
Contents
What is hypophosphatemia
Hypophosphatemia is a low level of phosphorus in the blood. Hypophosphatemia or phosphorus deficiencies may be seen with malnutrition, malabsorption, acid-base imbalances, increased blood calcium, and with disorders that affect kidney function. Someone with a mild to moderate hypophosphatemia often does not have any symptoms. Hypophosphatemia is typically asymptomatic and is present in up to 5% of patients. It is much more prevalent in alcoholism, diabetic ketoacidosis, or sepsis with a frequency of up to 80%. The morbidity of hypophosphatemia is highly dependent on its cause and severity. With a severe hypophosphatemia, symptoms may include muscle weakness and confusion. In the setting of severe hypophosphatemia, the phosphate contained in bone mineral provides a source of phosphorus for your metabolic needs 1.
Phosphorus is a mineral that combines with other substances to form organic and inorganic phosphate (Pi) compounds. The terms phosphorus and phosphate are often used interchangeably when talking about testing, but it is the amount of inorganic phosphate in the blood that is measured with a serum phosphorus/phosphate test.
Phosphates plays an important role in growth, development, bone formation, maintain the body’s acid-base balance, and cellular metabolism. Phosphates are vital for energy production, muscle and nerve function, and bone growth.
You get the phosphorus you need through the foods you eat. A typical, nutritious diet provides 1000 to 2000 mg of phosphate daily. Of this 600 mg to 1200 mg is absorbed via the intestines. Phosphates are found in many foods and are readily absorbed by the digestive tract. Most of the body’s phosphates combine with calcium to help form bones and teeth. Smaller amounts are found in muscle and nerve tissue. The rest is found within cells throughout the body, where they are mainly used to store energy.
In general, phosphate is upregulated through absorption in the intestines and decreased through renal excretion. Excesses are stored in the bones which act as a buffer to maintain a relatively stable total body content. Normal serum levels of phosphate should be 4 to 7 mg/dL in children and 3 to 4.5 mg/dL in adults 2.
Normally, only about 1% of total body phosphates are present in the blood. A wide variety of foods, such as beans, peas and nuts, cereals, dairy products, eggs, beef, chicken, and fish, contain significant amounts of phosphorus. The body maintains phosphorus/phosphate levels in the blood by regulating how much it absorbs from the intestines and how much it excretes via the kidneys. Phosphate levels are also affected by the interaction of parathyroid hormone (PTH), calcium, and vitamin D.
Inorganic phosphorus (Pi) exists primarily as the critical structural ion, phosphate (PO4), which serves as a constituent of hydroxyapatite, the mineral basis of the vertebrate skeleton, and at the molecular level, providing the molecular backbone of DNA 1. Phosphates chemical properties allow its use as a biological energy store as adenosine triphosphate (ATP). Additionally, phosphorus influences a variety of enzymatic reactions (e.g. glycolysis) and protein functions (e.g. the oxygen-carrying capacity of hemoglobin by regulation of 2,3-diphosphoglycerate synthesis). Finally, phosphorus is an important signaling moiety, as phosphorylation and dephosphorylation of protein structures serves as an activation signal. Indeed, phosphorus is one of the most abundant components of all tissues and disturbances in its homeostasis can affect almost any organ system. Most phosphorus within the body is in bone (600-700 g), while the remainder is largely distributed in soft tissue (100-200 g). The plasma contains 11-12 mg/dL of total phosphorus (in both organic and inorganic states) in adults. Inorganic phosphorus (Pi) primarily exists as phosphate (PO4), and is the commonly measured fraction, found in plasma at concentrations averaging 3-4 mg/dl in older children and adults. Plasma inorganic phosphorus (Pi) concentrations values in children are higher, not infrequently as high as 8 mg/dl in small infants, gradually declining throughout the first year of life, and further in later childhood to adult values. The organic phosphorus component is primarily found in phospholipids. Although this fraction is not routinely assessed clinically, it comprises approximately two-thirds of the total plasma phosphorus 3. Thus, the term “plasma phosphorus” generally is used when referring to plasma inorganic phosphorus (Pi) concentrations and because plasma inorganic phosphorus is nearly all in the form of the PO4 ion, the terms phosphorus and phosphate are often interchangeably used in the clinical chemistry laboratory. It should be noted that this terminology can be confusing when using mass units (i.e., mg/dl) as the weight of the phosphorus content of the phosphate is reported, yet “serum phosphate” is often used in the clinic setting. When using molar units, the concentration of the phosphate and of the phosphorus are equivalent, and less confusion may arise.
Elaborate mechanisms have evolved to maintain phosphate balance, reflecting the critical role that phosphorus plays in cell and organism physiology. Adaptive changes are manifest by a variety of measurable responses, as modified by metabolic phosphorus need and exogenous phosphorus supply. Such regulation maintains the plasma and extracellular fluid phosphorus within a relatively narrow range and depends primarily upon gastrointestinal absorption and renal excretion as adjustable mechanisms to effect homeostasis. Although investigators have recognized a variety of hormones and transporter proteins which influence these various processes, in concert with associated changes in other metabolic pathways, the sensory system, the messenger and the mechanisms underlying discriminant regulation of phosphorus balance remain incompletely understood.
While long-term changes in phosphorus balance depend on these variables, short-term changes in phosphorus concentrations can occur due to redistribution between the extracellular fluid and either bone or cell constituents. Such redistribution results secondary to various mechanisms including: elevated levels of insulin and/or glucose; increased concentrations of circulating catecholamines; respiratory alkalosis; enhanced cell production or anabolism; and rapid bone remineralization.
Because of the diverse presentation of hypophosphatemia, the condition is best managed by a multidisciplinary team that consists of an internist, endocrinologist, intensivist, general surgeon and a nurse practitioner. The effects of hypophosphatemia are broad and impact nearly every system. Symptoms of this deficiency become apparent below 0.32 mmol/L. In general, all symptomatic patients need treatment with phosphate; asymptomatic patients may be observed and monitored. The outlook for patients depends on the primary condition causing the hypophosphatemia. If the cause is benign, then treatment outcomes are excellent 4.
Hypophosphatemia causes
Hypophosphatemia is most commonly induced by one of three causes: (1) Inadequate phosphate intake, (2) increased phosphate excretion, and (3) shift from extracellular phosphate into the intracellular space 2.
The following may cause hypophosphatemia:
- Alcoholism
- Antacids
- Certain medicines, including insulin, acetazolamide, foscarnet, imatinib, pentamidine, and sorafenib
- Fanconi syndrome
- Starvation
- Too little vitamin D
- Overactive parathyroid gland (hyperparathyroidism)
- Burns
- Familial hypophosphatemia
Hypophosphatemia secondary to inadequate intake of phosphate occurs in the setting of prolonged poor dietary sources of phosphate, intestinal malabsorption, and intestinal binding by exogenous agents. As stated above, almost all diet types contain a surplus of phosphate sufficient to maintain needs. Additionally, renal adaptations typically can compensate for the short-term deficiency. Intestinal malabsorption may be due to a variety of causes. Notably, chronic diarrhea has been shown to increase phosphate losses through the intestines. Certain medications are known to bind with phosphate, decreasing the available free ion to be absorbed via the small intestines into circulation. Aluminum and magnesium antacids are notoriously associated with a net loss of phosphate from the body by binding to both ingested and excreted phosphate. This chemical reaction creates aluminum- or magnesium-bound phosphate salts which are nonabsorbable by the body.
Increased excretion of phosphate occurs primarily in the renal system. The proximal renal tubule reabsorbs up to 70% of filtered phosphate normally, and the distal tubule reabsorbs up to 15% of filtered phosphate. Resorption is regulated by serum phosphate concentration with mild phosphate depletion which directly triggers increased phosphate reabsorption via the sodium-phosphate cotransporters of the proximal tubule and increases expression and formation for new sodium-phosphate cotransporters. Conversely, parathyroid hormone functions to increase phosphate excretion by inhibiting the activity of sodium-phosphate cotransporters. Additionally, fibroblast growth factor 23, fibroblast growth factor 7, extracellular matrix phosphoglycoprotein, and secreted frizzled-related protein-4 decrease phosphate reabsorption by sodium-phosphate cotransporters. Therefore, any increase in parathyroid hormone has the potential for inducing hypophosphatemia.
Primary or secondary hyperparathyroidism are the most likely causes where primary hyperparathyroidism is due to hypercalcemia and secondary hyperparathyroidism is induced by any of the causes that lead to vitamin D deficiency. Primary renal phosphate-wasting syndromes also exist where there is a direct failure of the renal system without coexisting systemic failure. These include a wide variety of genetic malformations, leading to faulty sodium-phosphate cotransporters. One of the larger examples of this includes X-linked hypophosphatemic rickets where a mutation in the PHEX gene leads to increased levels of fibroblast growth factor 23 (FGF 23) and directly decreases resorption of phosphate in the proximal renal-collecting tubules. Mutations in the sodium-phosphate cotransporter gene SLC34A3 causes type 2c sodium-phosphate cotransporter failure. The SLC34A1 gene is responsible for encoding the type 2a sodium-phosphate cotransporter and has been associated with mutations. The sodium-hydrogen exchanger regulatory factor 1 is responsible for creating the sodium gradient which powers most ion reabsorption. Mutations here lead to pan-ionic losses. Fanconi syndrome is another classic cause of renal losses. It is a generalized impairment in proximal tubular function leading to urinary wasting most often due to illnesses such as multiple myeloma where immunoglobulin light chains induce renal tubular damage and Wilson disease with copper accumulation in children. Anything that increases urine production also will lead to increased phosphate loss, including glucosuria, alcohol, lithium, and diuretics such as acetazolamide and thiazides, rapid fluid volume expansion from oral or intravenous fluids. In patients with renal failure, hypophosphatemia can be seen as a result of dialysis therapy removing phosphate in bulk.
Intracellular shifting of phosphate stores may occur in a variety of clinical scenarios. Refeeding syndrome occurs when a patient who has been starved of nutrition suddenly is replenished with carbohydrates, proteins, and lipids. Insulin and glucose assist in driving phosphate intracellularly. The net body stores of phosphate necessary to perform basic metabolism such as glycolysis are depleted. The body begins to process the newfound foods to produce ATP for energy. Cells uptake all available free phosphate, leading to profound hypophosphatemia. Hungry bone syndrome occurs after correction of hyperparathyroidism where osteopenic bones begin to reabsorb and store phosphate and calcium. This leads to increased bone demand for these ions and hypophosphatemia. Acute respiratory alkalosis induces hypophosphatemia via changes in cellular pH. Increased pH stimulates phosphofructokinase, thus stimulating glycolysis to produce ATP thus consuming phosphate from the cellular space. Serum phosphate is shifted intracellularly to meet this demand. While typically mild, extreme hyperventilation with subsequent PCO2 changes to less than 20 mmHg can lower phosphate concentrations to below 0.32 mmol/L. This is thought to be the most common cause of marked hypophosphatemia in hospitalized patients.
Familial hypophosphatemia
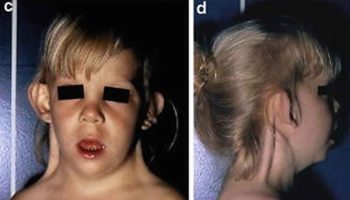
X-linked hypophosphatemic rickets/osteomalacia was initially recognized as a form of “vitamin D resistant” and later, as disorder of renal phosphate wasting 1. The disorder is inherited in X-linked dominant fashion and is manifest biochemically by a low renal threshold maximum for renal tubular phosphate reabsorption, consequent hypophosphatemia, and low, or inappropriate circulating levels of 1,25(OH)2D 1. Known biochemical characteristics of X-linked hypophosphatemic rickets/osteomalacia and other hypophosphatemic disorders are shown in Table 2. Characteristic features of the disease include growth retardation, osteomalacia and rickets in growing children. The clinical expression of the disease is widely variable, ranging from a mild abnormality, the apparent isolated occurrence of hypophosphatemia, to severe bone disease. Most would agree that a wide spectrum of phenotypic severity occurs in both males (with a mutated gene on their only X chromosome) and females (who are heterozygous for the defective X-linked gene), although anecdotal experience suggests that females with some mutations may express less severe disease 5. Evidence of disease may be detected at or shortly after birth or may not become apparent until age 12 months or older 6. The most common clinically evident manifestations of X-linked hypophosphatemic rickets/osteomalacia are short stature and limb deformities. Growth abnormalities and limb deformities are both more evident in the lower extremities, since they represent the fastest growing body segment before puberty.
The majority of affected children exhibit clinical evidence of rickets, varying from enlargement of the wrists and/or knees to severe malalignment defects such as bowing or knock-knee deformities. Such defects may result in waddling gait and leg length abnormalities 7. X-ray examination reveals expanded areas of non-mineralized cartilage in epiphyseal regions and lateral curvature of the femora and/or tibia. The severe secondary hyperparathyroidism that occurs in vitamin D deficiency is not present, however mildly elevated circulating levels of parathyroid hormone (PTH) occur in many patients naive to therapy. Other non-specific but typical findings include elevated serum alkaline phosphatase activity and osteocalcin levels. Serum alkaline phosphatase activity, although usually elevated to 2-3 times the upper limit of normal in childhood, is generally less than the levels observed in overt vitamin D- and calcium-deficiency rickets.
Additional signs of the disease may include delayed dentition and dental abscesses 8, which are thought to arise from the limited mineralization of the dentine compartment of the tooth. An enlarged pulp chamber is evident on dental radiographs. Strikingly absent are features commonly observed in vitamin D deficiency rickets, such as muscle weakness, tetany and convulsions.
Adults with X-linked hypophosphatemia (XLH) may be asymptomatic or present with severe bone pain. On clinical examination they often display evidence of post-rachitic deformities, such as bowed legs or short stature. However, overt biochemical changes such as elevated serum alkaline phosphatase activity or other biomarkers of bone turnover are often not evident. On occasion, some adult patients may present with “active” osteomalacia, characterized radiographically by pseudofractures, coarsened trabeculation, rarified areas and/or non-union fractures, and may have elevated serum alkaline phosphatase activity. Symptoms at presentation may reflect the end-result of chronic changes, and may not correlate with apparent current activity of the disease. In spite of marked variability in the clinical presentation of the disease, bone biopsy in affected children and adults nearly always reveals low turnover osteomalacia without osteopenia. Histomorphometry of biopsy samples invariably demonstrates a reduced rate of formation, diffuse patchy hypomineralization, a decrease in mineralizing surfaces and characteristic areas of hypomineralization of the periosteocytic lacunae 9.
Osteophytes, enthesopathy 10 and craniosynostosis are not uncommon. A great deal of the morbidity of X-linked hypophosphatemia (XLH) in adults arises from the high incidence of arthritis, calcified entheses, and osteophytes. Enthesopathy generally is first detectable radiographically by late in the second decade, or early in the third decade. Older subjects have more sites of involvement, and generally increasing involvement with age; the frequency of involvement is greater in males. With progressive enthesopathy and bony overgrowth, excruciating pain may occur, particularly with fusion of the sacroiliac joint(s) and spinal stenosis 11. These manifestations do not appear to be affected for the better or worse with respect to exposure to currently available therapies 12. It is peculiar that X-linked hypophosphatemia (XLH) represents a deficiency of mineralization at many skeletal sites, and pathologic ectopic mineralization elsewhere. This paradoxical situation raises the possibility that aberrant humoral factors, in addition to the ambient hypophosphatemia, may play a role in the discordant mineralization abnormalities observed.
Table 1. Familial hypophosphatemia
| Hypophosphatemia- FGF23 Mediated | ||
|---|---|---|
| X-linked hypophosphatemia (XLH) | PHEX | LOF* |
| Autosomal dominant hypophosphatemic rickets (ADHR) | FGF23 | GOF* |
| Autosomal recessive hypophosphatemic rickets 1 (ARHR1) | DMP1 | LOF |
| Autosomal recessive hypophosphatemic rickets 2 (ARHR2) | ENPP1 | LOF |
| McCune-Albright syndrome/fibrous dysplasia | GNAS1 | GOF |
| Osteoglophonic dysplasia | FGFR1 | GOF |
| Jansen metaphyseal chondrodysplasia | PTH1R | GOF |
| Klotho overexpression | 9;13 translocation | GOF |
| Epidermal nevus syndrome (ENS) | HRAS, NRAS | |
| Raine syndrome related hypophosphatemia | FAM20C | LOF |
| Opsismodysplasia | ||
| Tumor-induced osteomalacia (TIO) | ||
| Hypophosphatemia-Non FGF-mediated | ||
| Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) (NaPi-IIc deficiency) | SLC34A3 | LOF |
| Dent’s disease | CLCN5 | LOF |
| NaPi-IIa deficient Fanconi syndrome | SLC34A1 | LOF |
Abbreviations: FGF23 = fibroblast growth factor 23; GOF = gain of function; LOF = loss of function
[Source 1 ]Table 2. Biochemical Characteristics of Hypophosphatemic Disorders
| CALCIUM METABOLISM | PHOSPHATE METABOLISM | VITAMIN D METABOLISM | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Serum Ca | Urine Ca | Serum PTH | GI Ca Absorption | Serum Pi | TmP/ GFR | GI Pi Absorption | Serum 25(OH) | Serum 1,25(OH)2 | |
| X-linked hypophosphatemia | N | ↓ | N, | ↓ | ↓ | ↓ | ↓ | N | (↓) |
| Autosomal dominant hypophosphatemic rickets | N | ↓ | N | ↓ | ↓ | ↓ | ↓ | N | (↓) |
| Autosomal recessive hypophosphatemic rickets | N | ↓ | N | ? | ↓ | ↓ | ? | N | (↓) |
| Tumor-induced osteomalacia | N | ↓ | N | ↓ | ↓ | ↓ | ↓ | N | ↓ |
| X-linked recessive hypophosphatemia (Dent’s Disease) | N | ↑ | N, ↓ | ↑ | ↓ | ↓ | ↑ | N | ↑ |
| Hereditary Hypophosphatemic Rickets with Hypercalciuria | N | ↑ | N, ↓ | ↑ | ↓ | ↓ | ↑ | N | (↑) |
Footnote: Hereditary hypophosphatemic rickets with hypercalciuria.
Abbreviations: N = normal; , decreased = ↓; increased = ↑; decreased relative to the serum phosphorus concentration = (↓); ? = unknown.
[Source 1 ]Familial hypophosphatemia diagnosis
Clinical Biochemistry
The primary biochemical abnormality of X-linked hypophosphatemia (XLH) is hypophosphatemia due to increased urinary phosphate excretion. Moreover, mild gastrointestinal phosphate malabsorption is present in the majority of patients, which may contribute to the evolution of the hypophosphatemia (Table 2).
In contrast, the serum calcium concentration in affected subjects is normal despite gastrointestinal malabsorption of calcium. However, as a consequence of this defect, urinary calcium is often decreased. As noted above, circulating PTH levels may be normal to modestly elevated in naïve patients, but treatment with phosphate salts often aggravate this tendency such that persistent secondary hyperparathyroidism may occur. As noted above, serum alkaline phosphatase activity is usually elevated in children, although to lesser levels than seen in nutritional forms of rickets. Because of variability in adulthood, this measure is not a reliable marker of disease involvement in the older age group. Prior to the initiation of therapy, serum 25-OHD levels are normal, and serum 1,25(OH)2D levels are in the low normal range 13. The paradoxical occurrence of hypophosphatemia and normal serum calcitriol levels in affected subjects is consistent with aberrant regulation of both synthesis and clearance of this metabolite (due to increased 25-OHD-24-hydroxylase activity) 14. Circulating levels of FGF23 are generally elevated in individuals with X-linked hypophosphatemia (XLH). FGF23 levels do not appear to differ between normal adults and older children, nor between children and adults affected with X-linked hypophosphatemia (XLH). Caution should be used in using this measure as a strict diagnostic criterion for the diagnosis of X-linked hypophosphatemia (XLH), as some subjects have been shown to have normal FGF23 levels, and commercially available assays (which recognize “intact” species or both intact and C-terminal species) do not always provide concordant results.
Genetics
With the recognition that hypophosphatemia is the definitive marker for X-linked hypophosphatemia (XLH), Winters et al 15 and Burnett et al 16 discovered that this disease is transmitted as an X-linked dominant disorder. Analysis of data from 13 multigenerational pedigrees identified PHEX (for phosphate regulating gene with homologies to endopeptidases located on the X chromosome) as the gene mutated in X-linked hypophosphatemia (XLH) 17. PHEX is located on chromosome Xp22.1, and encodes a 749-amino acid protein with three putative domains: 1) a small aminoterminal intracellular tail; 2) a single, short transmembrane domain; and 3) a large carboxyterminal extracellular domain, containing ten conserved cysteine residues and a HEXXH pentapeptide motif, which characterizes many zinc metalloproteases. Further studies have revealed that PHEX is homologous to the M13 family of membrane-bound metalloproteases, or neutral endopeptidases. M13 family members, including neutral endopeptidase 24.11 (NEP), endothelin-converting enzymes 1 and 2 (ECE-1 and ECE-2), the Kell blood group antigen (KELL), neprilysin-like peptide (NL1), and endothelin converting enzyme-like 1 (ECEL1), degrade or activate a variety of peptide hormones. In addition, like other neutral endopeptidases, immunofluorescent studies have revealed a cell-surface location for PHEX in an orientation consistent with a type II integral membrane glycoprotein 18. It has been demonstrated that certain missense mutations in PHEX that substitute a highly conserved cysteine residue will interfere with normal trafficking of the molecule to the plasma membrane 19. Thus, it appears that one mechanism associated with the pathophysiology of X-linked hypophosphatemia (XLH) is to prevent PHEX from locating to the cell membrane.
Phex is predominantly expressed in bones (in osteoblasts/osteocytes) and teeth (in odontoblasts/ameloblasts) 20; mRNA, protein or both have also been found in lung, brain, muscle, gonads, skin and parathyroid glands. Subcellular locations appear to be the plasma membrane, endoplasmic reticulum and Golgi organelle. Immunohistochemistry studies suggest that Phex is most abundant on the cell surface of the osteocyte. In sum, the ontogeny of Phex expression suggests a possible role in mineralization in vivo.
The work of several groups has documented PHEX mutations in >160 patients 21. Mutations are scattered throughout the 749-amino acid extracellular domain, encoded by exons 2-22, and are diverse, consisting of deletions, insertions and duplications, as well as splice site, nonsense and missense mutations.
The location of Phex expression in bone cells have led to the hypothesis that diminished PHEX/Phex expression in bone initiates the cascade of events responsible for the pathogenesis of X-linked hypophosphatemia (XLH). In order to confirm this possibility, several investigators have used targeted over-expression of Phex in attempts to normalize osteoblast mineralization, in vitro, and rescue the Hyp phenotype in vivo 22. Results from these studies have not resulted in a complete skeletal rescue, raising questions as to the role of early developmental expression of PHEX, or at least the success of expression when targeted with osteocalcin or type I collagen promoters. Nevertheless, partial rescue of the mineralization defect in Hyp mice occurs, suggesting that local effects of the PHEX mutation may play some role in the mineralization process, but cannot completely restore the skeleton to normality. Of note, this partial rescue occurs in concert with a reduction in FGF23 levels, although not lowered to a truly normal range 22.
In sum, although a physiologic substrate for PHEX has not been identified, the consequence of loss-of-function of PHEX is an elevation in the circulating FGF23 level. Failure of targeted osteoblastic PHEX overexpression to completely rescue Hyp mice may reflect that critical sites (or developmental timing) for PHEX expression are not effectively generated with these models to effectively rescue the skeletal phenotype; this effect may be dependent upon the resultant capacity in these transgenic models of normal PHEX to reduce FGF23 production in mutant cells. ASARM peptides, fragments of SIBLING (small integrin binding ligand N-glycated) proteins, have been shown to inhibit mineralization and potentially play a role in modulation of renal P transport; these peptides have also been shown to be degraded by PHEX 23.
Familial hypophosphatemia treatment
Decades ago, physicians employed pharmacological doses of vitamin D as the cornerstone for treatment of X-linked hypophosphatemia (XLH). However, long-term observations indicate that this therapy fails to cure the disease and poses the serious problem of recurrent vitamin D intoxication and renal damage. Indeed, such treatment results only in incomplete healing of the rachitic abnormality, while hypophosphatemia and impaired growth remain. Similar unresponsiveness is typical with use of 25(OH)D.
With the recognition that phosphate depletion is an important contributor to impaired skeletal mineralization, physicians began to devise treatment strategies that employed oral phosphate supplementation to compensate for the renal phosphate wasting and thereby increasing the available Pi to the mineralizing skeleton. This strategy was somewhat successful in terms of improving skeletal lesions, although it was soon realized that pharmacologic amounts of vitamin D were necessary in combination with phosphate supplements to counter the exacerbation of hyperparathyroidism observed in this setting. Such combination therapy was found to be more effective than either administering vitamin D or phosphate alone. With the recognition that circulating 1,25(OH)2D levels are not appropriately regulated in X-linked hypophosphatemia (XLH), the use of this metabolite in combination with phosphate was subsequently used to treat the disease 24. The current treatment strategy directly addresses the combined calcitriol and phosphorus deficiency characteristic of the disorder. Although this combination therapy has become the conventional therapy for X-linked hypophosphatemia (XLH), complete healing of the skeletal lesions is usually not the case, and late complications of the disease are persistent and often debilitating.
In children the goal of therapy is to improve growth velocity, normalize any lower extremity defects, and heal the attendant bone disease. Generally, the treatment regimen includes a period of titration to achieve a maximum dose of 1,25(OH)2D3 (Rocaltrol® or calcitriol), 20-50 ng/kg/day in two divided doses, and phosphorus, (20-50 mg/kg/day, to a maximum of 1-2 gms/day) in 3-5 divided doses.
Use of 1,25(OH)2D3/phosphorus combination therapy involves a significant risk of toxicity. Hypercalcemia, hypercalciuria, renal calcinosis, and hyperparathyroidism can be sequelae of unmonitored therapy. Detrimental effects on renal function were particularly common prior to the frequent monitoring now generally employed with this therapy. Indeed, hypercalcemia, severe nephrocalcinosis, and/or diminished creatinine clearance necessitates appropriate dose adjustment, and in some cases discontinuation of therapy. Throughout the treatment course careful attention to renal function, as well as serum and urine calcium is extremely important. Nevertheless, in spite of these varied complications of therapy, treatment of X-linked hypophosphatemia (XLH) often proceeds with limited interruptions. Moreover, the improved outcome of this therapeutic intervention, compared to that achieved by previous regimens, justifies the aggressive approach that constitutes this current therapy.
While such combined therapy often improves growth velocity, refractoriness to the growth-promoting effects of treatment can be encountered in children who present with markedly short stature prior to 4 years of age. For that reason, the use of recombinant growth hormone as additional treatment has been suggested 25, however this approach has not been universally recommended in view of the lack of definitive benefits in controlled studies, and a risk of resultant worsening of the disproportional stature 26, although others have not identified significant concerns in this regard 27.
Indications for combined therapy in adults with X-linked hypophosphatemia (XLH) are less clear. The occurrence of intractable bone pain and refractory non-union fractures often respond to treatment with calcitriol and phosphorus 28. However, data remain unclear regarding the effects of treatment on fracture incidence (which may not be increased in untreated patients), There does not appear to be any effect of this therapy on enthesopathy, however superior dentition appears to occur in the setting of higher medication exposure through adulthood as well as the entire life span 29. Muscle weakness and general well-being may occur with therapy in some adults. In sum, the decision to treat affected adults must be individualized. In general, it is beneficial to offer adults with significant symptomatology a trial of this therapy, but only if routine biochemical monitoring can be performed. A detailed strategy for the management of children and adults with X-linked hypophosphatemia (XLH) is available 30.
Given the limitations of currently advised treatment for X-linked hypophosphatemia (XLH), the quest for new and better therapies for X-linked hypophosphatemia (XLH) continues. Correction of serum P levels and improved bony growth in Hyp mice treated with a neutralizing antibody to FGF23 have been shown 31, and subsequent short-term trials of an antibody to the human FGF23 protein, burosumab (KRN23) have been conducted 32. In a single dose study in adults with X-linked hypophosphatemia (XLH), both intravenous and subcutaneous dosing resulted in increased TmP/GFR, serum P and serum 1,25(OH)2D levels 32. A trial of monthly dosing in affected adults has been shown to be effective and well-tolerated 33. Most recently, subcutaneous administration of burosumab resulted in improvement of radiographic features of rickets in concert with correction of abnormal biochemical indices in children with X-linked hypophosphatemia (XLH) previously treated with conventional phosphate and active vitamin D therapy 34. Steady and stable correction of hypophosphatemia was attained with administration of the antibody every 2 weeks. These findings, together with a favorable safety profile hold promise for improving outcomes observed with current approaches to treating this disorder.
Hypophosphatemia signs and symptoms
Signs and symptoms of hypophosphatemia may include:
- Bone pain
- Confusion
- Muscle weakness
Most patients with hypophosphatemia are asymptomatic, and it is an incidental finding 2. Those with mild hypophosphatemia may complain of generalized mild to moderate weakness. The history of presenting illness will rarely indicate possible hypophosphatemia. For this reason, a clinician should have suspicion for phosphate abnormalities whenever cause is present that is associated with hypophosphatemia. Conditions to consider possible hypophosphatemia include poor nutritional status, symptoms or history of intestinal malabsorption, history of antacid use, frequent or recurrent bone pain, fractures, history of or suspicion for multiple myeloma, parenteral nutrition supplementation, medication use including chronic glucocorticoids, cisplatin, or pamidronate, current treatment for diabetic ketoacidosis, and any hospitalization requiring an Intensive care unit setting.
Mild hypophosphatemia will not be clinically apparent. Severe hypophosphatemia may have the clinical presence of altered mental status, neurological instability including seizures and focal neurologic findings such as numbness or reflexive weakness, a cardiac manifestation of possible heart failure, muscle pain, and muscular weakness.
Hypophosphatemia complications
The effects of hypophosphatemia are broad and impact nearly every system. Symptoms of this deficiency become apparent below 0.32 mmol/L. Effects primarily are due to intracellular depletion; however, chronic effects can be seen in the bone structures. Prolonged hypophosphatemia leads to osteopenia, osteoporosis, rickets, or osteomalacia due to decreased bone mineralization. The central nervous system may manifest with metabolic encephalopathy as a result of ATP depletion and may include altered mental state, irritability, paresthesias, numbness, seizures, or coma. Cardiac function is impacted by ATP depletion. In addition to possible systolic heart failure, the myocytes become less stable, and arrhythmias are possible. The decreased diaphragmatic function impacts pulmonary function with subsequent hypoventilation. Ventilator dependent patients have been shown to have longer hospital courses and worse outcomes when hypophosphatemia is present. Gastrointestinal dysfunction occurs as a result of ATP deficiency also with dysphagia or ileus possible. Generalized muscular weakness can occur. Rhabdomyolysis may occur resulting in renal injury and increased creatinine phosphokinases; however, this is typically only seen in acute or chronic hypophosphatemia such as in acutely ill persons with alcoholism. The hematology systems are rarely impacted, but depletion of ATP may result in increased erythrocyte rigidity, predisposing to hemolysis, reduced phagocytosis and granulocyte chemotaxis by white blood cells, and thrombocytopenia 35.
Hypophosphatemia diagnosis
Your health care provider will examine you.
The following tests may be done:
- Kidney function tests
- Vitamin D blood test
Exam and testing may show:
- Anemia due to too many red blood cells being destroyed (hemolytic anemia)
- Heart muscle damage (cardiomyopathy)
Hypophosphatemia is diagnosed with a simple serum measurement. Etiology is typically evident from the history. However, if unknown it is essential to determine renal phosphate excretion. Renal phosphate excretion can be measured either from a 24-hour urine collection or by calculation of the fractional excretion of filtered phosphate (FEPO4) from a random urine specimen. Excretion of filtered phosphate (EPO4) is calculated as follows where U is urine values and P is plasma values of phosphate (PO4) and creatinine (Cr):
- FEPO4 = (UPO4 x PCr x 100) / (PPO4 x UCr)
A 24-hour urine phosphate excretion less than 100 mg or fractional excretion of filtered phosphate (FEPO4) less than 5% shows decreased phosphate excretion, indicating hypophosphatemia is from a redistribution within the body or decreased intestinal absorption. A 24-hour urine phosphate excretion greater than 100 mg or fractional excretion of filtered phosphate (FEPO4) greater than 5% indicates renal phosphate wasting. Hypophosphatemia in this scenario is likely due to by hyperparathyroidism or vitamin D deficiency.
Hypophosphatemia treatment
Treatment depends on the cause of your hypophosphatemia. Phosphate can be given by mouth or through a vein (IV). How well you do depends on what has caused your hypophosphatemia.
While symptoms are not clinically present in mild cases, it is important to address and replace phosphate whenever abnormalities are noted. The appropriate regimen for replacement is determined depending on clinical symptoms. Mild, asymptomatic cases with a serum phosphate less than 0.64 mmol/L should receive oral phosphate therapy of 30 to 80 mmol of phosphate per day, depending on the severity of deficiency. Severe, symptomatic cases are appropriate for intravenous phosphate if the serum phosphate is less than 0.32 mmol/L and should be changed to oral replacement when the serum phosphate exceeds 0.48 mmol/L and there are no contraindicating reasons for oral replacement.
- Carpenter TO. Primary Disorders of Phosphate Metabolism. 2018 Oct 23. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279172[↩][↩][↩][↩][↩][↩]
- Sharma S, Castro D. Hypophosphatemia. [Updated 2019 Feb 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK493172[↩][↩][↩]
- Yanagawa N, Nakhoul F, Kurokawa K, Lee DBN. Physiology of phosphorus metabolism. In: Narins RG, ed. Clinical disorders of fluid and electrolyte metabolism. 5th ed. New York: McGraw Hill,1994; 307-371[↩]
- Al-Wassia H, Lyon AW, Rose SM, Sauve RS, Fenton TR. Hypophosphatemia is Prevalent among Preterm Infants Less than 1,500 Grams. Am J Perinatol. 2019 Jan 21[↩]
- Mumm S, Huskey M, Cajic A, Wollberg V, Zhang F, Madson KL, Wenkert D, McAlister WH, Gottesman GS, Whyte MP. J Bone Miner Res. 2015;30(1):137–43[↩]
- Harrison HE, Harrison HC, Lifshitz F, Johnson AD. Growth disturbance in hereditary hypophosphatemia. Am J Dis Child. 1996;112:290–297.[↩]
- Williams TF, Winters RW. Familial (hereditary) vitamin D-resistant rickets with hypophosphatemia. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, eds. The metabolic basis of inherited disease. 3rd ed. New York: McGraw-Hill. 1983; 1465-1485[↩]
- Shields ED, Scriver CR, Reade T, Fujiwara TM, Morgan K, Ciampi A, Schwartz S. X-linked hypophosphatemia: the mutant gene is expressed in teeth as well as in kidney. Am J Human Gen. 1990;46:434–442[↩]
- Marie PJ, Glorieux FH. Relation between hypomineralized periosteocytic lesions and bone mineralization in vitamin D-resistant rickets. Calcif Tissue Int. 1983;35:443–448.[↩]
- Polisson RP, Martinex S, Khoury M, Harrell RM, Lyles KW, Friedman N, Harrelson JM, Reisner E, Drezner MK. Calcificantion of entheses associated with X-linked hypophosphatemic osteomalacia. N Engl J Med. 1985;313:1–6.[↩]
- Pierce DS, Wallace WM, Herndon CH. Long term treatment of vitamin D-resistant rickets. J Bone Joint Surg Am. 1964;46:978–997[↩]
- Connor J, Olear EA, Insogna KL, Katz L, Baker SD, Kaur RD, Simpson CA, Sterpka J, Dubrow R, Zhang JH, Carpenter TO. Conventional therapy in adults with X-linked hypophosphatemia: effects on enthesopathy and dental disease. J Clin Endocrinol Metab. 2015;100:3625–3632.[↩]
- Drezner MK, Lyles KW, Haussler MR, Harrelson JM. Evaluation of a role for 1,25-dihydroxyvitamin D3 in the pathogenesis and treatment of X-linked hypophosphatemic rickets and osteomalacia. J Clin Invest. 1980;66:1020–1032.[↩]
- Roy S, Martel J, Ma S, Tenenhouse HS. Increased renal 25-hydroxyvitamin D3-24-hydroxylase messenger ribonucleic acid and immunoreactive protein in phosphate-deprived Hyp mice: a mechanism for accelerated 1,25-dihydroxyvitamin D3 catabolism in X-linked hypophosphatemic rickets. Endocrinology. 1994;134:1761–1767.[↩]
- Winters RW, Graham JB, Williams TF, McFalls VW, Burnett CH. A genetic study of familial hypophosphatemia and vitamin D-resistant rickets with a review of the literature. Medicine (Baltimore). 1958;37:97–142.[↩]
- Burnett CH, Dent CE, Harper C, Warland BJ. Vitamin D resistant rickets: analysis of 24 pedigrees and hereditary and sporadic cases. Am J Med. 1964;36:222–232[↩]
- Francis F, Henning S, Korn B, Reinhardt R, de Jong P, Poustka A, Lehrach H, Rowe PSN, Goulding JN, Summerfield T, Mountford R, Read AP, Popowska E, Pronicka E, Davies KE, O’Riordan JLH, Econs MJ, Nesbitt T, Drezner MK, Oudet C, Hanauer A, Strom TM, Meindl A, Lorenz B, Cagnoli M, Mohnike KL, Murken J, Meitinger T. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet. 1995;11:130–136.[↩]
- Lipman ML, Dibyendu P, Hugh PJ, Bennett JE, Henderson ES, Yingnian S, Goltzman D, Daraplis AC. Cloning of human Pex cDNA: expression subcellular localization and endopeptidase activity. J Biol Chem. 1998;273:13729–13737.[↩]
- Thompson DL, Roche PC, Drezner MK, Salisbury JL, Sabbagh Y, Tenenhouse HS, Grande JP, Poeschlia EM, Kumar R. Ontogeny of PHEX/PEX expression in the mouse embryo and studies on the subcellular localization of PHEX/PEX in osteoblasts. J Bone Miner Res. 2002;17:311–320[↩]
- Ruchon AF, Marcinkiewicz M, Siegfried G, Tenenhouse HS, DesGroseillers L, Crine P, Boileau G. Pex mRNA is localized in developing mouse osteoblasts and odontoblasts. J Histochem Cytochem. 1998;46:459–468[↩]
- Christie PT, Harding B, Nesbit MA, Whyte MP, Thakker RV. X-linked hypophosphatemia attributable to pseudoexons of the PHEX gene. J Clin Endocrinol Metab. 2001;86:3840–3844[↩]
- Boskey A, Frank A, Fujimoto Y, Spevak L, Verdelis K, Ellis B, Philbrick W, Carpenter T. The PHEX transgene corrects mineralization defects in 9-month-old hypophosphatemic mice. Calcif Tiss Int. 2009;84:126–137.[↩][↩]
- David V, Martin A, Hedge AM, Drezner MK, Rowe PS. Am J Physiol Renal Physiol. 2011;300(3):F783–91.[↩]
- Drezner MK, Lyles KW, Haussler MR, Harrelson JM. Evaluation of a role for 1,25-dihydroxyvitamin D3 in the pathogenesis and treatment of X-linked hypophosphatemic rickets and osteomalacia. J Clin Invest. 1980;66:1020–1032[↩]
- Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G. 2001 Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr. 1985;138:236–243[↩]
- Haffner D, Wuhl E, Blum WF, Schaefer F, Mehls O. Disproportionate growth following long-term growth hormone treatment in short children with X-linked hypophosphataemia. Eur J Pediatr. 1995;154:610–613.[↩]
- Zivicnjak M, Schnabel D, Staude H, et al. Three-Year Growth Hormone Treatment in Short Children with X-Linked Hypophosphatemic Rickets: Effects on Linear Growth and Body Disproportion. J Clin Endocrinol Metab. 2011;96(12):E2097–E2105[↩]
- Sullivan W, Carpenter TO, Glorieux F, Travers R, Insogna K. A prospective trial of phosphate and 1,25-dihydroxyvitamin D3 therapy on symptomatic adults with X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 1992;75:879–885[↩]
- Connor J, Olear EA, Insogna KL, Katz L, Baker SD, Kaur RD, Simpson CA, Sterpka J, Dubrow R, Zhang JH, Carpenter TO. Conventional therapy in adults with X-linked hypophosphatemia: effects on enthesopathy and dental disease. J Clin Endocrinol Metab. 2015;100:3625–3632[↩]
- Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381–1388[↩]
- Aono Y, Shimada T, Yamazaki Y, Hino R, Takeuchi Y, Fujita T, Fukumoto S, Nagano N, Wada M, Yamashita T. The neutralization of FGF-23 ameliorates hypophosphatemia and rickets in Hyp mice. J Bone Min Res. 2003;18:S16.[↩]
- Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, Kawakami T, Ito T, Zhang X, Humphrey J, Insogna KL, Peacock M. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124:1587–1597[↩][↩]
- Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab. 2015; 100, 2565-2573[↩]
- Carpenter TO, Imel E, Boot A, Hogler W, Linglart A, Padidela R, Van’t Hoff W, Whyte M, Zhou Y, Shrinar A, San Martin J, Portale A. Randomized, open-label, dose-finding, phase 2 Study of KRN23, a human monoclonal anti-FGF23 antibody, in children with X-linked hypophosphatemia (XLH). Endocrine Society Annual Meeting ENDO2016. 2016.[↩]
- Nasir M, Zaman BS, Kaleem A. What a Trainee Surgeon Should Know About Refeeding Syndrome: A Literature Review. Cureus. 2018 Mar 28;10(3):e2388.[↩]




{kind=link}