
Additional triggers can include cold, stress, excitement, fear, salt intake, prolonged immobility, glucocorticoids use, alcohol, and anesthetic procedures 75, 6.
Hypokalemic periodic paralysis complications
Health problems that may be due to hypokalemic periodic paralysis include:
- Irregular heartbeat during attacks or life-threatening cardiac arrhythmias due to hypokalemia (are uncommon but have been reported during attacks of muscle weakness) 93
- Respiratory insufficiency due to respiratory muscle paralysis
- Difficulty breathing, speaking, or swallowing during attacks (rare)
- Muscle weakness that worsens over time. Many patients can have muscle weakness during the interictal period (i.e., between paralytic attacks), but its frequency and the risk for long-lasting weakness are unknown 21. It is believed that it is the result of permanent sodium intake, which results from the cation leak through the gating pore current 6. This may respond to potassium administration or acetazolamide 92.
- Myopathy. Most patients develop progressive proximal myopathy; however, the frequency is unknown. Myopathy usually manifests after age 50, is less fluctuating, and less sensitive to medications, which suggest there is muscle degeneration, a fixed myopathy 92, 94, 21. It may be evident early on muscle biopsy before manifesting clinically. The myopathy is more profound in pelvic girdle muscles and proximal upper and lower limbs 92, 94. The severity of myopathy varies among individuals, and some develop only mild weakness, which does not affect normal daily activities, while some may develop severe myopathy enough to make them wheelchair-bound. There is little evidence to support the correlation between the development of myopathy and the frequency or severity of paralytic attacks 94, 22.
- Kidney stones (a side effect of acetazolamide). A report showed an occurrence of renal stones in up to 15% of patients taking acetazolamide for the long term. The treatment of acetazolamide induced renal stones is the removal of stone without stopping acetazolamide therapy 21
Hypokalemic periodic paralysis differential diagnosis
The differential diagnosis of primary hypokalemic periodic paralysis includes 7:
- Hyperkalemic periodic paralysis (HyperPP) or normokalemic periodic paralysis (NormoKPP). Generally, the distinction between hypokalemic periodic paralysis and normo/hyperkalemic periodic paralysis can be made by potassium level during attacks, EMG, and genetic testing. Normokalemic and Hyperkalemic Periodic Paralysis differ from the hypokalemic periodic paralysis in following ways:
- Normal or elevated serum potassium levels during attacks
- Absence of some precipitating factors for hypokalemic periodic paralysis, e.g., carbohydrate-rich meals
- Younger age of onset of attacks with high penetrance 75.
- EMG shows myotonic discharges between attacks, but the EMG findings in a short exercise test and long exercise test are difficult to distinguish from that in hypokalemic periodic paralysis 95.
- The response to oral potassium might differ from that to hypokalemic periodic paralysis, it can reduce or can even worsen the symptoms 6
- Thyrotoxic periodic paralysis (TPP). Thyrotoxic periodic paralysis also called thyrotoxic hypokalemic periodic paralysis (Thyrotoxic HypoKPP), is a rare neuromuscular disorder characterized by recurrent episodes of paralysis and hypokalemia (fall in blood potassium levels) during a high levels of thyroid hormone in their blood (hyperthyroidism or thyrotoxicosis). Hyperthyroidism is often called overactive thyroid is a condition in which the thyroid gland makes too much thyroid hormone. Hyperthyroidism or overactive thyroid happens specifically when your thyroid gland both produces and releases too much thyroid hormone. Thyrotoxicosis happens when you have too much thyroid hormone in your body in general. You could have too much thyroid hormone from taking too much thyroid medication, for example. This would be thyrotoxicosis, not hyperthyroidism. However, most people who develop high thyroid hormone levels due to hyperthyroidism or thyrotoxicosis are not at risk of periodic paralysis.
- Andersen-Tawil syndrome. Andersen-Tawil syndrome (ATS) is a disorder that causes episodes of muscle weakness (periodic paralysis), changes in heart rhythm (arrhythmia), and developmental abnormalities 96. The episodes of muscle weakness (periodic paralysis) begins early in life, and episodes last from hours to days. These episodes may occur after exercise or long periods of rest, but they often have no obvious trigger. Muscle strength usually returns to normal between episodes. However, mild muscle weakness may eventually become permanent. In Andersen-Tawil Syndrome (ATS) the potassium shifts during attacks of paralysis are inconsistent. Potassium may rise during one attack (hyperkalemia) and fall during another (hypokalemia). The traditional classifications of Hypokalemic Periodic Paralysis or Hyperkalemic Periodic Paralysis cannot be applied. Patients also tend to have generalized weakness between attacks. In addition, Andersen-Tawil Syndrome patients experience irregular heart rhythms including a prolonged QT interval 96. Some have unusual facial and hand characteristics, such as short stature, clinodactyly (an inward curvature of the 5th fingers), fused or webbed second and third toes, scoliosis (crooked spine), widely spaced eyes, low-set ears, a broad forehead, and a small jaw 96. These signs may be absent or very subtle, or they may exist in other family members who do not experience muscle weakness or paralysis 96. Andersen-Tawil Syndrome is inherited in an autosomal dominant pattern. All Andersen-Tawil Syndrome mutations identified so far have been on the potassium channel.
- Secondary hypokalemia. The most common symptom of hypokalemia is muscle weakness. A low potassium level between attacks is a clue to secondary hypokalemia. The patient should be carefully examined to look for the systemic manifestation of chronic hypokalemia secondary to renal, gastrointestinal, endocrine, and iatrogenic causes, and a careful interpretation of the patient’s blood pressure finding, urine potassium, and blood bicarbonate level should be performed to rule out the possibility of any secondary cause of hypokalemia. Following conditions can cause chronic hypokalemia:
- Diuretics use
- Type 4 renal tubular acidosis
- Hyperaldosteronism
- Hyperglucocorticoidism
- Gitelman syndrome
- Bartter syndrome
- Liddle syndrome
- Myasthenia gravis. Myasthenia gravis is an autoimmune disease where your body’s immune system makes antibodies that block or destroy acetylcholine receptor sites or muscle-specific receptor tyrosine kinase (MuSK) or change some of the nerve signals to your muscles. Research studies have found other antibodies and the number of antibodies involved will likely grow over time. This makes your muscles weaker. Muscle weakness caused by myasthenia gravis gets worse when the affected muscle is used. Because symptoms usually get better with rest, muscle weakness can come and go. However, the symptoms tend to progress over time. They usually reach their worst within a few years after the disease begins. Myasthenia gravis can affect people of any age, but it’s more common in women younger than 40 and in men older than 60. There’s no cure for myasthenia gravis. Your treatment will depend on your age, how severe your disease is and how fast it’s progressing.
- Paramyotonia congenita (PMC). Paramyotonia Congenita is a congenital disorder of muscle weakness and muscle stiffness (myotonia), induced by cold and aggravated with continued exercise. The patients develop prolonged myotonia (muscle stiffness) or weakness in a localized group of muscles. Paramyotonia Congenita (PMC) comes in two forms, one in which attacks are always associated with a rise in potassium (hyperkalemia) and a form called Paramyotonia von Eulenburg in which attacks can be associated with a fall in blood potassium levels or hypokalemia. Both result from mutations in the sodium channel. Both can accompany hyperkalemic periodic paralysis (HyperPP) or can occur alone. Paramyotonia Congenita (PMC) mainly affects eyelids, neck, and upper limb muscles. Patients characteristically present in their childhood complaining of inability to open their eyes following rapid, forceful successive closures. Weakness and myotonia last for minutes to hours. Even after the immediate rewarding of the muscles, cold-induced weakness usually persists for several hours. The disease is non-progressive, does not cause muscle wasting or hypertrophy.
- Metabolic Myopathies. Patients usually complain of fatigue, exercise intolerance, myalgia rather than muscle weakness. Rhabdomyolysis is common and may result from strenuous exercise, stress, illness, cold exposure. Muscle biopsy is required for the diagnosis of metabolic myopathies.
These conditions are associated with either recurrent episodes of hypokalemia, episodic attacks of muscle weakness, and weakness or stiffness associated with exercise. Thus they should be in the mind of the treating physician as they can mimic hypokalemic periodic paralysis. Nevertheless, they can be differentiated based on their clinical features and laboratory tests findings as they differ in several ways.
The following signs and symptoms suggest a diagnosis other than hypokalemic periodic paralysis (hypoPP) 6:
- Associated sensory symptoms, including pain or tenderness
- Sensory loss could suggest polyneuropathy such as Guillain-Barré syndrome.
- Pain could suggest myositis; however, some individuals with hypoPP report paralytic episodes as painful.
- Urinary retention or constipation, which may be observed in other causes of acute or subacute paralysis, but can occur rarely in hypoPP
- Associated symptoms that suggest myasthenia or involvement of the neuromuscular junction, including:
- Ptosis
- Diplopia
- Dysphagia
- Dysarthria
- Alteration or loss of consciousness
- Abnormal movement
- History of fever days before an attack, which could suggest poliomyelitis or other virus-caused paralysis
- History of back pain days before an attack, which could suggest acute transverse myelitis
- History of tick bite, which could suggest tick paralysis
Hypokalemic periodic paralysis (hypoPP) is the most common cause of periodic paralysis. The four major differential diagnoses are normokalemic potassium-sensitive periodic paralysis (normoPP), hyperkalemic periodic paralysis (hyperPP), thyrotoxic periodic paralysis (TPP), and Andersen-Tawil syndrome (ATS) (see Table 1).
Table 1. Different Categories of Periodic Paralyses and Associated Findings
| Hypokalemic periodic paralysis (HypoPP) | Normokalemic periodic paralysis (NormoPP) | Hyperkalemic periodic paralysis (HyperPP) | Thyrotoxic periodic paralysis (TPP) | Andersen-Tawil syndrome | |
|---|---|---|---|---|---|
| Main clinical features | Weakness episodes lasting hrs to days w/concomitant hypokalemia | Weakness episodes lasting hrs to days w/concomitant normokalemia | Weakness episodes lasting mins to hrs w/concomitant normo- or hyperkalemia | Identical to that of the paralytic episodes of hypoPP | Episodic PP, ventricular arrhythmias, prolonged QT interval, characteristic anomalies 2 |
| Age at first attacks | Late in 1st decade or in 2nd decade | Late in 1st decade or in 2nd decade | 1st years of life | Variable, dependent on onset of thyrotoxicosis | Late in 1st decade or in 2nd decade (usually after cardiac events) |
| Main triggers | Rest after exercise, carbohydrate-rich meal, salt intake, stress, cold | Rest after exercise, carbohydrate-rich meal, salt intake, stress, cold | Cold; rest after exercise, stress, & fatigue; alcohol; hunger; changes in activity level; potassium in food; specific foods | Thyrotoxicosis | Prolonged rest, rest after exertion |
| EMG: myotonic discharges | No | Some | Some | No | No |
| EMG tests | Late decrement w/LET (pattern IV, V) | Late decrement w/LET (pattern IV, V) | Pattern IV, V | Initial CMAP ↑ + ↓ | Variable (CMAP ↑ + ↓, normal CMAP + ↓, etc) |
| Extramuscular expression | None | None | None | Possible manifestations of thyrotoxicosis | Cardiac arrhythmia, dysmorphy |
| Prevention of paralysis attacks | ACZ, DCP | ACZ, DCP | ACZ, DCP | Normal thyroid function | ACZ, DCP |
| Curative treatment | None | None | None | Treatment of thyroid disorder | None |
| Known causative or susceptibility gene(s) 3 | CACNA1S; SCN4A | SCN4A | SCN4A | KCNJ18 | KCNJ2 |
| Defective ion channel(s) | Cav 1.1; Nav 1.4; Kir 6.2 | Nav 1.4 | Nav 1.4 | Kir 6.2 | Kir 2.1 |
Footnote:
- Thyrotoxic periodic paralysis (TPP) 97, 98, 99, 100, 101
- Andersen-Tawil syndrome anomalies include low-set ears, widely spaced eyes, small mandible, fifth-digit clinodactyly, syndactyly, short stature, and scoliosis.
- In a cohort of 60 Chinese individuals with primary periodic paralysis, 92.5% of those with a genetic diagnosis had pathogenic variants in CACNA1S, KCNJ2, or SCN4A 102.
Abbreviations: ACZ = acetazolamide; CMAP = compound muscle action potential; DCP = dichlorphenamide; LET = long exercise test
[Source 6 ]Hypokalemic periodic paralysis diagnosis
Your doctor may suspect hypokalemic periodic paralysis (HypoPP) based on a positive family history or previous personal history of similar attacks of muscle weakness. Other clues to hypokalemic periodic paralysis (HypoPP) are muscle weakness symptoms that come and go with normal or low results of a potassium on blood test (hypokalemia). When there is an established family history of hypokalemic periodic paralysis (HypoPP) , no further diagnostic investigations are required to confirm the diagnosis of an episode of a paralytic attack. Otherwise, a low serum potassium level (hypokalemia) during a typical attack of weakness establishes the diagnosis.
During an attack, muscle reflexes are decreased (hyporeflexia) or absent (areflexia). And muscles go limp rather than staying stiff. Muscle groups near the body, such as the shoulders and hips, are involved more often than the arms and legs. During an attack of muscle weakness, blood potassium level is low (hypokalemia) and this confirms the diagnosis. There is no decrease in total body potassium.
Between attacks, a physical examination shows nothing abnormal. Before an attack, there may be leg stiffness or heaviness in the legs. Blood potassium level is normal (normokalemia) between attacks.
Tests that may be done include:
- Electrocardiogram (ECG), which may be abnormal during attacks
- Electromyography (EMG), which is usually normal between attacks and abnormal during attacks.
- During attacks of weakness, electromyography (EMG) may demonstrate reduced amplitude of compound muscle action potential (CAMP) and may show electrical silence based on the degree of muscle weakness 75, 103.
- Between attacks, EMG techniques can be used to demonstrate the change in excitability of muscle fibers due to channelopathy, called the “exercise test.” In the long exercise test, an attack of focal muscle weakness is induced by vigorously exercising a single muscle for 2-5 minutes, and the change in postexercise compound muscle action potential (CMAP) in muscle fibers is measured by the EMG. The reduction of 40% or more in compound muscle action potential (CMAP) is considered abnormal and typical for periodic paralysis. The study showed no false-positive results when the reduction is more than 40% or more, and this change was present in greater than 70% of patients 21, 104, 105. The abduction range of the little finger measured postexercise, can be a possible alternative parameter to compound muscle action potential (CMAP) in a long exercise test for diagnosis of hypokalemic periodic paralysis between attacks of muscle weakness 106.
- Muscle biopsy, which may show abnormalities. Interattack muscle biopsy is usually not performed to confirm the diagnosis. It may show the presence of vacuolar changes or tubular aggregates, but are nonspecific findings to all periodic paralysis 75.
- Exercise test. In the exercise test, the patient vigorously exercises a single muscle for 2 to 5 minutes in an attempt to cause focal muscle weakness. Weakness is assessed by an electrophysiologic study called the compound muscle action potential (CMAP), which is done before and after exercise. A ≥ 40% decrease postexercise compound muscle action potential (CMAP) is abnormal and consistent with periodic paralysis.
- Genetic testing to identify heterozygous pathogenic variant in CACNA1S or SCN4A gene. However approximately 30% do not have a pathogenic variant identified in either of these known genes.
- Other tests may be ordered to rule out other causes. These include thyroid function test (TSH, T3, T4 level) to rule out hyperthyroidism, an electrocardiogram (ECG) to look for ECG changes consistent with hypokalemia, and an ECG may also show the feature of Andersen-Tawil syndrome, long QT interval 21.
- Provocative test. Administration of potassium or insulin and glucose can be used as a provocative test to diagnose hypokalemic periodic paralysis (HypoPP). However, provocative testing with potassium or glucose and insulin administration might be potentially dangerous as it can precipitate life-threatening arrhythmia or hypoglycemia. Thus they require intensive monitoring in a hospital setting and not necessary to establish the diagnosis 107. They have been largely replaced by the exercise test, which is relatively safer.
The diagnostic criteria for hypokalemic periodic paralysis (HypoPP) include the following 108:
- 2 or more episodes of muscle weakness with serum potassium less than 3.5 mmol/L or one relative had a similar attack.
- 3 or more of the following features should be present:
- Onset in the first or second decade, onset time longer than two hours,
- The presence of triggers (previous carbohydrate-rich meal, onset during rest after exercise, stress),
- Symptomatic relief with potassium intake,
- A family history of skeletal calcium or sodium channel mutation, and
- Positive long exercise tests.
- Other causes of hypokalemia (renal and adrenal disease, thyroid dysfunction, drug abuse) are excluded.
Neurological examination of the patient during attack shows generalized muscle weakness, usually proximal muscle involvement more than distal and when incomplete legs are more often involved than arms. Hyporeflexia (decreased deep tendon reflexes) or areflexia (absent deep tendon reflexes) is typical. Neurological examination findings are usually normal between attacks. Myotonia is uncommon, unlike in hyperkalemic periodic paralysis (HyperPP), where myotonic is a common finding 75, 86, 109.
The best diagnostic indicator is a history of typical episodes. If measured during an episode, serum potassium may be abnormal.
Previously, for diagnosis, an attempt was made to provoke episodes by giving dextrose and insulin (to cause the hypokalemic periodic paralysis) or potassium chloride (to cause the hyperkalemic periodic paralysis), but because these tests may cause respiratory paralysis or cardiac conduction abnormalities and are not needed to make the diagnosis, they have been replaced by a safer exercise test.
Diagnosis of the hyperkalemic periodic paralysis is based on clinical findings and/or the identification of a heterozygous pathogenic genetic variant in the alpha-subunit of the skeletal muscle sodium channel.
In a case series of 71 diagnosed patients of hypokalemic periodic paralysis, patients without genetic mutations, compared to patients with genetic mutations, were found to have disease presentation at old age, absence of diet as a precipitating factor, and muscle biopsy showed no vacuolar myopathy 86. Phenotypic variations were also noted in patients having mutations in this case series. Patients with sodium channel mutations had attacks of shorter durations, and vacuolar changes were more common on calcium channel mutation, while tubular aggregates were seen more in sodium channel mutations 86.
Hypokalemic periodic paralysis treatment
The primary goal of treatment is to alleviate the symptoms of acute attacks, prevention and management of immediate complications, and prevention of late complications and future attacks.
Table 2. Treatment principles for individuals with hypokalemic periodic paralysis
| Goal | Means | Practical Details |
|---|---|---|
| To avoid triggering or aggravating factors for paralytic attacks | Avoid: Strenuous effort; Prolonged immobility; Carbohydrate-rich diet; High sodium diet. | Monitor episodes of weakness noting time of day & specific triggers. Provide dietary review/counseling. |
| Treatment of paralytic attack: Shorten/prevent aggravation of the weakness episode. Normalize kalemia. | Provide potassium supplementation (oral, or IV if oral impossible or if potassium very low). Avoid glucose intake. | Do not use slow-release forms of potassium. Oral potassium: initially, 1 mEq/kg; add 0.3 mEq/kg after 30 min if no improvement IV potassium: 0.3 mEq/kg/h |
| Preventive treatment for paralytic attacks | Daily potassium supplementation | Slow-release forms of potassium may be used. |
| Acetazolamide | ||
| Dichlorphenamide | ||
| Potassium-sparing diuretics | ||
| Preventive treatment for late-onset myopathy | Acetazolamide? | |
| Medical precautions | Avoid corticosteroids if possible. Use alpha- or beta adrenergic drugs w/caution, even in local anesthesia or ophthalmology. | |
| Other elements of management | Kinesiotherapy in case of long-lasting pelvic deficit Adaptive measures: (1) at school & especially for sports; (2) in work setting |
Treatment of paralytic attack
The goal is to normalize the serum potassium level by administering oral potassium chloride, which is believed to be more readily absorbed compared to other oral potassium solutions, alleviates the symptoms of muscle weakness 7. Oral potassium chloride is administered in incremental dose, starting initially with 0.5 to 1 mEq/kg (i.e., 60 to 120 mEq of potassium for a 60 kg individual) is reasonable 7. If patients do not respond to the initial dose, then 30% of the initial dose (i.e., 0.3 mEq/kg) is repeated every 30 min 6, 110, 84, 107. If the patient requires the addition of more than 100 mEq of oral potassium, then close monitoring of serum potassium is needed, and the total dose of oral potassium should not be more than 200 mEq within the 24 hours of starting of the treatment 107. The starting dose of oral potassium may vary according to the severity of hypokalemia. Patients should be kept on ECG monitoring, and muscle strength should be examined periodically. Serum potassium level should be monitored for 24 hours after treatment as the posttreatment rise in serum potassium level can have an adverse effect on patients.
Intravenous (IV) potassium is not preferred initially and is reserved for cardiac arrhythmias due to hypokalemia or if the patient has swallowing difficulties or respiratory muscle paralysis 7. Intravenous (IV) potassium is preferentially administered with the mannitol, not with dextrose or saline as both carbohydrate and salt can itself trigger the muscle paralysis and thus may worsen the weakness 107, 111. IV potassium therapy requires inpatient, continuous ECG monitoring. 40 mEq/L in 5% of mannitol solution of IV potassium is infused at a rate not more than 20 mEq/hour, not exceeding 200 mEq in 24 hours 21.
Individuals having a milder form of attacks can also benefit from low-level exercises 21, 22.
Preventive treatment for paralytic attacks
Both pharmacological and nonpharmacological interventions can be used to prevent recurrent future attacks. Nonpharmacological interventions include educating patients about triggering factors and lifestyle modifications to avoid these factors. Pharmacologic interventions include medications like chronic potassium supplementation, carbonic anhydrase inhibitors, potassium-sparing diuretics that are used when lifestyle modifications become insufficient in reducing attack rates 7. The favored approach is to add one of the diuretics with the chronic potassium supplementation. The initial choice of diuretics is carbonic anhydrase inhibitor acetazolamide 7.
Carbonic anhydrase inhibitors seem to be potent in decreasing future attacks of muscle weakness, though the mechanism of carbonic anhydrase inhibitors in hypokalemic periodic paralysis is still unclear 7. Carbonic anhydrase inhibitors promote urine potassium loss and non-anion gap metabolic acidosis, which reduce the patient’s susceptibility to muscle paralysis. It is also suggested that carbonic anhydrase inhibitors increase the opening of the calcium-activated potassium channels. Furthermore, carbonic anhydrase inhibitors also reduce intracellular sodium accumulation, thus reducing the cellular toxicity and prevent muscle degeneration, which may be effective in the treatment of permanent weakness 21. 250 mg twice daily dose of acetazolamide has been effective in lessening the frequency of attacks 22, 75.
The genetic variation in response to acetazolamide treatment had been reported. Patients with SCN4A mutations show less response compared to patients with CACNA1S mutations 7. In a study of 74 identified cases of hypokalemic periodic paralysis, 56% (31/55) of patients with CACNA1S mutations, and only 16% (3/19) of patients with SCN4A mutations showed a response to acetazolamide therapy.[12] Patients with SCN4A mutations had reported the exacerbation of the hypokalemic periodic paralysis with acetazolamide therapy.[9][12] Overall, almost half of the hypokalemic periodic paralysis patients respond to treatment with acetazolamide 21.
FDA recently approved dichlorphenamide for the treatment of hypokalemic periodic paralysis. 50 mg twice daily dose of dichlorphenamide has been more effective than a placebo in reducing the occurrence, severity, and duration of future attacks 21, 107, 112, 113. Dichlorphenamide can be used as the first choice or as a substitute for patients who do not respond or are refractory to acetazolamide 107. Some patients also benefitted from the addition of a potassium-sparing diuretic, either spironolactone (100 mg daily) or triamterene (150 mg daily), to carbonic anhydrase inhibitors or when used as monotherapy 75. Electrolytes need to be monitored regularly in patients who are on diuretics therapy.
While no definitive therapy for the late-onset myopathy has been proven to date, but it is believed that reducing the attacks of muscle weakness helps to mitigate the resulting myopathy 114, 94.
A study also reported the improvement in severity and frequency of attacks with topiramate therapy in 11 years old twins with hypokalemic periodic paralysis, thus necessitates further study regarding the efficacy of topiramate in hypokalemic periodic paralysis 115.
Myopathy treatment
No curative treatment is known for fixed myopathy in hypokalemic periodic paralysis. The effects of muscle weakness are managed as in other disorders with similar manifestations.
- Physiotherapy may help to maintain strength and motor abilities, especially after 40 years of age, when long-lasting muscle weakness is more often seen.
- The physiotherapist must be aware of the following peculiarity of periodic paralysis: that sustained effort results in exacerbation of weakness. Therefore, self-managed exercise should be preferred to superimposed physiotherapy 116.
Pre- or postoperative paralysis
Because of the risk for paralysis preceding or following anesthesia, precautions should be taken during administration of anesthesia to individuals with hypokalemic periodic paralysis. Individuals with hypokalemic periodic paralysis with CACNA1S mutation are susceptible to malignant hyperthermia, as the CACNA1S gene is allelic to the gene that increases susceptibility to malignant hyperthermia 107. Individuals with hypokalemic periodic paralysis with CACNA1S mutation should be managed with a non-triggering anesthetic technique – although general anesthesia using volatile anesthetics and succinylcholine has been reported as safe in a small number of individuals with hypokalemic periodic paralysis.
Surgeons and anesthesiologists must be aware of this circumstance while using the inhalational anesthetics and muscle relaxants like succinylcholine during surgery and be ready to deal with it. Furthermore, the cold environment and the use of saline and dextrose during surgery, and stress due to surgery itself can act as a trigger and result in muscle weakness 107. Potassium monitoring is important in such patients during the peri-surgical period.
General guidelines for perioperative care include the following 6:
- Strict control of serum potassium concentration
- Avoidance of large glucose and salt loads
- Low-carbohydrate diet
- Maintenance of body temperature and acid-base balance
- Careful use of neuromuscular blocking agents and no depolarizing muscle relaxants
Pregnancy and hypokalemic periodic paralysis
During pregnancy, potassium management during the attacks should not differ from the pre-pregnancy state 7. However, drugs like acetazolamide and dichlorphenamide are FDA pregnancy category C so, their use during pregnancy is quite challenging, and risks and benefits of drug use should be weighed in them 7. Some pregnant women prefer not to take these medicines during pregnancy 107.
Hypokalemic periodic paralysis prognosis
The prognosis of hypokalemic periodic paralysis (HypoPP) varies among individuals. The attacks of muscle weakness responds well to oral potassium administration 7. Treatment may prevent, and even reverse, progressive muscle weakness. Although muscle strength starts out normal between attacks, repeated attacks may eventually cause worsening and permanent muscle weakness between attacks, which can cause significant sickness, increase hospital admissions, and therefore can affect the patient’s social and professional life 7. Slowly progressive, permanent weakness in the legs often develops after age 50. Men and women are equally affected. The deaths related due to muscle attacks are rare, but several deaths due to aspiration pneumonia have been reported 92.
- Hypokalemic periodic paralysis. https://rarediseases.info.nih.gov/diseases/6729/hypokalemic-periodic-paralysis[↩][↩][↩]
- Venkatesan PE, Gnanashanmugham G, Balakrishnan R, Ramadoss K. Hypokalemic periodic paralysis: Three rare secondary causes. Med J DY Patil Univ 2015;8:760-2. https://journals.lww.com/mjdy/pages/default.aspx/text.asp?2015/8/6/760/169918[↩]
- Velarde-Mejía Y, Gamboa-Cárdenas R, Ugarte-Gil M, et al.: Hypokalemic Paralysis: A Hidden Card of Several Autoimmune Diseases. Clin Med Insights Arthritis Musculoskelet Disord. 2017;10: 1179544117722763. 10.1177/1179544117722763[↩]
- Dormohammadi Toosi T, Naderi N, Movassaghi S, Seradj MH, Khalvat A, Shahbazi F. Secondary Sjogren’s syndrome presenting with hypokalemic periodic paralysis. Oxf Med Case Reports. 2014 Nov 3;2014(8):135-7. doi: 10.1093/omcr/omu052[↩]
- Hypokalemic periodic paralysis. https://www.uptodate.com/contents/hypokalemic-periodic-paralysis[↩][↩]
- Weber F, Lehmann-Horn F. Hypokalemic Periodic Paralysis. 2002 Apr 30 [Updated 2018 Jul 26]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1338[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Phuyal P, Nagalli S. Hypokalemic Periodic Paralysis. [Updated 2023 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559178[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Familial Periodic Paralyses. https://www.ninds.nih.gov/health-information/disorders/familial-periodic-paralyses[↩]
- Velarde-Mejía Y, Gamboa-Cárdenas R, Ugarte-Gil M, Asurza CP. Hypokalemic Paralysis: A Hidden Card of Several Autoimmune Diseases. Clin Med Insights Arthritis Musculoskelet Disord. 2017 Aug 3;10:1179544117722763. doi: 10.1177/1179544117722763[↩][↩]
- Koul PA, Wahid A. Distal renal tubular acidosis and hypokalemic paralysis in a patient with hypothyroidism. Saudi J Kidney Dis Transpl. 2011 Sep;22(5):1014-6.[↩][↩]
- Basak RC, Sharkawi KM, Rahman MM, Swar MM. Distal renal tubular acidosis, hypokalemic paralysis, nephrocalcinosis, primary hypothyroidism, growth retardation, osteomalacia and osteoporosis leading to pathological fracture: a case report. Oman Med J. 2011 Jul;26(4):271-4. doi: 10.5001/omj.2011.66[↩]
- Meregildo-Rodríguez ED, Failoc-Rojas VE. Case Report: Recurrent hypokalemic periodic paralysis associated with distal renal tubular acidosis (type 1) and hypothyroidism secondary to Hashimoto’s thyroiditis. F1000Res. 2018 Jul 30;7:1154. doi: 10.12688/f1000research.15662.3[↩]
- Meola G, Hanna MG, Fontaine B. Diagnosis and new treatment in muscle channelopathies. J Neurol Neurosurg Psychiatry 2009;80:360–5. doi:10.1136/jnnp.2008.164046[↩][↩][↩][↩]
- Jurkat-Rott K, Lehmann-Horn F. Muscle channelopathies and critical points in functional and genetic studies. J Clin Invest 2005;115:2000–9. doi:10.1172/JCI25525[↩][↩][↩][↩]
- Jurkat-Rott K, Weber MA, Fauler M, et al. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci USA 2009;106:4036–41. doi:10.1073/pnas.0811277106[↩]
- Goyal G, Kant R, Vajpayee A: Hypokalemic respiratory paralysis due to distal renal tubular acidosis as the presenting manifestation of Sjögren’s syndrome. Journal of Acute Medicine. 2014;4(1):49–52. 10.1016/j.jacme.2014.01.002[↩][↩]
- Sinha U, Sengupta N, Sinharay K, et al.: Recurrent hypokalemic paralysis: An atypical presentation of hypothyroidism. Indian J Endocrinol Metab. 2013;17(1):174–176. 10.4103/2230-8210.107880[↩]
- Singh ThK, Sangme N, Singh LN, et al.: Hypokalaemic Periodic Paralysis Associated with Hypothyroidism. Indian J Phys Med Rehabil. 2012;23(2):79–81.[↩]
- Gündüz E, Zengin Y, Dursun R, et al.: Hypokalemic Periodic Paralysis Due To Distal Renal Tubular Acidosis. Eur J Gen Med. 2015;12(2):164–166. 10.15197/sabad.1.12.33[↩]
- Finn BC, Young P, Bruetman JE, Forrester M, Lombi F, Campolo Girard V, Pereyra H, Trimarchi H. Hipokalemia, acidosis tubular distal y tiroiditis de Hashimoto [Hypokalemia, distal renal tubular acidosis, and Hashimoto’s thyroiditis]. Nefrologia. 2008;28(5):569-70. https://www.revistanefrologia.com/en-hypokalemia-distal-renal-tubular-acidosis-articulo-X2013251408004065[↩]
- Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, Sansone VA, Shieh PB, Tawil RN, Trivedi J, Cannon SC, Griggs RC. Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve. 2018 Apr;57(4):522-530. doi: 10.1002/mus.26009[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Fontaine B. Periodic paralysis. Adv Genet. 2008;63:3-23. doi: 10.1016/S0065-2660(08)01001-8[↩][↩][↩][↩][↩][↩][↩][↩]
- Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SC, Burge JA, Portaro S, Davis MB, Haworth A, Hanna MG. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. 2013 Apr 16;80(16):1472-5. doi: 10.1212/WNL.0b013e31828cf8d0[↩]
- Hypokalemic periodic paralysis. https://medlineplus.gov/genetics/condition/hypokalemic-periodic-paralysis[↩]
- Sur M, Mohiuddin SS. Potassium. [Updated 2022 Dec 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539791[↩][↩]
- Potassium. https://pubchem.ncbi.nlm.nih.gov/compound/potassium[↩]
- Unwin, R., Luft, F. & Shirley, D. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 7, 75–84 (2011). https://doi.org/10.1038/nrneph.2010.175[↩][↩][↩][↩]
- Stone MS, Martyn L, Weaver CM. Potassium Intake, Bioavailability, Hypertension, and Glucose Control. Nutrients. 2016 Jul 22;8(7):444. doi: 10.3390/nu8070444[↩][↩][↩][↩][↩]
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Food and Nutrition Board; Committee to Review the Dietary Reference Intakes for Sodium and Potassium; Oria M, Harrison M, Stallings VA, editors. Dietary Reference Intakes for Sodium and Potassium. Washington (DC): National Academies Press (US); 2019 Mar 5. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538102[↩][↩][↩]
- Brooks G. Potassium additive algorithm for use in continuous renal replacement therapy. Nurs Crit Care. 2006 Nov-Dec;11(6):273-80. doi: 10.1111/j.1478-5153.2006.00185.x[↩]
- Preuss HG, Clouatre DL. Sodium, chloride, and potassium. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:475-92.[↩][↩][↩][↩]
- Hinderling PH. The Pharmacokinetics of Potassium in Humans Is Unusual. J Clin Pharmacol. 2016 Oct;56(10):1212-20. doi: 10.1002/jcph.713[↩][↩]
- Palmer B.F. Regulation of potassium homeostasis. Clin. J. Am. Soc. Nephrol. 2015;10:1050–1060. doi: 10.2215/CJN.08580813[↩]
- Unwin R.J., Luft F.C., Shirley D.G. Pathophysiology and management of hypokalemia: A clinical perspective. Nat. Rev. Nephrol. 2011;7:75–84. doi: 10.1038/nrneph.2010.175[↩]
- Palmer BF, Clegg DJ. Physiology and Pathophysiology of Potassium Homeostasis: Core Curriculum 2019. Am J Kidney Dis. 2019 Nov;74(5):682-695. doi: 10.1053/j.ajkd.2019.03.427. Erratum in: Am J Kidney Dis. 2022 Nov;80(5):690.[↩][↩][↩][↩][↩]
- World Health Organisation (WHO) Guideline: Potassium Intake for Adults and Children. WHO; Geneva, Switzerland: 2012.[↩]
- Fulgoni VL 3rd, Keast DR, Bailey RL, Dwyer J. Foods, fortificants, and supplements: Where do Americans get their nutrients? J Nutr. 2011 Oct;141(10):1847-54. doi: 10.3945/jn.111.142257[↩][↩]
- O’Neil C.E., Keast D.R., Fulgoni V.L., Nicklas T.A. Food sources of energy and nutrients among adults in the us: Nhanes 2003–2006. Nutrients. 2012;4:2097–2120. doi: 10.3390/nu4122097[↩]
- DeSalvo K.B., Olson R., Casavale K.O. Dietary guidelines for americans. JAMA. 2016;315:457–458. doi: 10.1001/jama.2015.18396[↩]
- Weaver C.M. Potassium and health. Adv. Nutr. (Bethesda Md.) 2013;4:S368–S377. doi: 10.3945/an.112.003533[↩]
- He F.J., MacGregor G.A. Beneficial effects of potassium on human health. Physiol. Plant. 2008;133:725–735. doi: 10.1111/j.1399-3054.2007.01033.x[↩]
- Bailey JL, Sands JM, Franch HA. Water, electrolytes, and acid-based metabolism. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:102-32.[↩]
- Shils M.E., Shike M. Modern Nutrition in Health and Disease. Lippincott Williams & Wilkins; Baltimore, MD, USA: 2006.[↩]
- Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ. 2016 Dec;40(4):480-490. doi: 10.1152/advan.00121.2016[↩][↩][↩][↩][↩]
- Hené RJ, Koomans HA, Boer P, Dorhout Mees EJ. Adaptation to chronic potassium loading in normal man. Miner Electrolyte Metab. 1986;12(3):165-72.[↩]
- Rabelink TJ, Koomans HA, Hené RJ, Dorhout Mees EJ. Early and late adjustment to potassium loading in humans. Kidney Int. 1990 Nov;38(5):942-7. doi: 10.1038/ki.1990.295[↩]
- Potassium. https://ods.od.nih.gov/factsheets/Potassium-HealthProfessional[↩]
- Patrick J. Assessment of body potassium stores. Kidney Int 1977;11:476-90. https://www.kidney-international.org/article/S0085-2538(15)31757-9/pdf[↩][↩]
- Chatterjee R, Yeh HC, Edelman D, Brancati F. Potassium and risk of Type 2 diabetes. Expert Rev Endocrinol Metab. 2011 Sep;6(5):665-672. doi: 10.1586/eem.11.60[↩]
- Viera AJ, Wouk N. Potassium Disorders: Hypokalemia and Hyperkalemia. Am Fam Physician. 2015 Sep 15;92(6):487-95. https://www.aafp.org/pubs/afp/issues/2015/0915/p487.html[↩][↩]
- WHO. Diet, nutrition and the prevention of chronic disease. Report of a Joint WHO/FAO Expert Consultation. Geneva, World Health Organization (WHO), 2003. http://whqlibdoc.who.int/trs/WHO_TRS_916.pdf [↩]
- D’Elia L, Barba G, Cappuccio FP et al. Potassium intake, stroke, and cardiovascular disease a meta-analysis of prospective studies. Journal of the American College of Cardiology, 2011, 57(10):1210–1219. http://www.ncbi.nlm.nih.gov/pubmed/21371638[↩]
- Whelton PK, He J, Cutler JA et al. Effects of oral potassium on blood pressure. Meta-analysis of randomized controlled clinical trials. Journal of the American Medical Association, 1997, 277(20):1624–1632. http://www.ncbi.nlm.nih.gov/pubmed/9168293[↩]
- Geleijnse JM, Kok FJ, Grobbee DE. Blood pressure response to changes in sodium and potassium intake: a metaregression analysis of randomised trials. Journal of Human Hypertension, 2003, 17(7):471–480. http://www.ncbi.nlm.nih.gov/pubmed/12821954[↩]
- Dietary Guidelines Advisory Committee. The report of the Dietary Guidelines Advisory Committee on Dietary Guidelines for Americans. Washington, D.C., Department of Health and Human Services and Department of Agriculture, 2005. http://www.health.gov/dietaryguidelines/dga2005/report/default.htm[↩]
- World Health Organization. Guideline: Potassium intake for adults and children. http://apps.who.int/iris/bitstream/10665/77986/1/9789241504829_eng.pdf[↩][↩]
- American Heart Association. A Primer on Potassium. https://sodiumbreakup.heart.org/a_primer_on_potassium?utm_source=SRI&utm_medium=HeartOrg&utm_term=Website&utm_content=SodiumAndSalt&utm_campaign=SodiumBreakup[↩][↩][↩]
- Potassium. https://lpi.oregonstate.edu/mic/minerals/potassium[↩][↩][↩]
- Clausen T. Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: functional significance. J Gen Physiol. 2013 Oct;142(4):327-45. doi: 10.1085/jgp.201310980[↩]
- Larsen BR, Stoica A, MacAulay N. Managing Brain Extracellular K(+) during Neuronal Activity: The Physiological Role of the Na(+)/K(+)-ATPase Subunit Isoforms. Front Physiol. 2016 Apr 22;7:141. doi: 10.3389/fphys.2016.00141[↩]
- Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, Bridge JH, Chen-Izu Y, Clancy CE, Edwards A, Goldhaber J, Kaplan J, Lingrel JB, Pavlovic D, Philipson K, Sipido KR, Xie ZJ. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol. 2015 Mar 15;593(6):1361-82. doi: 10.1113/jphysiol.2014.282319[↩]
- DrugBank. Potassium. http://www.drugbank.ca/drugs/DB01345[↩]
- Brown IJ, Tzoulaki I, Candeias V, Elliott P. Salt intakes around the world: implications for public health. Int J Epidemiol2009;38:791-813.[↩]
- Kawasaki T, Itoh K, Kawasaki M. Reduction in blood pressure with a sodium-reduced, potassium- and magnesium-enriched mineral salt in subjects with mild essential hypertension. Hypertens Res1998;21:235-43. https://www.ncbi.nlm.nih.gov/pubmed/9877516?access_num=9877516&link_type=MED&dopt=Abstract[↩]
- Chang HY, Hu YW, Yue CS, Wen YW, Yeh WT, Hsu LS, et al. Effect of potassium-enriched salt on cardiovascular mortality and medical expenses of elderly men. Am J Clin Nutr2006;83:1289-96. http://ajcn.nutrition.org/content/83/6/1289?ijkey=52dc92efe22c9a7ff1bd4e7366b4fcecb7f45e7e&keytype2=tf_ipsecsha[↩]
- Sheng H-W. Sodium, chloride and potassium. In: Stipanuk M, ed. Biochemical and Physiological Aspects of Human Nutrition. Philadelphia: W.B. Saunders Company; 2000:686-710.[↩]
- Weiss JN, Qu Z, Shivkumar K. Electrophysiology of Hypokalemia and Hyperkalemia. Circ Arrhythm Electrophysiol. 2017 Mar;10(3):e004667. doi: 10.1161/CIRCEP.116.004667[↩]
- Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 8: advanced challenges in resuscitation: section 1: life-threatening electrolyte abnormalities. The American Heart Association in collaboration with the International Liaison Committee on Resuscitation. Circulation. 2000 Aug 22;102(8 Suppl):I217-22.[↩]
- Rosano GMC, Tamargo J, Kjeldsen KP, Lainscak M, Agewall S, Anker SD, Ceconi C, Coats AJS, Drexel H, Filippatos G, Kaski JC, Lund L, Niessner A, Ponikowski P, Savarese G, Schmidt TA, Seferovic P, Wassmann S, Walther T, Lewis BS. Expert consensus document on the management of hyperkalaemia in patients with cardiovascular disease treated with renin angiotensin aldosterone system inhibitors: coordinated by the Working Group on Cardiovascular Pharmacotherapy of the European Society of Cardiology. Eur Heart J Cardiovasc Pharmacother. 2018 Jul 1;4(3):180-188. doi: 10.1093/ehjcvp/pvy015[↩]
- Goyal A, Spertus JA, Gosch K, Venkitachalam L, Jones PG, Van den Berghe G, Kosiborod M. Serum potassium levels and mortality in acute myocardial infarction. JAMA. 2012 Jan 11;307(2):157-64. doi: 10.1001/jama.2011.1967[↩]
- Smyth A, Dunkler D, Gao P, Teo KK, Yusuf S, O’Donnell MJ, Mann JF, Clase CM; ONTARGET and TRANSCEND investigators. The relationship between estimated sodium and potassium excretion and subsequent renal outcomes. Kidney Int. 2014 Dec;86(6):1205-12. doi: 10.1038/ki.2014.214[↩]
- Potassium Blood Test. https://medlineplus.gov/lab-tests/potassium-blood-test[↩][↩]
- Bailey JL, Sands JM, Franch HA. Water, electrolytes, and acid — Base Metabolism In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease: Lippincott Williams & Wilkins; 2014:102-132.[↩]
- Food and Nutrition Board, Institute of Medicine. Potassium. Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate. Washington, D.C.: National Academies Press; 2005:186-268. https://nap.nationalacademies.org/read/10925/chapter/7[↩]
- Venance SL, Cannon SC, Fialho D, Fontaine B, Hanna MG, Ptacek LJ, Tristani-Firouzi M, Tawil R, Griggs RC; CINCH investigators. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006 Jan;129(Pt 1):8-17. doi: 10.1093/brain/awh639[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Groome JR, Moreau A, Delemotte L. Gating Pore Currents in Sodium Channels. Handb Exp Pharmacol. 2018;246:371-399. doi: 10.1007/164_2017_54[↩][↩][↩][↩][↩][↩]
- Jurkat-Rott K, Weber MA, Fauler M, Guo XH, Holzherr BD, Paczulla A, Nordsborg N, Joechle W, Lehmann-Horn F. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):4036-41. doi: 10.1073/pnas.0811277106[↩][↩][↩][↩]
- Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, Meola G, Schorge S, Kullmann DM, Davis MB, Hanna MG. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. 2009 May 5;72(18):1544-7. doi: 10.1212/01.wnl.0000342387.65477.46[↩][↩][↩]
- Francis DG, Rybalchenko V, Struyk A, Cannon SC. Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurology. 2011 May 10;76(19):1635-41. doi: 10.1212/WNL.0b013e318219fb57[↩][↩][↩][↩]
- CACNA1S gene. https://medlineplus.gov/genetics/gene/cacna1s[↩]
- Jurkat-Rott K, Holzherr B, Fauler M, Lehmann-Horn F. Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch. 2010 Jul;460(2):239-48. doi: 10.1007/s00424-010-0814-4[↩][↩][↩]
- Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, Lerche H, Nicole S, Vale-Santos J, Chauveau D, Fontaine B, Lehmann-Horn F. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci U S A. 2000 Aug 15;97(17):9549-54. doi: 10.1073/pnas.97.17.9549[↩][↩]
- SCN4A gene. https://medlineplus.gov/genetics/gene/scn4a[↩]
- Jurkat-Rott K, Lehmann-Horn F. State of the art in hereditary muscle channelopathies. Acta Myol. 2010 Oct;29(2):343-50. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3040592[↩][↩]
- Ke Q, Luo B, Qi M, Du Y, Wu W. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis. Muscle Nerve. 2013 Jan;47(1):41-5. doi: 10.1002/mus.23460[↩]
- Miller TM, Dias da Silva MR, Miller HA, Kwiecinski H, Mendell JR, Tawil R, McManis P, Griggs RC, Angelini C, Servidei S, Petajan J, Dalakas MC, Ranum LP, Fu YH, Ptácek LJ. Correlating phenotype and genotype in the periodic paralyses. Neurology. 2004 Nov 9;63(9):1647-55. doi: 10.1212/01.wnl.0000143383.91137.00[↩][↩][↩][↩]
- Jiang D, Gamal El-Din TM, Ing C, Lu P, Pomès R, Zheng N, Catterall WA. Structural basis for gating pore current in periodic paralysis. Nature. 2018 May;557(7706):590-594. doi: 10.1038/s41586-018-0120-4[↩]
- Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007 Mar 1;446(7131):76-8. doi: 10.1038/nature05598[↩]
- Lapie P, Goudet C, Nargeot J, Fontaine B, Lory P. Electrophysiological properties of the hypokalaemic periodic paralysis mutation (R528H) of the skeletal muscle alpha 1s subunit as expressed in mouse L cells. FEBS Lett. 1996 Mar 18;382(3):244-8. doi: 10.1016/0014-5793(96)00173-1[↩]
- Lehmann-Horn F, Jurkat-Rott K, Rüdel R; Ulm Muscle Centre. Diagnostics and therapy of muscle channelopathies–Guidelines of the Ulm Muscle Centre. Acta Myol. 2008 Dec;27(3):98-113. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2858942[↩]
- Sansone VA, Burge J, McDermott MP, Smith PC, Herr B, Tawil R, Pandya S, Kissel J, Ciafaloni E, Shieh P, Ralph JW, Amato A, Cannon SC, Trivedi J, Barohn R, Crum B, Mitsumoto H, Pestronk A, Meola G, Conwit R, Hanna MG, Griggs RC; Muscle Study Group. Randomized, placebo-controlled trials of dichlorphenamide in periodic paralysis. Neurology. 2016 Apr 12;86(15):1408-1416. doi: 10.1212/WNL.0000000000002416[↩][↩]
- Finsterer J. Primary periodic paralyses. Acta Neurol Scand. 2008 Mar;117(3):145-58. doi: 10.1111/j.1600-0404.2007.00963.x[↩][↩][↩][↩][↩][↩]
- Ober KP. Thyrotoxic periodic paralysis in the United States. Report of 7 cases and review of the literature. Medicine (Baltimore). 1992 May;71(3):109-20. doi: 10.1097/00005792-199205000-00001[↩]
- Links TP, Zwarts MJ, Wilmink JT, Molenaar WM, Oosterhuis HJ. Permanent muscle weakness in familial hypokalaemic periodic paralysis. Clinical, radiological and pathological aspects. Brain. 1990 Dec;113 ( Pt 6):1873-89. doi: 10.1093/brain/113.6.1873[↩][↩][↩][↩]
- Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B, Willer JC, Tabti N, Fontaine B. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. 2004 Nov;56(5):650-61. doi: 10.1002/ana.20241[↩]
- Andersen-Tawil syndrome. https://medlineplus.gov/genetics/condition/andersen-tawil-syndrome[↩][↩][↩][↩]
- Siddamreddy S, Dandu VH. Thyrotoxic Periodic Paralysis. [Updated 2022 Jul 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560670[↩]
- Lin C, Lin CS, Lee DJ, Lee CC, Chen SJ, Tsai SH, Kuo FC, Chau T, Lin SH. Artificial Intelligence-Assisted Electrocardiography for Early Diagnosis of Thyrotoxic Periodic Paralysis. J Endocr Soc. 2021 Jun 29;5(9):bvab120. doi: 10.1210/jendso/bvab120[↩]
- Verma V, Kumar Y, Kotwal N, Upreti V, Hari Kumar KVS, Singh Y, Menon AS. Thyrotoxic periodic paralysis: A retrospective, observational study from India. Indian J Med Res. 2020 Jan;151(1):42-46. doi: 10.4103/ijmr.IJMR_335_18[↩]
- Zhao SX, Liu W, Liang J, Gao GQ, et al. China Consortium for the Genetics of Autoimmune Thyroid Disease. Assessment of Molecular Subtypes in Thyrotoxic Periodic Paralysis and Graves Disease Among Chinese Han Adults: A Population-Based Genome-Wide Association Study. JAMA Netw Open. 2019 May 3;2(5):e193348. doi: 10.1001/jamanetworkopen.2019.3348[↩]
- Heydorn A, Bertelsen B, Nolsöe RLM, Eiken P, Kristensen PL. Thyrotoxic periodic paralysis in a Caucasian man without identifiable genetic predisposition: a case report. Thyroid Res. 2023 May 1;16(1):10. doi: 10.1186/s13044-023-00152-w[↩]
- Luo S, Xu M, Sun J, Qiao K, Song J, Cai S, Zhu W, Zhou L, Xi J, Lu J, Ni X, Dou T, Zhao C. Identification of gene mutations in patients with primary periodic paralysis using targeted next-generation sequencing. BMC Neurol. 2019 May 8;19(1):92. doi: 10.1186/s12883-019-1322-6[↩]
- Sharma CM, Nath K, Parekh J. Reversible electrophysiological abnormalities in hypokalemic paralysis: Case report of two cases. Ann Indian Acad Neurol. 2014 Jan;17(1):100-2. doi: 10.4103/0972-2327.128566[↩]
- McManis PG, Lambert EH, Daube JR. The exercise test in periodic paralysis. Muscle Nerve. 1986 Oct;9(8):704-10. doi: 10.1002/mus.880090805[↩]
- Tengan CH, Antunes AC, Gabbai AA, Manzano GM. The exercise test as a monitor of disease status in hypokalaemic periodic paralysis. J Neurol Neurosurg Psychiatry. 2004 Mar;75(3):497-9. doi: 10.1136/jnnp.2003.013870[↩]
- Zhang L, Niu J, Li Y, Guan Y, Cui L, Liu M. Abduction range: A potential parameter for the long exercise test in hypokalemic periodic paralysis during inter-attack periods. Muscle Nerve. 2020 Jan;61(1):104-107. doi: 10.1002/mus.26721[↩]
- Levitt JO. Practical aspects in the management of hypokalemic periodic paralysis. J Transl Med. 2008 Apr 21;6:18. doi: 10.1186/1479-5876-6-18. Erratum in: J Transl Med. 2014;12:198. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2374768[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Li J. Hypokalemic Periodic Paralysis Secondary to Medullary Sponge Kidney Complicated With Renal Tubular Acidosis. Cureus. 2022 Oct 10;14(10):e30160. doi: 10.7759/cureus.30160[↩]
- Sugiura Y, Makita N, Li L, Noble PJ, Kimura J, Kumagai Y, Soeda T, Yamamoto T. Cold induces shifts of voltage dependence in mutant SCN4A, causing hypokalemic periodic paralysis. Neurology. 2003 Oct 14;61(7):914-8. doi: 10.1212/01.wnl.0000086820.54065.a0[↩]
- Greig SL. Dichlorphenamide: A Review in Primary Periodic Paralyses. Drugs. 2016 Mar;76(4):501-7. doi: 10.1007/s40265-016-0559-2[↩]
- Griggs RC, Resnick J, Engel WK. Intravenous treatment of hypokalemic periodic paralysis. Arch Neurol. 1983 Sep;40(9):539-40. doi: 10.1001/archneur.1983.04050080039005[↩]
- Sansone V, Meola G, Links TP, Panzeri M, Rose MR. Treatment for periodic paralysis. Cochrane Database Syst Rev. 2008 Jan 23;(1):CD005045. doi: 10.1002/14651858.CD005045.pub2[↩]
- Tawil, R., McDermott, M.P., Brown, R., Jr, Shapiro, B.C., Ptacek, L.J., McManis, P.G., Dalakas, M.C., Spector, S.A., Mendell, J.R., Hahn, A.F., Griggs, R.C. and (2000), Randomized trials of dichlorphenamide in the periodic paralyses. Ann Neurol., 47: 46-53. https://doi.org/10.1002/1531-8249(200001)47:1<46::AID-ANA9>3.0.CO;2-H[↩]
- Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial Hypokalemic Periodic Paralysis. Muscle Nerve. 1983 Mar-Apr;6(3):182-6. doi: 10.1002/mus.880060303[↩]
- Fiore DM, Strober JB. Treatment of hypokalemic periodic paralysis with topiramate. Muscle Nerve. 2011 Jan;43(1):127-9. doi: 10.1002/mus.21854[↩]
- Cavel-Greant D, Lehmann-Horn F, Jurkat-Rott K. The impact of permanent muscle weakness on quality of life in periodic paralysis: a survey of 66 patients. Acta Myol. 2012 Oct;31(2):126-33. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3476862[↩]




{kind=link}