Contents
- What is hypoventilation
- Hypoventilation causes
- Hypoventilation symptoms
- Hypoventilation diagnosis
- Hypoventilation treatment
- Alveolar hypoventilation
- Congenital central hypoventilation syndrome
- Congenital central hypoventilation syndrome causes
- Congenital central hypoventilation syndrome inheritance pattern
- Congenital central hypoventilation syndrome symptoms
- Congenital central hypoventilation syndrome diagnosis
- Molecular Genetic Testing
- Will a person with a mutation in the PHOX2B gene definitely have or develop symptoms of congenital central hypoventilation syndrome?
- Congenital central hypoventilation syndrome treatment
- Obesity hypoventilation syndrome
- Obesity hypoventilation syndrome causes
- Obesity hypoventilation syndrome prevention
- Obesity hypoventilation syndrome symptoms
- Obesity hypoventilation syndrome possible complications
- Obesity hypoventilation syndrome diagnosis
- Obesity hypoventilation syndrome treatment
- Obesity hypoventilation syndrome prognosis
What is hypoventilation
Hypoventilation is breathing that is too shallow or too slow to meet the needs of the body. Hypoventilation is a medical term expressing an insufficient exchange of gases. This leads to increased levels of carbon dioxide in the blood, this causes a buildup of acid and too little oxygen in the blood and is especially dangerous when present in those with sleep apnea. A person will naturally adjust to the hypoventilation while awake, taking deeper and/or longer breaths as needed. During episodes of apnea though, where the body forcibly slows or shuts down breathing entirely in spurts, this can lead to dangerously high levels of carbon dioxide in the blood, and could lead to death in the most serious cases.
A person with hypoventilation might feel sleepy.
Hypoventilation causes
Hypoventilation can be caused by numerous factors. These may be caused by natural obstructions in the airway, or resistances to sufficient airflow. These are commonly brought on by other medical conditions that result in lower airway obstruction, including emphysema, cystic fibrosis and bronchitis. In other cases, the hypoventilation may be caused by another disease, or physical defect in the chest resulting in an abnormally shaped chest wall. People with muscular dystrophy run a higher risk of getting this disorder. People with diseases of the lung tissue are also at a high risk of developing this disorder. These are commonly caused by smoking, and smoking in general runs a high risk of development of a multitude of lower airway obstructing diseases. Any disease that inflames the lungs, and physically reduces the airflow passageway can also lead to this disorder. Sleep apnea may also cause this problem, though not through any physical means. The severely decreased intake of oxygen during some instances of sleep apnea can lead to the build up of carbon dioxide. Airway obstruction could also be the cause of environmental damage to the lungs, including exposure to, or breathing in of chemicals, pollution or other environmental factors.
Is some rare cases, the causes of hypoventilation may be unknown, as lungs and airways will appear normal under observation. This is termed as primary alveolar hypoventilation. A serious form of hypoventilation in infants called congenital central hypoventilation syndrome is thought to be the cause of some of the cases of sudden infant death syndrome or crib death, in which seemingly healthy infants unexpectedly pass away.
Hypoventilation symptoms
Common symptoms of hypoventilation will include headaches, heart problems, stomach problems and faintness. It may also lead to poor sleep due to common awakenings, and daytime sleepiness as a result.
Hypoventilation diagnosis
Successful diagnosis of this disorder will include taking an arterial blood gas test to measure the levels of carbon dioxide in your blood. You may also be asked to take an overnight sleep study to ensure that no sleep disorders are present that could lead to nocturnal hypoventilation, a dangerous condition that can be fatal if one or both of the offending conditions are not treated.
Reduction or narrowing of the airway can be caused by an over-consumption of alcohol, benzodiazepines and barbiturates, sleeping pills, sleep aids and medications, and the use of respiratory stimulants is the most common method for counteracting this reduction.
Hypoventilation treatment
When a sleeping disorder such as sleep apnea is present in any case involving hypoventilation, positive airway pressure (CPAP) or bilevel positive airway pressure (BiPAP) treatment will be often be used. These devices contain small masks which are placed over the mouth and nose during sleep, and continually feed a steady dose of oxygen into the lungs to ensure a proper level of oxygen and carbon dioxide. The level this will be set at will be determined after taking the above tests.
In extreme cases of severe hypoventilation, these treatments must be delivered day long, and not just while sleeping.
Alveolar hypoventilation
Primary alveolar hypoventilation is a rare disorder in which a person does not take enough breaths per minute. The lungs and airways are normal. Primary alveolar hypoventilation is also called Ondine’s curse, ventilatory failure, diminished hypoxic ventilator drive or diminished hypercapnic ventilator drive.
Alveolar hypoventilation causes
Normally, when the oxygen level in the blood is low or the carbon dioxide level is high, there is a signal from the brain to breathe more deeply or quickly. In people with primary alveolar hypoventilation, this change in breathing does not happen.
The cause of primary alveolar hypoventilation is unknown. Some people have a specific genetic defect.
Primary alveolar hypoventilation mainly affects men 20 to 50 years old. Primary alveolar hypoventilation may also occur in children.
Alveolar hypoventilation prevention
There is no known prevention. You should avoid using sleep medicines or other drugs that can cause drowsiness.
Alveolar hypoventilation symptoms
Symptoms are usually worse during sleep. Episodes of stopped breathing (apnea) often occur while sleeping. Often there is no shortness of breath during the day.
Alveolar hypoventilation symptoms include:
- Bluish coloration of the skin caused by lack of oxygen
- Daytime drowsiness
- Fatigue
- Morning headaches
- Swelling of the ankles
- Waking up from sleep unrested
- Waking up many times at night
People with this disease are very sensitive to even small doses of sedatives or narcotics. These drugs can make their breathing problem much worse.
Alveolar hypoventilation possible complications
Low blood oxygen level can cause high blood pressure in the lung blood vessels. This can lead to cor pulmonale (right-sided heart failure).
Alveolar hypoventilation diagnosis
Your health care provider will perform a physical exam and ask about symptoms.
Tests will be done to rule out other causes. For example, muscular dystrophy can make the rib muscles weak, and chronic obstructive pulmonary disease (COPD) damages the lung tissue itself. A small stroke can affect the breathing center in the brain.
Tests that may be done include:
- Measuring levels of oxygen and carbon dioxide in the blood (arterial blood gases)
- Chest x-ray or CT scan
- Hematocrit and hemoglobin blood tests tests to check oxygen carrying ability of red blood cells
- Lung function tests
- Overnight oxygen level measurements (oximetry)
- Blood gases
- Sleep study (polysomnography)
Alveolar hypoventilation treatment
Medicines that stimulate the respiratory system may be used but do not always work. Mechanical devices that assist breathing, particularly at night, may be helpful in some people. Oxygen therapy may help in a few people, but may worsen night symptoms in others.
Alveolar hypoventilation prognosis
Response to treatment varies.
Congenital central hypoventilation syndrome
Congenital central hypoventilation syndrome is a disorder of the autonomic nervous system that affects breathing. Congenital central hypoventilation syndrome causes a person to hypoventilate (especially during sleep), resulting in a shortage of oxygen and a buildup of carbon dioxide in the blood. Congenital central hypoventilation syndrome have two forms of presentation, a classic form that usually begin shortly after birth in newborns, and a milder later-onset presentaition in toddlers, children and adults.
Classic congenital central hypoventilation syndrome presents in newborns as:
- Apparent hypoventilation with monotonous respiratory rates and shallow breathing either during sleep only or while awake as well as asleep;
- Autonomic nervous system dysregulation; and
- In some individuals, altered development of neural crest-derived structures (i.e., Hirschsprung disease) and/or tumors of neural crest origin (neuroblastoma, ganglioneuroma, and ganglioneuroblastoma).
Affected infants hypoventilate upon falling asleep and exhibit a bluish appearance of the skin or lips (cyanosis). Other features may include difficulty regulating heart rate and blood pressure; decreased perception of pain; low body temperature; sporadic profuse sweating; Hirschsprung disease; constipation; learning difficulties; eye abnormalities; and a characteristic facial appearance (having a short, wide, somewhat flattened face). They can also have tumors of neural crest origin, such as neuroblastoma, ganglioneuroblastoma, and ganglioneuroma. The later-onset form is milder, and some cases may present as infants and children who die suddenly and unexpectedly (“SIDS” and “sudden unexplained death of childhood [SUDC]”) 1.
Congenital central hypoventilation syndrome is a relatively rare disorder. Approximately 1,000 individuals with this condition have been identified. Researchers believe that some cases of sudden infant death syndrome (SIDS) or sudden unexplained death in children may be caused by undiagnosed congenital central hypoventilation syndrome.
Life expectancy and the extent of any cognitive disabilities depend on the severity of the disorder, timing of the diagnosis, and the success of treatment.
Congenital central hypoventilation syndrome is caused by a variation (mutation) in the PHOX2B gene and is inherited in an autosomal dominant manner. However, over 90% of cases are due to a new mutation in the affected person and are not inherited from a parent. Diagnosis is made with the clinical symptoms and the genetic test showing the variation in the PHOX2B gene. Treatment typically includes mechanical ventilation or use of a diaphragm pacemaker 2. People who have been diagnosed as newborns and adequately ventilated throughout childhood may reach the age of 20 to 30 years, and can live independently. In the later-onset form, people who were diagnosed when they were 20 years or older have now reached the age of 30 to 55 years 1.
Congenital central hypoventilation syndrome causes
Mutations in the PHOX2B gene cause congenital central hypoventilation syndrome. The PHOX2B gene provides instructions for making a protein that acts early in development to help promote the formation of nerve cells (neurons) and regulate the process by which the neurons mature to carry out specific functions (differentiation). The protein is active in the neural crest, which is a group of cells in the early embryo that give rise to many tissues and organs. Neural crest cells migrate to form parts of the autonomic nervous system, many tissues in the face and skull, and other tissue and cell types.
Mutations are believed to interfere with the PHOX2B protein’s role in promoting neuron formation and differentiation, especially in the autonomic nervous system, resulting in the problems regulating breathing and other body functions that occur in congenital central hypoventilation syndrome.
Congenital central hypoventilation syndrome inheritance pattern
Congenital central hypoventilation syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
More than 90 percent of cases of congenital central hypoventilation syndrome result from new mutations in the PHOX2B gene. These cases occur in people with no history of the disorder in their family. Occasionally an affected person inherits the mutation from one affected parent. The number of such cases has been increasing as better treatment has allowed more affected individuals to live into adulthood.
About 5 to 10 percent of affected individuals inherit the mutation from a seemingly unaffected parent with somatic mosaicism. Somatic mosaicism means that some of the body’s cells have a PHOX2B gene mutation, and others do not. A parent with mosaicism for a PHOX2B gene mutation may not show any signs or symptoms of congenital central hypoventilation syndrome.
Congenital central hypoventilation syndrome symptoms
Symptoms of congenital central hypoventilation syndrome usually become apparent shortly after birth. Affected infants hypoventilate upon falling asleep and exhibit a bluish appearance of the skin or lips (cyanosis). Cyanosis is caused by lack of oxygen in the blood. In some milder cases, congenital central hypoventilation syndrome may be diagnosed later in life. In addition to the breathing problem, people with congenital central hypoventilation syndrome may have difficulty regulating their heart rate and blood pressure, for example in response to exercise or changes in body position. They may have abnormalities in the nerves that control the digestive tract (Hirschsprung disease), resulting in severe constipation, intestinal blockage, and enlargement of the colon. They are also at increased risk of developing certain tumors of the nervous system called neuroblastomas, ganglioneuromas, and ganglioneuroblastomas. Some affected individuals develop learning difficulties or other neurological problems, which may be worsened by oxygen deprivation if treatment to support their breathing is not completely effective.
Individuals with congenital central hypoventilation syndrome usually have eye abnormalities, including a decreased response of the pupils to light. They also have decreased perception of pain, low body temperature, and occasional episodes of profuse sweating.
People with congenital central hypoventilation syndrome, especially children, may have a characteristic appearance with a short, wide, somewhat flattened face often described as “box-shaped.”
Congenital central hypoventilation syndrome diagnosis
The American Thoracic Society has issued both an updated statement on the diagnosis and management of congenital central hypoventilation syndrome 3 and a lay summary 4.
Congenital central hypoventilation syndrome is diagnosed in newborns with the following:
- Hypoventilation with absent or attenuated ventilatory response to hypercarbia and/or hypoxemia when awake and asleep
- Generally adequate ventilation while awake and at rest and apparent hypoventilation with monotonous respiratory rate and shallow breathing (diminished tidal volume) during sleep OR apparent hypoventilation while both awake and asleep
- Absent perception of asphyxia (i.e., absent behavioral awareness of hypercarbia and/or hypoxemia) and absent arousal from sleep with development of physiologic compromise secondary to hypercarbia and/or hypoxemia
- No evidence of primary neuromuscular, lung, or cardiac disease or identifiable brain stem lesion that could account for the full constellation of signs and symptoms including autonomic nervous system dysregulation (ANSD)
- Presence of a congenital central hypoventilation syndrome-related PHOX2B variant
- Symptoms of autonomic nervous system dysregulation including but not limited to severe breath-holding spells; lack of physiologic responsiveness to the challenges of exercise and environmental stressors; diminished pupillary light response; esophageal dysmotility; severe constipation even in the absence of Hirschsprung disease; profuse sweating; reduced basal body temperature; and altered perception of anxiety
Later-onset congenital central hypoventilation syndrome is diagnosed in individuals with the following:
Same criteria as described above for congenital central hypoventilation syndrome of the newborn but with presentation after one month of life, often occurring in later childhood or adulthood.
Molecular Genetic Testing
Gene. PHOX2B is the only gene in which mutation is known to cause congenital central hypoventilation syndrome.
The two major types of PHOX2B variants observed in congenital central hypoventilation syndrome are polyalanine repeat expansion mutations (PARMs) and non-polyalanine repeat expansion mutations (NPARMs).
Polyalanine repeat expansion mutations (PARMs)
PHOX2B has two polyalanine repeat regions in exon 3, the second of which is the region of primary importance in congenital central hypoventilation syndrome. This polyalanine repeat comprises any one of four codon combinations — GCA, GCT, GCC, or GCG — as each one encodes the amino acid alanine. The term “GCN” has been used to designate these four codons.
Allele sizes. Allele sizes and categories are summarized here 5:
- Normal alleles. The unaffected individual has 20 alanines (GCN repeats) on both PHOXB alleles in the repeat region of exon 3.Though benign variants of 9, 13, 14, and 15 GCN repeats have been reported 6, individuals with alleles of this length have not been studied systematically to confirm they are entirely normal without any control of breathing deficit or autonomic dysregulation.
- Mutable normal alleles. Currently, this category of alleles is not known to occur in this disorder
- Reduced penetrance alleles. Individuals heterozygous for 24 alanine repeats (e.g., genotype 20/24) and a subset of individuals heterozygous for 25 alanine repeats (e.g., genotype 20/25) may have a very mild phenotype such that diagnosis is delayed and/or not manifest except when exposed to respiratory depressants or severe intercurrent pulmonary illness 7. Rarely a small NPARM will also have variable penetrance 8.
- Full penetrance alleles. Individuals with 25 alanine repeats who present in the newborn period (e.g., genotype 20/25), and those heterozygous for 26 to 33 alanine repeats (e.g., genotype 20/26 to 20/33) 5. The largest known repeat length is 33 alanines.
- Alleles of uncertain significance. Only one individual with such a small expansion allele has been described 6, in a study on schizophrenia; no clinical information relevant to congenital central hypoventilation syndrome phenotype is known about the individual.
Non-polyalanine repeat expansion mutations (NPARMs)
PHOX2B variants that are not specifically polyalanine expansions, including sequence alterations outside of the polyalanine repeat and frameshift variants affecting the region encoding the polyalanine repeat, are typically small out-of-frame deletions or duplications of approximately one to 38 nucleotides.
Note: Details of these variants from many published reports are summarized in Berry-Kravis et al 8 and Weese-Mayer et al 5.
Though individuals with NPARMs typically have a more severe phenotype than most individuals with PARMs, on rare occasion a small frameshift variant could have reduced but variable penetrance in a given family 8.
PHOX2B deletions ranging from 6,216 base pairs (involving only PHOX2B exon 3) to 2.6 megabases (involving all of PHOX2B and 12 other genes) have been observed in a small cohort of individuals with clinical findings that may include alveolar hypoventilation or Hirschsprung disease 9. Further study is necessary to elucidate the relationship between PHOX2B haploinsufficiency and the congenital central hypoventilation syndrome phenotype 9.
Will a person with a mutation in the PHOX2B gene definitely have or develop symptoms of congenital central hypoventilation syndrome?
Not necessarily. The genetics of congenital central hypoventilation syndrome is complex. The protein made from the PHOX2B gene has 2 regions where an amino acid called alanine is repeated multiple times. These stretches of alanines are known as polyalanine tracts or poly(A) tracts.[2] The second region is most important for congenital central hypoventilation syndrome 1.
There are 2 major types of PHOX2B variants (or mutations) in people with congenital central hypoventilation syndrome. They are called polyalanine repeat expansion mutations (PARMs) and non-polyalanine repeat expansion mutations (NPARMs) 1. Nucleotide repeats are short DNA sequences that are repeated a number of times in a row. A repeat expansion is a type of mutation that increases the number of times the short DNA sequence is repeated.[3] A polyalanine repeat expansion is a mutation in which extra alanines are added to the polyalanine tracts 10.
Congenital central hypoventilation syndrome treatment
Treatment of manifestations
Tracheostomy and home ventilator for individuals requiring ventilatory support 24 hours per day and for infants/children/adults requiring ventilatory support during sleep only. Diaphragm pacing by phrenic nerve stimulation can be considered in ambulatory children requiring mechanical ventilation 24 hours a day and potentially in older children and adults requiring nocturnal ventilation only, though tracheostomy removal for nocturnal diaphragm pacing is not assured. Mask ventilation or negative-pressure ventilation is a consideration in cooperative older children requiring ventilatory support during sleep; however, during intercurrent illnesses more aggressive ventilatory support such as intubation with continuous mechanical ventilation in an intensive care setting may be needed. A cardiac pacemaker may be required for prolonged sinus pauses. Hirschsprung disease is treated in the usual manner. Neuroblastomas are removed surgically; those beyond Stage 1 are treated with chemotherapy. Treatment of other tumors of neural crest origin is based on location and type, though surgical removal is typically recommended.
Children with hypercapnia and hypoxemia are at risk of developing progressive pulmonary hypertension. Supplemental oxygen alone is not sufficient for the treatment of hypoventilation, and it will not prevent pulmonary hypertension 11. Thus, congenital central hypoventilation syndrome patients require lifelong assisted ventilation. As children with congenital central hypoventilation syndrome do not have intrinsic severe lung disease, there are several options for assisted ventilatory support at home: 1) positive pressure ventilation (PPV) via tracheostomy; 2) noninvasive positive pressure ventilation (NIPPV); and 3) diaphragm pacing (DP) 12. Although negative pressure ventilation can be effective, it is not portable and is now rarely used.
All congenital central hypoventilation syndrome patients require assisted ventilation for life at least during sleep. Thus, weaning these patients off ventilatory support should not be attempted. congenital central hypoventilation syndrome patients are not like other children on home mechanical ventilation. They must be managed with vigilance due to their absence of objective and subjective responses to hypoxemia and hypercapnia. With acute respiratory infections, children with congenital central hypoventilation syndrome may not show a fever, increase their respiratory rate, or dyspnea. The use of pulse oximetry or end-tidal CO2 (PETCO2) monitoring are invaluable aids in addition to the highly skilled and consistent caregivers at home.
Positive pressure ventilation (PPV) via tracheostomy
There are two approaches to the long-term ventilation of infants: positive pressure ventilation (PPV) via tracheostomy and noninvasive ventilation when possible. There is a lack of current literature at present demonstrating that one mode of ventilation is superior, thus this is an area of needed research. The authors of the ATS statement believe that positive pressure ventilation (PPV) via tracheostomy in the first 6–8 years of life is associated with better oxygenation, and thus better neurologic development and function, than the use of noninvasive positive pressure ventilation (NIPPV) early in life 12.
For children receiving positive pressure ventilation (PPV) via tracheostomy, the authors prefer an uncuffed and relatively smaller tracheostomy tube, because it is less likely to cause tracheomalacia and it allows a large expiratory leak for phonation 13. Although small, uncuffed tracheostomy tubes are associated with leaks, which can be large and variable, the tracheostomy leak can be compensated for by using the ventilator in a pressure-control mode 13. Speaking valves may be used for phonation. It is preferable to initially ventilate infants for 24 h/day to ensure that oxygenation and ventilation remain adequate to optimize neurocognitive outcome. Ventilator settings are adjusted to achieve PETCO2 between 30–35 torr and SpO2 >95%. In our experience, children ventilated with these parameters have fewer complications, and they generally do better clinically. Allowing these children to be hypoxemic and/or hypercapnic defeats the purpose of chronic ventilatory support.
Ventilator settings should be assessed periodically to ensure adequate oxygenation and ventilation. This is best performed in a sleep laboratory. Periodic assessments of the airway and tracheostomy tube size should be conducted by an experienced pediatric otolaryngologist.
Between infancy and school age, some congenital central hypoventilation syndrome children gradually develop the ability to breathe adequately during wakefulness. These children, if stable, can be weaned from assisted ventilation while awake. This is best accomplished by sprint weaning. Sprint weaning is performed by removing the child from the ventilator for short periods of time. Between sprints, the child remains on full ventilatory support. Initially, these sprints may only last few minutes and supplemental oxygen may be required. The child must be carefully monitored noninvasively during sprints to prevent hypoxia or hypercapnia. These sprints are always started under medical observation, but may be gradually increased at home. However, congenital central hypoventilation syndrome children can never be weaned from ventilatory support during sleep.
Noninvasive positive pressure ventilation
Stable older children who require ventilatory support only during sleep can be ventilated with noninvasive positive pressure ventilation (NIPPV) delivered via nasal mask, nasal prongs, or face mask. Continuous positive airway pressure therapy should not be used in congenital central hypoventilation syndrome patients because it does not support ventilation. Spontaneous mode noninvasive positive pressure ventilation (NIPPV) cannot be used in congenital central hypoventilation syndrome because they cannot be trusted to increase their ventilatory rate in response to hypercapnia and hypoxemia on their own. noninvasive positive pressure ventilation (NIPPV) is provided via bi-level positive airway pressure or average volume assured pressure support 14 in the spontaneous/timed mode or timed mode with appropriate rate. Children with congenital central hypoventilation syndrome characteristically have diminutive tidal volumes and monotonous respiratory rates awake and asleep. Only the timed mode guarantees breath delivery in children who cannot generate adequate spontaneous breaths to trigger the ventilator. The spontaneous/timed mode could lead to a variable breathing pattern. Hence the authors use timed mode only 12. Inspiratory positive airway pressure and expiratory positive airway pressure are adjusted independently to achieve an inspiratory positive airway pressure to expiratory positive airway pressure difference that provides optimal tidal volume. Although most congenital central hypoventilation syndrome patients do not require supplemental oxygen with their noninvasive positive pressure ventilation (NIPPV), oxygen can be added during acute illness. The high flow of pressurized air with noninvasive positive pressure ventilation (NIPPV) can lead to the drying of nasal mucosa which may present as frequent nosebleeds or nasal congestion 15. Noninvasive positive pressure ventilation (NIPPV) can be used with the attached humidifier to circumvent this problem 16.
Mask ventilation has been associated with midface hypoplasia when instituted at an early age 15. Noninvasive positive pressure ventilation (NIPPV) should not be used routinely outside of sleep time as the mask interferes with daily activities, social interaction, and increases the risk of skin breakdown. positive pressure ventilation (PPV) via tracheostomy in children may avoid these complications.
During the first few years of life, congenital central hypoventilation syndrome patients can be very unstable. In general, noninvasive positive pressure ventilation (NIPPV) can be instituted in stable school-aged congenital central hypoventilation syndrome patients with a milder phenotype requiring ventilatory support only during sleep. In this group of patients, noninvasive positive pressure ventilation (NIPPV) allows for tracheostomy decannulation. When anticipating tracheostomy decannulation, we recommend an upper airway evaluation. Several congenital central hypoventilation syndrome centers prefer institution of noninvasive positive pressure ventilation (NIPPV) in the first few years of life. Some of the issues that may arise are finding an appropriate interface, prompt institution of noninvasive positive pressure ventilation (NIPPV) during daytime naps and development of midface hypoplasia. However, there are no comparative research studies on the effect of early noninvasive positive pressure ventilation (NIPPV) on brain development in congenital central hypoventilation syndrome.
Periodic assessment of noninvasive positive pressure ventilation (NIPPV) settings should be conducted in a sleep laboratory to ensure adequate oxygenation and ventilation 12. Polysomnography for adjustment of noninvasive positive pressure ventilation (NIPPV) settings is performed as per the American Academy of Sleep Medicine guidelines 17.
Diaphragm pacing
Diaphragm pacing is an attractive treatment option for some congenital central hypoventilation syndrome patients. It allows full-time ventilator-dependent patients to be free of positive pressure ventilation (PPV) during the day, allowing mobility. In those who are stable and ventilator-dependent only during sleep, diaphragm pacing may permit tracheostomy decannulation 18.
Diaphragm pacing generates breathing using the child’s own diaphragm as the respiratory pump. A diaphragm pacer system has four components: 1) electrodes that are surgically implanted on the phrenic nerves; 2) receivers that are subcutaneously implanted bilaterally in the abdomen; 3) antennae that are taped on the skin over the subcutaneous receivers when pacing; and 4) a battery operated external transmitter (Figure 1). The external transmitter generates a train of pulses that are transmitted via the external antenna. The subcutaneous receivers convert the radiofrequency signal into an electrical current that is then conducted to the phrenic nerve electrodes. Electrical stimulation of the phrenic nerve electrodes causes diaphragmatic contraction, which generates the breath. The amount of electrical voltage is proportional to the diaphragm contraction or tidal volume. Since diaphragm pacing uses the child’s own diaphragm as a ventilator, an ideal candidate should have normal diaphragm function and intact phrenic nerves. The patient must also have little or no lung disease.
Bilateral phrenic nerve electrodes are surgically implanted on the phrenic nerves thoracoscopically 19. Once implanted, the pacers are tested intraoperatively, but diaphragm pacing is not used right away. diaphragm pacing is initiated ~4–6 weeks later to allow for healing and stabilization of tissue reaction around the electrodes 20.
When diaphragm pacing is used for the first time, begin with the child awake in a friendly, supportive, and nonthreatening setting. The child is not on the ventilator when on diaphragm pacing. Begin with the lowest setting on each side, and increase the tidal volume settings until a reasonable diaphragm contraction is observed. Continuous pulse oximetry and PETCO2 monitoring are used to objectively assess SpO2 and PETCO2. The authors start pacing at 60–90 min/day because diaphragm pacing can result in fatigue (decreasing diaphragmatic contraction for the same electrical stimulus). The duration of diaphragm pacing is gradually increased by 30 min/day each week. “Aerobic training” of these muscle fibers is required to achieve full pacing. During this period, diaphragm pacing is used for a part of the day/night and baseline ventilator support for the rest of the day/night. With gradual diaphragm training, it takes ~2–3 months to establish full overnight pacing in those who are ventilator-dependent only during sleep 20. In general, diaphragm pacing is limited to 12–14 h/day. Diaphragm pacer equipment can probably last indefinitely, but can be damaged and may need to be replaced 21.
The greatest benefit of diaphragm pacing is portability and freedom from being tethered to a ventilator circuit in children who are full-time ventilator-dependent. Tracheostomy decannulation can be considered in children who are ventilator-dependent only during sleep after successful overnight diaphragm pacing for at least 3 months. Assessment of upper airway anatomy by a pediatric otolaryngologist and polysomnography with a capped tracheostomy tube should be performed prior to considering decannulation 18. Upper airway obstruction can occur with diaphragm pacing due to the absence of synchronous upper airway skeletal muscle contraction with diaphragm contraction. This can be surpassed by changes in diaphragm pacer settings and other medical management facilitating tracheostomy decannulation 18. Obesity is an obstacle to effective diaphragm pacing as the adipose tissue increases distance between the antenna and the receiver which may result in decreased diaphragm contractions 18. Therefore, the authors do not recommend diaphragm pacing in obese patients.
Once diaphragm pacers are placed, the patient cannot have magnetic resonance imaging due to the magnetic field. diaphragm pacing causes artifacts in transthoracic impedance-based cardiorespiratory monitors, and should not be used at home. Diaphragm pacing may not provide enough ventilatory support if congenital central hypoventilation syndrome patients become acutely ill or have major surgical procedures and these children may need other ventilatory support on a temporary basis in these situations. Diaphragm pacing in children with congenital central hypoventilation syndrome has been established as an effective method of mechanically assisted ventilation, and allows for maximum mobility. In one center, the authors have congenital central hypoventilation syndrome patients who have been using diaphragm pacing for 40 years without phrenic nerve or diaphragm damage 11.
Periodic noninvasive assessment and titration of diaphragm pacer settings should be conducted in a sleep lab to ensure adequate oxygenation and ventilation. This can be a challenge, since many sleep laboratories do not monitor PETCO2 and do not have experience adjusting diaphragm pacers. Congenital central hypoventilation syndrome centers with expertise in diaphragm pacing are available for management or consultation of these patients.
Figure 1. Diaphragm pacing for congenital central hypoventilation syndrome
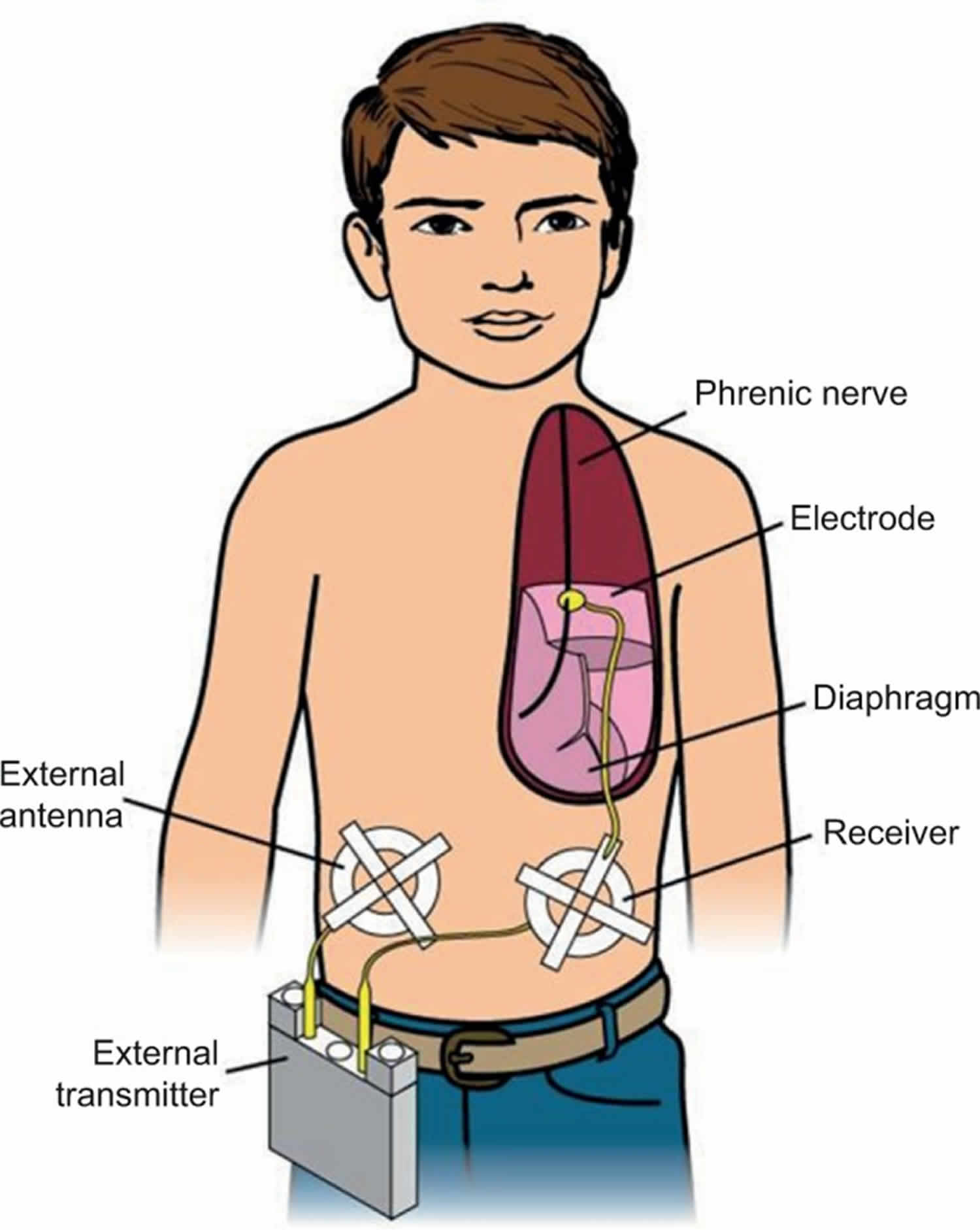
Footnote: A phrenic nerve electrode is surgically implanted. It is connected by a lead wire to a receiver implanted subcutaneously in the abdomen. A battery operated transmitter transmits electrical energy, via an antenna taped on the skin over the receiver, to the receiver. The receiver converts this to a standard electrical current, which causes contraction of the diaphragm.
[Source 11 ]Prevention of secondary complications
Mask ventilation in the infant and young child is strongly discouraged because it is not adequately stable as a life-sustaining support, with risk for repeated hypoxemia and neurocognitive compromise.
Surveillance
For all individuals with congenital central hypoventilation syndrome: at least yearly (every 6 months until age 3 years) comprehensive, multiple-day in-hospital physiologic evaluation to optimize ventilatory support awake and asleep and in varied levels of activity and concentration simulating activities of daily living; yearly 72-hour Holter recording to identify any prolonged sinus pauses; yearly echocardiogram to identify right ventricular hypertrophy or cor pulmonale; yearly hemoglobin, hematocrit, and reticulocyte counts to identify polycythemia; and yearly neurocognitive testing to evaluate the success of artificial ventilation. For children with specific PHOX2B variants placing them at higher risk: evaluate for Hirschsprung disease and tumors of neural crest origin.
Agents/circumstances to avoid
Swimming (asphyxia; death); breathholding contests (asphyxia; death); alcohol (respiratory depression), recreational drugs (varied effects including death), and prescribed as well as non-prescribed medications/sedatives/anesthetics that could induce respiratory depression.
Evaluation of relatives at risk
Both parents of children with a known PHOX2B pathogenic variant should be tested for the family-specific variant to determine their risk for later-onset congenital central hypoventilation syndrome or mosaicism.
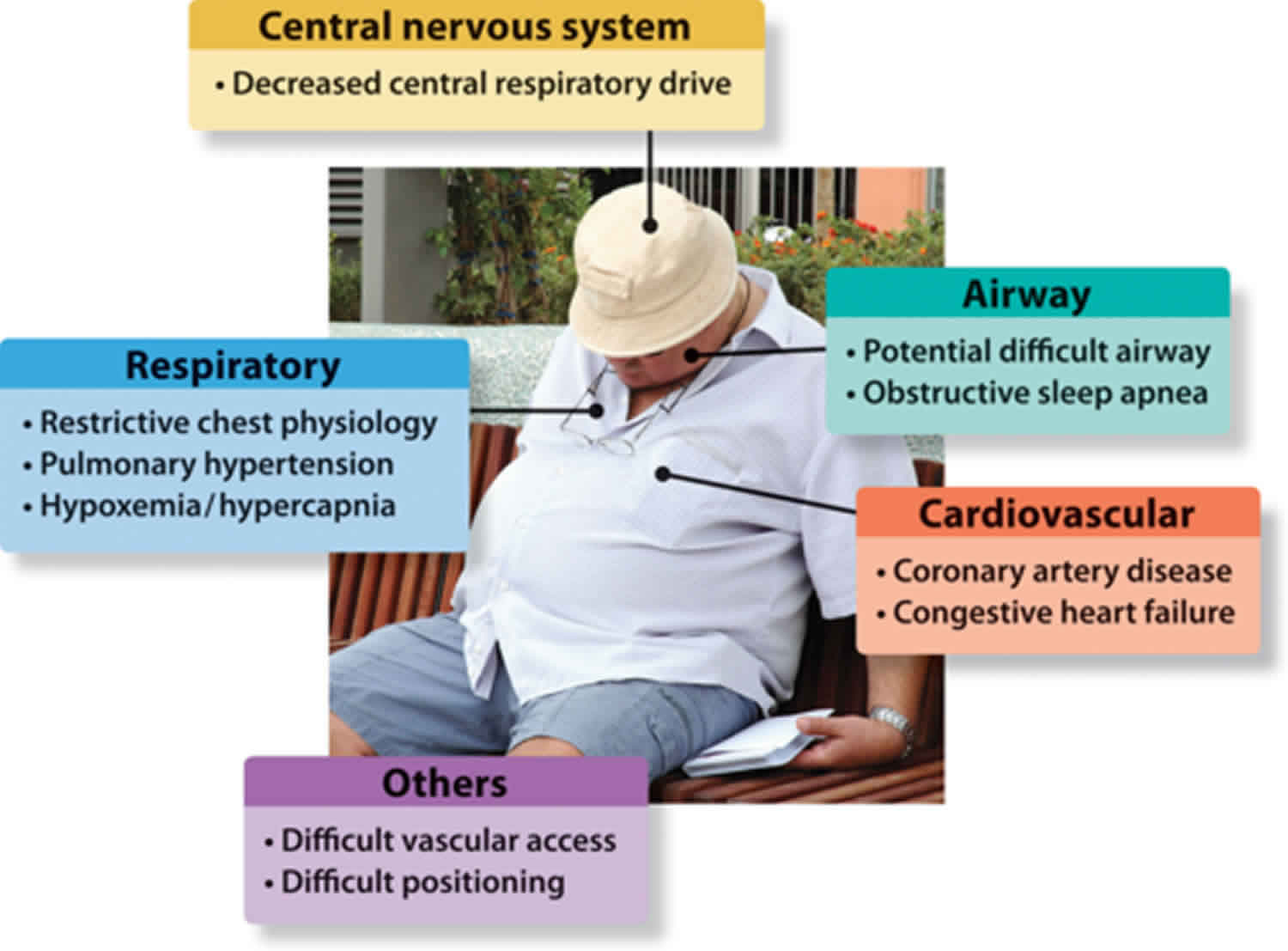
Obesity hypoventilation syndrome
Obesity hypoventilation syndrome is also called Pickwickian syndrome, is a condition in some obese people in which poor breathing leads to lower oxygen and higher carbon dioxide levels in the blood. It is not clear why obesity hypoventilation syndrome affects some people who have obesity and not others. Extra fat on your neck or chest or across your abdomen can make it difficult to breathe deeply and may produce hormones that affect your body’s breathing patterns. You may also have a problem with the way your brain controls your breathing. Most people who have obesity hypoventilation syndrome also have sleep apnea.
You can help prevent obesity hypoventilation syndrome by maintaining a healthy weight. If you have been diagnosed with obesity, your doctor may screen you for obesity hypoventilation syndrome by measuring your blood oxygen or carbon dioxide levels.
If you have obesity hypoventilation syndrome, you may feel sluggish or sleepy during the day, have headaches, or feel out of breath. You or a loved one may notice you often snore loudly, choke or gasp, or have trouble breathing at night. Your symptoms may get worse over time. Complications of obesity hypoventilation syndrome include pulmonary hypertension; right heart failure, also known as cor pulmonale; and secondary erythrocytosis.
To diagnose obesity hypoventilation syndrome, your doctor will perform a physical exam to measure your weight and height, calculate your body mass index (BMI), and measure your waist and neck circumference. Your doctor may perform other tests such as pulmonary function tests, sleep studies, a chest X-ray, or an arterial blood gas or serum bicarbonate test. Other blood tests may help rule out other causes or be used to plan your treatment. You may be diagnosed at the hospital if you have trouble breathing and go to the emergency room with respiratory failure. You may be diagnosed from tests routinely performed before a surgery.
If you are diagnosed with obesity hypoventilation syndrome, your doctor may recommend healthy lifestyle changes, such as aiming for a healthy weight and being physically active. You may also need a continuous positive airway pressure (CPAP) machine or other breathing device to help keep your airways open and increase blood oxygen levels. Other treatments may include weight loss surgery, medicines, or a tracheostomy.
To prevent complications, use your CPAP (continuous positive airway pressure) device as instructed and continue with your doctor’s recommended healthy lifestyle changes. Tell your doctor about new signs and symptoms, such as swelling around your ankles, chest pain, lightheadedness, or wheezing. Talk to your doctor if you will be flying or need surgery, as these situations can increase your risk for serious complications.
Obesity hypoventilation syndrome causes
The exact cause of obesity hypoventilation syndrome is not known. Researchers believe obesity hypoventilation syndrome results from a defect in the brain’s control over breathing. Excess weight against the chest wall also makes it harder for the muscles to draw in a deep breath and to breathe quickly enough. As a result, the blood contains too much carbon dioxide and not enough oxygen.
Obesity hypoventilation syndrome prevention
Maintain a healthy weight and avoid obesity. Use your continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BiPAP) treatment as your provider prescribed.
Obesity hypoventilation syndrome symptoms
The main symptoms of obesity hypoventilation syndrome are due to lack of sleep and include:
- Poor sleep quality
- Sleep apnea
- Daytime sleepiness
- Depression
- Headaches
- Tiredness
Symptoms of low blood oxygen level (chronic hypoxia) can also occur. Symptoms include shortness of breath or feeling tired after very little effort.
Obesity hypoventilation syndrome possible complications
Obesity hypoventilation syndrome complications related to a lack of sleep may include:
- Depression, agitation, irritability
- Increased risk of accidents or mistakes at work
- Problems with intimacy and sex
Obesity hypoventilation syndrome can also cause heart problems, such as:
- High blood pressure (hypertension)
- Right-sided heart failure (cor pulmonale)
- High blood pressure in the lungs (pulmonary hypertension)
Obesity hypoventilation syndrome diagnosis
People with obesity hypoventilation syndrome are usually very overweight. A physical exam may reveal:
- Bluish color in the lips, fingers, toes, or skin (cyanosis)
- Reddish skin
- Signs of right-sided heart failure (cor pulmonale), such as swollen legs or feet, shortness of breath, or feeling tired after little effort
- Signs of excessive sleepiness
Tests used to help diagnose and confirm obesity hypoventilation syndrome include:
- Arterial blood gas
- Chest x-ray or CT scan to rule out other possible causes
- Lung function tests (pulmonary function tests)
- Sleep study (polysomnography)
- Echocardiogram (ultrasound of the heart)
Health care providers can tell obesity hypoventilation syndrome from obstructive sleep apnea because a person with obesity hypoventilation syndrome has a high carbon dioxide level in their blood when awake.
Obesity hypoventilation syndrome treatment
Treatment involves breathing assistance using special machines (mechanical ventilation). Options include:
- Noninvasive mechanical ventilation such as continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BiPAP) through a mask that fits tightly over the nose or nose and mouth (mainly for sleep)
- Oxygen therapy
- Breathing help through an opening in the neck (tracheostomy) for severe cases
Treatment is started in the hospital or as an outpatient.
Other treatments are aimed at weight loss, which can reverse obesity hypoventilation syndrome.
Obesity hypoventilation syndrome prognosis
Untreated, obesity hypoventilation syndrome can lead to serious heart and blood vessel problems, severe disability, or death.
- Weese-Mayer DE, Marazita ML, Rand CM, et al. Congenital Central Hypoventilation Syndrome. 2004 Jan 28 [Updated 2014 Jan 30]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1427[↩][↩][↩][↩]
- Congenital central hypoventilation syndrome. https://ghr.nlm.nih.gov/condition/congenital-central-hypoventilation-syndrome[↩]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H, on behalf of the ATS Congenital Central Hypoventilation Syndrome Subcommittee. An official ATS clinical policy statement: congenital central hypoventilation syndrome: Genetic basis, diagnosis, and management. http://www.thoracic.org/statements/resources/respiratory-disease-pediatric/congenital-central-hypoventilation-syndrome.pdf[↩]
- Patwari PP, Lareau S, Sockrider M, Weese-Mayer DE. Congenital central hypoventilation syndrome (CCHS). American Thoracic Society Patient Information Series. 2010b. http://www.thoracic.org/patients/patient-resources/resources/congenital-central-hypoventilation-syndrome.pdf[↩]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H, on behalf of the ATS Congenital Central Hypoventilation Syndrome Subcommittee. An official ATS clinical policy statement: congenital central hypoventilation syndrome: Genetic basis, diagnosis, and management.[↩][↩][↩]
- Toyota T, Yoshitsugu K, Ebihara M, Yamada K, Ohba H, Fukasawa M, Minabe Y, Nakamura K, Sekine Y, Takei N, Suzuki K, Itokawa M, Meerabux JM, Iwayama-Shigeno Y, Tomaru Y, Shimizu H, Hattori E, Mori N, Yoshikawa T. Association between schizophrenia with ocular misalignment and polyalanine length variation in PMX2B. Hum Mol Genet. 2004;13:551–61.[↩][↩]
- Repetto GM, Corrales RJ, Abara SG, Zhou L, Berry-Kravis EM, Rand CM, Weese-Mayer DE. Later-onset congenital central hypoventilation syndrome due to a heterozygous 24-polyalanine repeat expansion mutation in the PHOX2B gene. Acta Pædiatr. 2009;98:192–5.[↩]
- Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med. 2006;174:1139–44.[↩][↩][↩]
- Jennings LJ, Yu M, Rand CM, Kravis N, Berry-Kravis EM, Patwari PP, Weese-Mayer DE. Variable human phenotype associated with novel deletions of the PHOX2B gene. Pediatr Pulmonol. 2012;47:153–61.[↩][↩]
- PHOX2B gene. https://ghr.nlm.nih.gov/gene/PHOX2B [↩]
- Kasi AS, Perez IA, Kun SS, Keens TG. Congenital central hypoventilation syndrome: diagnostic and management challenges. Pediatric Health Med Ther. 2016;7:99-107. Published 2016 Aug 18. doi:10.2147/PHMT.S95054 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5683295[↩][↩][↩]
- Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H, ATS Congenital Central Hypoventilation Syndrome Subcommittee An official ATS clinical policy statement: congenital central hypoventilation syndrome – genetic basis, diagnosis, and management. Am J Respir Crit Care Med. 2010;181(6):626–644.[↩][↩][↩][↩]
- Keens TG, Kun SS, Ward SLD. Chronic respiratory failure. In: Shaffner DH, Nichols DG, editors. Roger’s Textbook of Pediatric Intensive Care. 5th ed. Alphen aan den Rijn, the Netherlands: Wolters Kluwer; 2015. pp. 794–807.[↩][↩]
- Vagiakis E, Koutsourelakis I, Perraki E, Roussos C, Mastora Z, Zakynthinos S, Kotanidou A. Average volume-assured pressure support in a 16-year-old girl with congenital central hypoventilation syndrome. J Clin Sleep Med. 2010;6(6):609–612.[↩]
- Amin R, Al-Saleh S, Narang I. Domiciliary noninvasive positive airway pressure therapy in children. Pediatr Pulmonol. 2016;51(4):335–348.[↩][↩]
- Nilius G, Franke KJ, Domanski U, Schroeder M, Ruhle KH. Effect of APAP and heated humidification with a heated breathing tube on adherence, quality of life, and nasopharyngeal complaints. Sleep Breath. 2016;20(1):43–49.[↩]
- Berry RB, Chediak A, Brown LK, et al. NPPV Titration Task Force of the American Academy of Sleep Medicine Best clinical practices for the sleep center adjustment of noninvasive positive pressure ventilation (NPPV) in stable chronic alveolar hypoventilation syndromes. J Clin Sleep Med. 2010;6(5):491–509.[↩]
- Diep B, Wang A, Kun S, et al. Diaphragm pacing without tracheostomy in congenital central hypoventilation syndrome patients. Respiration. 2015;89(6):534–538.[↩][↩][↩][↩]
- Shaul DB, Danielson PD, McComb JG, Keens TG. Thoracoscopic placement of phrenic nerve electrodes for diaphragmatic pacing in children. J Pediatr Surg. 2002;37(7):974–978.[↩]
- Chen ML, Tablizo MA, Kun S, Keens TG. Diaphragm pacers as a treatment for congenital central hypoventilation syndrome. Expert Rev Med Devices. 2005;2(5):577–585.[↩][↩]
- Nicholson KJ, Nosanov LB, Bowen KA, Kun SS, Perez IA, Keens TG, Shin CE. Thoracoscopic placement of phrenic nerve pacers for diaphragm pacing in congenital central hypoventilation syndrome. J Pediatr Surg. 2015;50(1):78–81.[↩]




{kind=link}